
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reprogramming natural proteins using unnatural amino acids
Anup Adhikari†
 a,
Bibek Raj Bhattarai†a,
Ashika Aryalb,
Niru Thapaa,
Puja KCa,
Ashma Adhikaria,
Sushila Maharjana,
Prem B. Chandac,
Bishnu P. Regmid and
Niranjan Parajuli*a
a,
Bibek Raj Bhattarai†a,
Ashika Aryalb,
Niru Thapaa,
Puja KCa,
Ashma Adhikaria,
Sushila Maharjana,
Prem B. Chandac,
Bishnu P. Regmid and
Niranjan Parajuli*a
aBiological Chemistry Lab, Central Department of Chemistry, Tribhuvan University, Kritipur, 44618, Kathmandu, Nepal. E-mail: niranjan.parajuli@cdc.tu.edu.np
bDepartment of Chemistry, Birendra Multiple Campus, Tribhuvan University, Bharatpur, Chitwan, Nepal
cDepartment of Chemistry and Physics, Southeastern Louisiana University, Hammond, Louisiana 70402, USA
dDepartment of Chemistry, Florida Agricultural and Mechanical University, Tallahassee, Florida 32307, USA
First published on 26th November 2021
Abstract
Unnatural amino acids have gained significant attention in protein engineering and drug discovery as they allow the evolution of proteins with enhanced stability and activity. The incorporation of unnatural amino acids into proteins offers a rational approach to engineer enzymes for designing efficient biocatalysts that exhibit versatile physicochemical properties and biological functions. This review highlights the biological and synthetic routes of unnatural amino acids to yield a modified protein with altered functionality and their incorporation methods. Unnatural amino acids offer a wide array of applications such as antibody-drug conjugates, probes for change in protein conformation and structure–activity relationships, peptide-based imaging, antimicrobial activities, etc. Besides their emerging applications in fundamental and applied science, systemic research is necessary to explore unnatural amino acids with novel side chains that can address the limitations of natural amino acids.
1. Introduction
A vast majority of the physiological processes of cells require proteins that catalyse biological reactions, regulate gene expression and the immune response, provide a structural framework, and modulate transport systems, among other things.1 Most surprisingly, proteins that perform these diverse functions are composed of 20 building blocks—the natural amino acids. Unnatural amino acids (UAAs) are synthesized and can be incorporated into proteins to expand the domain of protein functions.2 UAAs are not genetically encoded by organisms.3 Since UAAs are not present in natural polypeptide chains,4 they are also called non-proteinogenic or non-canonical amino acids (ncAA).The structure of UAAs may resemble or differ significantly from natural amino acids and they are referred to as analogues or surrogates, respectively.3 The synthesis and applications of UAAs have received considerable attention, particularly in the field of enzymology and drug discovery. In protein engineering, the ability to substitute any natural amino acid with UAA at a particular point has emerged as a valuable molecular tool because such substitution allows the introduction of altered physicochemical and biological properties.5 UAAs can be incorporated into many structural units to develop potential leads in peptidic and non-peptidic complexes. The incorporation of UAAs into protein expands its functional diversity in biophysics, spectroscopy and optical probes, bio-orthogonal chemistry and protein labelling, post-translational modifications; their mimetics, signal transduction, protein interactions mapping, and photoactivated motifs.6 Furthermore, UAAs dramatically expand the possibilities for using chiral building blocks and molecular scaffolds to construct combinatorial libraries.7
UAAs can be synthesized chemically or they are produced naturally as secondary metabolites in several organisms, such as bacteria, fungi, plants, or marine organisms.8 Over the past few years, by constructing engineered microbial strains, various metabolic pathways have aided in the production of UAAs. Modern discoveries allow promising methods to produce sufficient amounts of proteins in vitro and in vivo using UAAs. The site-specific incorporation of UAAs into enzymes has enabled structural modification, accepting a wide range of substrates such as ketone, azide, alkyne, alkene, tetrazine, etc. Various probes, designed using UAAs, can be used for diverse applications, such as evaluating the effects of small molecule proteasome stimulators in live cells and comparing proteasome activity in different cancer cell types.9 Here, in this review, we have focused on the synthesis, incorporation approaches, applications of UAAs, as well as their limitations and prospects. We believe this review would provide useful information that helps researchers to reprogram natural proteins by incorporating UAAs.
2. Synthesis of UAAs
2.1 Biological synthesis
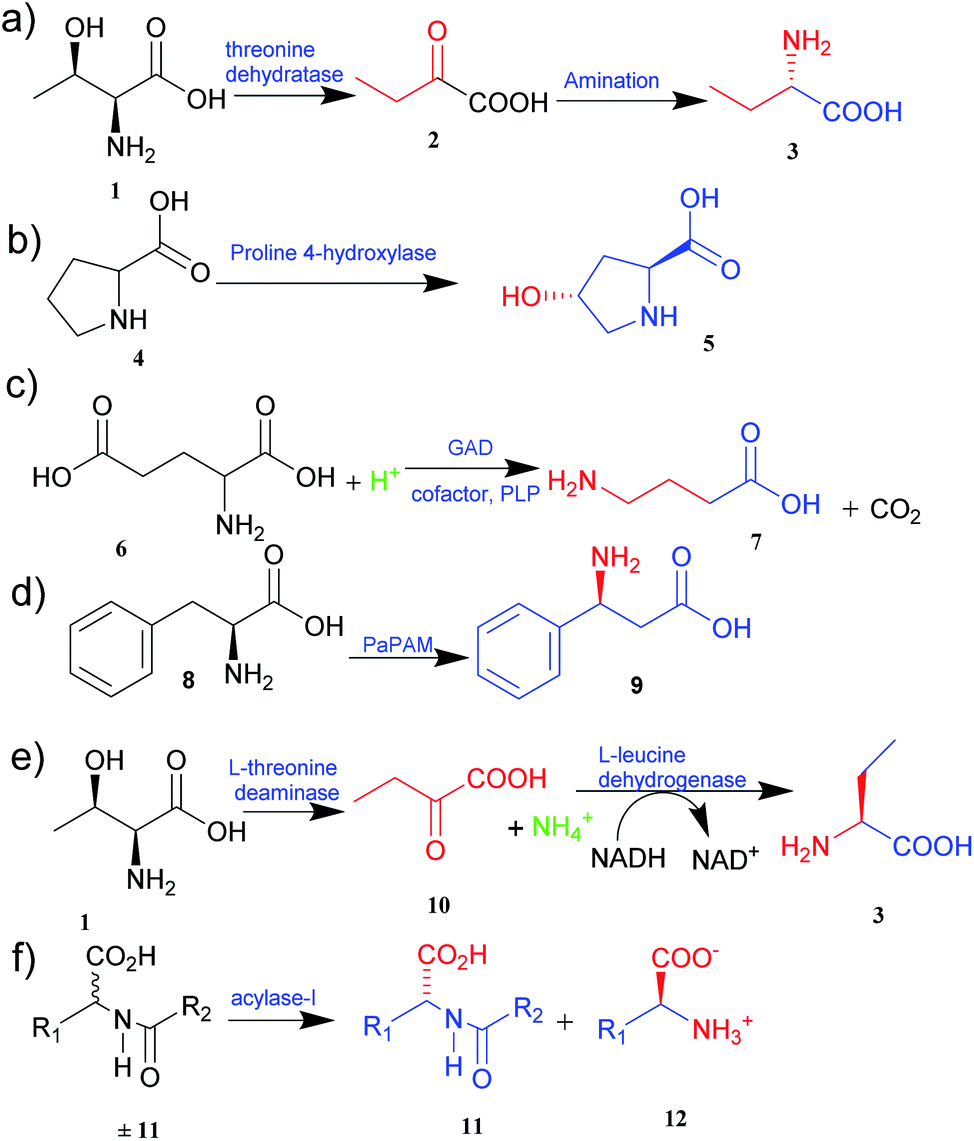 | ||
| Fig. 1 Synthesis of UAAs (a) synthesis of L-homoalanine 3 (b) synthesis of trans-4-hydroxy-L-proline 5 (c) synthesis of GABA 7 (d) synthesis of β-phenylalanine 9 (e) biosynthesis of L-ABA 3 (f) acylase-I for kinetic resolution of UAAs. | ||
The two primary biocatalytic methods for the synthesis of UAAs are kinetic resolution and asymmetric synthesis. While the kinetic resolution is limited by a maximum theoretical yield of 50%, asymmetric synthesis provides a theoretical yield of 100%.19,20
Asymmetric synthesis is an effective process that occurs without any protective group(s) and is based on straightforward reaction steps to synthesize new UAAs. Substrates are asymmetrically converted into their optically pure form by using transaminase and dehydrogenase.21 Deamination of L-threonine 1 followed by hydrogenation gives a better theoretical yield than other processes (Fig. 1e).22 Various amino acid dehydrogenases have been developed and utilized for enzymatic chiral amino acid synthesis.20,23 Even naturally occurring amine dehydrogenases have been engineered to improve their substrate specificity and catalytic efficiency. Cai et al.24 have synthesized γ-aminobutyric acid from biobased lignocellulosic waste using an engineered amine dehydrogenase.
Similarly, asymmetric synthesis by transaminase is much simpler and effective since the substrates like keto acids can easily be converted into amino acids. Displaying broad substrate specificities, transaminase also plays a vital role in nitrogen metabolism as an industrial biocatalyst. It exhibits a rapid reaction rate and requires no external cofactor regeneration. Hence, α-transaminases like tyrosine, aspartate, valine–alanine, branched-chain, and aromatic amino acid transaminases are widely studied.25 Approximately 50% of UAAs successfully incorporated into proteins are derived from tyrosine or phenylalanine.26 Synthetically easily accessible α-keto acid is taken as a target intermediate that can be easily converted into an amino acid with the help of enzymes in a single step. The keto group is replaced with an amino group in a stereospecific manner. In this process, aminotransferase (transaminase), which uses pyridoxal 5′-phosphate as a cofactor, reversibly catalyses the transamination reaction.26 The transaminases possessing relaxed substrate specificity and high enantioselectivity are widely employed for the biosynthesis of many UAAs. L-Homophenylalanine was synthesized using an enzyme of transaminase class, namely aspartate aminotransferase isolated from Paracoccus denitrifican.27 Similarly, several L-thienylalanines28 and L-phosphinothricin29 have been synthesized by employing tyrosine aminotransferase.
The biological synthesis of UAAs by the kinetic resolution uses different enzymes such as lipases and nitrilases during the synthetic process.30 Acylase I (aminoacylase; N-acyl amino acids amidohydrolase), the most widely used and applicable enzymatic catalyst, has been employed for the kinetic resolution of UAAs and α-amino acids. This enzyme accepts the substrate with a broad range of functionality and structure. Chenault et al.31 have studied this approach to synthesize UAAs by utilizing acylase from Aspergillus oryzae and porcine kidney. In this method, acylase-I catalysed the enantioselective hydrolysis of N-acyl-L-amino acids ±11 to 11 and 12. D-Amino acid and L-amino acid products having a variety of uses were obtained with high enantiomeric excess (Fig. 1f).
2.2 Chemical synthesis
Various synthetic methods have been developed to gain access to amino acid-based structures. Chemical routes are more profound than biocatalytic routes, possibly due to versatile synthetic ways. For the preparation of UAAs, a variety of chemical synthesis methods are available (Fig. 2–5).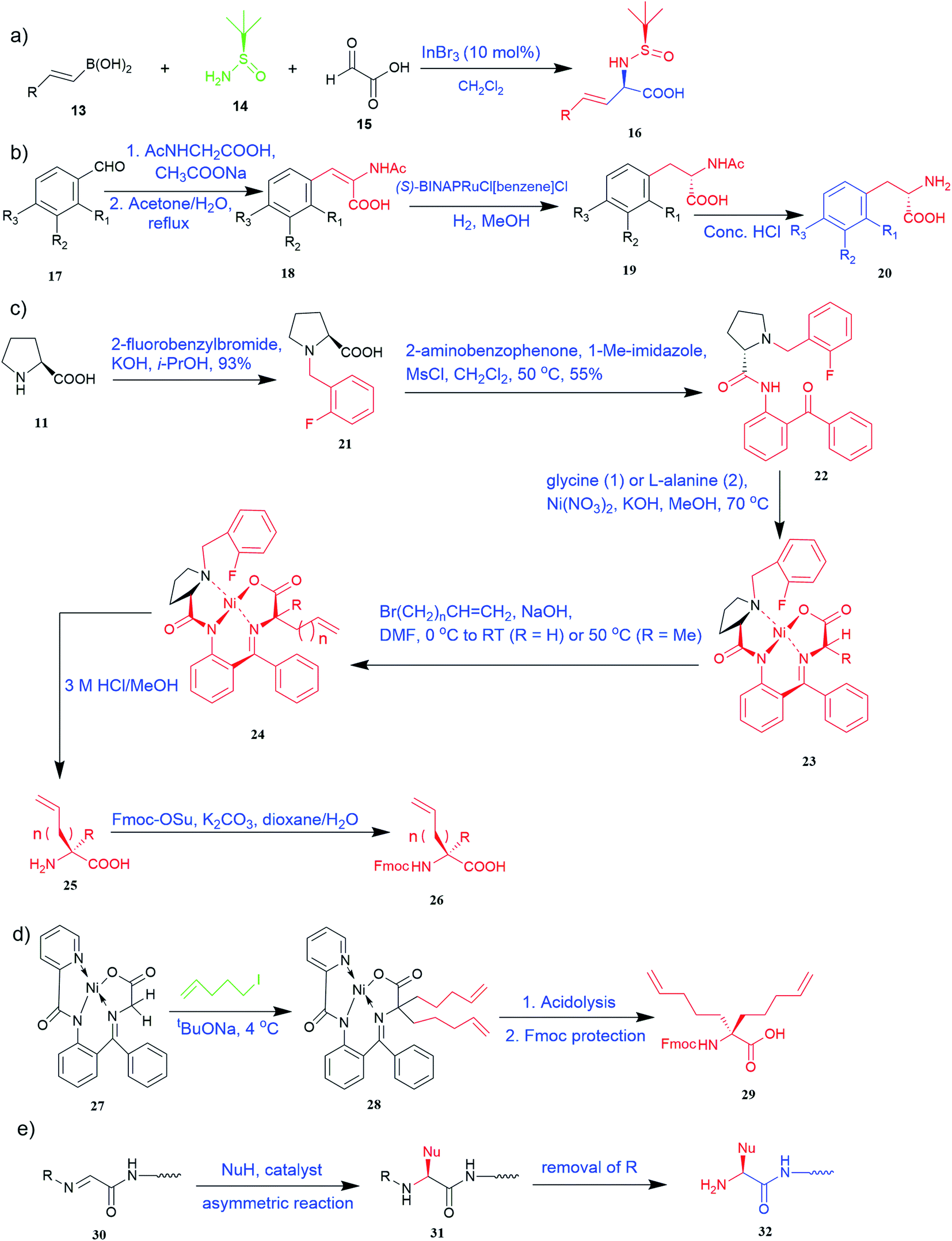 | ||
| Fig. 2 Synthesis of UAAs (a) Petasis borono–Mannich access to UAA 16 (b) synthesis from N-acetylamino phenyl acrylic acid 20 (c) synthetic route for unnatural alkenyl amino acids (25, 26) (d) synthesis of α-bisalkenyl substituted glycine 29 (e) asymmetric synthesis of UAA containing peptide 32. | ||
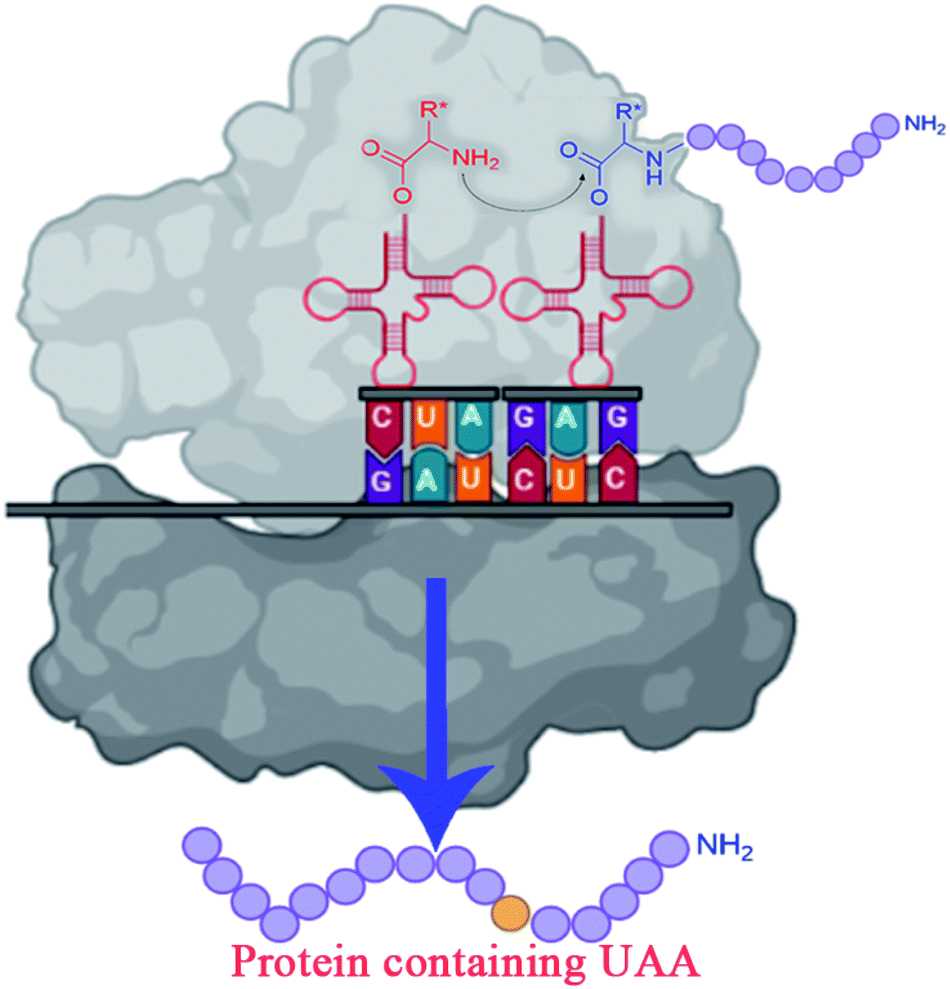 | ||
| Fig. 3 Incorporating unnatural amino acids into proteins using nonsense codon suppression. | ||
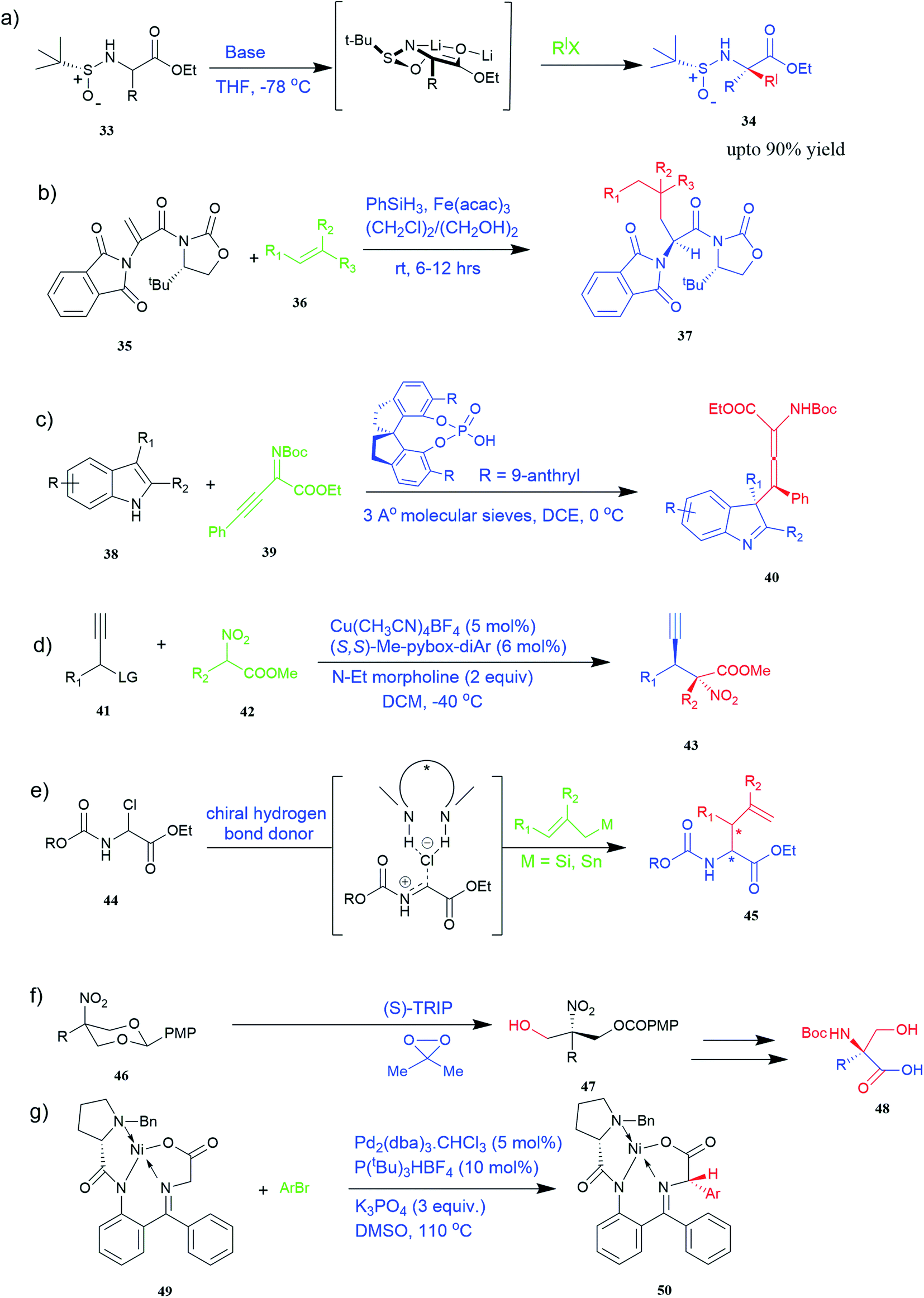 | ||
| Fig. 4 Synthesis of UAAs (a) diastereoselective synthesis of UAA by α-tert-butanesulfinamide 33 (b) iron-catalysed diastereoselective synthesis of unnatural chiral (S)-α-amino acid 37 (c) synthesis of tetrasubstituted α-amino allenoate 40 (d) synthesis of quaternary α-amino acid 43 (e) synthesis of α-allyl amino ester 45 (f) synthesis of unnatural α-substituted serine 48 (g) synthesis of arylglycine derivative 50. | ||
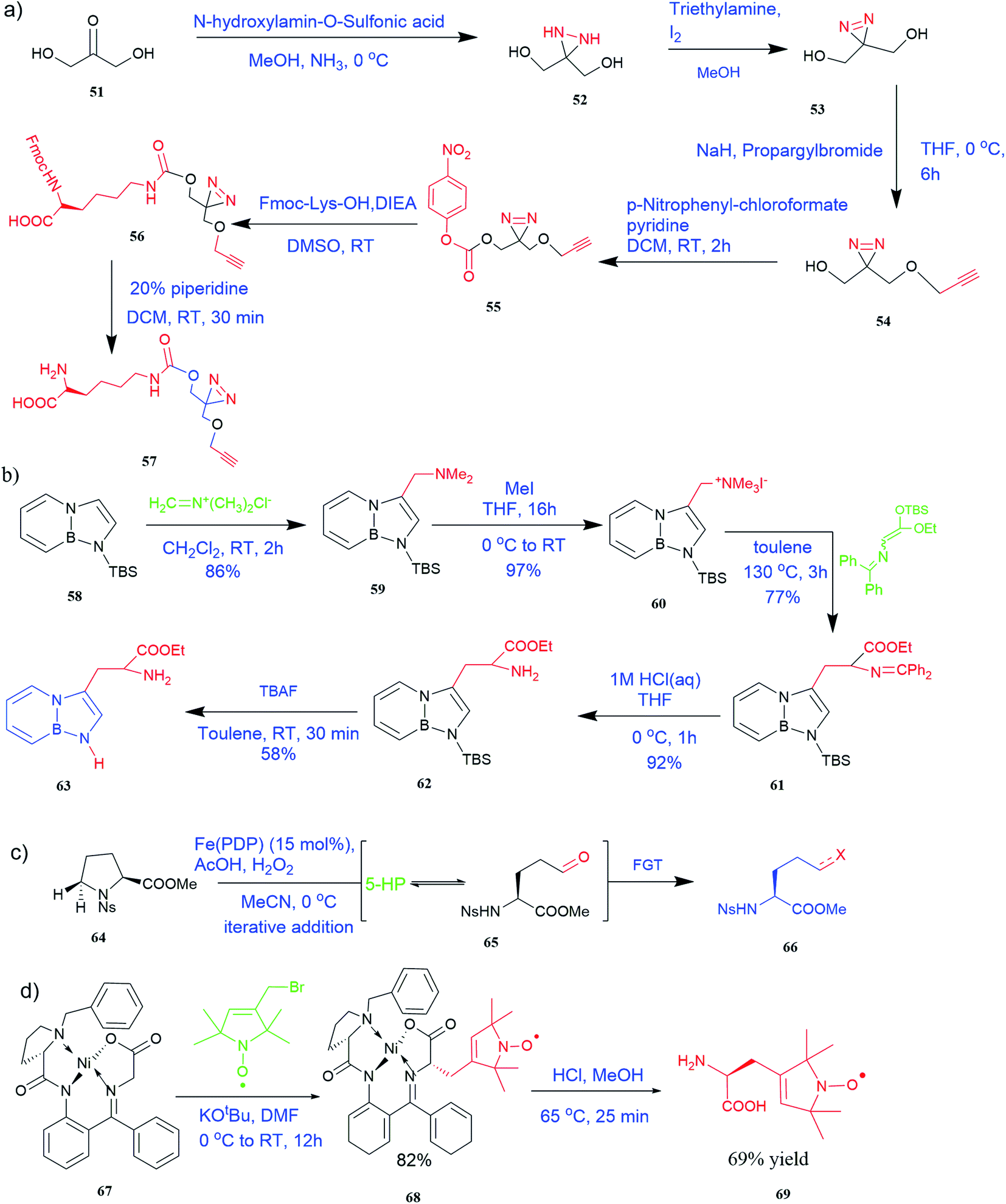 | ||
| Fig. 5 Synthesis of UAAs (a) synthesis of PrDiAzk 57 (b) synthesis of BN-tryptophan ester 63 (c) synthesis of UAA from oxidation of proline (d) synthesis of chiral spin-labeled amino acid 69. | ||
Of specific interest are catalytic methods with high levels of enantioselective control. A ruthenium catalyst was used for asymmetric hydrogenation of N-acetylamino phenyl acrylic acids 18 to yield chiral acids 20 (Fig. 2b), which are expected to be important in pharmaceuticals.34,35 Conformationally constrained peptides have a lower penalty for folding, which facilitates bioactive conformation. A new generation of chemical and drug discovery tools has emerged from conformationally constrained peptides. Asymmetric alkylation of a fluorine-modified Ni(II) Schiff base complex (23, 24) yielded an unnatural alkenyl amino acid (25, 26) required for peptide ‘stapling’ (Fig. 2c).36 The most apparent technique for producing UAAs is the alkylation of glycine.34 Moving to a more economical and environmentally suitable chiral phase transfer catalyst, the asymmetric alkylation of glycine has a yield between 60% and 90%. However, these synthetic routes depend on toxic reagents such as methylsulfonyl chloride.37,38 To minimize the effect of toxic reagents, 1-ethyl-3-(3-dimethyl aminopropyl)carbodiimide/4-dimethylaminopyridine (EDCI/DMAP) as a coupling agent, a chiral intermediate i.e. derivative of glycine non-proline 27 was synthesized starting with proline and then alkylating in the presence of sodium tert-butoxide which obtained 28 (Fig. 2d). By using this method, five UAAs for peptide stapling 29 were prepared with high enantioselectivity >99% ee and yields ranging from 60% to 70%.37 Furthermore, a new methodology for the asymmetric synthesis of UAAs-containing peptides 32 was also developed using imino peptides 30 such as α-imino perfluro alkylesters, imides, or thioesters (Fig. 2e).39
Known as an inexpensive and abundant metal, iron is commonly utilized as a catalyst in organic synthesis. Developing an effective and practical diastereoselective method to synthesize unnatural chiral α-amino acids using iron as a catalyst is highly desired. Toward this end, an iron-catalysed diastereoselective method has been developed; the method employs widely accessible iron salts, 2-phthaloyl acrylamide 35, and alkenes 36 as starting materials, and phenyl silanes as a reductant for unnatural chiral (S)-α-amino acids with γ-quaternary carbon centres 37 (Fig. 4b).41 This protocol has several benefits, including simple and broad substrates, moderate conditions, excellent diastereoselectivity, and simple workup methods.
It has been demonstrated that bifunctional UAAs called PrDiAzK 57 can be inserted into the protein interface, using genetic code expansion, with a minimum structural perturbation. Hoffmann et al.47 demonstrated bifunctionality for UAAs based on Z-lysine, benzophenone-alkyne “BPKyne,” and γ-selenolysine, in which a lysine-based bifunctional amino acid combines a photoreactive diazirine group with a terminal alkyne handle for reaction with azides using copper-catalysed click chemistry. The starting material was commercially available dihydroxyacetone 51. In liquid ammonia, the ketone was converted into diazirine (52, 53) in a two-step reaction. Propargyl bromide was used to alkylate one of the two hydroxyl groups. The single-substituted product 54 was isolated, converted into a reactive carbonate, and bound to lysine (56, 57) (Fig. 5a).47 In the hope to utilize this UAA in further protein studies and classify its properties concerning the natural substrate, boron and nitrogen-containing unnatural analogues of tryptophan 63 have also been synthesized through the functionalization of BN-indole 58. In this case, TBS-BN-indole 58 was subjected to a substitution reaction with dimethyliminium chloride. Iodomethane was used to methylate the resulting product 59, which was then displaced with silyl-ketene-acetal. The Schiff base protecting group of 62 can be removed in aqueous acidic conditions. Following this, the silyl protecting group was deprotected to yield BN-tryptophan ethyl ester 63 (Fig. 5b).48 More notably, these studies demonstrated the first example of an azaborine containing amino acid being introduced into proteins.
2.3 Other methods
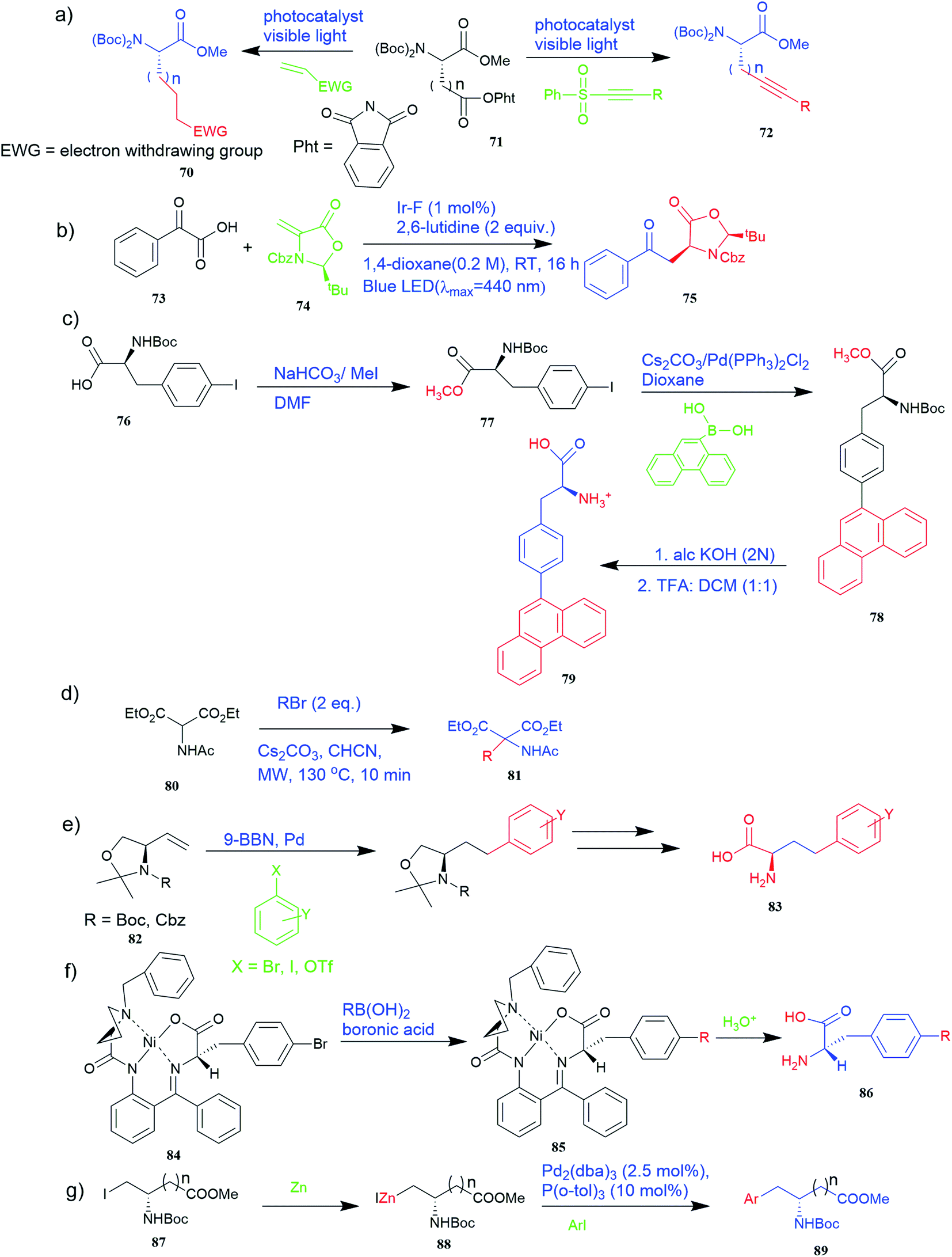 | ||
| Fig. 6 Synthesis of UAAs (a) synthesis of unnatural chiral α-amino acid using visible-light assistance approach (b) light-mediated protocol for the synthesis of UAA via the radical decarboxylative process (c) synthesis of fluorescent α-amino acid 79 that emits greenish-blue light (d) synthesis of UAA precursor from diethyl acetamidomalonate 80 (e) synthetic route for various R-amino alcohols and R-amino acids using Pd-mediated Suzuki cross-coupling reaction. (f) Synthesis of enantiomerically enriched non-protein (S)-α-amino acid from Schiff base Ni(II) complex 84 of the chiral auxiliary (S)-BPB. (g) Synthesis of unnatural aspartic and glutamic acid derivative 89. | ||
Furthermore, the production of genetically encoded fluorescent α-amino acids that produce greenish-blue light has widespread applications in research, biotechnology, and the pharmaceutical sector. The amino acid 4-phenanthren-9-yl-L-phenylalanine (Phen-AA) 79 that emits greenish-blue light in the visible region has good stability with a 75% quantum yield. As shown in Fig. 6c, its production was initiated with the synthesis of N-(tert-butoxycarbonyl)-4-iodo-L-phenylalanine methyl ester intermediate (Boc-Phe(4-I)-OMe) 77 where the carboxylate group through methylation of the commercially available N-(tert-butoxycarbonyl)-4-iodo-L-phenylalanine 76. The next step involved Suzuki–Miyaura cross-coupling reaction which coupled α-amino acids bearing vinyl or aryl halide side-chains with polyaromatic boronic acids and resulted in N-(Boc)-4-(9-phenanthracenyl)-L-phenylalanine methyl ester 78 intermediate. De-protection of the methyl and Boc protecting groups of 4 produced the final L-α-amino acid 4-phenanthracen9-yl-L-phenylalanine 79 i.e. novel fluorescent α-amino acid that emits greenish-blue fluorescence.53
Also, a synthetic method for new enantiomerically enriched non-protein (S)-α-amino acids 86 was developed to generate new carbon–carbon bonds in the side chain group of the amino acid moiety with high chemical yields and optical purity using the reaction of Suzuki. In this method, as the initial synthon, the Schiff base Ni(II) complex 84 of the chiral auxiliary (S)-BPB (N-benzyl proline benzophenone) and 4-bromo-L-phenylalanine amino acid were employed. The final Ni(II) complexes 85 were decomposed with aqueous HCl, and the amino acids were isolated with excellent enantioselectivities (>99% ee). The chiral auxiliary ligand (S)-BPB can be recycled and reused for the synthesis of starting Ni(II) complex (Fig. 6f).57
The Negishi cross-coupling is a powerful C–C bond-forming reaction widely utilized in many areas of organic synthesis. The Negishi cross-couplings were used to synthesize various UAAs like aromatic, heteroaromatic, and complex amino acid derivatives such as aspartic and glutamic acid derivatives 89, aryl glycine derivatives, phenylalanine derivatives, amino acids containing metal-coordinating side chains, β-amino acids, aryl bromides with an iodo serine derivative, naphthyl-appended amino acid, bis(amino acid) derivative, fluoroaromatic amino acid, p-(C-glucopyranosyl) phenylalanine derivative, cycloalkenyl-protected amino acid derivatives (Fig. 4g and 6g).58
Derivatization of natural amino acids including cysteine, threonine, serine, and tyrosine containing heteroatoms such as N, S, O could open the door to synthesizing many possible UAAs. Undoubtedly, novel synthetic methods to construct UAAs will continue to provide new ways for peptidomimetics.
2.4 Mutagenesis and incorporation of UAAs
The traditional enzyme engineering approach constitutes the selective substitution of one amino acid by any other 19 natural amino acids (NAAs) to alter the functionalities of enzymes. However, incorporating UAAs provides a better approach to engineering the enzymes for designing efficient biocatalysts that can display novel physicochemical properties and perform biological functions.59,60UAA(s) placed in proteins at strategically selected locations offer a new and fascinating variety of protein research and engineering options.61 In vivo incorporation of UAAs can be carried out in two ways: in site-specific approach and residue-specific approach. The site-specific incorporation uses an exogenously evolved tRNA or synthetase pair, and residue-specific incorporation uses the misacylation of the endogenous tRNA.60 In vivo incorporation methods are marked with many engineering challenges like UAAs transportation across the cell membrane, cytotoxicity, and low incorporation efficiency.59
To overcome the challenges of in vivo approach, strategies for in vitro incorporation of UAAs62 have received considerable attention to gain broader specificity and increase efficiency.59 The cell-free incorporation of UAAs results in the formation of complex proteins like integral membrane proteins, physiologically toxic proteins, and large protein complexes.63 The cell-free incorporation of UAAs can be achieved by global suppression methods and orthogonal translation systems. The former utilizes natural biological mechanisms while later reengineers tRNA, aminoacyl-tRNA synthetase, ribosome, elongation factor, and release factor via directed evolution or rational design to incorporate UAAs. The orthogonal translation system is commonly used because of its effectiveness towards site-specific UAAs incorporation in comparison to the global suppression method.59 Other ways to incorporate UAAs include solid-phase synthesis and chemical ligation methods. Solid-phase synthesis allows the direct transformation of peptides' side-chains into novel peptides on the solid support. Here, the accumulation of side products prevails because of incomplete deprotection or coupling reactions. This drawback is overcome by chemical ligation methods which permits peptide fragments to the couple in aqueous solutions.64–66 Chemical alteration of a synthetic fluorophore protein after translation and the use of chemically mis-attached tRNAs suppressors to insert UAAs are the most straightforward methods. These methods, however, give limited yields of proteins and are restricted to readily available protein locations.45 The incorporation of UAAs via codon reassignment offers novel chemical and biological functions to translated products.67 The amber stop codon (UAG), showing a low level of amino acids misincorporation, is most commonly targeted to reassign and incorporate UAAs during translation in comparison to opal (UGA) and ochre (UAA) stop codons.63,67 The UAA mutagenesis was found to enhance the efficiency and regioselectivity of CYP P450 oxidation catalysts.68 The detailed mechanism on UAA incorporation is well explained by Young and Schultz,2 and Lang and Chin.66
3. Applications of UAAs
3.1 Catalytic functions
Enzymes are potent bio-catalysts that have participated in promoting chemical reactions with remarkable efficiency and selectivity. The effect of incorporating UAAs into enzymes with potential applications in biocatalysis has recently received much attention. Incorporating UAAs into target proteins can improve enzyme activity69 and increase the stability of proteins under harsh conditions, such as the resistance to organic solvents.70 There are different methods for incorporating UAAs into target proteins with potential biocatalysis applications. One technique is a coupling methodology, which was utilized to design industrially relevant transaminase (ω-TA) since ω-TA shows promising activity in producing optically pure amine compounds.70Furthermore, incorporating UAAs, such as replacing methionine with norleucine, into enzymes may protect proteins from methionine oxidation. This approach is useful for biocatalysts that become inactive in oxidative conditions or require oxidizing substrates or cofactors.17 Another method was genetic code expansion (GCE) technology that has been used to introduce small labelling sites in the form of uniquely reactive ncAAs in a target protein. Low incorporation efficiency of UAAs and high background fluorescence limit have super-resolution microscopy (SRM) applications. SRM benefits immensely from the ability to mount photographic fluorescent labels on proteins.71 By choosing a sequential allylic C–H amination/vinylic C–H arylation, which began with inexpensive commercially available α-olefins and boronic acids, UAAs precursors were also obtained using Pd(II)/sulfoxide catalysis. Using coupled enzyme reactions, which are a novel biocatalytic method for synthesizing L-homoalanine from L-threonine consisting of a threonine deaminase (TD) and ω-TA. TD catalyses the dehydration/deamination of L-threonine, asymmetrically converting to L-homoalanine through transamination with benzylamine executed by ω-TA.72
N-Alkylated-α-amino acids produced via enantioselective methods are valuable building blocks for pharmaceutical and fine chemical industries. Therefore, they are treasured and widely investigated. While there are many chemical methods for their synthesis, biocatalytic approaches can give a greener and cleaner alternative to current practices. Alternative processes such as methylation in other proteins and peptides, including N-α-methylation, have biological activity of peptides.73 For example, cypemycin is a naturally occurring bacterial peptide with post-translational changes, including N-α-dimethylation, and it has decisive antibacterial action and in vitro efficacy against murine leukemia cells.74 Also, N-arylated α-amino acids and pyrazolidin-3-ones are widely used in pharmaceuticals and agrochemicals as chiral building blocks. They are biocatalytically produced by utilizing ethylenediamine-N,N′-disuccinic acid lyase (EDDS lyase) as a biocatalyst. This enzyme has a broad substrate range and high conversions, resulting in high isolated yields and enantiomeric excess of the relevant N-arylated aspartic acids.75 The use of biocatalyst engineering has greatly aided enzyme discovery and applications in industrial and pharmaceutical applications. The service and advancement of enzyme engineering techniques have grown tremendously in recent years. Importantly, engineering techniques incorporating UAAs successfully produce enzymes with more excellent stability, selectivity, and altered catalytic properties.
In recent decades, applications of enzymes have increased significantly, especially in scientific methodology, pharmaceutical science, food alteration, laundry, biofuel production, agro-industry, and many others. The increasing demand for biocatalysts as a replacement for traditional chemical catalysts has grown progressively. The engineering strategies of enzymes rely on nature's genetically encoded alphabet of twenty canonical amino acids. In the past few years, the emergence of genetic code expansion methods that allow several structurally diverse amino acids to be introduced into the proteins have been observed.76
UAA incorporation in enzymes produces enzyme resistance towards temperature and organic solvents, resulting in effective catalytic properties. Various UAAs with different side chains overcome the limitations encountered by NAAs in enzymes, thereby improving their potential applications. The residue-specific method and site-specific method are favoured for producing enzymes with improved and modified functions. Also, it can be done with the coupling of residue-specific and site-specific incorporation methods. Multifunctional green fluorescent proteins (GFP) were constructed through site-specific incorporation of L-3,4-dihydroxyphenylalanine and residue-specific incorporation of (2S,4S)-4-fluoroproline (4S-FP) or L-homopropargylglycine (hpg).77 Site-specific methods have attracted considerable attention as proteins or enzymes that bear multi-UAAs display improved functionalities. Interestingly, it has been discovered that integrating two chemically distinct UAAs into GFP by employing two orthogonal pairs in a single expression shows no mutual cross-reactivity and thus can be developed for efficient double labelling. In addition, a highly efficient suppressor plasmid pUltra has been generated, exhibiting higher suppression activity, enabling the efficient suppression of three different amber stop codons in GFP.78 In the residue-specific incorporation method, incorporation of 3-fluorotyrosine into ω-transaminase showed a 2 fold higher half-life with enhanced catalytic activity.79 Furthermore, the global incorporation of UAAs into β-galactopyranoside resulted in a two-fold increase in Vmax for ortho-nitrophenyl-β-D-galactopyranoside at pH 7.0 and a 4–5-fold increase in Vmax toward phenyl-β-D-galactopyranoside at the same pH.60 Global substitution using Klentaq DNA polymerase resulted in comparable activity in the same range and similar deoxyribonucleoside triphosphate conversion (dNTP). The addition of (4R)-fluoroproline to the DNA polymerase resulted in a fluorinated enzyme that was highly active.80
Cytochromes P450 (CYP) are biocatalysts that catalyse the transfer of an oxygen atom from molecular oxygen to an organic substrate upon a donation of electrons by coenzymes, such as NAD(P)H. In P450 BM-3, methionine residue was replaced with the isoteric methionine analogue norleucine to test whether the enzyme stability was hampered by Met oxidation throughout the reaction; a two-fold increase in peroxygenase activity along with a significant reduction in thermal stability has been demonstrated.81 Kolev et al.82 reported P450 BM-3 variant that oxidizes (+)-nootkatone as a representative substrate (since (+)-nootkatone has a various number of different C–H bonds (primary, secondary, tertiary, aromatic) as well as many functional groups such as carbonyl, ester and olefinic group) resulted with the higher turnover number (kcat) for an engineered CYP on a complex molecule. A five-fold increase in the turnover number (kcat) by the substitution of Leu75 with para-amino phenylalanine (pAmF) by stop codon suppression (SCS) in the active site of the enzyme was observed.82
Transaminases catalyse the transfer of an amino group between amino acids and α-keto acids, and they act as biocatalysts in the production of optically pure α-amines. The incorporation of meta-fluorotyrosine enhanced thermostability and organic solvent tolerance in ω-transaminase by using the selective pressure incorporation (SPI) method.79 N-Terminally truncated version of DNA polymerase I from Thermus aquaticus (Klentaq) was generated. Klentaq is a highly thermostable polymerase with no nuclease activity. Replacing 32 proline residues by (4R)-fluoroproline in Klentaq DNA polymerase by SPI method was 92% efficient with a highly active fluorinating enzyme.80 The substitution of the critical Tyr309 of phosphotriesterase from Agrobacterium radiobacter (arPTE) by the SCS method, i.e. arPTE variant containing L-(7-hydroxycoumarin-4-yl)ethyl glycine (HCEtG) in place of Tyr results in an eight-fold increase in kcat values. Phosphotriesterase hydrolyses organophosphates, such as pesticides with target residues located in the substrate-binding site. The electrostatic repulsion between the HCEtG and the product, which are both negatively charged, contributes to the increased rate-limiting product release step of substrate turnover. The 7-hydroxyl group of L-(7-hydroxycoumarin-4-yl)ethylglycine promoted the hydrolysis of pesticide paraoxon due to interactions between the bacterial phosphotriesterase enzyme and substrate during the Michaelis complex and product release.83 Disruption of Asp–His hydrogen bond by replacing the proximal histidine ligand with the unnatural structural analogue N-methylhistidine (NMH) by SCS into an engineered ascorbate peroxidase (APX2) achieved significantly increased turnover numbers. Heme peroxidases catalyse a range of oxidative transformations for various biotechnological applications.84 Introducing 3-fluorotyrosine to organophosphate hydrolase showed extended optimal pH activity and improved thermal stability at alkaline pH.85 Global fluorination of aromatic residues of lipase B from Candida antarctica enhanced stability with 4-fluorophenylalanine residue-specific substitution.86 Enhanced protein foldability and increased thermostability were shown by the fluorination of phosphotriesterase (PTE) with UAA 4-fluorophenylalanine. Approximately 30% of the enzymes' native structure was maintained.86 Replacement of hydrophobic amino acids, such as methionine, proline, and phenylalanine from Thermoanaerobacter thermohydrosulfuricus lipase related synthetic analogues like lipase analogues, in a residue-specific manner, resulted in a 25% increase in catalytic activity, substrate tolerance by up to 40%, changes in optimal temperature by up to 20 °C and pH by up to 3.69 A change in substrate specificity of the enzyme, N-acetylneuraminic acid lyase (NAL) mutants was obtained after incorporating various UAAs, thereby increasing activity towards aldol reactions of erythrose and pyruvate.87 Incorporation of UAAs in proteins leads to the mutation of amino acids intimately involved in an enzyme's catalytic mechanism, thereby overcoming challenges such as the substrate or product inhibition, organic solvent tolerance, catalytic efficiency, turnover number, thermal and chemical stability.
3.2 Therapeutic applications
UAAs find many applications ranging from being a chiral building block to protein probing to site-specific drug carriers and many pharmacological and therapeutic uses (Tables 1 and 2). UAAs display structural versatility and functional stability so that they can be modified or conjugated to incorporate some target-release drugs. Naphthalene-tripeptides containing alpha-aminoisobutyric acid or alanine have shown various potential uses in combating cancer and bacterial infections.88 The UAA p-acetyl phenylalanine (pAcF) can be integrated with an EGFR targeted, elastin-like polypeptide nanomaterials in the presence of reactive amino acids to produce bio-orthogonal ketone for attachment of doxorubicin, and thus produced nanomaterials are proven to have considerably higher cytotoxicity than non-target controls in several lines of cancer cells.61| Enzymes | Name of UAA | Structure of UAA | Characteristics | Incorporation method | References |
|---|---|---|---|---|---|
| ω-Transaminase | 3-Fluorotyrosine | 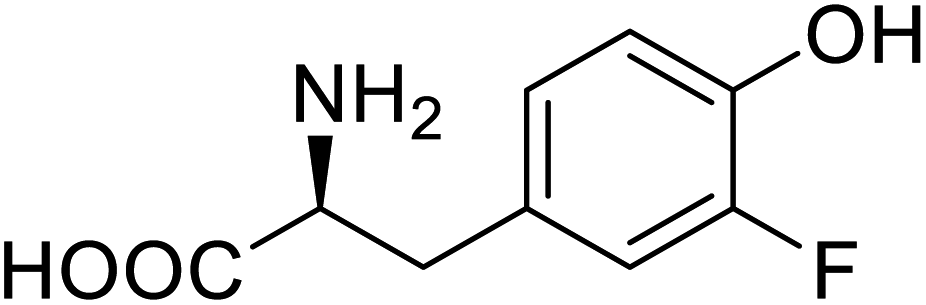 |
Enhanced catalytic activity and thermostability | Residue-specific incorporation of 4-fluoroproline | 79 |
| Organophosphate hydrolase | 3-Fluorotyrosine | 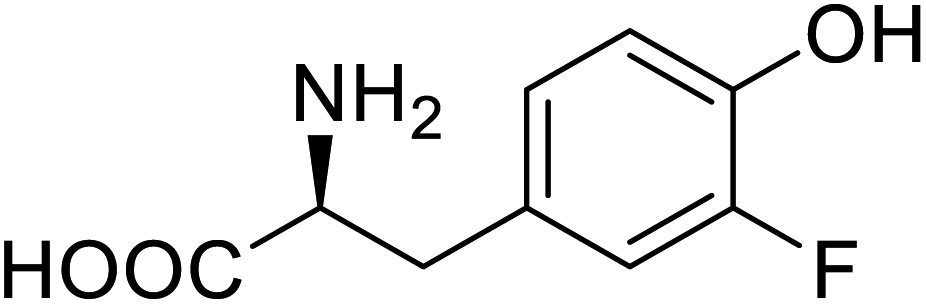 |
Thermal stability increment at alkaline pH | Residue-specific incorporation | 85 |
| P450 | Nor-leucine | 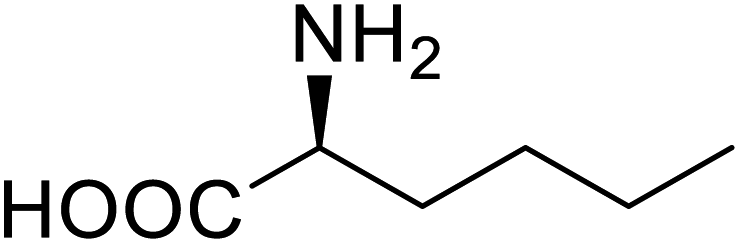 |
A two-fold increase in peroxygenase activity | Residue-specific incorporation | 81 |
| Lipase B | 4-Fluorophenylalae | 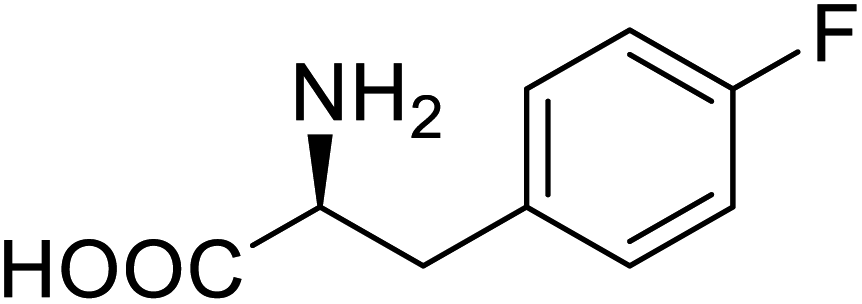 |
Prolonged the shelf life of lipase activity | Residue-specific incorporation of 4-fluorophenylalanine | 86 |
| Phospotriesterase (PTE) | 4-Fluorophenylalanine | 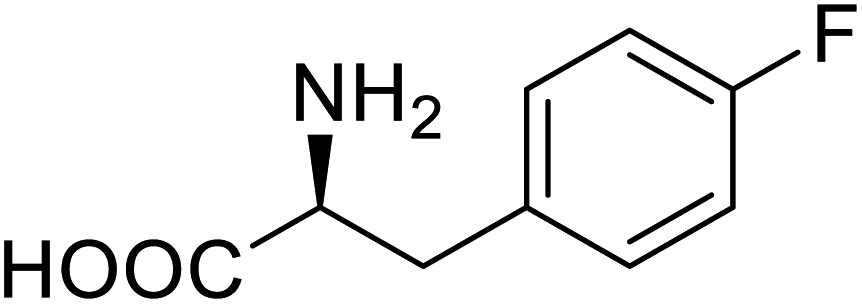 |
Enhanced protein refolding | Residue-specific incorporation of 4-fluorophenylalanine | 115 |
| Green fluorescent protein (GFP) | L-3,4-Dihydroxyphenylalanine | 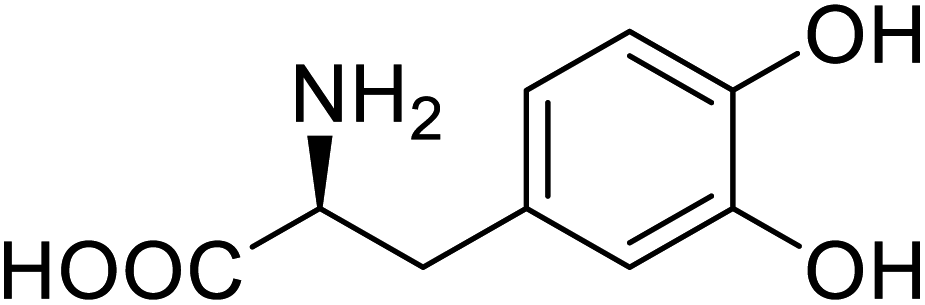 |
Forming protein–chitosan complexes enhancing stability | Site-specific incorporation of L-DOPA and residue-specific incorporation of 4-fluoroproline | 77 |
| Glutathione S-transferase (GST) | pNCSF | 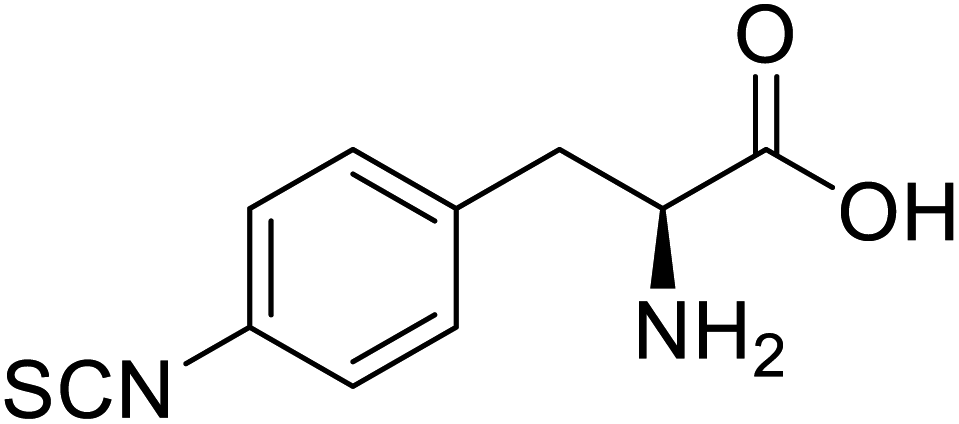 |
Formation of stable thiourea crosslinks | Site-specific incorporation | 83 |
| Lipase | Nor-leucine | 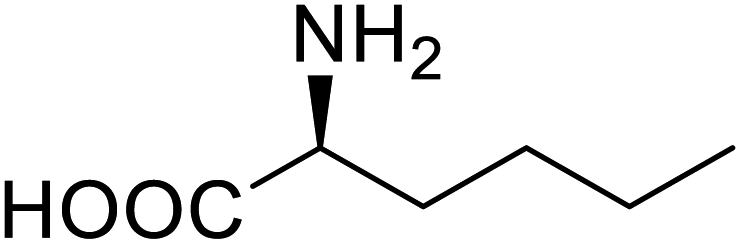 |
A 10-fold increase in catalytic activity | Residue-specific incorporation | 69 |
| Phosphotriester (PTE) | L-(7-Hydroxycoumarin-4-yl)ethylglycine | 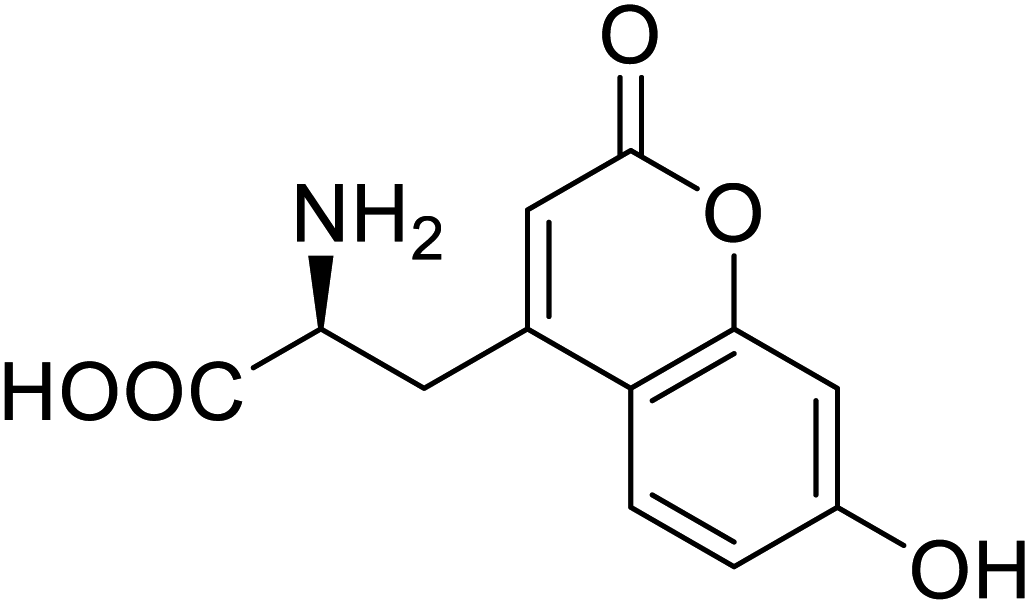 |
A 8-fold increase in turnover number; promotion of Michaelis complex formation | Site-specific incorporation | 83 |
| NAL | 2,3-Dihydroxypropyl cysteine | 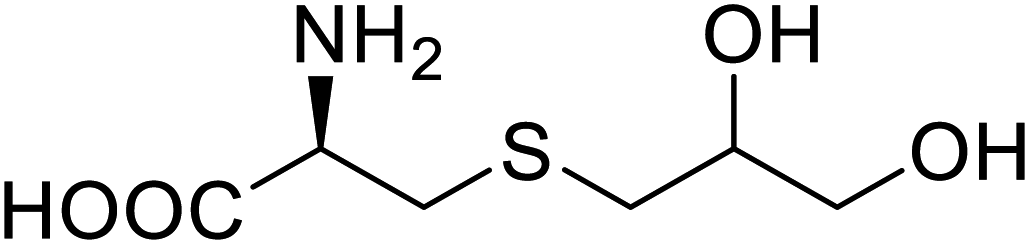 |
Alters substrate selectivity | Site-specific incorporation | 87 |
| UAA | Structure | Method of synthesis | Application | Mode of use | References |
|---|---|---|---|---|---|
| Substituted arylglycine | 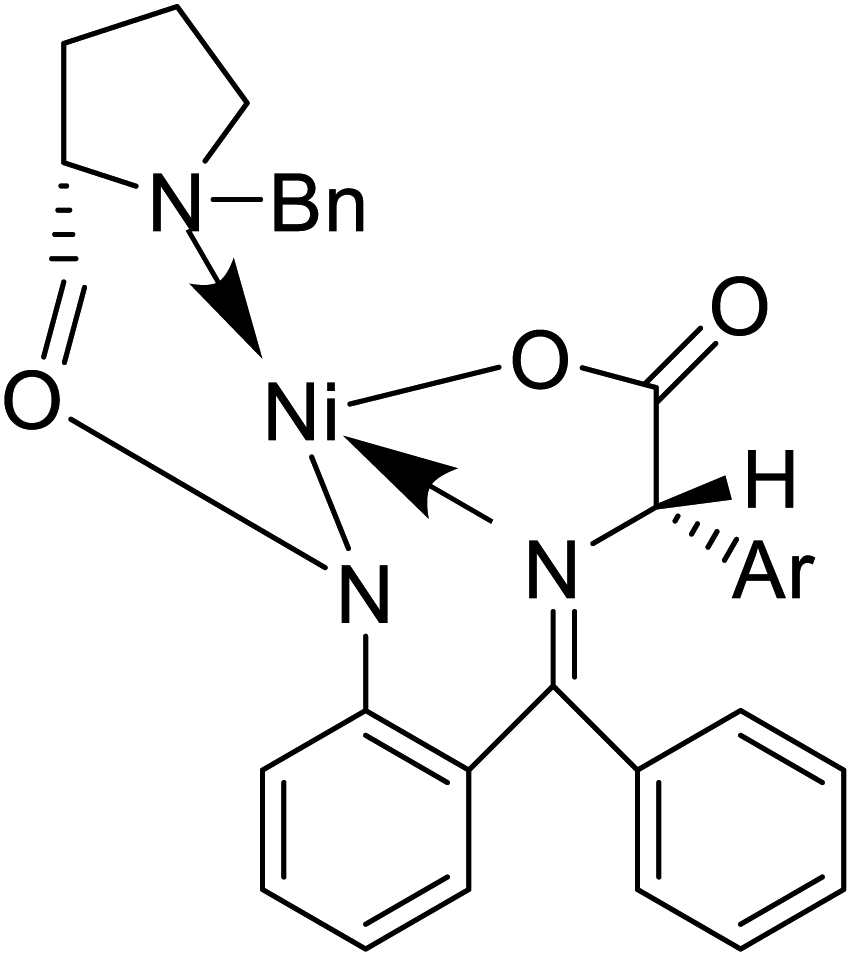 |
Chemical synthesis | Building blocks in many bioactive compounds and natural products | Assist in drug discovery | 46 |
| PrDiAzK | 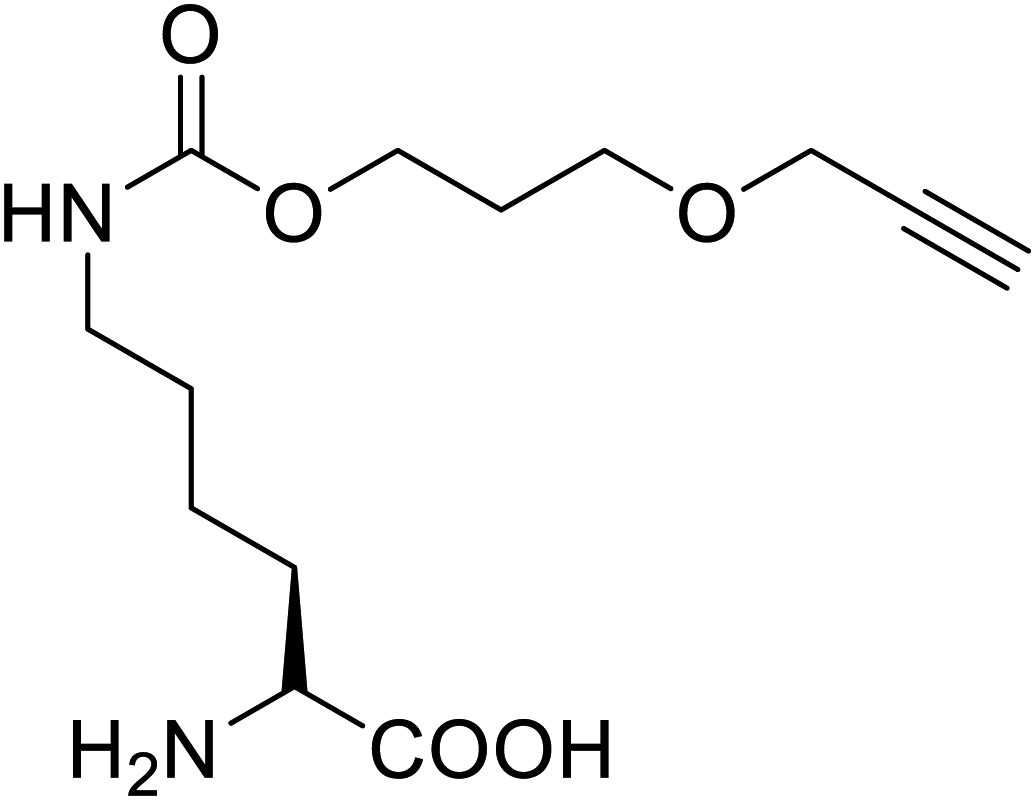 |
Chemical synthesis | Potential application in the system-wide mapping of protein–protein interaction | Site-specifically incorporated into proteins in both bacterial and mammalian cell culture | 47 |
| α-Substituted glutamic acid derivatives | 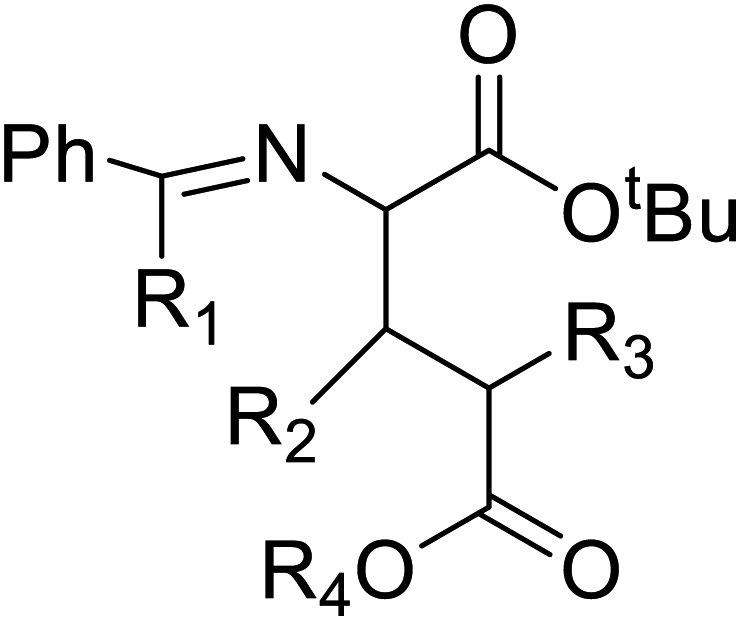 |
Chemical synthesis | Serves as a new pathway to open new route to biologically active molecules | Shortcut to active biomolecule | 124 |
| 2-Indolylglycine derivatives | 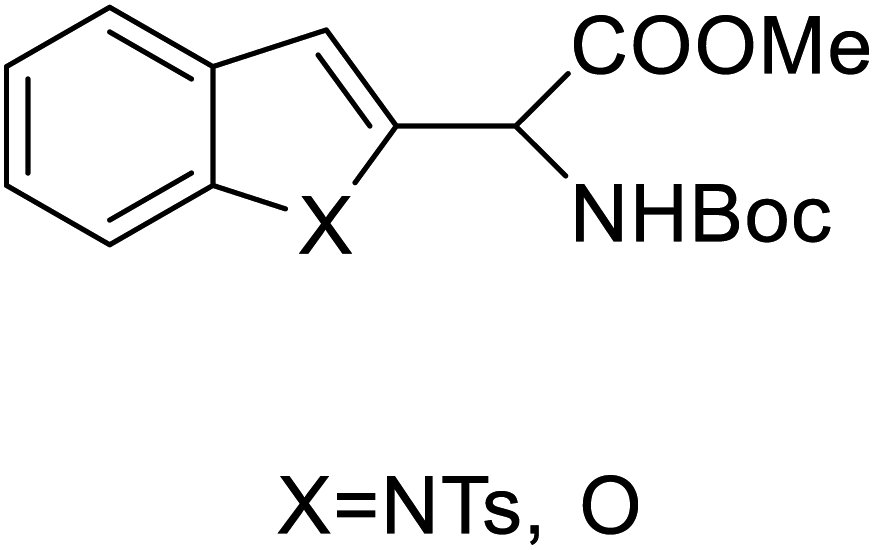 |
Chemical synthesis | Building blocks in biomolecules | Assist in drug development | 34 |
| (S)-N-(2-Benzoylphenyl)-1-(2-flurobenzyl)-pyrolidine-2-carboxamide | 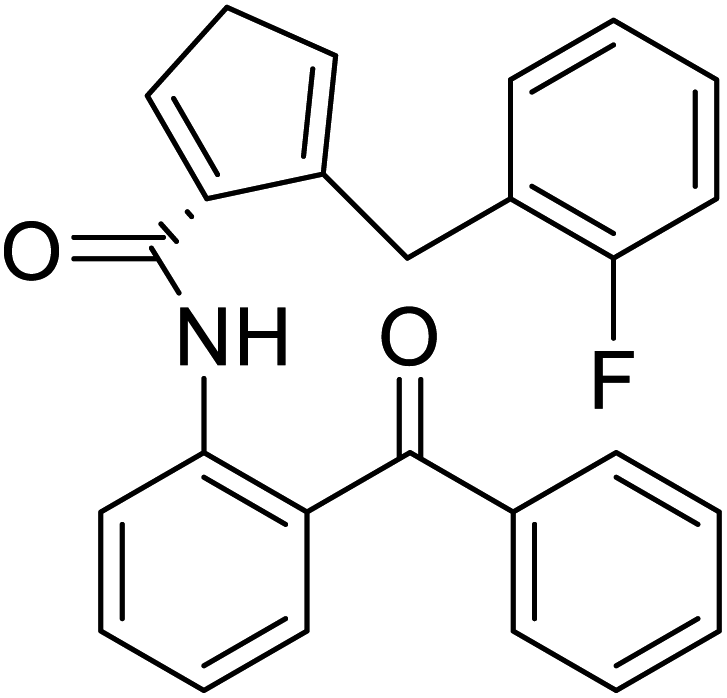 |
Chemical synthesis | Brings ease and efficiency to stapled peptide research | Peptide stapling | 37 |
| 4-Phenanthracen-9-yl-L-phenylalanine | 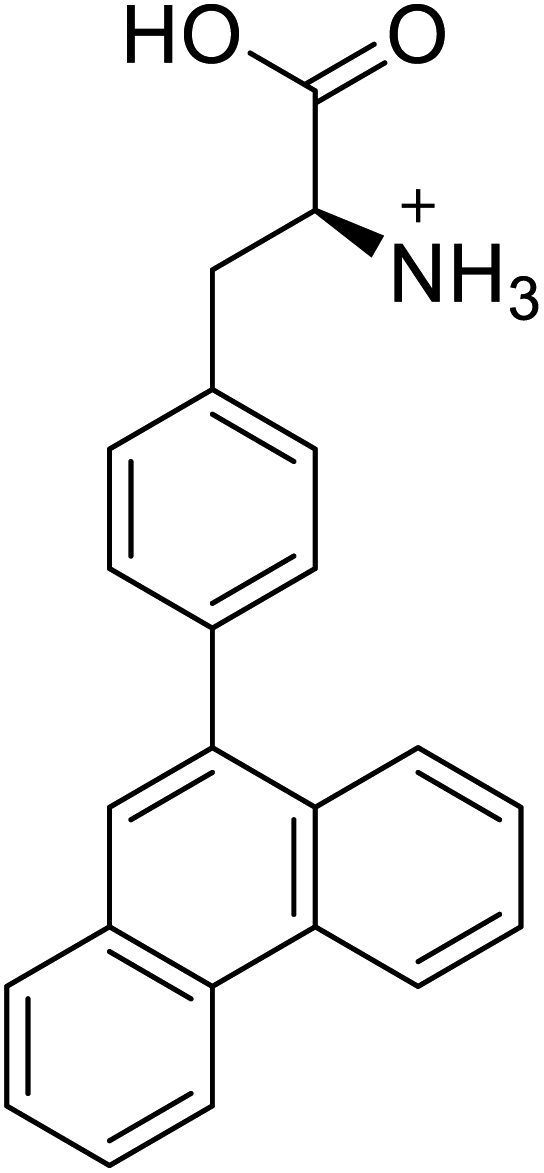 |
Chemical synthesis | Might find broad application in research, biotechnology, and pharmaceutical industry | Gets into human cells, being visible upon 450 nm laser excitation | 53 |
| BN (boron and nitrogen) tryptophan analog | 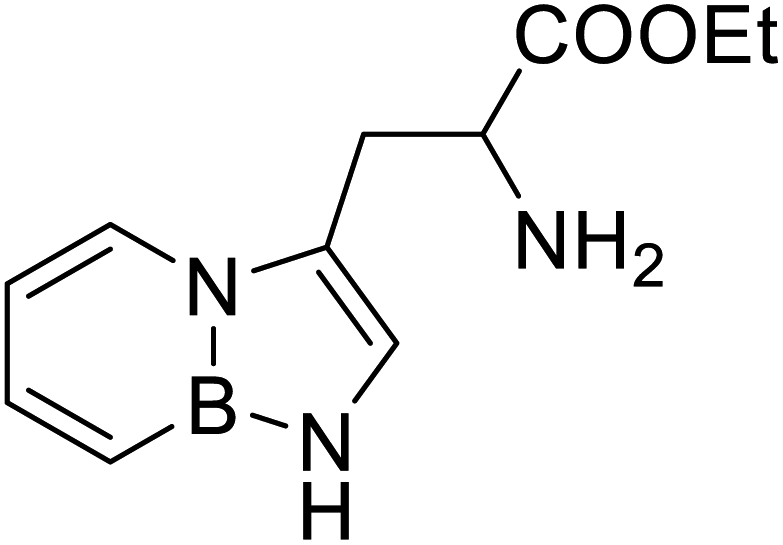 |
Chemical synthesis | Incorporation with proteins | Employ BN isoterism of arenes in a biological context where tryptophanyl-tRNA synthetase can recognize and azaborine containing amino acid | 48 |
| p-Acetylphenylalanine | 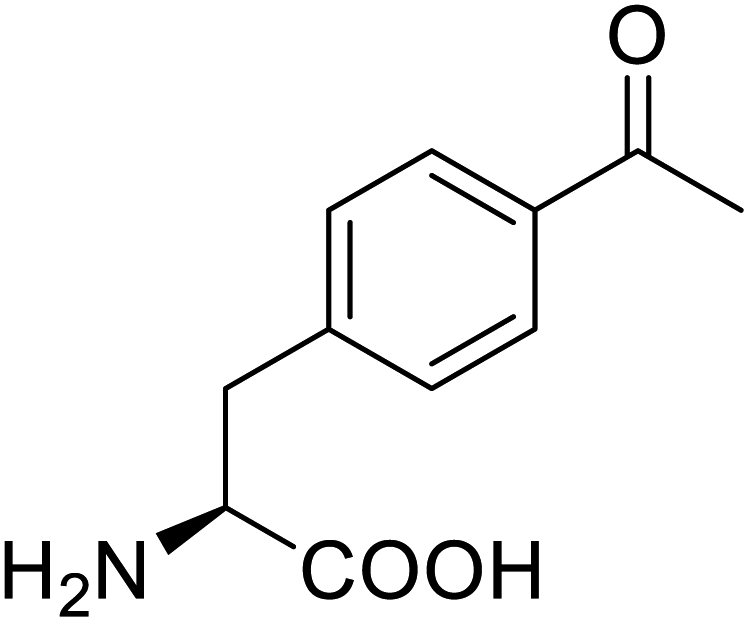 |
Biological synthesis | Antibody drug conjugates | Part of antibody-drug conjugates | 61 |
| γ-Aminobutyric acid (GABA) | 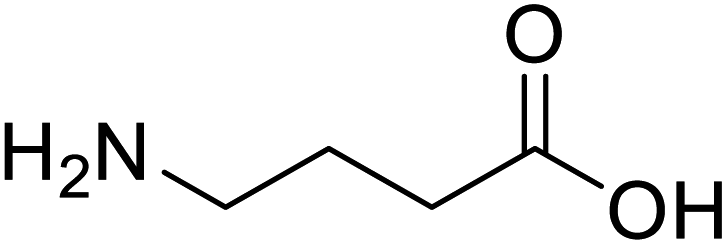 |
Biological synthesis | In metabolic engineering | To develop engineered strain | 14 |
| L-Azidohomoalanine | 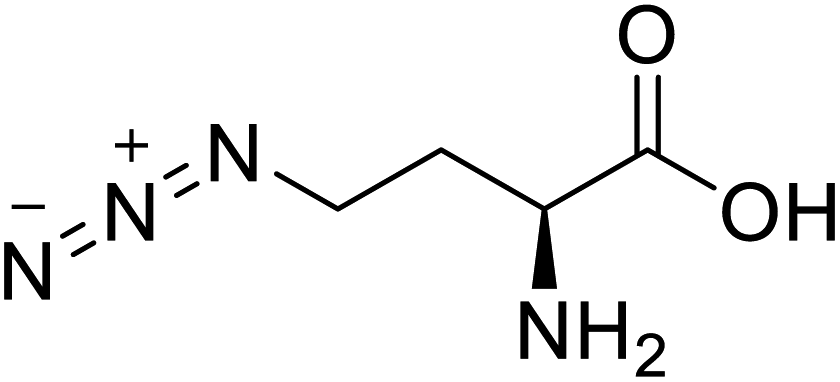 |
Biological synthesis | Investigate more engineering approaches and metabolic pathways | Biosynthesis of other pharmaceutically valuable UUAs | 125 |
| p-Azidomethyl-L-phenylalanine | 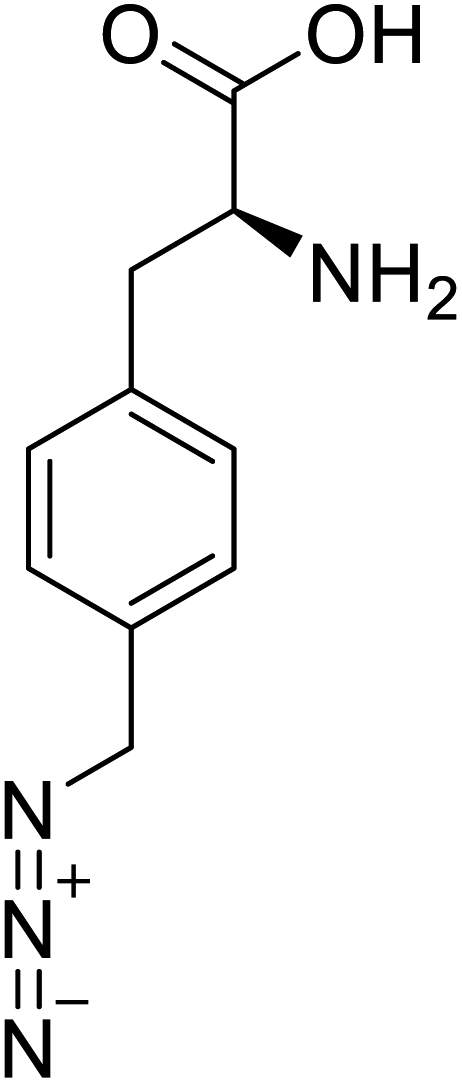 |
Chemical synthesis | Antibody drug conjugates | Part of antibody-drug conjugates | 93 |
| O-[18F]-Fluoroethyl-L-tyrosine | 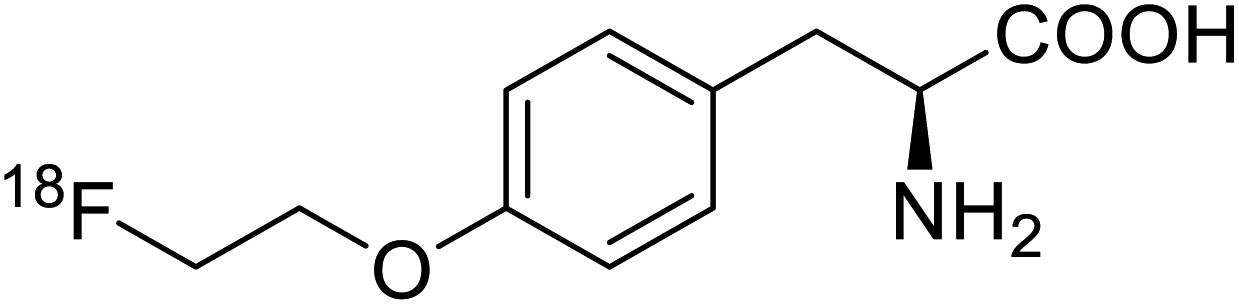 |
Radiochemical synthesis | Tumor imaging agent for PET | Radiolabeled tracer | 74 |
| 18F-FDOPA | 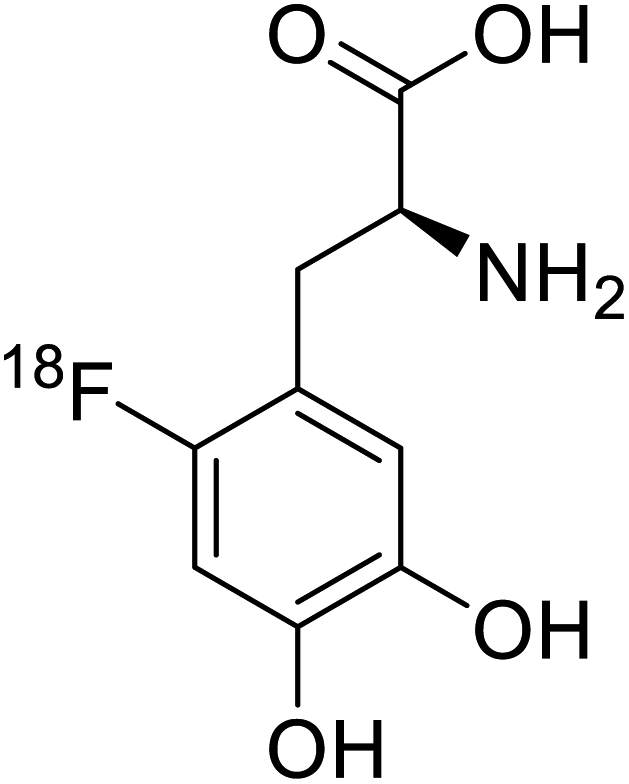 |
Chemical synthesis | Tumor imaging agent, PET | Radiolabeled tracer | 100 |
| 18F-Fluciclovine | 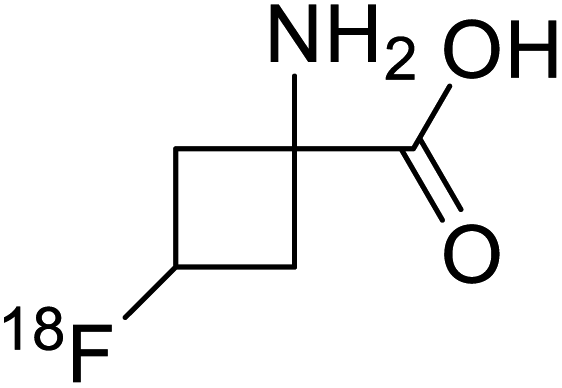 |
Chemical synthesis | PET tumor imaging agent | Radiolabeled tracer | 101 |
| L-(7-Hydroxycoumarin-4-yl)ethylglycine | 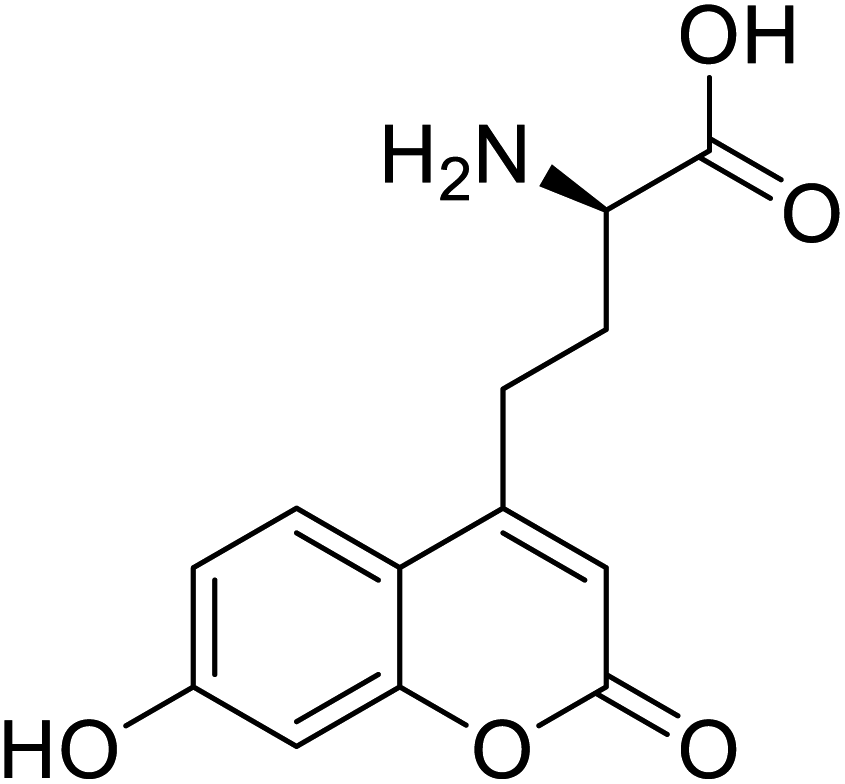 |
Chemical synthesis | Preparation of fluorescently labeled protein | Incorporated to improve catalytic activity | 126 |
| N-Acetylated fluorophenylalanine | 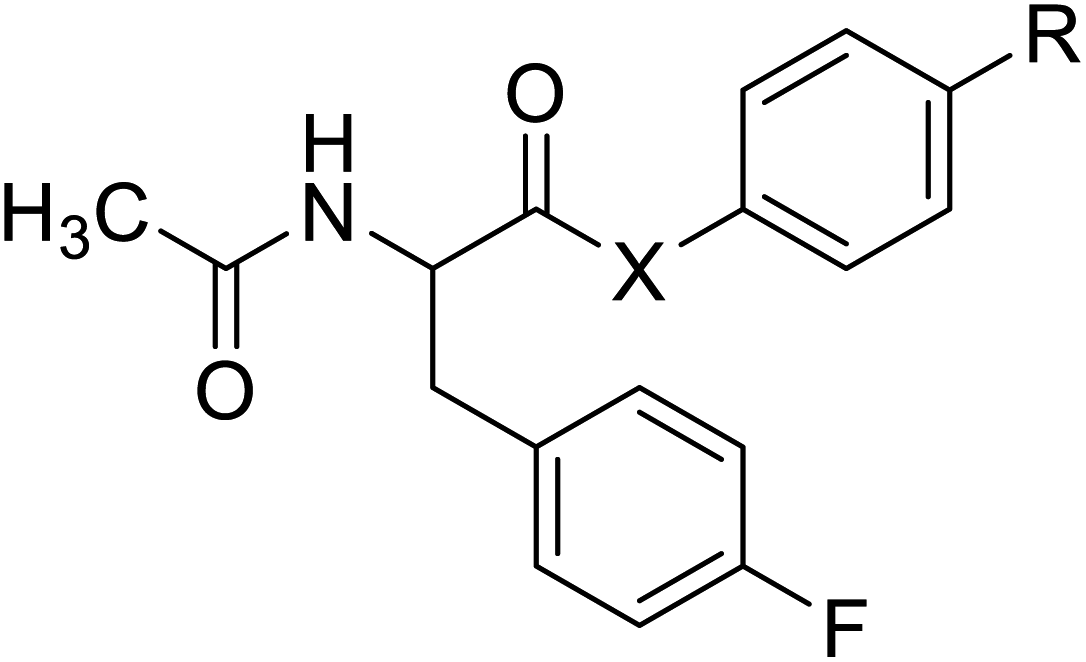 |
Chemical synthesis | Antiviral, anticancer drug conjugates | Part of antibody-drug conjugates | 127 |
| 4-Fluoroproline | 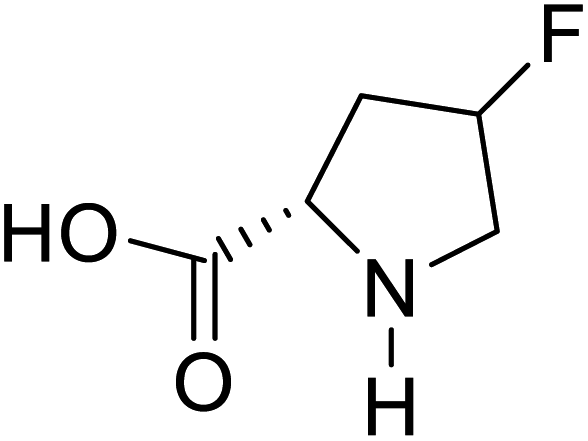 |
Chemical synthesis | Protein design and engineering | Enhances conformational stability upon proteins | 128 |
| Sarcosine (N-methyl-α-L-glycine) | 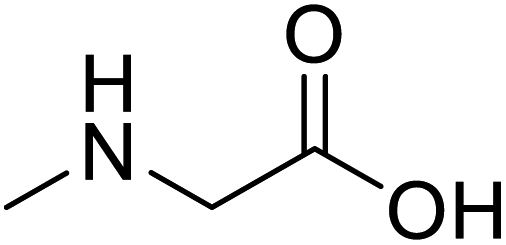 |
Biocatalytic synthesis | Used as a dietary supplement and as a non-specific glycine transport | Building blocks for the pharmaceutical | 73 |
| N,N-Dimethylglycine | 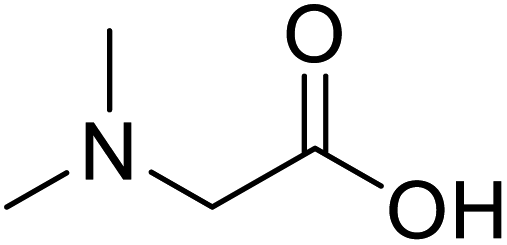 |
Biocatalytic synthesis | Acts as an athletic performance enhancer, displays anticonvulsant activity inhibitor | Building blocks for the pharmaceutical | 73 |
| Betaine (glycine betaine) | 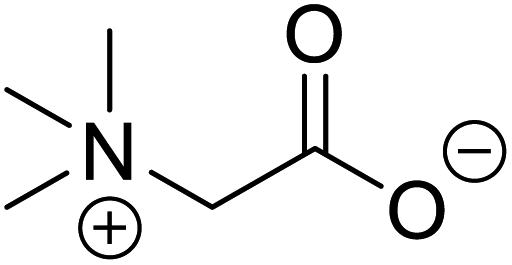 |
Biocatalytic synthesis | Treatment for homocystinuria and can offer benefits to human health, attenuation of liver injury | Display antioxidant and antihistaminic activity | 73 |
3.3 Antibody-drug conjugates
Antibody-drug conjugates (ADCs) have the potential to become the next generation of cancer therapeutics. UAAs are widely used in ADCs and result in better therapeutics due to their unique orthogonal coupling, site-specificity, and reduced toxicity. Among many UAAs, p-acetyl phenylalanine has been widely used in ADCs. Incorporating pAcF in human growth hormone (hGH) at specific locations allowed site-specific polyethylene glycol (PEG) conjugation to produce variants of hGH that are more effective and safer than hGh produced by mammalian cells as demonstrated in clinical trials of GH deficient adults.89 Also, when p-AcF was site-specifically incorporated into anti-CD11a IgG and then conjugated into aminooxy-modified LXR agonists, an ADC that demonstrated three-fold higher activation in humans THP-1 monocyte/macrophage cells in vitro than conventional LXR agonists have resulted.90 Thus produced ADC could be a better therapeutic agent for atherosclerosis. Following a similar strategy, conjugation of human antibody CD11 to highly potent PDE4 inhibitors resulted in a novel ADC with better therapeutic potential as an anti-inflammatory drug due to improved drug specificity.91Genetically encoded ncAA with orthogonal chemical reactivity are used for the synthesis of ADCs. Such produced ADCs have precise control over linkage site and stoichiometry, facilitating optimization of ADCs as therapeutic carriers and coupling agents.92 Another ADC with greater effect in vitro cell cytotoxic assays was produced through strain-promoted alkyne–azide cycloaddition (SPACC) copper-free click chemistry when unnatural amino acid, para-azidomethyl-L-phenylalanine (pAmF), was site-specifically incorporated, facilitating almost complete conjugation of DBCO-PEG-MMEF drug to the tumour-specific Her2-binding IgG trastuzumab.93
These days the main challenge in applying peptide therapeutics is their relatively short plasma half-life and the corresponding in vivo stability.94 A new molecule is thus introduced, called cell-penetrating peptides (CPPs), a short-chain of 5–30 amino acid residues that are widely used for the effective intracellular transportation of huge varieties of biologically active drug molecules, drug delivery vectors as well as cargos via biological membranes.95 CPPs can be a crucial aspect of therapeutic vaccines and drugs. A bio-reducible CPP, a branched m-R9 (B-mR9), was synthesized from nona-arginine (mR9) modification using disulfide bonds whose unusual polyplex B-mR9/siVEGF efficiently inhibited tumour sites and tumour growth as well as showed high accumulation and strong retention in comparison to other groups.96 Some of the restricted secondary structures of the peptides due to the presence of UAAs make them enzyme resistant compared to those containing natural amino acids.60 This greatly benefits the strength of CPPs.
3.4 Imaging agent
Positron emission tomography (PET) is an imaging technique where radioactive tracers scan metabolic and physiological activities. The UAA O-[18F]-fluoromethyl-L-tyrosine acts as a superior PET imaging agent for tumour detection and cancer imaging by amino acid transport97 as well as for differentiating tumour and inflamed tissues.98 This method is found more effective than conventional imaging techniques such as MRI. The [18F]-fluoro-ethyl-tyrosine PET shows high accuracy in diagnosing brain tumour patients and glioma in early stages, and it is also efficient in differentiating low grade from high-grade tumours.99 Radiolabeled amino acids are quickly becoming a choice as PET tracers because normal brain tissues poorly absorb such radiolabeled amino acids. Thus, brain tumours could be differentiated from normal brain tissues with high contrast.18F-FDOPA(6-[18F]-fluoro-L-3,4-dihydroxyphenylalanine) based PET/CT imaging was found to be very sensitive to post-treatment diagnosis of head and neck paragangliomas (HNPGLs),100 and diagnosis of pheochromocytoma-paraganglioma induced tumour.99 These treatments are found to be more effective than conventional MRI/CT by many such experimental trials. It could be a useful tool in the diagnosis of many neuroendocrine tumours. 18F-Fluciclovine (anti-1-amino-3-18F-fluorocyclobutane-1-carboxylic acid (FACBC)) PET/CT is an effective tool for imaging breast cancers even malignant ones such as invasive ductal carcinoma (IDC) and invasive lobular carcinoma (ILC), and also detection of extra-axillary nodular metastases.101 It also distinguishes between benign tissues and malignant tumours.102,103 PET/CT scans have been proved to be up-and-coming tools for imaging and a more precise diagnosis of nodal disorders in preclinical investigations. These studies imply that many of the flaws in traditional diagnostic approaches may be overcome.
3.5 Localization of proteins
Fluorescent UAAs have opened an efficient way of studying protein functions, conformational changes, and their localizations. Unnatural fluorescent amino acids (FIAAs) can label macromolecules fluorescently without altering their biomolecular properties, thereby facilitating the study of complex molecular processes.104A coumarin-based fluorescent amino acid (S)-1-carboxy-3-(7-hydroxy-2-oxo-2H-chromen-4-yl)propan-1-aminium (CouAA) was incorporated into E. coli FtsZ using an evolved Mj tRNATyr/TyrRS pair for the visualization of in vivo localization of particular proteins.105 Tryptophan is the most widely used amino acid as a fluorescent reporter for studying protein dynamics and functions. It has high sensitivity towards the electrostatic environment and greater quantum field.106 Various modifications are performed on tryptophan to synthesize more suitable and better-functioning fluorophores. A deep blue coloured tryptophan analogue, beta-(1-azulenyl)-L-alanine, having characteristic fluorescent and photophysical properties, was synthesized using Neighisi coupling. Upon incorporating this UAA into argyrin C, the group showed great potential in the localization and visualization of targeted proteins.107 Another tryptophan analogue, 4CN-Trp being a small fluorescent UAA possessing unique photochemical properties, is a potential candidate for its use in vitro and in vivo spectroscopic and microscopic studies of proteins.108
3.6 Antimicrobial peptides
Antimicrobial peptides (AMP) are amphipathic and cationic molecules with 10–50 amino acid residues that constitute key parts of the innate immune system and are considered potential alternatives to antibiotics as new therapeutic agents. To address these restrictions, certain changes have been made, and these include the use of peptidomimetic backbones,109 technical modification110 truncations from parental peptides, and incorporation of UAAs.109,111Due to dominating activity that is ineffective and low serum stability, systematic Cbf-14 modification by incorporating UAAs (ornithine [Orn], norleucine [Ile], and D-amino acids) results in a positively charged peptide mutant Cbf-14-2 with similar stability, negligible toxicity, and superior antimicrobial activities against penicillin-resistant bacteria both in vivo/in vitro.112 An in vitro study hinted toward a selective anti-cancer effect of Cbf-K16 against human non-small cell lung carcinoma H460 cells.113 Antibacterial peptides (ABPs) incorporated with UAAs (uABPs) are essential for host defence against microbial infections.
UAAs can be utilized to improve bio-redox processes. For example, the abiotic nicotinamide dinucleotide flucytosine cofactor can assist in identifying protein structures and modifying them after translation.114 A novel medication type, which can replace or be an alternative to treatments now utilized, has been found by UAAs synthesizers with its unique advantages. Further therapeutic research uses UAAs to aid unriddle emerging and unsolved pathogenic diseases.
3.7 Probes for changes in protein conformation
The ability to insert UAAs into proteins on a site-specific basis has significantly impacted protein engineering. In 2020, according to Dangerfield, although bacteriophage T7 DNA polymerase undergoes significant substrate-induced conformational changes that are considered to account for high replication fidelity, previous studies were hampered by mutations necessary to create a Cys-lite variation for site-specific fluorescence labelling. Dangerfield and Johnson employed orthogonal amber suppression machinery in E. coli to optimize the direct integration of fluorescent artificial amino acid (7-hydroxy-4-coumarin-yl)-ethylglycine.116 While screening a large number of random natural variants, site-directed incorporation of L-(7-hydroxycoumarin-4-yl)ethylglycine UAA into a bacterial phosphotriesterase improved catalytic activity significantly as compared to any natural mutation.117 Several UAAs have been introduced at a similar range of sites in dozens of proteins, demonstrating the generality of the nonsense suppression method. The majority of applications have used caged side chains, where an o-nitrobenzyl group shields a heteroatom. Photolysis eliminates the nitrobenzyl after a UAA is incorporated into a protein with caged side chains, revealing the previously caged functionality.118 The use of genetic code expansion to site-specifically incorporate UAAs into POIs is a potent option. The amber codon suppression approach can be used to integrate various functional groups such as azides, alkynes, alkenes, and tetrazines, which are then changed using appropriate chemicals. For example, integrated UAAs azides or alkynes can conduct a copper-catalysed azide–alkyne cycloaddition (CuAAC).119 When calcium binds to calmodulin protein (CaM), conformational changes occur, allowing Ca2+/CaM to recognize and bind a variety of target proteins with high affinity. Incorporation of p-azido-phenylalanine (AzF) in various positions of CaM, followed by infrared (IR)-spectroscopy of the azido stretching vibration. It distinguishes between distinct binding motifs based on the azido stretching modes spectrum properties.120 The use of vibrational probes containing unnatural amino acids with distinct altered side chains (e.g., nitrile, thiocyanate, azide) has substantially increased the application of vibrational spectroscopies to investigate local dynamics and conformational changes with residue-specific resolution. The stretching vibrations of the ester carbonyl side chain of numbers of UAAs is not only localized, but its frequency also varies linearly with an electrostatic field in both hydrogen bonding and non-hydrogen bonding environments.121 The addition of unnatural moieties, including fluorophores, affinity labels, spin-label probes, isotope, and bioorthogonal functional groups, introduced to proteins for several in vitro and in vivo processes and potential applications of such fusion proteins in molecular imaging.122 Genetic incorporation of UAA such as ketone-bearing amino acid p-acetyl phenylalanine (pAcPhe) into recombinantly expressed proteins has provided an elegant alternative to cysteine-based labelling methods where traditional thiol-based SDSL is not feasible.1234. Conclusions, limitations, and future perspectives
UAAs are non-proteinogenic amino acids that can be found in nature or can be synthesized chemically. Admittedly, the biological synthesis of UAAs has been economically feasible for large-scale production than the chemical synthesis. They are commonly utilized as blocks of chiral construction. Though D- or aberrant amino acids can be utilized to replace L-amino acids in physiologically active peptides, too much modification or replacement can cause cytotoxicity, immunogenicity, and other problems.Whereas UAA incorporation into proteins, being a powerful tool to modify the properties of proteins, this approach is becoming more attractive for biotechnological uses. Some biotechnological tools include molecular imaging probes, protein–protein interaction probes, therapeutic proteins, enzymes, fluorescent probes, biocontainment, biomaterials, and biocatalysts with novel functional and structural properties which can be prepared via UAA incorporation into proteins. Although the results obtained are eye-catching and promising, the potential is still far from achieved. It has been demonstrated that methyl ester form of UAAs against free acid form showed much better yield; the improved yield is possibly due to the improved membrane permeability of the methyl ester forms of the UAAs to E. coli cells as compared to the free acid forms, even though the same tRNA synthetases and E. coli strains were used for incorporation.129 A study in 2020 optimized the direct incorporation of a fluorescent UAA (7-hydroxy-4-coumarin-yl)-ethyl glycine, where MS methods confirm that the UAA is introduced at only one site and that there is limited precedent.116
The biochemical implications of altering the cellular translation machinery, as employed in GCE-based tagging, have yet to be extensively explored. First, cells were not examined or had an impact on ribosomal function in the cellular response to an exogene tRNA/tRNA synthetase combination. Second, the physiological effects of protein reading induced by UAAs are not yet fully examined in TAG endogenous codons.130 The integration of UAAs into proteins has been an area of active interest for several fundamental and applicable research sciences. Despite the various types of synthetic UAAs, due to the limited number of studies available, further studies are highly recommended. In the field of amino acids and peptide/protein chemistry, directing groups (e.g., 8-aminoquinoline) and protecting groups (e.g., Phth) are uncommon. The introduction and removal of these protecting and directing groups necessitate additional steps, lowering the atom and phase efficiency of the modification process. Even with the aforementioned current challenges, we predict that C–H functionalization would become a cost-effective solution for synthesizing many UAAs as the repertoire of the C–H functionalization technique grows.
E. coli is primarily utilized for large-scale protein synthesis. On the other hand, this prokaryotic workhorse is less frequent and incapable of producing post-translational modifications, including glycosylation, ubiquitination, phosphorylation, and other eukaryotic and proteolytic protein maturation. Furthermore, eukaryotic and mammalian cell systems are prone to contamination, frequently requiring specific growth sites and bypassing or disabling glycosylation systems to produce humanized therapeutic proteins without introducing additional factors that may induce immunogenicity. Both stationary phase biology and post-translational modifications in prokaryotic systems are still active research areas with many unanswered questions. Synthesizing appropriate UAAs and assembling UAAs and scaffolds to produce functional catalysts are distinct synthetic problems that cannot be overlooked or obscured by similar research aimed at uses other than catalysis. The absence of library techniques for iterative scaffold optimization is perhaps the biggest roadblock to creating hybrid catalysts that use UAAs.131
When acidic conditions were adopted for deprotection at the oxazolidinone centre, 96 percent of the free amino acid was produced. In addition, oxazolidinone species I and II may be easily derived to get more complex UAAs. Control data indicates that to introduce alkylation reactions under normal circumstances, there should not be the presence of any light or photocatalyst.52 Recent primary and secondary C(sp3)–H bond functionalization processes have selectively changed natural amino acids and peptides. Sequential C–H activation and C–X (X = C, B, O, N, F, etc.) bond formation reactions have successfully changed the side chains of amino acids. Even though substantial progress has been made in this area, there are still several obstacles to overcome. The reactivity of unactivated C(sp3)–H bonds, especially secondary C(sp3)–H bonds, is still low, which can be overcome by strategically developing new ligands and directing groups.132 ncAA-mediated methods broaden the range of things identified and generated, such as conjugates, vaccinations, and cell-based treatments. It is also becoming clear how important it is to apply medicinal chemistry ideas to bigger proteins, i.e., protein medicinal chemistry. Given the potency of channels for discovering therapies that employ just canonical amino acids, granting them full access to the chemical cabinet would very definitely result in totally new therapeutic groupings.131
The asymmetric synthesis of UAAs is a significant step to incorporate optically pure UAAs in natural proteins during the reprogramming of natural proteins by using UAAs. Therefore, an extensive study on enantioselective, catalytic, and chiral pool synthesis of optically pure UAAs has been reported in recent years.43–51 Further study in the design of novel catalysts and chiral auxiliaries, as well as proper selection of chiral precursors for chiral pool synthesis, could enhance the synthesis of chiral UAAs with an excellent enantiomeric excess (ee) and high yield. Consideration of the green chemistry approach towards the asymmetric synthesis of UAAs is highly recommended.
There are a limited number of UAAs in the current defined metabolic pathways, and in some cases, essential pieces of information are missing, including the enzymes involved. During the biosynthesis process, some UAAs inhibit the growth of chassis strain and may cause trouble in fermentation processes. Hence, the prospect needs the involvement of crucial enzyme representation and clear metabolic pathways. For balanced metabolism and increased resistance, more chassis strain is needed to be metabolically engineered or screened. After examining secondary structures of polypeptides containing proteinogenic amino acid residues, it isn't easy to anticipate the conformation of contemporary, non-natural polypeptide materials based on their side-chain structures. To elucidate their effects on secondary structures, fundamental understandings of side-chain interactions, such as charge, H-bonding, hydrophobic, and dipole–dipole interactions, are needed. Ultimately, synthetic polypeptides can be used as standalone therapeutics in addition to using polypeptide products as nano-carriers. Glatiramer acetate (GA), a statistical copolypeptide with four natural amino acid residues, is one noteworthy example.133
The full promise of protein medicinal chemistry and other ncAA applications in biotherapeutics will be realized only when high-performance ncAA integration systems in bacterial and eukaryotic expression systems are available. Given the strength of biological platforms using only canonical amino acids, full access to the chemical cabinet provides these platforms with completely new therapeutic classes.134 Future research should concentrate on reaction designation, enzyme property enhancement, and process optimization to develop commercially feasible platforms for derivatizing amino acids. Biocatalysts will likely be used to manufacture additional fine chemicals from amino acids on a commercial scale in the future.135 Scopes of UAAs in biotechnology, synthetic biology, and traditional tools should be upgraded by well-designed algorithms and computer-aided enzyme/protein molecular prediction, which supports the practical implementation of UAAs and helps to design different artificial enzymes.
Author contributions
A. Adhikari, B. R. Bhattarai, A. Aryal, N. Thapa, P. KC, Ashma Adhikari, and S. Maharjan reviewed the literature. P. B. Chanda, and B. P. Regmi executed critical revision. A. Adhikari, B. R. Bhattarai, and N. Parajuli drafted the manuscript. All authors accepted the submission for publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Mr Uttam Lamichhane, Mr Sijan Gautam, and Mr Sandeep Thapa for providing constructive suggestions to prepare this manuscript.Notes and references
- I. o. M. U. C. o. M. Nutrition, The Role of Protein and Amino Acids in Sustaining and Enhancing Performance, National Academies Press, US, 1999 Search PubMed.
- T. S. Young and P. G. Schultz, J. Biol. Chem., 2010, 285, 11039–11044 CrossRef CAS PubMed.
- F. Agostini, J.-S. Völler, B. Koksch, C. G. Acevedo-Rocha, V. Kubyshkin and N. Budisa, Angew. Chem., Int. Ed., 2017, 56, 9680–9703 CrossRef CAS PubMed.
- T. Narancic, S. A. Almahboub and K. E. O'Connor, World J. Microbiol. Biotechnol., 2019, 35, 67 CrossRef PubMed.
- W. H. Zhang, G. Otting and C. J. Jackson, Curr. Opin. Struct. Biol., 2013, 23, 581–587 CrossRef CAS PubMed.
- L. Davis and J. W. Chin, Nat. Rev. Mol. Cell Biol., 2012, 13, 168–182 CrossRef CAS PubMed.
- I. Drienovská and G. Roelfes, Nat. Catal., 2020, 3, 193–202 CrossRef.
- M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS PubMed.
- B. L. Zerfas, R. A. Coleman, A. F. Salazar-Chaparro, N. J. Macatangay and D. J. Trader, ACS Chem. Biol., 2020, 15, 2588–2596 CrossRef CAS PubMed.
- M. Chartrain, P. M. Salmon, D. K. Robinson and B. C. Buckland, Curr. Opin. Biotechnol., 2000, 11, 209–214 CrossRef CAS PubMed.
- K. Zhang, H. Li, K. M. Cho and J. C. Liao, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6234–6239 CrossRef CAS PubMed.
- J.-M. Xu, J.-Q. Li, B. Zhang, Z.-Q. Liu and Y.-G. Zheng, Microb. Cell Fact., 2019, 18, 43 CrossRef PubMed.
- T. Shibasaki, S. Hashimoto, H. Mori and A. Ozaki, J. Biosci. Bioeng., 2000, 90, 522–525 CrossRef CAS PubMed.
- K.-B. Park and S.-H. Oh, Biotechnol. Lett., 2006, 28, 1459–1463 CrossRef CAS PubMed.
- Y. Ma, H. Biava, R. Contestabile, N. Budisa and M. L. Di Salvo, Molecules, 2014, 19, 1004–1022 CrossRef PubMed.
- N. D. Ratnayake, C. Theisen, T. Walter and K. D. Walker, J. Biotechnol., 2016, 217, 12–21 CrossRef CAS PubMed.
- N. Anderhuber, P. Fladischer, M. Gruber-Khadjawi, J. Mairhofer, G. Striedner and B. Wiltschi, J. Biotechnol., 2016, 235, 100–111 CrossRef CAS PubMed.
- E. M. Brustad and F. H. Arnold, Curr. Opin. Chem. Biol., 2011, 15, 201–210 CrossRef CAS PubMed.
- J. Rudat, B. R. Brucher and C. Syldatk, AMB Express, 2012, 2, 11 CrossRef PubMed.
- Y.-P. Xue, C.-H. Cao and Y.-G. Zheng, Chem. Soc. Rev., 2018, 47, 1516–1561 RSC.
- P. P. Taylor, D. P. Pantaleone, R. F. Senkpeil and I. G. Fotheringham, Trends Biotechnol., 1998, 16, 412–418 CrossRef CAS PubMed.
- R. Tao, Y. Jiang, F. Zhu and S. Yang, Biotechnol. Lett., 2013, 36, 835–841 CrossRef PubMed.
- D. Zhang, X. Chen, R. Zhang, P. Yao, Q. Wu and D. Zhu, ACS Catal., 2015, 5, 2220–2224 CrossRef CAS.
- R.-F. Cai, L. Liu, F.-F. Chen, A. Li, J.-H. Xu and G.-W. Zheng, ACS Sustainable Chem. Eng., 2020, 8, 17054–17061 CrossRef CAS.
- B.-K. Cho, J.-H. Seo, T.-W. Kang and B.-G. Kim, Biotechnol. Bioeng., 2003, 83, 226–234 CrossRef CAS PubMed.
- J.-E. Jung, S. Y. Lee, H. Park, H. Cha, W. Ko, K. Sachin, D. W. Kim, D. Y. Chi and H. S. Lee, Chem. Sci., 2014, 5, 1881–1885 RSC.
- M. Senuma, K. Nakamichi, K. Nabe, S. Nishimoto and T. Tosa, Appl. Biochem. Biotechnol., 1989, 22, 141–150 CrossRef CAS.
- J. Meiwes, M. Schudok and G. Kretzschmar, Tetrahedron: Asymmetry, 1997, 8, 527–536 CrossRef CAS.
- K. Bartsch, R. Dichmann, P. Schmitt, E. Uhlmann and A. Schulz, Appl. Environ. Microbiol., 1990, 56, 7–12 CrossRef CAS PubMed.
- L. Martínková and V. Kren, Curr. Opin. Chem. Biol., 2010, 14, 130–137 CrossRef PubMed.
- H. K. Chenault, J. Dahmer and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 6354–6364 CrossRef CAS.
- F. Parmeggiani, N. J. Weise, S. T. Ahmed and N. J. Turner, Chem. Rev., 2018, 118, 73–118 CrossRef CAS PubMed.
- Y. Li and M.-H. Xu, Org. Lett., 2012, 14, 2062–2065 CrossRef CAS PubMed.
- A. Stevenazzi, M. Marchini, G. Sandrone, B. Vergani and M. Lattanzio, Bioorg. Med. Chem. Lett., 2014, 24, 5349–5356 CrossRef CAS PubMed.
- V. R. Arava, S. R. Amasa, B. K. G. Bhatthula, L. S. Kompella, V. P. Matta and M. C. S. Subha, Synth. Commun., 2013, 43, 2892–2897 CrossRef CAS.
- B. Aillard, N. S. Robertson, A. R. Baldwin, S. Robins and A. G. Jamieson, Org. Biomol. Chem., 2014, 12, 8775–8782 RSC.
- B. Li, J. Zhang, Y. Xu, X. Yang and L. Li, Tetrahedron Lett., 2017, 58, 2374–2377 CrossRef CAS.
- Z. Wang, D. Huang, P. Xu, X. Dong, X. Wang and Z. Dai, Tetrahedron Lett., 2015, 56, 1067–1071 CrossRef CAS.
- T. Inokuma, Yakugaku Zasshi, 2018, 138, 1371–1379 CrossRef CAS PubMed.
- N. C. Dwulet, T. A. Zolfaghari, M. L. Brown and J. S. Cannon, J. Org. Chem., 2018, 83, 11510–11518 CrossRef CAS PubMed.
- H. Zhang, H. Li, H. Yang and H. Fu, Org. Lett., 2016, 18, 3362–3365 CrossRef CAS PubMed.
- J. Yang, Z. Wang, Z. He, G. Li, L. Hong, W. Sun and R. Wang, Angew. Chem., Int. Ed., 2020, 59, 642–647 CrossRef CAS PubMed.
- Q. Zhu, B. Meng, C. Gu, Y. Xu, J. Chen, C. Lei and X. Wu, Org. Lett., 2019, 21, 9985–9989 CrossRef CAS PubMed.
- A. J. Bendelsmith, S. C. Kim, M. Wasa, S. P. Roche and E. N. Jacobsen, J. Am. Chem. Soc., 2019, 141, 11414–11419 CrossRef CAS PubMed.
- S.-S. Meng, W.-B. Tang and W.-H. Zheng, Org. Lett., 2018, 20, 518–521 CrossRef CAS PubMed.
- F. Zhang, H. Sun, Z. Song, S. Zhou, X. Wen, Q.-L. Xu and H. Sun, J. Org. Chem., 2015, 80, 4459–4464 CrossRef CAS PubMed.
- J.-E. Hoffmann, D. Dziuba, F. Stein and C. Schultz, Biochemistry, 2018, 57, 4747–4752 CrossRef CAS PubMed.
- K. Boknevitz, J. S. Italia, B. Li, A. Chatterjee and S.-Y. Liu, Chem. Sci., 2019, 10, 4994–4998 RSC.
- T. J. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan and M. C. White, Nature, 2016, 537, 214–219 CrossRef CAS PubMed.
- W. Vuong, F. Mosquera-Guagua, R. Sanichar, T. R. McDonald, O. P. Ernst, L. Wang and J. C. Vederas, Org. Lett., 2019, 21, 10149–10153 CrossRef CAS PubMed.
- M. Jiang, Y. Jin, H. Yang and H. Fu, Sci. Rep., 2016, 6, 26161 CrossRef CAS PubMed.
- K. Merkens, F. J. A. Troyano, J. Djossou and A. Gómez-Suárez, Adv. Synth. Catal., 2020, 362, 2354–2359 CrossRef CAS.
- A. Gupta, B. P. Garreffi and M. Guo, Chem. Commun., 2020, 56, 12578–12581 RSC.
- D. D. Young, J. Torres-Kolbus and A. Deiters, Bioorg. Med. Chem. Lett., 2008, 18, 5478–5480 CrossRef CAS PubMed.
- D. Astruc, Anal. Bioanal. Chem., 2011, 399, 1811–1814 CrossRef CAS PubMed.
- M. Sabat and C. R. Johnson, Org. Lett., 2000, 2, 1089–1092 CrossRef CAS PubMed.
- A. S. Saghyan, A. F. Mkrtchyan, Z. Z. Mardiyan, L. A. Hayriyan, Y. N. Belokon and P. Langer, ChemistrySelect, 2019, 4, 4686–4688 CrossRef CAS.
- W. D. G. Brittain and S. L. Cobb, Org. Biomol. Chem., 2017, 16, 10–20 RSC.
- W. Gao, E. Cho, Y. Liu and Y. Lu, Front. Pharmacol., 2019, 10, 611 CrossRef CAS PubMed.
- Y. Ravikumar, S. P. Nadarajan, T. H. Yoo, C. Lee and H. Yun, Trends Biotechnol., 2015, 33, 462–470 CrossRef CAS PubMed.
- S. A. Costa, D. Mozhdehi, M. J. Dzuricky, F. J. Isaacs, E. M. Brustad and A. Chilkoti, Nano Lett., 2019, 19, 247–254 CrossRef CAS PubMed.
- K.-H. Lee, C. Catherine and D.-M. Kim, J. Ind. Eng. Chem., 2016, 37, 90–94 CrossRef CAS.
- J. Adachi, K. Katsura, E. Seki, C. Takemoto, M. Shirouzu, T. Terada, T. Mukai, K. Sakamoto and S. Yokoyama, Int. J. Mol. Sci., 2019, 20, 492 CrossRef CAS PubMed.
- T. Kimmerlin and D. Seebach, J. Pept. Res., 2005, 65, 229–260 CrossRef CAS PubMed.
- M. Koprowska-Ratajska, A. Kluczyk, P. Stefanowicz, H. Bartosz-Bechowski and Z. Szewczuk, Amino Acids, 2008, 36, 309 CrossRef PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- Z. Cui, S. Mureev, M. E. Polinkovsky, Z. Tnimov, Z. Guo, T. Durek, A. Jones and K. Alexandrov, ACS Synth. Biol., 2017, 6, 535–544 CrossRef CAS PubMed.
- J. N. Kolev, J. M. Zaengle, R. Ravikumar and R. Fasan, ChemBioChem, 2014, 15, 1001–1010 CrossRef CAS PubMed.
- M. G. Hoesl, C. G. Acevedo-Rocha, S. Nehring, M. Royter, C. Wolschner, B. Wiltschi, N. Budisa and G. Antranikian, ChemCatChem, 2011, 3, 213–221 CrossRef CAS.
- K. Deepankumar, S. P. Nadarajan, S. Mathew, S.-G. Lee, T. H. Yoo, E. Y. Hong, B.-G. Kim and H. Yun, ChemCatChem, 2015, 7, 417–421 CrossRef CAS.
- I. Nikić, G. EstradaGirona, J. H. Kang, G. Paci, S. Mikhaleva, C. Koehler, N. V. Shymanska, C. Ventura Santos, D. Spitz and E. A. Lemke, Angew. Chem., Int. Ed., 2016, 55, 16172–16176 CrossRef PubMed.
- E. Park, M. Kim and J.-S. Shin, Adv. Synth. Catal., 2010, 352, 3391–3398 CrossRef CAS.
- J. F. Hyslop, S. L. Lovelock, A. J. B. Watson, A. J. B. Hutchins, P. W. Sutton and G.-D. Roiban, J. Biotechnol., 2019, 293, 56–65 CrossRef CAS PubMed.
- J. Claesen and M. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16297–16302 CrossRef CAS PubMed.
- H. Fu, A. Prats Luján, L. Bothof, J. Zhang, P. G. Tepper and G. J. Poelarends, ACS Catal., 2019, 9, 7292–7299 CrossRef CAS.
- J.-M. Choi, S.-S. Han and H.-S. Kim, Biotechnol. Adv., 2015, 33, 1443–1454 CrossRef CAS PubMed.
- K. Deepankumar, N. S. Prabhu, J.-H. Kim and H. Yun, Biotechnol. Bioprocess Eng., 2017, 22, 248–255 CrossRef CAS.
- A. Chatterjee, S. B. Sun, J. L. Furman, H. Xiao and P. G. Schultz, Biochemistry, 2013, 52, 1828–1837 CrossRef CAS PubMed.
- K. Deepankumar, M. Shon, S. P. Nadarajan, G. Shin, S. Mathew, N. Ayyadurai, B.-G. Kim, S.-H. Choi, S.-H. Lee and H. Yun, Adv. Synth. Catal., 2014, 356, 993–998 CrossRef CAS.
- B. Holzberger and A. Marx, J. Am. Chem. Soc., 2010, 132, 15708–15713 CrossRef CAS PubMed.
- P. C. Cirino, Y. Tang, K. Takahashi, D. A. Tirrell and F. H. Arnold, Biotechnol. Bioeng., 2003, 83, 729–734 CrossRef CAS PubMed.
- J. N. Kolev, J. M. Zaengle, R. Ravikumar and R. Fasan, ChemBioChem, 2014, 15, 1001–1010 CrossRef CAS PubMed.
- W. Xuan, J. Li, X. Luo and P. G. Schultz, Angew. Chem., Int. Ed., 2016, 55, 10065–10068 CrossRef CAS PubMed.
- C. L. Windle, K. J. Simmons, J. R. Ault, C. H. Trinh, A. Nelson, A. R. Pearson and A. Berry, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2610–2615 CrossRef CAS PubMed.
- Y. A. Votchitseva, E. N. Efremenko and S. D. Varfolomeyev, Russ. Chem. Bull., 2006, 55, 369–374 CrossRef CAS.
- N. Budisa, W. Wenger and B. Wiltschi, Mol. BioSyst., 2010, 6, 1630–1639 RSC.
- I. N. Ugwumba, K. Ozawa, Z.-Q. Xu, F. Ely, J.-L. Foo, A. J. Herlt, C. Coppin, S. Brown, M. C. Taylor, D. L. Ollis, L. N. Mander, G. Schenk, N. E. Dixon, G. Otting, J. G. Oakeshott and C. J. Jackson, J. Am. Chem. Soc., 2011, 133, 326–333 CrossRef CAS PubMed.
- A. F. Nahhas, R. Chang and T. J. Webster, J. Biomed. Nanotechnol., 2018, 14, 987–993 CrossRef CAS PubMed.
- H. Cho, T. Daniel, Y. J. Buechler, D. C. Litzinger, Z. Maio, A.-M. H. Putnam, V. S. Kraynov, B.-C. Sim, S. Bussell, T. Javahishvili, S. Kaphle, G. Viramontes, M. Ong, S. Chu, B. Gc, R. Lieu, N. Knudsen, P. Castiglioni, T. C. Norman, D. W. Axelrod, A. R. Hoffman, P. G. Schultz, R. D. DiMarchi and B. E. Kimmel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9060–9065 CrossRef CAS PubMed.
- R. K. V. Lim, S. Yu, B. Cheng, S. Li, N.-J. Kim, Y. Cao, V. Chi, J. Y. Kim, A. K. Chatterjee, P. G. Schultz, M. S Tremblay and S. A. Kazane, Bioconjugate Chem., 2015, 26, 2216–2222 CrossRef CAS PubMed.
- S. Yu, A. D. Pearson, R. K. Lim, D. T. Rodgers, S. Li, H. B. Parker, M. Weglarz, E. N. Hampton, M. J. Bollong, J. Shen, C. Zambaldo, D. Wang, A. K. Woods, T. M. Wright, P. G. Schultz, S. A. Kazane, T. S. Young and M. S. Tremblay, Mol. Ther., 2016, 24, 2078–2089 CrossRef CAS PubMed.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed.
- E. S. Zimmerman, T. H. Heibeck, A. Gill, X. Li, C. J. Murray, M. R. Madlansacay, C. Tran, N. T. Uter, G. Yin, P. J. Rivers, A. Y. Yam, W. D. Wang, A. R. Steiner, S. U. Bajad, K. Penta, W. Yang, T. J. Hallam, C. D. Thanos and A. K. Sato, Bioconjugate Chem., 2014, 25, 351–361 CrossRef CAS PubMed.
- S. Schuster, B. Biri-Kovács, B. Szeder, L. Buday, J. Gardi, Z. Szabó, G. Halmos and G. Mező, Pharmaceutics, 2018, 10, 223 CrossRef CAS PubMed.
- S. Rehmani and J. E. Dixon, Peptides, 2018, 100, 24–35 CrossRef CAS PubMed.
- J. Yoo, D Lee, V. Gujrati, N. S. Rejinold, K. M. Lekshmi, S. Uthaman, C. Jeong, I.-K. Park and Y.-C. Kim, J. Controlled Release, 2017, 246, 142–154 CrossRef CAS PubMed.
- R. Iwata, S. Furumoto, C. Pascali, A. Bogni and K. Ishiwata, ChemCatChem, 2015, 7, 555–556 Search PubMed.
- M. Suzuki, K. Yamaguchi, G. Honda, R. Iwata, S. Furumoto, M. Jeong, H. Fukuda and M. Itoh, Ann. Nucl. Med., 2005, 2019, 589–595 CrossRef PubMed.
- I. Ibbett, G. Schembri, R. Cook and J. Parkinson, World Neurosurg., 2019, 121, e712–e715 CrossRef PubMed.
- C. Heimburger, F. Veillon, D. Taïeb, B. Goichot, S. Riehm, J. Petit-Thomas, G. Averous, M. Cavalcanti, F. Hubelé, G. Chabrier, I. J. Namer, A. Charpiot and A. Imperiale, Eur. J. Nucl. Med. Mol. Imaging, 2017, 44, 979–987 CrossRef PubMed.
- G. A. Ulaner, D. A. Goldman, M. Gönen, H. Pham, R. Castillo, S. K. Lyashchenko, J. S. Lewis and C. Dang, J. Nucl. Med., 2016, 57, 1350–1356 CrossRef CAS PubMed.
- M. Elschot, K. M. Selnæs, E Sandsmark, B. Krüger-Stokke, Ø. Størkersen, M.-B. Tessem, S. A. Moestue, H. Bertilsson and T. F. Bathen, Eur. J. Nucl. Med. Mol. Imaging, 2017, 44, 695–703 CrossRef CAS PubMed.
- F. I. Tade, M. A. Cohen, T. M. Styblo, O. A. Odewole, A. I. Holbrook, M. S. Newell, B. Savir-Baruch, X. Li, M. M. Goodman, J. A. Nye and D. M. Schuster, J. Nucl. Med., 2016, 57, 1357–1363 CrossRef CAS PubMed.
- Z. Cheng, E. Kuru, A. Sachdeva and M. Vendrell, Nat. Rev. Chem., 2020, 4, 275–290 CrossRef CAS.
- G. Charbon, E. Brustad, K. A. Scott, J. Wang, A. Løbner-Olesen, P. G. Schultz, C. Jacobs-Wagner and E. Chapman, ChemBioChem, 2011, 12, 1818–1821 CrossRef CAS PubMed.
- P. R. Callis, J. Mol. Struct., 2014, 1077, 22–29 CrossRef CAS.
- E. Stempel, R. F.-X. Kaml, N. Budisa and M. Kalesse, Bioorg. Med. Chem., 2018, 26, 5259–5269 CrossRef CAS PubMed.
- M. R. Hilaire, I. A Ahmed, C.-W. Lin, H. Jo, W. F. DeGrado and F. Gai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6005–6009 CrossRef CAS PubMed.
- M. Ahn, P. Gunasekaran, G. Rajasekaran, E. Y. Kim, S.-J. Lee, G. Bang, K. Cho, J.-K. Hyun, H.-J. Lee, Y. H. Jeon, N.-H. Kim, E. K. Ryu, S. Y. Shin and J. K. Bang, Eur. J. Med. Chem., 2017, 125, 551–564 CrossRef CAS PubMed.
- S. Azmi, S. Srivastava, N. N. Mishra, J. K. Tripathi, P. K. Shukla and J. K. Ghosh, J. Med. Chem., 2013, 56, 924–939 CrossRef CAS PubMed.
- L. M. Johnson, S. Barrick, M. V. Hager, A. McFedries, E. A. Homan, M. E. Rabaglia, M. P. Keller, A. D. Attie, A. Saghatelian, A. Bisello and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 12848–12851 CrossRef CAS PubMed.
- W. Kang, H. Liu, L. Ma, M. Wang, S. Wei, P. Sun, M. Jiang, M. Guo, C. Zhou and J. Dou, Eur. J. Pharm. Sci., 2017, 105, 169–177 CrossRef CAS PubMed.
- Y. Tian, H. Wang, B. Li, M. Ke, J. Wang, J. Dou and C. Zhou, Oncol. Rep., 2013, 30, 2502–2510 CrossRef CAS PubMed.
- X. Xu, H. Zhong, W. Liu and Y. Tao, Front. Bioeng. Biotechnol., 2020, 8, 145 CrossRef PubMed.
- P. J. Baker and J. K. Montclare, ChemBioChem, 2011, 12, 1845–1848 CrossRef CAS PubMed.
- T. L. Dangerfield and K. A. Johnson, J. Biol. Chem., 2020, 295, 17265–17280 CrossRef CAS PubMed.
- C. Jackson, S. P. Duffy, K. R. Hess and R. A. Mehl, J. Am. Chem. Soc., 2006, 128, 11124–11127 CrossRef PubMed.
- D. A. Dougherty, Curr. Opin. Chem. Biol., 2000, 4, 645–652 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- A. Creon, I. Josts, S. Niebling, N. Huse and H. Tidow, Struct. Dyn., 2018, 5, 064701 CrossRef PubMed.
- H. Li, R. Lantz and D. Du, Molecules, 2019, 24, 186 CrossRef PubMed.
- H. Wang and X. Chen, Front. Biosci., 2008, 13, 1716–1732 CrossRef CAS PubMed.
- E. G. B. Evans and G. L. Millhauser, in Methods in Enzymology, ed. P. Z. Qin and K. Warncke, Academic Press, 2015, vol. 563, pp. 503–527 Search PubMed.
- T. Tsubogo, Y. Kano, K. Ikemoto, Y. Yamashita and S. Kobayashi, Tetrahedron: Asymmetry, 2010, 21, 1221–1225 CrossRef CAS.
- E. Szliszka, Z. P. Czuba, M. Domino, B. Mazur, G. Zydowicz and W. Krol, Molecules, 2009, 14, 738–754 CrossRef CAS PubMed.
- T. Koopmans, M. van Haren, L. Q. van Ufford, J. M. Beekman and N. I. Martin, Bioorg. Med. Chem., 2013, 21, 553–559 CrossRef CAS PubMed.
- M. Krátký, Š. Štěpánková, K. Vorčáková, L. Navrátilová, F. Trejtnar, J. Stolaříková and J. Vinšová, Bioorg. Chem., 2017, 71, 244–256 CrossRef PubMed.
- M. S. Chorghade, D. K. Mohapatra, G. Sahoo, M. K. Gurjar, M. V. Mandlecha, N. Bhoite, S. Moghe and R. T. Raines, J. Fluorine Chem., 2008, 129, 781–784 CrossRef CAS PubMed.
- H. Zhou, J. W. Cheung, T. Carpenter, S. K. Jones, N. H. Luong, N. C. Tran, S. E. Jacobs, S. A. Galbada Liyanage, T. A. Cropp and J. Yin, Bioorg. Med. Chem. Lett., 2020, 30, 126876 CrossRef CAS PubMed.
- N. Elia, FEBS J., 2021, 288, 1107–1117 CrossRef CAS PubMed.
- J. C. Lewis, Curr. Opin. Chem. Biol., 2015, 25, 27–35 CrossRef CAS PubMed.
- X. Lu, B. Xiao, R. Shang and L. Liu, Chin. Chem. Lett., 2016, 27, 305–311 CrossRef CAS.
- Z. Song, Z. Tan and J. Cheng, Macromolecules, 2019, 52, 8521–8539 CrossRef CAS.
- A. Rezhdo, M. Islam, M. Huang and J. A. Van Deventer, Curr. Opin. Biotechnol., 2019, 60, 168–178 CrossRef CAS PubMed.
- W. Song, X. Chen, J. Wu, J. Xu, W. Zhang, J. Liu, J. Chen and L. Liu, Biotechnol. Adv., 2020, 40, 107496 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |