
Introduction to hybrid catalysis
Motomu
Kanai
 *a and
Matthias
Beller
*b
*a and
Matthias
Beller
*b
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kanai@mol.f.u-tokyo.ac.jp
bLeibniz-Institut für Katalyse e.V., Albert-Einstein Straße 29a, Rostock, 18059, Germany. E-mail: matthias.beller@catalysis.de
Hybrid catalysis is a current trend in catalyst development, which integrates multiple functions of independent catalysts as a system. Hybrid catalysis promotes new reactions or known reactions with previously impossible efficiencies. Here, we are happy to edit the web themed issue Hybrid Catalysis in Organic & Biomolecular Chemistry. The catalyst integration patterns described in this collection can be classified into six categories (Fig. 1): (1) a catalyst activating another catalyst, (2) more than one catalyst simultaneously activating a substrate, (3) stepwise activation of a substrate by more than one catalyst, (4) tandem catalysis,2 (5) heterogeneous catalysis comprising more than one component, (6) combinations of two distinct modalities, such as electrochemical/transition metal catalysis and enzymatic/chemical catalysis.
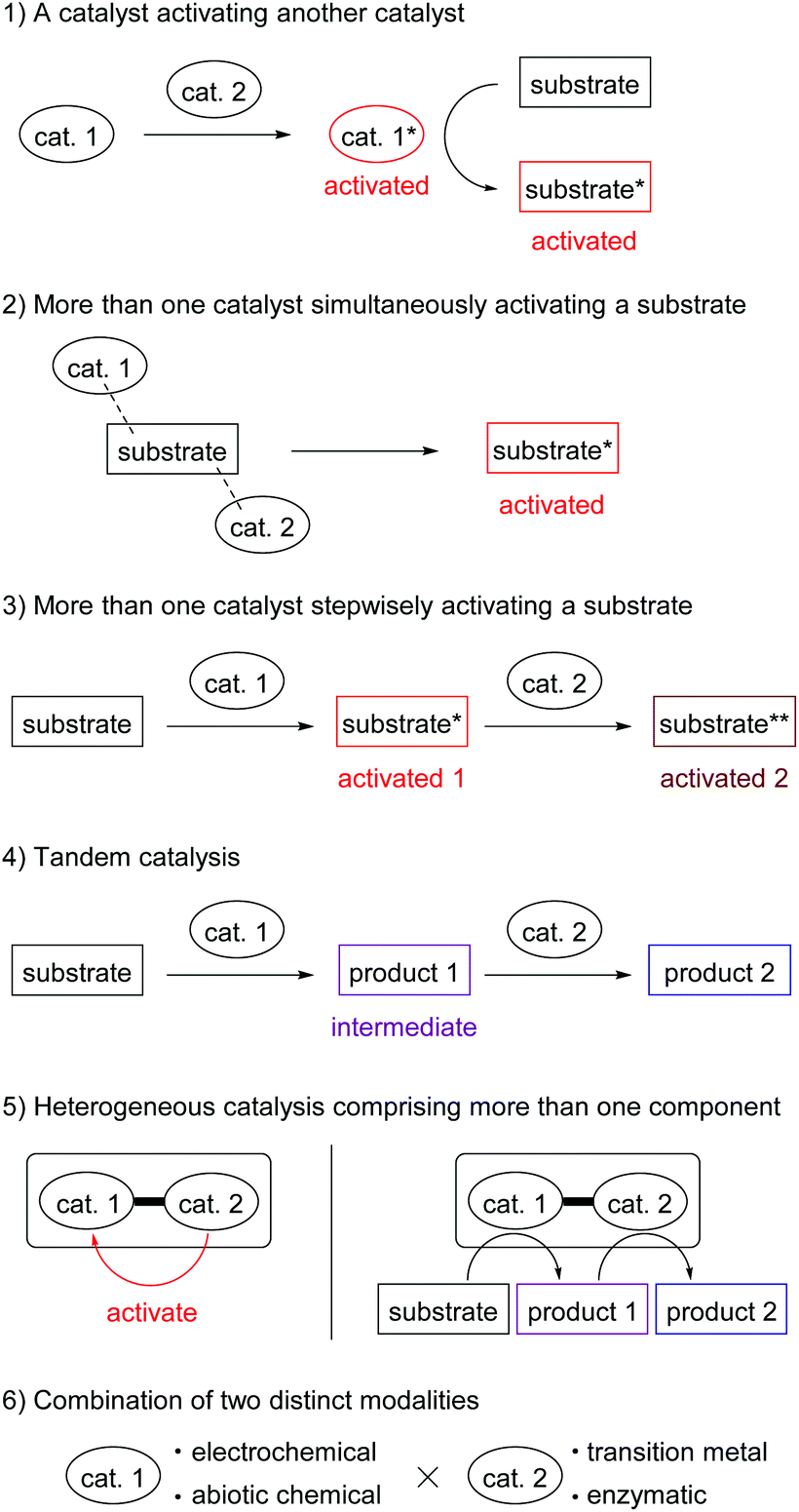 | ||
| Fig. 1 Six categories of catalyst integration in the hybrid catalysis themed issue. | ||
In category (1), Ooi and co-workers report a Mannich-type allylic C–H functionalization of enol silyl ethers catalysed by a combination of an iridium-based photosensitizer and a thiol catalyst (Org. Biomol. Chem., 2021, 19, 141–145, DOI: 10.1039/d0ob01862g). The iridium photosensitizer corresponding to cat. 2 in Fig. 1-1 abstracts a single electron from a thiol catalyst (cat. 1) to generate a thiyl radical (cat. 1*), which then furnished hydrogen atom abstraction at the allylic C–H bond of enol silyl ethers. The resulting nucleophilic allyl radical adds to imines. Since enol silyl ethers are generated from ketones, the overall process is an umpolung reaction3 between the β-position of ketones and electrophilic imines.
Byrne and Albrecht report an oxidative dimerization of primary benzylamines to imines under aerobic conditions using a triazolium iodide catalyst (Org. Biomol. Chem., 2020, 18, 7379–7387, DOI: 10.1039/d0ob01472a). The authors propose that the azolinium π system polarizes molecular diiodine, which is generated through aerobic oxidation of iodide anion, and the thus-generated activated diiodine acts as an active oxidant for conversion of amines to imines.
In category (2), Paradies and co-workers identified for the first time that amidines form frustrated Lewis pairs (FLPs)4 with B(C6F5)3. The FLP activates molecular hydrogen and generates borohydride species, thus promoting catalytic hydrogenation of electron-deficient C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, an imine, and a dehydroalanine derivative (Org. Biomol. Chem., 2020, 18, 7321–7325, DOI: 10.1039/d0ob01492c).
C double bonds, an imine, and a dehydroalanine derivative (Org. Biomol. Chem., 2020, 18, 7321–7325, DOI: 10.1039/d0ob01492c).
Extending the utility of FLPs from hydrogen splitting to enolization of ketones, Wasa and co-workers report a Conia-ene-type cyclization of alkynyl ketones through hybrid FLP and ZnI2 catalysis. The resulting alkenylzinc intermediate after addition of an enolate to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond is trapped by aryl iodide through Pd-mediated Negishi coupling (Org. Biomol. Chem., 2020, 18, 7090–7093, DOI: 10.1039/d0ob01678k). Thus, Wasa's work can be also categorized as an interesting example of domino catalysis.5
C triple bond is trapped by aryl iodide through Pd-mediated Negishi coupling (Org. Biomol. Chem., 2020, 18, 7090–7093, DOI: 10.1039/d0ob01678k). Thus, Wasa's work can be also categorized as an interesting example of domino catalysis.5
Matsuda and co-workers report reductive N–O bond cleavage of N-methoxyamides using a catalyst system consisting of i-Bu3Al and Pd(dba)2 (Org. Biomol. Chem., 2020, 18, 7545–7548, DOI: 10.1039/d0ob01815e). External reductants are not necessary in this reaction. The aluminium compound activates the substrates by forming N-methoxy imidates, which undergo facile oxidative addition at the palladium catalyst. Subsequent β-hydride elimination of the palladium methoxide intermediate with extrusion of formaldehyde, followed by reductive elimination of the palladium hydride affords the desired amide products.
Although the active catalyst structure and precise reaction mechanism are not clear, Kondoh, Ishikawa, and Terada report that a combination of NaOt-Bu and a chiral urea acts as an efficient asymmetric Brønsted base catalyst system for conjugate addition of diarylphosphine oxides to 1-alkenyl(diaryl)phosphine oxides (Org. Biomol. Chem., 2020, 18, 7814–7817, DOI: 10.1039/D0OB01778G). The products are valuable precursors of chiral 1,2-diphosphines, which are useful ligands in asymmetric transition metal catalysis.
In category (3), Sumida, Ohmiya, and co-workers disclose a cross coupling reaction between boracene-based alkylborates and aryl halides or triflates using a combination of an iridium photoredox catalyst and a Ni(acac)2 catalyst (Org. Biomol. Chem., 2020, 18, 6598–6601, DOI: 10.1039/D0OB01610A). Because photoexcited borates are oxidatively quenched with the photoexcited iridium catalyst, generation of an alkyl radical from the borates proceeds under low irradiance. The generated alkyl radical is trapped by the nickel catalyst for the subsequent coupling reaction.
In category (4), Hoshino and Mori show a one-pot, two-step synthesis of 3-indolyl-1-trifluoromethyl-isobenzofurans from trifluoromethyl ketone-containing benzylamines and indoles (Org. Biomol. Chem., 2020, 18, 6602–6606, DOI: 10.1039/D0OB01582B). The first step is a Yb(OTf)3-catalysed [1,4]-hydride shift from a benzylic C–H bond to the trifluoromethyl ketone moiety, affording cyclic aminal intermediates. Then, Friedel–Crafts reaction with indoles proceeds by the addition of an acid promoter, Tf2NH, in the same reaction vessel. Tokuyama and co-workers previously reported a cationic gold complex-catalysed N-acyl pyrrole synthesis from β-amido acetals and terminal alkynes through tandem acetylide addition to the acetals and intramolecular hydroamidation. The group has introduced electron-deficient internal alkynes as a third component in this procedure (Org. Biomol. Chem., 2021, 19, DOI: 10.1039/D0OB02018D). The gold complex further promotes Diels–Alder reaction between N-acyl pyrroles and electron-deficient alkynes, followed by C–N bond cleavage and aromatization, producing aniline derivatives in one pot.
In category (5), metal-based heterogeneous catalysts contain multiple electronically coupled catalytic sites, and are thus a unique group of hybrid catalysts. Specifically, metal–organic framework (MOF)-based catalysts are notable for their porosity and structural diversity/designability. In their excellent review, Xu and co-workers surveyed the wide applicability and future potential of MOF-based hybrid catalysis (Org. Biomol. Chem., 2020, 18, 8508–8525, DOI: 10.1039/D0OB01729A). Furthermore, Ferrer, Navalón, García, and co-workers report that a MOF-based catalyst, UiO-66(Zr)-NH2, promotes hydrogenation of 4-nitrophenol to 4-aminophenol by evolving hydrogen gas from methanol under photoirradiation (Org. Biomol. Chem., 2021, 19, DOI: 10.1039/d0ob01686a). The MOF catalyst allows for hydrogenation through a FLP mechanism. Importantly, the catalyst can be recycled without significant loss of activity, which constitutes an advantage over most homogeneous catalysts. Mitsudome and co-workers show the first deoxygenation of sulfoxides under ambient hydrogen pressure using a non-noble metal-based heterogeneous catalyst, a TiO2-supported Ni2P nanoalloy (Org. Biomol. Chem., 2020, 18, 8827–8833, DOI: 10.1039/D0OB01603A). The high catalyst activity is due to functional coupling between the TiO2 support and nano-Ni2P in the solid-state hybrid catalyst system. As with homogeneous catalysis, the activity and selectivity of metal-based heterogeneous catalysts can be enhanced by organic ligands. In this respect, Cui and co-workers report that product yield and terminal selectivity are both improved by the presence of a Xantphos ligand in hydrosilylation of alkynes catalysed by Ni/Al2O3 nanoparticles (Org. Biomol. Chem., 2020, 18, 7554–7558, DOI: 10.1039/D0OB01693D).
In addition to original papers, this themed collection contains three review articles, including Xu's article on MOF-based catalysts as mentioned above. Catalytic formation of C–N bonds is a central topic in organic synthesis. Thus, Roy and co-workers review the cutting edge of transition metal/electro hybrid catalysis, mainly focusing on C–H amination reactions (Org. Biomol. Chem., 2020, 18, 8994–9017, DOI: 10.1039/D0OB01874K). Remarkably, reaction conditions are much milder than traditional transition metal-catalysed C–H aminations. Finally, Adamson and Kanai review an emerging field of hybrid catalysis comprising abiotic and enzymatic catalysts in living cells (Org. Biomol. Chem., 2021, 19, 37–45, DOI: 10.1039/D0OB01898H). While this field is still in its infancy, exciting opportunities are suggested.
To summarise, the concept of hybrid catalysis is an avenue to many new horizons, such as new reaction patterns, applicability to stable, unactivated starting materials, and facile increase of molecular complexity. Hybrid catalysis requires sophisticated matching and integration of plural catalysts. This constitutes a fundamental challenge in catalysis.
References
- R. Noyori, Nat. Chem., 2009, 1, 5 CrossRef CAS.
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365 CrossRef CAS.
- D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239 CrossRef.
- D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535 RSC.
- For a review of domino reactions, see: L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |