
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceJourney of anthraquinones as anticancer agents – a systematic review of recent literature
M. Shaheer Malik
 *a,
Reem I. Alsantalib,
Rabab S. Jassasc,
Abdulrahman A. Alsimareed,
Riyaz Syede,
Meshari A. Alsharifa,
Kulkarni Kalpanaf,
Moataz Morada,
Ismail I. Althagafia and
Saleh A. Ahmed
*ag
*a,
Reem I. Alsantalib,
Rabab S. Jassasc,
Abdulrahman A. Alsimareed,
Riyaz Syede,
Meshari A. Alsharifa,
Kulkarni Kalpanaf,
Moataz Morada,
Ismail I. Althagafia and
Saleh A. Ahmed
*ag
aDepartment of Chemistry, Faculty of Applied Sciences, Umm Al-Qura University, Makkah 21955, Saudi Arabia. E-mail: msmalik@uqu.edu.sa; saahmed@uqu.edu.sa
bDepartment of Pharmaceutical Chemistry, College of Pharmacy, Taif University, P. O. Box 11099, Taif 21944, Saudi Arabia
cDepartment of Chemistry, Jamoum University College, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
dDepartment of Basic Science (Chemistry), College of Science and Humanities, Shaqra University, Afif, Saudi Arabia
eCentalla Discovery, JHUB, Jawaharlal Nehru Technological University Hyderabad, Kukatpally, Hyderabad 500085, India
fDepartment of Humanities and Sciences (Chemistry), Gokaraju Rangaraju Institute of Engineering and Technology, Bachupally, Hyderabad 500090, India
gDepartment of Chemistry, Faculty of Science, Assiut University, 71516 Assiut, Egypt
First published on 5th November 2021
Abstract
Anthraquinones are privileged chemical scaffolds that have been used for centuries in various therapeutic applications. The anthraquinone moiety forms the core of various anticancer agents. However, the emergence of drug-resistant cancers warrants the development of new anticancer agents. The research endeavours towards new anthraquinone-based compounds are increasing rapidly in recent years. They are used as a core chemical template to achieve structural modifications, resulting in the development of new anthraquinone-based compounds as promising anticancer agents. Mechanistically, most of the anthraquinone-based compounds inhibit cancer progression by targeting essential cellular proteins. Herein, we review new anthraquinone analogues that have been developed in recent years as anticancer agents. This includes a systematic review of the recent literature (2005–2021) on anthraquinone-based compounds in cell-based models and key target proteins such as kinases, topoisomerases, telomerases, matrix metalloproteinases and G-quadruplexes involved in the viability of cancer cells. In addition to this, the developments in PEG-based delivery of anthraquinones and the toxicity aspects of anthraquinone derivatives are also discussed. The review dispenses a compact background knowledge to understanding anthraquinones for future research on the expansion of anticancer therapeutics.
 M. Shaheer Malik | M. Shaheer Malik is currently Assistant Professor in chemistry at Umm Al-Qura University, Makkah (Saudi Arabia). He carried out his doctoral studies in organic chemistry at the Indian Institute of Chemical Technology, Hyderabad, and was awarded a PhD from Osmania University, India. Subsequently, he moved for a postdoctoral assignment to Yonsei University, Seoul (Korea), working in biocatalysis. His research interest deals with the design and synthesis of novel chemical compounds as pharmaceutical agents and the application of enzymes in organic transformations. He regularly contributes and reviews research findings towards the advancement of science in his area of interest. |
 Saleh A. Ahmed | Prof. Dr Saleh A. Ahmed was born in Assiut, Egypt in 1968 where he undertook undergraduate and postgraduate studies. He received his Bachler and MSc from Assiut University, Egypt and PhD in photochemistry (photochromism) under the supervision of Prof. Heinz Dürr at Saarland University, Saarbrücken, Germany. He worked as postdoctoral fellow, senior researcher and visiting professor in France (CNRS fellow), Japan (JSPS fellow), Germany (AvH, DFG and DAAD fellows), Italy (TEMPUS fellow) and USA (Arab fund fellow). His current research interests include synthesis and photophysical properties of novel organic compounds, electronic devices and solar energy conversion, photocatalysis, nanomaterials and their applications, fluorophores and biologically active molecules. |
1. Introduction
Anthraquinone (1), a quinone-containing chemical compound, is enriched with a myriad of interesting biological profiles that can be harnessed for multiple therapeutic applications with stepwise iterations. Quinones, a cyclic diketone structural compound, form the basis for the subgroup 9,10-anthraquinones (a.k.a 9,10-dioxoanthracenes, anthracene-9,10-diones, anthradiones, dioxoanthracenes, 9,10-anthrachinons and anthracene-9,10-quinones).1 The carbonyl function is present on the 9th and 10th carbon positions of the quinone moiety. The rigidity, planarity, and aromaticity of the anthraquinone system have been studied widely with respect to its pharmacological properties. In particular, the planarity of this molecule provides the advantage of embedding in the DNA double helix, thus acting as a DNA intercalator. An interesting historical journey of anthraquinones in anticancer drug development is depicted in Fig. 1. The anthraquinone moiety can be found in nature, for example in emodin (2), aloe-emodin (3), rhein (4), and chrysophanol (5) or utilized as a starting material in the development of many anticancer agents (Fig. 2).2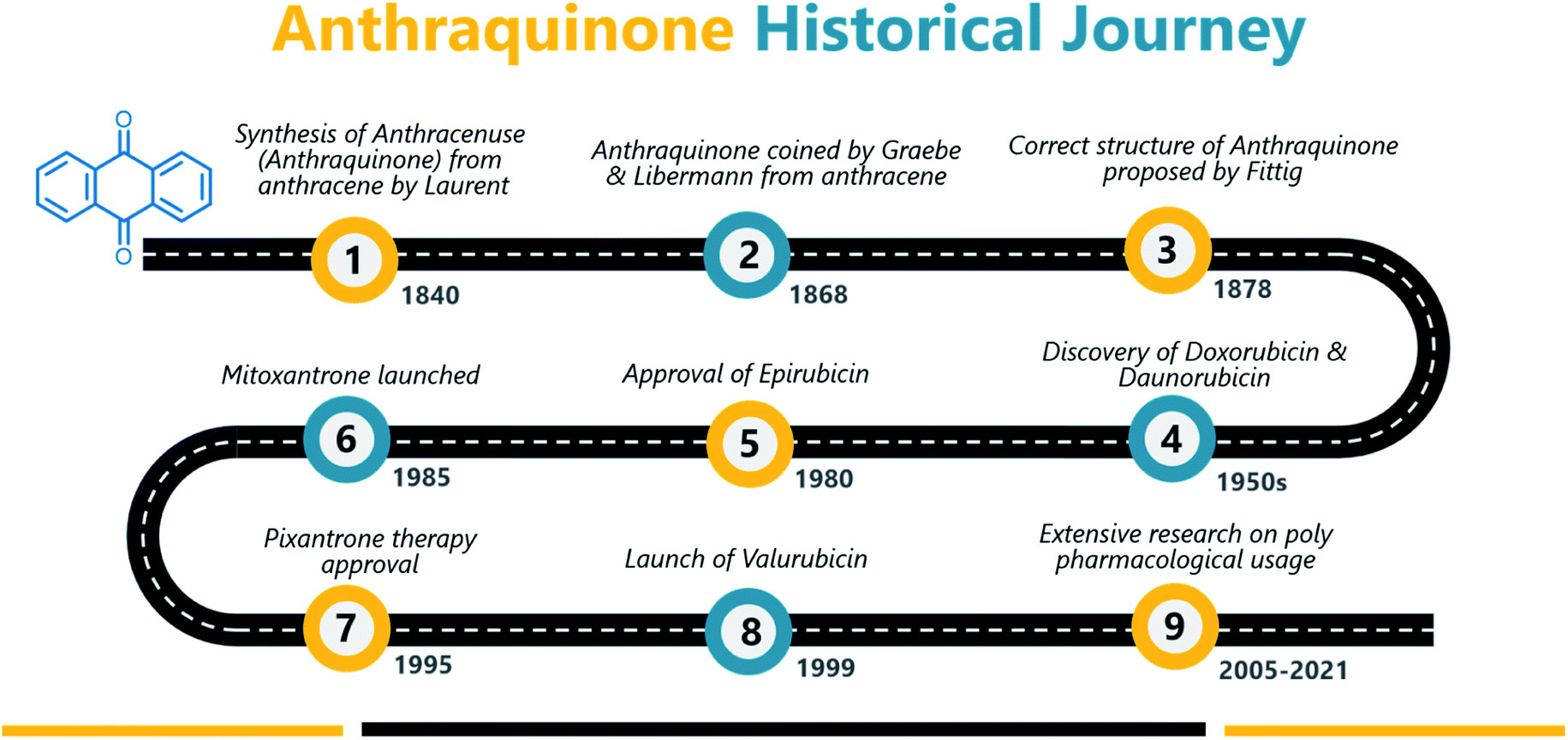 | ||
| Fig. 1 Milestones in anthraquinone journey as an anticancer agent. | ||
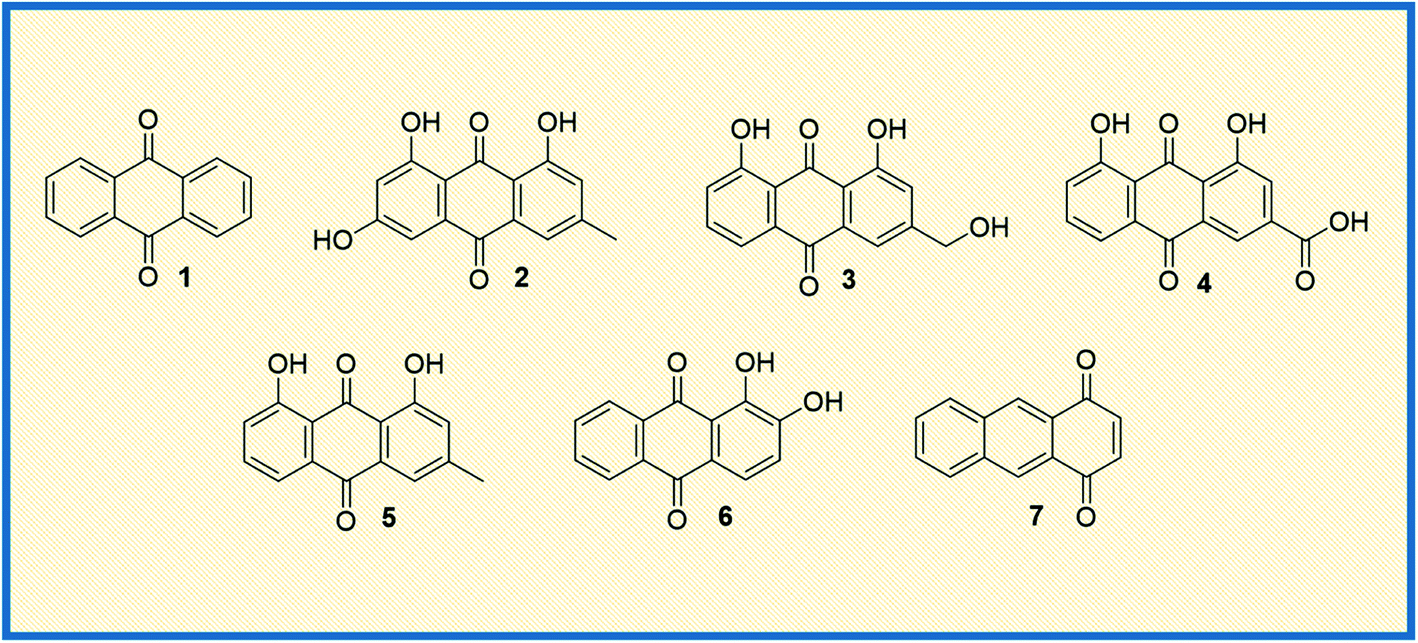 | ||
| Fig. 2 Naturally occurring anthraquinone molecules from different sources. | ||
A brief historical account of anthraquinones as a chemical class is provided here. The earliest recorded discovery of anthraquinone was in 1840 when Laurent oxidized anthracene to synthesize anthraquinone. He named the chemical “anthracenuse”, while it was termed “oxanthracen” by Anderson independently.3 Interestingly, the presently used name “anthraquinone” was proposed by Graebe and Libermann in the year 1868, almost three decades later to earlier nomenclature.4 Similar to Laurent, Fritsche reported the synthesis of anthraquinone via oxidation of anthracene with chromic acid.5 In a stimulating development, Graebe and Liebermann proposed the structural formula of anthracene and successfully established the relationship between anthraquinone and alizarin (6) by synthesizing alizarin from anthracene.4,6 Finally, in 1873, Fittig proposed the correct diketone structure of anthraquinone, which is widely used today,7 and more than 75 natural anthraquinones were identified from various sources like lichens, marine sources, fungi, and medicinal plants of various families.8,9
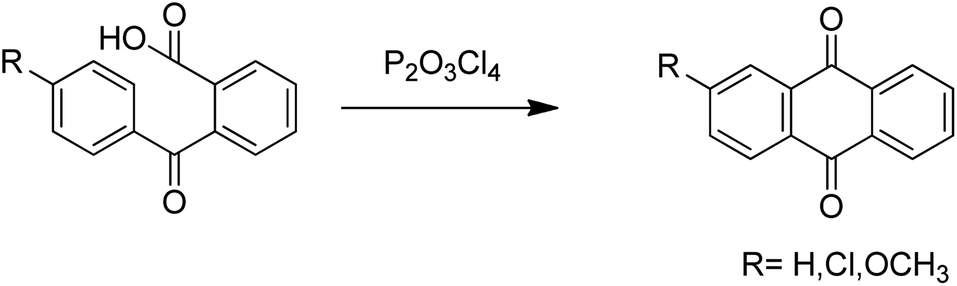
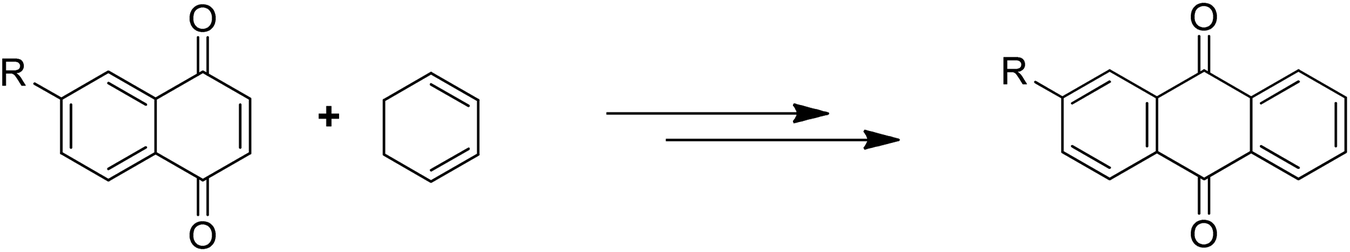
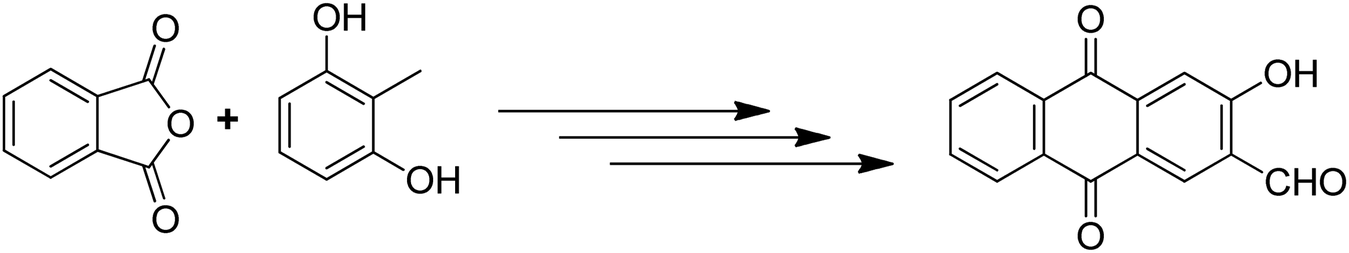
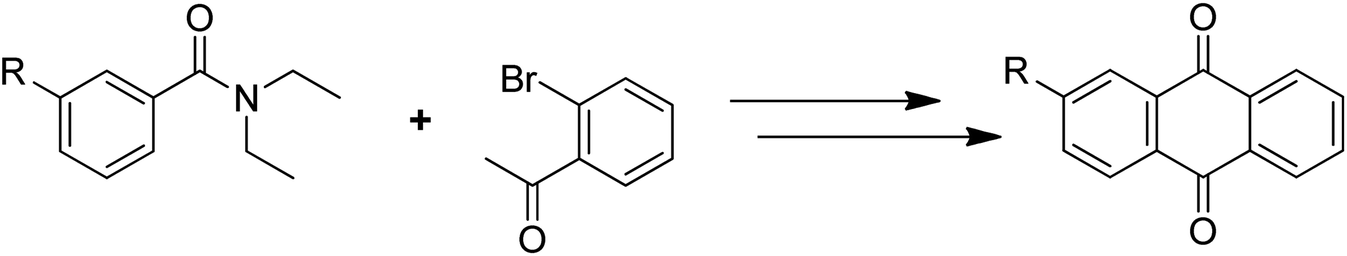
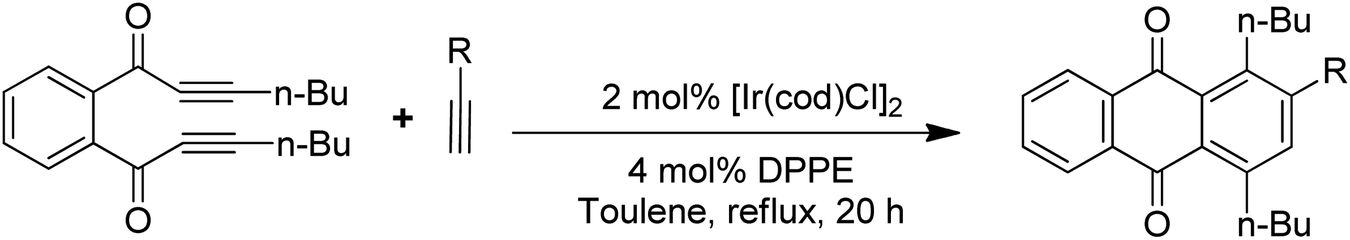
Anthraquinones have drawn the interest of chemists to access diversely substituted derivatives via different synthetic protocols. In recent years, most of the anthraquinone derivatives synthesized as potential anticancer agents utilize commercially available anthraquinones as starting material. However, several synthetic approaches are available to synthesize substituted anthraquinones, and a few of them are summarised here (Table 1). A classical approach to access anthraquinone ring is the intramolecular cyclization of 2-(4-alkyllbenzoyl)benzoic acid derivatives in pyrophosphoryl chloride to afford 7-substituted anthraquinone analogs (entry 1). Another classical strategy of Diels–Alder cycloaddition was improved by treating napthaquinone with cyclohexadiene to give anthraquinone moiety in the presence of ionic liquid (entry 2). In another approach to synthesize 2-substituted 1,3-dihydroxy anthraquinones, phthalic anhydride was treated with 2-methyl resorcinol to afford damnacanthal and nordamnacanthal (entry 3). Similarly, the 7-substituted anthraquinones were reported to be obtained from reacting appropriate N,N-diethyl benzamide with ortho-bromo benzaldehyde (entry 4). In an iridium catalysed route, the 1,4-dibutyl anthraquinones were synthesized from 1,1′-(1,2-phenylene)bis(hept-2-yn-1-one) and ethyne derivatives in the presence of organophosphorus compounds under refluxing toluene (entry 5). In addition to this, anthracene and anthracene-based compounds were converted to anthraquinones by utilizing different catalytic strategies.10–13
Because of their plethora of biological properties, anthraquinones have become an important class of compounds in drug discovery. In traditional medicines, anthraquinone compounds have been used for many centuries, and aloe is one of the classic examples.19 Many anthraquinone derivatives have also been identified in bacteria, fungi, and insects.20 In addition to their biological properties, many natural and synthetic anthraquinones are well known for their uses in the textile industry, paints, imaging photocleavable protecting groups, devices and biochips, foods, cosmetics, and pharmaceuticals.21–26 Moreover, they are useful catalysts in several chemical and biogeochemical processes like the degradation of contaminants exploiting the redox potential.27,28 More prominently, the scaffold is widely recognized for its diverse pharmacological profiles. A quick glance at the interesting activities of anthraquinone reveals its applications in antifungal,29 antiviral,30–32 antimalarial,33 antimicrobial,34,35 antiplatelet,36 antidiabetic,37,38 neuroprotective,39 laxative,40 and many more therapeutic settings. To discuss a few, antibacterial anthraquinone, emodin from the roots of Rheum ribes, displayed a MIC value of 39 μg mL−1 against Staphylococcus aureus.41 Antithrombotic compound PSB0702, exhibited potent activity in binding studies with Ki value of 21 nM,42 and the dehydration of gallic acid yielded a potent antimalarial agent rufigallol, active against Plasmodium falciparum with an IC50 value of 35 nM.43 The planarity and rigidity of anthraquinones, discussed at the start of this review, has attracted many medicinal chemists to explore their anticancer potential. The 1,4-anthraquinone (7) exhibits anticancer activity by inhibiting crucial proteins and nucleic acid synthesis in cellular machinery.44 There are a plethora of molecules that have been derived from core anthraquinones scaffold aimed at various cancer targets. This immense interest stems from the marketed anticancer drugs such as mitoxantrone, doxorubicin and epirubicin with anthraquinone ring structure. The new-age drug delivery techniques have further enhanced the target and site-specific delivery of these derivatives. In addition, the research towards the design and development of new anthraquinone derivatives is rising day by day owing to their profound biological activity.
\In specific, anthraquinone-based compounds plays a significant role in the treatment of cancer by chemotherapeutics agents. Some of the anthraquinone scaffold containing drugs such as daunorubicin (8), idarubicin (9), doxorubicin (10), epirubicin (11), valrubicin (12), pixantrone (13), mitoxantrone (14) are currently in clinical use for various types of cancer treatments (Fig. 3).45
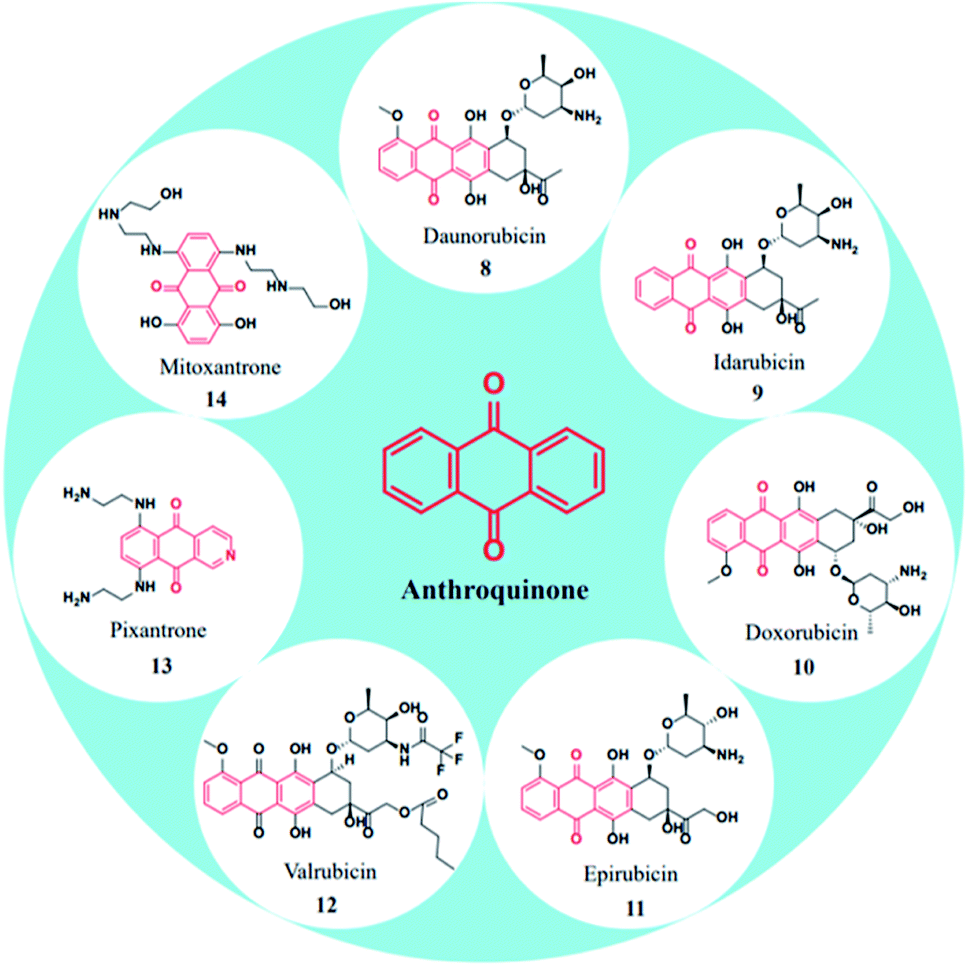 | ||
| Fig. 3 FDA approved drugs containing anthraquinone nucleus. | ||
In the past, several research groups reported the anticancer potential of anthraquinones and their derivatives against different cancer cell lines as well as cancer targets. Cancer, a complex disease, is characterized by the uncontrolled growth of abnormal cells that can be invasive or metastatic and is the second leading cause of human deaths worldwide.46 The majority of the anticancer drugs have failed at the clinical level due to non-selectivity, toxicity, low therapeutic window, and drug resistance.47 Hence, the design and development of novel anticancer drugs with fewer side effects are the primary focus of cancer drug discovery. Anthraquinones are potential anticancer agents which are easily metabolized and excreted renally after conversion to more hydrophilic glucuronides by uridine diphosphate glucuronosyl transferase (UGT) enzymes in the human body.48 The amount and percentage of each glucuronide formed from each anthraquinone in the liver and intestinal microsomes differ and is not always equal to the total glucuronides formed from each anthraquinone.49 The historical evidence of the therapeutic application of anthraquinones can be seen in the plant herb Compendium of Materia Medica, which is frequently used in Chinese traditional medicine.50 Later, the laxative effect of rhubarb roots rich in aloe-emodin, emodin, rhein, chrysophanol, and subsequent glucopyranosides was reported. Not only the phyto-based anthraquinones, the synthetic derivatives of anthraquinones were also found to be promising therapeutic agents against a wide array of diseases. The antitumour activities of anthraquinones include inhibition of cancer cell proliferation, invasion, migration, metastasis, induction of cellular apoptosis and tumour angiogenesis, regulation of the host immune response, antioxidant, anti-inflammatory, reversing tumour cell multidrug resistance, and so on. Furthermore, different research groups described the anticancer potential of anthraquinones by the inhibition of various targets like protein kinases topoisomerases, telomerase, ecto-nucleotidases, matrix metalloproteinases (MMPs), DNA quadruplex and many more. The developments in the field of anthraquinone-based anticancer agents is reviewed based primarily based on the biological targets.2,20,51
Herein, based on the abundance of literature from 2005 to date, we reviewed and recapitulated the developments in anthraquinones research in the context of anticancer agents to serve as a source and guiding tool for further investigation. The article provides insights into the systematic improvements to develop anthraquinone compounds towards anticancer therapeutics from 2005 onwards. This is followed by a critical discussion on the target protein-specific anthraquinone derivatives. The studies on the selective delivery of anthraquinones at specific sites exploiting the PEG-based approach and the toxicity profile of the anthraquinones are highlighted. Finally, a structure–activity relationship of the anthraquinone moiety on the antitumour potency is also discussed. This approach will allow readers to get a diverse and holistic perspective on the potential of anthraquinones and can guide them in designing novel anthraquinone-derived anticancer agents.
2. Development of non-specific cytotoxic anthraquinone derivatives
In recent decades, extensive research has been carried out to explore the anticancer potency of new anthraquinone derivatives. However, most of these reports deal with cell-based assays that assess the cytotoxicity of new anthraquinone derivatives against selected cancer cell lines. In these studies, the biochemical assays to determine the mechanism of action are not generally undertaken. For the purpose of better understanding, the research from 2005 to date are reviewed in a year wise manner.2.1 2005–2008
In general, anthraquinones and their derivatives show a unique antiproliferative activity. However, several researchers have made significant modifications in the structure from their initial discovery, resulting in the development of new anthraquinone derivatives with promising anticancer activity against various cancer types. Teng et al. reported the cytotoxic potency of 1,3-dihydroxy-9,10-anthraquinone compounds against different human cancer cell lines like HepG2, Hep3B, HT-29, and MCF-7. Among all the compounds synthesized, the anthraquinone derivative (15) showed selective cytotoxicity towards HepG2 cells with an ED50 value of 1.23 μM. Also, another derivative, 16, exhibited good activity against MCF-7 cells (Fig. 4). Further, the mechanistic studies revealed that compound 16 induces cell cycle arrest in G2/M phase and causes cell death via apoptosis.52 In another investigation, Dzieduszycka et al. described the anticancer potential of tetracyclic anthraquinone fused pyridine conjugates. The cytotoxic potential of the synthesized derivatives was examined on human cell lines such as human promyelocytic leukaemia (HL-60) and vincristine resistant (HL-60/VINC) and doxorubicin-resistant (HL-60/DX) cell lines. The anthraquinone analogue 17 exhibited decent activity against sensitive as well as resistant cell lines. It showed cytotoxicity activity of 311 nM, 1012 nM, and 667 nM against HL-60 cells, HL-60/VINC, and HL-60/DX. In addition, other derivatives of the series 18 and 19 displayed good activity towards HL-60 with IC50 values of 146 nM and 327 nM, respectively. The same compounds showed good to moderate activity against drug-resistant cell lines as well.53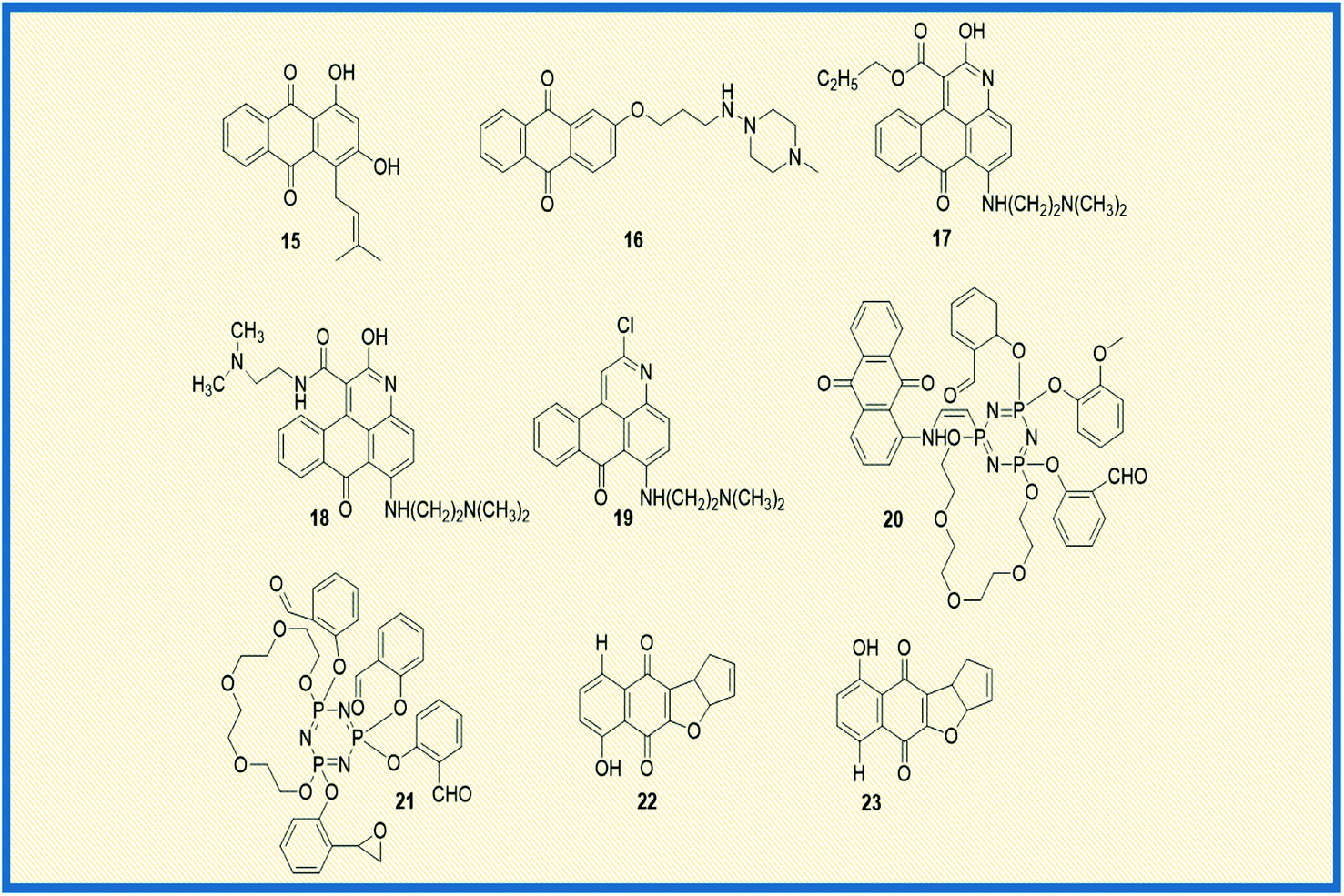 | ||
| Fig. 4 Structures of anthraquinone derivatives reported in the year 2005–2006. | ||
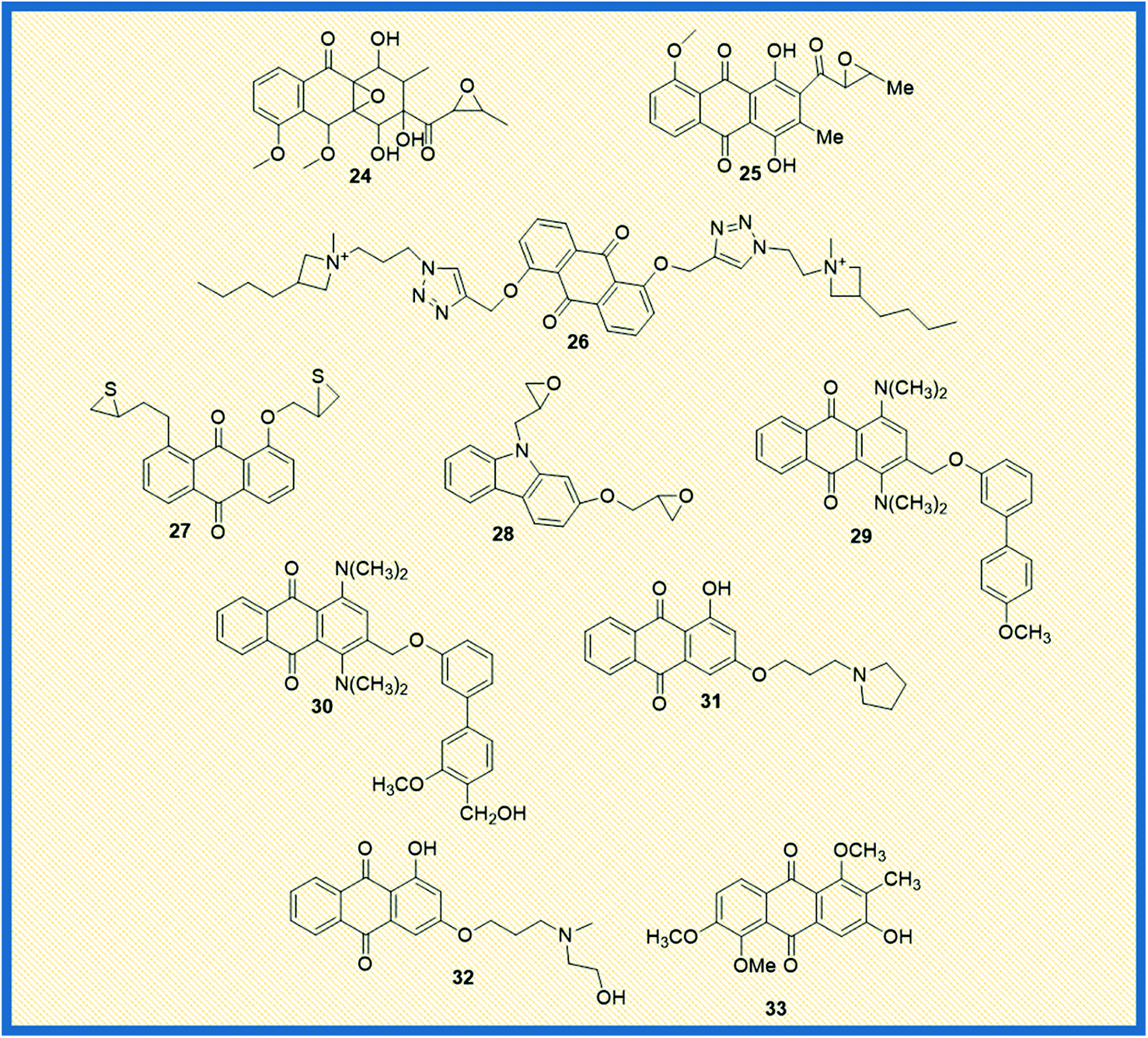
Similarly, Siwy et al. synthesized a series of 1,3-(oxytetraethylenoxy)cyclotriphosphazene derivatives and examined their antileukemic activity against MOLT4, L 1210, HL-60, and P388 cell lines. The derivatives 20 and 21 unveiled promising antiproliferative activity against MOLT4 cells with ID50 values of 2.1 and 1.14 μg mL−1, respectively.53 Valderrama et al. designed and examined the biological activity of anthraquinone epoxides and their isomerization products. The biological activity was investigated against human cancer gastric epithelial cells (AGS). Further, the non-toxic nature of the synthesized derivatives was studied on normal human lung cells (MRC-5). Among all the derivatives, 22 and 23 showed potential anticancer activity against AGS cells with IC50 values of 4.1 and 4.9 μM, respectively (Fig. 4).54 Likewise, Tietze et al. reported the synthesis of anthraquinones analogues that are derived from the natural products mensacarcin, islandicin, and chrysophanol. Later, the cytotoxicity of synthesized derivatives was studied against human lung carcinoma cell lines (A549). The compound mensacarcin and its analogue 24 displayed promising antitumour activity with ED50 values of 1.6 and 11.6 μM, respectively (Fig. 5).55
 | ||
| Fig. 5 Structures of anthraquinone analogs reported in the year 2007–2012. | ||
In another study, Huang et al. synthesized a series of 34 analogues of 2,7-bis-substituted amido-anthraquinone (25) and evaluated their effects on cancer cell proliferation and telomerase activity. Most of the derivatives showed promising anticancer and telomerase inhibitory activity.56 Click chemistry approach was used by Wang et al. to design and synthesize water-soluble anthraquinone derivatives. The anticancer activity evaluation of the same was performed on BGC gastric cancer cells along with mechanistic studies such as generation of reactive oxygen species, loss of mitochondrial membrane potential, transition of mitochondrial permeability, cell cycle arrest, and the release of cytochrome C. The derivative 26 exhibited promising antiproliferative activity against BGC cells with an IC50 of 4.02 μM. Further, the same derivative induced cancer cell death via apoptosis. At a concentration of 25 μM, the majority of the cells (39.4%) entered into the apoptotic phase.57
2.2 2009–2012
Yang et al. synthesized and reported the anticancer activity of oxiranyl and thiiranyl phenolic compounds. They investigated the cytotoxic potential of synthesized derivatives against a panel of different human cancer cells such as MDA-MB-231, LNCaP, DU145, and PC3 cells. The derivatives 27 and 28 demonstrated good activity towards PC3 cells with IC50 values of 2.5 and 4.0 μM, respectively. In addition, derivative 27 showed significant topoisomerase activity at 10 μM.58 In yet another study, Jin et al. reported the synthesis of – 1,4-bis(dimethylamino)-9,10-anthraquinone derivatives and assessed their biological evaluation against mouse leukemic tumour cells (p388). Most of the synthesized derivatives exhibited good to moderate activity against p388 cells. The analogue 29 showed an ED50 value of 1.3 μg mL−1 while analogue 30 exhibited 1.5 μg mL−1 against the tested tumour cells.59Tu et al. synthesized different anthraquinones derivatives that include 3-(3-alkylaminopropoxy)-9,10-anthraquinone and 1-hydroxy-3-(3-alkylaminopropoxy)-9,10-anthraquinone and evaluated their cytotoxicity towards different human cancer cell lines such as human urothelial carcinoma cells (NTUB1) and human prostate cancer cells (PC3). The derivatives 31 and 32 showed good anticancer activity against PC3 cells with IC50 values of 7.64 and 8.89 μM, respectively. Further, various mechanistic studies like increased ROS production, cell cycle arrest, immuno-fluorescent staining, and gene expression of p21, p53, Bax, and cyclin B1 were investigated. The studies revealed that compound 31 induces apoptotic cell death by arresting the cell growth in G2/M phase with up-regulation of p21, p53, Bax, and cyclin B1.60
2.3 2012–2014
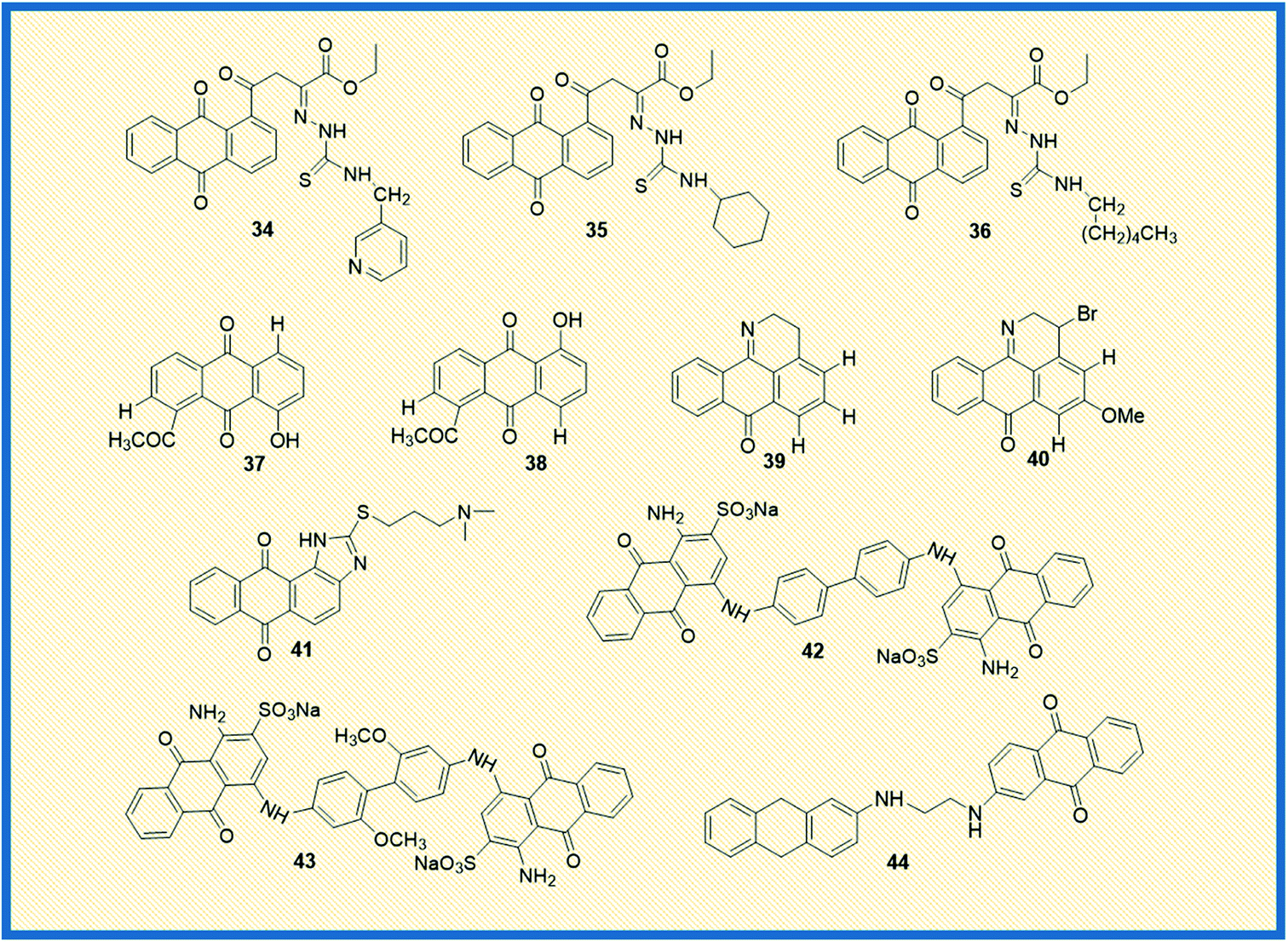
Feng et al. reported the antiproliferative activity of phytochemical-based anthraquinones such as 3-hydroxy-1,5,6-trimethoxy-2-methyl-9,10- anthraquinone, also referred to as PCON6 (33) (Fig. 5). The authors studied the anticancer potential of PCON6 against a panel of 15 different cancer cell lines that includes a group of 11 non-lung cancer cell lines and four NSCLC cell lines. The most active compound arrested the cell growth in almost all the tested cell lines. However, non-small cell lung cancer (H520) and breast cancer (MCF7) were more sensitive to PCON6 treatment. The compound exhibited IC50 values of 7.8 and 10.2 μM against H520 and MCF7 cells, respectively. Other mechanistic studies revealed that the compound arrested the cell growth in the S-phase of the cell cycle by inducing apoptosis-mediated cell death in tested cell lines.61In another study, Marković et al. synthesized anthraquinone-thiosemicarbazone derivatives and tested their anticancer potency against different human cancer cells such as HeLa, A549, K562, MDA-MB-453, MDA-MB-361. Almost all the compounds exhibited good cytotoxicity against the tested cell lines. Most of the derivatives showed good specific activity towards K562 cells. The derivatives 34 and 35 showed promising anticancer activity against K562 cells with IC50 values of 2.17 and 2.35 μM, respectively (Fig. 6). Notably, derivative 36 displayed good activity against HeLa cells with an IC50 value of 7.66 μM. Further, 36 induced cell cycle arrest in the sub-G1 phase and promoted apoptosis in HeLa cells.62
 | ||
| Fig. 6 Structures of anthraquinone derivatives reported in the year 2013. | ||
Bhasin et al. reported the synthesis of a series of substituted anthraquinones as well as 1,4-naphthoquinones and examined their biological activity against human prostate cancer cells (DU-145) and colon cancer (HT-29). Among the synthesized, anthraquinone 37 showed good antiproliferative activity against DU145 and HT-29 cells with IC50 values of 10.2 μM and 8.5 μM, respectively, while its analogue 38 exhibited 11.5 μM IC50 value towards DU-145 and 10.4 μM IC50 value against HT-29 cells.63 Castro et al. synthesized a series of 1-azabenzanthrone analogues, their 2,3-dihydro derivatives, and substituted 9,10-anthracenediones. Later, the authors examined their anticancer potential towards four different human cancer cells such as gastric adenocarcinoma (AGS), lung cancer cells (SK-MES-1), bladder cancer cells (J82), and myelocytic leukaemia cells (HL-60). Among the synthesized, compounds 39 and 40 exhibited promising antiproliferative activity against gastric adenocarcinoma cells with IC50 values of 1.5 and 3.3 μM, respectively.64
Similarly, Lee et al. examined the anticancer potential of seven anthraquinones derived small molecules which are previously synthesized in their laboratory and screened against the NCI's 60 panel of human cancer cells comprising colon cancer, NSCLC, ovarian cancer, breast cancer, leukaemia, renal cancer, prostate cancer, CNS, and melanoma cancer. Amongst the series, seven derivatives showed promising anticancer activity against all the tested cell lines. However, compound 41 exhibited promising activity and inhibited PARP enzyme (65%) at 10 μM in a dose-dependent manner.65 In another investigation, Liang et al. reported the synthesis and biological examination of new anthraquinone analogues. Afterward, they examined the c-Met kinase inhibition activity in A549 cells. Derivatives such 42 and 43 elicited good c-Met kinase inhibitory activity with IC50 values of 1.2 and 4.0 μM, respectively.66
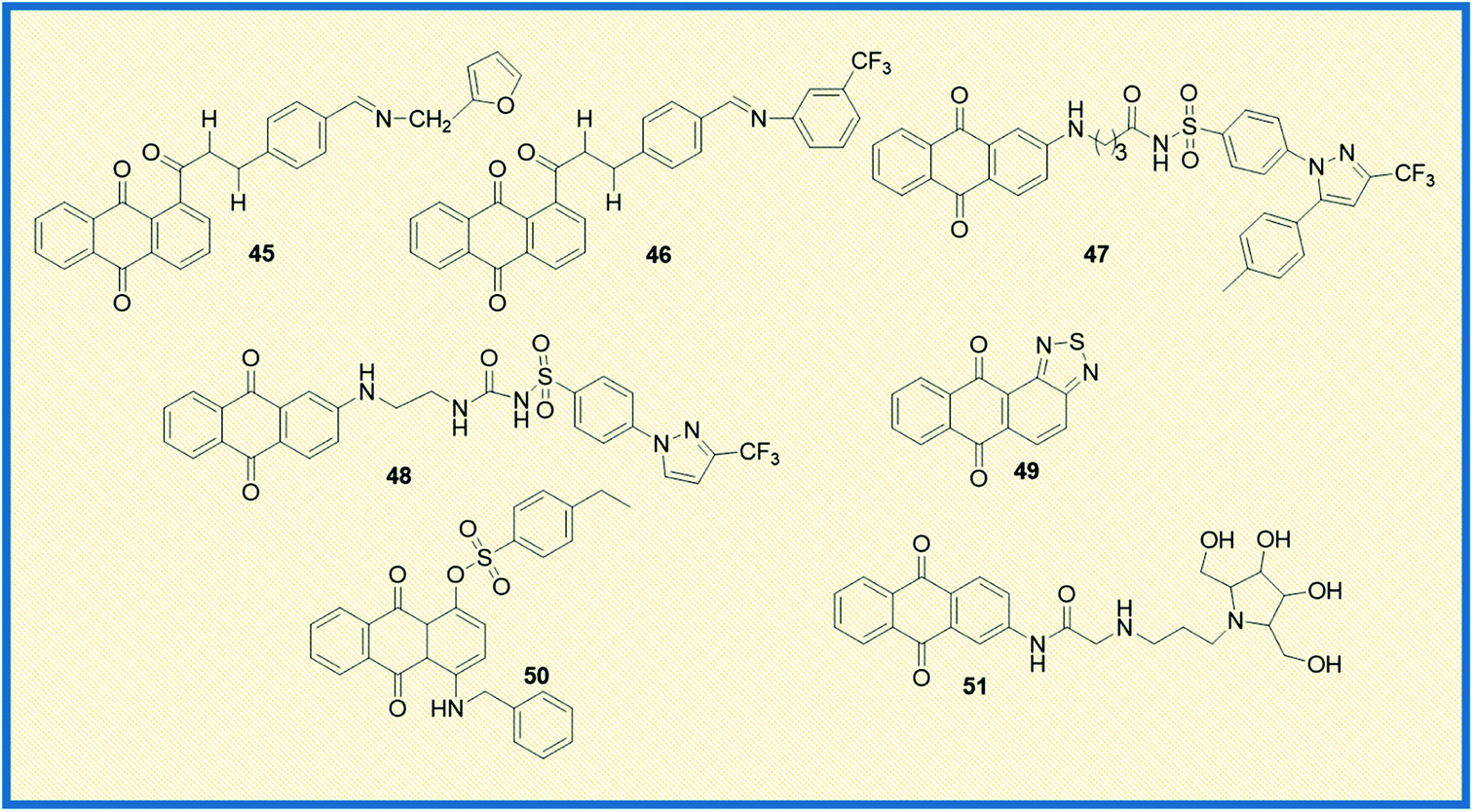
Taher et al. synthesized two series of bis-anthraquinone derivatives and evaluated their biological potential against different human cancer cell lines. Among the synthesized, five derivatives were selected for studying the anticancer potential towards a panel of 60 NCI human cancer cell lines. The derivative 44 showed potent activity against all the tested cell lines (Fig. 6). It also showed very good anticancer activity against colon cancer cells (HCT-116) with a GI50 value of 0.3 μM.67 Kolundžija et al. designed and synthesized a class of imine derivatives of hybrid chalcones containing anthraquinone derivatives and examined their in vitro cytotoxicity against HeLa, LS174, and A549 cancer cell lines. The derivatives 45 and 46 inhibited the proliferation of HeLa cells at concentrations of 1.45 μM and 1.82 μM, respectively (Fig. 7). Further, the compounds in this series elevated the levels of caspase-3 and -8 and showed strong anti-angiogenic activity.68
 | ||
| Fig. 7 Structures of anthraquinone derivatives reported in the year of 2014. | ||
Almutairi et al. reported the synthesis of hybrids of celecoxib and 2-amino anthraquinone derivatives and carried out cytotoxicity profile against hepatic carcinoma cells (HepG2). The hybrid molecules 47 and 48 displayed good activity against HepG2 cells with IC50 values of 3.74 and 3.92 μg mL−1, respectively.69 In another investigation, Chen et al. studied the anticancer potential of NSC745885 (49) in oral squamous carcinoma cells. The mechanistic studies such as annexin V staining, caspase expression, and other xenograft mouse model studies revealed that compound 49 is a potential therapeutic drug for treating oral squamous cell carcinoma.70
Sangthong et al. reported the synthesis of anthracene-9,10-dione derivatives, and their anticancer potential against human papillomavirus (HPV) positive cancer cell line, CaSki. The derivative 50 showed good activity against the tested cell line with an IC50 value of 0.3 μM. Further studies demonstrated that the derivative arrested the cell growth in G2/M phase of the cell cycle and up-regulated the expression of p53 while down-regulating Bcl-2 gene.71 Similarly, Zhang et al. synthesized a series of azasugar-modified 2-mono substituted, 2,6- and 2,7-bis substituted anthraquinone analogs and examined the anticancer activity against human breast cancer cells (MCF-7). The azasugar-anthraquinone derivative 51 showed superior activity against MCF-7 cells with an IC50 of 17.3 μM.72
2.4 2015–2016
From the literature, it was observed that most anticancer drugs either interact with DNA or inhibit DNA synthesis.73 In this context, Zuravka et al. synthesized bis-3-chloropiperidine tethered anthraquinone (52) nucleus (Fig. 8).74 The compound 52 was tested for its reactivity towards DNA using chlorambucil as a positive control. Results indicated that the compound binds at the guanine sites of supercoiled plasmid and duplex oligonucleotide and causes DNA cleavage. It was further concluded that the anthraquinone nucleus of the compound was more effective in driving DNA intercalation than its naphthalene derivative.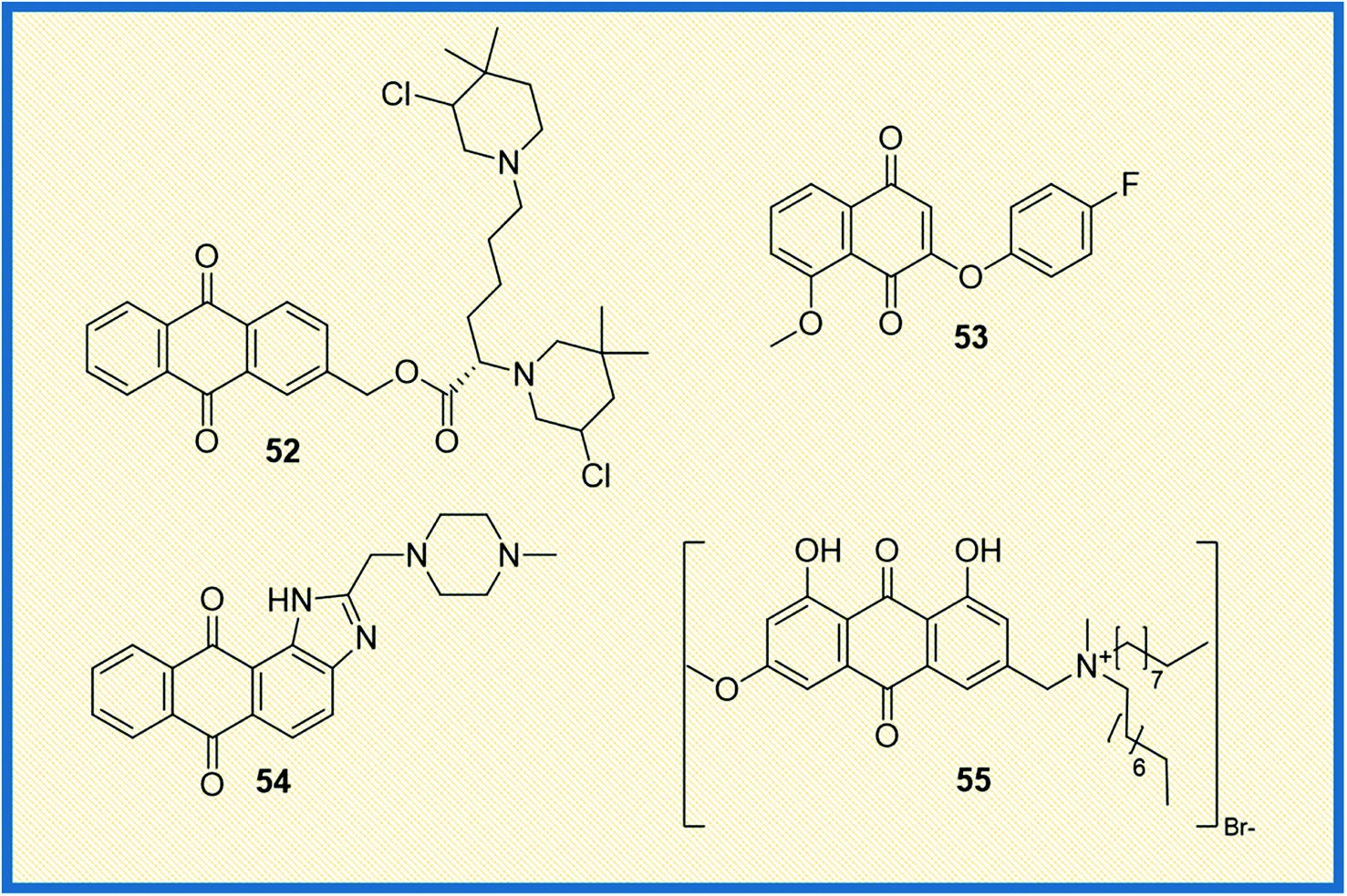 | ||
| Fig. 8 Structures of anthraquinone derivatives reported in the year 2015–2017. | ||
Similarly, Prati et al. synthesized 2-phenoxy-1,4-naphthoquinones derivatives and tested their activity against HT-29 and IGROV-1 cancer cell lines along with HDF non-cancerous cell line. The most active compound 53 was found to inhibit the tested cell lines at an IC50 of 2.70 and 1.43 μM, respectively. Further, the compounds that showed promising antitumour activity were analysed for their reactive oxygen consumption in bovine heart mitochondria.75 The main focus of a study reported by Lin et al. was to explore the binding mechanism of compound NSC749235 (54) to human telomeric G-quadruplex DNA, one of the vital targets in cancer progression. The enzymatic assay was evaluated by measuring the thermodynamic stability and affinity of telomeric G-quadruplex DNA via FRET melting assay. The results indicated that the ligands selectively stabilized the potassium form of human telomeric G-quadruplex DNA compared to the other forms. Further, the cytotoxic activity of the compounds was evaluated in HeLa and A549 cell lines using daunorubicin as a reference compound. The derivative 54 exhibited effective inhibitory activity against HeLa cell line. The stabilization of potassium-containing telomeric G-quadruplex DNA by 54 led to ΔTm values ranging from 3.66 to 8.04 °C. Compound 54 exhibits inhibitory activity against the HeLa and A549, with IC50 values ranging from 5.54 to 14.54 μM. The results indicated that HeLa cell line was much more sensitive to the compound 54 than the A549 cell line. All the studies collectively showed that NSC794235 (54) serves as a scaffold for designing new anticancer chemical entities.76
2.5 2017–2019
Zheng et al. synthesized quaternary ammonium salts of anthraquinone and tested their antiproliferative activities against A375, BGC-823, HepG2, and 8-HELF cancer cells. Among the tested compounds, the derivative 55 induced apoptosis and significantly increased ROS levels in A375 cells (Fig. 8). The derivative 55 exhibited IC50 values of 1.39 μM, 2.79 μM and 4.12 μM concentrations on A375, BGC-823 and HepG2 cell lines, respectively. Furthermore, the apoptotic capability was validated by tracing the indicator proteins caspase-3 and P53 using the western blot technique.77 Okumura et al. synthesized compounds intending to establish and understand the relation between redox properties and antitumour activity of anthraquinones with a hydroxyl and methoxy group (56) (Fig. 9). The synthesized derivatives with different substitutions like hydroxy and methoxy at the 4th, 5th, and 8th positions of anthraquinone were studied for their redox behaviour using cyclic voltammograms (CVs). The redox studies indicated that the oxidative behaviours are different for each derivative. To further understand the pattern behind it, the authors performed molecular orbital energy calculations. It was found that the LUMO energies of the compounds were identical, while the HOMO energies varied depending on the position of the substituent. Cytotoxic studies against HL-60 and HP100 cells (using LDH activity assay) indicated that oxidized radicals played a significant role in inducing cell death.78 The toxicity towards HL-60 increased with the increase in the concentration of 56. However, the toxicity in relation to HP100 was less than half the toxicity to HL-60, indicating that H2O2 is involved in the process leading to cell death. Specifically, the cytotoxicity observed against HL-60 could be ascribed to reactive oxygen species (ROS) originating from electron transfer to oxygen accompanying the formation of reduced or oxidized compound 56 radicals.78 In another investigation, Roa-Linares et al. synthesized 27 terphenyl-1,4-naphthoquinone (NQ), 1,4-anthraquinone (AQ), and heterocycle-fused quinone (HetQ) derivatives to evaluate their cytotoxicity against HeLa and Jurkat tumour cell lines. Compound 57 was found to be the most active against the tested cell lines, and the IC50 values of 57 observed to be cell lines 0.010 μM, 1.4 μM, and 231.9 μM against HeLa ATCC CRL-1958, Jurkat ATCC TIB-152, and Vero ATCC CCL-81, respectively.79Phosphoglycerate mutase 1 is one of the critical enzymes that supports cancer cell proliferation. Since it regulates glycolysis and biosynthesis, developing an inhibitor that regulates this enzyme is of therapeutic importance. In this study, Huang et al. synthesized anthraquinone derivatives and established the structure–activity relationship (SAR) of the 31 compounds synthesized. Compound 58 was the most effective, with an IC50 value of 0.27 μM. It also exhibited antiproliferative activity in different cancer cell lines such as H1299, A549, and PC9 with IC50 values 6.9 ± 1.2 μM; 12.7 ± 2.7 μM and 13.8 ± 1.0 μM, respectively. A deep look at SAR evaluation suggested that 3-sulfonamide substituents of the anthraquinone scaffold played a crucial role in determining the potency of the compounds.80
Celik et al. synthesized anthraquinone derivates for evaluation of cytotoxicity potential. Further, the density functional theory (DFT) B3LYP method was used to determine the most stable molecular structure. The stable piperazinyl anthraquinone derivative 59 exhibited potential cytotoxicity against the A549 cell line. However, it was found that high doses of the compound were lethal to healthy human cells, while low dose was ineffective in cancer cells.81 The existing body of research on PGAM1 inhibitor PGMI-004A suggests that anthraquinone regulates the key pathways like glycolysis and serine synthesis, essential for tumour growth. Based on this information, novel anthraquinone derivatives were synthesized to evaluate their PGAM1 inhibiting activity. Of all the compounds synthesized, compound 60 exhibited good PGAM1 inhibiting activity with an IC50 value of 0.25 ± 0.07 μM. It was further tested to evaluate its in vitro cytotoxic activity against H1299, A549, and PC9 cell lines and in vivo activity in H1299 xenografts models. The experimental results suggested that the efficacy of the compounds containing phenyl substituents was more active than the compound with dimethylamino and morpholine substituents. To further understand the site of binding, the crystal structure of the 60 and PGAM1 complex was evaluated.82
Literature studies suggest that structurally novel sulfonamide derivatives show pronounced antitumour activity. Taking this into account, Awasthi et al. synthesized 1-substituted anthraquinone sulfonamide derivatives and tested their cytotoxic, antibacterial, and antifungal properties. Of all the compounds synthesized, compound (61) displayed better cytotoxic activity in HeLa cell lines than the reference compound mitoxantrone. Compound 61 arrested the cell cycle progression at G1 and G2 phases. Docking studies between the synthesized compounds and telomeric sequence revealed that all the synthesized compounds could be suitable i-motif inhibitors.83 Another study involved the synthesis of 9,10-anthraquinone hooked piperidine units to evaluate their antiproliferative activity. The synthesized compounds were tested in drug-sensitive human cancer lines HL-60, LoVo, and drug-resistant cancer cell line HL60/MX2, LoVo/Dx. Later, the compounds were also evaluated in BALB/3T3 normal mouse fibroblasts cell lines (selectivity) using cisplatin, mitoxantrone and doxorubicin as reference compounds. Results suggested that all the compounds were effective against drug-resistant cell lines, with 1-(piperidin-1-yl)-9,10-anthraquinone (62) being the most potent of all (Fig. 9). Since all the compounds showed strong potency against drug-resistant HL60/MX2 cell line, it was concluded that piperidine substituted anthraquinone derivatives can be developed as anticancer agents.
 | ||
| Fig. 9 Structures of anthraquinone analogs reported in the year 2019. | ||
2.6 2020–2021
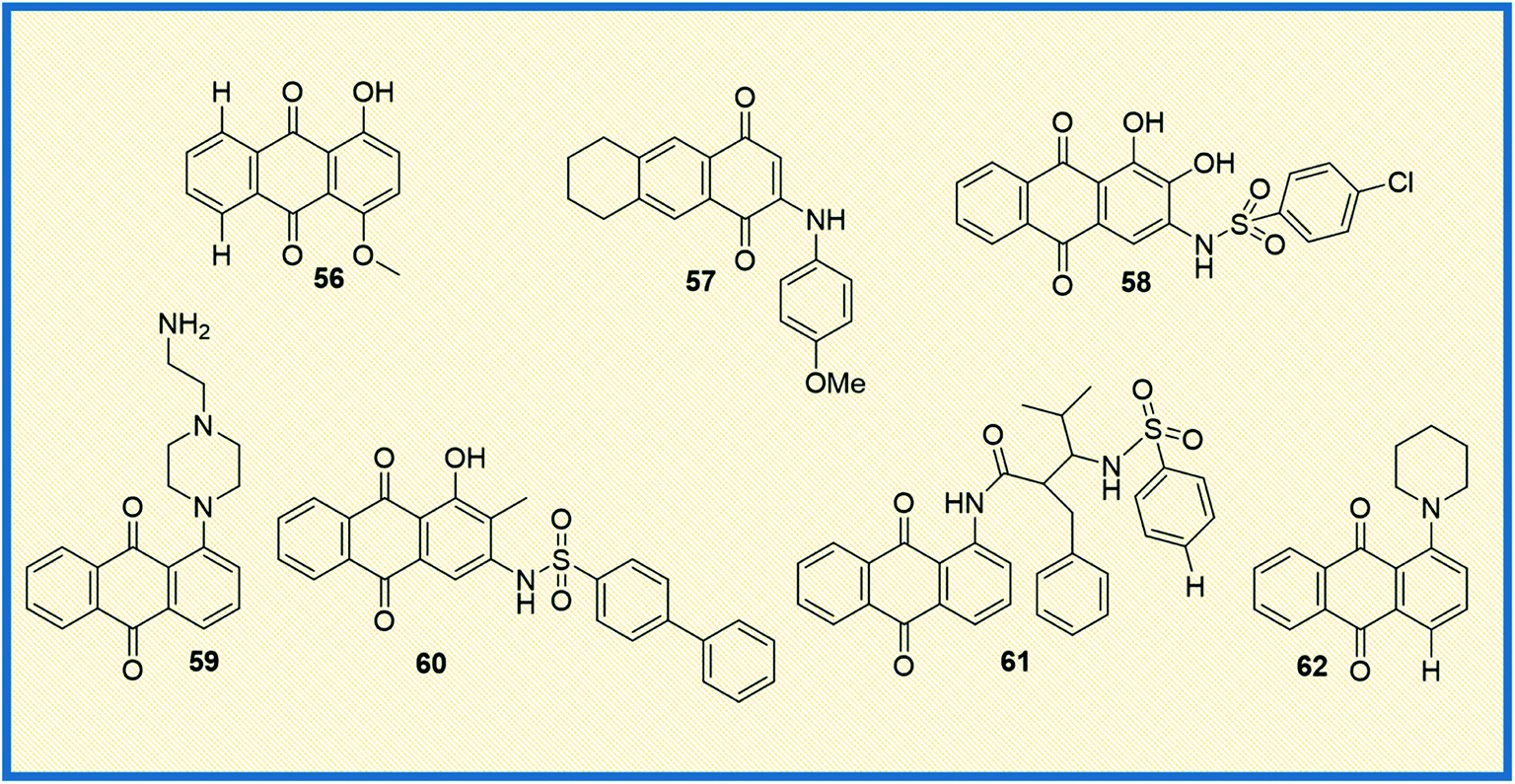
In the last couple of years, several anthraquinone-based compounds were synthesized and investigated for anticancer properties. The specific objective of the study carried out by Li et al. was to synthesize emodin anthraquinone derivatives using microwave-assisted one-step process. The synthesized compounds were examined for their antiproliferative activity in cancer cells. Among all the tested compounds, compound 63 exhibited antitumour effect in HCT116 cells, with an IC50 value of 108.1 μM (Fig. 10). Moreover, it displayed good apoptosis induction by G0/G1 cell cycle arrest and increased the reaction oxygen species at an intracellular level.84 In another study, Li et al. evaluated S. lycopersici. Associated with D. gemmacea and isolated two new anthraquinone derivatives, alterporriol Y and macrosporin 2-O-α-D-glucopyranoside. Apart from this, few other known compounds like altersolanol B and altersolanol A were also isolated. All the isolated compounds were evaluated for their antitumour activity in various cancer cells. Altersolanol A (64) exhibited significant inhibitory activity with IC50 values of 9.0 and 7.2 μM in HCT-116 and MCF-7 cells, respectively. Altersolanol A also showed growth inhibitory activity in Huh7 cancer stem cell-like cells making it a promising candidate for anticancer agents.85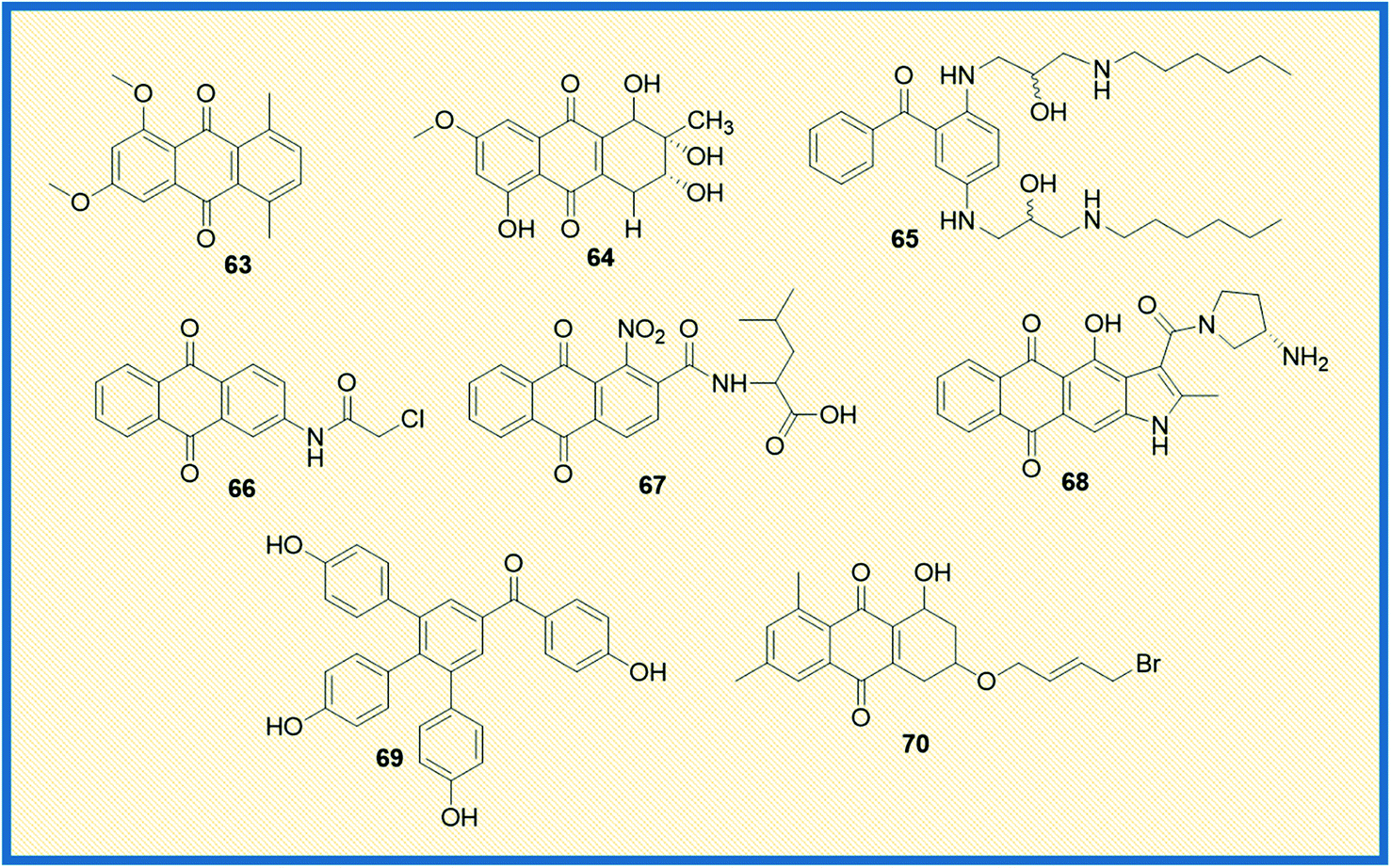 | ||
| Fig. 10 Structures of anthraquinone derivatives reported in the year 2020–2021. | ||
Although mitoxantrone is an established anthraquinone analog to treat cancer, its cardiotoxicity and other serious side effects are less desirable. Hence, Oliveira et al. synthesized N-alkylated and O-alkylated anthraquinone derivatives to overcome this limitation. The compounds synthesized were evaluated for their antiproliferative activity in MCF-7, HeLa, M059J tumour cells, and GM07492A non-cancerous human cells. Among all the synthesized, compound (65) showed the highest cytotoxic activity with IC50 values of 13.6, 14.1, and 14.8 μM on MCF-7, HeLa, and MO59J cells, respectively.86 Another compound from the series also exhibited antitumour activity with a selectivity index of 1.66 in HeLa and 1.87 in MCF-7 cells. Upon evaluating the structure–activity relationship, it was concluded that the cancer cell selectivity was dependant on the lipophilic nature of the substituents at the 1st and 4th positions of anthraquinone.
Anifowose et al. synthesized structural analogues of AQ-101 to evaluate the structural activity relationship of the compounds in acute lymphoblastic leukemia (ALL) cells. Among the synthesized compounds, 66 displayed significant cytotoxic activity against the leukemia cell line with an IC50 value of 0.74 μM. The biological activity of these compounds was evaluated using WST-8 assay. It was deciphered that the active compound 66 exhibited cytotoxicity through a different mechanism than its reference compound. Unlike the reference compound AQ-101, it up-regulated p53 expression but did not induced MDM2 degradation. The structural evaluation suggested that adding a methylene moiety by replacing the –NH in chloroacetamide group decreased cytotoxic activity but did not subdue the activity.87
Since chemotherapy causes several undesirable side effects, the research for new anticancer drugs continues to be a lucrative area of research. In a recent study, Li et al. focused on synthesizing amide anthraquinone derivatives to target the proteins in cancer cells specifically. Among the compounds synthesized, 67 showed significant efficacy with an IC50 value of 17.80 μg mL−1 in HCT116 cells. Upon evaluating the biological activity, it is found that the synthesized compounds induce tumour cell apoptosis by activation of ROS-JNK, which in turn releases cytochrome C into the cytoplasm. This reaction further sets off the e-cysteine protease pathway.88 Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Index Analysis (CoMSIA) models were used to analyze the structure–activity relationship of the compounds. From the studies, it was interpreted that the activity of the compounds greatly depended on the electron-withdrawing capacity of the nitro group at the C-1 position; the higher the electron-withdrawing capacity, the more the inhibitory activity of the compound.
A considerable amount of research has been published on the potential of heteroarene-fused anthraquinones as antitumour drugs. In this direction, Tikhomirov et al. examined the role of heterocyclic moiety tethered anthraquinones in regulating cancer. They synthesized a series of naphtho[2,3-f]indole-3- and anthra[2,3-b]thiophene-3-carboxamides that showed anticancer activity similar to the reference compound doxorubicin. Among all the compounds, naphtho[2,3-f]indole-3-carboxamide (68) exhibited anti-proliferative activity with IC50 values of 0.5 ± 0.2 μM; 0.9 ± 0.1 μM; 0.9 ± 0.2 μM; 0.9 ± 0.1 μM; 0.8 ± 0.1 μM against Capan-1, HCT116, NCIeH460, HL60 and K562 cancer cell lines respectively. The compound 68 also showed better DNA affinity as compared to its furan and thiophene counterparts. From in vivo studies, it was concluded that the compound enhanced the lifespan of mice carrying P388 lymphoma transplants hinting at tumour inhibition.89 Selaginella tamariscina is a traditional Chinese herb used to treat diseases like cancer, diabetes, and hepatitis. Rui Liu et al. isolated four new anthraquinone compounds selaginones A, selaginones B, triarylbenzophenone analogue, selagibenzophenone B from S. tamariscina herb. Among the compounds isolated, compound 69 showed antiproliferative activity against SMMC-7721 and MHCC97-H cell lines with IC50 values of 39.8 and 51.5 μM, respectively. The antiproliferative activity was tested using the CCK-8 method.90 Lin et al. synthesized 13 anthraquinone derivatives and tested them against a known reference compound cisplatin. Among the synthesized derivatives, 70 showed significant cytotoxicity in NTUB1 and PC3 cells with IC50 values of 1.51 ± 0.31 μM, and 12.78 ± 1.46 μM, respectively (Fig. 10). They further established the efficacy of the compounds using autophagy and MTT assays. Compound 70 at 1 and 3 μM concentrations induced DNA damage and triggered apoptosis in NTUB1 cells. The structural evaluation of 70 suggested that the hydroxy group at C-1 significantly enhanced the antiproliferative activity. Simultaneously, replacing the bromo atom in the side chain of C-3 significantly reduced the cytotoxicity.91
3. Development of target-specific cytotoxic anthraquinone derivatives
Apart from the above discussed year-wise non-target specific literature, the following section highlights the specific enzyme targeting ability of anthraquinone. Targets such as topoisomerases,92 telomerase,56 protein kinases,93 MMPs,94 and DNA95 are the major enzymes with which anthraquinones are known to exert their action.3.1 Topoisomerase inhibitors
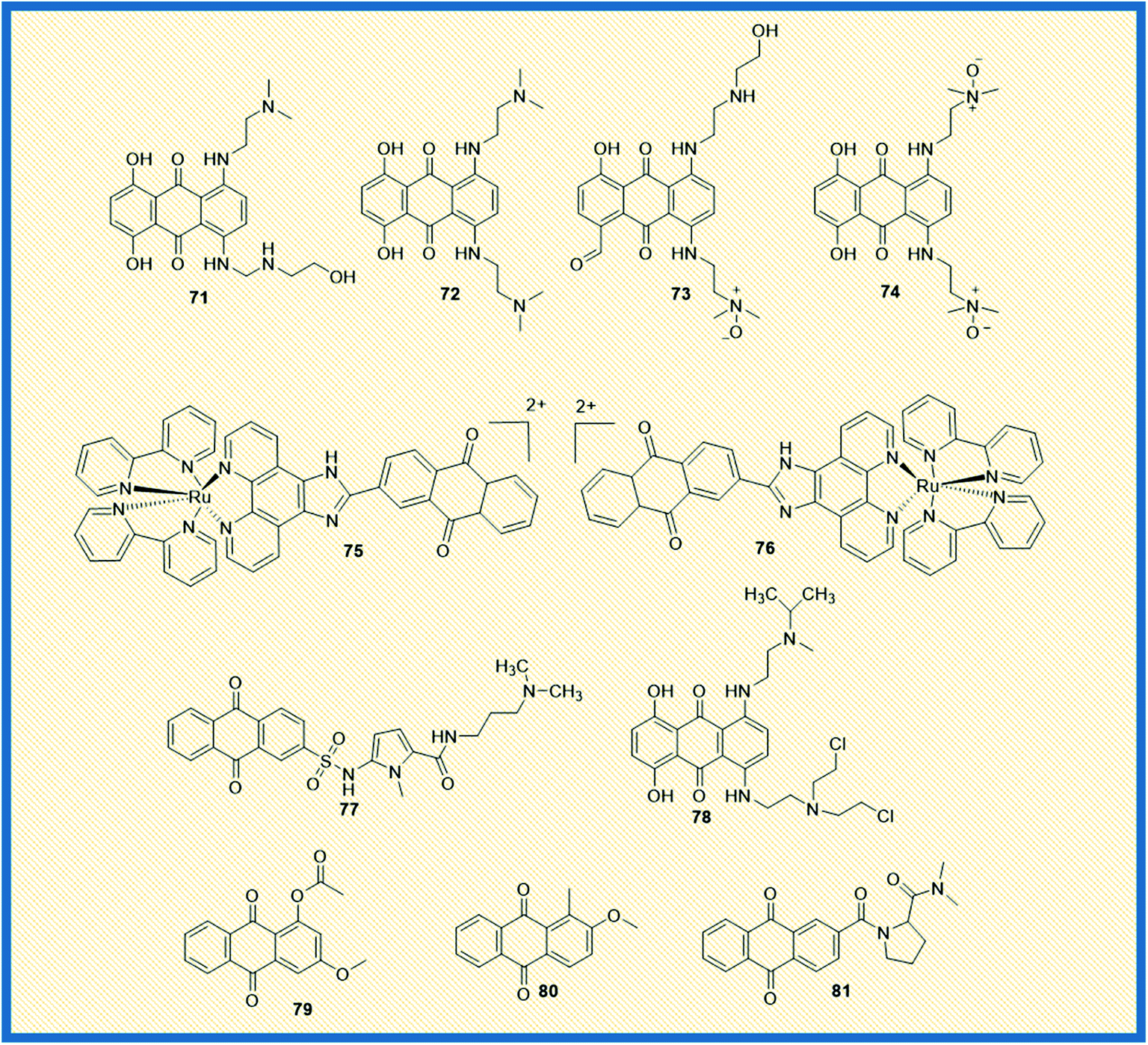
Topoisomerases remain an attractive chemotherapeutic drug target for the discovery and development of novel anticancer agents. Different types of topoisomerase enzymes play a crucial role in DNA replication and transcription within the cells. In addition, the enzymes are involved in the relaxation of positive or negatively supercoiled DNA, the introduction of positive or negative supercoils into the DNA, and catenation or decatenation of circular and linear DNA, which are vital for cell survival. The enzymes are also accountable for regulating cellular processes other than replication and transcription, DNA repair, chromosomal condensation/segregation, and so on.96 Type I and type II topoisomerases are the subfamilies of DNA topoisomerases. In general, Type I topoisomerases interrupt DNA topology by creating a transient single-strand DNA break followed by passage of the opposing single strand in duplex DNA using tyrosine residue in the active site to cleave the DNA strand and form a phosphodiester bond. On the other hand, double-stranded breaks are generated by type II topoisomerase using tyrosine residues in the active site.97 Functionally, type I topoisomerases are non-ATP-dependent proteins; hence they depend on the intrinsic strain energy of the supercoiled DNA. In contrast, type II topoisomerases are ATP-dependent proteins and possess a DNA-binding domain and ATP-binding domain. Two biochemically and genetically different topoisomerase II (topo II) forms exist in mammals and are named topo IIα and topo IIβ. Topo IIα plays a significant role in mitotic processes and is present in only proliferating cells. At the same time, topo IIβ is present in all the tissues and expressed abundantly in post-mitotic neuronal cells.98 Topoisomerase I, IIα, and IIβ are the principal targets for several marketed cancer drugs. Anthracyclines are the derivatives of anthraquinones, which are the first recognized class of topoisomerase inhibitors used in cancer chemotherapy.99 Doxorubicin (9), epirubicin (10), valrubicin (11), daunorubicin (7), and idarubicin (8) are clinically marketed anthracycline derivatives (Fig. 2). Further, emodin (2), a naturally occurring anthraquinone obtained from plants and fungi, inhibits DNA topoisomerase II. It generates DNA double-strand breaks through stabilization of topoisomerase II–DNA cleavage complex, thereby inhibiting ATP hydrolysis.97McKeown et al. and Smith et al. reported the topoisomerase activity of the alkylamino anthraquinones with their mono-N-oxide structures. Almost all the compounds exhibited good anticancer activity and also elevated the levels of topo IIα. Compound 71 displayed greater cytotoxicity and inhibited DNA synthesis in the S-phase of the cell cycle and was more active than the marketed drug mitoxantrone (Fig. 11). Though IC50 value of compound 71 is not reported, the molecular modelling investigations demonstrated that the compound could form stable, intercalated complexes with DNA. Other derivatives such as 72 showed similar cytotoxicity as that of mitoxantrone. The compound 72, when bound to DNA, inhibits topoisomerase II. It was found that any diffusion of compound 72 would result in toxicity to cells irrespective of the level of oxygenation. Further, the analogues like 73 (mono-N-oxide) and 74 (di-N-oxide) are presently in in vivo preclinical evaluation to establish their potential role as bio reductive agents in radiotherapy.100,101
 | ||
| Fig. 11 Anthraquinone derivatives effect the activity of topoisomerases. | ||
In addition, two Ru(II) chiral anthraquinone complexes 75 and 76 showed dual inhibition against topo I and II enzymes. The complexes were intercalated with DNA nucleotide base pairs by strong binding affinity.102 The propylamine oligopyrrole carboxamides linked with anthraquinones revealed promising anticancer activity by inhibiting topoisomerase I to enhance the biological activity of combilexins (77).103 Alchemix (78), a novel alkylating anthraquinone, displayed effective anticancer activity in both in vitro and in vivo in drug-resistant (doxorubicin and cisplatin) ovarian cancer cells. The molecule specifically inhibited topo IIα as compared to topo IIβ.104 The anthraquinone derivatives extracted from the roots of Rubia cordifolia, 79 and 80 exhibited maximum inhibition of topoisomerase I at 100 μM concentration.105 Further, a series of proline derivatives of anthraquinone-2-carboxylic acid displayed good cytotoxic activity against MCF-7 cells. The analogue 81 inhibited the catalytic activity of both topoisomerase I and II at 30 and 60 μM concentrations, respectively (Fig. 11).106
3.2 Matrix metalloproteinase (MMP) inhibitors
Matrix metalloproteinases (MMPs) are Zn and Ca-dependent neutral endopeptidases that play a crucial role in the physiological and pathological remodelling of the extracellular matrix.107 Based on the substrate specificity, MMP enzymes are categorized into five main groups: gelatinases, stromelysins, collagenases, membrane type enzymes, and others. The activity of the MMPs was evaluated in different disease areas like cancer, cardiovascular diseases, atherosclerosis, and arthritis. In cancer, the MMPs are mainly involved in invasion and metastasis.108 Among different types of MMPs, gelatinase B (MMP-9) has been associated with the invasive stage of carcinomas. MMP-9 destroys extracellular matrix components such as type I and IV collagen, a major component of the membrane.109Naturally occurring aloe-emodin (3) acts as a potent antitumour agent which inhibits MMP9 enzyme. The treatment of 3 with B16-F10 melanoma cell decreased proliferation in a time-dependent manner, with negligible cell toxicity. Antimetastatic capability of 3 was reported to be involved in induction of cell differentiation, increase in homotypic aggregation, reduction of both cell motility, and shape fickleness. The gelatin-zymographic analysis showed that aloe-emodin inhibits the secretory MMPs in B16-F10 cells. Compared to the untreated sample, the compound reduced 33% of the MMP-9 activity after 48 h, while a 29% reduction in the enzymatic activity occurred after 72 h. In contrast, MMP-2 activity was slightly increased after 48 h and was back to the control value after 72 h of aloe-emodin treatment.110 Further, aloe-emodin inhibited the nuclear translocation and DNA binding of NF-κB, a crucial transcription factor that controls MMP-2/9 and VEGF gene expression. Aloe-emodin successfully inhibited MMP-2/9 expression at both mRNA as well as protein levels.111
3.3 Telomerase inhibitors
Telomerase, a reverse transcriptase enzyme that stabilizes the telomere length and maintains the chromosome integrity, plays a vital role in cellular immortalization. In general, telomerase is expressed in germline cells and highly expressed in cancer cells (85%), whereas in normal somatic cells, the enzyme expression is not significant.112 When telomerase is repressed or inhibited, the cells can divide only a few times.113 Due to the differential expression of the telomerase, the enzyme is considered an essential molecular and specific drug target for cancer therapy. The expression of the catalytic subunit of telomerase, such as human telomerase reverse transcriptase (hTERT), seems to be a significant determinant for telomerase activity. In addition, stabilized secondary DNA structures such as G-tetraplexes are also active targets for drugs that bind directly to the telomerase and disrupt the telomere structure.562,6-Diamidoanthraquinone (82) showed promising anticancer activity as compared to mitoxantrone (14) and also inhibited the telomerase activity with an IC50 value of 0.1 μM (Fig. 12).114 Anthrax[1,2-d]imidazole-6,11-dione tetracyclic analogue (54) (NSC749235), a new telomerase inhibitor, exhibited good cytotoxicity against HeLa and A549 cell lines. Further, DNA binding and molecular modelling studies revealed that the analogue targets the potassium form of human telomeric G-quadruplex DNA at micromolar concentrations.76 Further, a series of 2,7-diamidoanthraquinone analogues exhibited a better inhibitory effect on telomerase activity, hTERT expression, and cell proliferation. Among all, anthraquinone derivative 83 revealed better telomerase inhibition by activating the expression of hTERT and secreted embryonic alkaline phosphatase levels (SEAP) without affecting the cell proliferation in the range of 1–20 μM.115 Further, another set of anthra[1,2-d]imidazole-6,11-dione derivatives showed promising anticancer activity against all the tested cell lines. Few of the compounds in the series exhibited promising anticancer activity towards NCI-60 panel cell lines. In addition, the analogues 84–86 affected the expression of SEAP without affecting cell proliferation. Also, the compounds selectively repressed the expression of hTRET and inhibited the telomerase activity. It was reported that small sidechain extension might have better cytotoxic effects on PC-3 cells. Among the synthesized, 84, 85, and 86 displayed moderate potency against PC-3 cells with IC50 values of 10.3 μM, >30 μM and 16 μM respectively. Similarly, compounds 84, 85, and 86 showed IC50 values of <1 μM against the inhibition of PhTERT-SEAP cells of H1299 in MTT assay. All three compounds showed IC50 at ∼100 μM towards IMR90 cells, and suggesting that they did not affect the growth of normal cells.116 The di-amino substituted anthraquinone derivative (87) displayed better telomerase inhibition (43.3%) at a 10 μM concentration, and the compound also intercalated in the DNA.117 Perry et al. synthesized a series of 1,4- and 2,6-di functionalized amido anthracene-9,10-diones and examined their cytotoxicity along with telomerase inhibitory activity. Among the synthesized, piperidine 2, 6-anthraquinone derivative (88) and N,N′-dimethiodide derivative (89) elicited good enzymatic inhibition of telomerase with IC50 values of 4.5 and 9.4 μM, respectively.118 Huang et al. reported the synthesis of 1,5-bisthioanthraquinones and 1,5-bisacyloxyanthraquinones and examined their telomerase activity along with the expression of telomerase. The most active compounds 90, 91, and 92 exhibited relative secreted embryonic alkaline phosphatase (SEAP) activity at concentrations of 2.8, 2.4, and 1.8 μM against PhTERT-SEAP (H1299) and PhTERT-SEAP (hTERT-BJ1) cell lines. The 1,5-bisacyloxyanthraquinones (90, 91, 92) demonstrated good telomerase inhibitory activity and activated hTERT expression without affecting the cell viability (Fig. 12).119
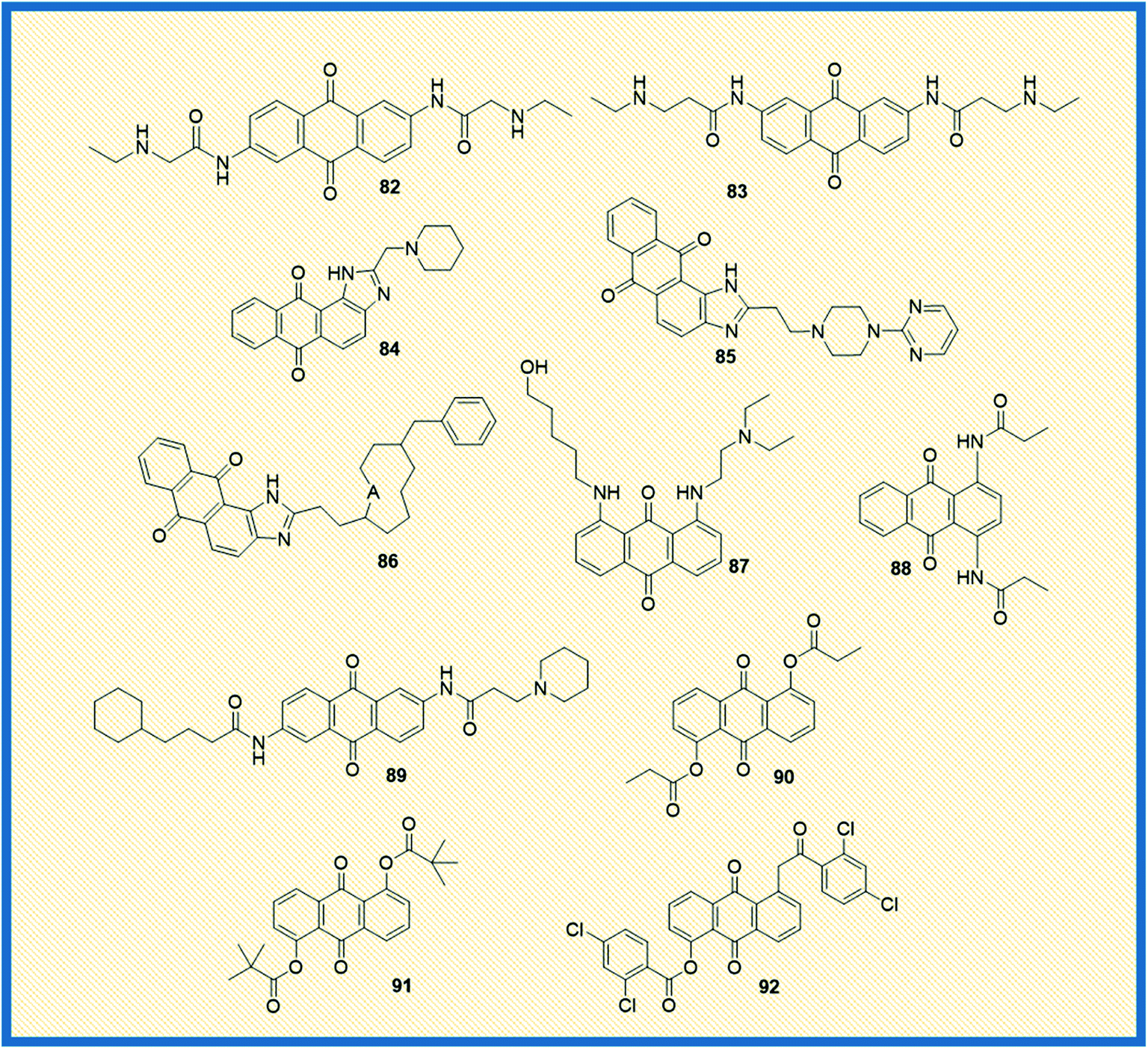 | ||
| Fig. 12 Structures of anthraquinone derivatives that target telomerases. | ||
DNA can acquire a range of alternative conformations based on specific sequence motifs and interactions with several proteins. Among these conformations, G-quadruplex structures are a form of non-canonical nucleic acid structures that can form within specific repetitive G-rich DNA or RNA regions.120 These G-quadruplex structures are unique and extensively involved in the regulation of several biological processes. In general, G-rich repeat sequences with the capability to form G-quadruplex structures are present and overrepresented in telomeres, transcriptional start sites, and double-strand break sites.121 The presence of G-quadruplex structures in telomeres is capable of inhibiting the activity of telomerase, an enzyme that is overexpressed in cancer cells.122 Hence, targeting the G-quadruplex structure is one of the promising strategies in developing anticancer therapeutics.
Due to the structural diversity and promising therapeutic activity, some of the anthraquinones derivatives are significantly bound and involved in stabilizing G-quadruplex structures. Das and Dutta reported the promising anticancer activity of anthraquinone-based natural compounds like aloe emodin, aloe emodin-8-glucoside, and aloin. Further, the authors investigated the binding affinity of these compounds against a set of six different quadruplex structures like c-KIT, c-MYC, HUMTEL, BCL-2, KRAS, and VEGF. Among all the examined structures, aloe emodin (3) exhibited significant binding affinity, i.e. (2.11 ± 0.33) × 105 M−1 towards c-KIT as compared to aloe emodin-8-glucoside ((9.70 ± 0.50) × 105 M−1). In contrast, aloin was not capable of targeting the quadruplex structures.123 In addition, Wang et al. reported the G-quadruplex structure stabilization activity of aloe emodin. Aloe emodin reduced the transcription of hTERT gene in the three different breast cancer cell lines such as MDA-MB-453, MDA-MB-231 and MCF-7. The results unveiled that aloe emodin binds and stabilizes the G-quadruplex DNA with a binding affinity of 2.55 × 106 M−1 and subsequently inhibits the enzymatic activity of the telomerase.124
In another study, Mei et al. reported the synthesis of a ruthenium(II) complex of emodin and the biological activity of the compound against c-myc G4 DNA. The compound showed good binding affinity with c-myc G-quadruplex DNA with binding affinity of 6.7 ± 0.19 × 104 mol L−1.125 Similarly, Elvira et al. synthesized nitrogen substituted 1-(3-aminoprop-1-ynyl)-4-hydroxyanthraquinone derivatives and studied their anticancer potential in a panel of cancer cells like MCF-7, U-87 MG, DU-145, SNB-19, and hTERT lung fibroblasts. Further, the molecular binding studies of the synthesized compounds towards DNA G-quadruplex revealed that almost all the derivatives showed good binding affinities towards DNA motifs.126 In yet another research study, 2,6-disubstituted amido anthracene-9,10-dione dimeric distamycin derivatives were designed and synthesized. Among all the synthesized derivatives, the disubstituted anthraquinone with tri-N-methylpyrrole side chain (ANTP) was found to be more promising and exhibited good activity towards c-Myc G-quadruplex DNA with a binding affinity of 3.8 ± 0.01 × 106 M−1.127
3.4 Kinase inhibitors
Protein kinases are the enzymes that phosphorylate protein by transferring γ-phosphate group to the protein, whereas phosphatase removes the phosphate group from protein. Phosphorylation is the most common form of reversible post-translational modifications of the protein.128 Approximately 50% of all proteins undergo phosphorylation, and specific kinases, as well as phosphatases tightly, control this process. Almost 538 known kinases are identified in the human genome. These kinases maintain cellular functions by switching the protein function on most protein kinases involved in signalling networks that employ phosphorylation, which modulate target protein activities. The kinases are critically involved in almost all cellular processes that promote cell survival, proliferation, metabolism, and migration.129 The abnormal expression of kinases leads to oncogenesis and other diseases. Several kinases are identified that are involved in cancer cell signalling pathways, angiogenesis, proliferation, and metastases of various types of cancer.130 Due to widespread clinical applications, kinases are considered promising drug targets for anticancer therapeutics.3-(Azidomethyl)-1,8-dihydroxy-6-methoxy anthracene-9,10-dione (93) is a phyto-based emodin derivative isolated from giant knotweed. The compound exhibited potent anticancer activity in both in vitro and in vivo models (Fig. 13). Further studies revealed that the compound inhibits the overexpression of Her2/neu in lung cancer and breast cancer through proteasomal degradation of Her2/neu.115 Another study demonstrated that the anthraquinone derivative 94 was more effective compared to emodin. It inhibited cell proliferation and transformation of HER-2/neu, which is overexpressed in human breast cancer cells via blocking the tyrosine phosphorylation of p185neu. The IC50 of 94 was found to be 17 μM and 1 μM against tyrosine phosphorylation of HER-2/neu and MDA-MB-453 cells, respectively.131 Further, the combination of emodin and paclitaxel synergistically inhibited the anchorage-dependent and -independent growth of HER-2/neu overexpressing breast cancer cells (MDA-MB-361) by 70% in in vitro assay along with inhibition of the tumour growth.93 Muto et al. reported that emodin extracted from the root and rhizome of Rheum palmatum L., induced apoptosis in myeloma cells. In addition, emodin down-regulated the Mcl-1(induced myeloid leukaemia cell differentiation protein), leading to the apoptotic cell death of cancer cells.132 Damnacanthal (95) is a potent natural anthraquinone molecule that selectively inhibits p56lck tyrosine kinase with an IC50 value of 17 nM. Further, the compound also has therapeutic efficacy in treating T-cell malignancies and autoimmune diseases.133 In a separate study, Shi et al. isolated several antiproliferative anthraquinone derivatives from Hedyotis diffusa. Among the isolated compounds, 2-hydroxy-3-methylanthraquinone (96) induced apoptotic mediated cell death in malignant cells via mitochondrial pathway by inhibiting receptor Src tyrosine kinase. Compound 96 displayed IC50 values of 33 μM and 67 μM against protein tyrosine kinases activities of pp60-src (3 U mL−1), active GST-v-src protein (0.1 mg mL−1) and natural SPCA-1 cell lysate (0.5 mg mL−1) prepared as target proteins. Further, compound 96 exhibited an IC50 value of 51 μM against HepG-2 cell lines.134 1-Deoxyrhodoptilometrin (97), another anthraquinone derivative, is a marine metabolite described to act as a potential lead for anticancer activity by inhibiting various distinct protein kinases such as EGFR, ERBB-2,4 and IGF-1. Compound 97 showed inhibitory activity against 23 protein kinases and was found to be the most potent inhibitor of Aurora-A, Aurora-B, EGF-R, SRC, and VEGF-R2 at IC50 of 3, 1.8, 4, 3.7, and 1.8 μM, respectively.135 Similarly, quinalizarin (98), a potent kinase inhibitor, possessed the ability to selectively inhibit CK2 (Ser/Thr protein kinase) comparable to emodin. It induced apoptosis more effectively than other CK2 inhibitors, which are commonly used like 4,5,6,7-tetra bromo-1H-benzotriazole and 2-dimethylamino-4,5,6,7-tetra bromo-1H-benzimidazole (Fig. 13). The IC50 value of quinalizarin was found to be 0.11 μM inhibiting HEK-293T cells. Jurkat cells upon treatment with compound 98 (5 μM) for 4 h treatment showed a 48% fall in CK2 activity in the cell lysates. Compound 98 (quinalizarin) is structurally very similar to emodin, a quite promiscuous inhibitor of CK2 and of several other protein kinases as well. It exhibited potency toward PIM3 (IC50 of 0.08 μM) that is 30-fold higher than that of CK2 (IC50 of 2.50 μM).136
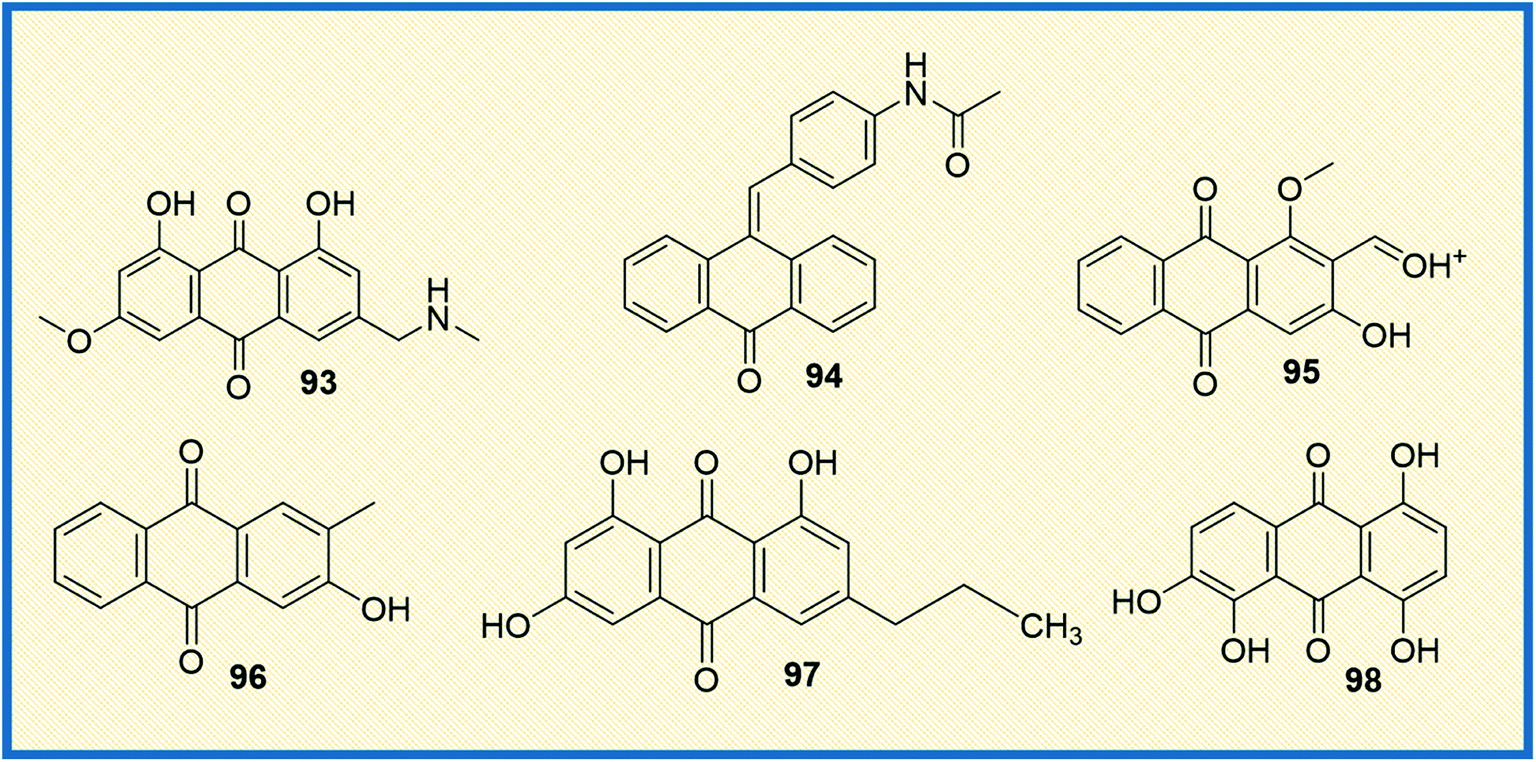 | ||
| Fig. 13 Anthraquinone derivatives targeting kinases involved in cancer progression. | ||
3.5 Miscellaneous cancer targets
Ecto-nucleotidases are the enzymes that hydrolyse the extracellular nucleotides to nucleosides and control nucleoside (P1) and nucleotide (P2) receptor-mediated signaling.137 The enzyme alters the adenosine level that in turn increases or decreases P1 and P2 receptor activity. Hence, the inhibition of adenosine production in the tumour cell environment, through inhibiting the enzyme activity, might be a promising and novel strategy for anticancer therapy.138,139 Baqi Y reported that some of the anthraquinone derivatives 99 and 100 were found to be potent inhibitors of ecto-nucleotidase with inhibitory constant (Ki) values of 150 and 260 nM, respectively (Fig. 14).140 Similarly, physcion (101), a naturally occurring anthraquinone derivative, is a promising anticancer agent primarily used to treat human nasopharyngeal cancer. It induced apoptosis and autophagy in human nasopharyngeal cancer cells by the downregulation of transcription factor Sp1. Compound 101, also known as parietin, upon treatment with physcion (5, 10, and 20 μmol L−1) in a dose-dependent manner suppressed the cell viability and colony formation in CNE2 cells. Physcion (10 and 20 μmol L−1) dose-dependently blocked cell cycle progression at G1 phase and induced both caspase-dependent apoptosis and autophagy in CNE2 cells. Similarly, 101 induced apoptosis and autophagy in human nasopharyngeal carcinoma cells by targeting Sp1, which was mediated by ROS/miR-27a/ZBTB10 signaling.141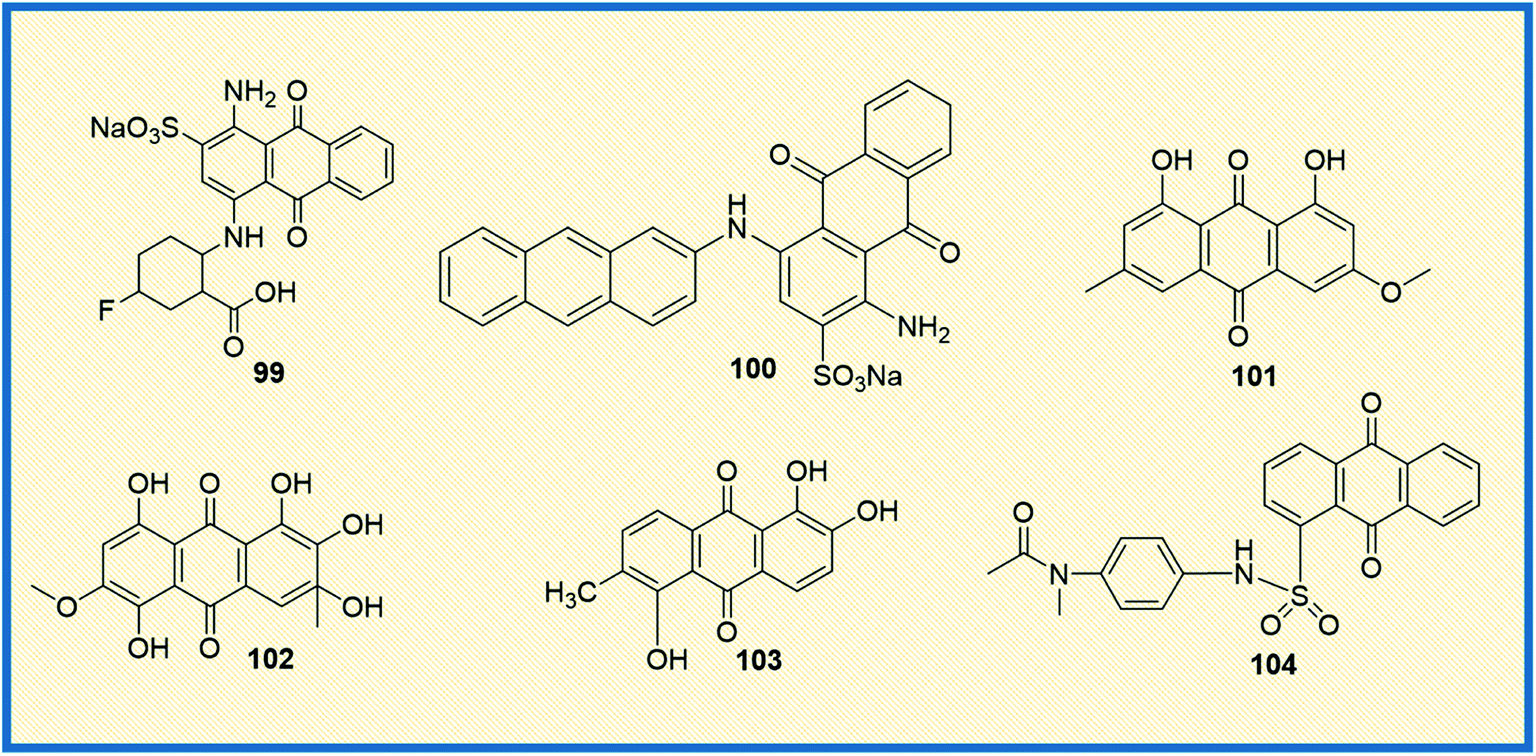 | ||
| Fig. 14 Anthraquinone derivatives that target other miscellaneous proteins involved in cancer growth. | ||
Apart from the mentioned targets, emodin exhibited selective activity towards human nasopharyngeal cancer cells (CNE-2Z). It reduced cell viability and induced cell cycle arrest and apoptotic cell death by targeting the chloride channels in CNE-2Z cells compared to positive control tamoxifen.142 In addition, emodin exhibited an anti-metastatic effect by the downregulation of CXC chemokine receptor type 4. The CXC chemokine receptor type 4 plays a crucial role in cancer invasion and metastasis.143 Furthermore, emodin also inhibited Vascular Endothelial Growth Factor Receptor (VEGFR) and MMPs in connotation with downregulation of runt-related transcription factor 2 (Runx2), which controls both VEGF signalling and transcriptional activity.144 Further, emodin has a structural similarity with ATP Citrate Lyase (ACL) inhibitors. ACL plays a significant role in de novo fatty acid and cholesterol biosynthesis. Moreover, the enzyme is highly expressed in some of the cancer cells. Hence it can act as a promising anticancer agent by inhibiting the ACL enzyme.145
Another naturally occurring anthraquinone 102, isolated from endophytic fungi, was reported as an anticancer agent that induced caspase-mediated apoptosis and also suppressed phosphorylation of Akt kinase. Compound 102 displayed activation of caspases (8, 9 and 3) and poly (ADP-ribose) polymerase (PARP) in MCF-7 and MDA-MB-435 breast cancer cells significantly at concentrations of 3.75 μM and 3 μM, respectively.146 Similarly, Kamiya et al. isolated ten anthraquinones from Morinda citrifolia roots and examined their anticancer potential against human colon cancer (HCT116) cells and DNA polymerase activity. One of the compounds, morindone (103), showed significant polymerase inhibition properties, thereby suppressing the growth of HCT116 cells. It induced cell growth suppression with LD50 value 32.2 μM on human DNA polymerase γ.147 Finally, the sulphonamide-anthraquinone derivative 104 was reported to inhibit cancer cell progression in several types of cancer cells. It significantly inhibited the proliferation of HCT116, HCT116 p53—/— and p53 mutant HT29 (R273H) cells at a level comparable to that of the positive control 5-FU (10 μM) in MTT assay. The derivative 104 induced cell cycle arrest at the S and G2/M phases (38.5, 42.2, and 33.4; 28.1, 29.5, and 49.0%, respectively) compared to that induced by the vehicle control (30.2, 36.3 and 21.2; 23.4, 27.1, and 20.5%, respectively). Treatment with 10 μM of 104 increased caspase-3 cleavage in all three cell lines (HCT116, HCT116 P53—/— and HT29). These results indicated that 104 induced both apoptotic and necrotic cell death in a p53-independent manner (Fig. 14).148
4. Toxicity of anthraquinones
Toxicity or safety assessment of a potential drug molecule is the primary area of concern before clinical usage.149 Apart from the favourable pharmaceutical applications, some anthraquinone derivatives may induce potential damage to cells due to their close resemblance to anthracene, a toxic analogue.150 A few in vitro studies describe the toxicity behavior of anthraquinone derivatives with limited information on in vivo studies of anthraquinones. Liu et al. investigated the in vitro and in silico hepatotoxicity of different anthraquinones and their derivatives. Among all the studied, rhein was identified as a potential liver toxicant against HuH-7 cells with an EC50 value of 93.9 μM upon repeated treatment.151 Chen et al. examined the photoinduced acute toxicity of a series of fourteen anthraquinone derivatives against Daphnia magna, an important model organism for toxicity assessment. Almost all the compounds exhibited no observable toxicity in the presence of visible light at the maximum concentration of the compounds used in the study. However, chloro and dihydroxy substituted anthraquinones exhibited apparent toxicity in presence of visible light with EC50 values of 837.0 and 959.3 nmol L−1, respectively.152Further, Viljoen et al. summarized the toxicity of emodin against mouse and rat foetuses. The study revealed that emodin displayed adverse effects against mice and rats at 17 and 60 mg kg−1 or higher doses. In addition, emodin was noticed to be toxic towards brine shrimp and exhibited lethality with an LC50 value of 0.19 μM.153 Oshida et al. examined the toxicological effects of emodin on differential gene expression profiles of the testis in mice models. The compound producing testicular toxicity in male mouse models via IGF-1 receptor signalling pathway.154 He et al. investigated the potential toxicity effects of emodin on zebrafish embryos. The compound displayed adverse effects on embryo survival as well as hatching success in zebrafish at a concentration of 0.93 μM and higher. In addition to this, it induced many abnormalities in zebrafish embryos, including oedema, abnormal morphogenesis, and crooked trunk.155
Anthraquinones present in plant species such as Rhamnus species (aloe-emodin, physcion, rhein, chrysophanol, emodin) were suspected to be involved in diseases such as renal failure, rhabdomyolysis, nephrotoxicity (regular intake), dehydration, anorexia (long-term exposure), genotoxicity in mammalian cells, mutation, tumour promotion, chromosomal aberration and liver enlargement. In addition to this, the long-term use of anthranoid laxatives is reported to exhibit symptoms associated with melanosis coli which is characterized by dark pigmentation of the colonic mucosa, and in few cases, it is also believed to result in morphological changes of the colonic myenteric system.156 This is because anthraquinone-based drugs contain chromophores which impart a bright yellow colour to colonic epithelial cells and is reversible in most of cases. However, they are also suspected to cause permanent physical change to colonic tissue as well as permanent damage of renal tubular cells resulting in more serious conditions such as Chron's disease or ulcerative colitis.
The anthraquinone derivatives, especially those derived from Rhubarb have shown neuroprotective effects. Emodin exerted a neuroprotective effect in cerebral ischemic stroke (CIS) by maintaining the integrity of blood–brain barrier (BBB), ameliorating inflammation, and controlling the apoptosis process. Li et al. attributed this effect of emodin to the inhibition of connexin 43 (Cx43) and aquaporin 4 (AQP4).157 The neuroprotective effect of chrysophanol (CHR) was reported to be associated with oxidative/antonitrosative, anti-inflammatory and by inhibiting apoptosis. Similarly, Zhao et al. reported neuroprotective effect of rhein is by oxidative stress and apoptosis.158
5. Polyethylene glycol (PEG) based anthraquinone drug delivery
Anthraquinones are one of the key chemotherapeutic agents used in the early management of cancer. Anthraquinones are potent anti-cancer agents with many beneficial effects; however, few unwanted properties such as low polarity and structural instability in in vivo conditions are also observed in some cases. Current research studies mainly focus on enhancing the biological efficacy of naturally occurring and chemically synthesized anthraquinone derivatives. However, few drug delivery approaches are reported to address the undesired properties of low polarity and stability. It is imperative to develop drug delivery vehicles for anthraquinone or build multi-drug delivery systems to administer two or three anti-cancer agents in one dose. Polymer based control release formulations are gaining importance as they are very efficient in loading multiple drugs in layers for attaining programmed release kinetics under in vivo conditions.159 One such example was a report where a DNA-damaging anthracycline agent doxorubicin (dox) (10) and a phosphatidylinositol-3 kinase inhibitor wortmannin (wor) were conjugated. These drugs were used alone or in combination with poly(ethylene glycol)-poly(aspartate hydrazide) block copolymers through a hydrazone bond.160 The polymer formed unimodal micelle structure encapsulating the conjugated anti-cancer drugs (dox and wor). The polymer-drug conjugated system developed in this way has particle size of <100 nm. The drug mixing ratios between dox/wor were precisely controlled in this delivery system. This polymer-drug conjugated system reported better drug release properties and also reduced the amount of drug (wor) required for cytotoxicity while maintaining the biological activity of the independent polymeric micelles.Other PEG conjugated systems reported in recent literature include poly(ethylene glycol) PEG-doxorubicin (dox) conjugates with polymers of linear or branched architecture (molecular weight 5000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), and different peptidyl linkers (GFLG, GLFG, GLG, GGRR, and RGLG). GFLG linker showed ∼30% release of dox at 5 h irrespective of PEG molecular weight or architecture. All PEG conjugates prepared by this method were more than 10-fold less toxic (IC50 values >2 μg mL−1) than free Dox (IC50 value of 0.24 μg mL−1). PEG-dox showed greater tumour-targeting than free Dox in radiolabelled studies. The radio iodinated PEGs showed a clear relationship between molecular weight, blood clearance, and tumour accumulation.161
000 g mol−1), and different peptidyl linkers (GFLG, GLFG, GLG, GGRR, and RGLG). GFLG linker showed ∼30% release of dox at 5 h irrespective of PEG molecular weight or architecture. All PEG conjugates prepared by this method were more than 10-fold less toxic (IC50 values >2 μg mL−1) than free Dox (IC50 value of 0.24 μg mL−1). PEG-dox showed greater tumour-targeting than free Dox in radiolabelled studies. The radio iodinated PEGs showed a clear relationship between molecular weight, blood clearance, and tumour accumulation.161
Other more advanced conjugate systems of anthraquinones reported in the literature are polymer enzyme liposome therapy (PELT),162 triple block nanocarrier (TBN) platforms,163 and amphiphilic core cross-linked star (CCS) polymers164 that showed excellent pharmacokinetic profile in in vivo models. In a TBN platform, hydrophilic polyethylene glycol (PEG) was used as an outer shell, and a hydrophobic biodegradable polycaprolactone (PCL) block for encapsulating anthracycline anticancer drug was utilized as the middle layer. The carboxylic-functionalized polycaprolactone (CPCL) based inner shell was filled with non-anthracycline anti-cancer drug for inducing synergistic effect. The dual drug-loaded TBN exhibited superior synergistic cell death at much lower drug concentrations.163
These findings represent the critical role of PEG-conjugated anthracycline-based effective anti-cancer drug delivery methodology that might reduce the effective dose and toxicity in vivo compared to the conventional drug formulations.
6. Structure–activity relationship of anthraquinones
Structure–activity relationship (SAR) studies of the anthraquinone ring structure provide an idea about the primary structural considerations that result in maximum biological effect. Moreover, it is essential to assess the substituents which undergo enzymatic degradation due to endogenous enzymes, thereby eliminating the molecule before it exerts its activity. SAR studies help understand the groups that improve the synthesized molecule's pharmacokinetics and substituents resistant to enzymatic degradation. Few of the SAR observations are discussed here.Studies showed that the sulphonamide and methyl ketone substituent at 1st position of anthraquinone would be advantageous in exerting superior anticancer activity. Similarly, carboxylic acid substitution at 1st position would result in activity loss.63 Another report suggested that when anthraquinone monomers are considered, the position of hydroxyl (C-5 and C-8) is very crucial for antitumour efficacy. The hydroxylation at the C-1 position was reported to drastically enhance its cytotoxic activity, which indicates that the phenolic hydroxyl groups are essential for the antitumour activity of anthraquinones.165 Emodin is identified as the most abundantly existing anthraquinone in plant species having a wide range of anticancer properties on various cancers. Dong et al. identified that C-1 and C-3 are the most important functional sites for antitumour activities of anthraquinone molecules.50 Zhou et al. reported six anthraquinones from Xanthophytum attopvensis pierre and studies revealed that a large steric hindrance due to glycosyl substituents was the reason for the weakening of the bond between drug and target cells. The substituents such as hydroxyl or hydroxymethyl groups present at only the C-1 position, then the anthraquinones exhibit similar cytotoxic activity. In one of the comparative studies between emodin (anthraquinone) and cassiamin (bianthraquinones), bianthroquinones showed lower anti-cancer activities than anthraquinones, and the reason ascribed to this observation was the steric hindrance of bianthroquinones with a distorted confirmation.166 Additionally, the atomic charge at C-10 position and the number of hydroxyl groups present on the benzene ring of monomer increased its anti-cancer potency compared to its dimer. Similarly, chlorination was found to decrease the efficacy of these compounds.167
7. Future perspectives & conclusion
Cancer, characterized by unbridled cell proliferation, is a leading cause of death because of high incidence and mortality, which is exacerbated by the emergence of drug resistance. Therefore, there is a need for the development of new anticancer agents. Anthraquinones have attracted the medicinal chemist's attention for their diverse biological potential. They are typical anticancer fragments that have a broad scope for chemical modifications to be exploited to develop new anticancer agents. A handful of anticancer drugs based on anthraquinone moiety are marketed and used in chemotherapy in stand-alone regimens or widely used in combination therapy alongside radiation treatment to mitigate the metastasis of cancer cells. However, the emergence of resistance is increasingly being reported, and new explorations were undertaken to develop new anthraquinone-based anticancer agents with some success.Unfortunately, the majority of the research being pursued in academia, leading to the generation of a library of anthraquinones, is not intensively explored towards understanding their mechanistic biological pathways and target-specific activity in in vitro and in vivo models. Moreover, the toxicity and safety assessment of the anthraquinone derivatives to be used as potential drug molecules needs to be thoroughly investigated at the academic research level as limited research is available in the public domain. The toxicity and efficacy studies such as pharmacokinetics and pharmacodynamics could help better understand the profile of new anthraquinones. Hence, it is highly desirable to explore the potential of these anthraquinone libraries to identify hit/lead-like molecules that can be taken up for further development.
Several opportunities are ready to be fully exploited, which include selective drug delivery, combination with other anticancer drugs, computational techniques (e.g., ligand-based drug designing), drug repurposing, etc. Some of the issues critical to few anthraquinone derivatives, such as low polarity and structural instability in in vivo conditions, could be successfully addressed by employing polyethylene glycol-based drug delivery vehicles. There is ample scope for utilizing anthraquinone-based anticancer drugs in combination therapies with newly approved anticancer drugs targeting different biological receptors. This can be achieved through a detailed understanding of the mechanistic pathways of the intended combination. The development of modern age tools and computational techniques such as ligand-based drug designing (LBDD) and scaffold hopping led to newer molecules with improved efficacies. The in silico drug designing tools tremendously boosted the field of anthraquinone-anticancer drug discovery in the diversification of targets, from kinases to genes and immune responses. By leveraging the new age bio/cheminformatics tools, one can design anthraquinone-based new chemical entities with much accuracy in terms of safety & efficacy. Further, the emergence of drug repurposing techniques could be utilized to explore this scaffold for new therapeutic applications, and there is development in this direction, as can be construed from literature.
In view of the ongoing challenge of drug resistance of the existing marketed drugs, concerted efforts are required to address the shortcomings in academia research (limited advanced studies) and utilize the aforementioned approaches. This could lead to the development of new anthraquinone-based anticancer agents that can eventually be translated into safer and effective chemotherapeutics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Deanship of the Scientific Research (DSR), Umm Al-Qura University, for the financial support through the project code: 19-SCI-1-01-0019.References
- H. Hussain, A. Al-Harrasi, A. Al-Rawahi, I. R. Green, R. Csuk, I. Ahmed, A. Shah, G. Abbas, N. U. Rehman and R. Ullah, Expert Opin. Ther. Pat., 2015, 25, 1053–1064 CrossRef CAS PubMed.
- W. Tian, C. Wang, D. Li and H. Hou, Future Med. Chem., 2020, 12, 627–644 CAS.
- Demselben, Justus Liebigs Ann. Chem., 1840, 34, 287–296 CrossRef.
- C. Graebe, Ber. Dtsch. Chem. Ges., 1890, 23, 3739–3740 CrossRef.
- M. Phillips, Chem. Rev., 1929, 6, 157–174 CrossRef CAS.
- C. Graebe and C. Liebermann, Ber. Dtsch. Chem. Ges., 1869, 2, 14 CrossRef.
- R. Fittig, Ber. Dtsch. Chem. Ges., 1873, 6, 167–169 CrossRef.
- A. R. Burnett and R. H. Thomson, J. Chem. Soc. C, 1967, 2100–2104, 10.1039/J39670002100.
- R. Singh and S. M. Chauhan, Chem. Biodiversity, 2004, 1, 1241–1264 CrossRef CAS PubMed.
- M. Klaper, P. Wessig and T. Linker, Chem. Commun., 2016, 52, 1210–1213 RSC.
- J. J. Dong, D. Unjaroen, F. Mecozzi, E. C. Harvey, P. Saisaha, D. Pijper, J. W. deBoer, P. Alsters, B. L. Feringa and W. R. Browne, ChemSusChem, 2013, 6, 1774–1778 CrossRef CAS PubMed.
- Y. Xu, Z. Yang, J. Hu and J. Yan, Synthesis, 2013, 45, 370–374 CrossRef CAS.
- R. Kumar, N. Sharma, N. Sharma, A. Sharma and A. K. Sinha, Mol. Diversity, 2011, 15, 687–695 CrossRef CAS PubMed.
- S. El-Sayrafi and S. Rayyan, Molecules, 2001, 6, 279–286 CrossRef CAS.
- H. Kaper, M. Antonietti and F. Goettmann, Tetrahedron Lett., 2008, 49, 4546–4549 CrossRef CAS.
- M. N. Akhtar, S. Zareen, S. K. Yeap, W. Y. Ho, K. M. Lo, A. Hasan and N. B. Alitheen, Molecules, 2013, 18, 10042–10055 CrossRef CAS PubMed.
- X. Wang and V. Snieckus, Synlett, 1990, 1990, 313–316 CrossRef.
- T. Sawano, Y. Toyoshima and R. Takeuchi, Inorganics, 2019, 7(11), 138 CrossRef CAS.
- T. J. Monks, R. P. Hanzlik, G. M. Cohen, D. Ross and D. G. Graham, Toxicol. Appl. Pharmacol., 1992, 112, 2–16 CrossRef CAS PubMed.
- E. M. Malik and C. E. Müller, Med. Res. Rev., 2016, 36, 705–748 CrossRef CAS PubMed.
- Y. Caro, L. Anamale, M. Fouillaud, P. Laurent, T. Petit and L. Dufosse, Nat. Prod. Bioprospect., 2012, 2, 174–193 CrossRef CAS.
- D. W. Shaw, Dermatitis, 2009, 20, 292–295 CrossRef PubMed.
- L. Dufossé, Food Res. Int., 2014, 65, 132–136 CrossRef.
- E. M. Malik, Y. Baqi and C. E. Müller, Beilstein J. Org. Chem., 2015, 11, 2326–2333 CrossRef CAS PubMed.
- T. Furuta, Y. Hirayama and M. Iwamura, Org. Lett., 2001, 3, 1809–1812 CrossRef CAS PubMed.
- S. Patnaik, A. Swami, D. Sethi, A. Pathak, B. S. Garg, K. C. Gupta and P. Kumar, Bioconjugate Chem., 2007, 18, 8–12 CrossRef CAS PubMed.
- J. C. Forti, R. S. Rocha, M. R. V. Lanza and R. Bertazzoli, J. Electroanal. Chem., 2007, 601, 63–67 CrossRef CAS.
- M. Uchimiya and A. T. Stone, Chemosphere, 2009, 77, 451–458 CrossRef CAS PubMed.
- G. Rath, M. Ndonzao and K. Hostettmann, Int. J. Pharmacogn., 1995, 33, 107–114 CrossRef CAS.
- D. L. Barnard, D. W. Fairbairn, K. L. O'Neill, T. L. Gage and R. W. Sidwell, Antiviral Res., 1995, 28, 317–329 CrossRef CAS PubMed.
- R. F. Schinazi, C. K. Chu, J. R. Babu, B. J. Oswald, V. Saalmann, D. L. Cannon, B. F. Eriksson and M. Nasr, Antiviral Res., 1990, 13, 265–272 CrossRef CAS PubMed.
- D. O. Andersen, N. D. Weber, S. G. Wood, B. G. Hughes, B. K. Murray and J. A. North, Antiviral Res., 1991, 16, 185–196 CrossRef CAS PubMed.
- C. P. Osman, N. H. Ismail, R. Ahmad, N. Ahmat, K. Awang and F. M. Jaafar, Molecules, 2010, 15, 7218–7226 CrossRef CAS PubMed.
- M. Y. Fosso, K. Y. Chan, R. Gregory and C. W. Chang, ACS Comb. Sci., 2012, 14, 231–235 CrossRef CAS PubMed.
- W. Wang, R. Chen, Z. Luo, W. Wang and J. Chen, Nat. Prod. Res., 2018, 32, 558–563 CrossRef CAS PubMed.
- K. H. Gan, C. H. Teng, H. C. Lin, K. T. Chen, Y. C. Chen, M. F. Hsu, J. P. Wang, C. M. Teng and C. N. Lin, Biol. Pharm. Bull., 2008, 31, 1547–1551 CrossRef CAS PubMed.
- S. Z. Choi, S. O. Lee, K. U. Jang, S. H. Chung, S. H. Park, H. C. Kang, E. Y. Yang, H. J. Cho and K. R. Lee, Arch. Pharmacal Res., 2005, 28, 1027–1030 CrossRef CAS PubMed.
- A. Arvindekar, T. More, P. V. Payghan, K. Laddha, N. Ghoshal and A. Arvindekar, Food Funct., 2015, 6, 2693–2700 RSC.
- T. C. Jackson, J. D. Verrier and P. M. Kochanek, Cell Death Dis., 2013, 4, e451 CrossRef CAS PubMed.
- B. A. van Gorkom, E. G. de Vries, A. Karrenbeld and J. H. Kleibeuker, Aliment. Pharmacol. Ther., 1999, 13, 443–452 CrossRef CAS PubMed.
- A. M. Alaadin, E. H. Al-Khateeb and A. K. Jäger, Pharm. Biol., 2007, 45, 688–690 CrossRef.
- Y. Baqi, K. Atzler, M. Köse, M. Glänzel and C. E. Müller, J. Med. Chem., 2009, 52, 3784–3793 CrossRef CAS PubMed.
- R. W. Winter, K. A. Cornell, L. L. Johnson, L. M. Isabelle, D. J. Hinrichs and M. K. Riscoe, Bioorg. Med. Chem. Lett., 1995, 5, 1927–1932 CrossRef CAS.
- E. M. Perchellet, M. J. Magill, X. Huang, D. M. Dalke, D. H. Hua and J. P. Perchellet, Anti-Cancer Drugs, 2000, 11, 339–352 CrossRef CAS PubMed.
- M. B. Martins-Teixeira and I. Carvalho, ChemMedChem, 2020, 15, 933–948 CrossRef CAS PubMed.
- Y. Lazebnik, Nat. Rev. Cancer, 2010, 10, 232–233 CrossRef CAS PubMed.
- L. A. Torre, R. L. Siegel, E. M. Ward and A. Jemal, Cancer Epidemiol., Biomarkers Prev., 2016, 25, 16–27 CrossRef PubMed.
- D. Wang, X.-H. Wang, X. Yu, F. Cao, X. Cai, P. Chen, M. Li, Y. Feng, H. Li and X. Wang, Front. Pharmacol., 2021, 12, 638993 CrossRef CAS PubMed.
- W. Wu, N. Hu, Q. Zhang, Y. Li, P. Li, R. Yan and Y. Wang, Chem.-Biol. Interact., 2014, 219, 18–27 CrossRef CAS PubMed.
- X. Dong, J. Fu, X. Yin, S. Cao, X. Li, L. Lin and J. Ni, Phytother. Res., 2016, 30, 1207–1218 CrossRef CAS PubMed.
- S. Siddamurthi, G. Gutti, S. Jana, A. Kumar and S. K. Singh, Future Med. Chem., 2020, 12, 1037–1069 CrossRef CAS PubMed.
- C. H. Teng, S. J. Won and C. N. Lin, Bioorg. Med. Chem., 2005, 13, 3439–3445 CrossRef CAS PubMed.
- M. Siwy, D. Sek, B. Kaczmarczyk, I. Jaroszewicz, A. Nasulewicz, M. Pelczyñska, D. Nevozhay and A. Opolski, J. Med. Chem., 2006, 49, 806–810 CrossRef CAS PubMed.
- J. A. Valderrama, O. Espinoza, M. F. González, R. A. Tapia, J. A. Rodríguez, C. Theoduloz and G. Schmeda-Hirschmann, Tetrahedron, 2006, 62, 2631–2638 CrossRef CAS.
- L. F. Tietze, K. M. Gericke and I. Schuberth, Eur. J. Org. Chem., 2007, 2007, 4563–4577 CrossRef.
- H. S. Huang, K. F. Huang, C. L. Li, Y. Y. Huang, Y. H. Chiang, F. C. Huang and J. J. Lin, Bioorg. Med. Chem., 2008, 16, 6976–6986 CrossRef CAS PubMed.
- S. Wang, Q. Wang, Y. Wang, L. Liu, X. Weng, G. Li, X. Zhang and X. Zhou, Bioorg. Med. Chem. Lett., 2008, 18, 6505–6508 CrossRef CAS PubMed.
- Z. Yang, J.-A. Kang, W.-H. Kim, A.-Y. Park, H.-S. Kim, J.-S. Kim, J.-A. Kim, P. Gong, L.-S. Jeong and H.-R. Moon, Bull. Korean Chem. Soc., 2009, 30, 1463–1469 CrossRef CAS.
- G. Z. Jin, H. S. Jin and L. L. Jin, Arch. Pharmacal Res., 2011, 34, 1071–1076 CrossRef CAS PubMed.
- H. Y. Tu, A. M. Huang, C. H. Teng, T. C. Hour, S. C. Yang, Y. S. Pu and C. N. Lin, Bioorg. Med. Chem., 2011, 19, 5670–5678 CrossRef CAS PubMed.
- S. X. Feng, Q. Guan, T. Chen and C. Du, Arch. Pharmacal Res., 2012, 35, 1251–1258 CrossRef CAS PubMed.
- V. Marković, A. Janićijević, T. Stanojković, B. Kolundžija, D. Sladić, M. Vujčić, B. Janović, L. Joksović, P. T. Djurdjević, N. Todorović, S. Trifunović and M. D. Joksović, Eur. J. Med. Chem., 2013, 64, 228–238 CrossRef PubMed.
- D. Bhasin, J. P. Etter, S. N. Chettiar, M. Mok and P. K. Li, Bioorg. Med. Chem. Lett., 2013, 23, 6864–6867 CrossRef CAS PubMed.
- V. Castro-Castillo, C. Suárez-Rozas, N. Castro-Loiza, C. Theoduloz and B. K. Cassels, Eur. J. Med. Chem., 2013, 62, 688–692 CrossRef CAS PubMed.
- Y. R. Lee, D. S. Yu, Y. C. Liang, K. F. Huang, S. J. Chou, T. C. Chen, C. C. Lee, C. L. Chen, S. H. Chiou and H. S. Huang, PLoS One, 2013, 8, e56284 CrossRef CAS PubMed.
- Z. Liang, J. Ai, X. Ding, X. Peng, D. Zhang, R. Zhang, Y. Wang, F. Liu, M. Zheng, H. Jiang, H. Liu, M. Geng and C. Luo, ACS Med. Chem. Lett., 2013, 4, 408–413 CrossRef CAS PubMed.
- A. T. Taher and G. H. Hegazy, Arch. Pharmacal Res., 2013, 36, 573–578 CrossRef CAS PubMed.
- B. Kolundžija, V. Marković, T. Stanojković, L. Joksović, I. Matić, N. Todorović, M. Nikolić and M. D. Joksović, Bioorg. Med. Chem. Lett., 2014, 24, 65–71 CrossRef PubMed.
- M. S. Almutairi, G. H. Hegazy, M. E. Haiba, H. I. Ali, N. M. Khalifa and M. Soliman Ael, Int. J. Mol. Sci., 2014, 15, 22580–22603 CrossRef CAS PubMed.
- Y. W. Chen, H. S. Huang, Y. S. Shieh, K. H. Ma, S. H. Huang, D. Y. Hueng, H. K. Sytwu and G. J. Lin, PLoS One, 2014, 9, e104703 CrossRef PubMed.
- S. Sangthong, N. Sangphech, T. Palaga, N. Ngamrojanavanich, S. Puthong, T. Vilaivan and N. Muangsin, Eur. J. Med. Chem., 2014, 77, 334–342 CrossRef CAS PubMed.
- P.-Z. Zhang, H.-L. Yang, C.-C. Li, Z.-C. Xia, X.-M. Wang, H. Wei, R.-X. Rong, Z.-R. Cao, K.-R. Wang, H. Chen and X.-L. Li, Chin. Chem. Lett., 2014, 25, 1057–1059 CrossRef CAS.
- L. Kelland, Idrugs, 1998, 1, 397–398 CAS.
- I. Zuravka, A. Sosic, B. Gatto and R. Göttlich, Bioorg. Med. Chem. Lett., 2015, 25, 4606–4609 CrossRef CAS PubMed.
- F. Prati, C. Bergamini, M. T. Molina, F. Falchi, A. Cavalli, M. Kaiser, R. Brun, R. Fato and M. L. Bolognesi, J. Med. Chem., 2015, 58, 6422–6434 CrossRef CAS PubMed.
- Y. H. Lin, S. M. Chuang, P. C. Wu, C. L. Chen, S. Jeyachandran, S. C. Lo, H. S. Huang and M. H. Hou, Sci. Rep., 2016, 6, 31019 CrossRef CAS PubMed.
- Y. Zheng, L. Zhu, L. Fan, W. Zhao, J. Wang, X. Hao, Y. Zhu, X. Hu, Y. Yuan, J. Shao and W. Wang, Eur. J. Med. Chem., 2017, 125, 902–913 CrossRef CAS PubMed.
- N. Okumura, H. Mizutani, T. Ishihama, M. Ito, A. Hashibe, T. Nakayama and B. Uno, Chem. Pharm. Bull., 2019, 67, 717–720 CrossRef CAS PubMed.
- V. C. Roa-Linares, Y. Miranda-Brand, V. Tangarife-Castaño, R. Ochoa, P. A. García, M. Castro, L. Betancur-Galvis and A. San Feliciano, Molecules, 2019, 24, 1279 CrossRef CAS PubMed.
- K. Huang, L. Jiang, H. Li, D. Ye and L. Zhou, Molecules, 2019, 24, 806 CrossRef PubMed.
- S. Celik, F. Ozkok, A. E. Ozel, Y. Müge Sahin, S. Akyuz, B. D. Sigirci, B. B. Kahraman, H. Darici and E. Karaoz, J. Biomol. Struct. Dyn., 2020, 38, 756–770 CrossRef CAS PubMed.
- K. Huang, L. Jiang, R. Liang, H. Li, X. Ruan, C. Shan, D. Ye and L. Zhou, Eur. J. Med. Chem., 2019, 168, 45–57 CrossRef CAS PubMed.
- P. Awasthi, M. Vatsal and A. Sharma, J. Biomol. Struct. Dyn., 2019, 37, 4465–4480 CrossRef CAS PubMed.
- Y. Li, F. Guo, T. Chen, L. Zhang, Z. Wang, Q. Su and L. Feng, Chem. Biodiversity, 2020, 17, e2000328 CAS.
- J. Li, Y. B. Zheng, T. Kurtán, M. X. Liu, H. Tang, C. L. Zhuang and W. Zhang, Nat. Prod. Res., 2020, 34, 2116–2123 CrossRef CAS PubMed.
- L. A. Oliveira, H. D. Nicolella, R. A. Furtado, N. M. Lima, D. C. Tavares, T. A. Corrêa and M. V. Almeida, Med. Chem. Res., 2020, 29, 1611–1620 CrossRef CAS.
- A. Anifowose, A. A. Agbowuro, R. Tripathi, W. Lu, C. Tan, X. Yang and B. Wang, Med. Chem. Res., 2020, 29, 1199–1210 CrossRef CAS PubMed.
- Y. Li, F. Guo, Y. Guan, T. Chen, K. Ma, L. Zhang, Z. Wang, Q. Su, L. Feng, Y. Liu and Y. Zhou, Molecules, 2020, 25, 1672 CrossRef CAS PubMed.
- A. S. Tikhomirov, V. A. Litvinova, D. V. Andreeva, V. B. Tsvetkov, L. G. Dezhenkova, Y. L. Volodina, D. N. Kaluzhny, I. D. Treshalin, D. Schols, A. A. Ramonova, M. M. Moisenovich, A. A. Shtil and A. E. Shchekotikhin, Eur. J. Med. Chem., 2020, 199, 112294 CrossRef CAS PubMed.
- R. Liu, H. Zou, Z. X. Zou, F. Cheng, X. Yu, P. S. Xu, X. M. Li, D. Li, K. P. Xu and G. S. Tan, Nat. Prod. Res., 2020, 34, 2709–2714 CrossRef CAS PubMed.
- K. W. Lin, W. H. Lin, C. L. Su, H. Y. Hsu and C. N. Lin, Bioorg. Chem., 2021, 107, 104395 CrossRef CAS PubMed.
- Y. Li, Y. Luan, X. Qi, M. Li, L. Gong, X. Xue, X. Wu, Y. Wu, M. Chen, G. Xing, J. Yao and J. Ren, Toxicol. Sci., 2010, 118, 435–443 CrossRef CAS PubMed.
- L. Zhang, Y. K. Lau, W. Xia, G. N. Hortobagyi and M. C. Hung, Clin. Cancer Res., 1999, 5, 343–353 CAS.
- Y. Y. Chen, S. Y. Chiang, J. G. Lin, Y. S. Ma, C. L. Liao, S. W. Weng, T. Y. Lai and J. G. Chung, Int. J. Oncol., 2010, 36, 1113–1120 CrossRef CAS PubMed.
- Y. Y. Chen, S. Y. Chiang, J. G. Lin, J. S. Yang, Y. S. Ma, C. L. Liao, T. Y. Lai, N. Y. Tang and J. G. Chung, Anticancer Res., 2010, 30, 945–951 Search PubMed.
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 CrossRef CAS PubMed.
- K. Hevener, T. A. Verstak, K. E. Lutat, D. L. Riggsbee and J. W. Mooney, Acta Pharm. Sin. B, 2018, 8, 844–861 CrossRef PubMed.
- G. Capranico, J. Marinello and G. Chillemi, J. Med. Chem., 2017, 60, 2169–2192 CrossRef CAS PubMed.
- M. N. Drwal, K. Agama, L. P. Wakelin, Y. Pommier and R. Griffith, PLoS One, 2011, 6, e25150 CrossRef CAS PubMed.
- P. J. Smith, N. J. Blunt, R. Desnoyers, Y. Giles and L. H. Patterson, Cancer Chemother. Pharmacol., 1997, 39, 455–461 CrossRef CAS PubMed.
- S. R. McKeown, M. V. Hejmadi, I. A. McIntyre, J. J. McAleer and L. H. Patterson, Br. J. Cancer, 1995, 72, 76–81 CrossRef CAS PubMed.
- J. F. Kou, C. Qian, J. Q. Wang, X. Chen, L. L. Wang, H. Chao and L. N. Ji, J. Biol. Inorg. Chem., 2012, 17, 81–96 CrossRef CAS PubMed.
- C. Hotzel, A. Marotto and U. Pindur, Eur. J. Med. Chem., 2003, 38, 189–197 CrossRef CAS PubMed.
- K. Pors, Z. Paniwnyk, P. Teesdale-Spittle, J. A. Plumb, E. Willmore, C. A. Austin and L. H. Patterson, Mol. Cancer Ther., 2003, 2, 607–610 CAS.
- J. K. Son, S. J. Jung, J. H. Jung, Z. Fang, C. S. Lee, C. S. Seo, D. C. Moon, B. S. Min, M. R. Kim and M. H. Woo, Chem. Pharm. Bull., 2008, 56, 213–216 CrossRef CAS PubMed.
- A. Bielawska, K. Kosk and K. Bielawski, Pol. J. Pharmacol., 2001, 53, 283–287 CAS.
- S. M. Wojtowicz-Praga, R. B. Dickson and M. J. Hawkins, Invest. New Drugs, 1997, 15, 61–75 CrossRef CAS PubMed.
- B. E. Bachmeier, C. M. Iancu, M. Jochum and A. G. Nerlich, Expert Rev. Anticancer Ther., 2005, 5, 149–163 CrossRef CAS PubMed.
- A. R. Farina and A. R. Mackay, Cancers, 2014, 6, 240–296 CrossRef PubMed.
- C. Tabolacci, A. Lentini, P. Mattioli, B. Provenzano, S. Oliverio, F. Carlomosti and S. Beninati, Life Sci., 2010, 87, 316–324 CrossRef CAS PubMed.
- P. Suboj, S. Babykutty, D. R. Valiyaparambil Gopi, R. S. Nair, P. Srinivas and S. Gopala, Eur. J. Pharm. Sci., 2012, 45, 581–591 CrossRef CAS PubMed.
- E. H. Blackburn, Cell, 2001, 106, 661–673 CrossRef CAS PubMed.
- J. W. Shay and S. Bacchetti, Eur. J. Cancer, 1997, 33, 787–791 CrossRef CAS PubMed.
- H. S. Huang, I. B. Chen, K. F. Huang, W. C. Lu, F. Y. Shieh, Y. Y. Huang, F. C. Huang and J. J. Lin, Chem. Pharm. Bull., 2007, 55, 284–292 CrossRef CAS PubMed.
- Y. Y. Yan, L. S. Zheng, X. Zhang, L. K. Chen, S. Singh, F. Wang, J. Y. Zhang, Y. J. Liang, C. L. Dai, L. Q. Gu, M. S. Zeng, T. T. Talele, Z. S. Chen and L. W. Fu, Mol. Pharmaceutics, 2011, 8, 1687–1697 CrossRef CAS PubMed.
- C. L. Chen, D. M. Chang, T. C. Chen, C. C. Lee, H. H. Hsieh, F. C. Huang, K. F. Huang, J. H. Guh, J. J. Lin and H. S. Huang, Eur. J. Med. Chem., 2013, 60, 29–41 CrossRef CAS PubMed.
- D. Cairns, E. Michalitsi, T. C. Jenkins and S. P. Mackay, Bioorg. Med. Chem., 2002, 10, 803–807 CrossRef CAS PubMed.
- P. J. Perry, S. M. Gowan, A. P. Reszka, P. Polucci, T. C. Jenkins, L. R. Kelland and S. Neidle, J. Med. Chem., 1998, 41, 3253–3260 CrossRef CAS PubMed.
- H. S. Huang, J. F. Chiou, Y. Fong, C. C. Hou, Y. C. Lu, J. Y. Wang, J. W. Shih, Y. R. Pan and J. J. Lin, J. Med. Chem., 2003, 46, 3300–3307 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- N. Kosiol, S. Juranek, P. Brossart, A. Heine and K. Paeschke, Mol. Cancer, 2021, 20, 40 CrossRef CAS PubMed.
- J. Blasiak, Curr. Med. Chem., 2017, 24, 1488–1503 CrossRef CAS PubMed.
- A. Das and S. Dutta, ACS Omega, 2021, 6, 18344–18351 CrossRef CAS PubMed.
- S. Wang, W. W. Yan, M. He, D. Wei, Z. J. Long and Y. M. Tao, Pharmacol. Rep., 2020, 72, 1383–1396 CrossRef CAS PubMed.
- Z. Zhang, X.-H. Wu, F.-Q. Sun, F. Shan, J.-C. Chen, L.-M. Chen, Y.-S. Zhou and W.-J. Mei, Inorg. Chim. Acta, 2014, 418, 23–29 CrossRef CAS.
- N. S. Sirazhetdinova, V. A. Savelyev, D. S. Baev, T. S. Golubeva, L. S. Klimenko, T. G. Tolstikova, J. Ganbaatar and E. E. Shults, Med. Chem. Res., 2021, 30, 1541–1556 CrossRef CAS.
- S. Roy, A. Ali, M. Kamra, K. Muniyappa and S. Bhattacharya, Eur. J. Med. Chem., 2020, 195, 112202 CrossRef CAS PubMed.
- E. De Moliner, S. Moro, S. Sarno, G. Zagotto, G. Zanotti, L. A. Pinna and R. Battistutta, J. Biol. Chem., 2003, 278, 1831–1836 CrossRef CAS PubMed.
- G. Maurer, B. Tarkowski and M. Baccarini, Oncogene, 2011, 30, 3477–3488 CrossRef CAS PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- L. Zhang, Y. K. Lau, L. Xi, R. L. Hong, D. S. Kim, C. F. Chen, G. N. Hortobagyi, C. Chang and M. C. Hung, Oncogene, 1998, 16, 2855–2863 CrossRef CAS PubMed.
- A. Muto, M. Hori, Y. Sasaki, A. Saitoh, I. Yasuda, T. Maekawa, T. Uchida, K. Asakura, T. Nakazato, T. Kaneda, M. Kizaki, Y. Ikeda and T. Yoshida, Mol. Cancer Ther., 2007, 6, 987–994 CrossRef CAS PubMed.
- C. R. Faltynek, J. Schroeder, P. Mauvais, D. Miller, S. Wang, D. Murphy, R. Lehr, M. Kelley, A. Maycock and W. Michne, et al., Biochemistry, 1995, 34, 12404–12410 CrossRef CAS PubMed.
- Y. Shi, C. H. Wang and X. G. Gong, Biol. Pharm. Bull., 2008, 31, 1075–1078 CrossRef CAS PubMed.
- W. Wätjen, S. S. Ebada, A. Bergermann, Y. Chovolou, F. Totzke, M. H. G. Kubbutat, W. Lin and P. Proksch, Arch. Toxicol., 2017, 91, 1485–1495 CrossRef PubMed.
- G. Cozza, M. Mazzorana, E. Papinutto, J. Bain, M. Elliott, G. di Maira, A. Gianoncelli, M. A. Pagano, S. Sarno, M. Ruzzene, R. Battistutta, F. Meggio, S. Moro, G. Zagotto and L. A. Pinna, Biochem. J., 2009, 421, 387–395 CrossRef CAS PubMed.
- S. C. Robson, J. Sévigny and H. Zimmermann, Purinergic Signalling, 2006, 2, 409–430 CrossRef CAS PubMed.
- V. Umansky, I. Shevchenko, A. V. Bazhin and J. Utikal, Cancer Immunol. Immunother., 2014, 63, 1073–1080 CrossRef CAS PubMed.
- S. S. Virtanen, A. Kukkonen-Macchi, M. Vainio, K. Elima, P. L. Härkönen, S. Jalkanen and G. G. Yegutkin, Mol. Cancer Res., 2014, 12, 1863–1874 CrossRef CAS PubMed.
- Y. Baqi, Drug discovery today, 2016, 21, 1571–1577 CrossRef CAS PubMed.
- M. J. Pang, Z. Yang, X. L. Zhang, Z. F. Liu, J. Fan and H. Y. Zhang, Acta Pharmacol. Sin., 2016, 37, 1623–1640 CrossRef CAS PubMed.
- L. Ma, Y. Yang, Z. Yin, M. Liu, L. Wang, L. Chen, L. Zhu and H. Yang, Biomed. Pharmacother., 2017, 90, 615–625 CrossRef CAS PubMed.
- K. A. Manu, M. K. Shanmugam, T. H. Ong, A. Subramaniam, K. S. Siveen, E. Perumal, R. P. Samy, P. Bist, L. H. Lim, A. P. Kumar, K. M. Hui and G. Sethi, PLoS One, 2013, 8, e57015 CrossRef CAS PubMed.
- J. Ma, H. Lu, S. Wang, B. Chen, Z. Liu, X. Ke, T. Liu and J. Fu, Int. J. Oncol., 2015, 46, 1619–1628 CrossRef CAS PubMed.
- S. K. Koerner, J. I. Hanai, S. Bai, F. E. Jernigan, M. Oki, C. Komaba, E. Shuto, V. P. Sukhatme and L. Sun, Eur. J. Med. Chem., 2017, 126, 920–928 CrossRef CAS PubMed.
- G. Xie, X. Zhu, Q. Li, M. Gu, Z. He, J. Wu, J. Li, Y. Lin, M. Li, Z. She and J. Yuan, Br. J. Pharmacol., 2010, 159, 689–697 CrossRef CAS PubMed.
- K. Kamiya, W. Hamabe, S. Tokuyama, K. Hirano, T. Satake, Y. Kumamoto-Yonezawa, H. Yoshida and Y. Mizushina, Food Chem., 2010, 118, 725–730 CrossRef CAS.
- H. K. Choi, H. Ryu, A. R. Son, B. Seo, S. G. Hwang, J. Y. Song and J. Ahn, Biomed. Pharmacother., 2016, 79, 308–314 CrossRef CAS PubMed.
- A. P. Li, Chem.-Biol. Interact., 2004, 150, 27–33 CrossRef CAS PubMed.
- L. E. Sendelbach, Toxicology, 1989, 57, 227–240 CrossRef CAS PubMed.
- Y. Liu, M. S. T. Mapa and R. L. Sprando, Food Chem. Toxicol., 2020, 140, 111313 CrossRef CAS PubMed.
- Y. Wang, J. Chen, L. Ge, D. Wang, X. Cai, L. Huang and C. Hao, Dyes Pigm., 2009, 83, 276–280 CrossRef CAS.
- R. B. Semwal, D. K. Semwal, S. Combrinck and A. Viljoen, Phytochemistry, 2021, 190, 112854 CrossRef CAS PubMed.
- K. Oshida, M. Hirakata, A. Maeda, T. Miyoshi and Y. Miyamoto, J. Appl. Toxicol., 2011, 31, 790–800 CrossRef CAS PubMed.
- Q. He, K. Liu, S. Wang, H. Hou, Y. Yuan and X. Wang, Drug Chem. Toxicol., 2012, 35, 149–154 CrossRef CAS PubMed.
- N. Yang, M. Ruan and S. Jin, Gastroenterol. Hepatol., 2020, 43, 266–272 CrossRef PubMed.
- Y. Li, Q. Q. Xu, C. S. Shan, Y. H. Shi, Y. Wang and G. Q. Zheng, Front. Pharmacol., 2018, 9, 943 CrossRef PubMed.
- Q. Zhao, X. Wang, A. Chen, X. Cheng, G. Zhang, J. Sun, Y. Zhao, Y. Huang and Y. Zhu, Int. J. Mol. Med., 2018, 41, 2802–2812 CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- Y. Bae, T. A. Diezi, A. Zhao and G. S. Kwon, J. Controlled Release, 2007, 122, 324–330 CrossRef CAS PubMed.
- F. M. Veronese, O. Schiavon, G. Pasut, R. Mendichi, L. Andersson, A. Tsirk, J. Ford, G. Wu, S. Kneller, J. Davies and R. Duncan, Bioconjugate Chem., 2005, 16, 775–784 CrossRef CAS PubMed.
- E. L. Ferguson, A. Scomparin, H. Hailu and R. Satchi-Fainaro, J. Drug Targeting, 2017, 25, 818–828 CrossRef CAS PubMed.
- B. Surnar and M. Jayakannan, Biomacromolecules, 2016, 17, 4075–4085 CrossRef CAS PubMed.
- D. Gu, K. Ladewig, M. Klimak, D. Haylock, K. M. McLean, A. J. O'Connor and G. G. Qiao, Polym. Chem., 2015, 6, 6475–6487 RSC.
- S.-X. Feng, J. Hao, T. Chen and S. X. Qiu, Helv. Chim. Acta, 2011, 94, 1843–1849 CrossRef CAS.
- J. Koyama, I. Morita, K. Tagahara, Y. Nobukuni, T. Mukainaka, M. Kuchide, H. Tokuda and H. Nishino, Cancer Lett., 2002, 182, 135–139 CrossRef CAS PubMed.
- J. Koyama, I. Morita, K. Tagahara, M. Ogata, T. Mukainaka, H. Tokuda and H. Nishino, Cancer Lett., 2001, 170, 15–18 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |