
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NIR laser scanning microscopy for photophysical characterization of upconversion nanoparticles and nanohybrids†
Juan
Ferrera-González
 *a,
Laura
Francés-Soriano
ab,
Nestor
Estébanez
a,
Enrique
Navarro-Raga
c,
María
González-Béjar
*a and
Julia
Pérez-Prieto
*a
*a,
Laura
Francés-Soriano
ab,
Nestor
Estébanez
a,
Enrique
Navarro-Raga
c,
María
González-Béjar
*a and
Julia
Pérez-Prieto
*a
aInstituto de Ciencia Molecular (ICMol), Departamento de Química Orgánica, University of Valencia, C/Catedrático José Beltrán, 2, Paterna, Valencia 46980, Spain. E-mail: juan.ferrera@uv.es; maria.gonzalez@uv.es; julia.perez@uv.es; Web: http://jperezprieto-prg.com/
bnanoFRET.com, Laboratoire COBRA (Chimie Organique, Bioorganique, Réactivité et Analyse), Université de Rouen Normandie, CNRS, INSA, 76821 Mont-Saint-Aignan Cedex, France
cServicio Central de Soporte a la Investigación Experimental (SCSIE). University of Valencia, Burjassot, Valencia 46100, Spain
First published on 18th May 2021
Abstract
Photophysical characterization of upconversion nanoparticles (UCNPs) and nanohybrids (UCNHs) is more challenging than that of down-conversion nanomaterials. Moreover, it is still difficult to gain knowledge about the homogeneity of the sample and colocalization of emissive chromophores and nanoparticles in nanohybrids. Near infrared laser scanning microscopy (NIR-LSM) is a well-known and useful imaging technique, which enables excitation in the NIR region and has been extensively applied to optical fluorescence imaging of organic fluorophores and nanomaterials, such as quantum dots, which exhibit a short-lived emission. NIR-LSM has recently been used to determine the empirical emission lifetime of UCNPs, thus extending its application range to nanomaterials with a long lifetime emission. Here, we review our previous findings and include new measurements and samples to fully address the potential of this technique. NIR-LSM has proved to be extraordinarily useful not only for photophysical characterization of UCNHs consisting of UCNPs capped with a fluorophore to easily visualize the occurrence of the resonance energy transfer process between the UCNH constituents and their homogeneity, but also to assess the colocalization of the fluorophore and the UCNP in the UCNH; all this information can be acquired on the micro-/nano-meter scale by just taking one image.
1. Introduction
Traditionally, near infrared laser scanning microscopy (NIR-LSM) has been used as a bioimaging technique consisting of scanning a surface and registering its emission, pixel by pixel. This technique has the advantages of common confocalmicroscopes due to multiphoton excitation – such as reduction of autofluorescence of biological tissues and out-of-focus light, and the multi-z scan (3D images). Moreover, a near-infrared (NIR) excitation laser allows higher penetration depths in biological samples.1–4 It was initially developed to excite multi-photonically organic fluorophores in biological media (multi-photon excitation microscopy, MPM) and, later on, its use was extended to excite nanomaterials, such as quantum-dots5 and perovskites.6,7On the other hand, UCNPs are novel photoactive nanomaterials that can upconvert light, specifically NIR light, into ultraviolet, visible and/or NIR light. Usually, UCNPs are co-doped with two different lanthanide ions, one of them acting as a sensitizer and the other one as an activator. The sensitizer (typically Yb3+) absorbs light to lead to its excited state (Yb3+*) with an extraordinarily long-lifetime (hundreds of μs).
Unlike coherent two-photon absorption of conventional chromophores, which occurs through “virtual” states, multiphoton sensitization in UCNPs8,9 occurs via stepwise population of real long-lived states of the activator (e.g., Er3+, Tm3+, Ho3+), from which light is emitted.9,10
In addition to their upconversion luminescence (UCL) feature, UCNPs display overall, unique optical and chemical properties, such as narrow emission bands, no photoblinking, no photobleaching, long emission lifetimes, high chemical and thermal stability and low cytotoxicity.11,12 Owing to their exceptional properties, UCNPs are promising in a wide range of applications e.g., (bio)sensing, solar cells, theranostics, and bioimaging.13–17
Although NIR-LSM was not initially conceived to be used with UCNPs, this technique has been adapted perfectly to imaging them since their absorption and the NIR-LSM excitation occur in the same region (specifically, in the NIR).18,19 Moreover, the long lifetime of UCNP emissions makes it possible to avoid the undesirable auto-fluorescence from the sample by using a simple time-gating technology.20,21 Different studies have demonstrated that single particle imaging of UCNPs is feasible,22,23 even for those measuring 10 nm, with a conventional multiphoton microscope24 and that sub-diffraction imaging is achievable when measuring UCNPs with super-resolution techniques, like NIR emission saturation (NIRES) nanoscopy25 and the stimulated emission depletion (STED) approach.26–28 These new results show the power in bioimaging when combining UCNPs and NIR-LSM.
UCL kinetic profiles of UCNPs show a characteristic slow rise and, in general, a long emission decay, which unfortunately cannot be used to easily obtain the intrinsic decay constants of a given emitting state from the UCL time dependence.29,‡
Our group has demonstrated the possibility of using the NIR-LSM as a technique for the determination of “empirical” UCNP emission lifetimes.30–32 The ability of the NIR-LSM to scan samples at different scan speeds by controlling the dwell-time has proved to be an appropriate solid-state approach to measure such decay times. The key step of the procedure is the transformation of the space-resolved image to a time-resolved profile, from which the empirical decay lifetime of the emissions can be obtained.
This approach has also been useful to detect the lengthening of the organic dye emission present in upconversion nanohybrids as a consequence of an energy transfer process from the UCNP emissive lanthanide to the organic dye.31,32 In this paper, recent findings with novelties only touched upon in previous works are gathered together and the scope of the NIR-LSM technique is explored in much more detail to offer a complete vision of the versatility of the NIR-LSM technique which is extremely useful for the photophysical characterization of long-lived emissive (nano)hybrids. To this end, four samples of UCNPs have been prepared with different types and concentration of dopants, namely oleate-capped NaYF4:Yb3+(20%),Er3+(2%),UCEr,2; NaYF4:Yb3+(20%),Er3+(20%),UCEr,20; NaYF4:Yb3+(20%),Tm3+(0.1%),UCTm; and NaYF4:Yb3+(20%),Ho3+(1%),UCHo. Also, two nanohybrids consisting of rhodamine dyes grafted to UCTm (specifically, UCTm@Rh101 and UCTm@Rh110) were prepared to study the co-localization and the homogeneity of the UCNHs.
Our results demonstrate that NIR-LSM can be used as a characterization technique to obtain empirical luminescence lifetimes, information on the homogeneity of the sample and the occurrence of energy transfer, as well as the co-occurrence of dyes and UCNPs in upconversion nanohybrids (UCNHs). All this considered, NIR-LSM has proved to be more than an imaging technique as it enables detailed photophysical characterization and visualization of upconversion nanoparticles and nanohybrids on the micro-/nano-meter scale. This information is associated with close-lying nanoparticles or an assemblage of nanoparticles (agglomerate) and it cannot be ascertained with a conventional NIR-excitation fluorometer, which is better suited to macroscopic and solvent-dependent measurements.
2. Experimental
The chemicals used for the synthesis of the nanoparticles were: lanthanide chlorides (YCl3·6H2O, YbCl3·6H2O, ErCl3·6H2O, TmCl3·6H2O, HoCl3·6H2O, and TbCl3·6H2O (>99.9%, all of them), 1-octadecene (95%), oleic acid (70%), NaOH, and NH4F (99.99%). All these chemicals were purchased from Merck and used as received without previous purification.2.1. UCNPs synthesis and characterization
Four oleate-capped UCNPs with different composition were synthesized following a previously described protocol (UCEr,2; UCEr,20; UCTm and UCHo).33 The amount and type of lanthanide dopants were adjusted in the synthesis in order to obtain the desired UCNPs (see ESI† for further details). The synthesized UCNPs composition was elucidated by energy dispersive X-ray spectroscopy (EDX, Fig. S1 and Table S1 in the ESI†). The ratio between different lanthanide ions matched with the initial amount of each lanthanide in the synthesis, i.e., NaYF4:Yb3+(20%),Er3+(2%) for UCEr,2, NaYF4:Yb3+(20%),Er3+(20%) for UCEr,20, NaYF4:Yb3+(20%),Tm3+(0.1%) for UCTm, and NaYF4:Yb3+(20%),Ho3+(1%) for UCHo. Their size and shape were obtained by transmission electron microscopy (TEM). All samples showed a hexagonal prism shape with comparable sizes (Fig. S2 in the ESI†). The obtained sizes were (16.3 ± 1.0) × (23.5 ± 0.9) nm for UCEr,2, (14.7 ± 1.0) × (22.6 ± 1.1) nm for UCEr,20, (15.2 ± 1.3) × (24.7 ± 1.1) nm for UCTm, and (21.4 ± 1.0) × (24.8 ± 1.1) nm for UCHo. X-Ray diffraction (XRD) diffractograms confirmed a β-NaYF4 crystal structure (Fig. S3 in the ESI†).2.2. UCNH preparation
Two UCNHs were prepared by grafting rhodamine dyes, rhodamine 101 and rhodamine 110, to bare-UCTm. The oleate ligands of UCTm were removed by following a previously described treatment with nitrosonium tetrafluoroborate.34 After that, bare-UCTm NPs were redispersed in N,N-dimethylformamide (DMF). The functionalization with the dyes was performed by stirring for 24 hours an aqueous solution containing the dye in excess and an aliquot of the bare-UCTm solution (mdye > 5 × mUCNP). The mixture was centrifugated (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 15′) and the precipitate was redispersed in water. Five centrifugation–redispersion cycles were performed with 10 mL of water until the supernatant did not show any absorption. Finally, the UCTm@dye nanohybrids were redispersed in DMF.
000g, 15′) and the precipitate was redispersed in water. Five centrifugation–redispersion cycles were performed with 10 mL of water until the supernatant did not show any absorption. Finally, the UCTm@dye nanohybrids were redispersed in DMF.
2.3. Sample preparation for NIR-LSM
This technique requires immobilization of samples, so the influence of solvent on the luminescence dynamics (although interesting)35 is out of the scope for this study. Therefore, samples were prepared by spin-coating a diluted aliquot of the UCNP or UCNH dispersion onto a 25 × 25 mm microscope glass slides. The spin-coating process was optimized in order to obtain reproducible and suitable time-resolved images on the microscope. Once the solvent had evaporated, the samples were covered with a 22 × 22 mm glass slide, the joints between glass slides sealed, and their emission was evaluated by microscopy. Interestingly, scanning electron microscopy (SEM) of emissive samples showed the presence of close-lying UCNPs, agglomerates and free UCNPs (Fig. S5 in the ESI†).2.4. Spin coating
Samples were prepared by spin-coating (spin-coater KW-4A, Chemat technology) at low concentrations producing a discontinuous nanoparticle monolayer with a thickness of a few nanometres. This procedure was optimized by using different rotation speeds, solution concentrations and volumes. Finally, 100 μL of UCNP dispersion in toluene (4 mg mL−1) or 100 μL aqueous dispersions of UCNHs (1 mg mL−1) were deposited on a 25 × 25 mm microscope glass slide. Then, the spin-coater was turned on first at 400 rpm for 3 seconds and secondly at 1500 rpm for 40 seconds. After that, the solvent was evaporated, and the sample was covered with a 22 × 22 mm glass slide (for time-resolved measurements).2.5. NIR-LSM time-resolved photoluminescence
NIR-LSM was performed on a commercial multi-photon excitation microscope (FV100MPE) comprising a BX61WI upright microscope with motorised focus and equipped with a XLPLN25 × WMP 1.05 NA (working distance 2 mm) full water immersion lens. The excitation source was a Mai-Tai HP Deep See (Spectra Physics) Ti-Sapphire pulsed tuneable laser with a range of 690 to 1040 nm, an average power of 2.0 W (pulse width: 100 fs; frequency: 80 MHz; peak power (800 nm): >266 kW) controlled by an acousto-optic modulator (AOM). The emission was detected via four photomultiplier tubes with specific filter cubes covering the whole visible range: detection channel 1 (C1), range 420–500 nm; channel 2 (C2), range 515–580 nm; channel 3 (C3), range 590–650 nm and channel 4 (C4), range 660–740 nm. Comparative measurements always have to be performed under identical excitation conditions (including dwell times).A computer-controlled imaging software (FV10-ASW) was used to select the laser excitation power and wavelength, and also to acquire the emission images. The experimental data were treated, either by the software or open source software Image J,36 to obtain intensity profiles in the spatial scale and convert them to kinetic profiles by using the Origin program. Only the clear and non-affected tails were analysed. Under certain conditions, even when we used diluted samples, some of the time profiles still overlapped. This issue can be solved just by changing the scan direction or by increasing the dwell time (thereby shortening the tails), but consequently sacrificing resolution in the time scale. The estimation of spatial resolution, lifetime window and energy density delivered per dwell time (Fd) is explained in the ESI.†
2.6. Steady-state photoluminescence
Steady-state photoluminescence spectra were recorded at room temperature with a 2 nm slit width and 5 nm s−1 speed scan using a SML AmingoBowmann series 2 (AB2) fluorometer (Microbeam, S.A.). The fluorometer was coupled to continuous wave (CW) 975 nm diode laser (Thorlabs L975P1WJ) as excitation source. Irradiance (I) of the laser was obtained by measuring the output power, with a thermal power meter (S470C ThorLabs), and the beam profile (D4σM and D4σm definitions), with a camera (SP920s Ophir). The AB2 software (v. 5.5) was used to register the data. All data were acquired using triangular quartz cuvettes under air atmosphere.3. Results and discussion
3.1. Evaluation of the empirical photoluminescence lifetime of UCNPs by using NIR-LSM
Full characterization of nanosystems requires interpretation and understanding of their photophysical mechanisms and is necessary for an optimal design of nanomaterials containing UCNPs for further applications.37,38 In this way, the emission properties and lifetimes of UCNPs here obtained are a fundamental source of information.Fig. 1(a)–(c) shows the scheme usually used to explain the production of the typical emissions of Er3+, Ho3+, and Tm3+-doped UCNPs with Yb3+ as the sensitizer. Giving rise to the characteristic emission features (Fig. 1(d)–(f)).
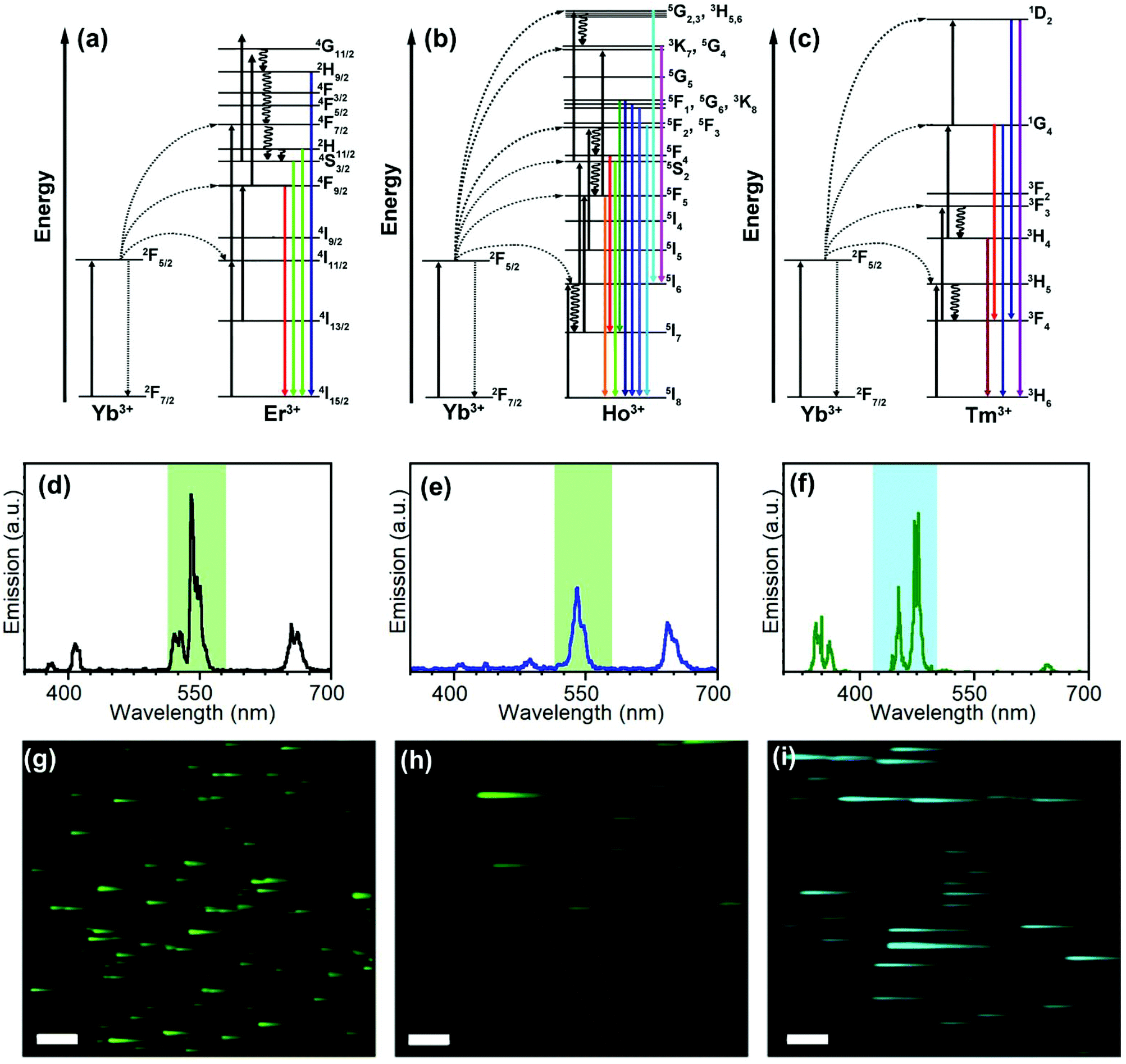 | ||
| Fig. 1 Analysis of the UCNP emissive properties. (a–c) Energy transfer processes involved in the emission of UCEr (a), UCHo (b) and UCTm (c). (d–f) Luminescence emission spectra of colloidal UCNPs (1 mg mL−1) in toluene (d) UCEr,2, (e) UCHo and (f) UCTm (λexc = 975 nm; I = 93 W cm−2). The colored columns indicate the detection channel range (C1, blue: 420–500 nm; C2, green: 515–580 nm) which was used to register the corresponding (g–i) NIR-LSM images (λexc = 975 nm and Fd = 0.2 J cm−2). Scale bar: 50 μm. | ||
The energy of photons absorbed by Yb3+ ions is transferred to the Er3+ excited states which, eventually, leads to the 4S3/2 → 4I15/2 (ca. 550 nm), 2H11/2, 4S3/2 → 4I15/2 (ca. 520 nm), and 4F9/2 → 4I15/2 (670 nm) emissive transitions (Fig. 1(a) and (d)). Similarly, the emission of NaYF4:Yb3+,Ho3+ UCNPs involves the transfer of two photons from Yb3+ to Ho3+ (Fig. 1(b)). The red emission was attributed to the transition from 5F5 to 5I8. Two processes may contribute to populating the 5F5 state; the first involves the intermediary level 5I6 of Ho3+ relaxing to 5I7 by nonradiative transition and then populating the 5F5 state by absorbing another photon via excited state absorption (ESA). The second involves a nonradiative relaxation process from the 5S2/5F4 state to the 5F5 state.8 It has been suggested that the green emission (from 5F4 and 5S2) is likely to be populated at a much faster rate than the red one due to the direct two-step energy transfer from Yb3+.8 As a result, the relaxation from the excited states of Ho3+ (5S2/5F4 → 5I8 and the 5F5 → 5I8) results in green (ca. 545 nm) and red (ca. 670 nm) emissions, respectively.39
The emission of UCTm is more complex than the others and involves more than two photons due to the discrete and ladder-like arrangement of Tm3+ excited states (Fig. 1(c)).40 Emissions in the UV (362 nm, 1D2 → 3H6), visible (450, 475 y 644 nm, 1D2 → 3F4, 1G4 → 3H6, and 1G4 → 3F4, respectively) and NIR (800 nm, 3H4 → 3H6) regions were observed.41 Mechanistic studies have proved that the transition 1G4 → 3H6 (475 nm) is due to a three-photon absorption process, whereas the transition 1D2 → 3F4 (450 nm) corresponds to a four-photon process.41
The spectral and time-resolved upconversion measurements are usually performed on specifically designed equipment (such as FluoTime 300®, Picoquant, Fluorolog-3, FLS-1000) or on adapted conventional fluorometers equipped with a cheap NIR-LED (CW) as the excitation source, coupled to a triggering system (e.g. a chopper) to pulse the pump signal.38 In both systems, the spectral resolution is achieved with a monochromator and the time resolution is determined by the detector response and the frequency and pulse width of the laser. This equipment allows for the macroscopic measurement of solid, liquid and film samples. Consequently, the sample response, although robust, is a macroscopic average of the entire excited sample. Therefore, it cannot be attributed to the signal of every NP unless other characterization techniques (such as TEM, HRTEM, DLS, EDX, ICP, etc.) demonstrate the homogeneity in size and dopant concentrations in the micro-/nano-meter scale.
By contrast, the time-resolved NIR-LSM approach to obtain the lifetimes was implemented in a commercial multi-photon microscope (MPM; FV1000-MPE), equipped with a femtosecond Ti-Sapphire laser, as excitation source, and a filtered photomultiplier detection system. It is worth mentioning that inexpensive, low-power laser diodes can also excite UCNPs in NIR-LSM systems,42,43 so logically, a NIR-LS microscope would also measure the emissive spectrum by just coupling a spectral detection system.
To show the temporal resolution versatility of NIR-LSM, the most emissive channels of UCEr,2, UCEr,20, UCTm, and UCHo were monitored after 975 nm excitation (Fig. 1(g)–(i) and Fig. S6 in the ESI†) with a multiphoton excitation imaging microscope by means of 25 × 1.05 N.A. Olympus dipping lens. An emission filter between 515 and 580 nm (channel 2) and a dwell time of 2 μs per pixel were used for UCEr,2, UCEr,20 and UCHo, whereas an emission filter between 420 and 500 nm (channel 1) and a dwell time of 4 μs per pixel were used for UCTm (Fig. 1(d)–(f)).
It is common knowledge that nanoparticles with long lifetimes are a drawback for imaging when using scanning microscopy techniques.30,44,45Fig. 1(g)–(i) shows images from sets of close-lying UCNPs and agglomerates; the clear tail in the fast-scan direction can be attributed to the detection of the off-axis emission once the laser beam has passed over an agglomerate in the x direction; this results in a loss of spatial resolution. This could be avoided by using a small pinhole in front of the detection system,18 or by using time-gated microscopy or long dwell times. However, we have previously demonstrated how this drawback is, in fact, advantageous for obtaining information about the evolution of the UCNP emission over time.30,32,46
The time-resolved NIR-LSM principle can be clearly explained by imaging an agglomerate of UCTm@Rh110 at 4 μs per pixel dwell time. The image of Fig. 2(a) is a composite of two images of the same region under 975 nm excitation in two different detection channels. The 475 nm – UCTm emission registered in C1 (420–500 nm) was artificially coloured in cyan blue. The emission of the dye attached to the UCNP surface produced via a two-photon excitation and collected in C2 (515–580 nm) was coloured in green. The excitation of the dye reveals information on the spatial location of the agglomerate and, therefore, indicates where the UCNP excitation is taking place (red profile in Fig. 2(c) and (d)). Consequently, excitation consists of a train of laser pulses (bursts) determined by laser repetition rate, the laser energy per pulse, the scanning rate (dwell time) and the length of the agglomerate (if longer than the pixel size). Even if the laser kept on shooting after the agglomerate, no species would be able to absorb it. Therefore, the rise and decay of the thulium off-axis emission of the previous close-lying nanoparticles/agglomerates would be observed; i.e. the detector would still be collecting the sample response since there was no pinhole (grey profile in Fig. 2(c)). Pixels right after the set of close-lying nanoparticles/agglomerates are acting as a consecutive “time gate” (temporal width equal to the dwell time) detection system (Fig. 2(c) inset). As a result, the kinetic profiles of the emission could be registered (Fig. 2(d)).
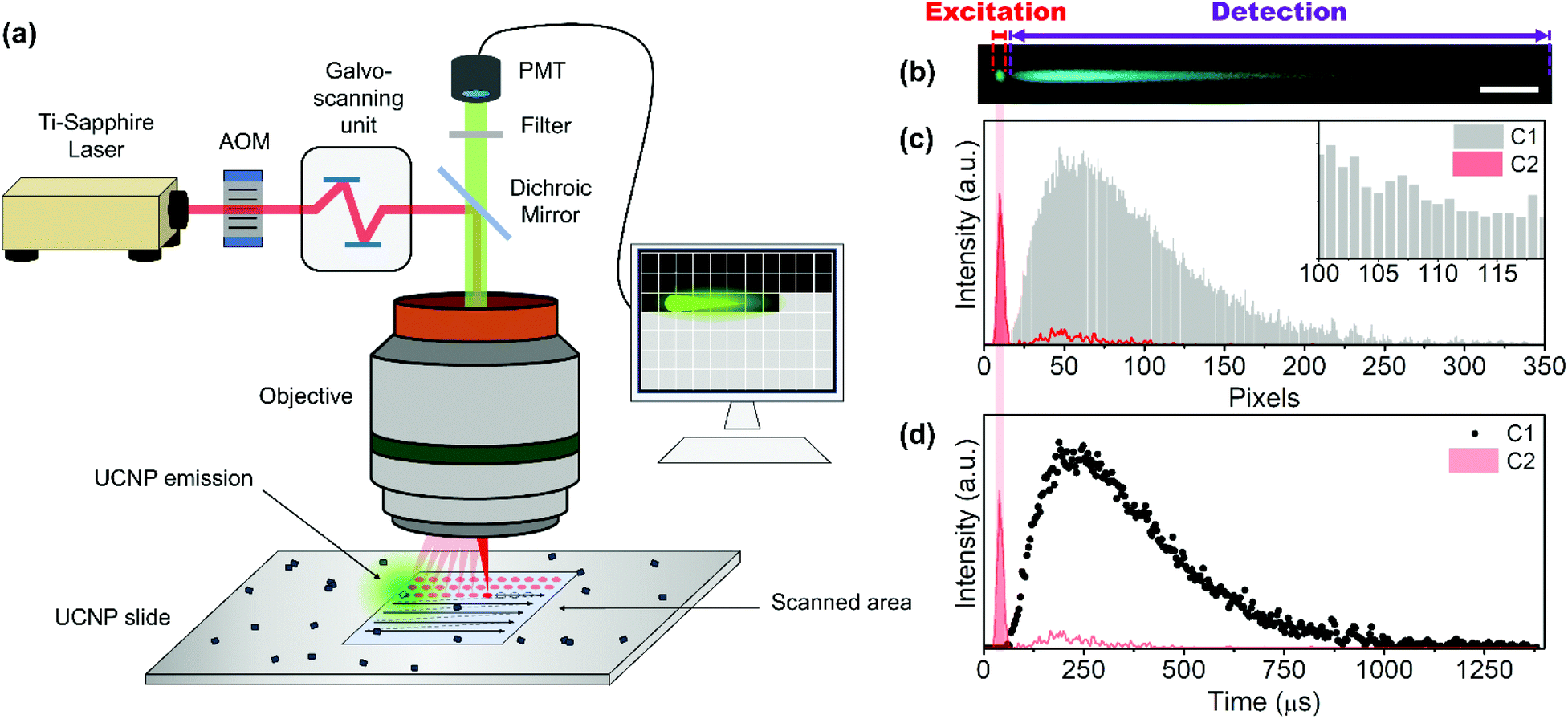 | ||
| Fig. 2 Scheme and visual representation of the time-resolved principle of NIR-LSM. (a) NIR-LSM scheme. (b) Composite image of image of the UCNP emission (blue, detection channel 1) and Rh110 two-photonic excitation emission (green, detection channel 2) at 975 nm excitation and 4 μs per pixel dwell time of the same agglomerate of UCTm@Rh110 UCNH (Fd = 10.4 J cm−2). Scale bar 20 μm. (c) Intensity profiles and (d) kinetic profiles obtained from image (a). | ||
Therefore, these images have to be understood as time-resolved data in the x-axis and not as conventional fluorescence images. After the “charging period” (excitation) of an agglomerate, when the beam has gone to other pixels, the system keeps the emission intensity value acquired at every dwell time (for example 2 μs) in different pixels. These pixels act as a storage site, which makes signal temporal contamination between pixels unfeasible in the x direction. Thus, the resolution limit affects the minimum excitation area (spot size) but not the temporal resolution. As expected, when increasing the dwell-time (i.e. decreasing the scanning speed) the UCL tail shortened (Fig. 3(a)) and the peak maximum shifted to a shorter x-axis value (Fig. 3(b)).
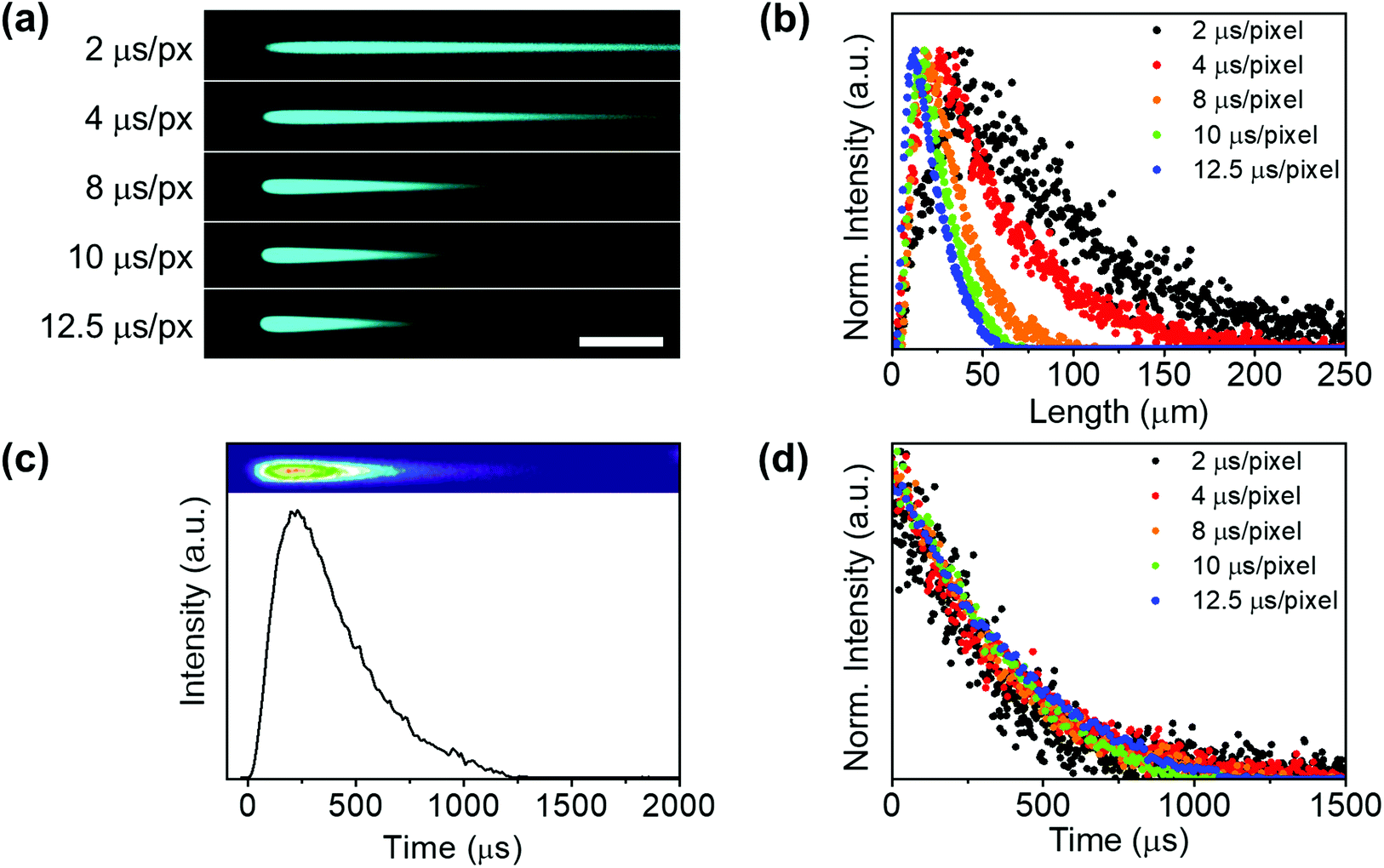 | ||
| Fig. 3 Analysis of the photoluminescence kinetic profile of UCTm agglomerate. (a) Images of the same UCTm agglomerate obtained at different dwell times in channel 1 (C1: 420–500 nm; λexc = 975 nm; Fd = 5.2–32.6 J cm−2). Scale bar 50 μm. (b) Intensity profiles obtained from readings in (a). (c) Thermal colored image and the intensity profile as a function of time of an UCTm agglomerate (10 μs per pixel; Fd = 26.0 J cm−2). (d) The decay profile as a function of time of the UCTm agglomerate obtained at different dwell times. | ||
A luminescence decay curve was easily obtained from the intensity profiles in the scan direction by transforming the spatial scale (μm) into a time scale (μs) using the dwell time value and the pixel length (Fig. 3(c)). Fig. 3(d) illustrates the kinetic profiles obtained from the tails registered at different dwell times (Fig. 3(a)). All decay kinetics showed a similar behaviour, thus corroborating that the emissive tails can certainly be ascribed to the intrinsic UCL agglomerate.
The UCL kinetic profiles of the different co-doped UCNPs after NIR light excitation showed the characteristic slow rise followed by a long decay (Fig. 4).
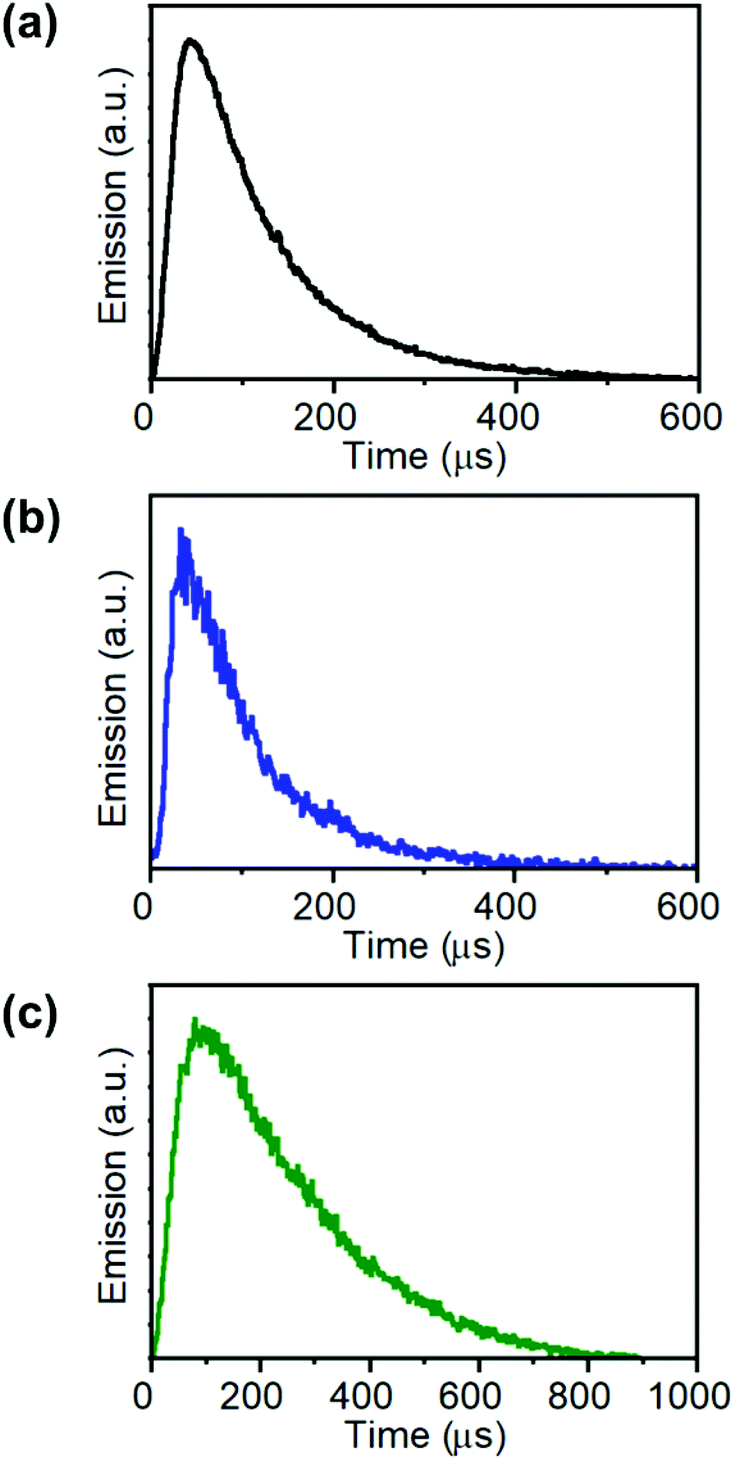 | ||
| Fig. 4 Kinetic profiles of the emission in channel 2 (C2: 515–580 nm) of (a) UCEr,2, (b) UCHo, and in channel 1 (C1: 420–500 nm) of (c) UCTm. (λexc = 975 nm; Fd = 0.2 J cm−2). | ||
However, while the rise times were found to be very sensible to the focus position between images and minimum changes produced a great divergence of values, the decay fitting values remained almost unaltered. Therefore, NIR-LSM can be used to obtain the decay lifetime of the UCL emissions as well as to compare the kinetic profiles of different UCNPs.
The UCL decay lifetimes of the UCNPs herein reported were estimated by fitting the temporal evolution of the UCNP emission to a mono-exponential equation (Fig. S7 and Table S2 in the ESI†). As expected, UCEr,20 displayed a shorter lifetime when compared to UCEr,2 (ca. 22 μs versus 68 μs, respectively), because of the luminescence concentration quenching effect. The longest lifetimes corresponded to Tm-doped UCNPs, followed by Ho-doped UCNPs (ca. 220 μs) and Er-doped UCNPs. These results are in agreement with previous studies.47–52
Equally important, this novel application of a NIR-LS microscope can also be applied to UCNP-based nanohybrids, e.g. nanoassemblies of UCNPs and other luminescent NPs or chromophores.30–32 Nanohybrids can be designed to lengthen the photoluminescence of a nearby acceptor (photoactive NPs, organic fluorophores) via resonant energy transfer (RET) from the UCNPs, which is of interest in (bio)sensing, photodynamic therapy, and bioimaging applications, and photonics.30–32 Previous studies performed by our group have demonstrated that when the RET process is highly efficient, only dots (instead of tails) are observed upon 975 nm excitation of the UCNHs due to a drastic quenching of the lifetime of the UCNPs.30 On the other hand, for moderately efficient RET, a shortening of the tail of the upconversion emission, together with a lengthening in the fluorophore lifetime, can be observed.32
Here, as an example to illustrate the visualization of a resonant energy transfer process by using this technique, an UCNH consisting of rhodamine 110 (Rh110) grafted onto UCTm was built. Rh110 absorption overlaps with the 475 nm emission band of UCTm (Fig. 5a). The presence of the dye in the UCNH was confirmed by UV-vis absorption spectroscopy (Fig. S4 in the ESI†). Consequently, RET from the UCNP to Rh110 was expected under 975 nm excitation. Moreover, Rh110 can be excited by two-photons at 975 nm. Therefore, two different events were expected: on the one hand, the fast-ordinary emission of the dye due to two-photon absorption, and on the other, a long-lived emission of Rh110 reflecting the long-lived UCTm transition involved in the RET. This dual emission was corroborated by following the emission of the UCTm@Rh110 in detection channel 2 (C2: 515–580 nm, Fig. 5(a)). The image (Fig. 5(b)) and the intensity profile (Fig. 5(c)) obtained showed the two-regime behaviour expected of the dye emission. Firstly, there was an instant emission which is observed as a dot, and right after the dot, there was a characteristic lengthened emission tail (C2 in Fig. 5(b)), whose rise and decay could be followed in C2 in Fig. 5(c).32 These results proved that it was possible to easily distinguish the direct emission of the dye separated from that of the sensitized emission of the dye whenever the RET efficiency was not very high. Moreover, this strategy made feasible to differentiate signals from the UCNP and dye in UCNHs. However, a limitation of NIR-LSM is that, in general, the decay times of the UCL cannot be used to calculate energy transfer efficiencies,29 which do require the actual knowledge of the lifetime of the donor emitting state. In certain cases, for example, energy transfer to an acceptor will not even affect the time profile of the emission of the donor state. This is because only one lanthanide emission is quenched, but the time profile remains unchanged due to the other non-affected emission. In fact, the energy transfer from 1G4 of Tm in the herein shown example does affect the overall time evolution of the UC emission, but it is unlikely that the changes in the 1G4 decay constant can easily be inferred from the UC emission. Despite that, the lengthening of the fast, intrinsic lifetime of the acceptor species is valid evidence that energy transfer has occurred.§
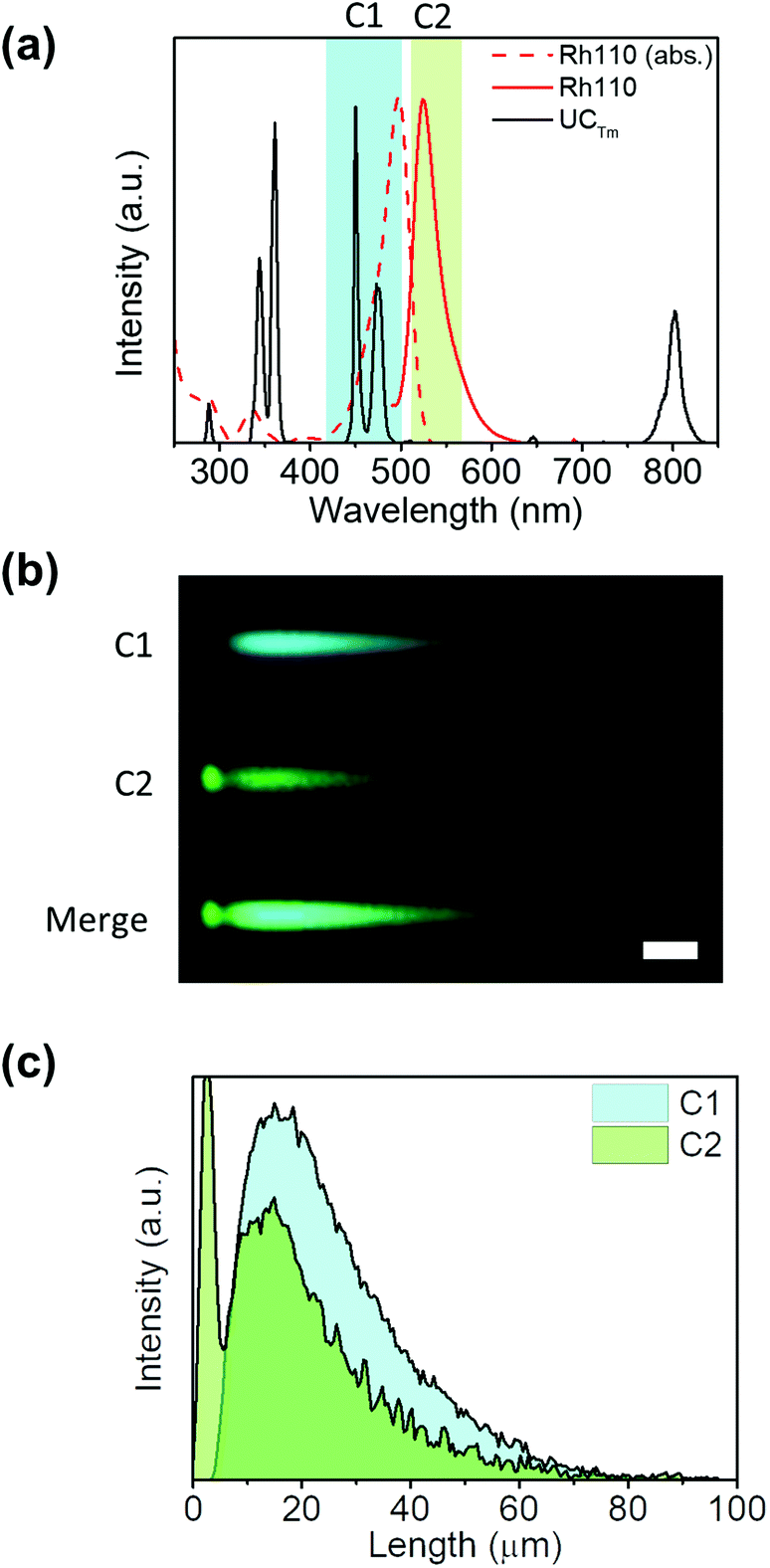 | ||
| Fig. 5 Resonance energy transfer recorded by NIR-LSM. (a) Absorption (dotted red line) and emission (red line) spectra of Rh110 in aqueous solution, emission spectrum of 2.0 mg mL−1 aqueous dispersion of UCTm (black line), and the detection ranges in channel 1 (C1: 420–500 nm, blue column) and channel 2 (C2: 515–580 nm, green column). (b) Detail of the NIR-LSM image obtained by exciting UCTm@Rh110 at 975 nm and 8 μs per pixel dwell time (Fd = 20.8 J cm−2); scale bar: 50 μm; the complete image is shown in Fig. S8.† (c) Intensity profiles of the signals obtained in channel 1 (520 V, blue) and channel 2 (600 V, green). | ||
Time-resolved NIR-LSM provided temporal information on the micrometre scale by scanning a 508.4 × 508.4 μm2 area of the microscope slide prepared by using a diluted sample (0.5 × 0.5 μm2 pixel size). The signal can originate from (i) a set of close-lying nanoparticles; (ii) an assemblage of nanoparticles (agglomerate) (Fig. S5 in the ESI†). It has to be taken into account that the formation of agglomerates is usually unavoidable when a colloid droplet, composed of nanoparticles, is deposited on a substrate and then dried; this occurs even if the initial solution is stable.53 The UCNP/UCNH colloids consist of nanoparticles with a narrow distribution of sizes as shown by the SEM images of the colloid. Although the photoluminescence intensity registered by NIR-LSM depends on the number of nanoparticles in an agglomerate, the kinetic profile reflects the degree of homogeneity in the response of its photoluminescence. Furthermore, this can be corroborated by analysing different agglomerates. Fig. 6 shows the similar temporal behaviour in channel 1 and channel 2 of different-sized micrometric agglomerates of UCTm@Rh110.
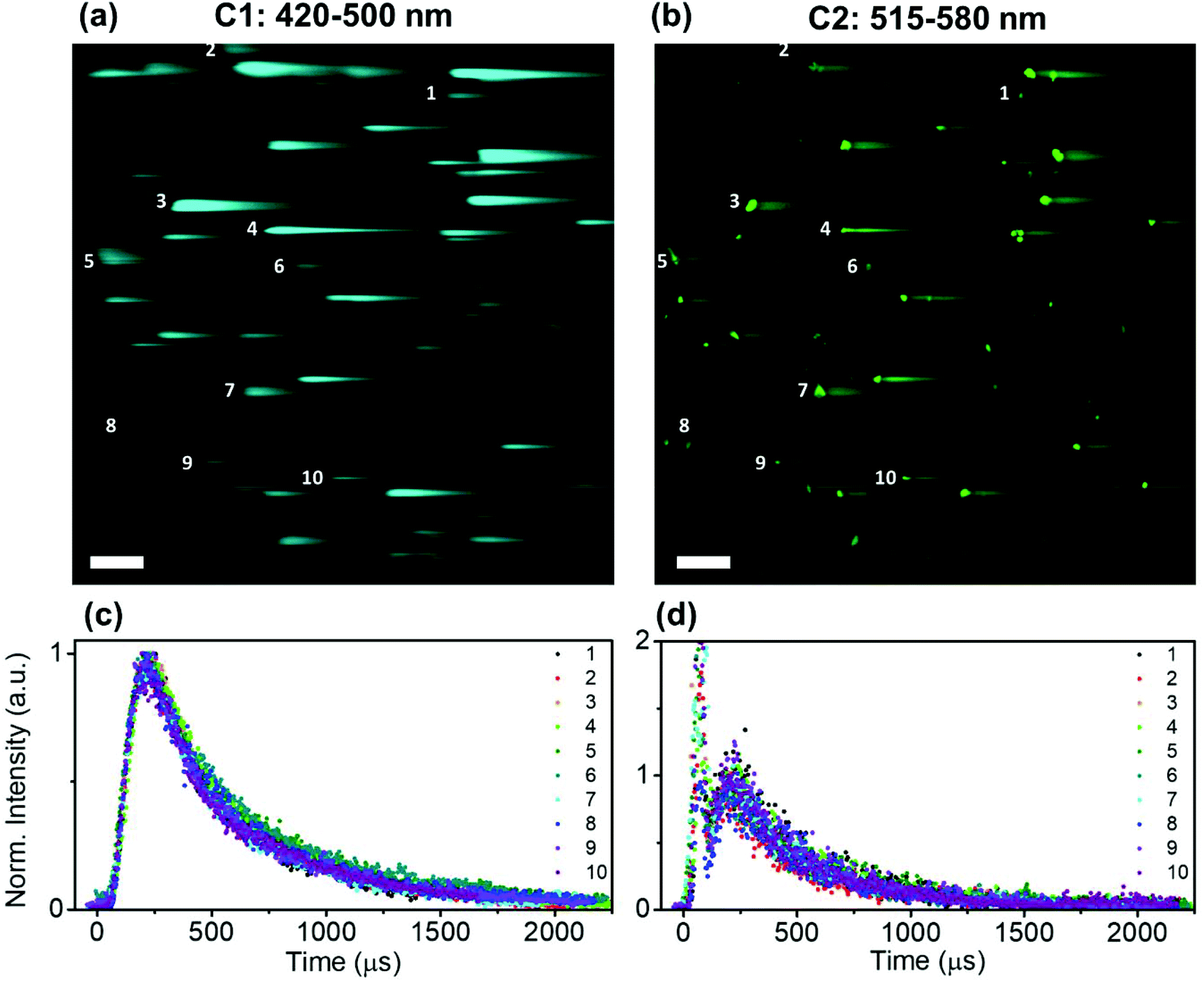 | ||
| Fig. 6 Equal temporal behavior of several agglomerates of UCTm@Rh110. NIR-LSM images of the same region of UCTm@Rh110 sample under 975 nm excitation (Fd = 0.9 J cm−2) in (a) channel 1 (C1: 420–500 nm; 560 V) and (b) channel 2 (C2: 515–580 nm; 635 V). Time-resolved normalized intensity profiles of numbered agglomerates in (c) channel 1 and (d) channel 2. Scale bar: 50 μm. | ||
3.2. Evaluation of the homogeneity/colocalization of the UCNHs
Colocalization is the analysis of the distribution of signals in fluorescence microscopy. It involves two components: co-occurrence, which is the spatial overlap of two probes, and correlation, which is the proportional intensity co-distribution of both species.54,55 Encouraged by the developments in microscopy image analysis over the last 20 years,54,56,57 we were interested in applying this knowledge to evaluate the homogeneity of UCNH samples. Several coefficients have been described to quantify colocalization as well as significance testing approaches. In this way, the most commonly used coefficients are the Pearson's correlation coefficient (PCC), which quantifies exclusively the correlation of two probes, and the Mander's Colocalization Coefficients (MCCs), which fundamentally measures co-occurrence independently of signal proportionality.54 However, other coefficients and approaches have also been described (Li, Van Steensel, Spearman, Lachmanovich, Boutté, Jaskolski).56 Different strategies have been proposed to test the statistical significance of colocalization measurements for a single experimental group. The most appropriate methods are the scrambling of Costes’ approach and the frame translation approach, since both are based on a comparison between the image of studying and a large number of random rearrangements.54,58,59 Nevertheless, the frame translation approach was found to yield more accurate random probability distributions.59 Therefore, PCC and MCCs were calculated to evaluate the homogeneity of the samples and the frame-translation approach was applied to test the statistical significance of the coefficients calculated.The NIR-LSM can be helpful for colocalization by making use of the femtosecond laser wavelength tunability from 690 to 1040 nm and the selective multichannel detection. Yb3+ absorption has a maximum at around 975 nm which diminishes drastically when the wavelength is tuned to higher energies. Therefore, the UCNP excitation is only possible in a narrow wavelength range at around 975 nm. In contrast, dyes can be excited by two-photons in a wider range, which allows us to excite them at a wavelength different from that of the UCNP (containing Yb3+). As a result, we conceived that the homogeneity of samples could be evaluated by comparing two images of the same region: one exciting UCNPs and the other exciting the fluorophore anchored to the UCNP. To show this versatility, the study was developed with two UCNHs consisting of UCTm and two rhodamine dyes (UCTm@Rh101 and UCTm@Rh110). Two dyes (Rh101 or Rh110) with different photophysical properties were used in order to show the flexibility of this instrument. The UCNHs were deposited onto the microscope slide and the images were collected after exciting the UCTm and the dye (either Rh101 or Rh110). It should be taken into account that images from the UCNP acquired at short dwell times are not suitable for colocalization analysis because the emission tail does not provide the definition of the UCNP agglomerate (Fig. S9 in the ESI†). Otherwise, the correlation and co-occurrence would be overestimated. Instead, a much more appropriate image for colocalization was obtained when the dwell time was set to 100 μs (Fig. 7(c) and (d)), although it was time-consuming, and the UCNP still exhibited some blurriness which increased its emission area compared to the dye emission area (Fig. S12 in the ESI†). However, well-defined dots were observed independently of the dwell time used when Rh101 and Rh110 were excited by two-photons (Fig. 7(e) and (f)). This is consistent with the nanosecond lifetime of the dye fluorescence, which is three orders of magnitude faster than the expected time resolution reached with this technique. Remarkably, the overlap of the images registered in the same channel obtained after exciting the UCNP (at 975 nm) and the fluorophore (at 1030 and 1020 nm for Rh101 and Rh110, respectively) provided a spatial sense of colocalization (Fig. 7(g) and (h)).
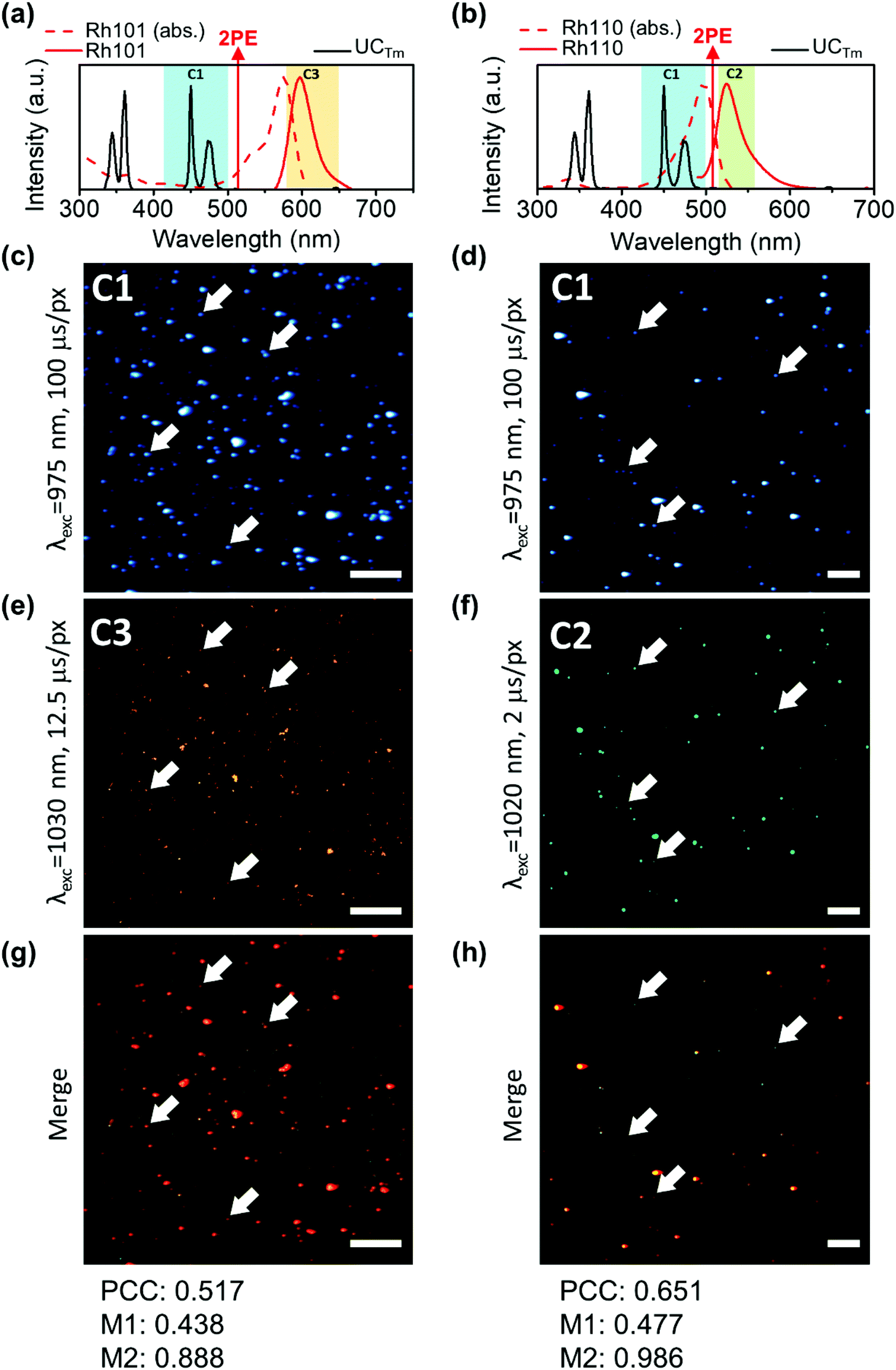 | ||
| Fig. 7 Colocalization analysis of the nanohybrids monitored with NIR-LSM. Spectral properties of (a) UCTm@Rh101 and (b) UCTm@Rh110 showing the absorption (dotted red line) and the emission (red line) of the dye in aqueous solution, as well as the emission of a 2.0 mg mL−1 aqueous dispersion of UCTm (black line), the wavelength used for the two-photon excitation (red arrows) and the detection channels (colored columns). Microscope images of the UCTm@Rh101 sample excited at (c) 975 nm (Fd = 218.5 J cm−2), (e) 1030 nm (Fd = 27.5 J cm−2), and (g) the combination of both images. Microscope images of the UCTm@Rh110 sample excited at (d) 975 nm (Fd = 260.5 J cm−2), (f) 1020 nm (Fd = 4.4 J cm−2), and (h) the combination of both images. Scale bar 50 μm. | ||
In order to quantify this visual colocalization, PCC and MCCs were calculated. In this way, the PCC values obtained show a reasonably good significant correlation (0.517 and 0.651 for UCTm@Rh101 and UCTm@Rh110, respectively). Usually in fluorescence imaging analysis, it is possible to calculate the ratio or stoichiometry of the probes, however this is not so trivial when comparing the emission of inorganic nanoparticles and dyes. To further demonstrate the sample homogeneity, although correlation inferred an important overlap, it was not as intuitive as co-occurrence which indicates the degree of overlap between the studied species. In this way, MCC significant values obtained for dyes (M2) reached 0.9–1.0 (0.888 and 0.986 for UCTm@Rh101 and UCTm@Rh110, respectively) indicating that approx. 90% of the dye signal co-occurred or overlapped with that of UCNP signals. This was a relevant result that clearly proved that the distribution of the dye on the microscope slide, and therefore of the UCNH samples, was due to a real interaction and not to a random distribution. Otherwise, the dye would have been distributed randomly on the microscope slide. In contrast, the co-occurrence fraction of UCNP emission with the dye signal (M1) was found to be between 0.4–0.5 (0.438 and 0.477 for UCTm@Rh101 and UCTm@Rh110, respectively), because M1 reflects that the area of each UCNH agglomerate in the image is higher when exciting UCNPs than when exciting the dye (Fig. S12 in the ESI†). Therefore, M1 seems to be useless for describing the UCNH sample homogeneity, unless a confocal technique is used to define the upconversion emission of the UCNH agglomerate (by adding a pinhole).18
All in all, we have demonstrated that PCC and MCC values can be a complementary source of information about the colocalization and the co-occurrence of the dye and the UCNP (M2). Note that a conventional spectrofluorometer gives an average signal, which cannot be attributed to the homogeneity of the sample.
4. Time-resolved luminescence spectroscopy and imaging techniques
A brief temporal resolution comparison between NIR-LSM and spectroscopic and imaging techniques is discussed in this section.4.1. Time-resolved spectroscopy techniques
Spectroscopic temporal resolution is usually achieved by using two different time-domain techniques: time-correlated single photon counting (TCSPC) and multi-channel scaling (MCS). Both are single photon counting techniques which require pulsed excitation sources and measure the arrival time of the photon at the detector. In the case of TCSPC, the delay time of START(excitation)-STOP(emission) pulses of multiple photons (millions) is measured to create a histogram of the luminescence decay profile, while the MCS works in a similar way to an oscilloscope by dividing the temporal window into small time gates (bins) and detects single photons reaching the detector gate-by-gate consecutively. TCSPC can measure lifetimes from ps to hundreds of ns efficiently and, although it could also determine long luminescence lifetimes, MCS is preferred to determine in the ns to ms timescale because of its versatility and fast acquisition. In both cases, the kinetic fitting is obtained by using an iterative deconvolution technique taking into account the time response of the instrument, usually termed instrument response function (IRF).60 However, when the emitter lifetime is far longer than the IRF, the deconvolution has a minimum effect on the recorded lifetime and is irrelevant.Similar to MCS, NIR-LSM acts as a consecutive time-gate in which the photons are collected in every pixel (Fig. 2(b)). It also presents a similar emitter lifetime resolution range, which is estimated to be from 2 μs (2 μs per pixel and assuming that 10 points, i.e. pixels, are needed to describe a one-decay profile) to 80 ms (200 μs per pixel, 4096 pixel line, assuming a one-decay profile); and it is only limited to the dwell time of the galvo-scanning unit of MPM. In our equipment the minimum dwell time is 2 μs per pixel and the maximum is 200 μs per pixel.
Spectroscopic techniques can be implemented in solid, liquid or films samples and register an average signal of the entire excited sample in a user-friendly fast way which only require the selection of appropriate excitation and detection conditions. In the case of organic photoactive compounds, the fact that the signal collected is an average of the excited sample is not relevant, while this is of paramount for nanoparticles. Nanoparticles emissive response may change drastically because of the sample size polydispersity or heterogenous doping between nanoparticles. So, spectroscopic measurements may reflect or neglect the lifetime of the longest-lived species and/or reveal the heterogeneity of the sample as a multi-exponential decay. As a consequence, the kinetics obtained in a spectrometer can only be attributed to the nanomaterial if other techniques corroborate the sample homogeneity in the nanoscale.
In contrast, NIR-LSM can record the luminescence of a microspace of the batch sample, which reflects more accurately the nano/micro-metric response of the UCNPs and UCNHs than a spectrometer. Genuinely, it is a micro-spectroscopy technique. Another important feature of this technique is that it enables an at-a-glance check of the lifetime or functionalization homogeneity of the sample by recording several images in different parts of the scanning area. However, it is not exempt of some drawbacks. The major one relies on the representativeness of the measurement. The sample observed in the NIR-LS microscope was prepared by spin-coating a very diluted microliter aliquot of the sample on a microscope slide, so how representative is the response observed in that slide? Representativeness is a common issue in all microscopy techniques (light and electron-based), and it can only be addressed in terms of repeatability and combination with other techniques. Therefore, NIR-LSM measurements are dependent on other characterization techniques in the same way that the macroscopic conventional measurement is. Moreover, the measurements can only be implemented in discontinuous few-layered 2D samples, where the solvent effect in the photophysical properties of the UCNPs cannot be studied (it is necessary to fix the sample). For the same reason, although preparing the sample was not difficult and just involved a few initial optimization steps, it required more time than a conventional lifetime measurement. Undoubtedly, the data processing was also time-consuming, but it could be automatized.
4.2. Time-resolved microscopy techniques
In microscopy, temporal resolution has also played an important role not only for imaging but also for material characterization. Note that, while intensity-based microscopy imaging is limited due to excitation source stability, autofluorescence background and emission overlapping; the lifetime-based microscopy is less sensitive to interferences since the contrast in the image is based on the lifetime. This makes time-resolved microscopy a unique powerful imaging technique. In this way, many different techniques have been proposed, such as fluorescence lifetime imaging (FLIM), phosphorescence/photoluminescence lifetime imaging (PLIM) and time-gated emission imaging microscopy, (TGM) among others. Detection of these techniques are frequency-domain-based and time-domain-based methods, such as TCSPC and time-gated detection. These detection methods can be implemented in widefield microscopes (WF), confocal microscopes (CM, laser scanning and disk-scanning) or multi-photon excitation microscopes (MPM).61Frequency domain methods measure the phase-shift and demodulation of the emission in respect of the excitation light, and they are related to the luminescence lifetime of the emitter. TCSPC technique works in the same way as for spectroscopy measurement, and time-gated detection uses one or more time channels (time gates) with a different time-offset with respect to the excitation pulse through mechanical choppers. In these time channels, photons can be detected by single photon counting or analog detection methods.61 These methodologies have been applied to lifetime measurements from short (ns) to long luminescence (μs-ms) lifetimes. In this way, FLIM and PLIM techniques register the whole emission decay within a pixel, during dwell time. In the FLIM case the maximum emission lifetime that can be acquired is above few μs. Therefore, it is not appropriate to analyse UCNPs. However, PLIM has a time scale that arrives up to 1 ms. This fact opens the possibility of characterizing UCNPs, but with huge dwell times that produce very long acquisition times.
Other approaches have been proposed to image the challenging long-lifetimes emitters in WF, CM and MP microscopes, by making use of either frequency-domain or time-domain detection techniques, and have been extensively reviewed by Zhang et al.21 and Baggaley et al.62 In general, long time kinetics require a low repetition rate excitation source (or a train pulses) followed by a sensitive detection with a long-temporal window to observe the decay emission between laser pulses (or train pulses).
In terms of comparison with the NIR-LSM here overviewed, we mainly focussed on the time-domain methodologies applied to MPM. Two approaches are used to circumvent the incompatibility between the high frequency laser source of MPM (80 MHz for Ti-Sapphire laser), and the long-lived emitters: (i) to reduce the repetition rate of the laser or (ii) to excite with a train of laser pulses followed by a detection time without laser excitation within the pixel dwell time.62 The first approach can be implemented by running the laser in a cavity dumped mode, which reduces the repetition rate but increases the energy per pulse. The second one needs a mechanical chopper63 or an (electro/acousto-) optical modulator (EOM or AOM)64,65 and the detection can be performed by TCSPC or by time gating. The second approach seems to be much more interesting for phosphors to avoid possible thermal or saturation effects, due to the high peak energy density, or the apparition of Moiré patterns (interference pattern) in the image and has enabled simultaneous fluorescence and phosphorescence imaging and oxygen and blood flow imaging.62,64,65 Remarkably two complementary works have been proposed to adapt a conventional CM: pinhole shifting lifetime imaging (PSLIM)66 and temporal sampling lifetime imaging (TSLIM).67 In PSLIM approach different emission times are recorded by a pinhole shift which enables the detection of emissions longer than the dwell time. This fact makes impossible a continuous acquisition of the emission decay, limiting the number of experimental points obtained and generating a lack of precision in the decay curve determination.
In sharp contrast, in TSLIM the emission curve decay is obtained by a single spot excitation, using the single spot bleaching option in the confocal, and then intensity kinetic profile is measured in a pixel. This technique can give temporal resolutions of few μs. Otherwise, the main drawback relies in the time-consuming spot by spot acquisition. Although this problem can be partially overcome using a detection line scanning confocal, analysing a row simultaneously.
All in all, although several approaches have been proposed for imaging (their aim), the acquisition time is usually the main drawback, since it is limited at least to 10× emitter lifetime (optimum temporal window to clearly show the decay profile) temporal units per pixel.
Similar to PSLIM technique, NIR-LSM is based on the detection of the off-axis emission. However, in this case, we do want to image the long tail of the long-lived upconversion emission.
Contrary to microscope imaging, NIR-LSM is a spectroscopic characterization technique in the microscale, in which the temporal resolution was not achieved by a time-domain or frequency-domain methodology but by scanning a microscopic area without a pinhole at a scanning rate faster than the luminescence lifetime of the emitter. As a result, the spatial resolution and acquisition times are not limiting factors but the detection of the off-axis emission fits in the scanning line. Interestingly, images in NIR-LSM can be obtained in a few seconds (from approx. 2 s at 2 μs dwell time and approx. 20 s at 200 μs dwell time for a 1024 × 1024 image), in contrast to imaging time-domain techniques for long-lived emitters. Even frequency-domain techniques require longer acquisition times, since it is necessary to register sets of images.67 Moreover, for NIR-LSM spectroscopy, the MPM does not require any modification or technical knowledge of electronics.
However, NIR-LSM strength is at the same time its weakness. Despite of being extremely useful for discontinuous few-layered 2D samples of long-lived UCNPs and UCNHs, NIR-LSM techniques suffer from poor lateral and axial resolution as compared to imaging ones and they cannot be used to image 3D specimens or to obtain the kinetics of 3D samples, so these parameters are out of the scope of the NIR-LSM.
5. Conclusions
Lifetime measurements of colloidal or solid nanoparticles are usually obtained with spectrofluorometers which measure the macroscopic average response of the sample; however, this does not mean that the sample is homogeneous. Nevertheless, the NIR-LSM response of upconversion nanoparticles is associated with close-lying nanoparticles or agglomerates, therefore it allows direct measurement on the micro-/nano-meter scale. Time-resolved microscope techniques, such as FLIM/PLIM, are based on TCSPC/MCS methodologies and, as imaging is their aim, usually require long measurement times, especially for luminescent μs-lifetime species.Our strategy consisted in rework the spatial-resolved image into a time-resolved spectrum by converting each value of distance (μm) into a value of time (μs), using the applied dwell time and the pixel size. The measurement time was short because imaging was not our aim, but undoubtedly, the data processing and analysis were more time-consuming. The strategy is limited to the scanning speed of the sample, consequently only the scanning speed limits its UCL lifetime (not the risetime); few μs to tens of ms can be measured because the minimum dwell time is 2 μs per pixel. In any case, the NIR-LSM technique showed the lengthening of the acceptor lifetime, indicating a RET mechanism.
NIR-LSM has also been revealed as a useful technique for evaluating the homogeneity of the sample by measuring the temporal behaviour of close-lying nanoparticles/agglomerates, as well as of the homogeneity in the UCNP functionalization of the UCNP-based nanohybrids, which could be also applied to other long-lived emitters. The homogeneity of the UCNP functionalization was evaluated in terms of colocalization of two different dyes on the UCNP surface, thereby demonstrating that the dyes were in the vast majority of the close-lying nanoparticles/agglomerates. Indeed, faster and more reliable co-occurrence results (M1, especially, and M2) would have been obtained if the microscope had confocal capabilities (pinhole) when exciting the UCNP. However, including a pinhole would prevent detecting the emission tails. In the case of UCNP, to fully characterize these nanosystems photophysically at the microscale the MPM would need a NIR detector, to detect the 1 μm Yb3+ emission; an aperture-controlled pinhole system, to perform more accurate colocalization studies; and a spectral resolution system (e.g. monochromator), instead of filter cubes.
All in all, these results prove that NIR-LSM is more than an imaging technique. Furthermore, it is a source of lifetime and functionalization information on the micro-/nano-meter scale for UCNP characterization, which together with the developments made with UCNP and NIR-LSM (subdiffraction imaging, power dependence of the emission and spectral information on the micro-/nano-meter scale) opens up new possibilities to researchers in this field.
Author contributions
Juan Ferrera-González: Investigation; conceptualization; writing original draft; visualization – Laura Francés-Soriano: Investigation – Nestor Estébanez: investigation – Enrique Navarro-Raga: Conceptualization; methodology; writing review and editing – María González-Béjar: Conceptualization; writing review and editing; supervision – Julia Pérez-Prieto: Conceptualization; supervision; writing review and editing; funding acquisition and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Ministerio de Economía, Industria y Competitividad (CTQ2017-82711-P, co-financed by FEDER; RYC-2013-14063; RyC grant M. G. B.), Ministerio de Ciencia e Innovación (Unit of Excellence “Maria de Maeztu” CEX2019-000919-M), and Ministerio de Educación, Cultura y Deporte (FPU grant J. F. G.). Generalitat Valenciana (IDIFEDER/2018/064 and PROMETEO/2019/080, co-financed by FEDER) and Fundación Ramón Areces are also acknowledged. We also thank to Marie Sklodowska Curie actions for L. F. S. financial support (Ref: 7944109).The TEM, SEM, HRTEM, and NIR-LSM images were performed in SCSIE (University of Valencia).
We also thank Álvaro Guadaño and Armando Izquierdo (Lasing S.A.) for their advice about laser beam quality and profile.
Notes and references
- S. Inoué, in Handbook of Biological Confocal Microscopy, ed. J. B. Pawley, Springer, New York, 3rd edn, 2006, pp. 1–20 Search PubMed.
- W. Denk, D. W. Piston and W. W. Webb, in Handbook of Biological Confocal Microscopy, ed. J. B. Pawley, Springer, New York, 3rd edn, 2006, pp. 535–549 Search PubMed.
- S. W. Paddock, T. J. Fellers and M. W. Davidson, Introductory Confocal Concepts, https://www.microscopyu.com/techniques/confocal, (accessed 24 April 2020).
- D. W. Piston, T. J. Fellers and M. W. Davidson, Multiphoton Microscopy, https://www.microscopyu.com/techniques/multi-photon/multiphoton-microscopy, (accessed 24 April 2020).
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- B. R. Watson, B. Yang, K. Xiao, Y. Z. Ma, B. Doughty and T. R. Calhoun, J. Phys. Chem. Lett., 2015, 6, 3283–3288 CrossRef CAS.
- Z. Gu, K. Wang, W. Sun, J. Li, S. Liu, Q. Song and S. Xiao, Adv. Opt. Mater., 2016, 4, 472–479 CrossRef CAS.
- X. Ge, J. Liu and L. Sun, Dalton Trans., 2017, 46, 16729–16737 RSC.
- L. Francés-Soriano, M. González-Béjar and J. Pérez-Prieto, in Upconverting Nanomaterials, CRC Press, 2016, pp. 101–138 Search PubMed.
- X. Li, F. Zhang and D. Zhao, Chem. Soc. Rev., 2015, 44, 1346–1378 RSC.
- X. Liu, C.-H. Yan and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1299–1301 RSC.
- B. del Rosal and D. Jaque, Methods Appl. Fluoresc., 2019, 7, 22001 CrossRef CAS PubMed.
- E. Andresen, U. Resch-Genger and M. Schäferling, Langmuir, 2019, 35, 5093–5113 CrossRef CAS PubMed.
- Y.-H. Chien, K. K. Chan, S. H. K. Yap and K.-T. Yong, J. Chem. Technol. Biotechnol., 2018, 93, 1519–1528 CrossRef CAS.
- X. Qin, J. Xu, Y. Wu and X. Liu, ACS Cent. Sci., 2019, 5, 29–42 CrossRef CAS PubMed.
- Y. Liu, X. Meng and W. Bu, Coord. Chem. Rev., 2019, 379, 82–98 CrossRef CAS.
- M. Liras, M. González-Béjar, E. Peinado, L. Francés-Soriano, J. Pérez-Prieto, I. Quijada-Garrido and O. García, Chem. Mater., 2014, 26, 4014–4022 CrossRef CAS.
- M. Yu, F. Li, Z. Chen, H. Hu, C. Zhan, H. Yang and C. Huang, Anal. Chem., 2009, 81, 930–935 CrossRef CAS PubMed.
- L. M. Maestro, E. M. Rodriguez, F. Vetrone, R. Naccache, H. L. Ramirez, D. Jaque, J. A. Capobianco and J. G. Solé, Opt. Express, 2010, 18, 23544 CrossRef CAS PubMed.
- D. Jin and J. A. Piper, Anal. Chem., 2011, 83, 2294–2300 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- A. D. Ostrowski, E. M. Chan, D. J. Gargas, E. M. Katz, G. Han, P. J. Schuck, D. J. Milliron and B. E. Cohen, ACS Nano, 2012, 6, 2686–2692 CrossRef CAS PubMed.
- D. Denkova, M. Ploschner, M. Das, L. M. Parker, X. Zheng, Y. Lu, A. Orth, N. H. Packer and J. A. Piper, Nat. Commun., 2019, 10, 1–12 CrossRef CAS PubMed.
- D. J. Gargas, E. M. Chan, A. D. Ostrowski, S. Aloni, M. V. P. Altoe, E. S. Barnard, B. Sanii, J. J. Urban, D. J. Milliron, B. E. Cohen and P. J. Schuck, Nat. Nanotechnol., 2014, 9, 300–305 CrossRef CAS PubMed.
- C. Chen, F. Wang, S. Wen, Q. P. Su, M. C. L. Wu, Y. Liu, B. Wang, D. Li, X. Shan, M. Kianinia, I. Aharonovich, M. Toth, S. P. Jackson, P. Xi and D. Jin, Nat. Commun., 2018, 9, 4–9 CrossRef PubMed.
- Y. Liu, Y. Lu, X. Yang, X. Zheng, S. Wen, F. Wang, X. Vidal, J. Zhao, D. Liu, Z. Zhou, C. Ma, J. Zhou, J. A. Piper, P. Xi and D. Jin, Nature, 2017, 543, 229–233 CrossRef CAS PubMed.
- X. Peng, B. Huang, R. Pu, H. Liu, T. Zhang, J. Widengren, Q. Zhan and H. Ågren, Nanoscale, 2019, 11, 1563–1569 RSC.
- Q. Zhan, H. Liu, B. Wang, Q. Wu, R. Pu, C. Zhou, B. Huang, X. Peng, H. Ågren and S. He, Nat. Commun., 2017, 8, 1–11 CrossRef CAS PubMed.
- P. S. May and M. Berry, Methods Appl. Fluoresc., 2019, 7, 1–14 Search PubMed.
- L. Francés-Soriano, S. Gonzalez-Carrero, E. Navarro-Raga, R. E. Galian, M. González-Béjar and J. Pérez-Prieto, Adv. Funct. Mater., 2016, 26, 5131–5138 CrossRef.
- L. Francés-Soriano, M. A. Zakharko, M. González-Béjar, P. A. Panchenko, V. Herranz-Pérez, D. A. Pritmov, M. A. Grin, A. F. Mironov, J. M. García-Verdugo, O. A. Fedorova and J. Pérez-Prieto, Chem. Mater., 2018, 30, 3677–3682 CrossRef.
- N. Estebanez, J. Ferrera-González, I. A. Cortez-Cevallos, M. González-Béjar and J. Pérez-Prieto, Adv. Opt. Mater., 2020, 8, 1902030 CrossRef CAS.
- Z. Li and Y. Zhang, Nanotechnology, 2008, 19, 16–21 Search PubMed.
- V. Muhr, S. Wilhelm, T. Hirsch and O. S. Wolfbeis, Acc. Chem. Res., 2014, 47, 3481–3493 CrossRef CAS PubMed.
- C. Wurth, M. Kaiser, S. Wilhelm, B. Grauel, T. Hirsch and U. Resch-Genger, Nanoscale, 2017, 9, 4283–4294 RSC.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- U. Resch-Genger and H. H. Gorris, Anal. Bioanal. Chem., 2017, 409, 5855–5874 CrossRef CAS PubMed.
- X. Liu, R. Deng, Y. Zhang, Y. Wang, H. Chang, L. Huang and X. Liu, Chem. Soc. Rev., 2015, 44, 1479–1508 RSC.
- O. Ehlert, R. Thomann, M. Darbandi and T. Nann, ACS Nano, 2008, 2, 120–124 CrossRef CAS PubMed.
- H. Dong, L.-D. Sun and C.-H. Yan, Chem. Soc. Rev., 2015, 44, 1608–1634 RSC.
- B. del Rosal, D. H. Ortgies, N. Fernández, F. Sanz-Rodríguez, D. Jaque and E. M. Rodríguez, Adv. Mater., 2016, 28, 10188–10193 CrossRef CAS PubMed.
- J. Hodak, Z. Chen, S. Wu and R. Etchenique, Anal. Chem., 2016, 88, 1468–1475 CrossRef CAS PubMed.
- B. Tian, A. Fernandez-Bravo, H. Najafiaghdam, N. A. Torquato, M. V. P. Altoe, A. Teitelboim, C. A. Tajon, Y. Tian, N. J. Borys, E. S. Barnard, M. Anwar, E. M. Chan, P. J. Schuck and B. E. Cohen, Nat. Commun., 2018, 9, 4–11 CrossRef PubMed.
- C. F. Gainer, U. Utzinger and M. Romanowski, J. Biomed. Opt., 2012, 17, 760031–760037 CrossRef PubMed.
- J. Pichaandi, J.-C. Boyer, K. R. Delaney and F. C. J. M. van Veggel, J. Phys. Chem. C, 2011, 115, 19054–19064 CrossRef CAS.
- N. Estebanez, J. Ferrera-González, L. Francés-Soriano, R. Arenal, M. González-Béjar and J. Pérez-Prieto, Nanoscale, 2018, 10, 12297–12301 RSC.
- W. Gao, X. Kong, Q. Han, Y. Chen, J. Zhang, X. Zhao, X. Yan, J. Liu, J. Shi and J. Dong, J. Lumin., 2018, 202, 381–387 CrossRef CAS.
- X. P. Chen, W. J. Zhang and Q. Y. Zhang, Physica B: Condens. Matter, 2011, 406, 1248–1252 CrossRef CAS.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2014, 8, 32–36 CrossRef CAS.
- R. Arppe, I. Hyppänen, N. Perälä, R. Peltomaa, M. Kaiser, C. Würth, S. Christ, U. Resch-Genger, M. Schäferling and T. Soukka, Nanoscale, 2015, 7, 11746–11757 RSC.
- M. Kaiser, C. Würth, M. Kraft, I. Hyppänen, T. Soukka and U. Resch-Genger, Nanoscale, 2017, 9, 10051–10058 RSC.
- Y. Wang, L. Tu, J. Zhao, Y. Sun, X. Kong and H. Zhang, J. Phys. Chem. C, 2009, 113, 7164–7169 CrossRef CAS.
- A. Delvallée, N. Feltin, S. Ducourtieux, M. Trabelsi and J. F. Hochepied, Meas. Sci. Technol., 2015, 26, 085601 CrossRef.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, 723–742 CrossRef PubMed.
- J. S. Aaron, A. B. Taylor and T. L. Chew, J. Cell Sci., 2018, 131, 1–10 CrossRef PubMed.
- S. Bolte and F. P. Cordelières, J. Microsc., 2006, 224, 213–232 CrossRef CAS PubMed.
- J. W. D. Comeau, S. Costantino and P. W. Wiseman, Biophys. J., 2006, 91, 4611–4622 CrossRef CAS PubMed.
- S. V. Costes, D. Daelemans, E. H. Cho, Z. Dobbin, G. Pavlakis and S. Lockett, Biophys. J., 2004, 86, 3993–4003 CrossRef CAS PubMed.
- O. Ramírez, A. García, R. Rojas, A. Couve and S. Härtel, J. Microsc., 2010, 239, 173–183 CrossRef PubMed.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Springer Science+Business Media, New York, 3rd edn, 2006, pp. 98–157 Search PubMed.
- H. C. Gerritsen, A. Draaijer, D. J. van den Heuvel and A. V. Agronskaia, in Handbook of Biological Confocal Microscopy, ed. J. B. Pawley, Springer Science+Business Media, New York, 3rd edn, 2006, pp. 516–534 Search PubMed.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, in Luminiscent and Photoactive Transition Metal Complexes as Biomolecular Probes and Cellular Reagents, ed. K. Kam-Wing Lo, Springer, Berlin Heidelberg, 2015, pp. 205–256 Search PubMed.
- G. Marriott, R. M. Clegg, D. J. Arndt-Jovin and T. M. Jovin, Biophys. J., 1991, 60, 1374–1387 CrossRef CAS PubMed.
- W. Becker, B. Su, A. Bergmann, K. Weisshart and O. Holub, in Multiphoton Microscopy in the Biomedical Sciences XI, ed. A. Periasamy, K. König and P. T. C. So, SPIE, 2011, vol. 7903, p. 790320 Search PubMed.
- J. Lecoq, A. Parpaleix, E. Roussakis, M. Ducros, Y. G. Houssen, S. A. Vinogradov and S. Charpak, Nat. Med., 2011, 17, 893–898 CrossRef CAS PubMed.
- V. K. Ramshesh and J. J. Lemasters, J. Biomed. Opt., 2008, 13, 064001 CrossRef PubMed.
- A. Grichine, A. Haefele, S. Pascal, A. Duperray, R. Michel, C. Andraud and O. Maury, Chem. Sci., 2014, 5, 3475–3485 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, EDX, XRD, UV-vis absorption of UCNHs, SEM, NIR-LSM images of UCNPs and UCNHs, kinetic profile fitting, kinetic parameters of UCNPs and UCNHs, colocalization significance test of UCNHs. See DOI: 10.1039/d1nr00389e |
| ‡ An exception is the time profile of the blue emission in the Er/Yb system because its intrinsic lifetime is relatively fast compared to the feeding processes and, therefore, the time dependence of the UC emission reflects the kinetics of the excited-state energy transfer between the donor and acceptor (ref. 29). |
| § Clarification made by one of the referees of this paper. |
| This journal is © The Royal Society of Chemistry 2021 |