
Recyclable, reprocessable, self-adhered and repairable carbon fiber reinforced polymers using full biobased matrices from camphoric acid and epoxidized soybean oil†
Weiwei
Zhang‡
a,
Jianqiao
Wu‡
b,
Liang
Gao
c,
Baoyan
Zhang
c,
Jianxin
Jiang
*a and
Jun
Hu
 *b
*b
aDepartment of Chemistry and Chemical Engineering, MOE Engineering Research Center of Forestry Biomass Materials and Bioenergy, National Forest and Grass Administration Woody Spices (East China) Engineering Technology Research Center, Beijing Forestry University, Beijing 100083, China. E-mail: jiangjx@bjfu.edu.cn
bBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: jhu@mail.buct.edu.cn
cDepartment of Resin & Prepreg, AVIC Manufacturing Technology Institute Composite Technology Center, Beijing 101300, China
First published on 19th March 2021
Abstract
Carbon fiber reinforced epoxy is the most commonly used carbon fiber composite, and it has superior performance. However, the growing demand for carbon fiber reinforced epoxy results in the huge consumption of petroleum products and intractable disposal problems, which have a heavy burden on the environment and affect the sustainable development of humans. To address this dilemma, in this work, recyclable carbon fiber composites were prepared using full biobased dynamic crosslinked matrices from natural camphoric acid (CPA) and epoxidized soybean oil (ESO) without additional chemical modification. As the dynamic CPA/ESO networks could be topologically rearranged via transesterification reactions (TERs), the composite laminates possessed usable performance at room temperature, and could be easily reprocessed, self-adhered, and repaired at elevated temperatures. In addition, the carbon fibers could be fully recycled via degrading the composites using ethylene glycol, where the recycled fibers maintained nearly 100% of the mechanical properties possessed by virgin samples. This work prepared recyclable carbon fiber composites using full biobased epoxy matrices for the first time, which offered a green and convenient strategy for designing recyclable eco-materials.
Introduction
Carbon fiber reinforced polymers (CFRPs) are a type of advanced composite with superior properties of light weight, strength, stiffness and thermal resistance. They have been applied extensively in many fields such as aerospace, the automobile industry and for sports equipment, and their use is still expanding rapidly into new advanced areas.1,2 With the increasing production of CFRPs, the consumption of carbon fibers (CFs) has increased dramatically and was expected to be 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons in 2020.3 Meanwhile, as the most commonly used composite, epoxy composite consists of epoxy resin matrices and fibers, and its global compound annual growth rate is estimated to reach 7.3% from 2015 to 2022.4 To date, most epoxy resins on the market are from non-renewable resources which leads to a huge consumption of petroleum products in the world.5 In addition, the disposal of large amounts of resin matrices and fibers after using CFRPs has a heavy burden on the environment.6 To overcome these issues, in recent years researchers and industries have started to focus on green chemistry to design eco-friendly composites.7–9
000 tons in 2020.3 Meanwhile, as the most commonly used composite, epoxy composite consists of epoxy resin matrices and fibers, and its global compound annual growth rate is estimated to reach 7.3% from 2015 to 2022.4 To date, most epoxy resins on the market are from non-renewable resources which leads to a huge consumption of petroleum products in the world.5 In addition, the disposal of large amounts of resin matrices and fibers after using CFRPs has a heavy burden on the environment.6 To overcome these issues, in recent years researchers and industries have started to focus on green chemistry to design eco-friendly composites.7–9
Taking this concept and using it in materials’ design, much effort has been devoted to developing biobased epoxy resins as substitutes for petroleum-based products for advanced composites.10–13 For example, Miyagawa et al. have constructed tough CFRPs using a biobased epoxy/clay matrix with 50 wt% epoxidized linseed oil, the flexural strength and modulus of which reached 1.3 and 130 GPa, respectively.10 Yi and co-workers have designed a rosin-based epoxy matrix for the preparation of glass fiber prepreg and composite laminates.11 Because of the rigid structure of rosin, their bending strength, modulus and glass transition temperature (Tg) increased by 44%, 73% and 70 °C, respectively, when compared with those composites constructed from a petroleum sourced matrix. Apparently, these biobased epoxy resins remained as a continuous phase and bonded well with fibers, thus leading to the high performance and superior interface property of the materials. However, they cannot be reshaped and reprocessed because of their irreversible crosslinked structures. Also, they cannot tackle the problem of the environmental problems: the difficulty in removing individual components from their structures to enable recycling at the end of their service life.7
Designing a matrix with dynamic covalent networks is an effective and available strategy to resolve the problems discussed previously, because dynamic covalent networks can be topologically rearranged at high temperatures by bond exchange reactions.14 Vitrimer, invented by Leibler and co-workers in 2011,15–17 is a typical dynamic covalent network which performs like thermosets at room temperature but it can be welded, reprocessed, repaired and recycled at elevated high temperatures.18–20 It has been used in repairing adhesives,21,22 soft actuators,23–25 shape memory polymers,26,27 and 3D printing thermosets,28,29 as well as in the matrix of fiber reinforced composites.30–40 For example, Zhang and co-workers synthesized a malleable polyimine network, and used it to prepare fully recyclable CFRPs by an imine exchange reaction.30 Whereas Qi and co-workers utilized an epoxy-acid matrix to prepare a repairable and recyclable CFRP promoted by transesterification reactions (TERs).31 Inspired by this concept, Zhu and co-workers recently reported vanillin-based dynamic epoxy matrices for preparing CFRPs, the mechanical properties of which were comparable with commercial products.33,34 As these vanillin-derived epoxy matrices can be topologically rearranged via an imine exchange, their CFRPs could be easily recycled, both for carbon fibers and polymer monomers. Nevertheless, some defects still remain in current reported materials: (1) the tedious chemical modification process for raw resin was not avoided, and (2) the imine and acetal networks were sensitive to pH and showed poor acid-resistance, which consequently limited their further applications.
Following this train of thought, the challenge in matrix design for preparing eco-friendly recycled CFRPs is how to construct the robust and general dynamic covalent networks without additional chemical modification of the bioresources. In this case, dynamic ester bonds are preferentially considered because of their thermal, pH, and solvent resistance. It also means that the resulting dynamic networks might be stable and robust at the operating temperatures, as little residual creep could be induced by such a hard bond exchange.41 Meanwhile, natural polybasic acids are the preferred curing agents to crosslink biobased epoxies to avoid chemical modification and to achieve a “general” dynamic network.42,43 Herein, in this research CFRPs were prepared by using a full biobased dynamic epoxy matrix derived from natural camphoric acid (CPA) and epoxidized soybean oil (ESO) via TERs in the presence of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as catalyst (Fig. 1, top). CPA is the oxidation product of bicyclic terpene from the camphor laurel tree, and contains a rigid five-membered ring with two carboxyl groups.44–46 The rigid skeleton of CPA can maintain the stiffness of the polymer chains47,48 whereas the carboxyl groups could react with the epoxy groups, making CPA an ideal epoxy curing agent without additional chemical modification. In addition, as another low-cost, non-toxic, and non-volatile biobased resource, ESO can react with amines,49 carboxylic acid50 and anhydrides51 to construct flexible crosslinked networks, especially for shape memory materials.42,52 By comprehensively studying the dynamic crosslinked networks of CPA/ESO with different stoichiometric ratios (R = carboxylic acid/epoxy group, Fig. 1, middle), a suitable system CPA/ESO (R = 1.25) was selected as the matrix for dip-coating the woven carbon fiber for preparing the CF/CPA/ESO composite (Fig. 1, bottom). Benefiting from the topological rearrangements of TER-based network, the resulting CF/CPA/ESO composite laminates not only possessed a usable performance at room temperature, but could also be easily reprocessed, self-adhered, and repaired at 200 °C. Furthermore, the CF/CPA/ESO can be easily chemical degraded by ethylene glycol (EG) with nearly 100% recycling of the CFs. This work aimed to prepare multi-functional CFRPs from low cost, renewable resources and to recycle CFs from CFRPs materials. It is the first recyclable CFRPs prepared by using full biobased epoxy matrices, which offers a green and convenient strategy to design recyclable eco-materials.
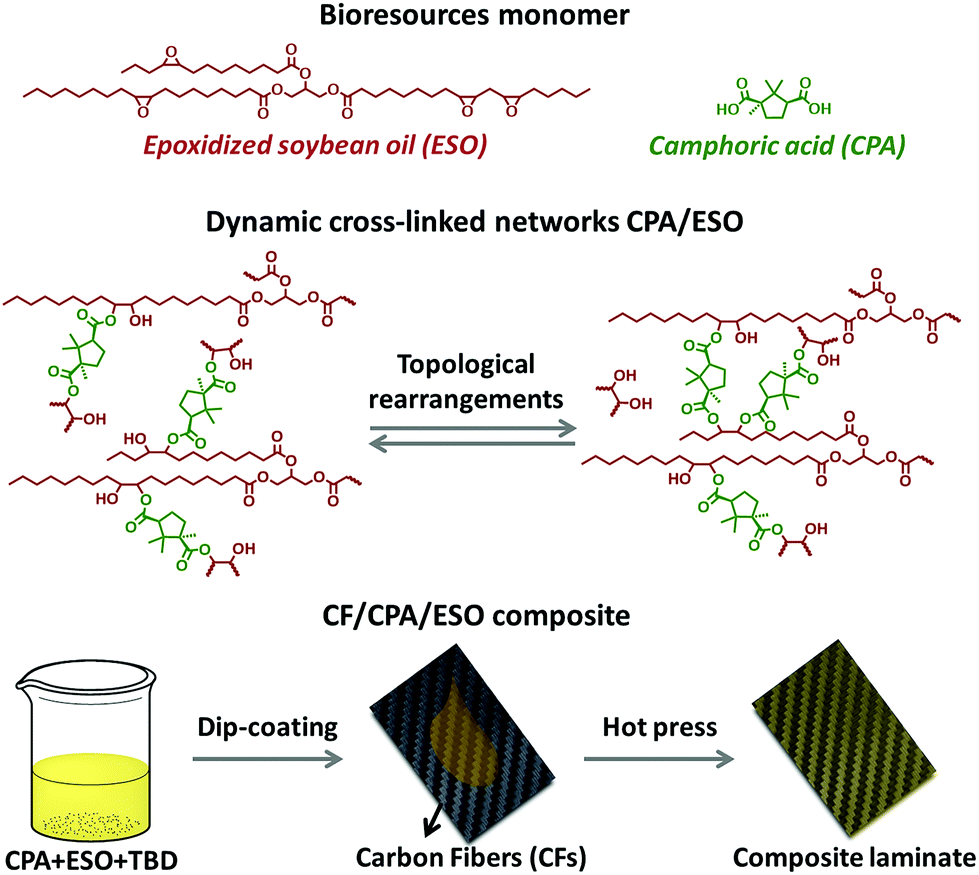 | ||
| Fig. 1 Illustrations of CPA and ESO structures (top), the topological rearrangements of dynamic crosslinked CPA/ESO networks (middle), and the preparation of a CF/CPA/ESO composite laminate (bottom). | ||
Experimental section
Materials
Epoxidized soybean oil (ESO, average functionality = four epoxides per triglyceride, Macklin Biochemical, AR), D-camphoric acid (CPA, Energy Chemical, 98%), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, Ark Pharm, 98%), woven carbon fiber T300 (GW3011, Weihai Tuozhan, China), acetonitrile, and EG were used as received without further purification.Synthesis of dynamic crosslinked networks CPA/ESO
Initially, CPA was added into a mixture of ESO and TBD (5% molar ratio to carboxylic acid) in a beaker at 100 °C. Next, the mixture was poured into a stainless mold (100 mm × 80 mm × 1 mm) at 100 °C, and then placed in a press vulcaniser. Lastly, the transparent, yellow coloured CPA/ESO vitrimer was obtained after curing at 140 °C for 1 h, then at 170 °C for 2 h, and at 200 °C for 4 h (pressure: 10 tons) in a press vulcaniser. The stoichiometric ratios (R = carboxylic acid/epoxy group) were 0.75, 1.00, and 1.25, respectively.Preparation of the CF/CPA/ESO composite
Initially, CPA (0.56 g, 2.78 mmol) was added into a mixture of ESO (1.07 g, 1.13 mmol) and TBD (0.04 g, 0.28 mmol) in a beaker at 100 °C to obtain a homogeneous resin solution. Next, four slices of woven CF were dip-coated in the resin solution layer-by-layer, while the volume fraction of CF was controlled at about 60%. The coated woven CF was then put into a stainless mold (100 mm × 80 mm × 1 mm) and cured at 140 °C for 1 h, then at 170 °C for 2 h, and finally at 200 °C for 4 h (pressure: 10 tons) in a press vulcaniser.Results and discussion
Synthesis and characterization of dynamic crosslinked CPA/ESO networks
The crosslinking of CPA and ESO at different stoichiometric ratios (carboxyl acid/epoxy group, R = 0.75, 1.00, 1.25) were explored in the presence of TBD, which was used to catalyze the TERs. Their curing processes were studied by using differential scanning calorimeter (DSC) with a heating rate of 5 °C min−1 from 50 to 200 °C. As shown in Fig. 2a, broad exothermic peaks at 168, 170, and 165 °C were clearly observed for the mixtures of R = 0.75, 1.00, and 1.25, respectively, which were different from the peaks of the ESO or CPA monomer (Fig. S1, ESI†), which indicated the occurrence of the curing reactions. Moreover, these exothermic peaks shifted to lower temperatures in comparison with those of CPA/ESO in the absence of TBD (Fig. S2, ESI†), which showed that TBD promoted the curing reactions.53 To better determine the curing conditions which should cover the temperature range of the curing reactions and avoid the degradation of the raw materials, 140, 170, and 200 °C were used as the precuring, curing, and post curing temperatures, respectively. The precuring process was used to provide an environment for preventing flash polymerization at the initial time of the crosslinked network formation, whereas the post curing process was utilized to ensure that the crosslinking was complete and to release the internal stress of the materials. Therefore, the curing process was set as “140 °C per 1 h + 170 °C per 2 h + 200 °C per 4 h” at a pressure of 10 tons. After curing, the CPA/ESO networks were confirmed by the FTIR spectra. As shown in Fig. 2b, the epoxy peaks at 823 and 843 cm−1 disappeared, indicating the complete consumption of epoxy groups on ESO.42,43 Moreover, the signal at 1700 cm−1 assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of carboxyl groups on CPA, shifted to 1736 cm−1 after curing, indicating the generation of ester bonds (Fig. S3, ESI†). Note that the peak of the hydroxyl group at 3440 cm−1 was enhanced with the loading of CPA. In the CPA/ESO networks, hydroxyl groups were generated upon the attack of carboxyl groups of CPA on the epoxy groups of ESO during the curing process, and these generated hydroxyl groups can also be consumed by reaction with epoxy to form ether bonds (Scheme S1, ESI†).43,54 Because epoxy was more accessible to react with carboxyl than hydroxyl groups,43 in this case the concentration of hydroxyl groups rose with the increase of CPA.
O of carboxyl groups on CPA, shifted to 1736 cm−1 after curing, indicating the generation of ester bonds (Fig. S3, ESI†). Note that the peak of the hydroxyl group at 3440 cm−1 was enhanced with the loading of CPA. In the CPA/ESO networks, hydroxyl groups were generated upon the attack of carboxyl groups of CPA on the epoxy groups of ESO during the curing process, and these generated hydroxyl groups can also be consumed by reaction with epoxy to form ether bonds (Scheme S1, ESI†).43,54 Because epoxy was more accessible to react with carboxyl than hydroxyl groups,43 in this case the concentration of hydroxyl groups rose with the increase of CPA.
 | ||
| Fig. 2 (a) The DSC curves of the CPA/ESO mixtures at a heating rate of 5 °C min−1. (b) The FTIR spectra of the ESO, CPA, and CPA/ESO networks. (c) The TGA curves of the CPA/ESO networks at a heating rate of 10 °C min−1. (d) Storage modulus (G′, squares) and tanδ (circles) values of the CPA/ESO networks. R = 0.75, 1.00, and 1.25. The concentration of TBD was 5 mol%. | ||
The formulations and thermal properties of CPA/ESO networks are summarized in Table 1. Similar gel contents from 93% to 96% were obtained for all the networks, indicating the formation of crosslinked networks. Their swelling ratios in acetonitrile were 13%, 8% and 4%, which revealed the high crosslinking density of the CPA/ESO networks. Their thermal stability was studied by using a thermogravimetric analysis (TGA), which showed that the temperature at 5% of weight loss (Td5) was 293, 301, and 295 °C for R = 0.75, 1.00 and 1.25, respectively, (Fig. 2c, Fig. S4 (ESI†), Table 1), which indicated their good thermal stability. The hydrolysis resistance is also an important property for materials. Here the hydrolysis properties of CPA/ESO (R = 1.25) were investigated both at room temperature and at 80 °C. The results showed that CPA/ESO (R = 1.25) maintained high gel contents (>95%) and low swelling ratios (<15%) in water both at room temperature and 80 °C, revealing the good hydrolysis resistance of the networks (Fig. S5, ESI†). On one hand, the dynamic crosslinked networks maintained a higher stability than traditional linear polyesters.55 On the other hand, the ester bonds were stable, and exhibited less water sensitivity than other dynamic bonds such as the boron ester bond.56
| Gel contenta (%) | Swelling ratioa (%) |
T
d5
(°C) |
T
g
(°C) |
|
|---|---|---|---|---|
|
a Using acetonitrile as solvent.
b Temperature at 5 wt% loss determined via TGA (Fig. 2c).
c Determined from the tanδ curve obtained from dynamic thermomechanical analysis (DMA) (Fig. 2d).
|
||||
| R = 0.75 | 96 | 13 | 293 | 40 |
| R = 1.00 | 96 | 8 | 301 | 45 |
| R = 1.25 | 93 | 4 | 295 | 48 |
The viscoelastic properties of the CPA/ESO networks were investigated by dynamic thermomechanical analysis (DMA). As shown in Fig. 2d, an definite transition was observed from the glass transition plateau to the rubber plateau for each CPA/ESO network. Note that the storage modulus (G′) ranged from 1800 MPa to 2400 MPa, which was much higher than the reported flexible citric acid curing ESO system (400 MPa to 600 MPa).42 Moreover, the peaks of tanδ at 40, 45, and 48 °C were assigned to their corresponding Tg values. Generally, the rigid skeleton of the monomer makes the polymer chains stiffer, which consequently results in a higher Tg of the polymer networks. In CPA/ESO systems, ESO gave flexible chains whereas CPA gave rigid segments for the polymer networks. As the amount of CPA increased, the CPA/ESO networks became stiffer as their Tg increased from 40 to 48 °C. In addition, the peak value of tanδ increased with the increase of CPA content, and eventually reached 2.3 for the CPA/ESO network with an R of 1.25. This demonstrated that the CPA/ESO networks exhibited excellent damping behavior, because tanδ can be used to evaluate the damping performance of materials, the higher the tanδ value, the better the damping property.57 Furthermore, the wide temperature region of the glass transition (from 2 to 72 °C for R = 0.75, from 5 to 83 °C for R = 1.00, and from 10 to 90 °C for R = 1.25) ensured it would take effect in reality.
Characterization of CPA/ESO network rearrangements
The rearrangements of the CPA/ESO network (R = 0.75, 1.00, and 1.25, TBD 5 mol%) based on TERs were investigated by stress relaxation tests using a rotational rheometer. Their stress relaxation values at 200 °C are shown in Fig. S6 (ESI†), where the modulus (G) dropped as the networks rearranging (Fig. 1, middle). The relaxation times (τ*), determined as the material relaxed to 1/e of the initial modulus (G/G0 = 1/e), were 7036 s, 8244 s, and 7245 s for R = 0.75, 1.00, and 1.25, respectively, which revealed the similar rate of exchange reactions of these three networks. It is known that both the flexibility of polymer chains and the density of hydroxyl groups can promote the TERs.43 In this system, the flexibility of CPA/ESO networks with different R values were closed to each other as their Tg was maintained between 40 to 48 °C (Fig. 2d and Table 1), which dominated the rate of TERs of the CPA/ESO networks.In order to investigate the kinetics of the network rearrangements, CPA/ESO (R = 1.25, TBD 5 mol%) was chosen to be relaxed at 200, 210, 220, 230, and 240 °C, respectively. As shown in Fig. 3a, the τ* was 7245 s (2 h) at 200 °C, 5560 s (1.5 h) at 210 °C, 2909 s (48 min) at 220 °C, 1953 s (33 min) at 230 °C, and 1560 s (26 min) at 240 °C. The drop in relaxation time meant that the TERs were accelerated at high temperatures. Furthermore, the relaxation followed a Maxwell model fitted by an Arrhenius-type equation:
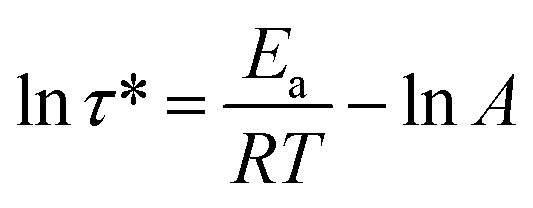 | (1) |
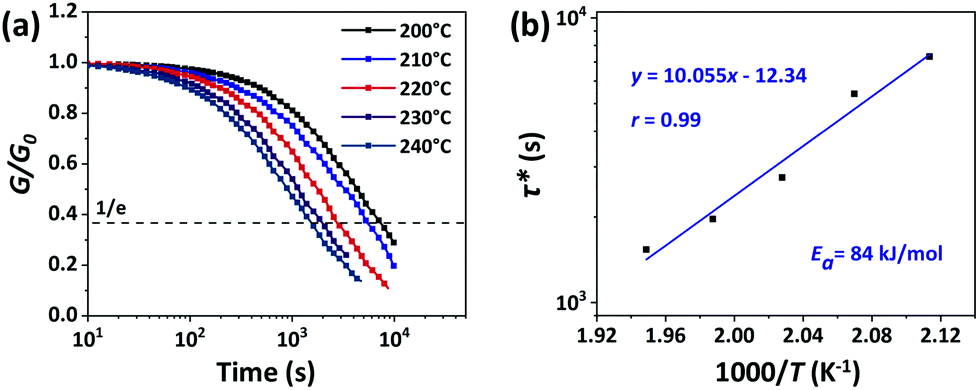 | ||
| Fig. 3 (a) The normalized stress relaxation of CPA/ESO networks (R = 1.25, TBD 5 mol%) at 200, 210, 220, 230, and 240 °C, respectively. (b) Fitting of the experimental values of relaxation times (τ*) derived from (a) to an Arrhenius-type equation. | ||
In general, most of the CFRPs are used for structural materials which often require high mechanical properties, and high thermal and dimensional stability. To obtain a dynamic network capable of acting as the matrix for CFRPs, the stability of matrix should be considered as a matter of priority. This “stability” could be interpreted by the following two features: (1) the ability to resist external stimuli, and (2) the degree of difficulty of starting the network rearrangement. It is known that the rate of stress relaxation is an important indicator for vitrimer materials as it can directly reflect the property of the network rearrangement. Although rapid stress relaxation at a lower temperature benefits the reprocessing of vitrimer materials, it also means poor dimensional stability or creep resistance.41,60,61 For CPA/ESO networks, they are more stable than other systems constructed by boron ester bonds56 or imine bonds33 which are sensitive to water or acid stimuli, because dynamic ester bonds are only sensitive to temperature (Table S1, ESI†). Moreover, the lower relaxation rate of the CPA/ESO networks further revealed their good dimensional and thermal stability (Table S1, ESI†). For better understanding of the slow rate in the CPA/ESO networks, two factors should be mainly considered: (1) transesterification was less thermally sensitive than other exchange reactions such as boron ester bonds and imine bonds, and (2) the insufficient hydroxyl groups on the polymer chains led to a low collision probability between reactants, thus causing the slow exchange rate. As stated previously, CPA/ESO (R = 1.25, TBD 5 mol%) was chosen as a suitable matrix for CFRPs because it maintains good dimensional and thermal stability.
The recycling, reprocessing, welding, and repairing of CPA/ESO networks
Next, the recycling, reprocessing, welding, and repair properties of CPA/ESO networks (R = 1.25, TBD 5 mol%) were demonstrated. To study the recycling properties, the CPA/ESO film was initially cut into pieces, and then reloaded into the mold (Fig. 4a). After a hot press at 200 °C with a pressure of 10 tons, these pieces were reconfigured to form an integral film which maintained a similar transparency to that of the initial CPA/ESO film. It should be noted that the weight of the CPA/ESO networks hardly decreased during the recycling process, which was also supported by TGA results (Fig. 2c, S4 (ESI†)). The efficiency of recycling was studied using the tensile test results for original and recycled samples. As shown in Fig. 4b, the tensile strength and tensile modulus of CPA/ESO materials were 0.56 MPa and 2.4 MPa, respectively, whereas the elongation of the materials was 152% which showed that the toughness of the materials was good. After recycling, the tensile strength, tensile modulus, and elongation of CPA/ESO (R = 1.25, TBD 5 mol%) were 0.46 MPa, 1.8 MPa, and 169%, respectively. That is to say, the recycle efficiency was over 75%, which indicated that the structure of the CPA/ESO network was hardly affected by the topological rearrangements.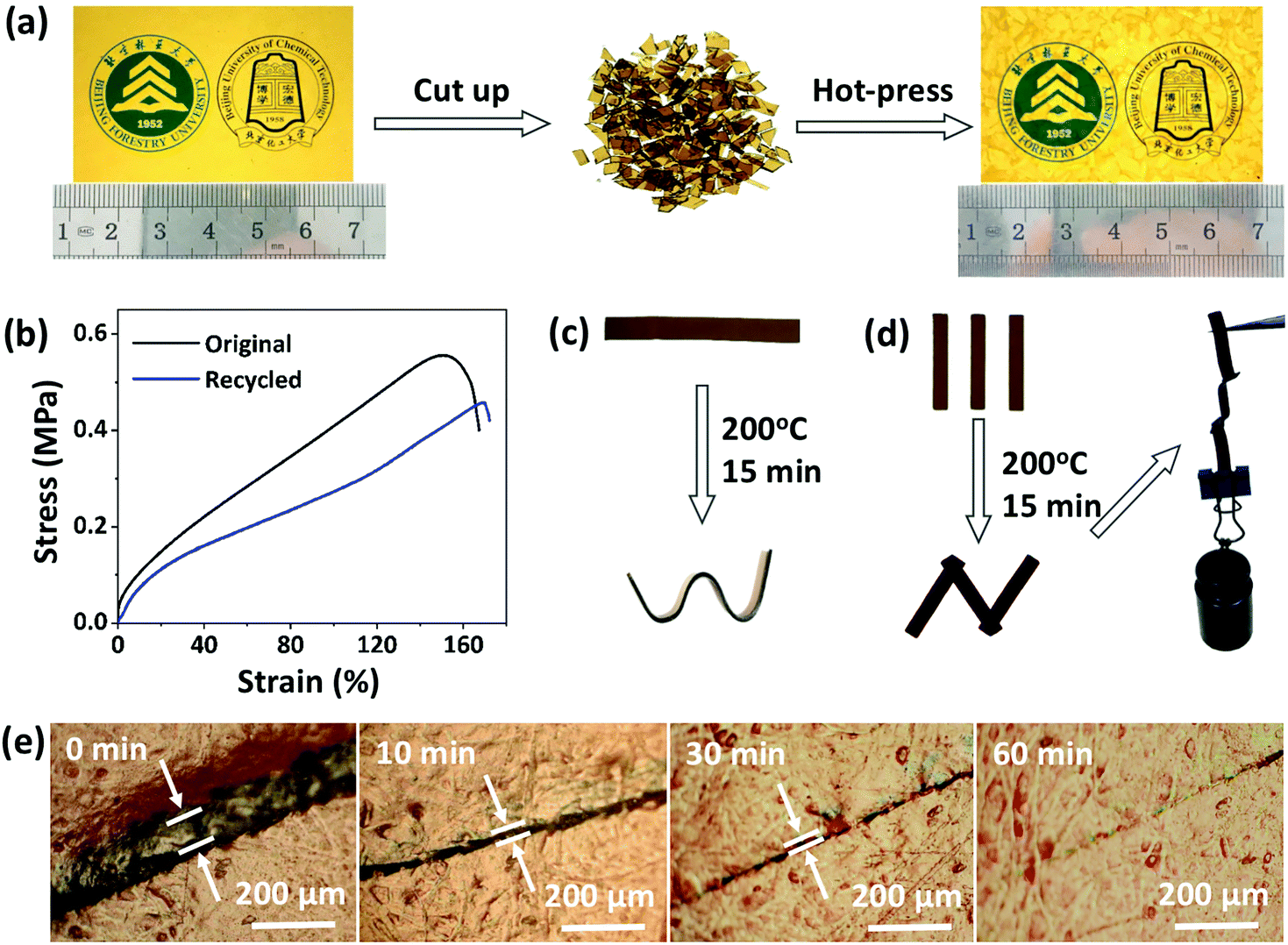 | ||
| Fig. 4 (a) Digital photographs of the recycling process of CPA/ESO networks. (b) Stress–strain curves of CPA/ESO networks at room temperature before and after recycling. (c) Reshaping and (d) welding testing of CPA/ESO networks. (e) Optical microscopy images of the crack repairing of CPA/ESO networks at 200 °C. For (a–e), R = 1.25 and TBD = 5 mol%. | ||
Fig. 4c demonstrates the direct visualization of the materials’ reprocessing. The flat ribbon was fixed in a silicone rubber mold to maintain a shape of “W” initially, followed by heating at 200 °C in an oven. After 15 min, a new shape “W” was obtained because the stress was relaxed by the topological network rearrangements. This permanent shape cannot recover the original shape because of the TER-induced “plasticity deformation”,23,24 exhibiting the “plastic” properties of CPA/ESO networks. The welding property of the CPA/ESO networks is shown in Fig. 4d. Because of the TERs of the networks at the interface, three pieces of CPA/ESO were easily welded to form an integral “N” structure by heating at 200 °C for 15 min without any pressure. This “N” structure could bear 110 g of weight, i.e., 250 times heavier than itself. For the repair property, a piece of CPA/ESO film was scratched so that it had a crack on the surface, followed by the heating at 200 °C in an oven. By observation using an optical microscope, the width of the crack decreased from 60 to 20 μm after 5 min, and then reached 5 μm after 30 min, and had nearly disappeared after 60 min (Fig. 4e), thus, showing the good repair ability of the CPA/ESO materials. Obviously, the topological rearrangement endowed the CPA/ESO materials with excellent recycling, reprocessing, welding, and repair properties, which would be inherited by the CF/CPA/ESO composites and give the multi-functionalities to the composites.
Preparation and characterization of CF/CPA/ESO composites
Carbon fiber composites were prepared layer-by-layer by using CPA/ESO networks (R = 1.25, TBD 5 mol%) as matrices and woven T300 as CFs (Fig. 1, bottom). It should be noted that the fiber volume fraction was controlled at about 60%. After curing, the composite laminates were obtained as shown in Fig. S8 (ESI†). The X-ray 3D microscopy was firstly used to study the inside structures of a 10 mm × 10 mm × 1 mm composite sample (Fig. 5a). The image in Fig. 5a1 is the integral material, whereas the cross profile images of XZ, XY, and YZ of the materials are shown in Fig. 5a2, a3 and a4, respectively. It is obvious that the braided structures can be seen in Fig. 5a1 and a2, which came from the woven carbon fiber. It is worth noting that the CF/CPA/ESO composites exhibit good interface binding, which can be seen from the XY cross profile (Fig. 5a3 and Movie S1, ESI†) and the YZ cross profile (Fig. 5a4 and Movie S2, ESI†). In addition, tensile and flexural tests were performed to study the mechanical properties of the CF/CPA/ESO composites. The tensile strength and modulus of the CF/CPA/ESO composites were 1.6 GPa and 16 GPa, respectively, (Fig. 5b), whereas the flexural strength and flexural modulus of the composites were 65 MPa and 20.6 GPa (Fig. S9, ESI†), respectively. These results indicated that the remarkable enhancement was achieved by the CFs reinforcing the CPA/ESO. Moreover, the thermomechanical tests showed that the storage modulus of CF/CPA/ESO was 24.3 GPa at the glass transition plateau with a Tg of 56 °C (Fig. S10, ESI†), which corresponded well with that of the CPA/ESO networks (R = 1.25, TBD 5 mol%).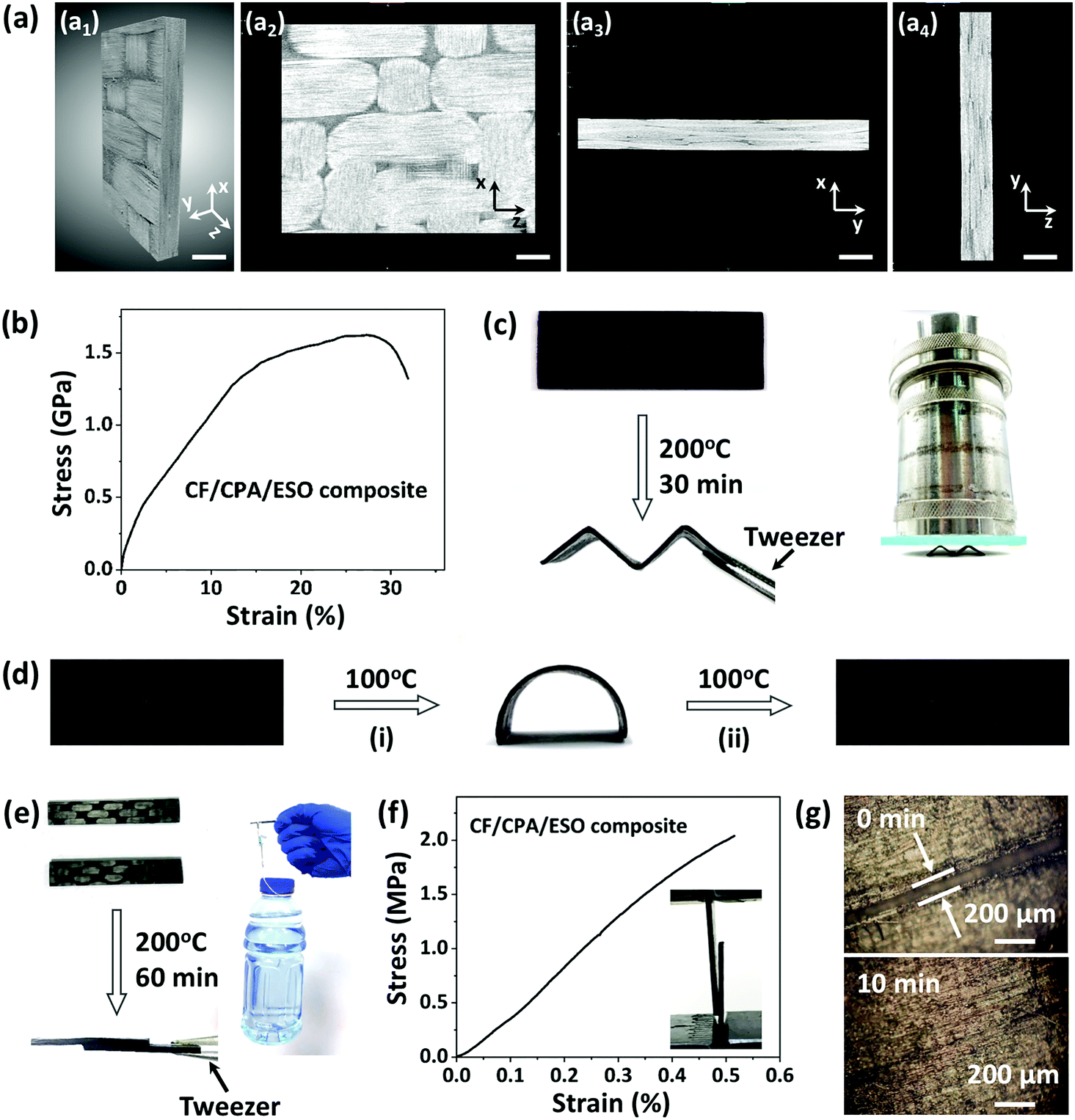 | ||
| Fig. 5 (a) X-ray 3D microscopy images of CF/CPA/ESO composites: (a1) an integral sample; (a2), (a3) and (a4) images of the XZ, XY, and YZ cross profiles, respectively. Scale bar = 1 mm. (b) Stress–strain curve of a CF/CPA/ESO composite obtained from tensile tests. (c) Reprocessing of CF/CPA/ESO composites. The reprocessed composite was loaded with a weight of 2.8 kg. (d) The shape memory of the CF/CPA/ESO composite: (i) a flat sample of CF/CPA/ESO was remoulded into a temporary arc at 100 °C for 60 s and this shape remained when cooled to RT; (ii) the flat shape was recovered upon heating at 100 °C for 150 s. (e) Digital photographs of CF/CPA/ESO composites before and after self-adhering tests. (f) The lap-shear test was used to obtain the stress–strain curve of a welded composite sample; the inset photograph shows the behavior of the welded sample at the end of tests. (g) Optical microscopy images of the crack repairing of CF/CPA/ESO composites at 200 °C. | ||
Traditional thermoset composites, cannot be reprocessed, self-adhered, or repaired during their service life, which limits their formation process and makes them susceptible to different forms of damage. Conversely, the dynamic crosslinked networks can endow the previously describer properties to composites because of their topological rearrangements. To investigate the multi-functionalities of the composites, the reprocessing, shape memory, self-adhering, and repair of the CF/CPA/ESO composites were demonstrated. For reprocessing, a composite laminate was heated and bent to an “M” shape, and was then fixed in a zig-zag mold and heated at 200 °C. After 30 min, the “M” was obtained because of the topological rearrangements of the dynamic networks (Fig. 5c), where it was strong enough to bear the weight of 2.8 kg (≈3500 times its own weight). It could also bear the weight of 1.2 kg (≈1500 times its own weight) for 1 h without deformation and for 4 h with a slight tilt (Fig. S11, ESI†). To quantitatively measure the maximum weight the reprocessed sample “M” could resist, compression tests were performed as shown in Fig. S12 (ESI†). The maximum force was 283 N which is roughly equivalent to 29 kg of weight, and the “M” shape could be recovered after stress removal. To explore the toughness contribution for CFRPs, compression tests for pure CPA/ESO (R = 1.25) were performed. As shown in Fig. S13 (ESI†), it could be compressed to 60% strain and this could also be recovered after withdrawing the stress. Moreover, the stress under 60% strain could recover to 71% after five cycles, showing the elastic properties of the matrices. That is to say, the recoverable behavior of the reprocessed composites was mainly due to the elasticity of the CPA/ESO matrices. It was apparent, that the reprocessable CF/CPA/ESO composites offered the possibility of preparing 3D CFRPs from previously manufactured 2D laminates.
In addition, the CF/CPA/ESO composites exhibited shape memory abilities which were attributed to the glass transition of the CPA/ESO networks. As shown in Fig. 5d, the original flat tape was heated at 100 °C (above the Tg) and then switched to a temporary arc shape. After cooling to room temperature, the arc was fixed because the mobility of the CPA/ESO chain segments decreased. Finally, the arc recovered to its original flat shape after heating at 100 °C again and the stored entropic energy was released. It is worth noting, that this is the first CFRP that simultaneously had a reprocessing ability and shape memory properties when compared with other reported CFRPs constructed with dynamic covalent networks.30–40 In some real scenarios, adhesives are normally used to bind each manufactured part of CFRPs when constructing large-scale components.62 Benefitting from the dynamic nature of CPA/ESO matrices, CF/CPA/ESO composites could self-adhere by applying heat and pressure, thus avoiding the use of adhesives in applications. As shown in Fig. 5e, two CF/CPA/ESO composite laminates were bound together, with half of the parts overlapping, by heating at 200 °C for 60 min. This new structure could bear 500 g of weight which was 1000 times heavier than itself. To further evaluate the effect of the welded CFRPs, lap-shear tests were performed as shown in Fig. 5f. The lap-shear strength was 2.2 MPa, revealing the good self-adhering property of CF/CPA/ESO composites. In addition, the repair property of CF/CPA/ESO composites was also investigated by using optical microscopy. Initially, the composite laminate was scratched so that there was a crack on the surface, and then a drop of EG was used to help the networks repair and then the sample was heated at 200 °C in an oven. The width of the crack was 100 μm initially, whereas it had nearly disappeared after 10 min (Fig. 5g), thus, exhibiting the repair property of CF/CPA/ESO composites. It is worth noting that the crack could also be repaired from 80 to 50 μm without the help of EG (Fig. S14, ESI†). However, the repair efficiency of CF/CPA/ESO composites was lower than that of pure matrices, which might be attributed to the difficulty of networks flowing after binding with carbon fibers. It should be pointed out that extra thermoset powders were often required for repairing CFRPs in reported dynamic matrices,31 which needed a tedious and difficult smashing process. However, the in situ repair was achieved in this system without any additional materials, which greatly avoided the previous complicated process.
Recycling of CF/CPA/ESO composites
To recycle CFs from composites, EG was selected as a reactant and solvent to degrade the networks of the CF/CPA/ESO composites by TERs at high temperatures.31 The mechanism of the degradation process was proposed and is shown in Fig. 6a. Initially, the matrices swelled as the EG entered into the polymer networks. After that, the hydroxyl groups of EG participated in TERs at 190 °C, resulting in the partial degradation of the CPA/ESO networks. Finally, all the ester bonds in the networks and the sizing agents were degraded by EG, which left clean CFs. It is worth noting that the sizing agents usually existed on the surface of the CFs. They bond with the matrices to supply good interfaces and could also be degraded by EG, whereas the limited contents of the sizing agents hardly affected the properties of the recycled CFs.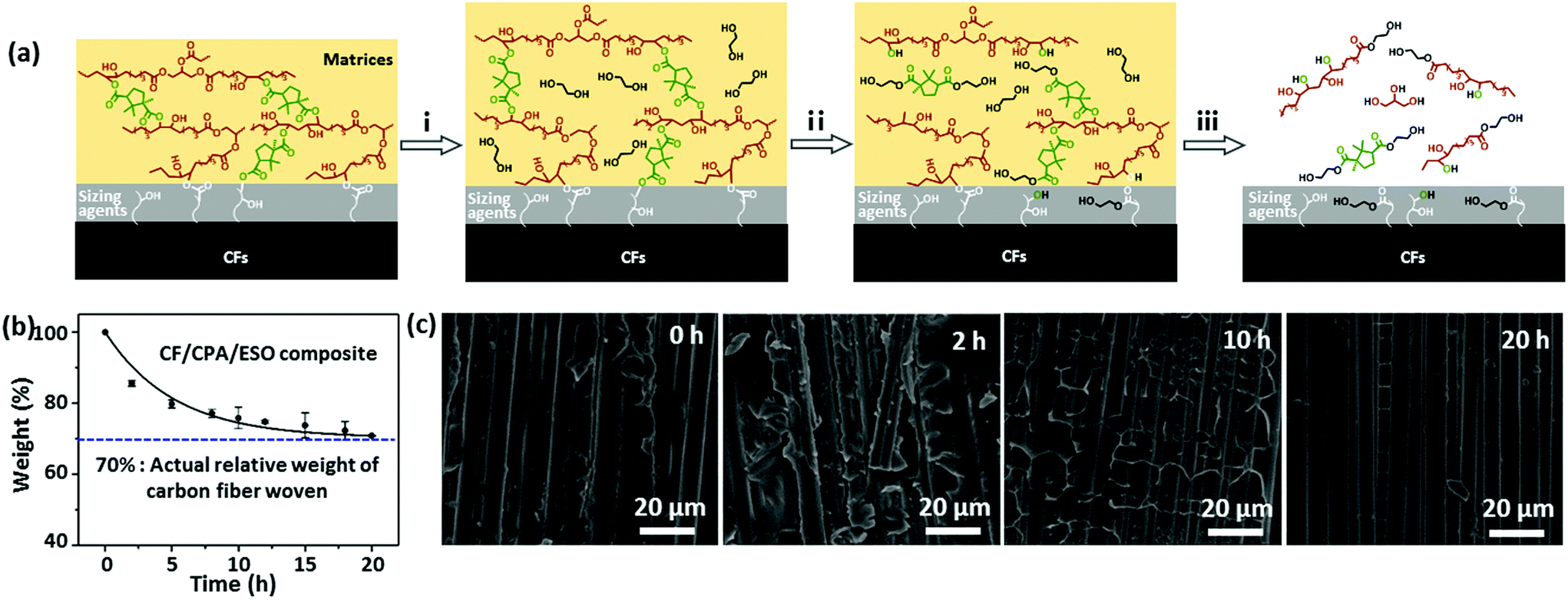 | ||
| Fig. 6 (a) The degradation mechanism of CF/CPA/ESO composites in EG at 190 °C: (i) CPA/ESO matrices swollen in EG at 190 °C; (ii) the CPA/ESO matrices and sizing agents partially degraded with EG, participating in TERs at 190 °C; (iii) the CPA/ESO matrices and sizing agents completely degraded in EG at 190 °C. (b) The degradation kinetics of CF/CPA/ESO composites in EG at 190 °C. The line is fitted with the exponential decay function mt = mf + mre−t/τ, where mt, mf, and mr represent the relative residual composite weight, the relative weight of CFs before degradation (0.70), and the relative weight of matrices before degradation (0.30), respectively, whereas τ (5.15 h) is the characteristic time when mt − mf decreases to 36.8% of mr. (c) SEM images of the cleavage plane of CF/CPA/ESO composites during the degradation process. The degradation times were 0 h, 2 h, 10 h, and 20 h, respectively. | ||
The degradation kinetics of the pure matrices and CF/CPA/ESO composites were investigated in detail by measuring their relative weights at different degradation times. For pure matrices (CPA/ESO, R = 1.25), the weight reduced rapidly to 60% within the first 5 h, and decreased to 15% after 24 h (Fig. S15, ESI†). For CF/CPA/ESO composites, the weight decreased to 86% after the first 2 h, and then reached 76% at 10 h and finally declined to 71%, which approached the actual relative weight (70%) of woven carbon fiber in the CFRPs (Fig. 6b). The faster degradation rate for matrices with CFs compared to the pure one was mainly because of its larger contact area with CFs. Although CF/CPA/ESO had a good interface between the matrices and the CFs, they also provided an interspace for the solvent to enter and react. Scanning electron microscopy (SEM) was used to further monitor the degradation process (Fig. 6c). The cleavage plane of CF/CPA/ESO exhibited a robust interface between the CPA/ESO matrices and the CFs, which corresponded well with the results from the X-ray 3D microscopy shown in Fig. 5a. After 2 h, a small part of the matrices degraded which exposed some of the CFs. Upon degradation for 10 h, a large volume of the matrices depolymerized which resulted in CFs being revealed. Finally, nearly all the matrices were removed from the CFs after 20 h. Following these results, the CFs could be recycled after 20 h.
Additionally, the digital photographs obtained gave direct evidence for the recycling process as shown in Fig. 7a. It was obvious that the solution color deepened as the incubation time increased, with the separation of the CFs layers. After 20 h, the CPA/ESO matrix was completely removed from the CFs. Finally, the CFs were removed from the solvent and cleaned with acetone. The microstructures of the recycled CFs and virgin CFs were studied by SEM and used for evaluating the effect of the recycling process (Fig. 7b). After recycling, the surface of the CFs was clean and smooth, and showed no visible difference to those of the virgin carbon fibers, which indicated the complete removal of the CPA/ESO matrices. To study the chemical structure of virgin and recycled CFs, their Raman spectra were obtained and are shown in Fig. 7c. It was apparent, there was no difference between the virgin CFs and the recycled CFs, revealing that the degradation hardly affected the graphitization structure of the CFs. The mechanical properties of the recycled CFs were measured by using fiber monofilament tensile tests. As shown in Fig. 7d, the strength, modulus, and elongation at break of the recycled CFs were 3.89 GPa, 217 GPa, and 1.60%, respectively, which were close to the values of virgin CF monofilament. In this case, these recycled CFs can be reused for CFRP preparation. In brief, CFs could be recycled from CF/CPA/ESO composites through TER-induced degradation, and these recycled CFs maintained similar original textile structures, surface morphology, chemical structure, and mechanical properties as the ones for the virgin CFs.
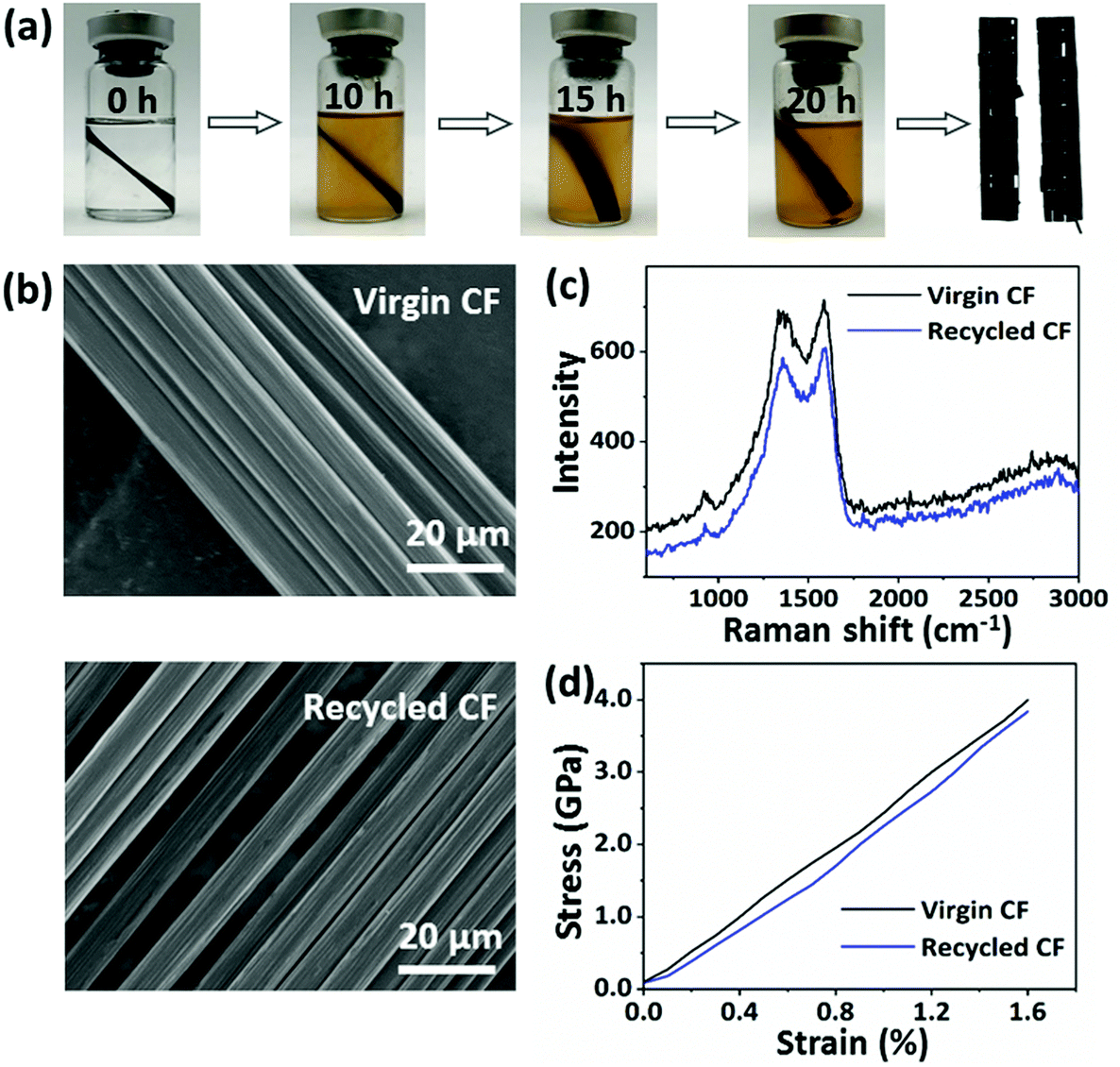 | ||
| Fig. 7 (a) Digital photographs of the recycling process of CF/CPA/ESO composites in EG at 190 °C. (b) SEM images of the surface morphologies of the virgin and recycled CFs. (c) Raman spectra of the virgin and recycled CFs. (d) Tensile stress–strain curves of the virgin and recycled CF monofilaments. | ||
Conclusions
In this work, carbon fiber composites (CF/CPA/ESO) were prepared using full biobased dynamic crosslinked matrices from natural CPA and ESO without additional chemical modification. The dynamic CPA/ESO networks had high crosslinking density and good thermal stability with a Tg range of 40 to 48 °C for different molar ratios of CPA and ESO. The stress relaxation of CPA/ESO at high temperatures revealed the occurrence of TERs, where the Ea value of the reaction was 84 kJ mol−1. The stress relaxation time of CPA/ESO (R = 1.25) was 7245 s at 200 °C and was much longer than those of other dynamic networks using matrices in composites, and revealed the high stability of the CPA/ESO networks. Taking advantage of the network topological rearrangements via TERs, both CPA/ESO matrices and CF/CPA/ESO composites exhibited good reprocessing, self-adhering, and repair properties. Furthermore, carbon fibers could be fully recycled via degrading CF/CPA/ESO composites using ethylene glycol. These recycled fibers maintained a similar original textile structure, surface microscopic morphology, chemical structure, and mechanical properties to virgin samples. In this work recyclable carbon fiber composites were prepared for the first time using full biobased epoxy matrices, which offered a green and convenient strategy for designing recyclable eco-materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Open Fund of National Key Laboratory of Science and Technology on Advanced Composites (KZ42191814) and the National Key R&D Program of China (no. 2017YFD0200302). The authors wish to thank the Composite Testing Technology Center of AVIC Composite Co., Ltd for their help with the X-ray 3D microscopy testing.References
- S. Yao, F. Jin, K. Y. Rhee, D. Hui and S.-J. Park, Composites, Part B, 2018, 142, 241–250 CrossRef CAS.
- N. Forintos and T. Czigany, Composites, Part B, 2019, 162, 331–343 CrossRef CAS.
- G. Oliveux, L. O. Dandy and G. A. Leeke, Prog. Mater. Sci., 2015, 72, 61–99 CrossRef CAS.
- G. M. Roudsari, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2017, 5, 9528–9541 CrossRef.
- N. Hernandez, R. C. Williams and E. W. Cochran, Org. Biomol. Chem., 2014, 12, 2834–2849 RSC.
- A. K. Mohanty, S. Vivekanandhan, J. M. Pin and M. Misra, Science, 2018, 362, 536–542 CrossRef CAS PubMed.
- R. J. Tapper, M. L. Longana, A. Norton, K. D. Potter and I. Hamerton, Composites, Part B, 2020, 184, 107665 CrossRef CAS.
- M. R. Sanjay, P. Madhu, M. Jawaid, P. Senthamaraikannan, S. Senthil and S. Pradeep, J. Cleaner Prod., 2018, 172, 566–581 CrossRef CAS.
- K. L. Pickering, M. G. A. Efendy and T. M. Le, Composites, Part A, 2016, 83, 98–112 CrossRef CAS.
- H. Miyagawaa, R. J. Jureka, A. K. Mohantyb, M. Misraa and L. T. Drzal, Composites, Part A, 2006, 37, 54–62 CrossRef.
- X. Zhang, Y. Wu, J. Wei, J. Tong and X. Yi, Sci. China: Technol. Sci., 2017, 60, 1318–1331 CrossRef CAS.
- N. Hosseini, D. C. Webster and C. Ulven, Eur. Polym. J., 2016, 79, 63–71 CrossRef CAS.
- A. Viretto and J. Galy, Macromol. Mater. Eng., 2018, 303, 1700521 CrossRef.
- B. Wang, S. Ma, S. Yan and J. Zhu, Green Chem., 2019, 21, 5781–5796 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. D. Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- J. Tang, L. Wan, Y. Zhou, H. Pan and F. Huang, J. Mater. Chem. A, 2017, 5, 21169–21177 RSC.
- Q. Zhao, W. Zou, Y. Luo and T. Xie, Sci. Adv., 2016, 2, e1501297 CrossRef PubMed.
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS PubMed.
- Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, J. Am. Chem. Soc., 2016, 138, 2118–2121 CrossRef CAS PubMed.
- X. Lu, S. Guo, X. Tong, H. Xia and Y. Zhao, Adv. Mater., 2017, 29, 1606467 CrossRef PubMed.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. J. Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831 CrossRef PubMed.
- P. Taynton, H. Ni, C. Zhu, K. Yu, S. Loob, Y. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2016, 28, 2904–2909 CrossRef CAS PubMed.
- K. Yu, Q. Shi, M. L. Dunn, T. Wang and H. J. Qi, Adv. Funct. Mater., 2016, 26, 6098–6106 CrossRef CAS.
- A. R. Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabanero, J. Rodriguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, W. Yuan, S. Zhou, S. You and J. Zhu, Green Chem., 2019, 21, 1484–1487 RSC.
- S. Ma, J. Wei, Z. Jia, T. Yu, W. Yuan, Q. Li, S. Wang, S. You, R. Liu and J. Zhu, J. Mater. Chem. A, 2019, 7, 1233–1243 RSC.
- H. Si, L. Zhou, Y. Wu, L. Song, M. Kang, X. Zhao and M. Chen, Composites, Part B, 2020, 199, 108278 CrossRef CAS.
- Y. Liu, J. He, Y. Li, X. Zhao and J. Zeng, Compos. Commun., 2020, 22, 100445 CrossRef.
- H. Memon, Y. Wei, L. Zhang, Q. Jiang and W. Liu, Compos. Sci. Technol., 2020, 199, 108314 CrossRef CAS.
- E. Chabert, J. Vial, J.-P. Cauchois, M. Mihalutac and F. Tournilhac, Soft Matter, 2016, 12, 4838–4845 RSC.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- T. Liu, C. Hao, L. Shao, W. Kuang, L. Cosimbescu, K. L. Simmons and J. Zhang, Macromol. Rapid Commun., 2021, 42, 2000458 CrossRef CAS PubMed.
- G. P. Kar, M. O. Saed and E. M. Terentjev, J. Mater. Chem. A, 2020, 8, 24137–24147 RSC.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Green Chem., 2013, 15, 3360–3366 RSC.
- J. Wu, X. Yu, H. Zhang, J. Guo, J. Hu and M.-H. Li, ACS Sustainable Chem. Eng., 2020, 8, 6479–6487 CrossRef CAS.
- H. Zhang, J. Li, Z. Tian and F. Liu, J. Appl. Polym. Sci., 2013, 129, 3333–3340 CrossRef CAS.
- P. Yang, X. He, M. Li, Q. Ye, J. Ge, Z. Wang, S. Zhu, M. Shao and H. Cai, J. Mater. Chem., 2012, 22, 2398–2400 RSC.
- S. Guo, Z. Geng, W. Zhang, J. Liang, C. Wang, Z. Deng and S. Du, Int. J. Mol. Sci., 2016, 17, 1836–1841 CrossRef PubMed.
- O. Nsengiyumva and S. A. Miller, Green Chem., 2019, 21, 973–978 RSC.
- C. Pang, X. Jiang, Y. Yu, L. Chen, J. Ma and H. Gao, ACS Macro Lett., 2019, 8, 1442–1448 CrossRef CAS.
- D. Ratna, Polym. Int., 2001, 50, 179–184 CrossRef CAS.
- X. Wang, L. Chen, J. Wu, T. Fu and Y. Wang, ACS Sustainable Chem. Eng., 2017, 5, 3353–3361 CrossRef CAS.
- A. E. Gerbase, C. L. Petzhold and A. P. O. Costa, J. Am. Oil Chem. Soc., 2002, 79, 797–802 CrossRef CAS.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, Mater. Des., 2020, 186, 108248–108257 CrossRef CAS.
- T. N. Tran, C. D. Mauro, A. Graillot and A. Mija, Macromolecules, 2020, 53, 2526–2538 CrossRef CAS.
- X. Xu, J. Dai, Z. Ma, L. Liu, X. Zhang, H. Liu, L. Tang, G. Huang, H. Wang and P. Song, Composites, Part B, 2020, 190, 107930 CrossRef CAS.
- S. Sun, L. Wang, P. Song, L. Ding and Y. Bai, Chem. Eng. J., 2017, 328, 406–416 CrossRef CAS.
- S. Wang, X. Xing, X. Zhang, X. Wang and X. Jing, J. Mater. Chem. A, 2018, 6, 10868–10878 RSC.
- X. Feng, J. Fan, A. Li and G. Li, ACS Sustainable Chem. Eng., 2020, 8, 874–883 CrossRef CAS.
- K. Yu, P. Taynton, W. Zhang, L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 48682–48690 RSC.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS.
- Q. Chen, Y. Li, Y. Yang, Y. Xu, X. Qian, Y. Wei and Y. Ji, Chem. Sci., 2019, 10, 3025–3030 RSC.
- Y. Yang, E. M. Terentjev, Y. Zhang, Q. Chen, Y. Zhao, Y. Wei and Y. Ji, Angew. Chem., Int. Ed., 2019, 58, 17474–17479 CrossRef CAS PubMed.
- S. Budhea, M. D. Baneaa, S. de Barrosa and L. F. M. da Silva, Int. J. Adhes. Adhes., 2017, 72, 30–42 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc00648g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |