
CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure: from electronic properties to product selectivity
Yaning
Wang
 a,
Lea R.
Winter
b,
Jingguang G.
Chen
*b and
Binhang
Yan
*a
a,
Lea R.
Winter
b,
Jingguang G.
Chen
*b and
Binhang
Yan
*a
aDepartment of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: binhangyan@tsinghua.edu.cn; Tel: +(86) 010-62797920
bDepartment of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: jgchen@columbia.edu; Tel: +(212) 854-6166
First published on 24th November 2020
Abstract
The production of chemicals and fuels via chemical reduction of CO2 by green H2 represents a promising means of mitigating CO2 emissions. The heterogeneous catalytic reaction of CO2 and H2 under atmospheric pressure primarily produces CO and CH4, while CH3OH and C2+ hydrocarbons are obtained at high pressure. Improving the catalytic selectivity improves the energy efficiency for a given yield and greatly reduces the downstream separation costs. In this work, we review the recent progress in tuning the selectivity of CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure. We describe fundamental insights into the relationships among the electronic properties of active metals, the binding strengths of key intermediates, and the CO2 hydrogenation selectivity. The manipulation of the electronic properties, and consequently the product selectivity, can be achieved mainly by controlling the particle size, bimetallic effects, and strong metal–support interactions. Finally, we discuss challenges and opportunities for the rational design of CO2 hydrogenation catalysts with high activity and desired selectivity.
1. Introduction
In recent decades, global warming has become an increasingly serious problem that affects human survival and development. Rising temperatures have led to many global environmental crises such as the melting of glaciers,1 worsening wildfires,2 and extreme weather.3 There is considerable evidence that the excessive emission of infrared-trapping gases such as carbon dioxide to the atmosphere is indeed the primary culprit for global warming.4,5 The CO2 emissions are mainly contributed by the burning of fossil fuels, and since CO2 remains in the atmosphere for hundreds of years, the concentration of atmospheric CO2 is increasing steadily.6,7 Thus, significant efforts are required to mitigate atmospheric CO2 emissions.Among the strategies for decreasing the concentration of atmospheric CO2, the most common methods are CO2 capture, storage, and chemical utilization. Compared with sequestration, transforming CO2 into commodity chemicals, especially with CO2-free H2,8 not only consumes CO2 but also reduces the use of fossil-derived raw materials. The research of Tackett et al.8 revealed that a process powered by electricity emitting less than 0.2 kg of CO2 per kW per h would achieve a net reduction in CO2, although the high price of renewable energy limits the large-scale application of the CO2 hydrogenation processes at present. Zhang et al.9 have demonstrated that the economic profit can be gained when a suitable carbon tax is implemented and the cost of renewable energy is greatly decreased in the future. Generally, a net CO2 reduction via the hydrogenation processes can be achieved with the availability of low-carbon or carbon-free energy. Nowadays, thermochemical and electrochemical reductions of CO2 represent the two main pathways of CO2 conversion; this perspective article focuses on thermochemical methods. Although the thermal reduction of CO2 is promising, the efficient activation of the thermodynamically stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in CO2 (bond energy = 806 kJ mol−1) poses a major challenge. The development of highly efficient catalysts is crucial to reduce the activation barrier and hence the energy required for converting CO2 into commodity chemicals.
O bond in CO2 (bond energy = 806 kJ mol−1) poses a major challenge. The development of highly efficient catalysts is crucial to reduce the activation barrier and hence the energy required for converting CO2 into commodity chemicals.
Under atmospheric pressure, the products of CO2 thermocatalytic reduction with H2 are CO and CH4, through the reverse water–gas shift (RWGS) reaction and methanation (also known as the Sabatier reaction), respectively:
| CO2 + H2 ↔ CO + H2O ΔH298 °C = +41.2 kJ mol−1 | (1) |
| CO2 + 4H2 ↔ CH4 + 2H2O ΔH298 °C = −165 kJ mol−1 | (2) |
Conversion of CO2 to either product with high selectivity is desired according to specific application requirements. CO can be further converted to valuable chemicals and fuels through well-developed synthesis gas conversion technologies, such as Fischer–Tropsch (FT) synthesis and methanol synthesis. The methanation reaction is widely explored as one of the potential Power-to-Gas (PtG) technologies to foster interaction of gas and electric grids, enabling the methane production (from anthropogenic captured CO2 and renewable generated hydrogen) to be injected into the gas grid or locally used (industrial process, heating system, vehicles).
Other hydrogenation products such as methanol and C2+ hydrocarbons are obtained under high pressure with low yields. There are many similarities between the atmospheric-pressure and high-pressure reactions in the reaction mechanism and catalyst design, although this perspective article mainly discusses the atmospheric-pressure reactions. Most research was aimed at increasing catalytic activity to boost production. However, the demand for selective heterogeneous catalysis has become more apparent in CO2 hydrogenation. It is of paramount importance that a molecular-level understanding of the factors that control the selectivity of a catalytic reaction from the phenomenological or “trial and error” mode into the rational design of catalysts.10 The activity and selectivity of CO2 hydrogenation have been demonstrated to be very sensitive to the catalyst structure.11 Therefore, understanding catalyst structure is paramount to the rational design of heterogeneous catalysts for selective CO2 hydrogenation.
Several articles have reviewed the progress in experimental and theoretical studies of CO2 hydrogenation. Porosoff et al.12 discussed the challenges and opportunities for CO2 hydrogenation into CO, methanol, and hydrocarbons. Aziz et al.13 summarized the progress in the development of various catalysts for CO2 methanation. Daza et al.14 compared RWGS catalysts and the corresponding reaction mechanisms. Chen et al.15 reviewed single-atom catalysts and their extraordinary performance during RWGS. In addition to these reviews of experimental findings, Li et al.16 provided a theoretical overview of CO2 hydrogenation. Several other reviews focus on recent advances in a single CO2 hydrogenation pathway, i.e., CO2 methanation,17–21 reverse water gas shift,22,23 or CO2 to methanol.24–27 In contrast, this perspective article aims to provide insight into recent developments in the competing atmospheric-pressure pathways of CO2 methanation and RWGS. We focus on providing guidance for fine-tuning the electronic properties of active metals in order to enable the design catalysts with high selectivity and activity for CO2 hydrogenation. Accordingly, the basic principles underlying the design and development of nanostructured catalysts also are discussed.
According to the Sabatier principle, the most active catalysts should bind adsorbates with a moderate strength: catalysts showing weak adsorbate binding energy are insufficient for activating reactants, but strong binding energy prevents product desorption. Research on reaction mechanisms has improved the development of a rationale for optimizing catalyst composition. Kattel et al.28 proposed a reaction mechanism based on density functional theory (DFT) calculations (Fig. 1), suggesting that the key intermediate of CO2 hydrogenation via the RWGS + CO-hydrogenation pathway or the direct C–O bond cleavage pathway is adsorbed CO. This mechanism has been verified through in situ characterization. For example, Heine et al.29 used ambient-pressure X-ray photoelectron spectroscopy (AP-XPS) to demonstrate that the methanation reaction over Ni(111) proceeded via dissociation of CO2, followed by reduction of CO to atomic carbon and its subsequent hydrogenation to methane. Zhao et al.30 unraveled the surface reaction mechanism of CO2 hydrogenation on Ru/Al2O3via in situ control of the individual formation and hydrogenation steps for each adsorbed species, using operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) combined with iterative Gaussian fitting. According to their research, CO2 → *HCO3− on the metal-support interface → *CO on the metal surface → CH4 is the dominant reaction step. Martin et al.31 observed the linearly-adsorbed CO species on reduced Rh (Rh–COlin) as the active intermediate in CO2 methanation over Rh/Al2O3. According to their results, the rate-determining step for CO2 reduction and RWGS was the conversion of *HCOO, whereas that for CH4 formation was the hydrogenation of *CO. As shown in Fig. 1, absorbed CO serves as the key intermediate of the CO2 hydrogenation pathways that lead to either CO or CH4 as the final products.
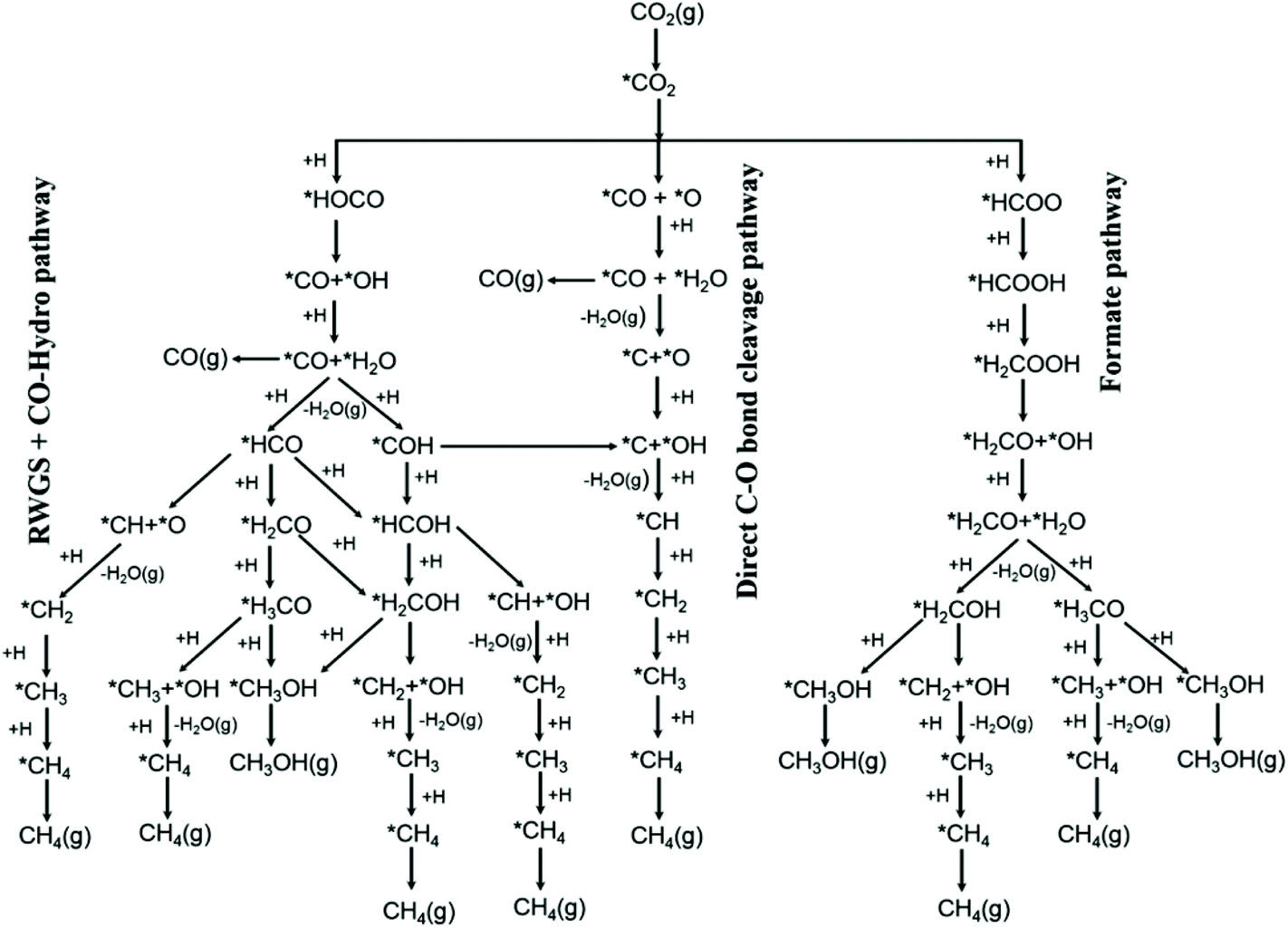 | ||
| Fig. 1 Possible reaction pathways of CO2 hydrogenation to CO, CH3OH, and CH4. *(X) indicates adsorbed species. Reproduced from ref. 28. | ||
In this perspective article, we focus on CO2 hydrogenation pathways for which adsorbed CO leads to selective CO or CH4 production. The key factor that determines the reaction pathway is the activation energy difference between CO desorption and further reaction of adsorbed CO. The adsorption strength of CO is determined by the electronic structure of the catalyst and in particular that of the active metals.32,33 Thus, understanding and controlling the intrinsic electronic properties of the active metal is crucial to the design of catalysts with high selectivity. The electronic properties of the active metal can be tuned through the electron transfer resulting from the formation of bimetallic bonds and interactions with catalyst supports. In this work, we review recent progress in CO2 hydrogenation at atmospheric pressure and discuss the relationship between CO binding energy and the catalytic selectivity. We summarize the effects of intrinsic electronic properties, particle sizes, bimetallic formation, supports, and other related factors on CO2 hydrogenation selectivity. Finally, challenges and opportunities for achieving the rational design of CO2 hydrogenation catalysts with high activity and selectivity will be discussed.
2. Intrinsic electronic properties of metal catalysts
The catalytic performance of supported catalysts is closely related to the adsorption strength and orientation of reactants, intermediates, and products, which are mainly determined by the intrinsic electronic properties of the active metal. For example, Mutschler et al.34 examined a series of pristine transition metals for CO2 hydrogenation and found that Co exhibited high activity and selectivity toward methane. Garbarino et al.35 have investigated the CO2 hydrogenation performance of unsupported Co and Ni nanoparticles. According to their research, unsupported metallic cobalt nanoparticles showed significant activity in CO2 methanation but deactivated upon time on stream due to fast sintering and the formation of encapsulating carbon. Unsupported Ni nanoparticles exhibited weak activity in either the RWGS reaction or the methanation reaction.36,37 In the research of Kim et al.,38 unsupported Fe–oxide NPs (10 to 20 nm) were tested at 873 K, showing 85% CO selectivity and medium CO2 conversion (∼30%). According to the research of Bersani et al.,39 copper nanoparticles yielded mixtures of CO and CH4 in the temperature range of 250 °C to 500 °C. As for precious metals, Panagiotopoulou40 revealed that the activity for CO2 conversion over TiO2-supported metal catalysts followed the order of Pd < Pt < Ru < Rh, where the Ru- and Rh-based catalysts favored methanation and the Pt- and Pd-based catalysts mainly produced CO. In general, the main product of atmospheric-pressure CO2 hydrogenation over Rh-,31,41 Ru-,34,42–44 Co- and Ni-based45–48 catalysts is methane, while the Pd-,49 Fe-,50–52 Pt-,53–55 and Cu-based catalysts favor the RWGS reaction, as shown in Fig. 2.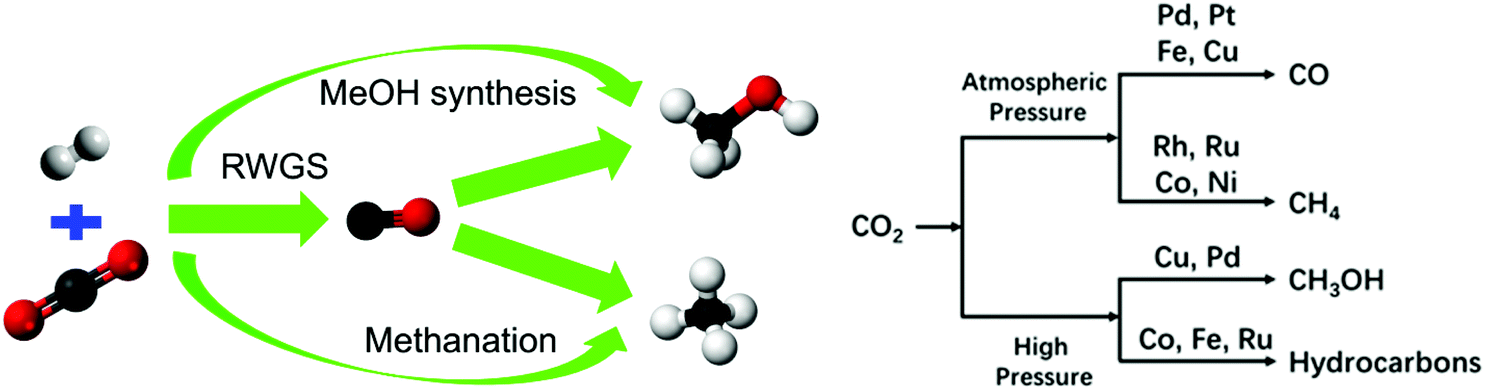 | ||
| Fig. 2 Schematic showing possible products and potential catalysts for CO2 hydrogenation. | ||
A summary of the catalytic performance of several monometallic catalysts is shown in Table 1, revealing that different transition metals exhibit widely different selectivity for CO2 hydrogenation. The electronic properties of metal catalysts, which affect the CO binding energy and the reaction selectivity, can be modified by various means in order to promote desired reaction pathways and inhibit side reactions. The methods for tuning the intrinsic catalyst performance are detailed in the following sections.
| Catalysts | Loading (wt%) | H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 ratio :CO2 ratio |
Temperature (K) | Conversion (%) | CO Selectivity (%) | CH4 Selectivity (%) |
|---|---|---|---|---|---|---|
| Ru/SiO256 |
5 | 9:1 |
473 | 3.2 | 0.6 | 99.4 |
| Ru/Al2O334 |
0.5 | 4:1 |
653 | 74.7 | 1.2 | 98.8 |
| Ru/TiO242 |
0.5 | 4:1 |
623 | ∼21 | 14 | 86 |
| Ru/CeO243 |
1.8 | 4:1 |
573 | ∼83 | 0 | 100 |
| Ru/MSN57 | 5 | 4:1 |
623 | 95.7 | 0 | 100 |
| Rh/SiO231 |
3 | 5:1 |
723 | ∼39 | ∼22 | ∼78 |
| Rh/Al2O341 |
0.2 | 4:1 |
473 | ∼98 | ∼0 | ∼100 |
| Rh/TiO240 |
0.5 | 4:1 |
623 | ∼75 | 0 | 100 |
| Rh/CeO249 |
3 | 5:1 |
623 | ∼46 | 0 | 100 |
| Rh/MSN57 | 5 | 4:1 |
623 | 99.5 | 0 | 100 |
| Ni34 | — | 4:1 |
793 | 54.7 | 20.1 | 79.9 |
| Ni/SiO247 |
6.2 | 4:1 |
723 | 36.8 | 18.2 | 81.8 |
| Ni/SiO245 |
5 | 4:1 |
623 | 27.6 | 11.6 | 85.5 |
| Ni/SiO246 |
10 | 4:1 |
573 | 42.4 | 3.4 | 96.6 |
| Ni/SiO249 |
3 | 5:1 |
723 | ∼45 | ∼54 | ∼46 |
| Ni/SiO256 |
8.2 | 9:1 |
473 | 1 | 0.2 | 99.8 |
| Ni/Al2O349 |
3 | 5:1 |
723 | ∼5 | ∼60 | ∼40 |
| Ni/Al2O356 |
8.2 | 9:1 |
473 | 2.6 | 0.6 | 99.4 |
| Ni/TiO248 |
10 | 4:1 |
623 | ∼42 | ∼24 | ∼76 |
| Ni/MSN57 | 5 | 4:1 |
623 | 85.4 | 0.1 | 99.9 |
| Ni/CeO249 |
3 | 5:1 |
623 | ∼44 | ∼0 | ∼100 |
| Pt/SiO254 |
1.7 | 2:1 |
623 | 9.0 | 98.4 | 1.6 |
| Pt/Al2O355 |
1.0 | 7:10 |
673 | 18 | ∼100 | ∼0 |
| Pt/TiO254 |
1.7 | 2:1 |
623 | 10.2 | ∼100 | ∼0 |
| Pt/CeO253 |
1 | 1:1 |
673 | ∼16 | ∼100 | ∼0 |
| Pd/SiO249 |
3 | 5:1 |
723 | ∼13 | ∼40 | ∼60 |
| Pd/Al2O349 |
3 | 5:1 |
723 | ∼25 | ∼50 | ∼50 |
| Pd/TiO240 |
0.5 | 4:1 |
623 | ∼7 | 100 | 0 |
| Pd/CeO249 |
3 | 5:1 |
723 | ∼51 | ∼87 | ∼13 |
| Fe34 | — | 4:1 |
813 | 26.7 | 84.1 | 15.9 |
| Fe/MSN57 | 5 | 4:1 |
623 | 4 | 8 | 92 |
| Cu/TiO258 |
5 | 3:1 |
493 | 0.5 | 83.6 | 16.4 |
| Cu/ZrO258 |
5 | 3:1 |
493 | 0.5 | 80.2 | 19.8 |
| Cu/MSN13 | 5 | 4:1 |
623 | 3.3 | 21 | 79 |
3. Particle size effect
As shown in Table 1, intrinsic catalytic performance depends on the particle size of the active metal, which correlates with metal loading (it is common for the particles to grow up with the increasing of loading). When the particles decrease to sub-nm size or even to the scale of single atoms, the methanation reaction is inhibited.59In recent years, single-atom and sub-nm catalysts have been studied intensively because of their unique chemical properties and extraordinary catalytic behavior.60 Many studies have revealed that metals that produce a mixture of methane and CO as nanoparticles show suppression of methane generation as single-atoms.61,62 The catalytic performance of single-atom catalysts and metal nanoparticles are listed in Table 2. Compared to metal particles, single-atom catalysts not only utilize the atom more efficiently but also benefit for CO selectivity in RWGS.11
| Catalysts | Loading (%) | Particle size (nm) | H2:CO2 ratio |
Temperature (K) | Conversion (%) | CO Selectivity (%) | CH4 Selectivity (%) |
|---|---|---|---|---|---|---|---|
| Pd/Al2O366 |
10 | ∼2 | 3:1 |
673 | ∼40 | ∼10 | ∼90 |
| Pd/Al2O366 |
0.5 | SA | 3:1 |
673 | ∼20 | ∼62 | ∼38 |
| Ru/Al2O365 |
2 | 2 | 3:1 |
573 | — | ∼12 | ∼88 |
| Ru/Al2O365 |
0.5 | SA | 3:1 |
573 | — | ∼84 | ∼16 |
| Ru/TiO279 |
10 | ∼2.5 | 4:1 |
473 | <5 | <2 | >98 |
| Ru/TiO279 |
0.5 | SA | 4:1 |
473 | <5 | 35 | 65 |
| Ir/CeO269 |
15 | ∼2.5 | 4:1 |
573 | 6.9 | 44 | 56 |
| Ir/CeO269 |
5 | <1 | 4:1 |
573 | 6.8 | >99 | <1 |
| Ir/TiO215 |
5 | ∼2 | 1:1 |
623 | ∼2.1 | 21.8 | 78.2 |
| Ir/TiO215 |
0.1 | SA | 1:1 |
623 | ∼2.1 | ∼100 | ∼0 |
| Pt/CeO270 |
2 | ∼1.5 | 12.5:1 |
723 | — | 0 | 100 |
| Pt/CeO270 |
0.5 | SA | 12.5:1 |
723 | — | 100 | 0 |
| Ni/CeO250 |
1.5 | 0.9 | 3:1 |
623 | 26.8 | 21.6 | 78.4 |
| Ni/CeO250 |
0.5 | 0.6 | 3:1 |
623 | 23.1 | 40.6 | 59.4 |
| Ni/SiO271 |
10 | ∼9 | 1:1 |
623 | ∼10 | ∼10 | ∼90 |
| Ni/SiO271 |
0.5 | SA | 1:1 |
623 | ∼10 | ∼40 | ∼60 |
The underlying properties of single-atom catalysts that give rise to these unique characteristics have been widely studied.63,64 It is generally accepted that the electron density of single-atom catalysts is changed due to the low-coordination environment and strong metal–support interaction. Single-atom catalysts are positively charged (usually noted as Mδ+), leading to a weaker interaction with CO. Kwak et al.65,66 prepared atomically-dispersed Pd/Al2O3 and Ru/Al2O3 catalysts to investigate the influence of particle size on the CO2 hydrogenation process. The results suggested that the RWGS reaction was the dominant reaction pathway over single-atom catalysts. The same trends were obtained using Rh/CeO2 single-atom catalysts.67 Matsubu et al.68 investigated CO2 hydrogenation over Rh/TiO2 with different Rh loadings. They observed a close correspondence between the turnover frequency (TOF) of RWGS and the fraction of isolated Rh sites, as shown in Fig. 3. A similar result was obtained by Li et al.69 As shown in Fig. 4, the CO selectivity increased as the Ir loading amount decreased. In addition to achieving high selectivity toward CO, single-atom catalysts also show extraordinary activity. Wang et al.70 compared the difference between 0.5 wt% Pt single-atom and 2 wt% Pt nanocluster catalysts on CeO2 during CO2 hydrogenation. The activity results indicated that the single-atom catalysts exhibited a more than 7-fold increase in rate compared with the nanoclustered catalyst and with 100% CO selectivity. The authors demonstrated that the weak binding between isolated Pt atoms and CO restricted the further hydrogenation of CO.
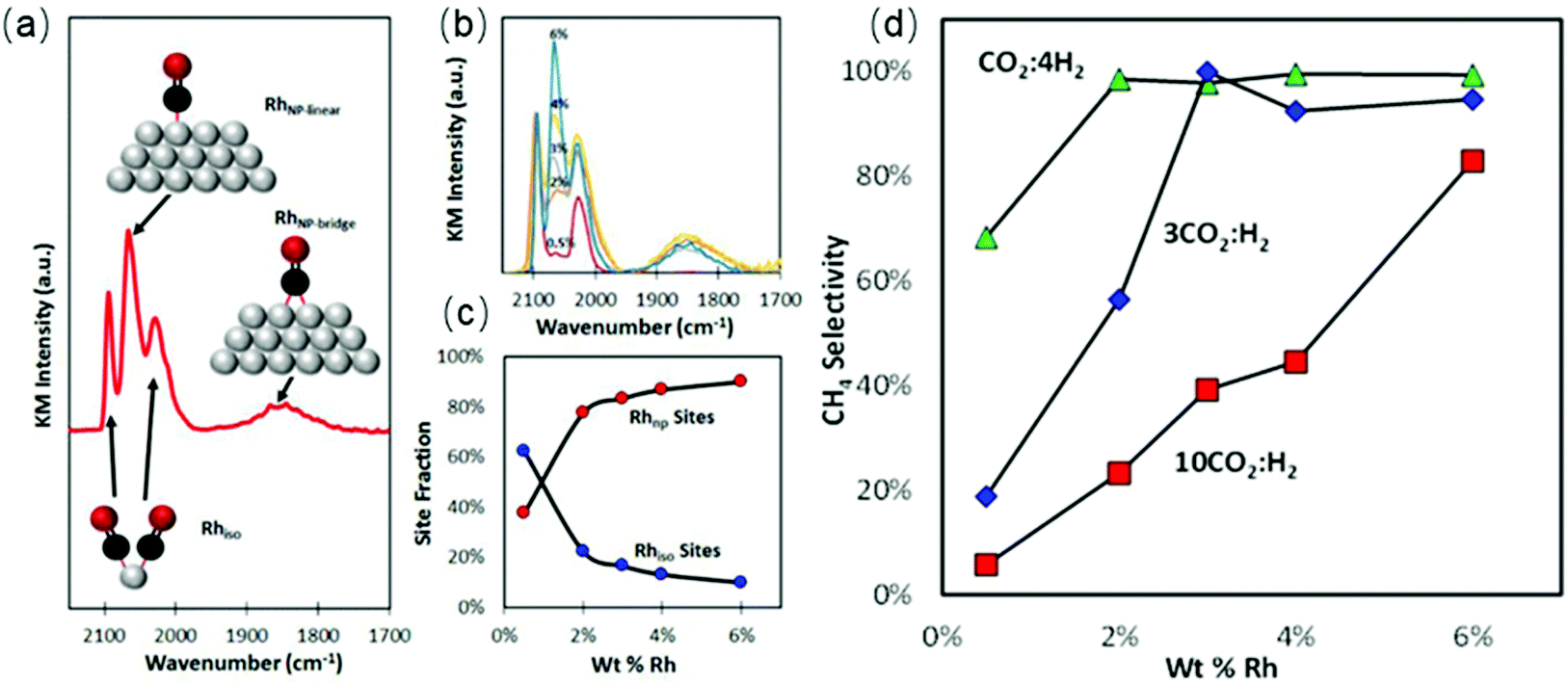 | ||
| Fig. 3 (a) DRIFT spectrum obtained from a saturated layer of CO adsorbed at 300 K on 4% Rh/TiO2. Insets show ball-and-stick models of assigned vibrational modes. (b) DRIFT spectra of CO on all five weight loadings of Rh/TiO2 catalysts. The spectra are displayed in Kubelka–Munk (KM) units and normalized by the symmetric gem-dicarbonyl peak (2097 cm−1) height to allow for comparison. (c) Site fraction (%) of isolated (Rhiso) and nanoparticle-based Rh sites (RhNP) as a function of wt% Rh. (d) CH4 selectivity as a function of wt% Rh for 0.25, 3, and 10 CO2:H2 feed ratios measured at 200 °C. | ||
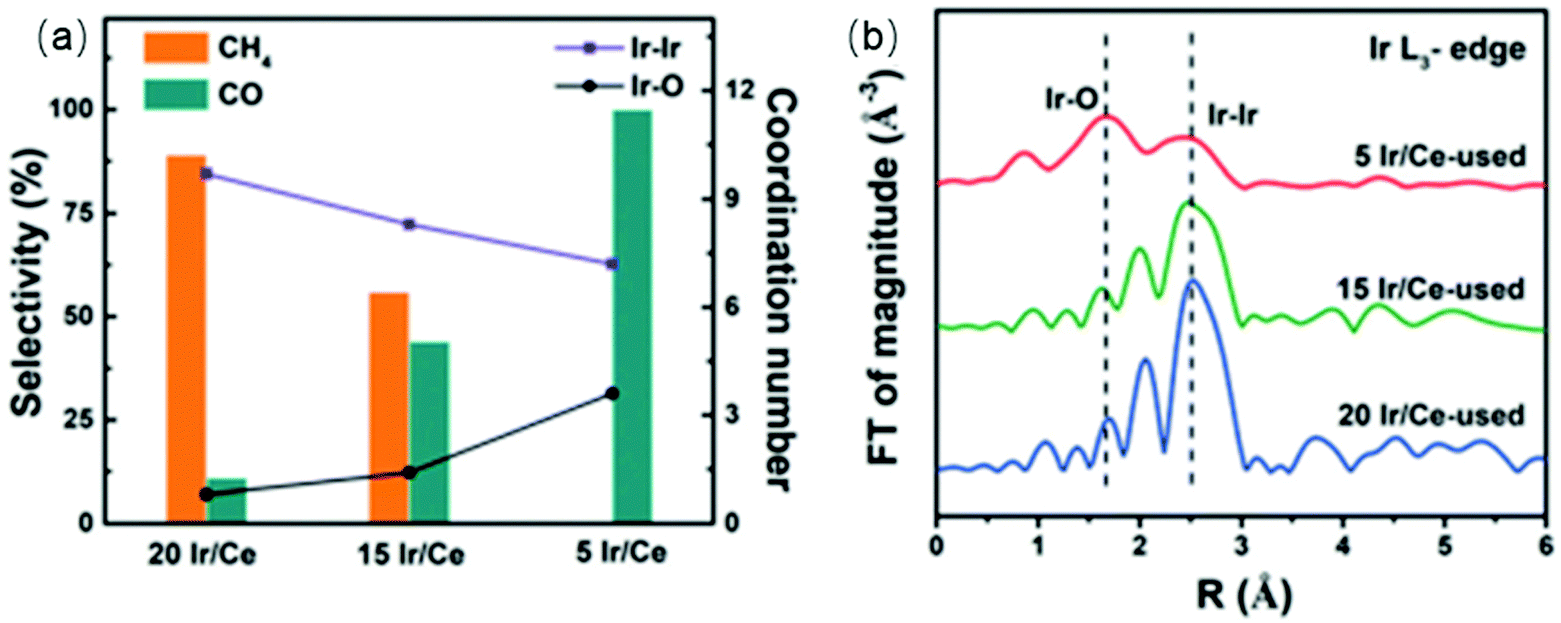 | ||
| Fig. 4 (a) The coordination number of Ir–Ir and Ir–O shells (line graph, right axis) relative to catalyst selectivity (bars, left axis) for CO2 hydrogenation over Ir/Ce catalysts with different Ir loadings. (b) Ir L3-edge Extended X-ray Absorption Fine Structure (EXAFS) of the Ir/Ce-used catalysts. Reproduced from ref. 69. | ||
The same trend that single-atom or sub-nanometer catalysts favor CO formation also has been observed over non-precious metal catalysts. However, the synthesis of highly-dispersed non-precious metal catalysts is much more difficult than the preparation of precious-metal single-atom catalysts. In general, metals are highly dispersed on the support at low metal loadings, whereas the metal particles aggregate at high metal loadings, forming large particles. Therefore, small clusters or single-atom catalysts are usually synthesized by reducing metal loading (≤0.5%).
Applying this method, Wu et al.71 compared the catalytic performance of 0.5 wt% Ni/SiO2 and 10 wt% Ni/SiO2. The authors proposed a consecutive reaction pathway (intermediates → CO → CH4) and a parallel pathway (intermediates → CO and intermediates → CH4). The reactants underwent consecutive pathways over small Ni particles and both consecutive and parallel pathways over larger particles. The consecutive pathway is associated with low H2 coverage on the Ni surface, which leads to the dissociation of formate intermediates and results in CO formation and high CO selectivity. Lu et al.72,73 demonstrated that monodispersed NiO/CeO2 and NiO/SBA-15 catalysts exhibited 100% CO selectivity regardless of the reaction temperature. Gonçalves et al.74 employed magnetron sputtering deposition to prepare small Ni nanoclusters on SiO2 and reached the same conclusions that small Ni particles tended to produce CO rather than methane. The relationship between selectivity and the Ni electronic properties was investigated by Vogt et al.75 Through operando quick X-ray absorption spectroscopy (Q-XAS), the authors suggested that sub-2 nm Ni particles exhibited lower d-band energy or higher electron localization than large nanoparticles, which had a considerable impact on the catalytic activity.
The enhanced CO selectivity using single-atom catalysts also was demonstrated by theoretical calculations. Yan et al.76 illustrated that the RWGS route was more energetically favorable over monolayer Ru sites with a relatively low energy barrier for CO2 activation and CO formation, while methanation was favored over 3D Ru nanoclusters. Chen et al.15 combined experimental data with DFT calculations to investigate single-atom catalysts for CO2 hydrogenation. Single-atom Ir/TiO2 exhibited 100% selectivity toward CO due to the low coordination number of Ir. The authors demonstrated that the desorption energy of CO was lower than the dissociation barriers of the intermediates for single-atom Ir/TiO2, Pt/TiO2, and Au/TiO2 catalysts. As displayed in Fig. 5, the DFT calculation results correlated well with the experimental data over a series of catalysts.
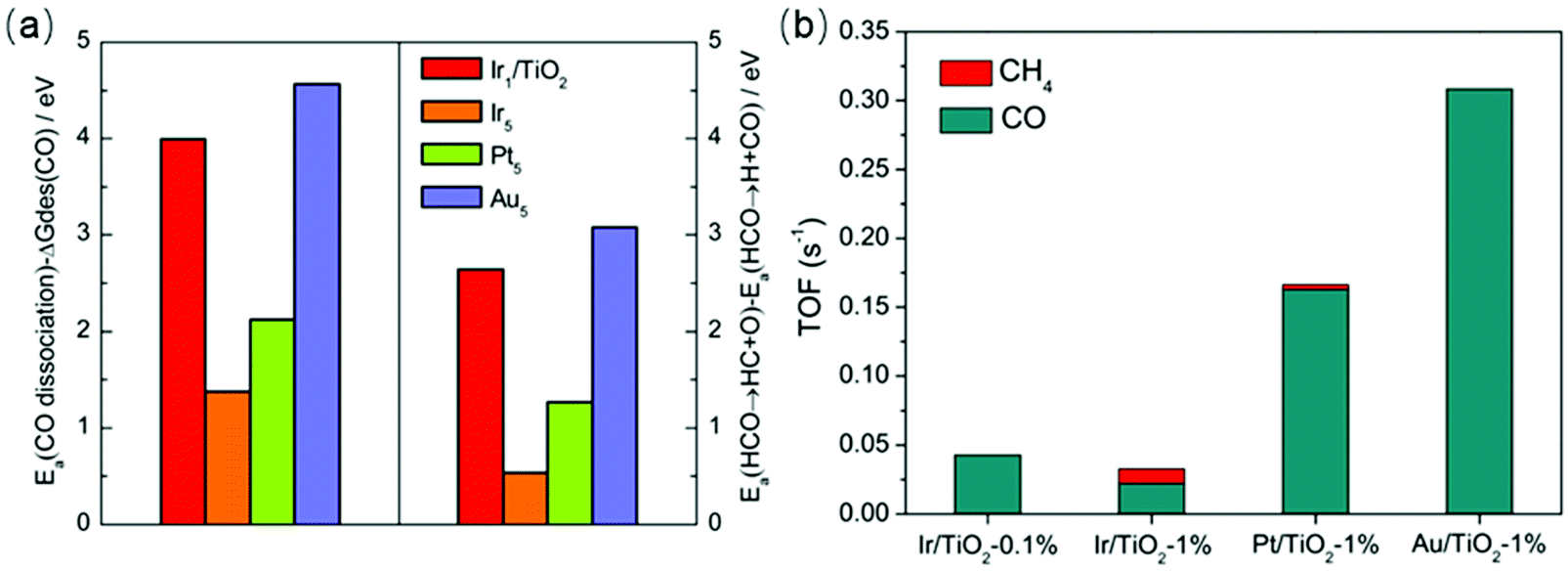 | ||
| Fig. 5 (a) (Left) Difference between activation energies Ea for CO dissociation and desorption free energies of CO. (Right) Difference in activation energies between HCO → HC + O and HCO → H + CO on Ir1/TiO2 and stepped Ir5, Pt5, and Au5 surfaces. (b) Calculated TOFs for RWGS and methanation reactions on Ir/TiO2-0.1%, Ir/TiO2-1%, Pt/TiO2-1%, and Au/TiO2-1% catalysts. Reproduced from ref. 15. | ||
While single-atom catalysts demonstrate promising activity and selectivity for CO2 hydrogenation, the instability of single atoms poses a major challenge for practical applications.76–78 Single-atom active metals tend to agglomerate into larger particles during the reaction, especially at high temperatures. Therefore, the observed decline of catalytic activity and CO selectivity is ascribed to the sintering of the single atoms. Currently, the synthesis of single-atom catalysts with high thermal stability and sintering resistance remains challenging.
Overall, the CO2 hydrogenation selectivity varies with the metal particle size. The methanation selectivity gradually decreases as the particle size decreases until the electronic properties of the metal become altered due to the electron transfer. Notably, when the particle size decreases to the sub-nm and even single-atom scale, the metals become positively charged and the exhibits high CO selectivity.
However, except for the electron properties, there are other factors that have impact on CO2 hydrogenation performance. Control of nanoparticle shape and size will ultimately determine the dominant surface-active terrace, edge, and corner sites. However, the geometric effect of catalysts in CO2 hydrogenation reaction is quite complicated, and different authors have drawn different conclusions. DFT calculations showed a stronger backdonation from the metal atoms to the 2π* antibonding of adsorbed CO on low coordinated surface atoms (corners and edges).80 Martins et al.81 found that high CH4 selectivity was favored in the presence of small particles with a majority of surface sites being edges and corners. However, Loveless et al.82 demonstrated that the corner sites were less active due to the lack of vacancies required for H-assisted hydrogenation paths. The catalytic investigation of Beierlein et al.83 showed that CO2 methanation on Ni–Al2O3 was a structure–insensitive reaction and terrace atoms were the active sites. Additionally, the H-spillover effect also should be taken into accounts. Guo et al.84 demonstrated the methanation activity was determined jointly by strong metal–support interactions and H-spillover effects, suggesting a more complex relationship between particle size and selectivity. The strong metal–support interactions between single Ru atoms and CeO2 suppressed the activation of carbonyls, while the significant H-spillover effects over large Ru nanoparticles greatly hindered the removal of H2O – conditions which are both unfavorable for methanation. As displayed in Scheme 1, the methanation activity reached a maximum over intermediate-sized Ru nanoparticles when both factors were taken into account.
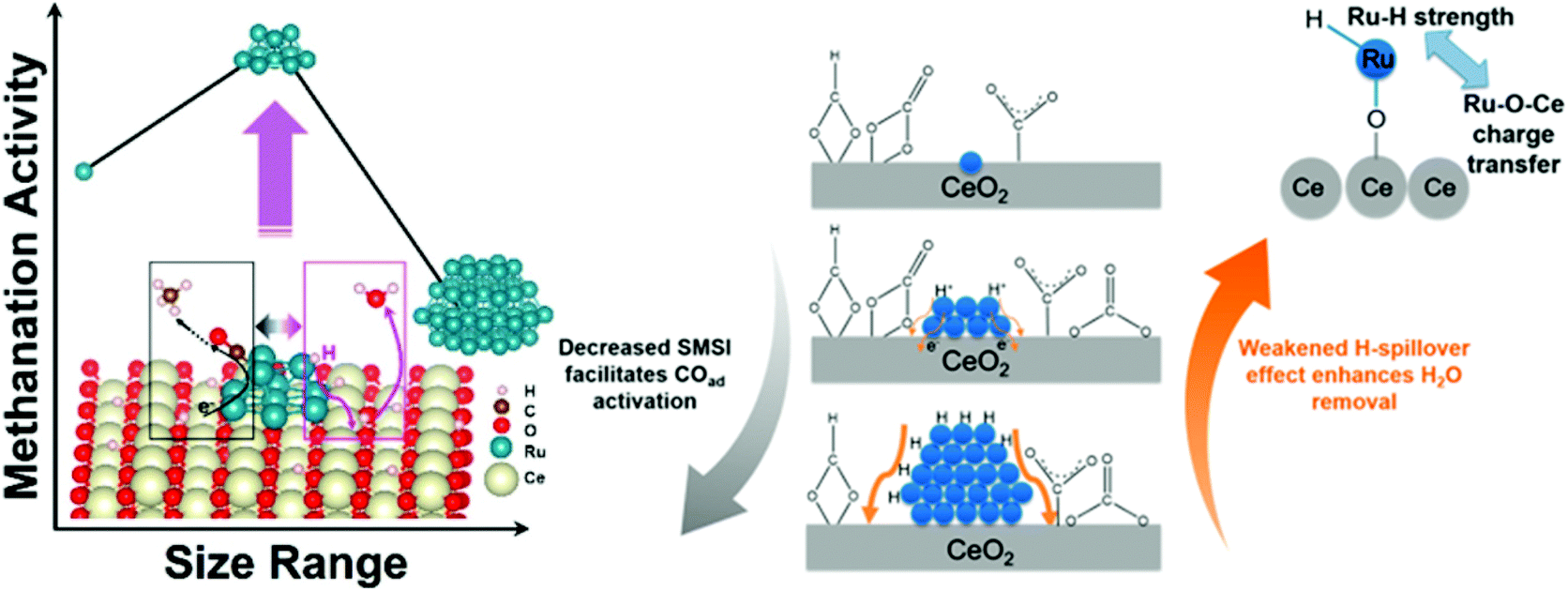 | ||
| Scheme 1 Competitive strong metal–support interactions (SMSI) and H-spillover effect lead to competing CO activation and surface dehydration for CeO2-supported single Ru atoms, Ru nanoclusters, and large Ru nanoparticles. Reproduced from ref. 84. | ||
4. Bimetallic effect
The introduction of a second metal is another effective way to tune the electronic properties of the active metal. The electron transfer between the two metals modifies the electronic properties of the active metal and changes the selectivity of the products. For example, the introduction of typical methanation metals such as Rh, Ru, Co, and Ni to a catalyst improves the methane selectivity. Liu et al.85 found that the bimetallic NiCo/Al2O3 catalysts exhibited enhanced methanation activity and stability compared to monometallic Ni/Al2O3. Xu et al.86 revealed that the addition of Co to Ni/Al2O3 decreased the activation energy of methanation. Bimetallic Ni–Rh/LaAlO3 achieved a 52% enhancement in the methanation turnover frequency [13.9 mol (mol h)−1] compared to Rh/LaNi0.08Al0.92O3 [9.16 mol (mol h)−1].87 Shang et al.88 and Liu et al.89 demonstrated that the addition of Ru boosted the methanation yield of Ni-based catalysts.According to the literature,37,50,90–94 the effect of Fe addition on Ni-based catalysts is quite complicated. Mebrahtu et al.92 reported that CH4 selectivity continuously decreased as the Fe content increased in FeNi bimetallic catalysts. Results from Winter et al.50 showed that a bimetallic FeNi catalyst with a 3:1 molar ratio of Ni to Fe exhibited comparable activity to a monometallic Ni catalyst but improved CO selectivity. The Fe K-edge X-ray absorption near edge structure (XANES) spectra in Fig. 6 revealed that Fe in the bimetallic Ni3Fe1 and Ni3Fe3 catalysts was less oxidized than that in the monometallic Fe3 catalyst. They elucidated that the metallic Ni enhanced H2 dissociation and facilitated spillover of atomic hydrogen, which in turn promoted the reduction of Fe oxides in the Ni–Fe catalysts. The synergetic effect of Fe and Ni in ZrO2-supported bimetallic catalysts was investigated through DFT calculations and in situ DRIFTS by Yan et al.94 As the scanning transmission electron microscopy with electron energy loss spectroscopy (STEM-EELS) analysis displayed in Fig. 7, Fe tended to precipitate on the surface of Ni particles. The theoretical calculations and experimental observations indicated that CH4 was formed mainly via the RWGS + CO-hydrogenation pathway with *CO as a key intermediate. The interaction of *CO with the Ni–ZrO2 interface was revealed to be strong enough to facilitate further *CO hydrogenation to CH4, while the weak interaction of *CO on the Ni–FeOx interface enabled CO desorption as the product.
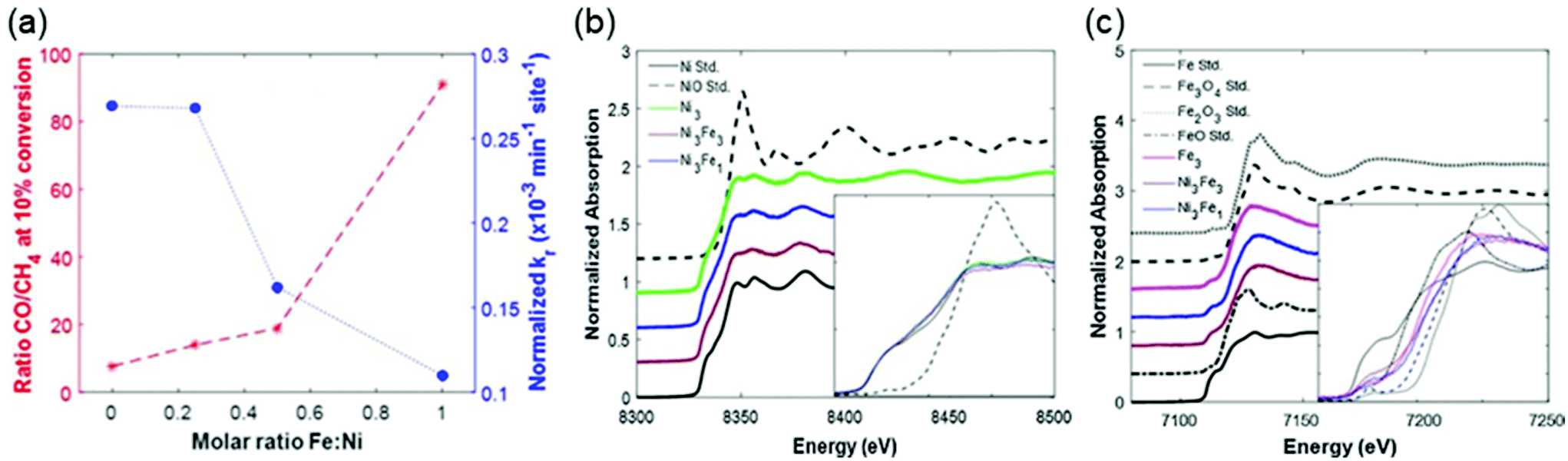 | ||
| Fig. 6 (a) Summary of activity based on reaction rate constants normalized by catalyst mass and number of active sites, and selectivity based on the ratio of CO/CH4 produced at the time of 10% conversion of CO2 by H2, for varying molar ratio of Fe:Ni in bimetallic FeNi/CeO2 catalysts. X-ray absorption near edge structure (XANES) spectra of (b) the Ni K-edge for CeO2-supported Ni3, Ni3Fe3, and Ni3Fe1 catalysts with metallic and oxidized Ni reference standards, and (c) the Fe K-edge for Fe3, Ni3Fe3, and Ni3Fe1 catalysts with metallic and oxidized Fe reference standards. Insets focus on the near edge region with the pre-edge normalized to zero for all spectra. Reproduced from ref. 50. | ||
 | ||
| Fig. 7 Annular dark-field (ADF)-STEM images and corresponding EELS elemental maps of the spent Ni3Fe3/ZrO2 and Ni3Fe9/ZrO2 samples: (a) ADF-STEM image of Ni3Fe3/ZrO2, (b–d) Ni (green), Fe (red), and mixed maps in Ni3Fe3/ZrO2; (e) ADF-STEM image of Ni3Fe9/ZrO2, (f–h) Ni (green), Fe (red), and mixed maps in Ni3Fe9/ZrO2.94 | ||
In addition to changing the electronic properties, the introduction of a second metal also plays an important role in enhancing the metal dispersion and contributes to the activation of the reactants. Mihet et al.95 concluded that the CO2 conversion of Al2O3-supported catalysts was enhanced in the series: Ni–Pd ≥ Ni–Pt > Ni and methane selectivity followed the trend: Ni–Pt ≥ Ni–Pd > Ni. The authors attributed the promotion effect to the enhancement of metal dispersion and the improvement of NiO reducibility. Beaumont et al.96 revealed that the individual Pt nanoparticles next to Co nanoparticles supported on silica enhanced the methanation activity. In situ XANES measurements during the reduction of the Co NPs showed that the addition of Pt NPs significantly enhanced the reduction of Co. The hydrogen atoms dissociated on Pt were transferred to the Co NPs via long-distance hydrogen atom spillover,97 as shown in Scheme 2. The results of DFT calculations conducted by Ou et al.98 provided theoretical insight into the promotion effects of Pt in bimetallic catalysts. The authors found that introducing Pt to Ni(111) facilitated the chemisorption of gaseous carbon dioxide.
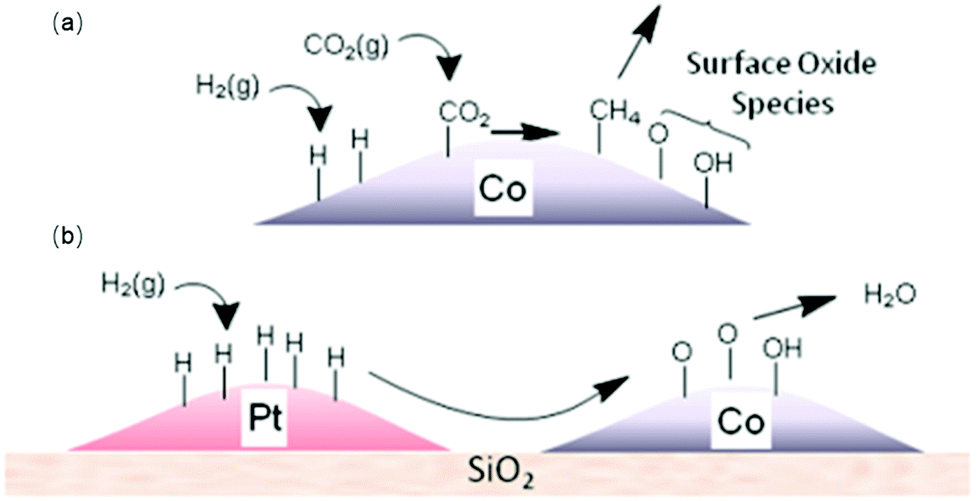 | ||
| Scheme 2 (a) Production of surface oxide species from CO2 hydrogenation and (b) transfer of adsorbed hydrogen from Pt to Co via a spillover mechanism to re-reduce the cobalt oxide surface, releasing water.96 | ||
Alkali and alkaline earth metals have promoter effects on CO2 hydrogenation as well. Bacariza et al.99 tested the effect of the addition of a series of alkaline cations into Ni/USY zeolites. The results showed that the order of improvement of the methanation activity was Cs+ > Na+ > Li+ > K+ for the monovalent cations and Mg2+ > Ca2+ > Ba2+ for the divalent cations. The impregnation of 0.9–2.5% Mg on a 4.8% Ni/USY considerably improved the dispersion of Ni0 and increased the CO2 conversion up to 15% higher than that for the unpromoted 4.8% Ni/USY. Büchel et al.100 investigated the effect of Ba and K addition to Rh/Al2O3 catalysts on CO2 hydrogenation. Below 440 °C, Rh/Al2O3 exhibited 100% CH4 selectivity while K-promoted catalysts shifted to 100% CO selectivity. The change in selectivity was ascribed to the effect of Ba and K on the Rh(0)/Rh(I) ratio and on the adsorption behavior, according to the DRIFTS and CO adsorption measurements. Heyl et al.101 revealed that the addition of K to Rh/Al2O3 affected the adsorption sites of CO on Rh and influenced the catalyst's ability to activate H2. Thus, more facile desorption of adsorbed CO from the catalyst surface was favored and the methanation of CO was hindered. Porosoff et al.102 modified Mo2C/γ-Al2O3 with a K promoter and observed a significant enhancement in CO selectivity and yield. Liang et al.103 and Yang et al.104 provided insights into the influence of K modification by comparing K-promoted Pt/mullite and Pt/L catalysts. Their results revealed that the formation of Pt–O(OH)–K interfacial sites promoted CO desorption, thus preventing the further hydrogenation of CO to methane.
5. Support effect
The catalytic performance of supported catalysts is not only determined by the active metal but also affected by the support materials. Catalyst supports provide certain structural and physicochemical properties that ensure that the active metals remain well-dispersed. During CO2 hydrogenation reactions, the support also participates in the adsorption and activation of CO2. Thus, the reducibility and basicity of the support play an important role in the catalytic behavior. Dreyer et al.105 selected Al2O3 as a representative irreducible support, ZnO and MnOx as representative neutral reducible oxides, and CeO2 as a representative basic reducible support to investigate the influence of support reducibility and basicity on catalytic behavior. The trend of CO2 conversion over the pristine supports followed the strength of the support basicity as determined with CO2 temperature-programmed desorption (TPD), following in the order of ZnO > CeO2 > MnOx > Al2O3. For pristine supports, the CO2 conversion was determined by the ability of the materials to activate CO2. Loading metals onto the supports changed the activity trend due to the interaction between the support and the active metal. After the addition of Ru, the CO2 conversion and CH4 selectivity followed the order of Ru/CeO2 > Ru/Al2O3 > Ru/MnOx > Ru/ZnO. Díez-Ramírez et al.106 studied the effect of the support on Co-catalyzed CO2 hydrogenation. The authors reported that methane selectivity followed the order of Co/CeO2 (96%) > Co/ZnO (54%) > Co/Ga2O3 (53%) ∼ Co/ZrO2 (53%) at 573 K. The enhanced methanation activity of Co/CeO2 catalysts was mainly ascribed to its superior reducibility linked to Co–ceria interactions. According to results from Muroyama et al.,107 CH4 yield over supported Ni catalysts at 523 K followed the trend of Ni/Y2O3 > Ni/Sm2O3 > Ni/ZrO2 > Ni/CeO2 > Ni/Al2O3 > Ni/La2O3 and the catalytic activity could be partly explained by the basic property of the catalysts.In general, reducible metal oxide supports, such as CeO2 and ZrO2, activate CO2 effectively through the metal oxide redox cycles. Since the oxygen vacancies generated by the reducible supports favor CO2 adsorption and activation, many studies have illustrated that employing a reducible metal oxide support improves the catalytic performance.45,109–112 The beneficial effect of Zr and Ce doping on the catalytic activity and stability has been ascribed to the enhancement of the concentration of oxygen vacancies and oxygen mobility.113,114 Winter et al.108 conducted isotope exchange studies using C18O2 in a batch reactor equipped with Fourier transform infrared spectroscopy and mass spectrometry to determine the involvement of surface and lattice oxygen of CeO2 in CO2 hydrogenation reaction. The results in Fig. 8 suggested that oxygen was exchanged by both simple heteroexchange (between one oxygen atom of a gas-phase molecule and one oxygen atom of the oxide support) and multiple heteroexchange (between an oxygen-containing gas-phase molecule and at least two oxygen atoms of the support) mechanisms. The exchange appeared to occur in two steps, where one oxygen atom in CO2 was exchanged rapidly, and the exchange of the second oxygen atom occurred subsequently for some of the singly-exchanged C16O18O molecules. Nie et al.115 provided theoretical insights into the effect of supports (ZrO2 and Al2O3) on the adsorption and activation of key species. DFT results showed that the binding energies of CO2, H2, and CO on Co4/ZrO2(111) are stronger than those on Co4/Al2O3(110), in agreement with experimental TPD results. The reducibility and oxygen mobility of the supports can be promoted further by using a CeO2–ZrO2 solid solution, which has exhibited enhanced activity with respect to CeO2 and ZrO2.116–118
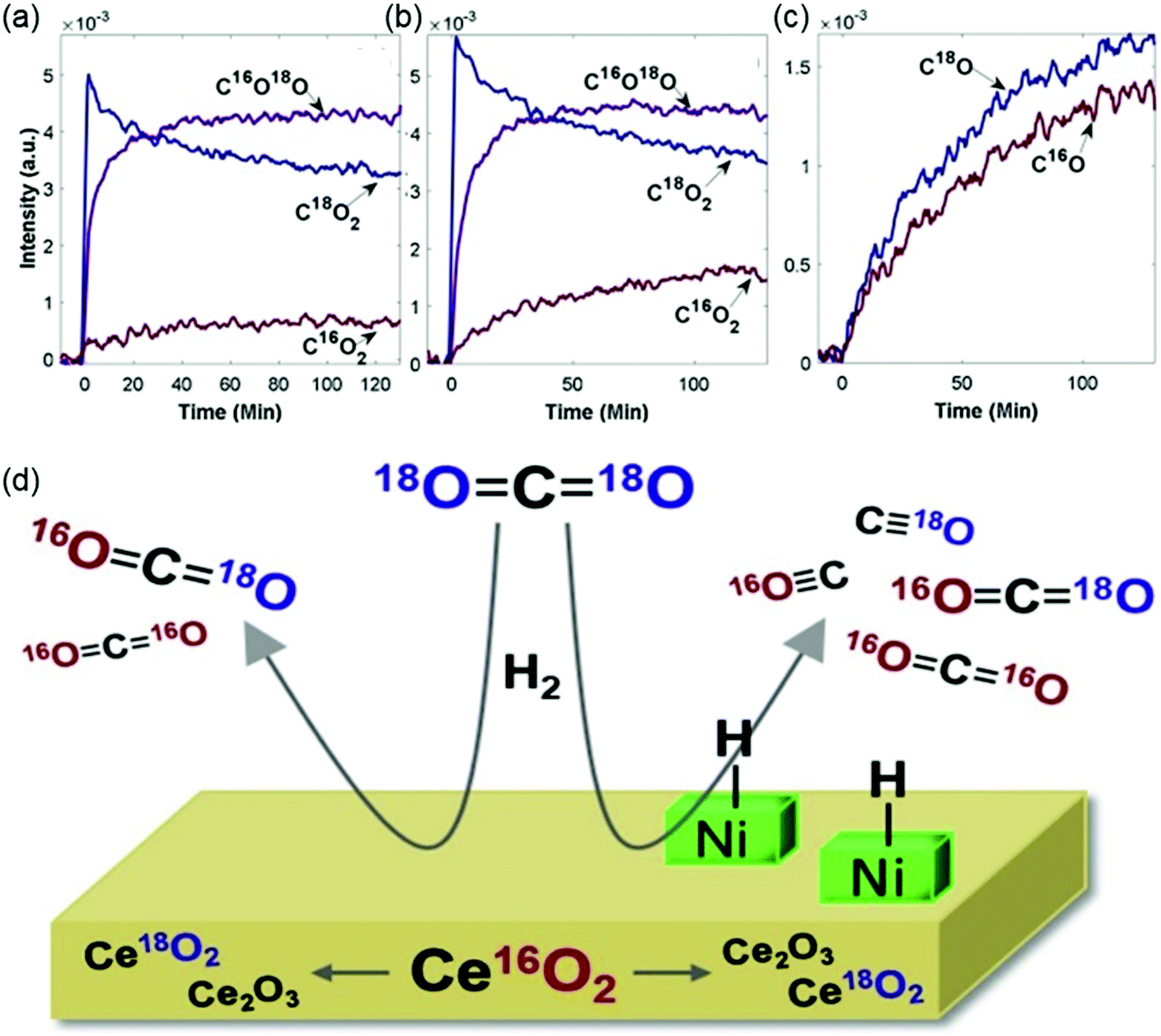 | ||
| Fig. 8 The evolution of CO2 isotope peaks over time during the reaction of C18O2 and H2 over (a) blank CeO2 and (b) 0.5% Ni/CeO2. The separable peaks for each isotope were monitored for C18O2 (3510 cm−1), C16O18O (3583 cm−1), and C16O2 (3729 cm−1). (c) The evolution of CO isotope gas concentrations (monitored for C18O at 2070 cm−1 and for C16O at 2170 cm−1) over time during the reaction of C18O2 and H2 over 0.5% Ni/CeO2. (d) Oxygen exchange between CO2 and CeO2 support (both the surface and lattice oxygen) under CO2 hydrogenation conditions. Reproduced from ref. 108. | ||
The chemical properties of the supports are influenced by many parameters, such as the morphology and the crystal phase. Different morphologies of a support with a specific crystal surface exposed exhibit distinct activity and selectivity.119,120 Sakpal et al.121 compared Ru/CeO2 catalysts with different morphologies in the methanation reaction. Compared to cubes and octahedras, Ru/CeO2 nanorods containing more oxygen vacancies showed higher activity in CO2 methanation. As reported by Lin et al.,122 Ru supported on rutile TiO2 exhibited higher activity than on anatase TiO2. Electron microscopy measurements revealed that RuO2 was prone to sintering on anatase TiO2 but became more dispersed on rutile TiO2.123 Therefore, rutile TiO2-supported catalysts exhibited much higher activity and thermal stability for CO2 methanation than those supported on anatase TiO2.124,125
The preparation methods used for the supports also affect methanation activity. The pretreatment temperature was found to affect the density and strength of the acid sites and methanation activity of Ni/Nb2O5.126 Over Ru/rutile TiO2, the TOF values increased significantly, from 0.26 s−1 to 1.59 s−1, with increasing pretreatment temperature.127 The enhanced methanation activity resulted from the increase in the extent of encapsulation of Ru particles by TiOx layers and in the amount of hydroxyl groups on the TiO2 surface, which facilitated CO2 dissociation. Zhang et al.128 observed that selectivity toward CO over Ir/TiO2 catalysts increased with the reduction temperature, as shown in Fig. 9. Ir/TiO2 catalysts reduced at temperatures lower than 300 °C exhibited complete methanation of CO2 conversion, while samples reduced beyond 600 °C led to complete CO formation. The reduced TiOx species that originated from SMSI played a crucial role in tuning the product selectivity. Moreover, catalysts derived from a metal–organic framework (MOF) template exhibited a specific activity towards methanation.129 Ru/ZrO2 derived from a Ru/UIO-66 material exhibited better methanation activity and selectivity than impregnated Ru/ZrO2 due to the high specific surface area of UIO-66.130
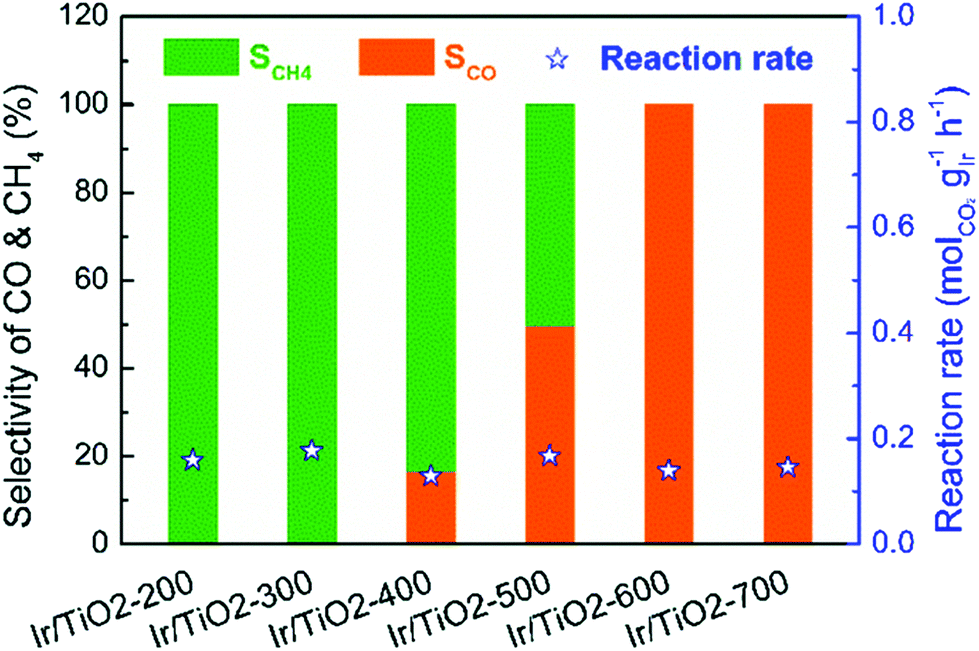 | ||
| Fig. 9 Catalytic performance of the series of Ir/TiO2-x catalysts for CO2 hydrogenation reactions. Reaction conditions: 280 °C, 0.1 MPa, space velocity = 9000 mL h−1 gcat−1, and H2/CO2/N2 = 70/20/10. X refers to the reduction temperature in °C.128 | ||
In summary, support materials influence CO2 hydrogenation activity and selectivity due to their role in the activation of CO2. The basic and reducible metal oxide supports usually show greater activity toward CO2 activation. The supports also contribute to tuning the electronic properties of the active metals through metal-support interactions: the electron transfer between the active metal and support modifies the binding strength of CO. In addition, the specific interfacial sites between the active metal and the support also modify the catalyst selectivity.131–133
6. Other effects
In addition to controlling the materials and structural characteristics of the active metals and supports, there exist other methods for modifying the electronic properties of the catalyst. Electrons generated by the photoelectric effect can improve the electron density. Kim et al.134 revealed that Rh- and Ru-based catalysts exhibited enhanced methane yields under light irradiation. As shown in Fig. 10, the CO2 conversion was 1.6% at 150 °C without light on Ru, but the conversion increased to 32.6% at 150 °C with light. Other metals, such as Pt, Ni, and Cu, showed no difference with or without light irradiation.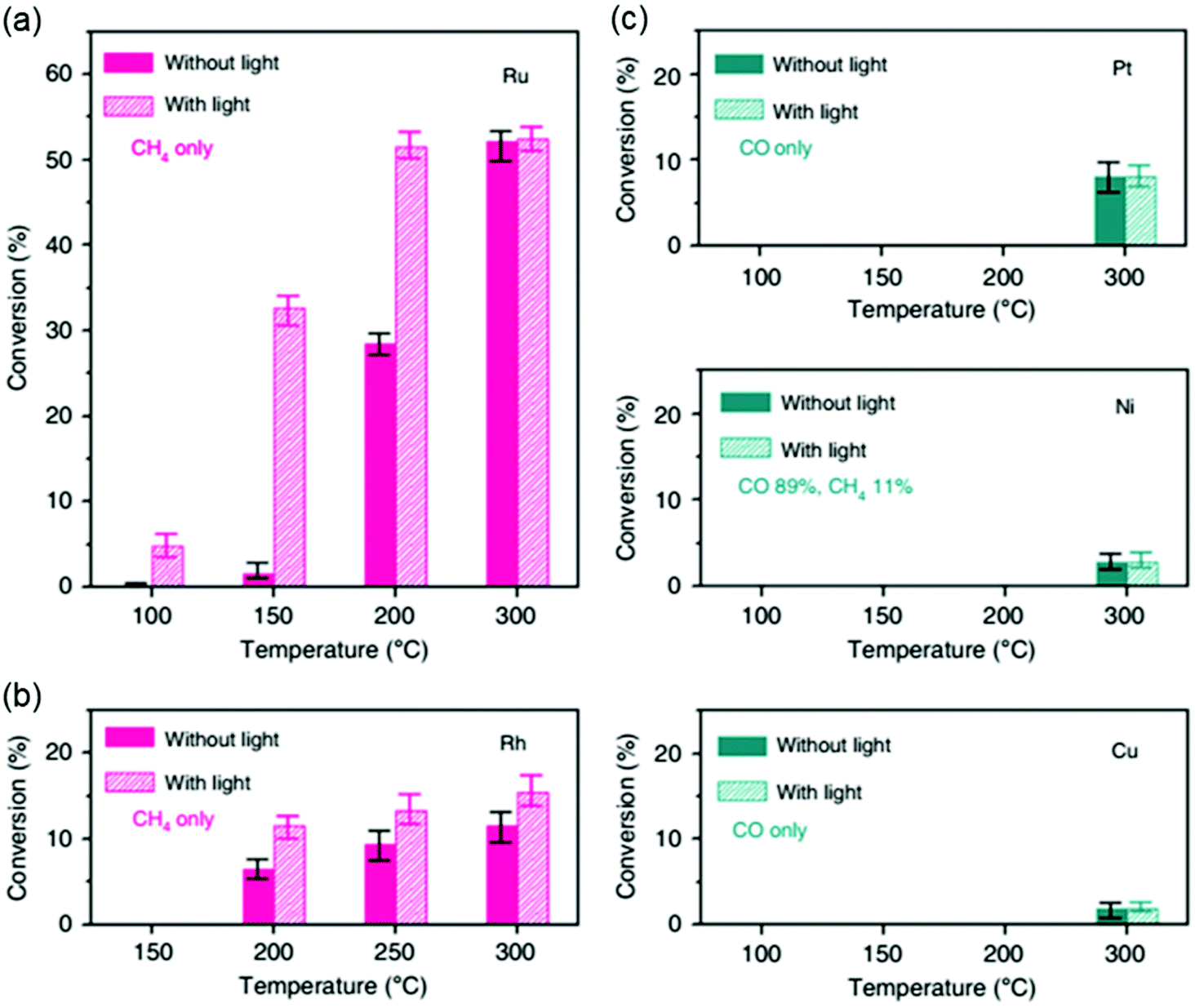 | ||
| Fig. 10 Light enhancement effects on the CO2 hydrogenation activity of metal catalysts. CO2 conversion on (a) Ru, (b) Rh, and (c) Pt, Ni, and Cu catalysts deposited onto silica with (solid bars) and without (hatched bars) light irradiation.134 | ||
In addition to generating emitted electrons from the active metals, the photoelectric effect also can induce electron emissions from the supports. Lin et al.135 exhibited the effect of visible light on methanation over Ru/TiO2. As a result of the photoelectric effect, the TiO2 support generated electrons that were transferred to the Ru particles, increasing the surface electron density of Ru and consequently promoting the activation of CO2. Doping nitrogen into TiO2 further improved the photoelectric effect, thus promoting methanation more effectively under visible light. However, this method is only applicable to materials with a significant photoelectric effect under visible light irradiation.
Another method of tuning the electronic properties is to derive the catalysts with higher valence state from the structured catalysts such as perovskites. Zhao et al.136 reported that the CO2 hydrogenation selectivity could be tuned by controlling the valence state of nickel using lanthanum–iron–nickel perovskites through a doping-segregation process. The higher valence states of nickel weakened the binding of CO, increasing the activation barrier for further CO hydrogenation and leading to a higher CO selectivity than that obtained with metallic nickel.
In addition to the structural and electronic properties of the catalyst, other species involved in the reaction, such as water, can affect the promotion or inhibition of CO desorption. The introduction of water vapor decreased the CO2 conversion because H2O inhibited CO2 activation, reducing the amount of CO adsorbed on Ru sites.137 Therefore, these results suggest that strategies for in situ water removal – such as sorption-enhanced processes or membrane reactors – may improve CO2 conversion and merit further investigation.
Changing the coordination environment of the active metal through targeted synthesis also influences the electronic properties. Arandiyan et al.138 synthesized mesoporous Rh (meso-Rh) nanoparticles by a soft template method to obtain a large distribution of atomic steps. In contrast with Rh nanoparticles prepared by a simple wet chemical reduction, the meso-Rh with low coordination atoms favored methanation over RWGS.
7. Challenges and opportunities
Thermocatalytic reduction of CO2 by green H2 represents a promising strategy for producing chemicals and fuels using CO2 as an environmentally–friendly carbon source. This perspective article presents recent advances in atmospheric-pressure CO2 hydrogenation to CO and CH4 with a focus on the electronic properties of heterogeneous catalysts. The key descriptor of CO2 hydrogenation selectivity is the binding energy of the key intermediate (e.g., CO), which is closely related to the electronic properties of the active metal. The intrinsic properties, particle size, bimetallic effects, and metal–support interactions are critical factors that influence the electronic properties of the active metal and, therefore, the hydrogenation selectivity. Generally, catalysts with high electron density adsorb CO strongly, leading to the further hydrogenation of CO into CH4, while catalysts with low electron density favor CO desorption.For the selective hydrogenation of CO2 over heterogeneous catalysts, significant challenges and research opportunities remain the following areas:
7.1. Identifying key reaction intermediates for tuning selectivity
As discussed earlier, CO is the most important intermediate in the selective catalytic hydrogenation of CO2 to CO and CH4. The product selectivity correlates well with the CO binding energy, which is determined by the electronic properties of the active metal. Catalysts with a weaker CO binding energy favor CO formation (e.g., NiFe/ZrO2, LaFe0.9Ni0.1O3, etc.) while those with a stronger CO binding energy favor CH4 synthesis (e.g., Ni/ZrO2, Ni/CeO2, etc.). For the selective hydrogenation of CO2 to methanol and other hydrocarbons, the identification of key reaction intermediates is critical for controlling the selectivity for the desired product. Therefore, future research efforts should focus on identifying the correct intermediate(s) for promising heterogeneous catalysts.7.2. Stabilizing key intermediates for CH4/CH3OH/hydrocarbons synthesis
For direct CH4/CH3OH/hydrocarbons synthesis at elevated pressure, the stabilization of key intermediates is necessary for increasing the selectivity of the desired product. Therefore, significant work remains for identifying key intermediates and developing catalysts whose interactions with those intermediates facilitate further hydrogenation to the desired products. Future research in this area might aim to identify optimized Mδ− catalysts that selectively promote CO2 to CH4, CH3OH, or long-chain hydrocarbons. Through electron transfers from supports and other metals to the active metal, the realization of Mδ− catalysts is potentially possible, which would improve the catalytic performance for the CO2-to-CH4/CH3OH/hydrocarbons processes. Recently, negatively charged Au species (Auδ−) were found in Au/TiO2 as a result of the electron migration of anatase–TiO2 onto the metal surface due to the SMSI effect.139,140 The Auδ− catalysts exhibited enhanced catalytic activity toward CO oxidation and the water–gas shift reaction. Electron-rich Niδ− species were also observed through electron transfer from Ti and O on the anatase TiO2 surface to atomically dispersed Ni.1417.3. Promoting CO desorption for RWGS
The determination and controlled synthesis of active sites are necessary to enable the development of active, selective, and stable catalysts for scaling up in industrial processes. For the reduction of CO2 by H2, dual-functional catalysts that are active for binding CO2 and for its subsequent hydrogenation are required. Strong metal–support interactions (SMSIs) and metal–oxide interfaces facilitate these mechanisms,152 where a support with strong oxygen affinity enhances CO2 adsorption, and well-dispersed transition metals dissociate H2, which then spill over onto the support to hydrogenate CO2.153–155 Therefore, efforts to improve CO2 hydrogenation catalysts may aim to characterize the active sites and roles of the metal and the support in order to enable controlled synthesis of more effective catalysts.
SMSIs and bimetallic formation can modify the electronic properties of a catalyst, but these properties may be affected by reaction gas atmosphere and temperature. Therefore, accurate characterization of catalysts under reaction conditions (in situ or operando characterization) is crucial for correctly describing the active sites. These accurate descriptions of active sites under reaction conditions can then be used in models (for example, using DFT calculations and Kinetic Monte Carlo simulations) to correlate fundamental descriptors with reaction trends. Furthermore, this characterization is necessary for the identification of appropriate descriptors, such as oxidation state, adsorbed intermediates, and oxygen, CO, or hydrogen binding energy, to be used for predicting more active catalysts. The identification of accurate descriptors through correlating DFT calculations with reaction trends can save significant amounts of catalyst screening time.
The effects of oxide support properties and oxygen exchange on CO2 activation are significant, but the specific mechanisms are not well understood. The oxygen storage capacity and oxygen mobility in reducible metal oxides, particularly ceria, may favor CO2 adsorption onto oxygen vacancies and promote C–O bond scission.156–161 Therefore, future studies may explore the effects of oxygen storage capacity of high surface area reducible metal oxide supports on CO2 reduction under reaction conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors from Tsinghua University acknowledge support from the National Natural Science Foundation of China (NSFC) under Grant No. 21978148 and 21673125. Authors from Columbia University acknowledge support from the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Catalysis Science Program, under Contract No. DE-SC0012704. We would like to thank Dr Qiyuan Wu from Stony Brook University for his suggestions on this perspective article.References
- A. A. Lacis, G. A. Schmidt, D. Rind and R. A. Ruedy, Atmospheric CO2: Principal Control Knob Governing Earth's Temperature, Science, 2010, 330(6002), 356 CrossRef CAS.
- J. Piñol, J. Terradas and F. Lloret, Climate Warming, Wildfire Hazard, and, Wildfire Occurrence in Coastal Eastern Spain, Clim. Change, 1998, 38(3), 345 CrossRef.
- M. M. Q. Mirza, Climate change and extreme weather events: can developing countries adapt?, Clim. Policy, 2003, 3(3), 233 CrossRef.
- D. A. Lashof and D. R. Ahuja, Relative contributions of greenhouse gas emissions to global warming, Nature, 1990, 344(6266), 529 CrossRef CAS.
- A. A. Lacis, G. A. Schmidt, D. Rind and R. A. Ruedy, Atmospheric CO2: principal control knob governing Earth's temperature, Science, 2010, 330(6002), 356 CrossRef CAS.
- P. R. Doose, in The Geochemical Society Special Publications, ed. R. J. Hill, J. Leventhal, Z. Aizenshtat, M. J. Baedecker, G. Claypool, R. Eganhouse, M. Goldhaber and K. Peters, Elsevier, 2004, vol. 9 Search PubMed.
- A. Montenegro, K. Zickfeld, M. Eby, D. Archer, K. J. Meissner and A. J. Weaver, Lifetime of Anthropogenic Climate Change: Millennial Time Scales of Potential CO2 and Surface Temperature Perturbations, J. Clim., 2009, 22(10), 2501 CrossRef.
- B. M. Tackett, E. Gomez and J. G. Chen, Net reduction of CO2 via its thermocatalytic and electrocatalytic transformation reactions in standard and hybrid processes, Nat. Catal., 2019, 2(5), 381 CrossRef CAS.
- J. Zhang, Z. Li, Z. Zhang, K. Feng and B. Yan, Can thermocatalytic transformations of captured CO2 reduce CO2 emissions?, Appl. Energy, 2021, 281, 116076 CrossRef CAS.
- G. A. Somorjai and C. J. Kliewer, Reaction selectivity in heterogeneous catalysis, React. Kinet. Catal. Lett., 2009, 96(2), 191 CrossRef CAS.
- X. Su, X. F. Yang, Y. Huang, B. Liu and T. Zhang, Single-Atom Catalysis toward Efficient CO2 Conversion to CO and Formate Products, Acc. Chem. Res., 2019, 52(3), 656 CrossRef CAS.
- M. D. Porosoff, B. Yan and J. G. Chen, Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities, Energy Environ. Sci., 2016, 9(1), 62 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, CO2 methanation over heterogeneous catalysts: recent progress and future prospects, Green Chem., 2015, 17(5), 2647 RSC.
- Y. A. Daza and J. N. Kuhn, CO2conversion by reverse water gas shift catalysis: comparison of catalysts, mechanisms and their consequences for CO2conversion to liquid fuels, RSC Adv., 2016, 6(55), 49675 RSC.
- X. Chen, X. Su, H.-Y. Su, X. Liu, S. Miao, Y. Zhao, K. Sun, Y. Huang and T. Zhang, Theoretical Insights and the Corresponding Construction of Supported Metal Catalysts for Highly Selective CO2 to CO Conversion, ACS Catal., 2017, 7(7), 4613 CrossRef CAS.
- Y. Li, S. H. Chan and Q. Sun, Heterogeneous catalytic conversion of CO2: a comprehensive theoretical review, Nanoscale, 2015, 7(19), 8663 RSC.
- M. Younas, L. Loong Kong, M. J. K. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Recent Advancements, Fundamental Challenges, and Opportunities in Catalytic Methanation of CO2, Energy Fuels, 2016, 30(11), 8815 CrossRef CAS.
- W. Wei and G. Jinlong, Methanation of carbon dioxide: an overview, Front. Chem. Sci. Eng., 2011, 5(1), 2 CrossRef.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Supported Catalysts for CO2 Methanation: A Review, Catalysts, 2017, 7(12), 59 CrossRef.
- W. J. Lee, C. Li, H. Prajitno, J. Yoo, J. Patel, Y. Yang and S. Lim, Recent trend in thermal catalytic low temperature CO2 methanation: A critical review, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.02.017.
- A. Solis-Garcia and J. C. Fierro-Gonzalez, Mechanistic Insights into the CO(2) Methanation Catalyzed by Supported Metals: A Review, J. Nanosci. Nanotechnol., 2019, 19(6), 3110 CrossRef CAS.
- M. Zhu, Q. Ge and X. Zhu, Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts, Trans. Tianjin Univ., 2020, 26(3), 172 CrossRef CAS.
- X. Su, X. Yang, B. Zhao and Y. Huang, Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: recent advances and the future directions, J. Energy Chem., 2017, 26(5), 854 CrossRef.
- I. U. Din, M. S. Shaharun, M. A. Alotaibi, A. I. Alharthi and A. Naeem, Recent developments on heterogeneous catalytic CO2 reduction to methanol, J. CO2 Util., 2019, 34, 20 CrossRef CAS.
- R. Guil-Lopez, N. Mota, J. Llorente, E. Millan, B. Pawelec, J. L. G. Fierro and R. M. Navarro, Methanol Synthesis from CO2: A Review of the Latest Developments in Heterogeneous Catalysis, Materials, 2019, 12(23), 3902 CrossRef CAS.
- X.-M. Liu, G. Q. Lu, Z.-F. Yan and J. Beltramini, Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2, Ind. Eng. Chem. Res., 2003, 42(25), 6518 CrossRef CAS.
- M. Bowker, Methanol Synthesis from CO2 Hydrogenation, ChemCatChem, 2019, 11(17), 4238 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface, J. Am. Chem. Soc., 2017, 139(29), 9739 CrossRef CAS.
- C. Heine, B. A. Lechner, H. Bluhm and M. Salmeron, Recycling of CO2: Probing the Chemical State of the Ni(111) Surface during the Methanation Reaction with Ambient-Pressure X-Ray Photoelectron Spectroscopy, J. Am. Chem. Soc., 2016, 138(40), 13246 CrossRef CAS.
- K. Zhao, L. Wang, M. Calizzi, E. Moioli and A. Züttel, In Situ Control of the Adsorption Species in CO2 Hydrogenation: Determination of Intermediates and Byproducts, J. Phys. Chem. C, 2018, 122(36), 20888 CrossRef CAS.
- N. M. Martin, F. Hemmingsson, X. Wang, L. R. Merte, U. Hejral, J. Gustafson, M. Skoglundh, D. M. Meira, A.-C. Dippel and O. Gutowski, et al., Structure–function relationship during CO2 methanation over Rh/Al2O3 and Rh/SiO2 catalysts under atmospheric pressure conditions, Catal. Sci. Technol., 2018, 8(10), 2686 RSC.
- J. Resasco, L. DeRita, S. Dai, J. P. Chada, M. Xu, X. Yan, J. Finzel, S. Hanukovich, A. S. Hoffman and G. W. Graham, et al., Uniformity Is Key in Defining Structure-Function Relationships for Atomically Dispersed Metal Catalysts: The Case of Pt/CeO2, J. Am. Chem. Soc., 2020, 142(1), 169 CrossRef CAS.
- L. Zhang, I. A. W. Filot, Y. Q. Su, J. X. Liu and E. J. M. Hensen, Understanding the Impact of Defects on Catalytic CO Oxidation of LaFeO3-Supported Rh, Pd, and Pt Single-Atom Catalysts, J. Phys. Chem. C, 2019, 123(12), 7290 CrossRef CAS.
- R. Mutschler, E. Moioli, W. Luo, N. Gallandat and A. Züttel, CO2 hydrogenation reaction over pristine Fe, Co, Ni, Cu and Al2O3 supported Ru: Comparison and determination of the activation energies, J. Catal., 2018, 366, 139 CrossRef CAS.
- G. Garbarino, T. Cavattoni, P. Riani and G. Busca, Support effects in metal catalysis: a study of the behavior of unsupported and silica-supported cobalt catalysts in the hydrogenation of CO2 at atmospheric pressure, Catal. Today, 2020, 345, 213 CrossRef CAS.
- G. Garbarino, P. Riani, L. Magistri and G. Busca, A study of the methanation of carbon dioxide on Ni/Al2O3 catalysts at atmospheric pressure, Int. J. Hydrogen Energy, 2014, 39(22), 11557 CrossRef CAS.
- D. Pandey, K. Ray, R. Bhardwaj, S. Bojja, K. V. R. Chary and G. Deo, Promotion of unsupported nickel catalyst using iron for CO2 methanation, Int. J. Hydrogen Energy, 2018, 43(10), 4987 CrossRef CAS.
- D. H. Kim, S. W. Han, H. S. Yoon and Y. D. Kim, Reverse water gas shift reaction catalyzed by Fe nanoparticles with high catalytic activity and stability, J. Ind. Eng. Chem., 2015, 23, 67 CrossRef CAS.
- M. Bersani, K. Gupta, A. K. Mishra, R. Lanza, S. F. R. Taylor, H.-U. Islam, N. Hollingsworth, C. Hardacre, N. H. de Leeuw and J. A. Darr, Combined EXAFS, XRD, DRIFTS, and DFT Study of Nano Copper-Based Catalysts for CO2Hydrogenation, ACS Catal., 2016, 6(9), 5823 CrossRef CAS.
- P. Panagiotopoulou, Hydrogenation of CO 2 over supported noble metal catalysts, Appl. Catal., A, 2017, 542, 63 CrossRef CAS.
- A. Beuls, C. Swalus, M. Jacquemin, G. Heyen, A. Karelovic and P. Ruiz, Methanation of CO2: Further insight into the mechanism over Rh/γ-Al2O3 catalyst, Appl. Catal., B, 2012, 113–114, 2 CrossRef CAS.
- A. Petala and P. Panagiotopoulou, Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: I. Effect of alkali additives on catalytic activity and selectivity, Appl. Catal., B, 2018, 224, 919 CrossRef CAS.
- S. Tada, O. J. Ochieng, R. Kikuchi, T. Haneda and H. Kameyama, Promotion of CO2 methanation activity and CH4 selectivity at low temperatures over Ru/CeO2/Al2O3 catalysts, Int. J. Hydrogen Energy, 2014, 39(19), 10090 CrossRef CAS.
- T. Avanesian, G. S. Gusmão and P. Christopher, Mechanism of CO2 reduction by H2 on Ru(0 0 0 1) and general selectivity descriptors for late-transition metal catalysts, J. Catal., 2016, 343, 86 CrossRef CAS.
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy, Catal. Today, 2013, 215, 201 CrossRef CAS.
- D. C. D. da Silva, S. Letichevsky, L. E. P. Borges and L. G. Appel, The Ni/ZrO2 catalyst and the methanation of CO and CO2, Int. J. Hydrogen Energy, 2012, 37(11), 8923 CrossRef CAS.
- J.-N. Park and E. W. McFarland, A highly dispersed Pd–Mg/SiO2 catalyst active for methanation of CO2, J. Catal., 2009, 266(1), 92 CrossRef CAS.
- R. Zhou, N. Rui, Z. Fan and C.-J. Liu, Effect of the structure of Ni/TiO2 catalyst on CO2 methanation, Int. J. Hydrogen Energy, 2016, 41(47), 22017 CrossRef CAS.
- N. M. Martin, P. Velin, M. Skoglundh, M. Bauer and P.-A. Carlsson, Catalytic hydrogenation of CO2 to methane over supported Pd, Rh and Ni catalysts, Catal. Sci. Technol., 2017, 7(5), 1086 RSC.
- L. R. Winter, E. Gomez, B. Yan, S. Yao and J. G. Chen, Tuning Ni-catalyzed CO2 hydrogenation selectivity via Ni-ceria support interactions and Ni-Fe bimetallic formation, Appl. Catal., B, 2018, 224, 442 CrossRef CAS.
- G. D. Weatherbee and C. H. Bartholomew, Hydrogenation of CO2 on group VIII metals: IV. Specific activities and selectivities of silica-supported Co, Fe, and Ru, J. Catal., 1984, 87(2), 352 CrossRef CAS.
- J. Zhu, G. Zhang, W. Li, X. Zhang, F. Ding, C. Song and X. Guo, Deconvolution of the Particle Size Effect on CO2 Hydrogenation over Iron-Based Catalysts, ACS Catal., 2020, 10(13), 7424 CrossRef CAS.
- X. Chen, X. Su, B. Liang, X. Yang, X. Ren, H. Duan, Y. Huang and T. Zhang, Identification of relevant active sites and a mechanism study for reverse water gas shift reaction over Pt/CeO 2 catalysts, J. Energy Chem., 2016, 25(6), 1051 CrossRef.
- S. Kattel, B. Yan, J. G. Chen and P. Liu, CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support, J. Catal., 2016, 343, 115 CrossRef CAS.
- S. S. Kim, H. H. Lee and S. C. Hong, A study on the effect of support's reducibility on the reverse water-gas shift reaction over Pt catalysts, Appl. Catal., A, 2012, 423–424, 100 CAS.
- S.-I. Fujita and N. Takezawa, Difference in the selectivity of CO and CO2 methanation reactions, Chem. Eng. J., 1997, 68(1), 63 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and S. M. Sidik, Methanation of carbon dioxide on metal-promoted mesostructured silica nanoparticles, Appl. Catal., A, 2014, 486, 115 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, Optimizing Binding Energies of Key Intermediates for CO2 Hydrogenation to Methanol over Oxide-Supported Copper, J. Am. Chem. Soc., 2016, 138(38), 12440 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2(6), 65 CrossRef CAS.
- M. Dhiman and V. Polshettiwar, Supported Single Atom and Pseudo-Single Atom of Metals as Sustainable Heterogeneous Nanocatalysts, ChemCatChem, 2018, 10(5), 881 CrossRef CAS.
- H. Zhao, S. Yao, M. Zhang, F. Huang, Q. Fan, S. Zhang, H. Liu, D. Ma and C. Gao, Ultra-Small Platinum Nanoparticles Encapsulated in Sub-50 nm Hollow Titania Nanospheres for Low-Temperature Water-Gas Shift Reaction, ACS Appl. Mater. Interfaces, 2018, 10(43), 36954 CrossRef CAS.
- X. Zhao, H. Xu, X. Wang, Z. Zheng, Z. Xu and J. Ge, Monodisperse Metal-Organic Framework Nanospheres with Encapsulated Core-Shell Nanoparticles Pt/Au@Pd@{Co2(oba)4(3-bpdh)2}4H2O for the Highly Selective Conversion of CO2 to CO, ACS Appl. Mater. Interfaces, 2018, 10(17), 15096 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis, Acc. Chem. Res., 2013, 46(8), 1740 CrossRef CAS.
- H. Zhang, G. Liu, L. Shi and J. Ye, Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis, Adv. Energy Mater., 2018, 8(1), 1701343 CrossRef.
- J. H. Kwak, L. Kovarik and J. Szanyi, CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity, ACS Catal., 2013, 3(11), 2449 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, Heterogeneous Catalysis on Atomically Dispersed Supported Metals: CO2 Reduction on Multifunctional Pd Catalysts, ACS Catal., 2013, 3(9), 2094 CrossRef CAS.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, Low-Temperature Restructuring of CeO2-Supported Ru Nanoparticles Determines Selectivity in CO2 Catalytic Reduction, J. Am. Chem. Soc., 2018, 140(42), 13736 CrossRef CAS.
- J. C. Matsubu, V. N. Yang and P. Christopher, Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity, J. Am. Chem. Soc., 2015, 137(8), 3076 CrossRef CAS.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge and C. Yang, et al., Tuning the Selectivity of Catalytic Carbon Dioxide Hydrogenation over Iridium/Cerium Oxide Catalysts with a Strong Metal-Support Interaction, Angew. Chem., Int. Ed., 2017, 56(36), 10761 CrossRef CAS.
- Y. Wang, H. Arandiyan, J. Scott, K.-F. Aguey-Zinsou and R. Amal, Single Atom and Nanoclustered Pt Catalysts for Selective CO2 Reduction, ACS Appl. Energy Mater., 2018, 1(12), 6781 CrossRef CAS.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: the influence of particle size on selectivity and reaction pathway, Catal. Sci. Technol., 2015, 5(8), 4154 RSC.
- B. Lu and K. Kawamoto, Preparation of monodispersed NiO particles in SBA-15, and its enhanced selectivity for reverse water gas shift reaction, J. Environ. Chem. Eng., 2013, 1(3), 300 CrossRef CAS.
- B. Lu and K. Kawamoto, Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction, Mater. Res. Bull., 2014, 53, 70 CrossRef CAS.
- R. V. Gonçalves, L. L. R. Vono, R. Wojcieszak, C. S. B. Dias, H. Wender, E. Teixeira-Neto and L. M. Rossi, Selective hydrogenation of CO 2 into CO on a highly dispersed nickel catalyst obtained by magnetron sputtering deposition: A step towards liquid fuels, Appl. Catal., B, 2017, 209, 240 CrossRef.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Unravelling structure sensitivity in CO2 hydrogenation over nickel, Nat. Catal., 2018, 1(2), 127 CrossRef CAS.
- Y. Yan, Q. Wang, C. Jiang, Y. Yao, D. Lu, J. Zheng, Y. Dai, H. Wang and Y. Yang, Ru/Al2O3 catalyzed CO2 hydrogenation: Oxygen-exchange on metal-support interfaces, J. Catal., 2018, 367, 194 CrossRef CAS.
- S. Liang, C. Hao and Y. Shi, The Power of Single-Atom Catalysis, ChemCatChem, 2015, 7(17), 2559 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang and T. Zhang, et al., Thermally stable single atom Pt/m-Al2O3 for selective hydrogenation and CO oxidation, Nat. Commun., 2017, 8, 16100 CrossRef CAS.
- P. Panagiotopoulou, Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: II. Effect of alkali additives on the reaction pathway, Appl. Catal., B, 2018, 236, 162 CrossRef CAS.
- P. Chou and M. A. Vannice, Calorimetric heat of adsorption measurements on palladium: I. Influence of crystallite size and support on hydrogen adsorption, J. Catal., 1987, 104(1), 1 CrossRef CAS.
- J. Martins, N. Batail, S. Silva, S. Rafik-Clement, A. Karelovic, D. P. Debecker, A. Chaumonnot and D. Uzio, CO2 hydrogenation with shape-controlled Pd nanoparticles embedded in mesoporous silica: Elucidating stability and selectivity issues, Catal. Commun., 2015, 58, 11 CrossRef CAS.
- B. T. Loveless, C. Buda, M. Neurock and E. Iglesia, CO chemisorption and dissociation at high coverages during CO hydrogenation on Ru catalysts, J. Am. Chem. Soc., 2013, 135(16), 6107 CrossRef CAS.
- D. Beierlein, D. Häussermann, M. Pfeifer, T. Schwarz, K. Stöwe, Y. Traa and E. Klemm, Is the CO2 methanation on highly loaded Ni-Al2O3 catalysts really structure-sensitive?, Appl. Catal., B, 2019, 247, 200 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, Low-Temperature CO2 Methanation over CeO2-Supported Ru Single Atoms, Nanoclusters, and Nanoparticles Competitively Tuned by Strong Metal–Support Interactions and H-Spillover Effect, ACS Catal., 2018, 8(7), 6203 CrossRef CAS.
- Q. Liu, B. Bian, J. Fan and J. Yang, Cobalt doped Ni based ordered mesoporous catalysts for CO2 methanation with enhanced catalytic performance, Int. J. Hydrogen Energy, 2018, 43(10), 4893 CrossRef CAS.
- L. Xu, X. Lian, M. Chen, Y. Cui, F. Wang, W. Li and B. Huang, CO2 methanation over Co Ni bimetal-doped ordered mesoporous Al2O3 catalysts with enhanced low-temperature activities, Int. J. Hydrogen Energy, 2018, 43(36), 17172 CrossRef CAS.
- H. Arandiyan, Y. Wang, J. Scott, S. Mesgari, H. Dai and R. Amal, In Situ Exsolution of Bimetallic Rh-Ni Nanoalloys: a Highly Efficient Catalyst for CO2 Methanation, ACS Appl. Mater. Interfaces, 2018, 10(19), 16352 CrossRef CAS.
- X. Shang, D. Deng, X. Wang, W. Xuan, X. Zou, W. Ding and X. Lu, Enhanced low-temperature activity for CO2 methanation over Ru doped the Ni/CexZr(1−)O2 catalysts prepared by one-pot hydrolysis method, Int. J. Hydrogen Energy, 2018, 43(14), 7179 CrossRef CAS.
- Q. Liu, S. Wang, G. Zhao, H. Yang, M. Yuan, X. An, H. Zhou, Y. Qiao and Y. Tian, CO2 methanation over ordered mesoporous NiRu-doped CaO-Al2O3 nanocomposites with enhanced catalytic performance, Int. J. Hydrogen Energy, 2018, 43(1), 239 CrossRef CAS.
- T. Burger, F. Koschany, O. Thomys, K. Köhler and O. Hinrichsen, CO2 methanation over Fe- and Mn-promoted co-precipitated Ni-Al catalysts: Synthesis, characterization and catalysis study, Appl. Catal., A, 2018, 558, 44 CrossRef CAS.
- H. Lu, X. Yang, G. Gao, J. Wang, C. Han, X. Liang, C. Li, Y. Li, W. Zhang and X. Chen, Metal (Fe, Co, Ce or La) doped nickel catalyst supported on ZrO2 modified mesoporous clays for CO and CO2 methanation, Fuel, 2016, 183, 335 CrossRef CAS.
- C. Mebrahtu, F. Krebs, S. Perathoner, S. Abate, G. Centi and R. Palkovits, Hydrotalcite based Ni–Fe/(Mg, Al)Ox catalysts for CO2 methanation – tailoring Fe content for improved CO dissociation, basicity, and particle size, Catal. Sci. Technol., 2018, 8(4), 1016 RSC.
- B. Mutz, M. Belimov, W. Wang, P. Sprenger, M.-A. Serrer, D. Wang, P. Pfeifer, W. Kleist and J.-D. Grunwaldt, Potential of an Alumina-Supported Ni3Fe Catalyst in the Methanation of CO2: Impact of Alloy Formation on Activity and Stability, ACS Catal., 2017, 7(10), 6802 CrossRef CAS.
- B. Yan, B. Zhao, S. Kattel, Q. Wu, S. Yao, D. Su and J. G. Chen, Tuning CO2 hydrogenation selectivity via metal-oxide interfacial sites, J. Catal., 2019, 374, 60 CrossRef CAS.
- M. Mihet and M. D. Lazar, Methanation of CO2 on Ni/γ-Al2O3: Influence of Pt, Pd or Rh promotion, Catal. Today, 2018, 306, 294 CrossRef CAS.
- S. K. Beaumont, S. Alayoglu, C. Specht, W. D. Michalak, V. V. Pushkarev, J. Guo, N. Kruse and G. A. Somorjai, Combining in situ NEXAFS spectroscopy and CO(2) methanation kinetics to study Pt and Co nanoparticle catalysts reveals key insights into the role of platinum in promoted cobalt catalysis, J. Am. Chem. Soc., 2014, 136(28), 9898 CrossRef CAS.
- S. K. Beaumont, S. Alayoglu, C. Specht, N. Kruse and G. A. Somorjai, A nanoscale demonstration of hydrogen atom spillover and surface diffusion across silica using the kinetics of CO2 methanation catalyzed on spatially separate Pt and Co nanoparticles, Nano Lett., 2014, 14(8), 4792 CrossRef CAS.
- Z. Ou, C. Qin, J. Niu, L. Zhang and J. Ran, A comprehensive DFT study of CO2 catalytic conversion by H2 over Pt-doped Ni catalysts, Int. J. Hydrogen Energy, 2019, 44(2), 819 CrossRef CAS.
- M. C. Bacariza, R. Bértolo, I. Graça, J. M. Lopes and C. Henriques, The effect of the compensating cation on the catalytic performances of Ni/USY zeolites towards CO2 methanation, J. CO2 Util., 2017, 21, 280 CrossRef CAS.
- R. Büchel, A. Baiker and S. E. Pratsinis, Effect of Ba and K addition and controlled spatial deposition of Rh in Rh/Al2O3 catalysts for CO2 hydrogenation, Appl. Catal., A, 2014, 477, 93 CrossRef.
- D. Heyl, U. Rodemerck and U. Bentrup, Mechanistic Study of Low-Temperature CO2 Hydrogenation over Modified Rh/Al2O3 Catalysts, ACS Catal., 2016, 6(9), 6275 CrossRef CAS.
- M. D. Porosoff, J. W. Baldwin, X. Peng, G. Mpourmpakis and H. D. Willauer, Potassium-Promoted Molybdenum Carbide as a Highly Active and Selective Catalyst for CO2 Conversion to CO, ChemSusChem, 2017, 10(11), 2408 CrossRef CAS.
- B. Liang, H. Duan, X. Su, X. Chen, Y. Huang, X. Chen, J. J. Delgado and T. Zhang, Promoting role of potassium in the reverse water gas shift reaction on Pt/mullite catalyst, Catal. Today, 2017, 281, 319 CrossRef CAS.
- X. Yang, X. Su, X. Chen, H. Duan, B. Liang, Q. Liu, X. Liu, Y. Ren, Y. Huang and T. Zhang, Promotion effects of potassium on the activity and selectivity of Pt/zeolite catalysts for reverse water gas shift reaction, Appl. Catal., B, 2017, 216, 95 CrossRef CAS.
- J. A. H. Dreyer, P. Li, L. Zhang, G. K. Beh, R. Zhang, P. H. L. Sit and W. Y. Teoh, Influence of the oxide support reducibility on the CO2 methanation over Ru-based catalysts, Appl. Catal., B, 2017, 219, 715 CrossRef CAS.
- J. Díez-Ramírez, P. Sánchez, V. Kyriakou, S. Zafeiratos, G. E. Marnellos, M. Konsolakis and F. Dorado, Effect of support nature on the cobalt-catalyzed CO2 hydrogenation, J. CO2 Util., 2017, 21, 562 CrossRef.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, Carbon dioxide methanation over Ni catalysts supported on various metal oxides, J. Catal., 2016, 343, 178 CrossRef CAS.
- L. R. Winter, R. Chen, X. Chen, K. Chang, Z. Liu, S. D. Senanayake, A. M. Ebrahim and J. G. Chen, Elucidating the roles of metallic Ni and oxygen vacancies in CO2 hydrogenation over Ni/CeO2 using isotope exchange and in situ measurements, Appl. Catal., B, 2019, 245, 360 CrossRef CAS.
- A. Lechkar, A. Barroso Bogeat, G. Blanco, J. M. Pintado and M. Soussi el Begrani, Methanation of carbon dioxide over ceria-praseodymia promoted Ni-alumina catalysts. Influence of metal loading, promoter composition and alumina modifier, Fuel, 2018, 234, 1401 CrossRef CAS.
- J. Lin, C. Ma, Q. Wang, Y. Xu, G. Ma, J. Wang, H. Wang, C. Dong, C. Zhang and M. Ding, Enhanced low-temperature performance of CO2 methanation over mesoporous Ni/Al2O3-ZrO2 catalysts, Appl. Catal., B, 2019, 243, 262 CrossRef CAS.
- F. Meng, Z. Li, F. Ji and M. Li, Effect of ZrO2 on catalyst structure and catalytic methanation performance over Ni-based catalyst in slurry-bed reactor, Int. J. Hydrogen Energy, 2015, 40(29), 8833 CrossRef CAS.
- W. Nie, X. Zou, X. Shang, X. Wang, W. Ding and X. Lu, CeO 2 -assisted Ni nanocatalysts supported on mesoporous γ-Al 2 O 3 for the production of synthetic natural gas, Fuel, 2017, 202, 135 CrossRef CAS.
- I. Iglesias, A. Quindimil, F. Mariño, U. De-La-Torre and J. R. González-Velasco, Zr promotion effect in CO2 methanation over ceria supported nickel catalysts, Int. J. Hydrogen Energy, 2019, 44(3), 1710 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu, Z. Jiao, Y. Liu and X. Zhang, Role of surface Ni and Ce species of Ni/CeO2 catalyst in CO2 methanation, Appl. Surf. Sci., 2016, 383, 248 CrossRef CAS.
- X. Nie, H. Wang, W. Li, Y. Chen, X. Guo and C. Song, DFT insight into the support effect on the adsorption and activation of key species over Co catalysts for CO2 methanation, J. CO2 Util., 2018, 24, 99 CrossRef CAS.
- Q. Pan, J. Peng, T. Sun, D. Gao, S. Wang and S. Wang, CO2 methanation on Ni/Ce0.5Zr0.5O2 catalysts for the production of synthetic natural gas, Fuel Process. Technol., 2014, 123, 166 CrossRef CAS.
- Q. Pan, J. Peng, S. Wang and S. Wang, In situ FTIR spectroscopic study of the CO2methanation mechanism on Ni/Ce0.5Zr0.5O2, Catal. Sci. Technol., 2014, 4(2), 502 RSC.
- F.-M. Sun, C.-F. Yan, Z.-D. Wang, C.-Q. Guo and S.-L. Huang, Ni/Ce–Zr–O catalyst for high CO2 conversion during reverse water gas shift reaction (RWGS), Int. J. Hydrogen Energy, 2015, 40(46), 15985 CrossRef CAS.
- X. Liu, C. Kunkel, P. Ramírez de la Piscina, N. Homs, F. Viñes and F. Illas, Effective and Highly Selective CO Generation from CO2Using a Polycrystalline α-Mo2C Catalyst, ACS Catal., 2017, 7(7), 4323 CrossRef CAS.
- H.-S. Na, J.-O. Shim, S.-Y. Ahn, W.-J. Jang, K.-W. Jeon, H.-M. Kim, Y.-L. Lee, K.-J. Kim and H.-S. Roh, Effect of precipitation sequence on physicochemical properties of CeO2 support for hydrogen production from low-temperature water-gas shift reaction, Int. J. Hydrogen Energy, 2018, 43(37), 17718 CrossRef CAS.
- T. Sakpal and L. Lefferts, Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation, J. Catal., 2018, 367, 171 CrossRef CAS.
- Q. Lin, X. Y. Liu, Y. Jiang, Y. Wang, Y. Huang and T. Zhang, Crystal phase effects on the structure and performance of ruthenium nanoparticles for CO2 hydrogenation, Catal. Sci. Technol., 2014, 4(7), 2058 RSC.
- A. Kim, C. Sanchez, G. Patriarche, O. Ersen, S. Moldovan, A. Wisnet, C. Sassoye and D. P. Debecker, Selective CO2 methanation on Ru/TiO2 catalysts: unravelling the decisive role of the TiO2 support crystal structure, Catal. Sci. Technol., 2016, 6(22), 8117 RSC.
- A. Kim, D. P. Debecker, F. Devred, V. Dubois, C. Sanchez and C. Sassoye, CO2 methanation on Ru/TiO2 catalysts: On the effect of mixing anatase and rutile TiO2 supports, Appl. Catal., B, 2018, 220, 615 CrossRef CAS.
- Y. Lin, Y. Zhu, X. Pan and X. Bao, Modulating the methanation activity of Ni by the crystal phase of TiO2, Catal. Sci. Technol., 2017, 7(13), 2813 RSC.
- E. S. Gnanakumar, N. Chandran, I. V. Kozhevnikov, A. Grau-Atienza, E. V. Ramos Fernández, A. Sepulveda-Escribano and N. R. Shiju, Highly efficient nickel-niobia composite catalysts for hydrogenation of CO2 to methane, Chem. Eng. Sci., 2019, 194, 2 CrossRef CAS.
- K. Zhao, W. Wang and Z. Li, Highly efficient Ni/ZrO2 catalysts prepared via combustion method for CO2 methanation, J. CO2 Util., 2016, 16, 236 CrossRef CAS.
- Y. Zhang, Z. Zhang, X. Yang, R. Wang, H. Duan, Z. Shen, L. Li, Y. Su, R. Yang and Y. Zhang, et al., Tuning selectivity of CO2 hydrogenation by modulating the strong metal–support interaction over Ir/TiO2 catalysts, Green Chem., 2020, 22(20), 6855–6861 RSC.
- R. Lippi, S. C. Howard, H. Barron, C. D. Easton, I. C. Madsen, L. J. Waddington, C. Vogt, M. R. Hill, C. J. Sumby and C. J. Doonan, et al., Highly active catalyst for CO2 methanation derived from a metal organic framework template, J. Mater. Chem. A, 2017, 5(25), 12990 RSC.
- Z.-W. Zhao, X. Zhou, Y.-N. Liu, C.-C. Shen, C.-Z. Yuan, Y.-F. Jiang, S.-J. Zhao, L.-B. Ma, T.-Y. Cheang and A.-W. Xu, Ultrasmall Ni nanoparticles embedded in Zr-based MOFs provide high selectivity for CO2 hydrogenation to methane at low temperatures, Catal. Sci. Technol., 2018, 8(12), 3160 RSC.
- E. Lam, J. J. Corral-Perez, K. Larmier, G. Noh, P. Wolf, A. Comas-Vives, A. Urakawa and C. Coperet, CO2 Hydrogenation on Cu/Al2 O3 : Role of the Metal/Support Interface in Driving Activity and Selectivity of a Bifunctional Catalyst, Angew. Chem., Int. Ed., 2019, 58(39), 13989 CrossRef CAS.
- F. Liao, Y. Huang, J. Ge, W. Zheng, K. Tedsree, P. Collier, X. Hong and S. C. Tsang, Morphology-dependent interactions of ZnO with Cu nanoparticles at the materials’ interface in selective hydrogenation of CO2 to CH3OH, Angew. Chem., Int. Ed., 2011, 50(9), 2162 CrossRef CAS.
- X. Yang, S. Kattel, S. D. Senanayake, J. A. Boscoboinik, X. Nie, J. Graciani, J. A. Rodriguez, P. Liu, D. J. Stacchiola and J. G. Chen, Low Pressure CO2 Hydrogenation to Methanol over Gold Nanoparticles Activated on a CeOx/TiO2 Interface, J. Am. Chem. Soc., 2015, 137(32), 10104 CrossRef CAS.
- C. Kim, S. Hyeon, J. Lee, W. D. Kim, D. C. Lee, J. Kim and H. Lee, Energy-efficient CO2 hydrogenation with fast response using photoexcitation of CO2 adsorbed on metal catalysts, Nat. Commun., 2018, 9(1), 3027 CrossRef.
- L. Lin, K. Wang, K. Yang, X. Chen, X. Fu and W. Dai, The visible-light-assisted thermocatalytic methanation of CO2 over Ru/TiO(2-x)Nx, Appl. Catal., B, 2017, 204, 440 CrossRef CAS.
- B. Zhao, B. Yan, Z. Jiang, S. Yao, Z. Liu, Q. Wu, R. Ran, S. D. Senanayake, D. Weng and J. G. Chen, High selectivity of CO2 hydrogenation to CO by controlling the valence state of nickel using perovskite, Chem. Commun., 2018, 54(53), 7354 RSC.
- A. M. Abdel-Mageed, S. Eckle and R. J. Behm, High Selectivity of Supported Ru Catalysts in the Selective CO Methanation-Water Makes the Difference, J. Am. Chem. Soc., 2015, 137(27), 8672 CrossRef CAS.
- H. Arandiyan, K. Kani, Y. Wang, B. Jiang, J. Kim, M. Yoshino, M. Rezaei, A. E. Rowan, H. Dai and Y. Yamauchi, Highly Selective Reduction of Carbon Dioxide to Methane on Novel Mesoporous Rh Catalysts, ACS Appl. Mater. Interfaces, 2018, 10(30), 24963 CrossRef CAS.
- N. Liu, M. Xu, Y. Yang, S. Zhang, J. Zhang, W. Wang, L. Zheng, S. Hong and M. Wei, Auδ−–Ov–Ti3+ Interfacial Site: Catalytic Active Center toward Low-Temperature Water Gas Shift Reaction, ACS Catal., 2019, 9(4), 2707 CrossRef CAS.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang and M. Haruta, et al., Classical strong metal–support interactions between gold nanoparticles and titanium dioxide, Sci. Adv., 2017, 3(10), e1700231 CrossRef.
- X. Zhang, P. Yan, B. Zhao and Z. C. Zhang, Identification of electron-rich mononuclear Ni atoms on TiO2-A distinguished from Ni particles on TiO2-R in guaiacol hydrodeoxygenation pathways, Catal. Sci. Technol., 2020 10.1039/d0cy01720e 10.1039/d0cy01720e.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, Remarkable performance of Ir1/FeO(x) single-atom catalyst in water gas shift reaction, J. Am. Chem. Soc., 2013, 135(41), 15314 CrossRef CAS.
- Y. Chen, S. Ji, W. Sun, W. Chen, J. Dong, J. Wen, J. Zhang, Z. Li, L. Zheng and C. Chen, et al., Discovering Partially Charged Single-Atom Pt for Enhanced Anti-Markovnikov Alkene Hydrosilylation, J. Am. Chem. Soc., 2018, 140(24), 7407 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes, Nat. Commun., 2014, 5, 5634 CrossRef CAS.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. T. Cui, Y. Tan, B. Qiao, L. Li and A. Wang, et al., Hydroformylation of Olefins by a Rhodium Single-Atom Catalyst with Activity Comparable to RhCl(PPh3)3, Angew. Chem., Int. Ed., 2016, 55(52), 16054 CrossRef CAS.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Ultrastable single-atom gold catalysts with strong covalent metal-support interaction (CMSI), Nano Res., 2015, 8(9), 2913 CrossRef CAS.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, Highly Active and Stable Single-Atom Cu Catalysts Supported by a Metal-Organic Framework, J. Am. Chem. Soc., 2019, 141(13), 5201 CrossRef CAS.
- A. Peremans, K. Fukutani, K. Mase and Y. Murata, CO and CO+ photodesorption from Pt(001) at 193 nm, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(7), 4135 CrossRef CAS.
- X. Guo, J. Yoshinobu and J. T. Yates, Photon–induced desorption of CO chemisorbed on the oxidized Ni(111) surface, J. Chem. Phys., 1990, 92(7), 4320 CrossRef CAS.
- H. Nakatsuji, H. Morita, H. Nakai, Y. Murata and K. Fukutani, Theoretical study on the photostimulated desorption of CO from a Pt surface, J. Chem. Phys., 1996, 104(2), 714 CrossRef CAS.
- H. Aizawa and S. Tsuneyuki, First-principles investigation of photo-induced desorption of CO and NO from Pt(111), Surf. Sci., 1996, 363(1), 223 CrossRef CAS.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, Hydrogenation of CO2 to Methanol: Importance of Metal–Oxide and Metal–Carbide Interfaces in the Activation of CO2, ACS Catal., 2015, 5(11), 6696 CrossRef CAS.
- D. Duprez, in Studies in Surface Science and Catalysis, ed. C. Li and Q. Xin, Elsevier, 1997, vol. 112 Search PubMed.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, Optimizing Binding Energies of Key Intermediates for CO2 Hydrogenation to Methanol over Oxide-Supported Copper, J. Am. Chem. Soc., 2016, 138(38), 12440 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, Mechanism of CO2 Hydrogenation on Pd/Al2O3 Catalysts: Kinetics and Transient DRIFTS-MS Studies, ACS Catal., 2015, 5(11), 6337 CrossRef CAS.
- T. Jin, T. Okuhara, G. J. Mains and J. M. White, Temperature-programmed desorption of carbon monoxide and carbon dioxide from platinum/ceria: an important role for lattice oxygen in carbon monoxide oxidation, J. Phys. Chem., 1987, 91(12), 3310 CrossRef CAS.
- C. E. Hori, H. Permana, K. Y. S. Ng, A. Brenner, K. More, K. M. Rahmoeller and D. Belton, Thermal stability of oxygen storage properties in a mixed CeO2-ZrO2 system, Appl. Catal., B, 1998, 16(2), 105 CrossRef CAS.
- C. E. Hori, A. Brenner, K. Y. Simon Ng, K. M. Rahmoeller and D. Belton, Studies of the oxygen release reaction in the platinum–ceria–zirconia system, Catal. Today, 1999, 50(2), 299 CrossRef CAS.
- F. Dong, A. Suda, T. Tanabe, Y. Nagai, H. Sobukawa, H. Shinjoh, M. Sugiura, C. Descorme and D. Duprez, Dynamic oxygen mobility and a new insight into the role of Zr atoms in three-way catalysts of Pt/CeO2–ZrO2, Catal. Today, 2004, 93–95, 827 CrossRef CAS.
- E. M. Sadovskaya, Y. A. Ivanova, L. G. Pinaeva, G. Grasso, T. G. Kuznetsova, A. van Veen, V. A. Sadykov and C. Mirodatos, Kinetics of Oxygen Exchange over CeO2–ZrO2 Fluorite-Based Catalysts, J. Phys. Chem. A, 2007, 111(20), 4498 CrossRef CAS.
- A. Bueno-López, K. Krishna and M. Makkee, Oxygen exchange mechanism between isotopic CO2 and Pt/CeO2, Appl. Catal., A, 2008, 342(1), 144 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |