
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThioxanthone: a powerful photocatalyst for organic reactions
Nikolaos F.
Nikitas†
,
Petros L.
Gkizis†
 and
Christoforos G.
Kokotos
*
and
Christoforos G.
Kokotos
*
Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, Athens 15771, Greece. E-mail: ckokotos@chem.uoa.gr
First published on 14th May 2021
Abstract
Photoorganocatalysis has been recognised by the organic chemistry community as an important part of photochemistry and catalysis. In general, aromatic ketones constitute key players in this type of catalysis as they are involved in a plethora of examples in the literature. Among the various aromatic ketones, thioxanthone (TX) seems to play a unique role in photochemistry. In comparison with other aromatic ketones, TX has a high triplet energy and a relatively long triplet lifetime, while it has the ability to participate successfully in merger reactions with metal complexes. In this review, we will discuss the photophysical properties of this small organic molecule, as well as the numerous examples of photochemical reactions, where it is employed as a mediator and more specifically in polymerisation reactions, and organic transformations.
 From left to right: Dr Petros L. Gkizis, Associate Professor Christoforos G. Kokotos and Nikolaos F. Nikitas | Nikolaos F. Nikitas is a PhD student at the Laboratory of Organic Chemistry of the Department of Chemistry, National and Kapodistrian University of Athens, under the supervision of Associate Professor Christoforos G. Kokotos. Nikolaos was born in Athens in 1993 and studied chemistry at the Department of Chemistry of the University of Patras (2015), where he also obtained his MSc in the field of synthetic organic chemistry (2017). He then moved to Athens to pursue PhD studies in the Kokotos group. His research interest includes photochemistry. In 2019, he made a short-term visit (STSM supported by Cost Action CA15106 CHAOS) to Professor Renaud's group at the University of Bern. |
Dr Petros L. Gkizis was born in Athens, Greece. He obtained his BSc, MSc and PhD from Aristotle University of Thessaloniki. In 2012, he was appointed as a postdoctoral fellow at Aristotle University of Thessaloniki in collaboration with the University of Crete (ERC-Grant). In 2014, he was appointed as a Technology Transfer Research Scientist at Pharmathen S.A. Pharmaceuticals, Thessaloniki, Greece. In 2020, he joined the group of Assoc. Prof. Christoforos Kokotos at the National and Kapodistrian University of Athens as a postdoctoral researcher. His research interest includes the development of photochemical reactions for the formation of C–C bonds. |
Prof. Christoforos G. Kokotos was born in Athens, Greece (1981) and studied chemistry at the National and Kapodistrian University of Athens, Greece (2003). He completed his PhD studies at the University of Bristol, UK (2007) under the supervision of Prof. Varinder Aggarwal. He then carried out postdoctoral work at Princeton University, USA in the group of Prof. David W. C. MacMillan. He was appointed as a Lecturer of Organic Chemistry in the Department of Chemistry of the National and Kapodistrian University of Athens, Greece and promoted to Assistant Professor (2016) and Associate Professor (2020). His research interests include asymmetric organocatalysis, photocatalysis and green methodologies on oxidation reactions and their application in the synthesis of pharmaceuticals, bioactive molecules and high-added-value chemicals. |
1. Introduction
Polar organic chemistry always had the lion's share in newly developed organic methodologies, especially since radical chemistry has always been considered complex, rendering it a field of limited attention. This has changed since the introduction of modern photocatalysis, where radicals are produced in a mild and controlled way. Through this new and powerful field, novel domains of reactivities have emerged. Especially in the last decade, photoredox catalysis has become a powerful tool in the hands of scientists, in order to forge new molecules by overcoming the limitations of the past.1 Photoredox catalysis is mainly based on the ability of various metal complexes to absorb light in the visible region and undergo a metal to ligand charge transfer to an excited state which can react with a reagent either by providing an electron (oxidative quenching) or by acquiring one (reductive quenching).1 Photoredox catalysis can also be achieved with the use of redox active small organic molecules, like acridinium salts, compounds with the oxidation potential higher than that of metal photocatalysts, making it a great example of the potential of photoorganocatalysts.2 The concept of photoorganocatalysis, the use of small organic molecules as photocatalysts to promote chemical transformations, has been highlighted by Albini, Fagnoni and coworkers,3 who reviewed examples of molecules that can catalyze reactions by hydrogen atom transfer (HAT), single electron transfer (SET) or energy transfer (EnT). Aryl ketones are an important category of photoorganocatalysts. Among the various aromatic ketones, thioxanthone (TX) seems to play a unique role in photochemistry. In comparison with other aromatic ketones, TX has a high triplet energy and a relatively long triplet lifetime, while it has the ability to participate successfully in merger reactions with metal complexes. In this review, we will discuss the photophysical properties of thioxanthone, as well as its applications in photochemical reactions.2. Photophysical properties of thioxanthone
Thioxanthone (TX, 1), like other aromatic ketones,4–8 is a well-studied example of photoactive compounds, capable of performing various chemical transformations.9–13 The conformation of TX is not fixed, but it interconverts from a planar (C2v) to a non-planar conformation.14,15 This interconversion has been characterized by Rubio-Pons as the “butterfly motion” of TX.16 The two discrete conformations possess different energy levels and exhibit two different absorption bands in the UV-Vis spectrum, one at 361 nm and the other at 376 nm, where the first corresponds to the non-planar conformation and the second to the planar conformation, respectively. Upon irradiation, TX is excited to the singlet state, followed by intersystem crossing (ISC) to the triplet state. In the case of TX, the intersystem crossing of an electron normally occurs from a non-bonding orbital (n) to an anti-bonding pi orbital (π*) (nπ* configuration) and occurs 103 times faster than in the case of migration from the same type of molecular orbital (ππ* migration) (Scheme 1).17 The configuration of the triplet state depends on the polarity of the solvent. In more polar solvents, the triplet state ππ* is stabilized as the lower lying triplet state, capable of reacting with hydrogen or electron donors (Scheme 1, breaking of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of TX to the C–O biradical).18,19 Polar solvents tend to stabilize the ππ* state and destabilize the nπ* state. Thus, in polar solvents, fluorescence is favored compared to phosphorescence. The latter triplet state ππ* of the aromatic ketone mostly participates in hydrogen atom abstraction events from a hydrogen donor, producing a ketyl radical, instead of an electron transfer event, which is favored in non-polar solvents, where the major configuration is nπ* (Scheme 1).20,21 Theoretical studies support that TX's low lying triplet state is close enough to the singlet state, favoring in this way internal conversion (IC) and ISC upon photoexcitation.9,22,23
O bond of TX to the C–O biradical).18,19 Polar solvents tend to stabilize the ππ* state and destabilize the nπ* state. Thus, in polar solvents, fluorescence is favored compared to phosphorescence. The latter triplet state ππ* of the aromatic ketone mostly participates in hydrogen atom abstraction events from a hydrogen donor, producing a ketyl radical, instead of an electron transfer event, which is favored in non-polar solvents, where the major configuration is nπ* (Scheme 1).20,21 Theoretical studies support that TX's low lying triplet state is close enough to the singlet state, favoring in this way internal conversion (IC) and ISC upon photoexcitation.9,22,23
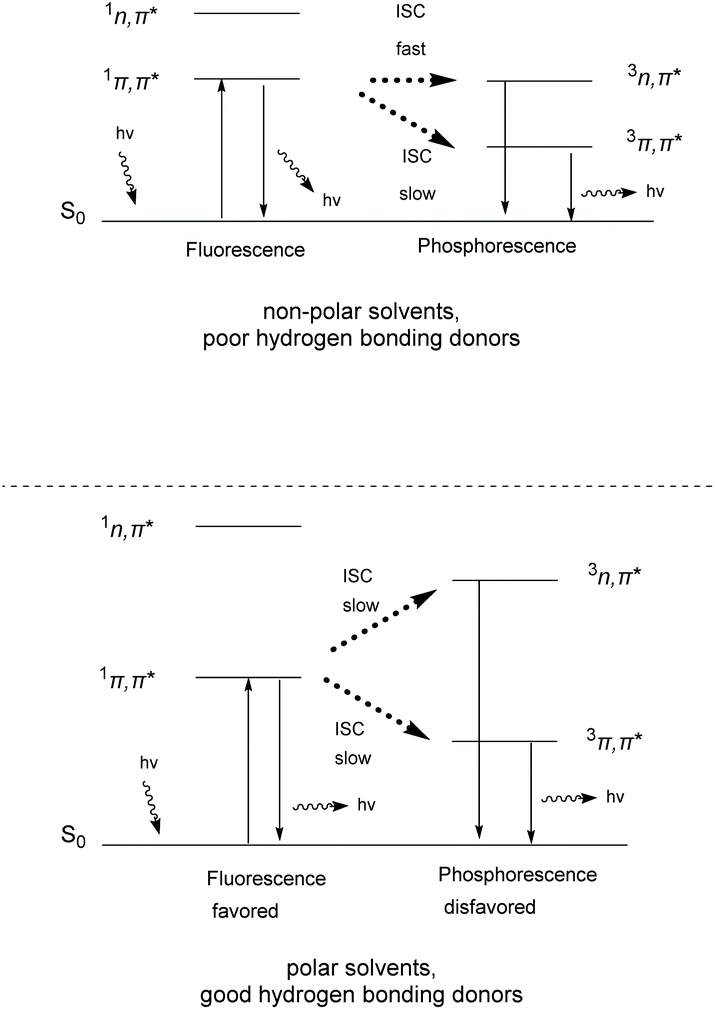 | ||
| Scheme 1 Relation between electron distribution in excited TX and solvent polarity. | ||
Except from its ability to participate in HAT events, triplet TX has a mild oxidative character (Ered = +1.18 V),2 compared to benzophenone (Ered = +1.28 V),2 xanthone (Ered = +1.57 V)2 and anthraquinone (Ered = +1.77 V).2 In protic polar solvents (such as MeCN and H2O), triplet TX leads to the TXH˙ radical (2), the product of the direct HAT from the solvent. However, in the presence of an electron donor, such as an easily oxidized amine, a radical reduction of the ketone can occur in a known two-step mechanism,5 which involves firstly an oxidation and secondly a protonation of the radical anion of TX˙−.24
Apart from electron or hydrogen abstraction, TX can participate in chemical transformations through energy transfer (EnT). Compounds upon excitation can pass their excited state energy to acceptors, compounds with lower energy levels. Along these lines, Engel et al. studied the decomposition of azo compounds using sensitizers with TX being a part of this study, which showed that TX is a capable triplet state energy sensitizer.25,26 TX with a high singlet state energy  ,20 a triplet state energy
,20 a triplet state energy 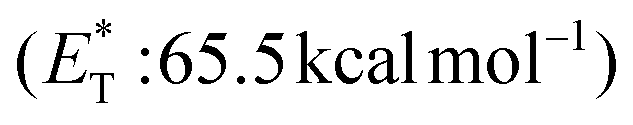
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 and a lifetime estimated at τ0 = 77 μs,28 a hundred times longer than that of benzophenone (τ0 = 0.7 μs),28 proved to be a potent energy transfer molecule. TX has a relatively high triplet energy compared to similar aromatic ketones, such as benzil (
27 and a lifetime estimated at τ0 = 77 μs,28 a hundred times longer than that of benzophenone (τ0 = 0.7 μs),28 proved to be a potent energy transfer molecule. TX has a relatively high triplet energy compared to similar aromatic ketones, such as benzil (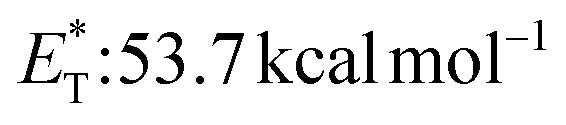 , triplet lifetime τ0 = 36 μs),29 benzophenone (
, triplet lifetime τ0 = 36 μs),29 benzophenone (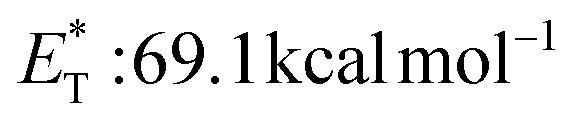 ),29 anthraquinone (
),29 anthraquinone (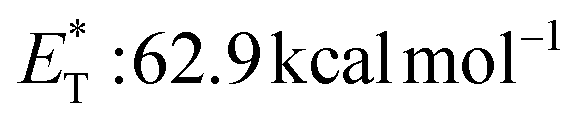 , triplet lifetime τ0 = 2280–3200 μs),29 acenaphthenequinone
, triplet lifetime τ0 = 2280–3200 μs),29 acenaphthenequinone 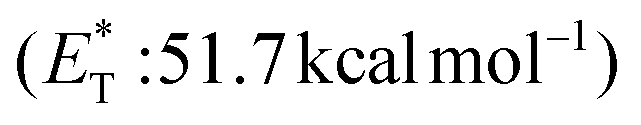 29 and fluorenone (
29 and fluorenone (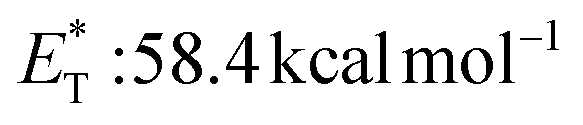 , triplet lifetime τ0 = 3 μs).29
, triplet lifetime τ0 = 3 μs).29
Due to its high triplet energy, TX is an efficient sensitizer for oxygen, leading to singlet oxygen,30–32 since the excitation of oxygen requires around 23 kcal mol−1.33
3. Thioxanthone as the photoinitiator in polymerisation reactions
The employed organic molecules as photoinitiators in polymerisation reactions are divided into two major groups: type I and type II (Scheme 2). In type I photoinitiators, upon light absorption and excitation, the corresponding excited state undergoes an α-cleavage, leading to new radical species that can participate in the initiation process (Scheme 2A). TX is a typical type II photoinitiator (the excited molecule requires interaction with a co-initiator) that has been used widely in polymerisation reactions (Scheme 2B). The study of TX as a photoinitiator goes back to 1981, when Amirzadeh and Schnabel studied the role of different substituents on TX and the use of various amines as co-initiators.34 Until then, TX had already been known for its role in photocuring and coating.35,36 This study was followed by the work of Timpe and Kronfeld, in which various aromatic ketones, including thioxanthone, were studied in the photopolymerisation of methyl methacrylate in the presence or absence of a hydrogen donor as a co-initiator.37 In the first case, TX can interact with the hydrogen donor, forming radical species, which can propagate the polymerisation chain. In the other case, the excited carbonyl compound reacts with electron-rich double bonds, forming an oxetane ring, a procedure that terminates the polymerisation route.37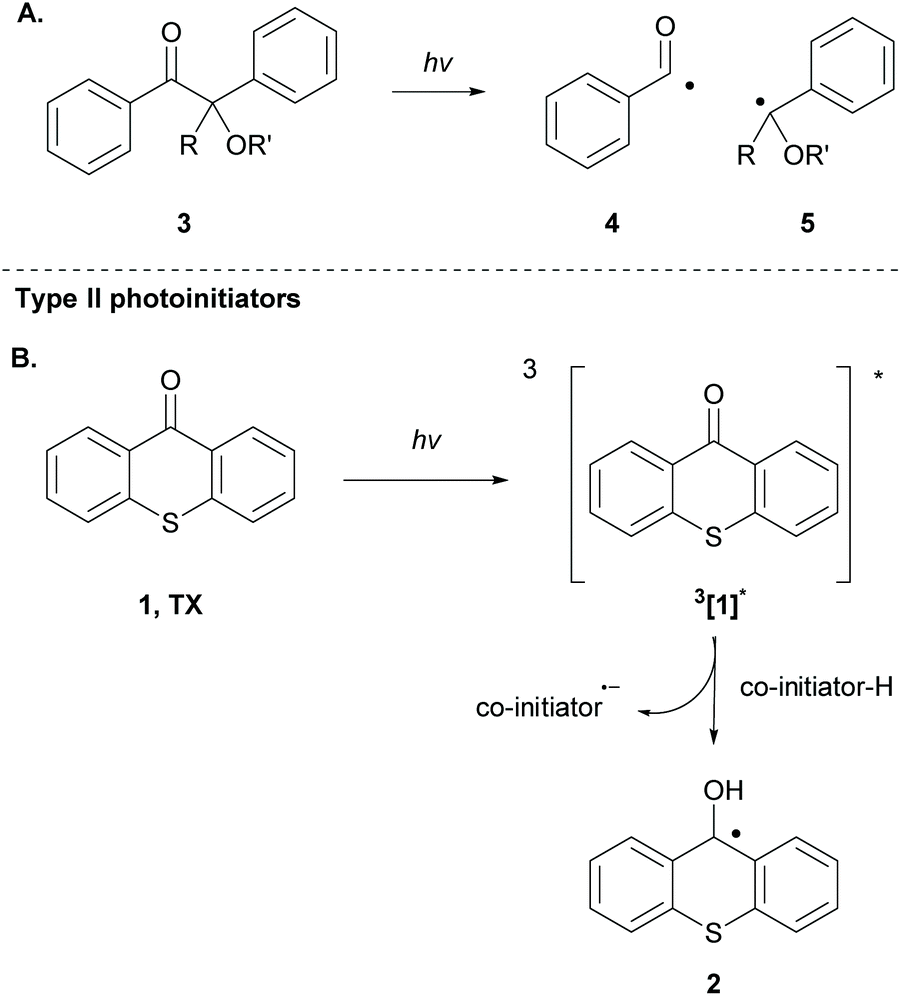 | ||
| Scheme 2 Distinct types of photoinitiators for polymerisation based on different types of photoinitiation. | ||
When an hydrogen donor is present, such as an amine, TXH˙ (2) is formed (Scheme 3).38 TXH˙ (2) can dimerize, react with another radical 2, affording pinacol product 6 (Scheme 3). Alternatively, disproportionation of radical 2 can lead to 1 and thioxanthole 7 (Scheme 3). In some cases, this radical can terminate the radical polymerisation by forming an adduct with the newly-formed polymer (Scheme 3, 8). Similarly, molecular oxygen can react with TXH˙ and the formed adduct 9 decomposes to TX and active radical oxygen species, regenerating in this way the photocatalyst.
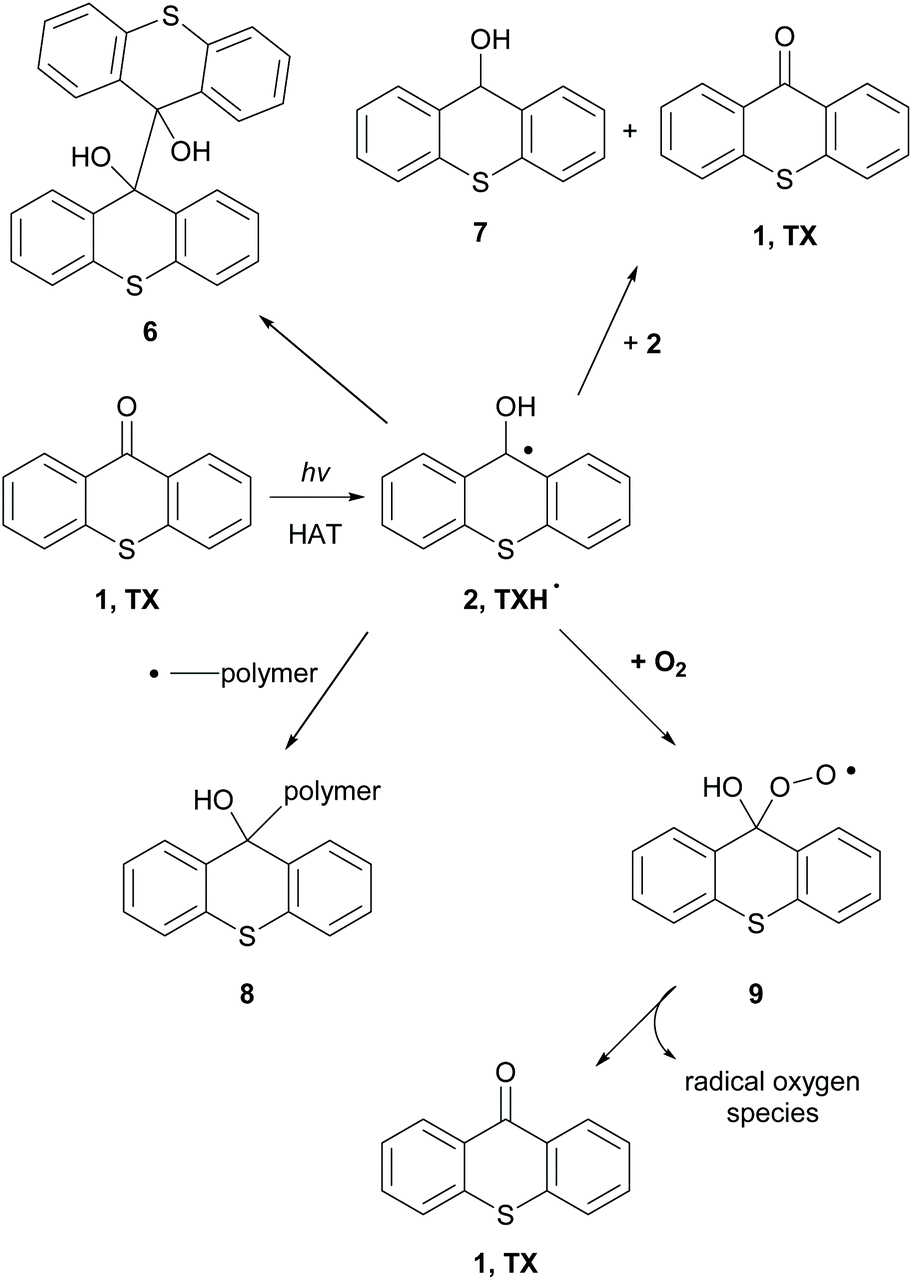 | ||
| Scheme 3 Possible pathways of the TX ketyl radical during photopolymerisation reactions. | ||
The great importance of type II initiators, especially thioxanthone, is highlighted in the great variety of co-initiators that can be employed. In the literature, the combination of TX 1 with triphenyl sulfonium anion 20 (co-initiator) for the polymerization of tetrahydrofuran (THF) 11 has been reported (Scheme 4).39 This type of initiation occurs through a single electron oxidation of the co-initiator from the sulfonium cation and subsequent hydrogen atom abstraction from THF to form the α-oxygen radical (12), which can participate in the propagation route. The authors cleverly exploited the propensity of excited TX to participate in HAT from weak C–H bonds.
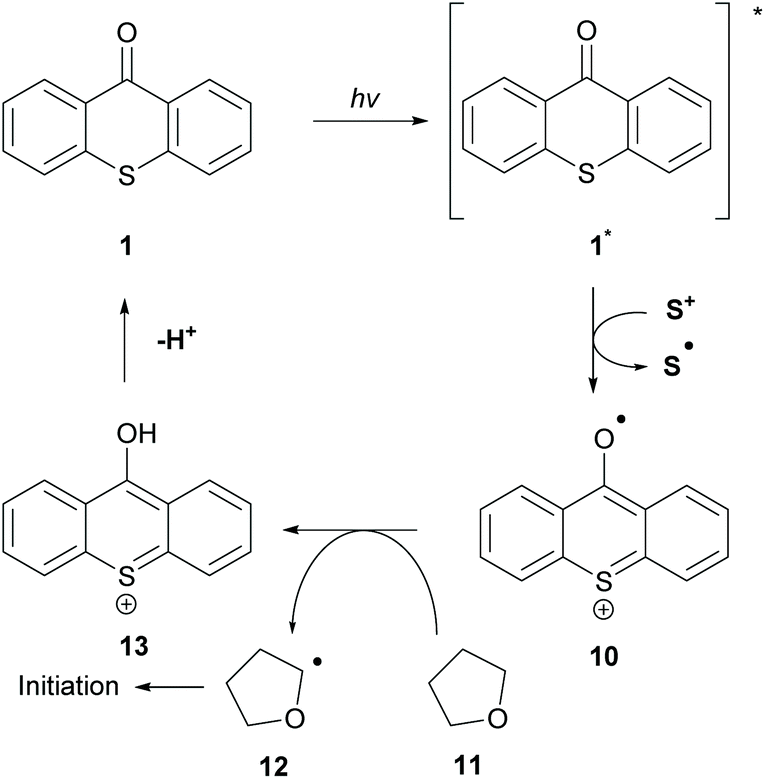 | ||
| Scheme 4 Thioxanthone initiation mechanism combining co-initiators through SET. | ||
There is a diversity of different co-initiators reported in the literature, such as molecules designed to act as hydrogen donors, for example, various amines like MDEA 14 (Scheme 5)40,41 or more specific molecules like benzoxamides 15 (Scheme 5).42 Apart from hydrogen atom donors, co-initiators are also chosen based on their potential to interact with the initiator through SET procedures (Scheme 6). Examples of this type of co-initiator are sulfonium ions,39,43 iodonium,44N-protected BDN or DBU,45 and NHC.46 In order to bypass the need for a co-initiator, researchers designed thioxanthone-type molecules, bearing different functionalities, that upon irradiation can be utilized in polymerisations without the use of an external co-initiator (Scheme 7). More specifically, molecules bearing a thioxanthone scaffold, such as 25–28, upon irradiation, photodecompose to radical species, which are capable of propagating the radical route of polymerisation.47–49
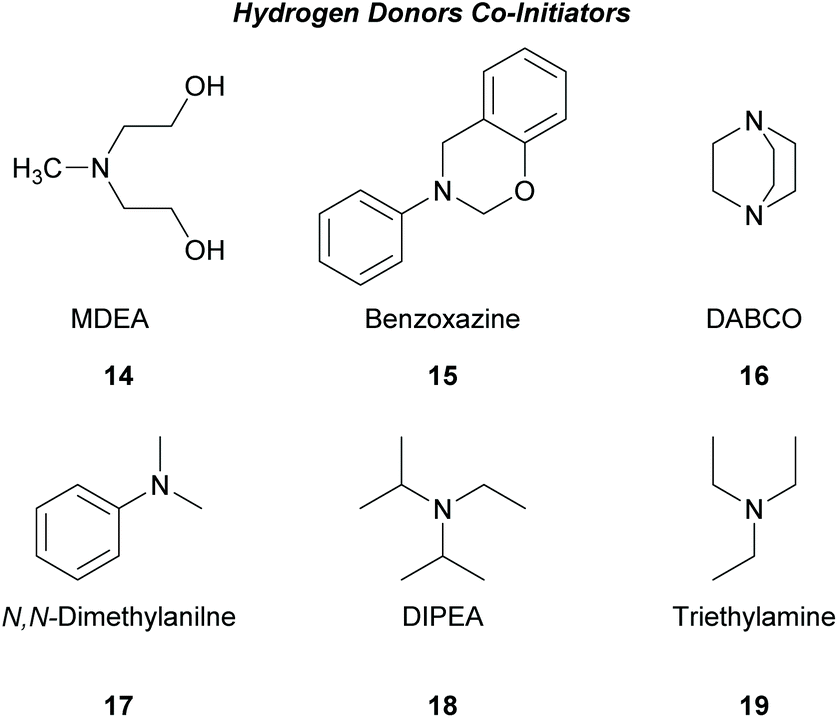 | ||
| Scheme 5 Commonly used hydrogen donors as co-initiators. | ||
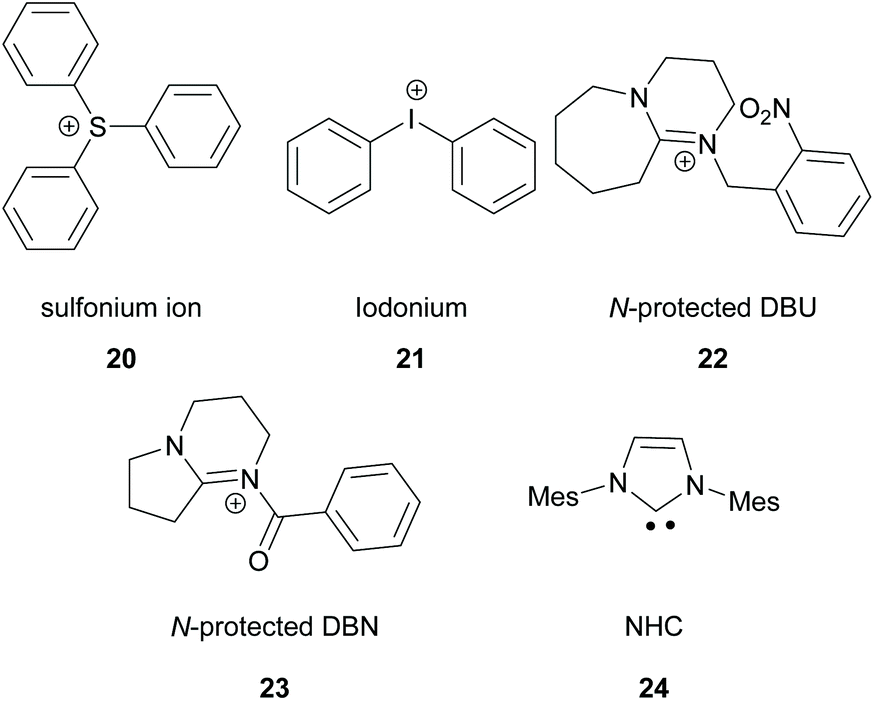 | ||
| Scheme 6 Commonly used molecules interacting with TX via SET. | ||
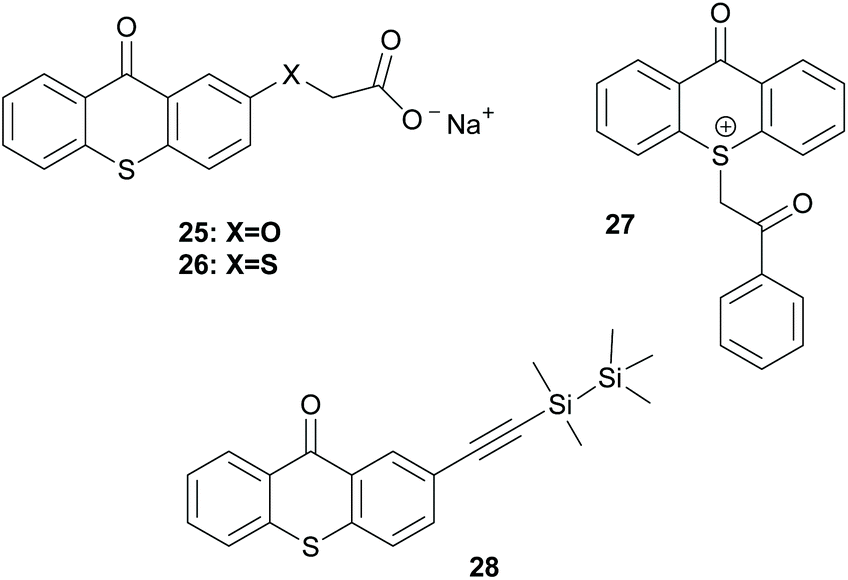 | ||
| Scheme 7 Examples of initiators bearing the thioxanthone chromophore that work via photodecomposition. | ||
Alternatively, thioxanthone-like molecules can promote the polymerisation via an intramolecular HAT (Scheme 8). Molecules like 29, 30 and oligopolymer 31 can promote the polymerisation through this intramolecular HAT mechanism, bearing the active carbonyl and the hydrogen donor in the same molecule. After irradiation, this intramolecular HAT leads to the key radical species that can promote the polymerisation reaction.50–52
 | ||
| Scheme 8 Examples of initiators bearing the thioxanthone chromophore that work via intramolecular HAT. | ||
Finally, there is also a third group of molecules that provides basically a combination of the two above discussed mechanisms, where intermolecular and intramolecular HAT can be performed by molecules 32 and 34 (Scheme 9) to promote the propagation route or interaction with oxygen, like molecule 33 (Scheme 9), forming radical oxygen species that can abstract hydrogen from the monomers.53–55 All the above three categories of photoinitiators were developed because the use of an external co-initiator can often lead to various undesired pathways.
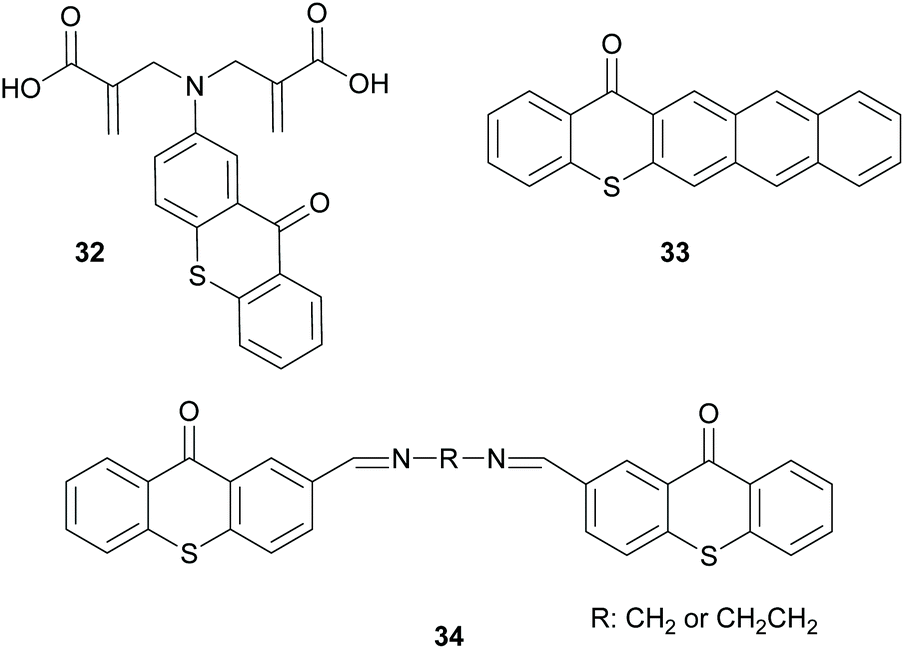 | ||
| Scheme 9 Other examples of initiators bearing the thioxanthone chromophore. | ||
4. Photochemical applications of thioxanthone in organic synthesis
Since thioxanthone (1) can mediate reactions via triplet energy transfer (EnT), hydrogen atom transfer (HAT) or single electron transfer (SET), it has attracted the interest of many scientists worldwide. Apart from the previously reported use in polymerisation chemistry, thioxanthone and its derivatives have been employed in a wide variety of chemical transformations, such as photoisomerisation reactions,56 photoinduced rearrangements,57 [2 + 2] photocycloadditions, Paterno–Buchi cycloadditions, photoredox catalysis and cross-coupling reactions leading to C–O, C–N, C–P and other bond formation. This review is intended to cover them, highlighting the role played by thioxanthone in the reaction mechanism.[2 + 2] Photocycloadditions
Generally, the [2 + 2] cycloaddition reaction constitutes a powerful tool in organic synthesis, since a four-membered ring system, which appears in many biologically active natural products, can be easily generated. Since 2008, photocatalysis has created new perspectives and scientists worldwide explore new methodologies to access the cyclobutane core. The reaction usually proceeds in the presence of a photosensitizer, such as thioxanthone 1, under milder conditions, hindering the possibility of the reverse reaction to occur.58 In 1980, Padwa and coworkers,59 during their mechanistic studies on the photochemical behavior of substituted cyclopropene derivatives, reported a [2 + 2] intramolecular photocycloaddition, which outcompetes diradical dimerisation, when the reaction was performed in the presence of a triplet state sensitizer, such as thioxanthone 1.Since then, a significant amount of effort has been put into the development of asymmetric versions of this transformation. This is not a trivial task, since along with the taming of radicals, the methodology should insert high levels of asymmetry. Bach and coworkers have developed several chiral sensitizers based on aryl ketones, such as benzophenone and xanthone, that were successfully applied towards intramolecular [2 + 2] cycloaddition reactions.60 In a milestone contribution in 2014, they developed the novel photosensitizer 35 based on thioxanthone (Scheme 10A).61 In this novel catalyst, a 1,3,5-trimethyl-3-azabicyclo[3.3.1]nonan-2-one moiety is attached to thioxanthone via an oxazole ring. The formation of two hydrogen bonds between the chiral thioxanthone catalyst and a substrate bearing a lactam moiety (36) resulted in a stable complex, as shown in Scheme 10A, in which one face is shielded, allowing the reaction to take place from the other face. This new catalyst was applied in the enantioselective intramolecular [2 + 2] photocycloaddition of quinolone derivatives 37, affording the desired tetracyclic products 38 in high yields with excellent enantioselectivities (Scheme 10B),61 and in the enantioselective intermolecular [2 + 2] photocycloaddition of various quinolones 39 with electron-deficient alkenes 40 (Scheme 10C).62 The complexation of chiral thioxanthone with the quinolone core, which occurs via the formation of hydrogen bonding, is responsible for the enantioselective approach of the alkene. Mechanistic and computational studies by Yu and coworkers63 revealed that the enantioselectivity is governed by the quick and stable formation of the complex between thioxanthone derivative 35 and the quinolone core of the substrate, in a similar fashion to that shown in Scheme 10A. The lower triplet energy of the thioxanthone derivative (263 kJ mol−1), compared to the previously reported xanthone derivative (316 kJ mol−1), increases the selectivity of the [2 + 2] photocycloaddition.
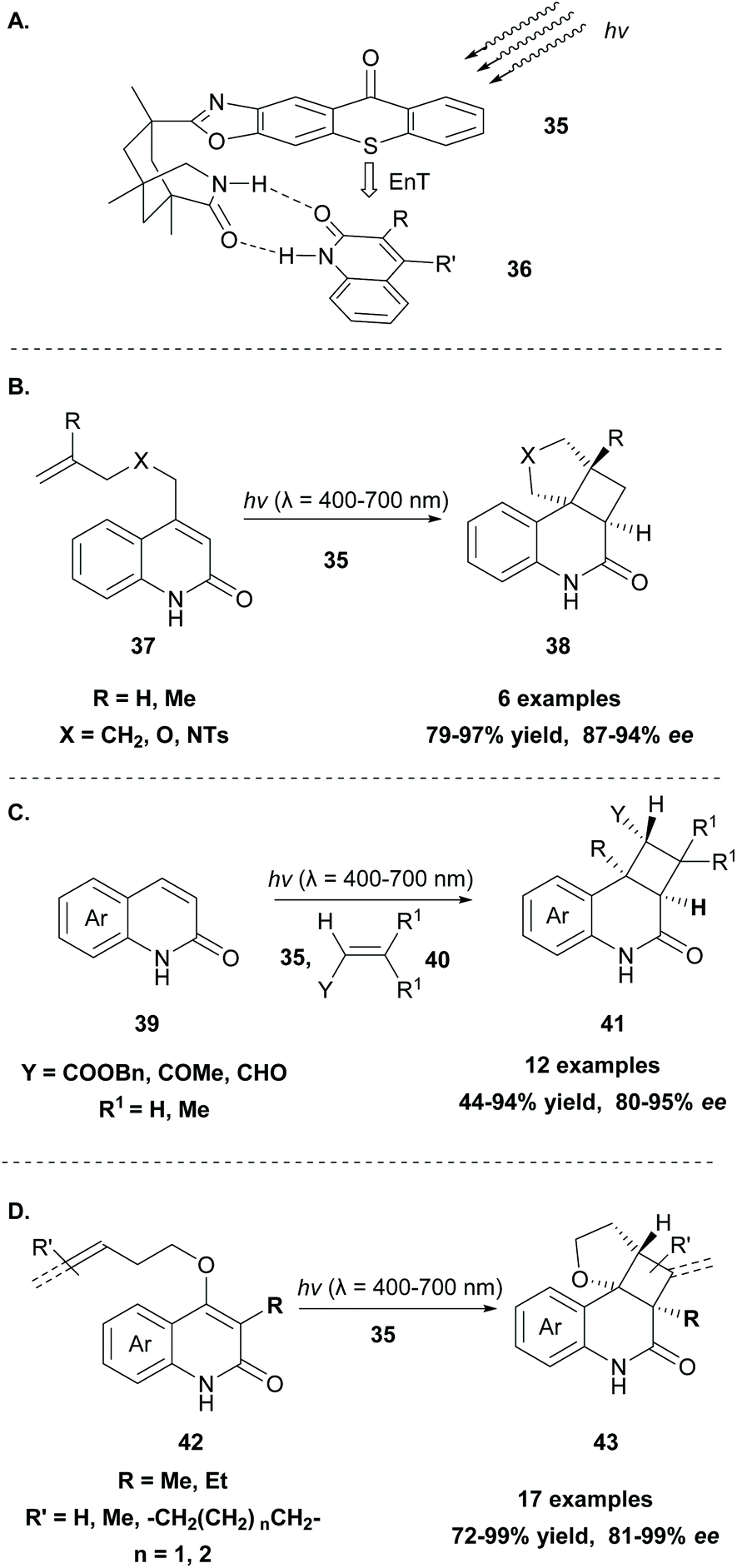 | ||
| Scheme 10 (A) Hydrogen bond complex formation between chiral thioxanthone 35 and general lactam substrate 36 and (B–D) enantioselective [2 + 2] photocycloadditions in the presence of chiral sensitizer 35. | ||
Recently, Bach and coworkers expanded their strategy for the construction of fused cyclobutane fragments, employing chiral catalyst 35 in the intramolecular enantioselective [2 + 2] photoaddition of hydroxyquinolones tethered with alkenes and allenes to form cyclobutene derivatives 43 (Scheme 10D).64 To their surprise, the intramolecular photocycloaddition reaction of quinolone 43 (R = H) in the presence of catalyst 35 failed. This was, later, attributed to the higher triplet energy of quinolone 43 (R = H) 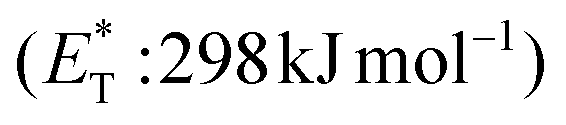 compared to the triplet state energy of the sensitizer
compared to the triplet state energy of the sensitizer 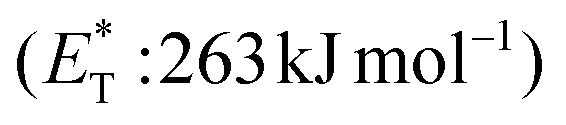 . Methyl substitution on the 3-position of the quinolone core lowered the triplet state energy
. Methyl substitution on the 3-position of the quinolone core lowered the triplet state energy 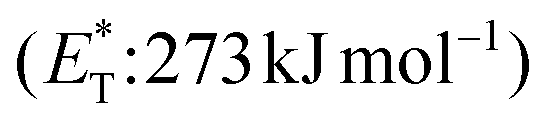 , facilitating the energy transfer from thioxanthone 35, to furnish the desired products with high enantioselectivities. Thus, subtle changes on the reagent backbone can tune the triplet energy required for the reaction to occur. In all cases, an energy transfer mechanism is responsible for the final outcome. Compared to similar approaches using chiral catalysts derived from xanthone and benzophenone, the chiral catalyst derived from thioxanthone offers the use of lower energy wavelength irradiation. In all cases, the major restriction appears to be the a priori existence of a lactam moiety on the substrate, which narrows down the catalyst's general application.
, facilitating the energy transfer from thioxanthone 35, to furnish the desired products with high enantioselectivities. Thus, subtle changes on the reagent backbone can tune the triplet energy required for the reaction to occur. In all cases, an energy transfer mechanism is responsible for the final outcome. Compared to similar approaches using chiral catalysts derived from xanthone and benzophenone, the chiral catalyst derived from thioxanthone offers the use of lower energy wavelength irradiation. In all cases, the major restriction appears to be the a priori existence of a lactam moiety on the substrate, which narrows down the catalyst's general application.
Having shown that the formation of hydrogen bonds between the lactam core of sensitizer 35 and an amide embedded on the substrate governs the enantioselectivity of the photocycloaddition reaction, Bach and coworkers probed other potential candidates. Based on the previous studies of Sivaguru and Beeler,65 several chiral bisthioureas 44 were prepared and applied towards the intramolecular [2 + 2] photocycloaddition reaction of dihydropyridone 45.66 Indeed, as expected, association of bisthiourea 44 with the substrate induced enantioselectivity. Thioxanthone 1 proved to be the ideal triplet state sensitizer, which apart from transferring energy to the complex formed, upon irradiation, also acted as a steric shield, inducing enantioselectivity (Scheme 11). Furthermore, they introduced a novel class of bifunctional photocatalysts,67 where a thiourea moiety was attached on thioxanthone through various chiral linkers. These catalysts were employed in the intramolecular [2 + 2] photocyclization of 2-aryloxycyclohex-2-enones, affording the desired product with low enantioselectivity, indicating that the hydrogen bond interaction between the sensitizer and the substrate is likely to occur.
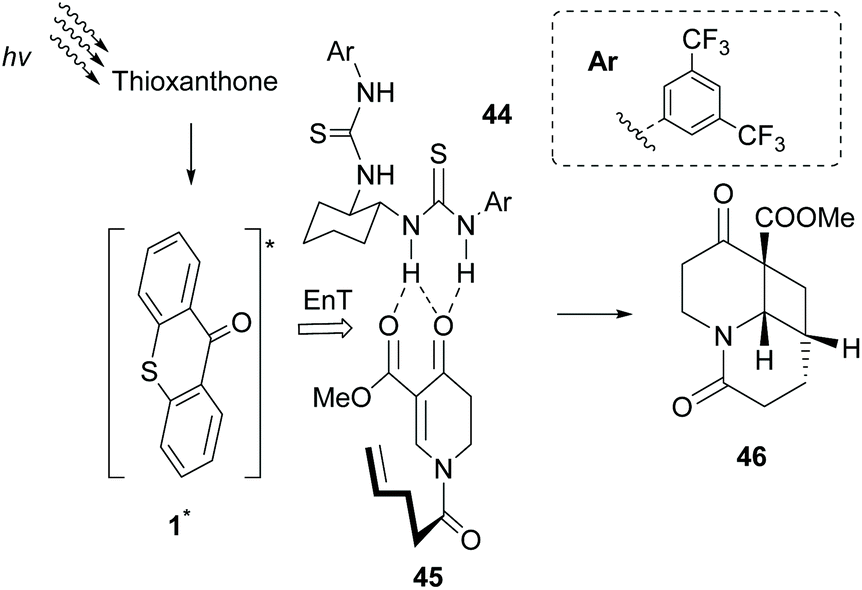 | ||
| Scheme 11 Thioxanthone acting as an energy transfer mediator and an one-face shield. | ||
In 2014, Sivaguru and coworkers68 followed a different strategy to access enantioenriched cyclobutane fragments. They postulated that molecular asymmetry on the chromophore or the substrate could enhance the enantioselectivity in [2 + 2] photocycloaddition reactions. Thus, they exploited various atropoisomeric maleimides in intramolecular [2 + 2] photocycloaddition reactions, using thioxanthone 1 as the sensitizer. For this reason, several o-substituted N-phenylmaleimides 47 were synthesized. Methyl substitution at the 6-position proved to be crucial as it restricts rotation around the C–N bond. The reaction took place upon irradiation at 420 nm, using 30 mol% of thioxanthone 1 as the triplet sensitiser, affording the desired products in moderate to high yields with excellent enantioselectivity (Scheme 12A). Based on their achievements, two years later, they expanded their strategy employing a series of atropisomeric enone imides and enone amides bearing N–Caryl bond rotation, demonstrating that the axial chirality could easily control the enantioselectivity in [2 + 2] photocycloaddition.69
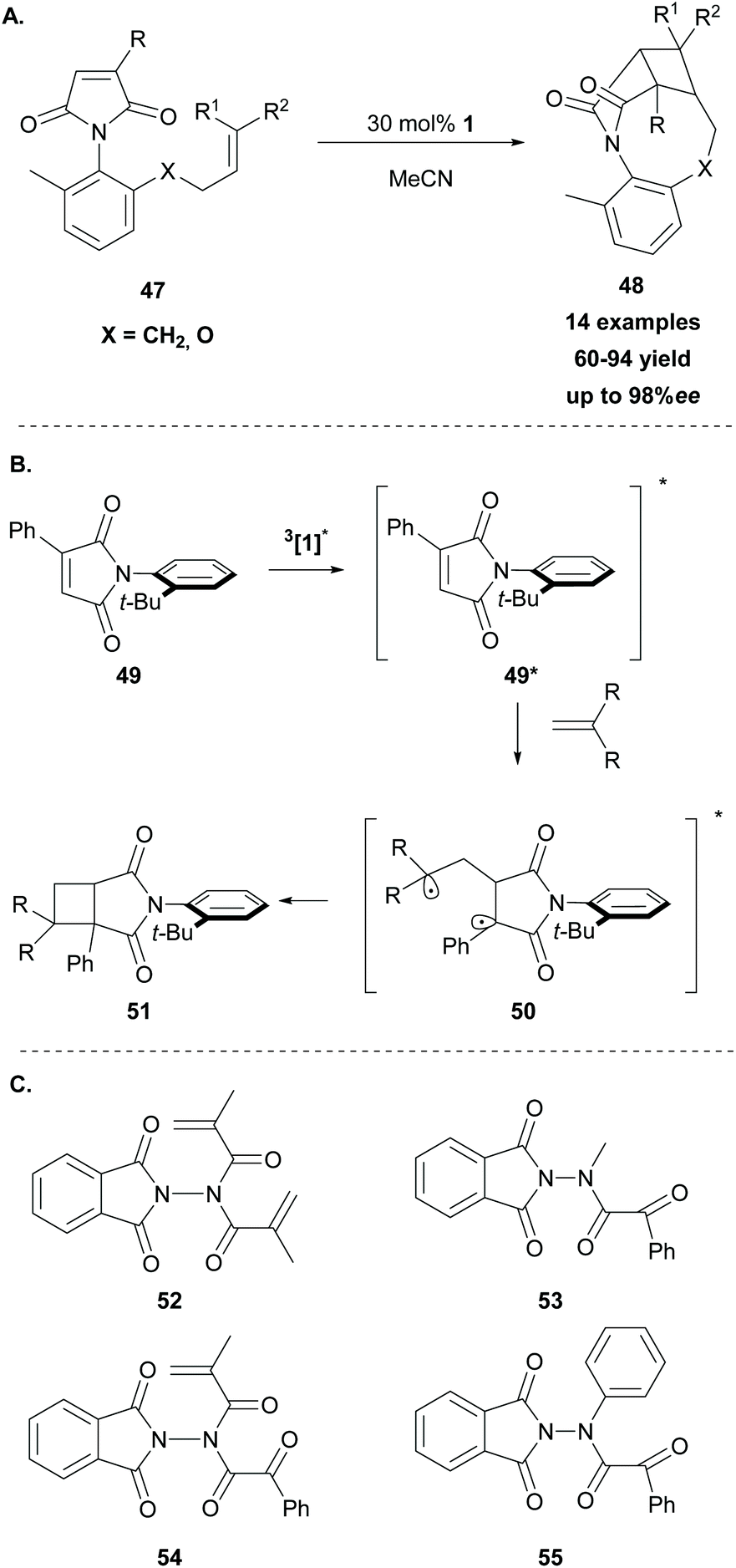 | ||
| Scheme 12 (A) Intramolecular [2 + 2] photocycloaddition induced by the axial chirality, (B) energy transfer mechanism of the intermolecular [2 + 2] cycloaddition, and (C) hydrazides derived from N-aminophthalimides. | ||
In 2019, Sivaguru employed thioxanthone 1 as the triplet sensitizer in the intermolecular [2 + 2] photocycloaddition of atropoisomeric maleimides with various substituted alkenes with complete control of the regioselectivity.70 Mechanistic studies revealed a triplet state energy transfer from excited thioxanthone to N-phenyl maleimide 49. The latter reacted with the alkene to furnish the more stable biradical 50, which crossed to the singlet state and upon cyclization formed the desired product 51, as shown in Scheme 12B. The phenyl group attached on the maleimide core plays a major role in the regioselectivity of the reaction, stabilizing the intermediate biradical. Following their interest in the field of enantioselective [2 + 2] photocycloaddition, Sivaguru and coworkers examined various hydrazides 52–55 derived from N-aminophthalimides as photoauxiliaries (Scheme 12C).71 The substrates bearing hydrazide-derived substituents were applied towards [2 + 2] photocycloadditions, Paterno–Buchi reactions, Norrish–Yang reactions and 6π-photocyclizations, using thioxanthone as the triplet state sensitizer. In all cases, the hydrazide chromophore, due to its specific conformation, acted as an excellent photoauxiliary. This previous achievement led them to the expansion of their strategy, employing hydrazides derived from oxazolidinones as potential reactants, towards enantioselective [2 + 2] photocycloadditions with comparable results.72 The mechanism of the reaction is substrate-dependent and involves either an energy transfer pathway or an electron transfer pathway.
In 2019, Gilmour and coworkers reported a biomimetic one-pot synthesis of fused dihydrocoumarins 58 from cinnamic acids 56, which comprised a photosensitized E to Z isomerization, followed by subsequent lactonization and an intramolecular [2 + 2] cycloaddition via two energy transfer events.73 The energy requirement for the E/Z isomerization is rather low, and the isomerization can be performed in the presence of various sensitizers. However, for the second energy transfer event, given the high triplet energy of the intermediate coumarin fragment 57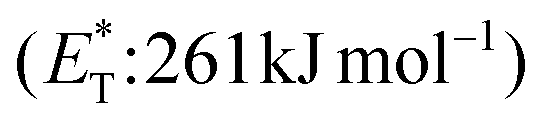 , thioxanthone (1) with a significantly higher triplet energy
, thioxanthone (1) with a significantly higher triplet energy 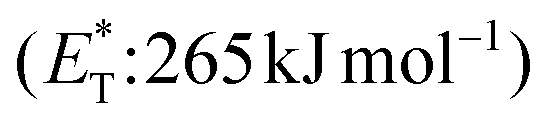 than that of (−)-riboflavin
than that of (−)-riboflavin 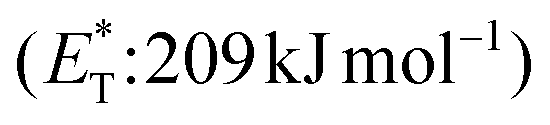 was the obvious choice (Scheme 13). The scope of the intermolecular [2 + 2] cycloaddition is unfortunately limited to the use of terminal alkenes.
was the obvious choice (Scheme 13). The scope of the intermolecular [2 + 2] cycloaddition is unfortunately limited to the use of terminal alkenes.
 | ||
| Scheme 13 Biomimetic one-pot synthesis of fused dihydrocoumarins. | ||
Recently, Kappe and coworkers, exploring the potential of flow technology in photochemistry, designed and optimized a scalable [2 + 2] photocycloaddition reaction between citraconic anhydride and ethylene.74 Thioxanthone (1) was compared to other aromatic ketones, such as benzophenone, as triplet energy sensitizers for the gaseous reactant ethylene. In this case, benzophenone worked better than 1. Apart from its use in synthesis, thioxanthone was also employed to promote DNA [2 + 2] photocycloaddition in 5-cinnamoyl-substituted cytidine, thus facilitating its conversion to thymine, providing an efficient method for the methylation profile of DNA.75
Another well-investigated [2 + 2] photocycloaddition reaction is the Paterno–Buchi reaction,76 which occurs between an excited state carbonyl group and an alkene leading to oxetanes, usually in the presence of a triplet state sensitizer. Recently, Bach and coworkers employed 3-substituted quinoxalinones 59 as imine scaffolds, towards an aza-Paterno–Buchi reaction (Scheme 14).77 To attain high enantioselectivity, chiral sensitizer 35 was employed. As shown in Scheme 10A, hydrogen bond interactions between the lactam core of the sensitizer and substrate 59 led to a complex, which allows the approach of alkene 60, only from the one side, leading to 61 in good to excellent yields with high enantioselectivities (Scheme 14). The authors examined the scope of the reaction employing various arylethylenes. Various substituents on the aryl ring did not alter the final outcome. Notably, in the absence of the substituent at the C-3 position of the quinoxalinone fragment, the reaction does not proceed. In this case, the substitution product at the C-3 position is formed in a low yield, probably due to the quick hydrogen abstraction.
 | ||
| Scheme 14 Enantioselective aza-Paterno–Buchi cycloaddition. | ||
Merger of photoredox catalysis with transition metal catalysis
During the last few years, many scientists have focused their attention on the merger of metal catalysis with photocatalysis. This strategy allows access to high oxidation state metal species using milder conditions. These light-triggered processes allow the synthesis of diverse organic structures in a rapid and precise manner. A very interesting approach was developed by Yagci and coworkers, incorporating photo-initiators onto CuII catalysts. They reported a ligand-free photocatalyst, a combination of the thioxanthone carboxylate moiety with CuII ions, to promote the CuAAC reaction.78Murakami and coworkers reported a nickel-catalysed homocoupling of aryl halides, introducing thioxanthone 1 as an efficient photoredox catalyst for single-electron transfer. The reaction takes place in the presence of a catalytic amount of complex 64 [(dtbbpy)NiCl2] (dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) and stoichiometric iPr2NEt, which acts as an electron donor (Scheme 15).79 A plausible mechanism for the homocoupling reaction of aryl halides proposed by Murakami consists of three redox cycles as shown in Scheme 15. The first photoredox cycle commences with the excitation of thioxanthone by visible light irradiation and its excited triplet state undergoes reduction with iPr2NEt to furnish the corresponding radical anion 1˙−. The latter reduces Ni(II) species to Ni(0) species regenerating thioxanthone. Redox cycle II begins with the oxidative addition of aryl halide 62 to Ni(0) species to produce complex 66, which either forms Ni(II)dibromide 64, following and completing cycle II, or forms diarylnickel(II) 67, when engaged to cycle III. Finally, diarylnickel(II) 67 species via reductive elimination delivers the desired product 63, regenerating Ni(0) and completing cycle III. A wide variety of substrates were examined by Murakami applying the optimal reaction conditions. Aryl iodides and bromides proved to be excellent substrates, affording the desired products in moderate to high yields, while the reaction using 4-chlorotoluene afforded a low yield, due to possible competing reduction. This homocoupling reaction displayed high functional group tolerance, even in the case of the potentially reactive boryl group. Less reactive heteroarenes (e.g. 3-bromothiophene) resulted in a lower yield. Furthermore, this coupling reaction was also implemented in a flow system affording comparable results.
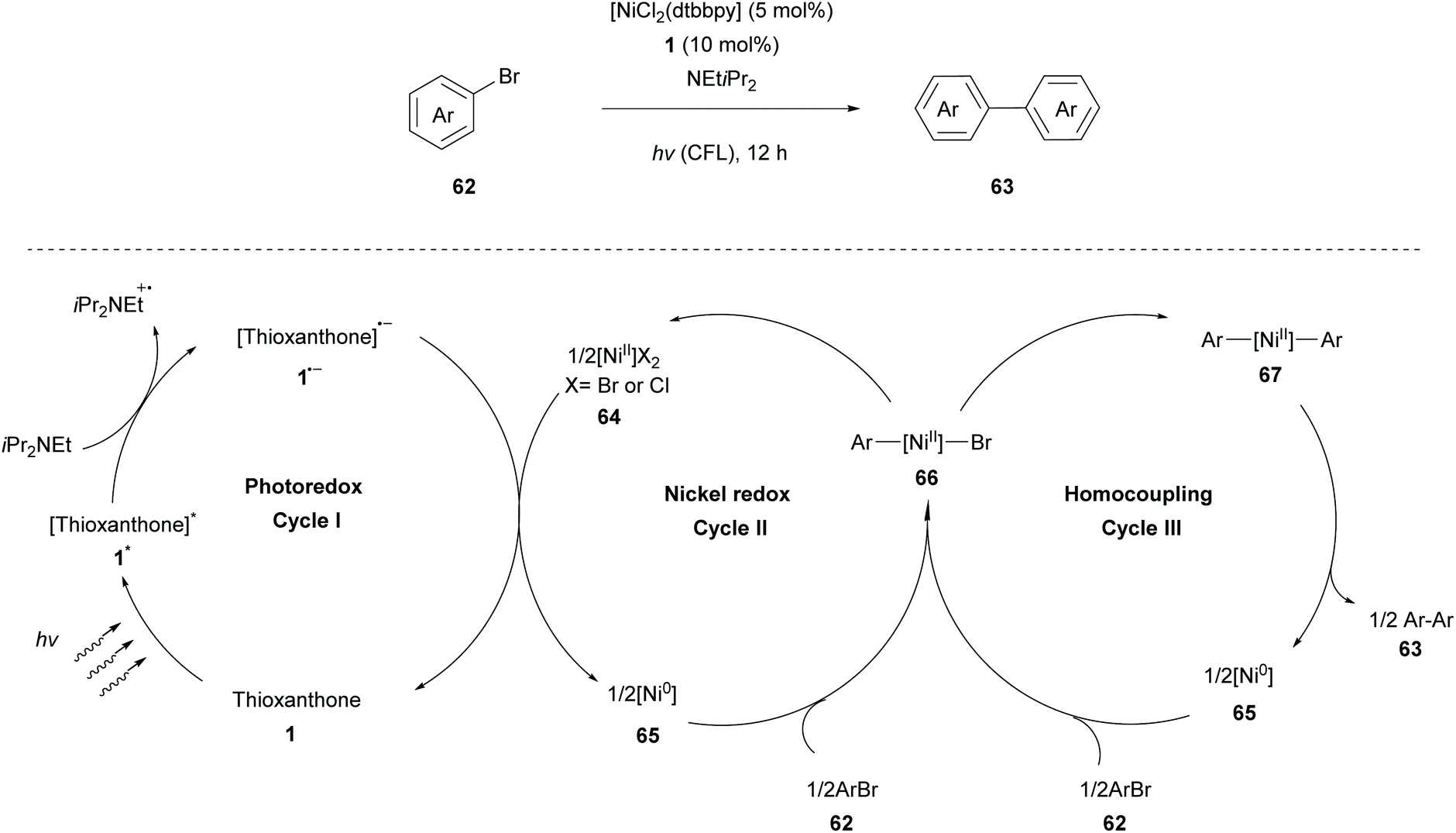 | ||
| Scheme 15 Dual photoredox and nickel catalysis mechanism. | ||
In 2019, Lang and coworkers reported a new nickel-catalysed esterification reaction between aromatic carboxylic acids and aryl halides, employing thioxanthone as the triplet energy transfer photosensitizer.80 Optimization trials revealed that the use of 20 mol% thioxanthone, 5 mol% NiBr2, 6 mol% dtbby, and 1.5 eq. of K3PO4 in DMSO promoted the esterification reaction of 4-bromobenzonitrile and benzoic acid, furnishing the desired ester in 93% yield after 24 h of irradiation with 45 W CFL. The authors applied the optimum reaction conditions in a variety of aryl bromides bearing electron-withdrawing groups on the aromatic ring. The reaction displayed excellent tolerance affording the desired esters in moderate to high yields (Scheme 16). In contrast, the esterification reaction did not proceed when bromobenzene or p-bromotoluene was used, while the corresponding esters in these two cases were prepared in low yields from iodoaryls. Lang and co-workers performed the reaction of 4-bromobenzonitrile and benzoic acid in the presence of various photosensitizers to shed light on the mechanism of the reaction. It appears that the reactivity was directly correlated with the triplet state energy 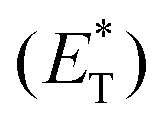 of the catalyst, proving that thioxanthone was the most effective. It must be mentioned that when the same reaction was carried out in the presence of catalysts with higher redox potentials than that of thioxanthone, such as CF3-thioxanthone or Cl-thioxanthone, decreased yields were observed, implying that the potential oxidation of Ni(II) to Ni(III) via a single electron transfer (SET) process is not applicable in this mechanism. Further mechanistic studies, comprising irradiation of (dtbbpy)-4-cyanophenylnickel(II)bromide and CsOAc in the presence of thioxanthone for 24 h, afforded 4-cyanophenyl acetate in 81% yield. When the same experiment was conducted in the absence of the photosensitizer, after 120 h of irradiation, the desired product was afforded in 15% yield. These results indicated that the reaction proceeds through triplet energy transfer from excited-state thioxanthone to the (dtbbpy)-4-cyanophenylnickel(II)carboxy intermediate 71. According to Lang, the reaction commences with the oxidative addition of aryl halide to nickel providing complex 70, followed by ligand exchange with a carboxylate nucleophile to form intermediate 71 (Scheme 16). Simultaneously, upon visible light irradiation, thioxanthone is excited to the triplet state. Intermediate 71 is excited upon triplet–triplet energy transfer from thioxanthone. The triplet excited state intermediate 71*via reductive elimination delivers the desired ester 69, completing the catalytic cycle (Scheme 16). Although the authors mainly employed aryl bromides, the use of iodoaryls was shown to lead to superior results. Also, acids bearing an aliphatic side chain were employed successfully.
of the catalyst, proving that thioxanthone was the most effective. It must be mentioned that when the same reaction was carried out in the presence of catalysts with higher redox potentials than that of thioxanthone, such as CF3-thioxanthone or Cl-thioxanthone, decreased yields were observed, implying that the potential oxidation of Ni(II) to Ni(III) via a single electron transfer (SET) process is not applicable in this mechanism. Further mechanistic studies, comprising irradiation of (dtbbpy)-4-cyanophenylnickel(II)bromide and CsOAc in the presence of thioxanthone for 24 h, afforded 4-cyanophenyl acetate in 81% yield. When the same experiment was conducted in the absence of the photosensitizer, after 120 h of irradiation, the desired product was afforded in 15% yield. These results indicated that the reaction proceeds through triplet energy transfer from excited-state thioxanthone to the (dtbbpy)-4-cyanophenylnickel(II)carboxy intermediate 71. According to Lang, the reaction commences with the oxidative addition of aryl halide to nickel providing complex 70, followed by ligand exchange with a carboxylate nucleophile to form intermediate 71 (Scheme 16). Simultaneously, upon visible light irradiation, thioxanthone is excited to the triplet state. Intermediate 71 is excited upon triplet–triplet energy transfer from thioxanthone. The triplet excited state intermediate 71*via reductive elimination delivers the desired ester 69, completing the catalytic cycle (Scheme 16). Although the authors mainly employed aryl bromides, the use of iodoaryls was shown to lead to superior results. Also, acids bearing an aliphatic side chain were employed successfully.
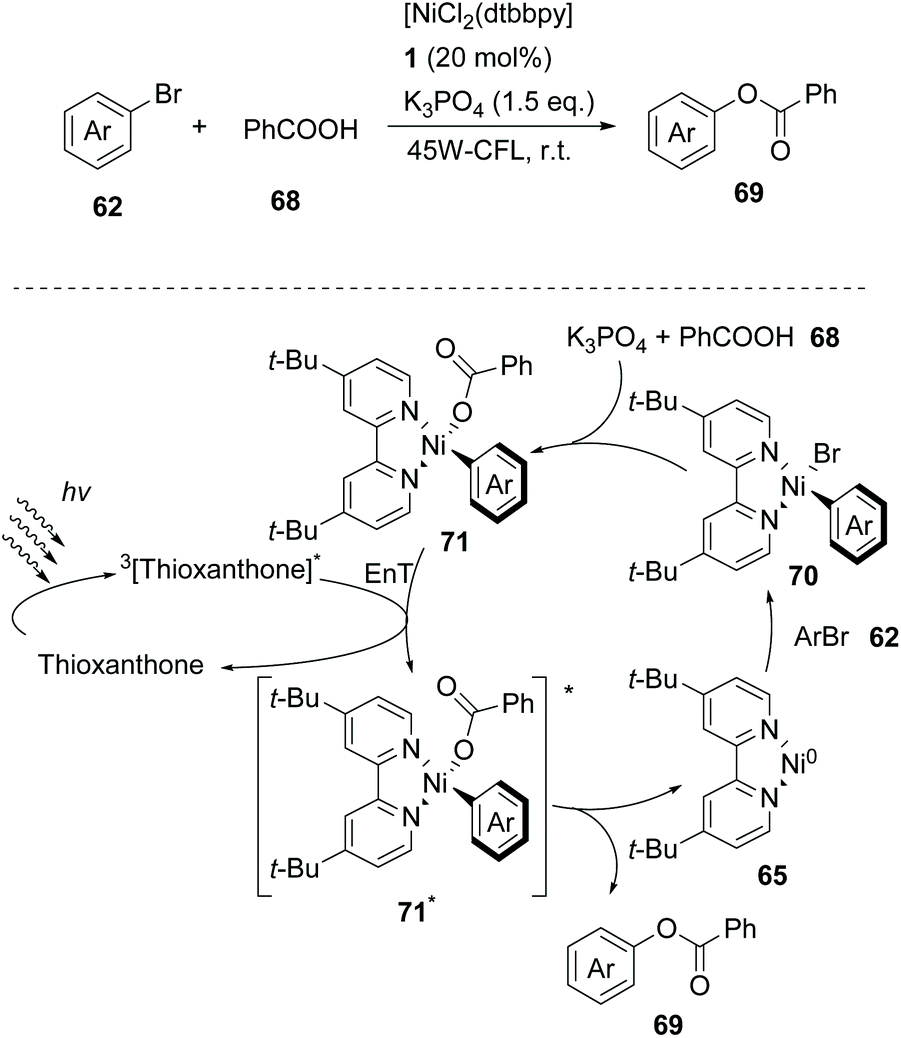 | ||
| Scheme 16 Mechanism of the nickel-catalyzed esterification reaction. | ||
Based on their gained experience, in 2020, Lang and coworkers exploited thioxanthone as the triplet state sensitizer in Csp–Csp2 cross-coupling reactions between aryl halides and alkynes.81 Parallel experiments performing the same reaction in the presence of various photocatalysts indicated that the reaction does not involve electron transfer but proceeds via energy transfer. As shown in Scheme 17, it was proposed that the reaction begins with an oxidative addition of [dtbbpyNi0] to aryl halide 62, affording complex 70. The latter reacts with the in situ-generated Zn(II) acetylide to afford complex 74, which is excited to its triplet state from the excited triplet state thioxanthone. The resulting excited state Ni(II) complex 74* generates 73 and 65 completing the catalytic cycle. Aryl- and alkyl-substituted terminal alkynes proved to be excellent substrates, while heteroaryl alkynes afforded the desired product in a moderate yield. Aryl- and heteroaryl-bromides bearing electron-withdrawing groups afforded the desired product in good to excellent yields, while aryl bromides bearing electron-donating groups afforded products in low yields.
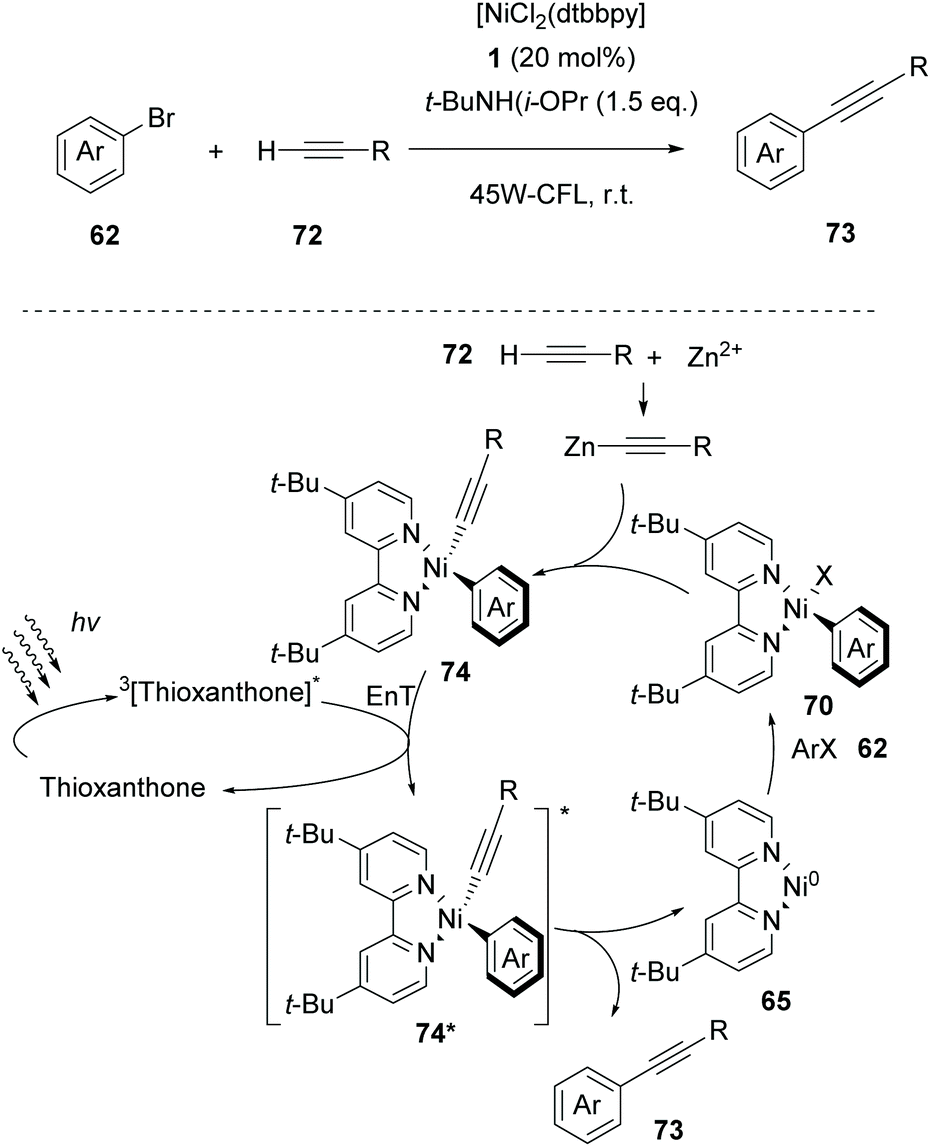 | ||
| Scheme 17 Nickel-catalyzed Sonogashira cross-coupling reaction. | ||
In 2021, Li and coworkers merged thioxanthone catalysis with nickel catalysis to perform an efficient visible-light photoredox arylation of H-phosphine oxides or H-phosphites with aryl halides (Scheme 18).82 Extensive mechanistic studies were performed, indicating that the cross-coupling reaction proceeds via a SET event between excited state thioxanthone and H-phosphine oxides or H-phosphites. This was further supported upon correlating the reactivity of the excited-state redox potentials of the photocatalyst. Thioxanthone 1, with the highest excited-state reduction potential (Ered = +1.18 V versus SCE), resulted in a higher yield compared to photocatalysts with lower excited-state potentials, such as fluoren-9-one (Ered = 0.95 V versus SCE) or Ru(bpy)32+ [E(RuII*/RuI) = 0.77 V versus SCE]. This protocol described an easy and economical access to arylphosphine oxides and arylphosphonates which can be further used for further functionalization.
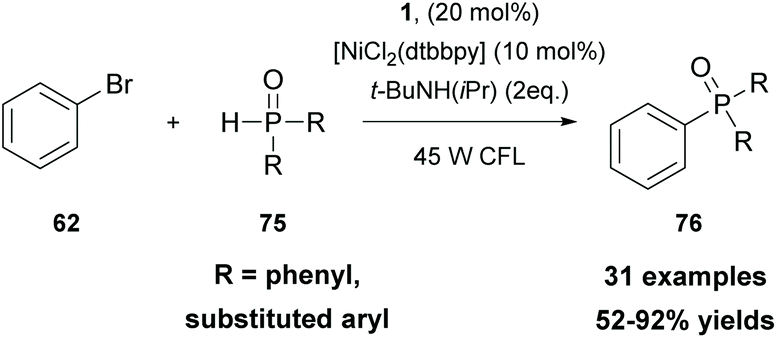 | ||
| Scheme 18 C–H functionalization using phosphine oxides and phosphonates. | ||
Singlet oxygen generation
In 2017, Xiao and coworkers developed a new series of bifunctional photocatalysts that can easily coordinate to a metal.83 This novel class of photocatalysts consisted of two parts. The first part was a ligand that coordinates with a metal. For this purpose, they employed bisoxalines (BOX), privileged ligands in the field of asymmetric catalysis. The second part was thioxanthone, which serves as a sensitizer. These two parts were combined via an ester linkage. The novel class of catalysts 79 was found to generate singlet oxygen upon excitation by visible light, promoting asymmetric oxidation of β-keto esters and β-ketoamides. Switching the ester moiety from methyl and isopropyl to the bulkier adamantly group increased the enantioselectivity of the reaction. Various substituted 1-indonanone derivatives were tested, producing the corresponding α-hydroxy derivatives 78 in high yields with excellent enantioselectivities (Scheme 19).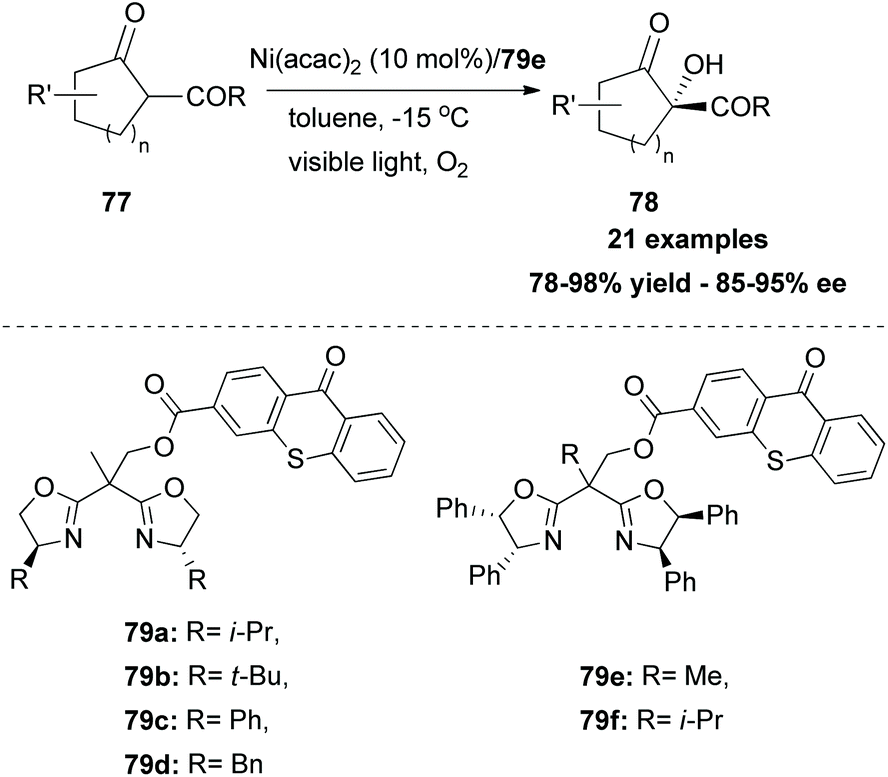 | ||
| Scheme 19 Aerobic oxidation: access to enantioenriched hydroxy ketoesters. | ||
In 2018, Guo and coworkers84 reported a metal-free aerobic oxidation of sulfides to sulfoxides, using 4-phenyl thioxanthone 82 as the visible light sensitizer (Scheme 20). Upon visible light irradiation, oxygen is sensitized to its singlet state, providing the oxidant source for the reaction. Several aromatic and dialkyl sulfides were tested under the optimal conditions delivering the corresponding sulfoxides in good to excellent yields. Unfortunately, the authors did not perform the reaction in the presence of other, prone to oxidation, moieties such as olefins, alkynes, etc.
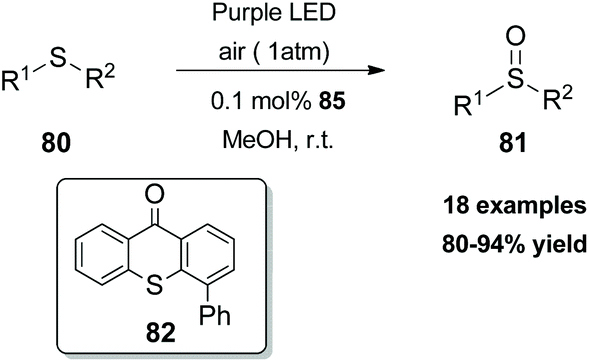 | ||
| Scheme 20 Aerobic oxidation of thioethers with generated singlet oxygen. | ||
In 2020, Kokotos and coworkers, following their continuous interest in photocatalysis, exploited thioxanthone, developing a green and sustainable methodology for the oxidation of alcohols to their corresponding carbonyl derivatives (Scheme 21).85 Experimental observation indicated that singlet oxygen appears to be the oxidant source. Extensive mechanistic studies were performed to distinguish whether the reaction involves an energy transfer event or a hydrogen atom transfer process. A plausible mechanism for this reaction involves excitation of thioxanthone by visible light and subsequent energy transfer to molecular oxygen, generating singlet oxygen. Then, two pathways are possible. Singlet oxygen abstracts a hydrogen atom from benzylic alcohol 83, furnishing the corresponding benzyl radical (trapped with TEMPO). This event is followed by fast radical recombination, leading to peroxy hemiacetal 86, which under further irradiation is decomposed the corresponding benzaldehyde 84 (Scheme 21). The second possibility is a direct insertion of singlet oxygen to 83 (Scheme 21). This methodology was applied to numerous aliphatic and aromatic alcohols, affording the corresponding carbonyl derivatives in moderate to excellent yields. Furthermore, it was also examined using natural light, affording comparable results. However, primary aliphatic alcohols proved to be poor substrates.
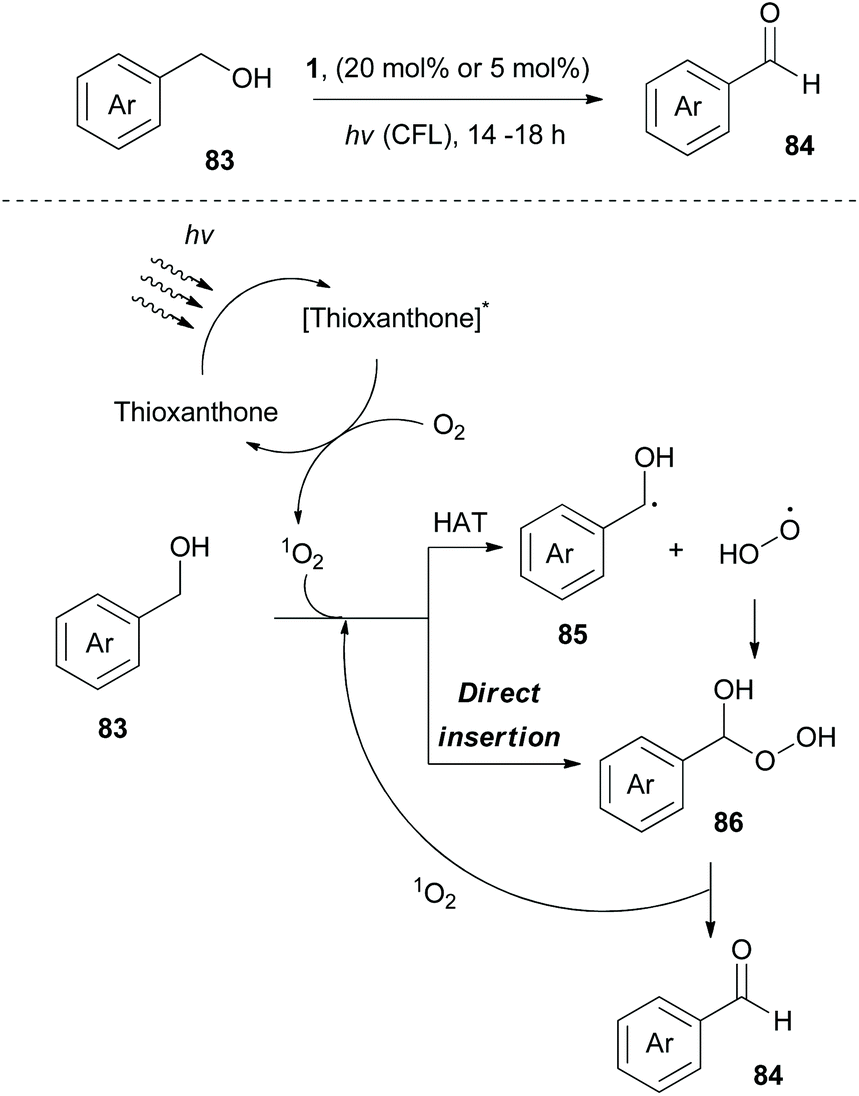 | ||
| Scheme 21 Aerobic oxidation of alcohols with generated singlet oxygen. | ||
Miscellaneous applications
While this review was in the process of writing, a seminal methodology from Glorius and coworkers was published.86 Investigating the synthetic utility of a novel oxyamination strategy, they reported a metal-free regioselective oxyimination addition to unactivated alkenes (Scheme 22). Based on their previous reports,87 Glorius and coworkers envisioned that oxime esters could be excellent candidates for the simultaneous generation of both N- and O-centered radicals. For this reason, they studied the reaction of several benzophenone oxime esters 87 with 1-octene. Oxime esters derived from acetic acid and benzoic acid failed to generate O-radicals due to the competitive decarboxylation reaction, which is kinetically favored. Phosphonate esters and carbamates also failed to afford the desired product. Remarkably, methyl and ethyl carbonate esters proved to be the optimal substrates, generating O-radicals. The reaction was performed in the presence of various photocatalysts. Among all, thioxanthone, having the highest triplet state energy, afforded the desired oxyimination product in the highest yield. Mechanistic studies supported the occurrence of an energy transfer event between the excited thioxanthone and the oxime ester (Scheme 22). The excited oxime ester undergoes a homolytic cleavage of the N–O bond to generate N- and O-centered radicals. Both radicals are added in the double bond to form the 1,2-oxyimination product 89 (Scheme 22). This oxyamination methodology was applied in a variety of mono-, di-, and tri-substituted alkenes, generating in all cases the desired addition products, while tetra-substituted alkenes were unreactive. The authors also examined the application of the reaction in more complex alkenes derived from (1S)-(−)-camphanic acid, camphene, diprogulic acid, (−)-menthol and cholesterol. Furthermore, the synthetic utility of this protocol was shown, applying it to the short synthesis of the amino alcohols leucinol and isoleucinol on a gram scale.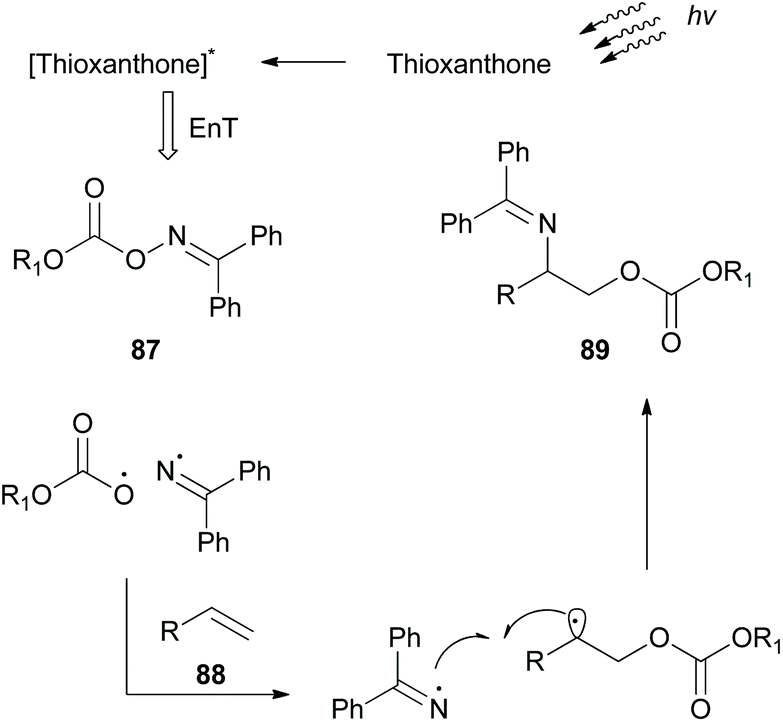 | ||
| Scheme 22 Oxyamination of unactivated alkenes. | ||
In the field of photoredox and enamine catalysis, Aleman and coworkers, inspired by Xiao,83 developed a series of bifunctional photoaminocatalysts from thioxanthone and various substituted imidazolidinones.88 The use of chiral imidazolidinones was expected to control the facile approach of the radical inducing enantioselectivity, while the attached thioxanthone would mediate an energy transfer event or an electron transfer event. Photophysical and electrochemical studies on the new photocatalysts pointed out their potential to act as visible light sensitizers. Photoaminocatalysts 90 and 91 were applied towards the α-alkylation of various aldehydes using a range of α-bromocarbonyls and bromonitriles as the radical precursors, affording excellent enantioselectivity and yield (Scheme 23).
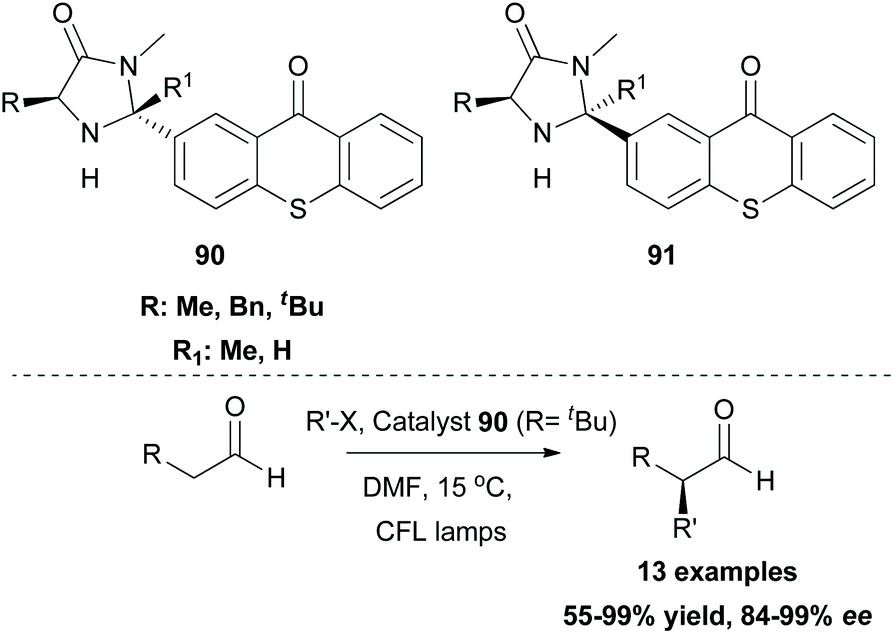 | ||
| Scheme 23 Enantioselective α-alkylation of aldehydes. | ||
Bach and coworkers employed chiral sensitizer 35 in the deracemization of allenes 92,89 cyclopropanes 9390 and sulfoxides 94 (Scheme 24).91 In all cases, the basic requirement is the coordination of the substrate to the catalyst prior to sensitization. Upon excitation of the catalyst, energy is transferred to the substrate, enriching one enantiomer.
 | ||
| Scheme 24 Deracemization using chiral sensitizer 35. | ||
In 2004, Steiner reported thioxanthone as the most efficient photocatalyst for the triplet sensitization of photolabile protecting groups (ppg), such as the 2-(2-nitrophenyl)propoxycarbonyl (NPPOC) group, which are widely used in photolithographic DNA chip synthesis.92 Three years later, following this observation, they developed a new class of ppg 95, where thioxanthone was linked on the NPPOC group, and studied the mechanism of the intramolecular triplet–triplet energy transfer (Scheme 25).93 They proposed that the mechanism for triplet–triplet energy transfer depends on the linker chain. In the case of long linkers or of inflexible short linkers, energy transfer takes place from the low triplet ππ* state of thioxanthone, whereas for short and flexible linkers, it occurs from the higher triplet nπ* state of thioxanthone. In 2010, Pirrung and coworkers94 reported that isopropylthioxanthone having a large two-photon absorption cross section can be used in the two-photon photochemical removal of NPPOC.
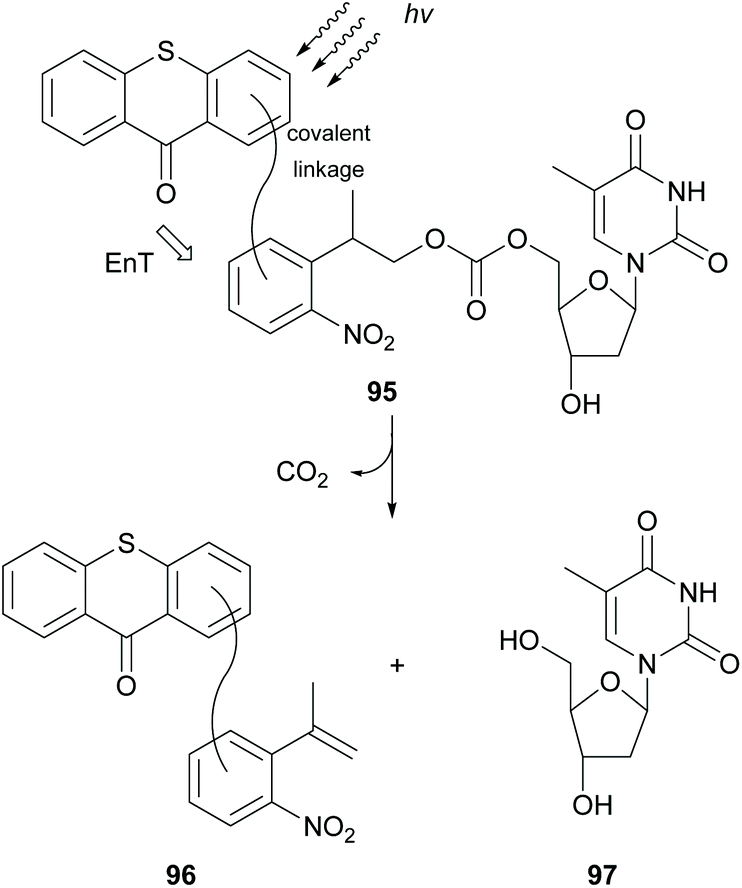 | ||
| Scheme 25 Novel class of thioxanthone-based NPPOC protecting groups. | ||
In 2017, Ooi and coworkers examined the ability of several photocatalysts derived from thioxanthone 98 to act as excited state reductants in their attempt to generate a nitrogen-based radical from N-acyloxyphthalimides 99 (Scheme 26).95 Interestingly, thioxanthone substituted with an electron-donating group under 365 nm LED irradiation proved to be an excellent catalyst for nitrogen-based radical generation. To distinguish whether the arene imidation process takes place through an energy transfer event or a single electron transfer event, Ooi and coworkers examined the triplet energy of all reactants. It was obvious that since thioxanthone derivatives exhibited lower triplet energy than the other reactants, energy transfer had to be excluded. On the other hand, examination of the triplet state oxidation potentials of the catalysts and the reduction potentials of the substrate corroborated the possibility of an electron transfer event. Based on their findings, they proposed a catalytic cycle that starts with the excitation of the substituted thioxanthone catalyst, followed by a subsequent intersystem crossing (ISC) to afford the triplet excited state. Excited substituted thioxanthone donates an electron to the imidating agent, forming radical anion 99˙−. The latter undergoes fragmentation to generate a phthalimidyl radical (PhthN˙) and a 3,5-bis(trifluoromethyl)benzoate anion. Addition of the phthalimide radical to the arene delivers neutral radical species that are oxidized by thioxanthone radical cation [98]˙+ to provide intermediate 102. Deprotonation of intermediate 102 by a 3,5-bis-(trifluoromethyl)benzoate anion produces the desired product 103, along with the corresponding carboxylic acid. Various arenes and heteroarenes underwent the reaction in good to excellent yields. However, in the case of arenes bearing electron-withdrawing groups, an increased amount of the catalyst was necessary for reaction completion. Ooi and coworkers extended this methodology exploiting thioxanthone derivatives to generate a carboxyl radical from peroxides to access acyloxy derivatives. Thioxanthone photocatalyst 98 was also examined towards C-centered radical generation from 1,3-dioxolane. This process involved a single-electron transfer event, followed by a proton transfer event. Ooi and coworkers reported the reaction of the 1,3-dioxolane radical with α,β-unsaturated carboxylic acids.96
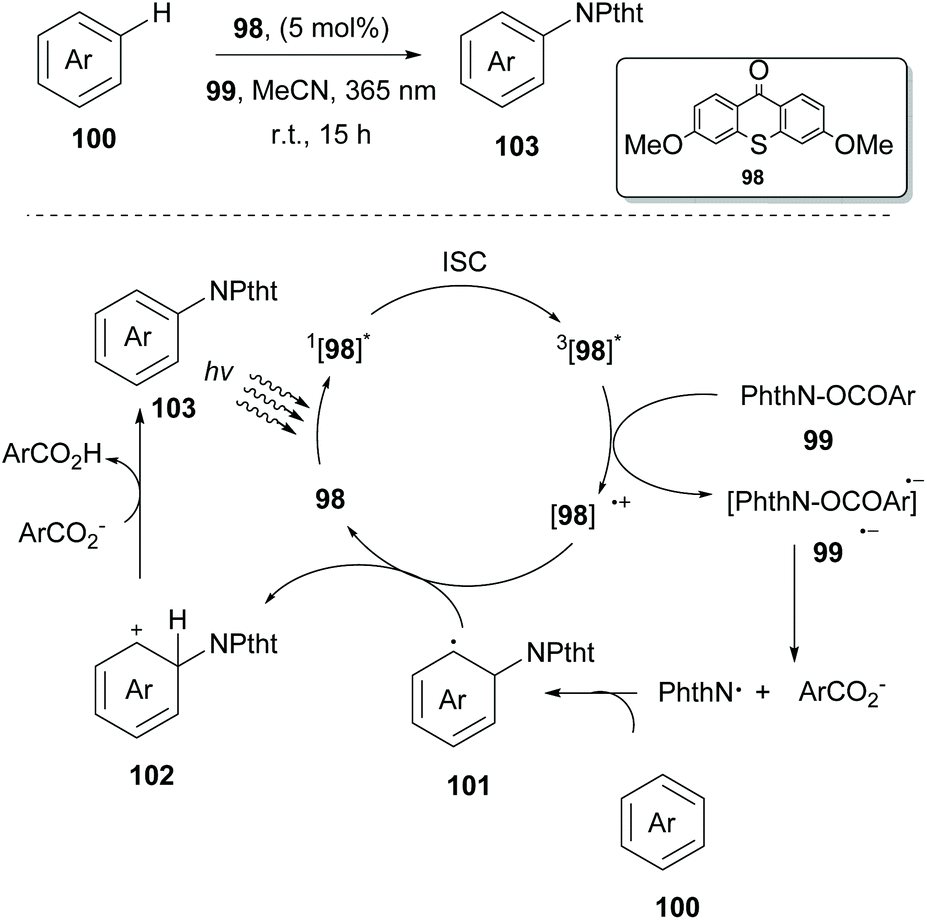 | ||
| Scheme 26 C–H imidation mechanism. | ||
In 2019, Guo and coworkers reported an iodine-atom transfer addition (I-ATRA) from aryl iodides 105 to alkenyl iodides 108 (Scheme 27).97 Among all photosensitizers, thioxanthone, having the highest triplet energy, was the one to promote the reaction under visible light irradiation. Upon switching the irradiation to purple LED irradiation, a significant increase in the yield was observed. Furthermore, electron-donating substituents on thioxanthone were found to enhance the catalytic activity. Mechanistic studies on the reaction indicated that it could take place involving both heterolysis and homolysis of the Caryl–I bond. As shown in Scheme 27, the reaction commences upon excitation of the photocatalyst in the singlet state and through intersystem crossing to its triplet state. Thereafter, an energy transfer event takes place between the excited catalyst 104 and the substrate to form triplet 105*. The latter may act reversibly in two ways. The reaction may proceed through homolysis of the Caryl–I bond, generating radical 106, which upon cyclization and coupling with an iodide radical furnishes the desired product 108via107. Alternatively, heterolysis of the Caryl–I bond leads to cation 109 and an iodine anion. An intermolecular nucleophilic attack on the triple bond, followed by coupling of the formed alkenyl cation with the iodine anion forms product 108via110. Incorporation of bromine in the final product upon addition of tetrabutylammonium bromide supported the existence of an ionic pathway. Several substrates bearing electron-donating or electron-withdrawing groups on aromatic rings Ar1 and Ar2 were examined, and the I-ATRA products were formed in moderate to excellent yields. Additionally, electron-donating and electron-withdrawing groups on the aromatic ring Ar3 in the alkyne terminal afforded the corresponding iodides in good yields. Furthermore, I-ATRA products were subjected to coupling reactions (Suzuki, Sonogashira, and Kumada cross-coupling reactions) affording the corresponding products in excellent yields.
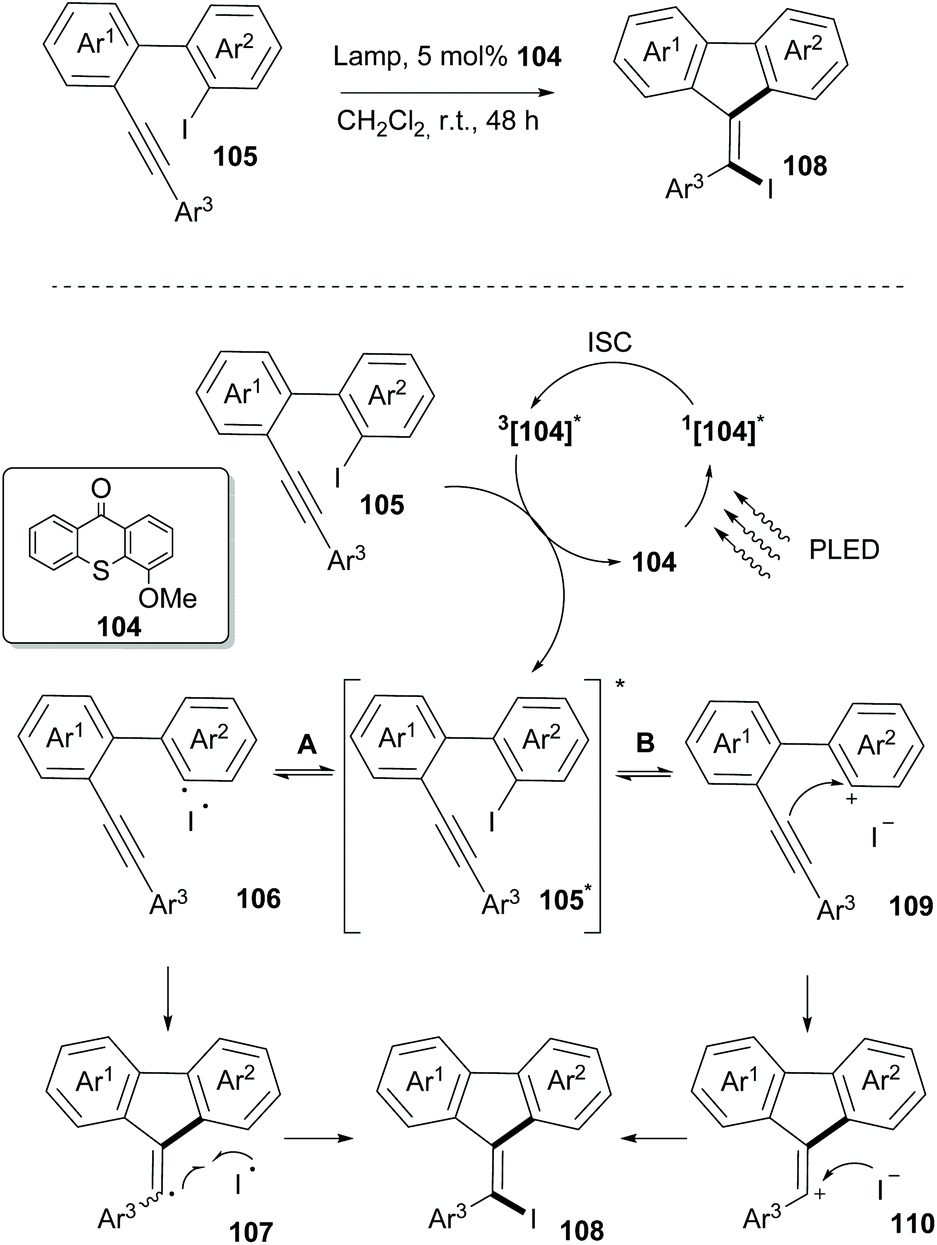 | ||
| Scheme 27 I-ATRA reaction. | ||
In 2019, Chemtob and coworkers reported the generation of N-heterocyclic carbenes (NHC) 24 from arylborate salts 112 and isopropyl thioxanthone 111.98 Previous reports have shown that arylborate salts upon irradiation may act as electron donors in the photoinduced electron transfer reaction (PET). Based on that, Chemtob and coworkers demonstrated that isopropylthioxanthone upon excitation can participate in an electron transfer event accepting an electron from a boranyl anion, forming excited isopropylthioxanthone radical anion 113. The latter can abstract a proton from the azolium cation forming the free NHC 24 (Scheme 28).
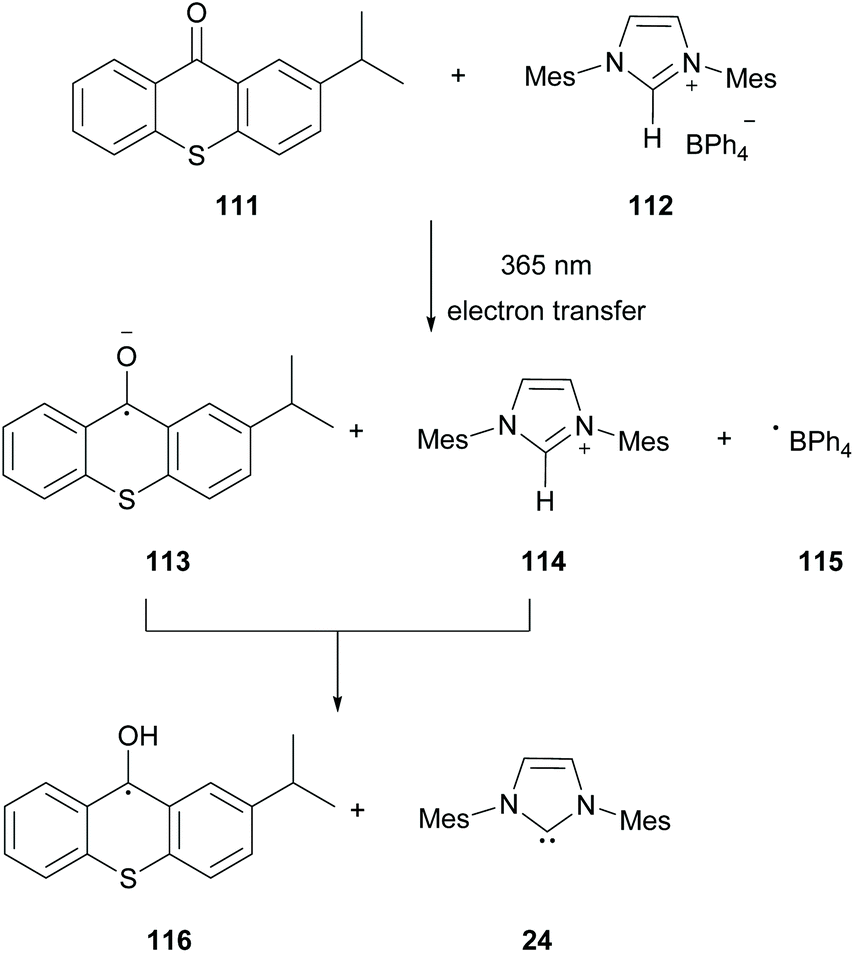 | ||
| Scheme 28 N-Heterocyclic carbenes (NHC). | ||
In 2019, Kunishima and coworkers employed thioxanthone as the photocatalyst to promote alkyne generation from UV labile amino-substituted cyclopropenone derivatives (Scheme 29).99 Based on previous reports where in situ-generated alkyne acted as an intermediate stepping stone for amide formation, they examined various photocatalysts to generate ynamine 121 from aminocyclopropenone 117. Among all photocatalysts, thioxanthone proved to be the catalyst of choice to promote decarbonylation of aminocyclopropenone. This indication was further supported after examination of the redox potential of both the substrate and thioxanthone. According to Kunishima's proposition, a plausible mechanism involves excitation of thioxanthone in its triplet state. Excited thioxanthone oxidizes aminocyclopropenone to produce radical cation 118, which subsequently undergoes ring opening to furnish cation 119. 119 receives one electron from a thioxanthone radical anion to form zwitterion 120. Decarbonylation of 120 delivers the desired ynamine 121. The desired ynamine 121 was also possible to be derived from cation 119 releasing CO. Having optimized the reaction conditions, aminocyclopropenone was examined in the condensation reaction between an acid and an amine to form the corresponding amide in moderate yields. Replacing aliphatic amines with aniline disturbed the reaction due to photodecomposition quenching of the photocatalyst.
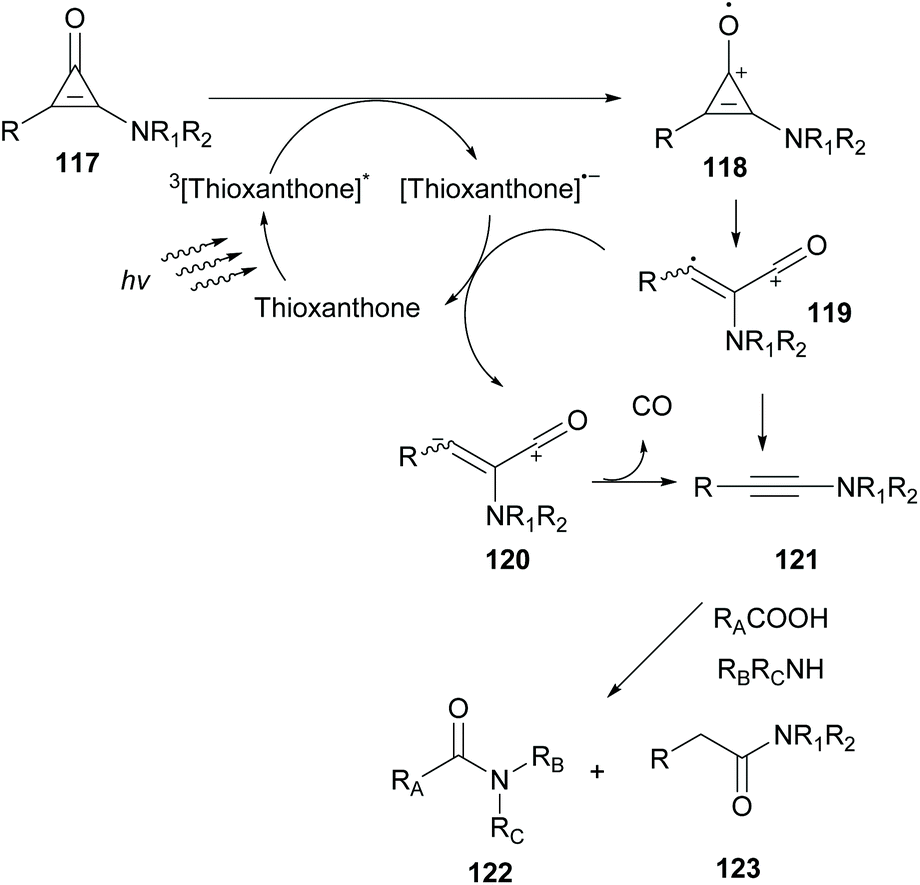 | ||
| Scheme 29 Cyclopropene fragmentation and direct access to alkynes. | ||
In 2019, Kokotos and coworkers developed a new, green, and mild photo-organocatalytic methodology for the conversion of aldehydes to the corresponding acetals.100 For this purpose, they employed thioxanthone as the photocatalyst. Mechanistic studies supported that excited thioxanthone can pass its excited triplet energy to the aldehyde via energy transfer. This energy transfer event facilitates the nucleophilic attack from the alcohol. This protocol was applied to a plethora of substrates bearing different functional groups delivering the corresponding acetals in excellent yields. CFL lamp irradiation was employed, which can be substituted by sunlight.
During the preparation of this review, Kokotos and coworkers reported the use of thioxanthone as the photocatalyst for the generation of acyl radicals from aromatic aldehydes.101 This acyl radical was trapped by N-chlorosuccinimide, leading to the corresponding acyl chloride, which is subsequently reacted with amine, a hydroxyamine or an alcohol leading to an amide, a hydroxamic acid or an ester, respectively. Aliphatic aldehydes could not be employed. Given the close BDEs of C–H in aliphatic chains, the in situ formed succinimidyl radical, which is responsible for the propagation, leads to complex mixtures. It is very interesting to note that depending on the concentration of N-chlorosuccinimide and the presence of acetonitrile as the solvent, a second mechanism was found to be also operable.
5. Conclusions
Thioxanthone, a well-known photocatalyst that mediates energy transfer (EnT), single electron transfer (SET) and hydrogen atom transfer (HAT) events, has been employed during the last 50 years in photochemical reactions. Recently, it has been found that it is also compatible with merger catalysis, adding extra options on the synthetic arsenal of scientists worldwide. Although there is a significant number of reports in the literature, this field seems to be rather unexplored, offering great opportunities for further development. Furthermore, the discovery of highly selective chiral and bifunctional photocatalysts based on thioxanthone that tune the regioselectivity and enantioselectivity is expected to take the baton from current studies as the rapid advancement in the field will continue to grow. The high versatility of thioxanthone in encompassing such different mechanisms of action has moved its employment from its traditional use to novel and more challenging organic transformations. For these reasons, thioxanthone will definitely play a decisive role in the future of photocatalysis by replacing expensive metal complexes and will offer opportunities for enantioselective photocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Hellenic Foundation for Research and Innovation (HFRI) for financial support through a grant, which is financed by 1st Call for H.F.R.I. Research Projects to Support Faculty Members & Researchers and the procurement of high-cost research equipment grant (grant number 655). N. F. N. would like to thank the State Scholarships Foundation (IKY) for financial support through a doctoral fellowship, which is co-financed by Greece and the European Union (European Social Fund – ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning” in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation (IKY).Notes and references
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (c) K. L. Scubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 17, 10035–10074 CrossRef; (d) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS; (b) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (c) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- (a) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC; (b) M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833–857 CrossRef CAS.
- S. F. Yates and G. B. Schuster, J. Org. Chem., 1984, 49, 3349–3356 CrossRef CAS.
- J. P. Fouassier, D.-J. Lougnot, I. Zuchowicz, P.-N. Green, H.-J. Timpe, K.-P. Kronfeld and U. Muller, J. Photochem., 1987, 36, 347–363 CrossRef CAS.
- D. G. Anderson, R. S. Davidson and J. J. Elvery, Polymer, 1996, 37, 2477–2484 CrossRef CAS.
- L. A. Linden, J. Paczkowski, J. F. Rabek and A. Wrzyszczynski, Polimery, 1999, 44, 161–176 CrossRef CAS.
- F. Scigalski and J. Paczkowski, Polimery, 2007, 52, 19–28 CrossRef CAS.
- X. Allonas, C. Ley, C. Bibaut, P. Jacques and J. P. Fouassier, Chem. Phys. Lett., 2000, 322, 483–490 CrossRef CAS.
- C. Ley, F. Morlet-Savary, J. P. Fouassier and P. Jacques, J. Photochem. Photobiol., A, 1989, 46, 253–267 CrossRef.
- F. Morlet-Savary, C. Ley, P. Jacques, F. Wieder and J. P. Fouassier, J. Photochem. Photobiol., A, 1999, 126, 7–14 CrossRef CAS.
- M. Goez and B. H. M. Hussein, Phys. Chem. Chem. Phys., 2004, 6, 5490–5497 RSC.
- L. T. Okano, T. C. Barros, D. T. H. Chou, A. J. Bennet and C. Bohne, J. Phys. Chem. B, 2001, 105, 2122–2128 CrossRef CAS.
- K. Iijima, I. Oonishi, S. Fujisawa and S. Shibata, Bull. Chem. Soc. Jpn., 1987, 60, 3887–3890 CrossRef CAS.
- O. Rubio-Pons, L. Serrano-Andres, D. Burget and P. Jacques, J. Photochem. Photobiol., A, 2006, 179, 298–304 CrossRef CAS.
- J. C. Dalton and F. C. Montgomery, J. Am. Chem. Soc., 1974, 18, 6230–6232 CrossRef.
- J. J. Cavareli, K. Prater and R. M. Bowman, Chem. Phys. Lett., 1996, 259, 495–502 CrossRef.
- M. G. Neumann, M. H. Gehlen, M. V. Encinas, N. S. Allen, T. Corrales, C. Peinado and F. Catalina, J. Chem. Soc., Faraday Trans., 1997, 93, 1517–1521 RSC.
- N. S. Allen, Photopolymerization and Photoimaging Science and Technology, Elsevier, New York, 1989 Search PubMed.
- J. P. Fouassier, Photoinitiation, Photopolymerization and Photocuring; Fundamentals of Applications, Munich Hanser Publishers, 1995 Search PubMed.
- V. Rai-Constapel, S. Salzmann and C. M. Marian, J. Phys. Chem. A, 2011, 115, 8589–8596 CrossRef CAS PubMed.
- G. Angulo, J. Grilj, E. Vauthey, I. Serrano-Andres and P. Jacques, ChemPhysChem, 2010, 11, 480–488 CrossRef CAS PubMed.
- L. Chen, Q.-H. Zhou, X. Liu, X.-Q. Zhou and S.-L. Liu, Chin. J. Chem. Phys., 2015, 28, 493–500 CrossRef CAS.
- P. S. Engel, J. Am. Chem. Soc., 1969, 25, 6903–6907 CrossRef PubMed.
- (a) P. S. Engel and P. D. Barlett, J. Am. Chem. Soc., 1970, 20, 5883–5891 CrossRef; (b) P. S. Engel, D. W. Horsey, J. N. Scholz, T. Karatsu and A. Kitamura, J. Phys. Chem., 1992, 96, 7524–7535 CrossRef CAS.
- Triplet energy from J. Calvert and J. Pitts, Photochemistry, John Wiley and Sons, Inc., New York, N. Y., 1966, p. 298 Search PubMed.
- I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1–250 CrossRef.
- A. Das and K. R. J. Thomas, Eur. J. Org. Chem., 2020, 7214–7218 CrossRef CAS.
- H. Zhu, W. Wang and S. Yao, Invest. New Drugs, 2006, 24, 465–470 CrossRef CAS.
- L. Shen and H.-F. Ji, Int. J. Mol. Sci., 2009, 10, 4284–4289 CrossRef CAS PubMed.
- K. Hirakawa, M. Yoshida, S. Oikawa and S. Kawanishi, Photochem. Photobiol., 2003, 77, 349–355 CrossRef CAS.
- G. Herzberg, Molecular spectra and molecular structure I: Spectra of diatomic molecules, VonNostrand, New York, 2nd edn, 1950 Search PubMed.
- G. Amirzadeh and W. Schnabel, Makromol. Chem., 1981, 182, 2821–2835 CrossRef CAS.
- M. J. Davis, J. Doherty, A. A. Godfrey, P. N. Green, J. R. Young and M. A. Parrish, J. Oil Colour Chem. Assoc., 1978, 68, 256 Search PubMed.
- A. Hult and B. Ranby, Polym. Degrad. Stab., 1984, 8, 89–105 CrossRef CAS.
- H. J. Timpe and K. P. Kronfeld, J. Photochem. Photobiol., A, 1989, 46, 253–267 CrossRef CAS.
- D. G. Anderson, R. S. Davidson and J. J. Elvery, Polymer, 1996, 37, 2477–2484 CrossRef CAS.
- M. R. Rodriquez and M. G. Neuann, Macromol. Chem. Phys., 2001, 202, 2776–2782 CrossRef.
- N. Karaca, D. K. Balta, N. Ocal and N. Arsu, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1012–1019 CrossRef CAS.
- G. Temel and N. Arsu, J. Polym. Sci., Part A: Polym. Chem., 2007, 191, 149–152 CAS.
- M. A. Tasdelen, B. Kiskan and Y. Yagci, Macromol. Rapid Commun., 2006, 27, 1539–1544 CrossRef CAS.
- J. Li, S. Li, Y. Li, R. Li, J. Nie and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2020, 389, 112225 CAS.
- G. Yilmaz, S. Beyazit and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1591–1596 CrossRef CAS.
- A. Kobayashi, Y. Endo, A. Shiraishi and T. Yamashita, J. Photopolym. Sci. Technol., 2018, 31, 107–112 CrossRef CAS.
- T. K. H. Trinh, J.-P. Malval, F. Morlet-Savary, J. Pinaud, P. Lacroix-Desmazes, C. Reibel, V. Heroquez and A. Chemtob, Chem. – Eur. J., 2019, 25, 9242–9252 CrossRef CAS.
- D. K. Balta, G. Temel, M. Aydin and N. Arsu, Eur. Polym. J., 2010, 46, 1374–1379 CrossRef CAS.
- K. Kaya, J. Kreutzer and Y. Yagci, ChemPhotoChem, 2019, 3, 1187–1192 CrossRef CAS.
- J. Lalevee, N. Blanchard, M. A. Tehfe, C. Fries, F. Morlet-Savary, D. Gigmes and J. P. Fouassier, Polym. Chem., 2011, 2, 1077–1084 RSC.
- G. Chen, X. Guan, R. Xu, J. Tian, F. Lu, M. He and J. Yang, RSC Adv., 2016, 6, 77093–77099 RSC.
- S. K. Dogruyol, Z. Dogruyol and N. Arsu, Polym. Chem., 2011, 49, 4037–4043 CrossRef CAS.
- T. N. Eren, N. Okte, F. Morlet-Savary, J. P. Fouassier, J. Lalevee and D. Avci, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3370–3378 CrossRef CAS.
- T. N. Eren, B. Graff, J. Lalevee and D. Avci, Prog. Org. Coat., 2019, 128, 148–156 CrossRef.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockush and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- J. Kreutzer and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3475–3482 CrossRef CAS.
- M. Sakamoto, T. Ishida, T. Fujita and S. Watanabe, J. Org. Chem., 1992, 57, 2419–2422 CrossRef CAS.
- H. E. Zimmerman and C. W. Wright, J. Am. Chem. Soc., 1992, 114, 6603–6612 CrossRef CAS.
- G. D. Reddy, O. Wiest, T. Hudlicky, V. Schapiro and D. Gonzalez, J. Org. Chem., 1999, 64, 2860–2863 CrossRef CAS.
- A. Padwa, T. J. Blacklock, R. Loza and R. Polniaszek, J. Org. Chem., 1980, 45, 2181–2189 CrossRef CAS.
- M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 7661–7664 CrossRef CAS PubMed.
- R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS PubMed.
- A. Troster, R. Alonso, A. Bauer and T. Bach, J. Am. Chem. Soc., 2016, 138, 7808–7811 CrossRef PubMed.
- Y. Yang, Y. Wen, Z. Dang and H. Yu, J. Phys. Chem. A, 2017, 121, 4552–4559 CrossRef CAS PubMed.
- X. Li, C. Jandl and T. Bach, Org. Lett., 2020, 22, 3618–3622 CrossRef CAS PubMed.
- (a) N. Vallanoju, S. Selvakumar, S. Jockusch, M. P. Sibi and J. Sivaguru, Angew. Chem., Int. Ed., 2014, 53, 5604–5608 CrossRef PubMed; (b) N. Vallanoju, S. Selvakumar, S. Jockusch, M. T. Prabhakaran, M. P. Sibi and J. Sivaguru, Adv. Synth. Catal., 2014, 356, 2763–2768 CrossRef; (c) R. Telmesani, S. H. Park, T. Lynch-Colameta and A. B. Beeler, Angew. Chem., Int. Ed., 2015, 54, 11521–11525 CrossRef CAS PubMed.
- F. Mayr, R. Brimioulle and T. Bach, J. Org. Chem., 2016, 81, 6965–6971 CrossRef CAS PubMed.
- F. Mayr, L.-M. Mohr, E. Rodriguez and T. Bach, Synthesis, 2017, 49, 5238–5250 CrossRef CAS.
- E. Kumarasamy, R. Raghunathan, S. Jockusch, A. Ugrinov and J. Sivaguru, J. Am. Chem. Soc., 2014, 136, 8729–8737 CrossRef CAS PubMed.
- A. Clay, N. Vallanoju, R. Krishnan, A. Ugrimov and J. Sivaguru, J. Org. Lett., 2016, 81, 7191–7200 CAS.
- S. Ahuja, S. Jockusch, A. Ugrinov and J. Sivaguru, Eur. J. Org. Chem., 2020, 1478–1481 CrossRef CAS.
- A. Iyer, S. Jockusch and J. Sivaguru, Chem. Commun., 2017, 53, 1692–1695 RSC.
- S. Ahuja, A. Iyer, S. K. Kandappa and J. Sivaguru, J. Photochem. Photobiol., A, 2019, 382, 111883–111888 CrossRef CAS.
- T. Nevesely, C. G. Daniliuc and R. Gilmour, Org. Lett., 2019, 21, 9724–9728 CrossRef CAS.
- J. D. Williams, M. Nakano, R. Gerardy, J. A. Rincon, O. De Frutos, C. Mateos, J.-C. M. Monbaliu and O. C. Kappe, Org. Process Res. Dev., 2019, 23, 78–87 CrossRef CAS.
- X. Li, E. Erturk, X. Chen, S. Kumar, C. Guo, S. Jockusch, J. J. Russo, T. H. Bestor and J. Ju, Photochem. Photobiol. Sci., 2018, 17, 1049–1055 CrossRef CAS PubMed.
- (a) H. J. Timpe, K. P. Kronfeld and M. Schiller, J. Photochem. Photobiol., A, 1992, 62, 245–259 CrossRef; (b) M. D'Auria, Photochem. Photobiol. Sci., 2019, 18, 2297–2362 CrossRef.
- X. Li, J. Groβkopf, C. Jandl and T. Bach, Angew. Chem., 2021, 60, 2684–2688 CrossRef CAS PubMed.
- S. Dadashi-Silab and Y. Yagci, Tetrahedron Lett., 2015, 56, 6440–6443 CrossRef CAS.
- Y. Masuda, N. Ishida and M. Murakami, Eur. J. Org. Chem., 2016, 5822–5825 CrossRef CAS.
- D.-L. Zhu, H.-X. Li, Z.-M. Xu, H.-Y. Li, D. J. Young and J.-P. Lang, Org. Chem. Front., 2019, 6, 2353–2359 RSC.
- D.-L. Zhu, R. Xu, Q. Wu, H.-Y. Li, J.-P. Lang and D. J. Young, J. Org. Chem., 2020, 85, 9201–9212 CrossRef CAS PubMed.
- D.-L. Zhu, S. Jiang, Q. Wu, H. Wang, L.-L. Chai, H.-Y. Li and H.-X. Li, Org. Lett., 2021, 23, 160–165 CrossRef CAS PubMed.
- W. Ding, L.-Q. Li, Q.-Q. Zhou, Y. Wei, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 63–66 CrossRef CAS.
- C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 2205–2210 CrossRef PubMed.
- N. F. Nikitas, D. I. Tzaras, I. Triandafillidi and C. G. Kokotos, Green Chem., 2020, 22, 471–477 RSC.
- T. Patra, M. Das, C. G. Danilluc and F. Glorius, Nat. Catal., 2021, 4, 54–61 CrossRef CAS.
- T. Patra, P. Bellotti, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., 2020, 59, 3172–3177 CrossRef CAS PubMed.
- T. Rigotti, A. Casado-Sanchez, S. Cabrera, R. Perez-Ruiz, M. Liras, V. A. de la Pena O'Shea and J. Aleman, ACS Catal., 2018, 8, 5928–5940 CrossRef CAS.
- A. Holzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–244 CrossRef.
- A. Troster, A. Bauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 3538–3541 CrossRef.
- L. Wimberger, T. Kratz and T. Bach, Synthesis, 2019, 51, 4417–4424 CrossRef CAS.
- D. Woll, S. Walbert, K.-P. Stengele, T. J. Albert, T. Richmond, J. Norton, M. Singer, R. D. Green, W. Pfleiderer and U. E. Steiner, Helv. Chim. Acta, 2004, 87, 28–45 CrossRef.
- D. Woll, S. Laimgruber, M. Galetskaya, J. Smimova, W. Pfleiderer, B. Heinz, P. Gilch and U. E. Steiner, J. Am. Chem. Soc., 2007, 129, 12148–12158 CrossRef PubMed.
- M. C. Pirrung, T. M. Dore, Y. Zhu and V. S. Rama, Chem. Commun., 2010, 46, 5313–5315 RSC.
- C. B. Tripathi, T. Ohtani, M. T. Corbett and T. Ooi, Chem. Sci., 2017, 8, 5622–5627 RSC.
- K. Kailong, T. Ohtani, C. B. Tripathi, D. Uraguchi and T. Ooi, Chem. Lett., 2019, 48, 715–717 CrossRef.
- Y. Zhang, J. Xu and H. Guo, Org. Lett., 2019, 21, 9133–9137 CrossRef CAS PubMed.
- T. K. H. Trinh, F. Morlet-Savary, J. Pinaud, P. Lacroix-Desmazes, C. Reibel, C. Joyeux, D. Le Nouen, R. Metivier, A. Brosseau, V. Heroguez and A. Chemtob, Phys. Chem. Chem. Phys., 2019, 21, 17036–17046 RSC.
- K. Mishiro, T. Kimura, T. Furuyama and M. Kunishima, Org. Lett., 2019, 21, 4101–4105 CrossRef CAS.
- N. F. Nikitas, I. Triandafillidi and C. G. Kokotos, Green Chem., 2019, 21, 669–674 RSC.
- N. F. Nikitas, M. K. Apostolopoulou, E. Skolia, A. Tsoukaki and C. G. Kokotos, Chem. – Eur. J., 2021, 27, 7915–7922 CrossRef CAS PubMed.
Footnote |
| † Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |