
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, properties and structural features of molybdenum(V) oxide trichloride complexes with neutral chalcogenoether ligands†
Danielle E.
Smith
,
William
Levason
 ,
James
Powell
and
Gillian
Reid
*
,
James
Powell
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 3rd March 2021
Abstract
Complexes of oxotrichloromolybdenum(V) with neutral group 16 donor ligands, [MoOCl3(L–L)] (L–L = RS(CH2)2SR, R = iPr, Ph; MeS(CH2)3SMe; MeSe(CH2)2SeMe; MeSe(CH2)3SeMe), [{MoOCl2(EMe2)}2(μ-Cl)2] (E = S, Se, Te), [(MoOCl3)2{o-C6H4(EMe)2}]n (E = Se or Te) and [(MoOCl3)2{MeTe(CH2)3TeMe}]n, have been obtained by reaction of the ligands with [MoOCl3(thf)2] or MoOCl3 in either CH2Cl2 or toluene, and characterised by microanalysis, IR and UV-visible spectroscopy and magnetic measurements. The telluroethers are the first examples containing Mo in a positive oxidation state. X-ray crystal structures are reported for the six-coordinate fac-[MoOCl3{MeS(CH2)3SMe}], mer-[MoOCl3{iPrS(CH2)2SiPr}] and mer-[MoOCl3{MeSe(CH2)2SeMe}], as well as the six-coordinate chloride-bridged dimers, [{MoOCl2(SMe2)}2(μ-Cl)2] and [{MoOCl2(SeMe2)}2(μ-Cl)2]. The structure of the mixed-valence decomposition product, [MoIVCl{o-C6H4(TeMe)2}2(μ-O)MoVOCl4], was also determined. In toluene solution MoOCl4 is reduced by MeS(CH2)3SMe to produce the Mo(V) complex, [MoOCl3{ MeS(CH2)3SMe}]. Crystal structures of the previously unknown diphosphine analogue, [MoOCl3{Me2P(CH2)2PMe2}], and the mixed-valence derivative [MoIVCl{Me2P(CH2)2PMe2}2(μ-O)MoVOCl4] are also reported for comparison and help to clarify earlier contradictory literature reports. In contrast to the dimeric EMe2 complexes, [{MoOCl2(EMe2)}2(μ-Cl)2], PMe3 forms the monomeric complex, fac-[MoOCl3(PMe3)2].
Introduction
The coordination chemistry of high oxidation state molybdenum halides and oxide halides was first explored in some detail in the 1970′s, with the emphasis on neutral N- and O-donor ligands1–4 and with much of the impetus coming from modelling of the metal sites in molybdenum enzymes and applications in catalysis.4–6 Interest in high oxidation molybdenum complexes bearing sulfur donor ligands stems in part from the presence of Mo–S coordination in the molybdenum-containing enzymes, nitrate reductase, sulfite oxidase and Fe–Mo nitrogenases which involve (anionic) cysteine or sulfide ligands.4–6 The chemistry with neutral P- and As-donor ligands with Mo(V) has also been investigated,7–11 but sulfur-based ligands were mostly represented by charged thiolate and dithiocarbamate ligands.2–4 More recent work has reported a series of extremely moisture sensitive Mo(VI) complexes [MoO2X2(dithioether)] (X = Cl or Br; dithioether = RS(CH2)2SR, R = Me, Et, iPr), which have distorted octahedral structures with the sulfur donor atoms trans to Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O;12,13 there are also some thia-macrocyclic analogues.14,15 Complexes of the type [MoOCl3(dithioether)] were briefly described in the 1970's, characterised only by microanalysis and IR spectroscopy, but the structures and isomer(s) present were not established.15,16 There is a single preliminary report of a selenoether complex of MoOCl3,17 but no known telluroether complexes.
O;12,13 there are also some thia-macrocyclic analogues.14,15 Complexes of the type [MoOCl3(dithioether)] were briefly described in the 1970's, characterised only by microanalysis and IR spectroscopy, but the structures and isomer(s) present were not established.15,16 There is a single preliminary report of a selenoether complex of MoOCl3,17 but no known telluroether complexes.
We have recently examined the complexes of WOCl4, WOCl3, WSCl4 and WSCl3 with mono- and di-thio- and -seleno-ethers, and found that W(VI) or W(V) complexes could be isolated depending upon the reaction conditions. We also showed that selected dithioether complexes, for example [(WSCl4)2(μ-iPrSCH2CH2SiPr)], can function as single source LPCVD (low pressure chemical vapour deposition) reagents for the growth of thin films of WS2, an important semiconducting material.18 In contrast, very little data on the molybdenum chalcogenide halides or their coordination complexes exists.19 The crystal structures of two forms of MoSCl3 obtained from crystals grown at high temperature found that both contain Mo(IV) as Mo2 units and disulfide groups, and not Mo(V).20 It is unclear if MoSCl3 prepared at low temperatures from MoCl5 and S(SiMe3)2 or Sb2S3 contains Mo(V),21,22 while MoSCl4 is unknown.19
In order to allow comparisons with the WOCl4, WOCl3, WSCl4 and WSCl3 chemistry, we have examined the chemistry of MoOCl3 with neutral chalcogenoethers and report here complexes of mono- and bi-dentate thio-, seleno- and telluro-ethers. Data on diphosphine analogues, which clarifies some of the (inconsistent) earlier studies,7–9 is also presented.
Results and discussion
Scheme 1 shows the range of chalcogenoether complexes of Mo(V) prepared in this study and the different structure types observed.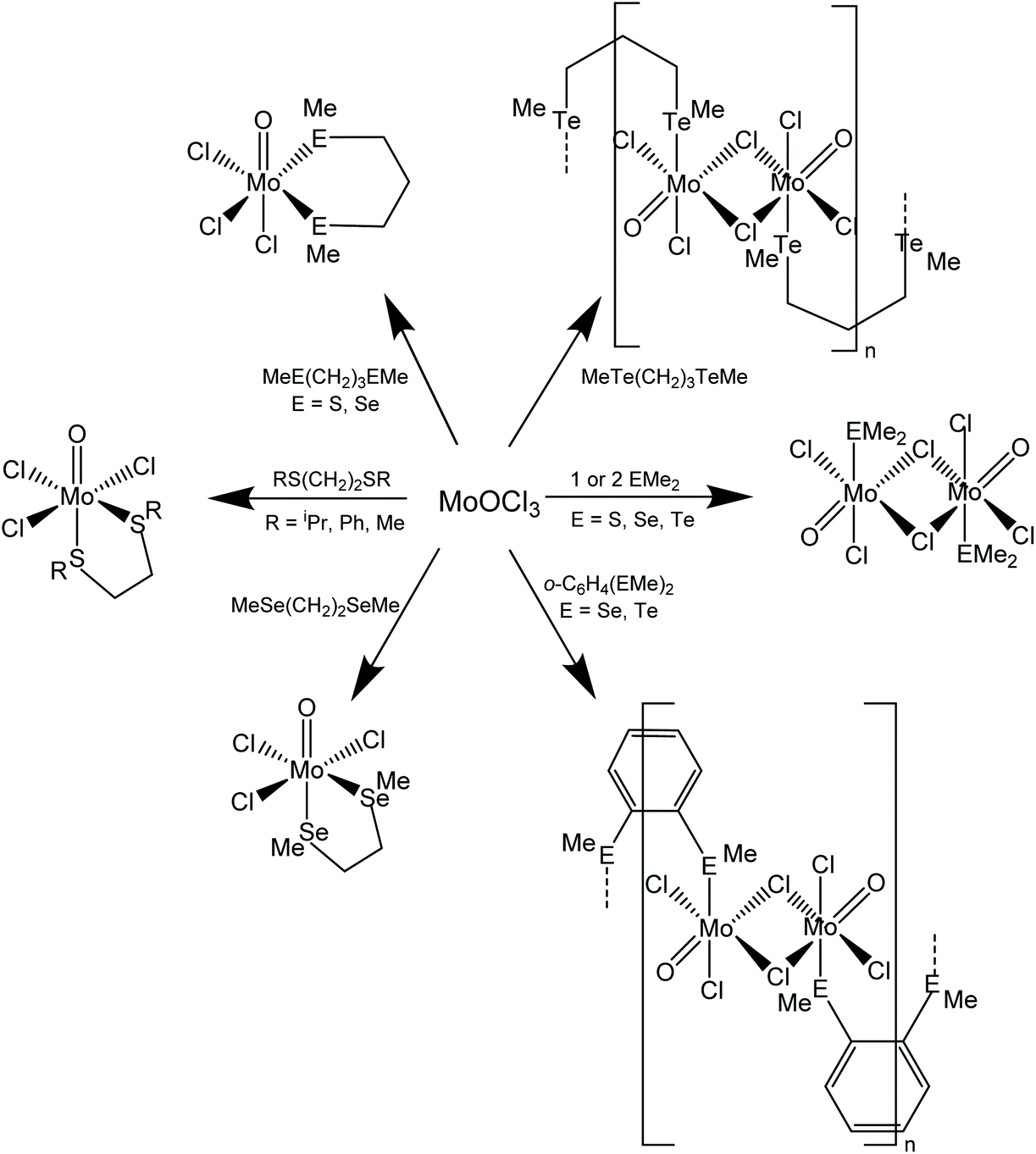 | ||
| Scheme 1 Methods for the synthesis of the Mo(V) chalcogenoether complexes obtained from MoOCl3. Note that for some of the alkyl-substituted dithioether and diselenoether complexes [MoOCl3(thf)2] was used as the Mo(V) source – see discussion below and Experimental. | ||
Dithio- and diseleno-ether complexes
The reaction of [MoOCl3(thf)2] with MeS(CH2)3SMe or iPrS(CH2)2SiPr in dry CH2Cl2 produced moisture sensitive. green [MoOCl3(dithioether)] complexes. Structures of both species were determined and revealed that [MoOCl3{MeS(CH2)3SMe}] (six-membered chelate ring) was the fac isomer, whilst [MoOCl3{iPrS(CH2)2SiPr}] (five-membered chelate ring) was the mer-isomer (Fig. 1). The reason for the different isomers with the five- and six-membered rings is uncertain, although the difference in the S–Mo–S chelate angles of ∼20° is notable. The behaviour replicates that found with the tungsten(V) analogues, fac-[WOCl3{MeS(CH2)3SMe}] and mer-[WOCl3{MeS(CH2)2SMe}].18 The bond lengths within the two structures show the expected short MoO of ∼1.67 Å and that the Mo–Cl and Mo–S trans to MoO are longer than the other bonds of each type, indicating the high trans-influence of the MoO bond.
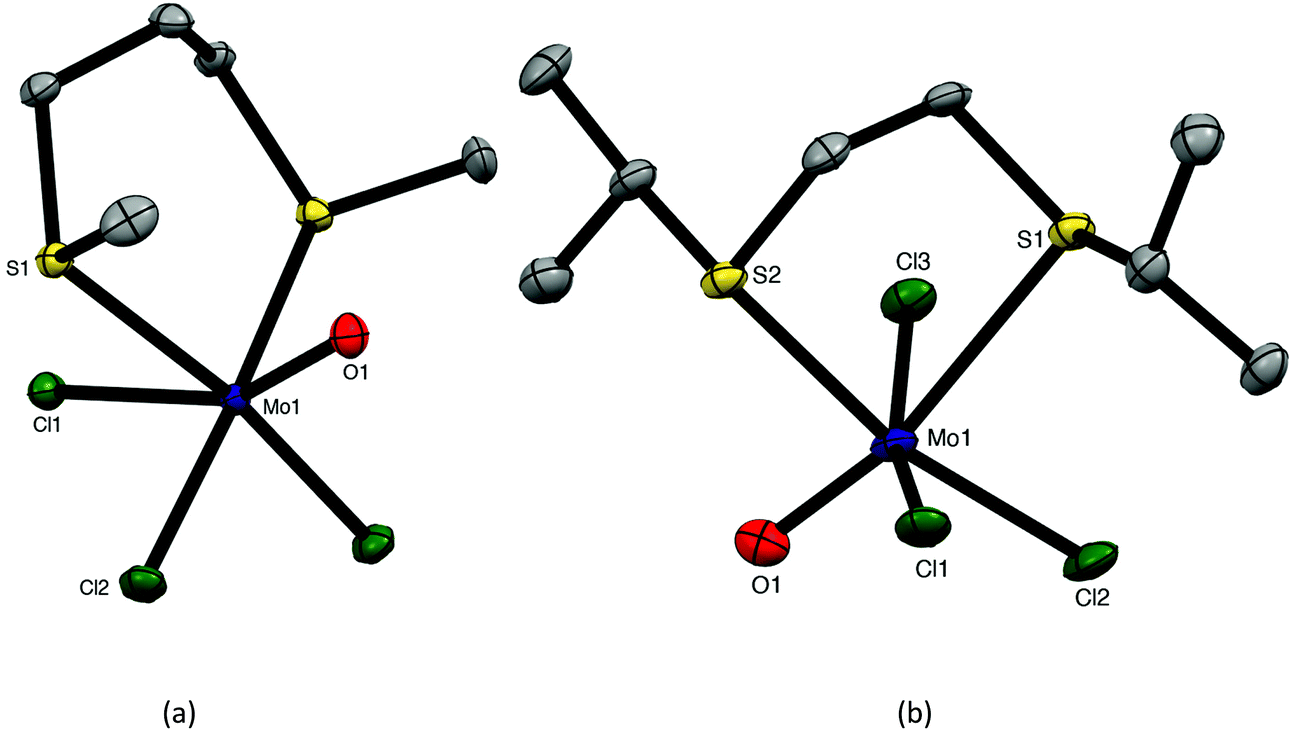 | ||
| Fig. 1 Crystal structures of fac-[MoOCl3{MeS(CH2)3SMe}] (a) and mer-[MoOCl3{iPrS(CH2)2SiPr}] (b) showing the atom numbering scheme. Ellipsoids are shown at 50% probability, hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): (a) Mo1–Cl1 = 2.4540(2), Mo1–Cl2 = 2.3451(2), Mo1–Cl3 = 2.3451(4), Mo1–O1 = 1.674(1), Mo1–S1 = 2.5388(3), Cl1–Mo1–Cl2 = 94.52(2), Cl2–Mo1–Cl3 = 92.55(3), Cl2–Mo1–O1 = 101.87(4), Cl3–Mo1–O1 = 101.87(4), S1–Mo1–S1 = 97.95(2); (b) Mo1–Cl1 = 2.3578(8), Mo1–Cl2 = 2.3378(8), Mo1–Cl3 = 2.3618(7), Mo1–O1 = 1.671(2), Mo1–S1 = 2.8298(8), Mo1–S2 = 2.5665(7), Cl1–Mo1–Cl2 = 91.25(3), Cl1–Mo1–O1 = 98.99(8), Cl1–Mo1–Cl3 = 89.69(3), Cl2–Mo1–O1 = 98.99(8), S1–Mo1–S2 = 78.83(2). | ||
The reaction of MoOCl4 with MeS(CH2)3SMe in dry toluene gave a green product with an identical IR spectrum to that of mer-[MoOCl3{MeS(CH2)3SMe}] and the X-ray structure determination of a crystal obtained from the MoOCl4 synthesis route (Method 2) indeed confirmed it to be the Mo(V) complex. The structural data were identical to that in Table S1,† and hence are not reported, but confirm that thioether ligands reduce MoOCl4 to MoOCl3 complexes, similar to the behaviour reported with some O- and N-donor ligands.23
The weaker σ-donor PhS(CH2)2SPh failed to displace the thf from [MoOCl3(thf)2], but it reacted with a suspension of MoOCl3 in CH2Cl2 to form brown [MoOCl3{PhS(CH2)2SPh}]. The crystal structure of this complex showed it to be the mer-isomer (Fig. 2), which suggests that the ability to form a five-membered chelate ring with a smaller chelate angle (S1–Mo1–S2 = 78.55(3)°) may be an important factor influencing the isomer formed. The structure also reveals a very markedly longer Mo–StransO = 2.911(1) Å, which compares with Mo–StransCl = 2.531(1) Å, showing the high trans-influence of the MoO bond on the weaker aryl thioether donor ligand.
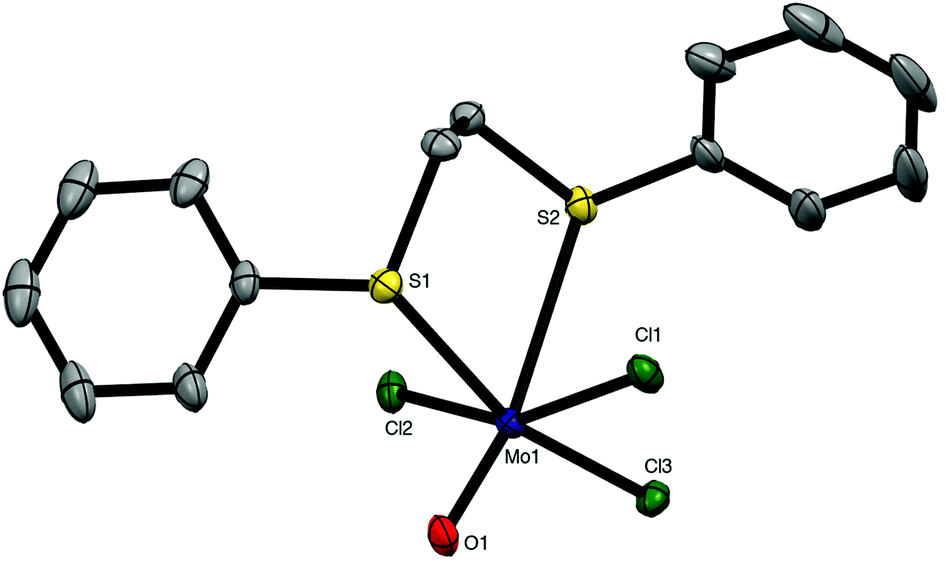 | ||
Fig. 2 Crystal structure of mer-[MoOCl3{PhS(CH2)2SPh}] showing the atom numbering scheme. Ellipsoids are shown at 50% probability and hydrogen atoms are omitted for clarity. Note that the O/Cl exhibited disorder, which was modelled with split atom sites, refined to occupancies of 0.53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.47. Only the major form is shown. Selected bond lengths (Å) and angles (°): Mo1–Cl1 = 2.324(1), Mo1–Cl2 = 2.394(1), Mo1–Cl3 = 2.311(3), Mo1–O1 = 1.706(2), Mo1–S1 = 2.531(1), Mo1–S2 = 2.911(1), Cl1–Mo1–Cl3 = 89.93(7), Cl1–Mo1–O1 = 102.2(5), O1–Mo1–Cl2 = 101.1(5), O1–Mo1–Cl3 = 106.6(3), Cl2–Mo1–Cl3 = 91.28(7), Cl2–Mo1–S1 = 88.39(4), Cl1–Mo1–S2 = 81.49(4), S1–Mo1–S2 = 78.55(3). :0.47. Only the major form is shown. Selected bond lengths (Å) and angles (°): Mo1–Cl1 = 2.324(1), Mo1–Cl2 = 2.394(1), Mo1–Cl3 = 2.311(3), Mo1–O1 = 1.706(2), Mo1–S1 = 2.531(1), Mo1–S2 = 2.911(1), Cl1–Mo1–Cl3 = 89.93(7), Cl1–Mo1–O1 = 102.2(5), O1–Mo1–Cl2 = 101.1(5), O1–Mo1–Cl3 = 106.6(3), Cl2–Mo1–Cl3 = 91.28(7), Cl2–Mo1–S1 = 88.39(4), Cl1–Mo1–S2 = 81.49(4), S1–Mo1–S2 = 78.55(3). | ||
Brownish diselenoether complexes, [MoOCl3(diselenoether)] (diselenoether = MeSe(CH2)2SeMe, MeSe(CH2)3SeMe), were obtained from reaction of the ligands with MoOCl3 or [MoOCl3(thf)2] in a 1:1 molar ratio, but o-C6H4(SeMe)2 did not displace thf from [MoOCl3(thf)2]. The reaction of MeSeCH2SeMe with MoOCl3 produced a black oily decomposition product. However, the 1:1 reaction of o-C6H4(SeMe)2 with MoOCl3 in CH2Cl2 gave a brown product for which the microanalytical data indicated a 2:1 MoOCl3:diselenoether stoichiometry. This is discussed along with the similar ditelluroether complexes below. The X-ray crystal structure of mer-[MoOCl3{MeSe(CH2)2SeMe}] was obtained (Fig. 3).
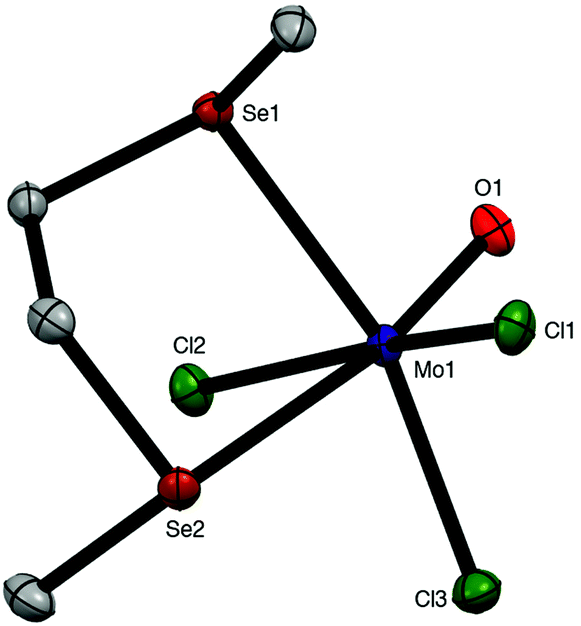 | ||
| Fig. 3 Crystal structure of mer-[MoOCl3{MeSe(CH2)2SeMe}] showing the atom numbering scheme. Ellipsoids are shown at 50% probability and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–Cl1 = 2.3553(5), Mo1–Cl2 = 2.3517(5), Mo1–Cl3 = 2.3453(5), Mo1–O1 = 1.673(1), Mo1–Se1 = 2.6564(2), Mo1–Se2 = 2.8937(3), Cl1–Mo1–Cl3 = 90.35(2), Cl1––Mo1–O1 = 98.98(5), Cl2–Mo1–Cl3 = 90.65(2), Cl3–Mo1–O1 = 107.36(5), Se1–Mo1–Se2 = 79.76(1). | ||
The five complexes described have room temperature magnetic moments of ∼1.7 B.M., similar to other MoOCl3 complexes,1,2,8,9 and close to the spin-only value expected for a d1 complex. This indicates that any orbital contribution is quenched by the very asymmetric field of the molybdenum environment.24 The IR spectra show very strong single bands due to ν(MoO) in the range 950–980 cm−1, as well as strong overlapping bands at 355–300 cm−1 assigned as Mo–Cl modes, but do not appear to readily distinguish the isomer present. The UV/visible spectra of the solids show a clear band at 13000–14000 cm−1 and a second band or shoulder at ∼19000–21000 cm−1. Assuming C4v symmetry (the actual metal centre symmetry is lower) and placing MoO as the dominant contribution along the four-fold axis, leads to the assignment as the d–d bands as 2B2 → 2E and 2B2 → 2B1, respectively.25 The intense absorptions >20000 cm−1, assigned as charge transfer bands, are less clearly resolved, but based upon the usual ligand electronegativities,25 we assign the first intense feature (∼21000–22000 cm−1) as S/Se(π) → Mo(d) and the broad overlapping features at ∼25000–30000 cm−1 as Cl(π) → Mo(d). The complexity of the electronic spectra in compounds of this type is shown by a combined UV/visible absorption, MCD and DFT study of [MoOCl3{Ph2P(CH2)2PPh2}];26 here we are using the spectra to confirm the presence of Mo(V) in the isolated complexes.
Dimethylchalcogenides (EMe2, E = S, Se, Te)
Neither SMe2 or SeMe2 was found to displace thf from [MoOCl3(thf)2]. However, reaction of a suspension of MoOCl3 in dry CH2Cl2 with 2 equivalents of EMe2 produced complexes with a 1:1 Mo:EMe2 empirical composition, MoOCl3(EMe2) (E = S, Se). There was no evidence for the formation of the 1:2 [MoOCl3(EMe2)2] complexes. Crystals of both MoOCl3(EMe2) (E = S, Se) complexes were obtained and the structures, which are isomorphous (Fig. 4), showed them to be chloride-bridged dimers, with six-coordinate Mo(V) centres, i.e. [{MoOCl2(EMe2)}2(μ-Cl2)] (E = S, Se).
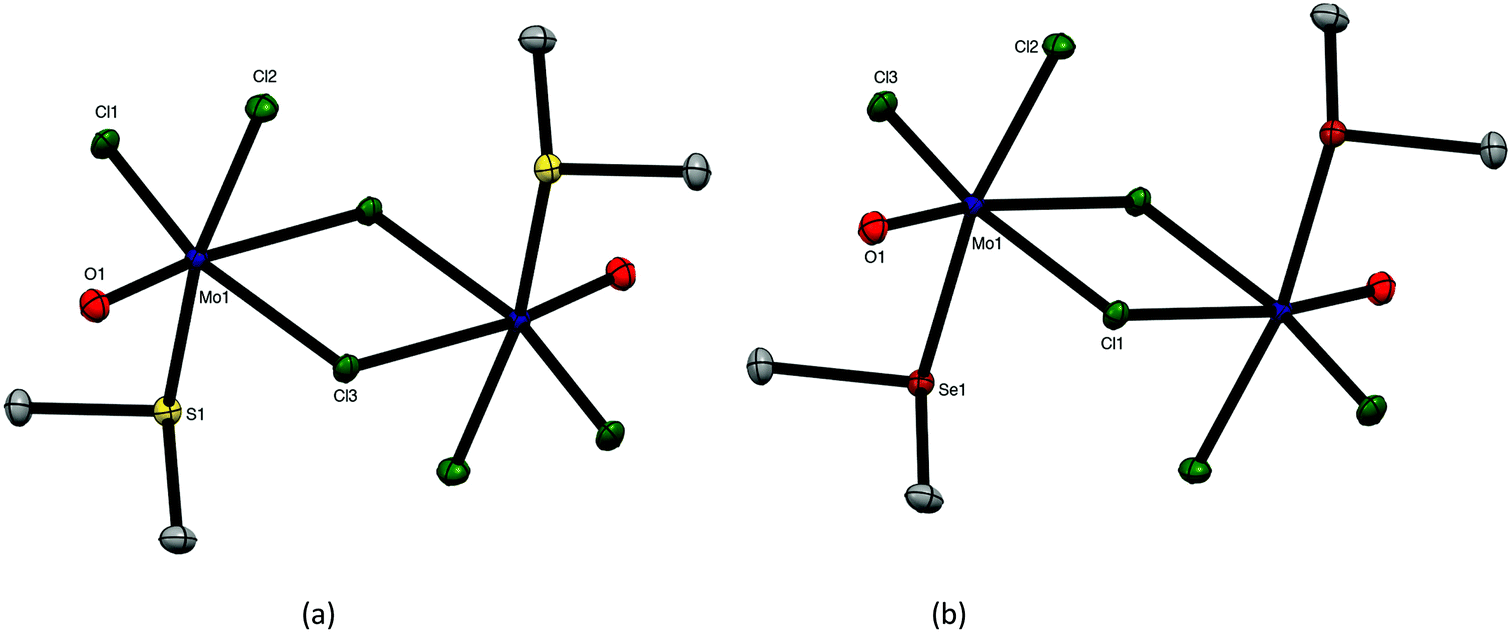 | ||
| Fig. 4 Crystal structures of [{MoOCl2(SMe2)}2(μ-Cl)2] (a) and [{MoOCl2(SeMe2)}2(μ-Cl)2] (b) showing the atom numbering scheme. Ellipsoids are shown at 50% probability and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): (a) Mo1–Cl1 = 2.3264(2), Mo1–Cl2 = 2.3341(3), Mo1–Cl3 = 2.3953(3), Mo1–Cl3′ = 2.7942(2), Mo1–O1 = 1.6515(8), Mo1–S1 = 2.5537(3), Cl1–Mo1–O1 = 102.48(1), Cl2–Mo1–O1 = 102.08(3), Cl3–Mo1–O1 = 99.55(3), Cl2–Mo1–Cl3 = 85.77(1), Cl1–Mo1–Cl3 = 92.340(9), Cl1–Mo1–S1 = 85.233(9), O1–Mo1–S1 = 92.67(3), Cl3–Mo1–Cl3 = 77.520(8); (b) Mo1–Cl1 = 2.4024(4), Mo1–Cl2 = 2.3385(4), Mo1–Cl3 = 2.3299(4), Mo1–Cl1′ = 2.7927(4), Mo1–O1 = 1.653(1), Mo1–Se1 = 2.6647(3), Cl1–Mo1–O1 = 98.89(4), Cl2–Mo1–O1 = 103.10(4), Cl3–Mo1–O1 = 102.38(4), Cl2–Mo1–Cl3 = 92.32(2), Se1–Mo1–Cl1 = 78.76(1), Se1–Mo1–Cl1 = 87.438(10), O1–Mo1–Se1 = 92.89(4). | ||
The MoO bonds (∼1.65 Å) are trans to asymmetrically bound (by ∼0.4 Å) bridging chlorides, with the EMe2 groups arranged anti and perpendicular to the Mo2Cl4O2 plane. The geometries are very similar to those found in [(MoOCl2L)2(μ-Cl)2] (L = OC(H)OMe, thf, O = CEt2).27–29
The reaction of MoOCl3 with TeMe2 in toluene produced brown [{MoOCl2(TeMe2)}2(μ-Cl)2], which is the first Mo(V) complex with a neutral tellurium donor ligand. Crystals were not obtained from this complex due to poor solubility and limited stability in solution, but spectroscopically it is very similar to the other EMe2 complexes. The failure to produce the six-coordinate monomers, [MoOCl3(EMe2)2], even in the presence of excess EMe2, shows that the molybdenum(V) prefers to bind a chloride from another molecule, creating the bridged dimer structure, and is consistent with the weak donor properties of the EMe2. The dimers are clearly distinguished from the [MoOCl3(dichalcogenoether)] monomers by their IR spectra, with the dimers showing a strong ν(MoO) vibration in the range at 985–1005 cm−1 (higher frequency than in the monomeric [MoOCl3(dichalcogenoether)] type) and terminal Mo–Cl modes 360–310 cm−1; weaker bands in the region ∼270–250 cm−1 and absent in the spectra of the [MoOCl3(dichalcogenoether)] monomers, may be due to the chloride bridges. The magnetic moments of ∼1.7 B.M./Mo confirm the Mo(V) assignment and the absence of any magnetic interactions between the molybdenum centres.
Ditelluroethers
The reaction of o-C6H4(SeMe)2, o-C6H4(TeMe)2 and MeTe(CH2)3TeMe (L–L) with MoOCl3 in a 1:1 molar ratio in CH2Cl2 failed to produce the expected [MoOCl3(L–L)] type complexes. Instead, dark brown complexes, identified by microanalysis as [(MoOCl3)2(L–L)], were obtained. Once isolated the compounds are very poorly soluble in CH2Cl2 and many attempts to produce crystals for an X-ray structure determination have been unsuccessful. However, the magnetic moments of ∼1.7 B.M./Mo and the UV-visible spectra of these solids are consistent with their formulation as six-coordinate oxo-molybdenum(V) complexes.
The UV-visible spectra of the ditelluroether complexes show a d–d band at ∼14000 cm−1 (2B2 → 2E); a second more intense feature 18000–20000 cm−1 may be the second d–d band (2B2 → 2B1), but given the lower electronegativity of Te24 is probably the Te(π) → Mo(d) charge transfer transition, which obscures the d–d band.
The IR spectra are significantly different to those of [MoOCl3(L–L)] (L–L = dithioether or diphosphine),8,9 but are similar to those of [{MoOCl2(EMe2)}2(μ-Cl)2]. In particular, the ν(MoO) vibrations are at higher frequency (985–1000 cm−1), and in addition to several terminal ν(Mo–Cl) modes ∼320–300 cm−1, also show a peak ∼250 cm−1, probably due to a chloride bridge. In the absence of a crystal structure, the geometries cannot be established unequivocally, but the spectroscopic data (and insolubility) are consistent with a structure type similar to those in [{MoOCl2(EMe2)}2(μ-Cl)2], with the EMe2 ligands replaced by bridging ditelluroethers, leading to the formulation as an oligomer, [(MoOCl2)2(μ-Cl)2(μ-ditelluroether)]n. There are several literature examples of Group 16 ligands with o-phenylene backbones adopting a bridging mode, authenticated by X-ray crystal structures.30–32
The brown solution from the preparation of [{MoOCl3}2{o-C6H4(TeMe)2}]n also deposited a few dark green crystals, which were shown by X-ray crystallographic analysis to be the mixed valence complex, [MoIVCl{o-C6H4(TeMe)2}2(μ-O)MoVOCl4] (Fig. 5). This complex contains a Mo(IV) centre coordinated to two chelating ditelluroethers, a terminal chloride and an MoO group, which forms a very asymmetric bridge to a square pyramidal MoOCl4− anion, with Mo1–O1 = 1.705(4) Å and Mo2–O1 = 2.368(4) Å. These bond distances may be compared with the terminal MoO bond distance (Mo2–O2 = 1.659(5) Å) in the latter. This complex appears to be the first structurally characterised molybdenum-ditelluroether complex in a positive formal oxidation state of the metal; all previously reported complexes are substituted carbonyls.33,34 Analogous complexes with some diphosphine and diarsine ligands have been reported,8,9 and the structure of (the previously unknown) [MoCl{Me2P(CH2)2PMe2}2(μ-O)(MoOCl4)] is discussed below. The crystals of [MoIVCl{o-C6H4(TeMe)2}2(μ-O)MoVOCl4] result from a redox reaction, and its structure is not consistent with the spectroscopic data on the bulk [{MoOCl3}2{o-C6H4(TeMe)2}]n. The failure to isolate mononuclear [MoOCl3(L–L)] complexes with chelating o-C6H4(SeMe)2, o-C6H4(TeMe)2 and MeTe(CH2)3TeMe, seems analogous to the case of [{MoOCl2(EMe2)}2(μ-Cl)2], where the Mo(V) centre prefers to form chloride bridges rather than coordinate to a second, weakly donating chalcogenenoether.
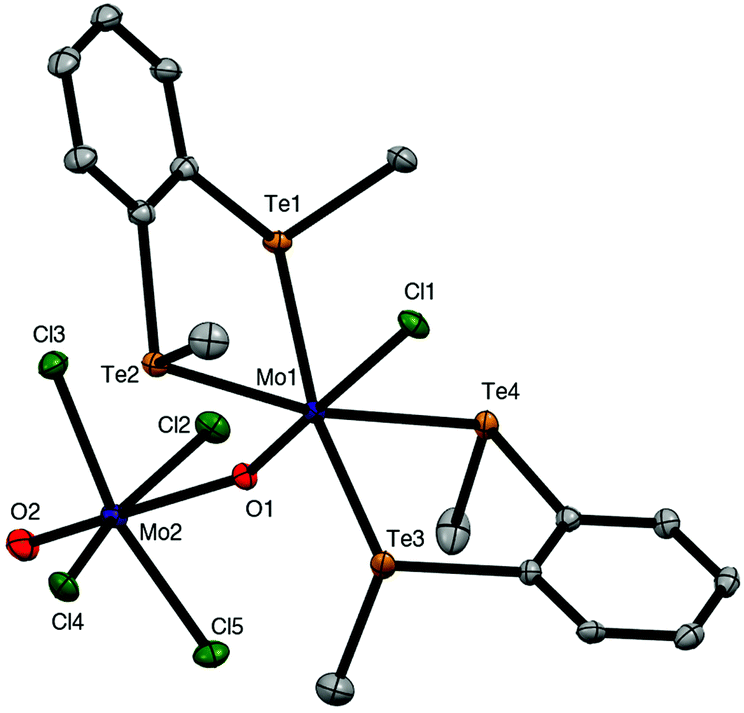 | ||
| Fig. 5 Crystal structure of [MoCl{o-C6H4(TeMe)2}2(μ-O)MoOCl4]·CH2Cl2 showing the atom numbering scheme. Ellipsoids are shown at 50% probability and hydrogen atoms and solvent are omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–Cl1 = 2.4486(7), Mo2–Cl2 – 5 = 2.3640(7) – 2.3930(7), Mo1–Te1 – 4 = 2.7432(3) – 2.7822(3), Mo1–O1 = 1.704(2), Mo2–O1 = 2.370(2), Mo2–O2 = 1.655(2), O2–Mo2–Cl2 – 5 = 78.4(1) – 82.2(1), Te–Mo1–Te(chelate) = 85.316(8), 86.185(8), Te1 – 4–Mo1–Cl1 = 79.647(18)–89.939(18), Cl1–Mo1–O1 = 177.91(7), Mo1–O1–Mo2 = 159.63.1(11). | ||
Phosphine complexes
The coordination behaviour of the chalcogenoether ligands to MoOCl3 has both significant analogies and differences to that of some phosphine ligands, making for informative comparisons. Pink or red complexes [MoOCl3(diphosphine)] (diphosphine = Ph2P(CH2)2PPh2, cis-Ph2PCH=CHPPh2, o-C6H4(PPh2)2,) were reported in the 1970s and confirmed by IR, UV/visible spectroscopy and magnetic measurements as Mo(V) compounds.7–9 No structures were obtained, but EPR spectra supported fac octahedral isomers.8,9 A second (brown) form with Ph2P(CH2)2PPh2 and cis-Ph2PCH = CHPPh2 obtained by refluxing the red form in alcohol for several hours, had similar, but not identical, spectroscopic properties; Isovitsch et al.,10 confirmed the crystal structure of the red form of the Ph2P(CH2)2PPh2 complex as the fac isomer. In the present work we prepared the new complex [MoOCl3{Me2P(CH2)2PMe2}] from [MoOCl3(thf)2] and confirmed the fac geometry by a crystal structure (Fig. 6). The spectroscopic data on this complex (Experimental section) are in good agreement with that of the red isomers with other diphosphines.7–9 Notably, the five-membered chelate ring diphosphine complexes are fac isomers, contrasting with the mer-[MoOCl3(dichalcogenoether)] described above.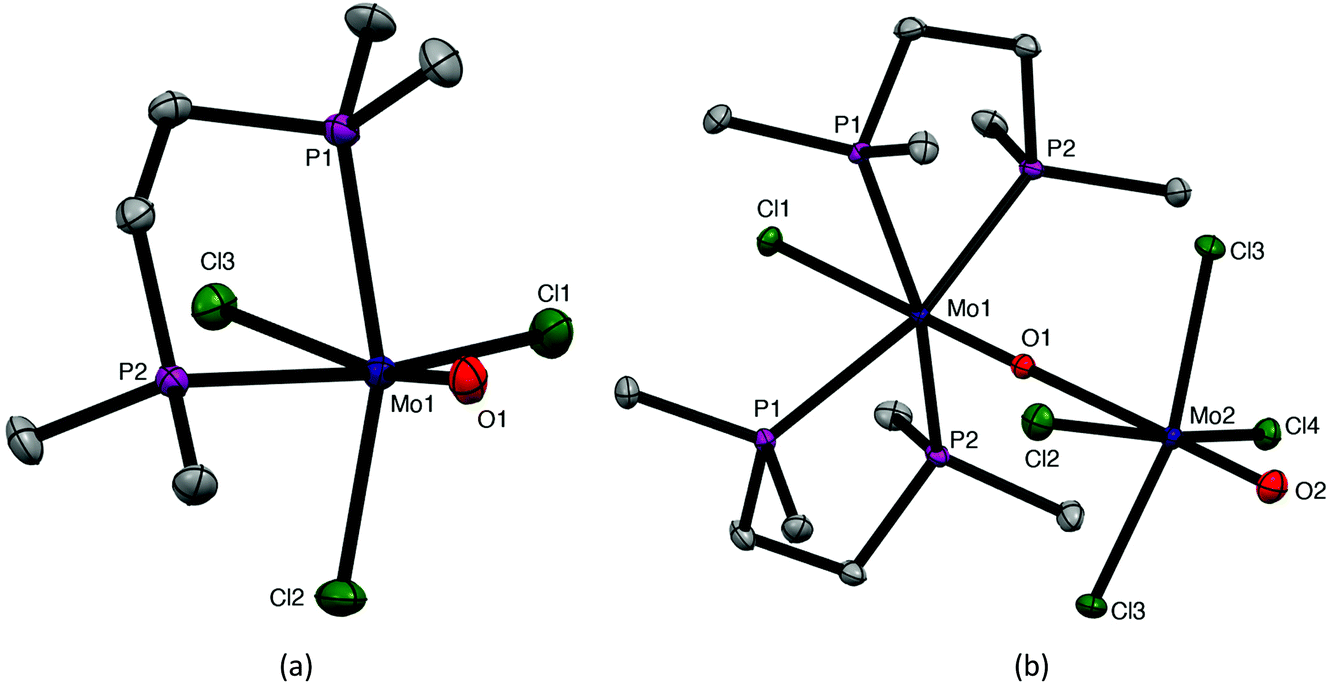 | ||
| Fig. 6 Crystal structures of [MoOCl3(Me2PCH2CH2PMe2)] (a) and [MoCl(Me2PCH2CH2PMe2)2(μ-O)MoOCl4] (b) showing the atom numbering scheme. Ellipsoids are shown at 50% probability and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): (a) Mo1–Cl1 = 2.3824(9), Mo1–Cl2 = 2.383(1), Mo1–Cl3 = 2.5011(8), Mo1–O1 = 1.680(2), Mo1–P1 = 2.5260(2), Mo1–P2 = 2.5250(8), Cl1–Mo1–Cl2 = 96.98(3), Cl1–Mo1–O1 = 100.19(8), Cl2–Mo1–Cl3 = 88.76(3), Cl2–Mo1–O1 = 104.66(8), P1–Mo1–P2 = 78.84(3); (b) Mo1–Cl1 = 2.5186(2), Mo1–O1 = 1.703(4), Mo1–P1 = 2.5131(2), Mo1–P2 = 2.5139(3), Mo2–O1 = 2.394(2), Mo2–O2 = 1.657(8), Mo2–Cl2 – 5 = 2.3573(4) – 2.3824(3), Cl1–Mo1–P1 = 78.92(2), Cl1–Mo1–P2 = 83.26(4), O2–Mo2–Cl2 – 5 = 97.57(2) – 98.89(4), P1–Mo1–P2 = 80.16(4), Mo1–O1–Mo2 = 178.05(8). | ||
The nature of the brown “isomers” is not entirely clear, but the original study8 of the red and brown forms of [MoOCl3{cis-Ph2PCH=CHPPh2}] showed they had identical EPR spectra with coupling to equivalent phosphorus donors, i.e. were both fac forms. Hence the brown form seems likely to be the red isomer co-crystallised with a second complex, probably an EPR silent Mo(IV) species. The presence of varying amounts of a co-crystallised second species would account for the various (small) differences reported by other workers.7,8,10 Similar problems, including X-ray structures with a variety of bond lengths for apparently the same complex, led to the proposal of bond-stretch or distortional isomerism in some other early d-block complexes, a concept subsequently considered to be erroneous.35
Pink or purple complexes with microanalyses indicating a [MoCl2.5O(diphosphine)] were isolated in some systems7–9 and were formulated as the ionic Mo(IV)–Mo(V) species [MoIVOCl(diphosphine)2][MoVOCl4], based upon spectroscopic data, and the observation that metathesis with NaBPh4 gave [MoIVOCl(diphosphine)2][BPh4].
During attempts to grow crystals of orange-yellow [MoOCl3{Me2P(CH2)2PMe2}], a few deep purple crystals were also isolated that were confirmed by an X-ray structure (Fig. 6) to be [MoCl{Me2P(CH2)2PMe2}2(μ-O)(MoOCl4)], analogous to [MoCl{o-C6H4(TeMe)2}2(μ-O)(MoOCl4)] described above. Both molybdenum centres are in a distorted octahedral geometry and linked by a very asymmetric oxide bridge, Mo1–O1 = 1.703(4), Mo2–O1 = 2.394(2) Å, which may be compared with Mo2–O2 = 1.657(8) Å for the terminal MoO unit. The original formulation7,8 was as ionic salts, [MoIVOCl(diphosphine)2][MoVOCl4]. The reformulation as neutral μ-oxido dimers in the solid state is likely to apply to all the reported examples, with the long Mo–O bond easily cleaved to give ions in solution.
Red fac-[MoOCl3(PMe3)2] was obtained by Limberg et al.11 as one product from reaction of the alkoxide complex, [Cl2OMo(μ-OEt)2(μ-EtOH)MoOCl2] with PMe3; we obtained the same complex directly from [MoOCl3(thf)2] and PMe3 in CH2Cl2. Our X-ray structure and the spectroscopy (Experimental section) are in good agreement with published data,7 and are not discussed further here. The interest lies in the formation of a discrete pseudo-octahedral 1:2 Mo:PMe3 monomer with the strong σ-donor alkyl phosphine, which contrasts with the formation of chloride-bridged dimers, [{MoOCl2(EMe2)}2(μ-Cl)2] (E = S, Se, Te), with the weaker donor chalcogenoethers discussed above.
Experimental
Syntheses were performed using standard Schlenk and glovebox techniques under a dry N2 atmosphere. Solvents were dried by distillation from CaH2 (CH2Cl2) or Na/benzophenone ketyl (toluene, n-hexane, diethyl ether). MoCl5 and O(SiMe3)2 were obtained from Sigma-Aldrich. The monodentate ligands (SMe2, PMe3, SeMe2) were obtained from Sigma-Aldrich or Strem and dried over molecular sieves. TeMe2 was made by the method of Kuhn et al.36 The dithioethers,37 diselenoethers38,39 and ditelluroethers40,41 were prepared as described or by minor modifications thereof. MoOCl3 was prepared from MoCl5 and O(SiMe3)2.42 and MoOCl4 obtained from Climax Molybdenum.Infrared spectra were recorded on a PerkinElmer Spectrum 100 spectrometer in the range 4000–200 cm−1, with samples prepared as Nujol mulls between CsI plates. UV/visible spectra were recorded on powdered solids using the diffuse reflectance attachment of a PerkinElmer 750S spectrometer. Magnetic measurements were made using a Johnson Matthey magnetic balance. Microanalyses on new compounds were undertaken by London Metropolitan University or Medac Ltd.
mer-[MoOCl3(thf)2]
Prepared following the literature method.43 Yield: 87%. IR spectrum (Nujol, v/cm−1): 982 s MoO, 1117 s, 833 s br thf, 342 s, 315 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 32550, 26200, 22000, 13250. μeff: 1.71 B.M.
fac-[MoOCl3{MeS(CH2)3SMe}]
O, 348 s, 327 s, 306 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 27400, 26000, 21150, 18350, 13700. μeff: 1.71 B.M.
mer-[MoOCl3{iPrS(CH2)2SiPr}]
[MoOCl3{iPrS(CH2)2SiPr}] was prepared similarly to Method 1 above, and isolated as a pale green solid. Yield: 62%. Required for C8H18Cl3MoOS2 (396.66): C, 24.22; H, 4.57. Found: C: 24.45; H, 4.15%. IR spectrum (Nujol, v/cm−1): 979 s MoO, 349 s, 312 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 32300, 30400, 27700, 23000 sh, 21500 sh, 13600. μeff: 1.69 B.M.
mer-[MoOCl3{PhS(CH2)2SPh}]
MoOCl3 (0.150 g, 0.69 mmol) was suspended in CH2Cl2 (3 mL) and a solution of PhS(CH2)2SPh (0.170 g, 0.69 mmol) in CH2Cl2 (2 mL) was added slowly and the resulting green solution left to stir for 1 h. The resulting brown solution was concentrated to 3 mL in vacuo and filtered, and the orange-brown solid dried in vacuo. Yield: 0.244 g, 76%. Required for C14H14Cl3MoOS2 (464.69): C, 36.19; H, 3.04. Found: C, 35.97; H, 3.18%. IR spectrum (Nujol, v/cm−1): 966 s MoO, 354 s, 319 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 32200, 26900, 22600, 21300, 18500 sh, 13000. μeff: 1.71 B.M.
[{MoOCl2(SMe2)}2(μ-Cl)2]
MoOCl3 (0.150 g, 0.69 mmol) was suspended in CH2Cl2 (3 mL) and a solution of SMe2 (0.085 g, 1.38 mmol) in CH2Cl2 (2 mL) was added slowly and the green solution left to stir for 1 h. The clear green solution was then concentrated to 3 mL in vacuo and layered with hexane (3 mL). The green crystals formed were isolated via filtration and dried in vacuo. Yield: 0.73 g, 38%. Required for C4H12Cl6Mo2O2S2 (560.86): C, 8.57; H, 2.16. Found: C, 8.98; H, 2.37%. IR spectrum (Nujol, v/cm−1): 1004 s MoO, 356 s, 319 s, 268 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 26000, 22300, 19600 sh, 13800. μeff: 1.72 B.M./Mo.
[{MoOCl2(SeMe2)}2(μ-Cl)2]
MoOCl3 (0.150 g, 0.69 mmol) was suspended in CH2Cl2 (3 mL) and a solution of SeMe2 (0.150 g, 1.38 mmol) in CH2Cl2 (2 mL) was slowly added and the green solution left to stir for 1 h. The red solution formed was concentrated to 3 mL in vacuo and layered with hexane (3 mL). The dark brown crystals were isolated via filtration, and dried in vacuo. Yield: 0.154 g, 68%. Required for C4H12Cl6Mo2O2Se2 (654.65): C, 7.34; H, 1.85%. Found: C, 7.43; H, 1.93%. IR spectrum (Nujol, v/cm−1): 1004 s MoO, 368 sh, 351 s, 313 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 32500, 26500, 20700, 14100. μeff: 1.68 B.M./Mo.
mer-[MoOCl3{MeSe(CH2)2SeMe}]
[MoOCl3(thf)2] (0.150 g, 0.41 mmol) was suspended in CH2Cl2 (3 mL) and a solution of MeSe(CH2)3SeMe (0.089 g, 0.41 mmol) in CH2Cl2 (2 mL) was added slowly and the green solution left to stir for 1 h. The resulting brown solution was concentrated to 3 mL in vacuo and filtered, then the solid dried in vacuo, isolating a dark brown solid. Crystals grown from CH2Cl2 were dark green. Yield: 0.160 g, 90%. Required for C4H10Cl3MoOSe2 (434.34): C, 11.06; H, 2.32. Found: C, 11.60; H, 2.50%. IR spectrum (Nujol, v/cm−1): 960 s MoO, 342 s, 310 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 25800, 21500, 19300, 14600. μeff: 1.71 B.M.
[MoOCl3{MeSe(CH2)3SeMe}]
MoOCl3 (0.150 g, 0.69 mmol) was suspended in CH2Cl2 (3 mL) and a solution of MeSe(CH2)3SeMe (0.158 g, 0.69 mmol) in CH2Cl2 (2 mL) was slowly added and the red/brown solution left to stir for 1 h. The brown solution was concentrated to 3 mL in vacuo and filtered and the dark brown solid isolated was dried in vacuo. A deep orange-brown crystalline solid was obtained from CH2Cl2 solution. Yield: 0.178 g, 58%. Required for C5H12Cl3MoOSe2·CH2Cl2 (533.30): C, 13.51; H, 2.65. Found: C, 13.96; H, 2.95%. IR spectrum (Nujol v/cm−1): 954 s MoO, 346 s vbr, Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 32000 sh, 2700 br, 21400, 19500, 14000. μeff: 1.70 B.M.
[(MoOCl3)2{o-C6H4(SeMe)2}]n
MoOCl3 (0.150 g, 0.69 mmol) was suspended in dichloromethane (3 mL) and a solution of o-C6H4(SeMe)2 (0.226 g, 0.69 mmol) in dichloromethane (2 mL) was added slowly and the dark red/brown solution left to stir for 1 h. The brown solution was then concentrated to 3 mL in vacuo, producing a brown precipitate, which was washed with OEt2 (3 × 5 mL), then the brown-pink solid was dried in vacuo. Yield: 0.170 g, 53%. Required for C8H10Cl6Mo2O2Se2 (700.68): C, 13.71; H, 1.44. Found: C, 13.43; H, 1.53%. IR spectrum (Nujol, v/cm−1): 999 s br MoO, 351 w, 302 s, 292 sh, 256 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 29500 sh, 24900, 20900, 14300. μeff: 1.69 B.M./Mo.
[{MoOCl2(TeMe2)}2(μ-Cl)2]
MoOCl3 (0.150 g, 0.69 mmol) was suspended in toluene (3 mL) and a solution of TeMe2 (0.217 g, 1.38 mmol) in toluene (2 mL) was added slowly and the purple solution left to stir for 1 h. The deep purple solution was concentrated to 3 mL in vacuo and filtered, then the dark brown solid was dried in vacuo. Yield: 0.203 g, 78%. Required for C4H12Cl6Mo2O2Te2 (751.93): C, 6.39; H, 1.61. Found: C, 6.76; H, 2.06%. IR spectrum (Nujol, v/cm−1): 985 s br MoO, 327, 302 s br, 256 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 34500, 29700 sh, 27150, 26100, 20900, 19500, 14400. μeff: 1.68 B.M./Mo.
[(MoOCl3)2{MeTe(CH2)3TeMe}]n
MoOCl3 (0.150 g, 0.69 mmol) was suspended in dichloromethane (3 mL) and a solution of MeTe(CH2)3TeMe (0.217 g, 0.69 mmol) in dichloromethane (2 mL) was added slowly and the brown solution left to stir for 1 h. The brown solution was concentrated to 3 mL in vacuo, producing a brown precipitate which was washed with OEt2 (3 × 5 mL), then the dark brown solid was dried in vacuo. Yield: 0.322 g, 61%. Required for C5H12Cl6Mo2O2Te2 (763.95): C, 7.86; H, 1.58. Found: C, 7.20; H, 1.38%. IR spectrum (Nujol, v/cm−1): 988 m MoO, 303 s, 292 m, 249 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 26500, 21700, 18600, ∼13000. μeff: 1.68 B.M./Mo.
[(MoOCl3)2{o-C6H4(TeMe)2}]n
MoOCl3 (0.150 g, 0.69 mmol) was suspended in dichloromethane (3 mL) and a solution of o-C6H4(TeMe)2 (0.249 g, 0.69 mmol) in dichloromethane (2 mL) was added slowly and the dark brown solution left to stir for 1 h. The brown solution was concentrated to 3 mL in vacuo, producing a brown precipitate which was washed with OEt2 (3 × 5 mL), and dried in vacuo. Yield: 0.285 g, 52%. Required for C8H10Cl6Mo2O2Te2 (797.96): C, 12.04; H, 1.26. Found: C, 12.27; H, 1.43%. IR spectrum (Nujol, v/cm−1): 992 s br MoO, 343 m, 328 m, 302 s, 254 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 32500, 25000 sh, 21000, 19200, 14500. μeff: 1.70 B.M./Mo.
fac-[MoOCl3(PMe3)2]
[MoOCl3(thf)2] (0.150 g, 0.41 mmol) was suspended in CH2Cl2 (3 mL) and a solution of PMe3 (0.063 g, 0.82 mmol) in CH2Cl2 (3 mL) was added slowly and the dark green solution left to stir for 1 h. The red solution produced was then concentrated to 3 mL in vacuo and filtered, and the red solid dried in vacuo. Yield: 0.047 g, 31%. Required for C6H18Cl3MoOP2 (370.45): C, 19.45; H, 4.90. Found: C, 19.28; H, 4.74%. IR spectrum (Nujol, v/cm−1): 957 s MoO, 352 sh, 324 s, 305 m Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 29600, 26500, 21600, 20500 sh, 15500.
fac-[MoOCl3{Me2P(CH2)2PMe2}]
[MoOCl3(thf)2] (0.150 g, 0.41 mmol) was suspended in CH2Cl2 (3 mL) and a solution of Me2PCH2CH2PMe2 (0.165 g, 0.41 mmol) in CH2Cl2 (3 mL) was added slowly and the solution left to stir for 1 h. The was concentrated to 3 mL in vacuo, filtered and then the solid was dried in vacuo. Yield: 0.131 g, 81%. Required for C6H16Cl3MoOP2 (368.44): C, 19.56; H, 4.38. Found: C, 19.83; H, 4.26%. IR spectrum (Nujol, v/cm−1): 951 s MoO, 362 m, 325 s, 306 s Mo–Cl. UV/Vis spectrum (diffuse reflectance) ν/cm−1: 29600, 26500 sh, 21600, 20000, 15500. μeff: 1.72 B.M.
X-ray experimental
Crystals were grown from slow evaporation of saturated solutions in CH2Cl2 or by liquid–liquid diffusion using CH2Cl2 and hexane. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 micron focus) with the crystal held at 100 K (N2 cryostream). Crystallographic parameters are presented in Table S1.† Structure solution and refinement were performed using SHELX(T)-2018/2, SHELX-2018/3 through Olex244 and were mostly straightforward. H atoms were added and refined with a riding model. Where additional restraints were required, details are provided in the cif file for each structure found on CCDC.Conclusions
A range of MoOCl3 complexes with thio- and seleno-ethers have been prepared from [MoOCl3(thf)2] and the ligands in anhydrous CH2Cl2 solution. The more weakly coordinating PhS(CH2)2SPh, SMe2 and SeMe2 fail to displace the thf, but complexes of these can be obtained using a suspension of MoOCl3 in CH2Cl2. The reaction of MoOCl4 with dithioethers results in reduction to Mo(V) as [MoOCl3(dithioether)], behaviour which contrasts with that of WOCl4 or WSCl4, where either W(VI) or W(V) complexes can be obtained depending upon the reaction conditions.18 The stabilising effect of two oxido-groups on molybdenum(VI) is shown by the successful isolation of [MoO2X2(dithioether)] (X = Cl or Br).12,13 The limited affinity of the hard MoOCl3 for the weaker donor monochalcogenoethers is reflected in the formation of 1:1 adducts, which achieve six-coordination by forming chloride bridges, as in [{MoOCl2(E'Me2)}2(μ-Cl)2] (E′ = S, Se), rather than by coordinating a second neutral donor ligand. The same explanation accounts for the formation of oligomeric complexes, [(MoOCl3)2(L–L)]n with o-C6H4(SeMe)2, o-C6H4(TeMe)2 and MeTe(CH2)3TeMe, postulated to have a structure with only one chalcogen donor atom on each molybdenum, and where six-coordination is achieved via bridging chlorides and bridging dichalcogenoethers (Scheme 1). Although bridging behaviour might seem unexpected for chelates with o-C6H4-backbones, the presence of aryl groups makes these ligands weaker donors to hard metal centres – compare PhS(CH2)2SPh and iPrS(CH2)2SiPr. There are several literature examples of o-phenylene-based dichalcogenoethers adopting a bridging coordination mode.30–32 The behaviour contrasts with that of o-C6H4-based group 15 ligands, where o-C6H4(PMe2)2 or o-C6H4(AsMe2)2 can produce seven- or eight-coordination in tungsten(VI) complexes, such as [WOCl4{o-C6H4(PMe2)2}] or [WF4{o-C6H4(PMe2)2}2]2+.45,46 The present work has also reported the first examples of Mo(V) telluroether complexes. Although the large soft tellurium centres are not usually thought to be good ligands for high valent d-block metals, a range of compounds has been reported in the last few years, including examples with NbCl4,47 NbCl548 and TaCl5,48 although the complexes reported here are the first examples in Group 6. Also notable is the X-ray structural characterisation of the mixed valence [MoIVCl{o-C6H4(TeMe)2}2(μ-O)MoVOCl4] and of the diphosphine analogue [MoIVCl{Me2P(CH2)2PMe2}2(μ-O)(MoVOCl4)]; complexes of the latter type were reported in the 1970s7,8 but this is the first structural authentication.
The work has provided detailed characterisation of MoOCl3-chalocogenoether complexes, and comparison with the W(VI) and W(V) analogues, and lays the basis for exploration of corresponding molybdenum sulfide chloride complexes,19 which may provide single source LPCVD reagents for deposition of MoS2 thin films. The sulfide chloride systems will form the basis of future work.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank the EPSRC for support (EP/P025137/1) and for a studentship to D. E. S. (EP/N509747/1).References
- E. I. Stiefel, Prog. Inorg. Chem., 1977, 22, 1 CAS.
- E. I. Stiefel, Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, ch. 36.5, pp. 1375 Search PubMed.
- C. D. Garner and J. M. Charnock, Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, ch. 36.4, pp. 1329 Search PubMed.
- C. G. Young, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 4, pp. 415 Search PubMed.
- C. D. Garner, Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, ch. 36.6, pp. 1421 Search PubMed.
- Molybdenum and Tungsten: Their Roles in Biological Processes, ed. A. Sigel and H. Sigel, Marcel Dekker, New York, 2002 Search PubMed.
- A. V. Butcher and J. Chatt, J. Chem. Soc. A, 1971, 2356 RSC.
- W. Levason, C. A. McAuliffe and B. J. Sayle, J. Chem. Soc., Dalton Trans., 1976, 1177 RSC.
- C. A. McAuliffe, B. J. Sayle and W. Levason, J. Chem. Soc., Dalton Trans., 1977, 2055 RSC.
- R. A. Isovitsch, F. R. Fronczec and A. W. Maverick, Polyhedron, 1998, 17, 1617 CrossRef.
- C. Limberg, M. Buechner, K. Heinze and O. Walter, Inorg. Chem., 1997, 36, 872 CrossRef CAS.
- M. D. Brown, M. B. Hursthouse, W. Levason, R. Ratnani and G. Reid, Dalton Trans., 2004, 2487 RSC.
- M. F. Davis, W. Levason, M. E. Light, R. Ratnani, G. Reid, K. Saraswat and M. Webster, Eur. J. Inorg. Chem., 2007, 1903 CrossRef CAS.
- D. Sevdic and I. Fekete, Inorg. Chim. Acta, 1982, 57, 111 CrossRef CAS.
- C. A. McAuliffe and B. J. Sayle, Inorg. Chim. Acta, 1978, 30, 35 CrossRef CAS.
- W. Levason, C. A. McAuliffe, F. P. McCullough, S. G. Murray and C. A. Rice, Inorg. Chim. Acta, 1977, 22, 227 CrossRef CAS.
- C. A. McAuliffe and B. J. Sayle, Inorg. Chim. Acta, 1982, 64, L19 CrossRef CAS.
- D. E. Smith, V. K. Greenacre, A. L. Hector, R. Huang, W. Levason, G. Reid, F. Robinson and S. Thomas, Dalton Trans., 2020, 49, 2496 RSC.
- V. K. Greenacre, W. Levason, G. Reid and D. E. Smith, Coord. Chem. Rev., 2020, 424, 213512 CrossRef CAS.
- S. M. Islam, K. S. Subrahmanyam, C. D. Malliakis and M. G. Kanatzidis, Chem. Mater., 2014, 26, 5151 CrossRef CAS.
- V. C. Gibson, T. P. Kee and A. Shaw, Polyhedron, 1990, 9, 2293 CrossRef CAS.
- G. W. A. Fowles, R. J. Hobson, D. A. Rice and K. J. Shanton, J. Chem. Soc., Chem. Commun., 1976, 552 RSC.
- M. I. Larson and F. W. Moore, Inorg. Chem., 1966, 5, 801 CrossRef CAS.
- B. N. Figgis and J. Lewis, Prog. Inorg. Chem., 1967, 6, 1 CrossRef.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1984 Search PubMed.
- A. Westphal, H. Broda, P. Kurz, F. Neese and F. Tuczek, Inorg. Chem., 2012, 51, 5748 CrossRef CAS.
- S. Dolci, F. Marchetti, G. Pampaloni and S. Zacchini, Eur. J. Inorg. Chem., 2013, 1371 CrossRef CAS.
- L. Favero, F. Marchetti, G. Pampaloni and S. Zacchini, Dalton Trans., 2014, 43, 495 RSC.
- F. Marchetti, G. Pampaloni and S. Zacchini, Dalton Trans., 2013, 42, 2477 RSC.
- J. R. Black, N. R. Champness, W. Levason and G. Reid, Inorg. Chem., 1996, 35, 1820 CrossRef CAS.
- W. Levason, S. Maheshwari, R. Ratnani, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2010, 49, 9036 CrossRef CAS PubMed.
- A. L. Hector, A. Jolleys, W. Levason and G. Reid, Dalton Trans., 2012, 41, 10988 RSC.
- W. Levason, S. D. Orchard and G. Reid, Coord. Chem. Rev., 2002, 225, 159 CrossRef CAS.
- A. J. Barton, W. Levason and G. Reid, J. Organomet. Chem., 1999, 597, 235 CrossRef.
- G. Parkin, Chem. Rev., 1993, 93, 887 CrossRef CAS.
- N. Kuhn, P. Faupel and E. Zauder, J. Organomet. Chem., 1986, 302, C4 CrossRef CAS.
- F. R. Hartley, S. G. Murray, W. Levason, H. E. Soutter and C. A. McAuliffe, Inorg. Chim. Acta, 1979, 265, 35 Search PubMed.
- D. J. Gulliver, E. G. Hope, W. Levason, S. G. Murray, D. M. Potter and G. L. Marshall, J. Chem. Soc., Perkin Trans. 2, 1984, 429 RSC.
- E. G. Hope, T. Kemmitt and W. Levason, J. Chem. Soc., Perkin Trans. 2, 1987, 487 RSC.
- T. Kemmitt and W. Levason, Organometallics, 1989, 8, 1303 CrossRef CAS.
- E. G. Hope, T. Kemmitt and W. Levason, Organometallics, 1988, 7, 78 CrossRef CAS.
- V. C. Gibson, A. Shaw and D. N. Williams, Polyhedron, 1989, 8, 549 CrossRef CAS.
- K. Feenan and G. W. A. Fowles, Inorg. Chem., 1962, 4, 310 CrossRef.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- W. Levason, G. Reid, D. E. Smith and W. Zhang, Polyhedron, 2020, 179, 114372 CrossRef CAS.
- W. Levason, F. M. Monzittu, G. Reid and W. Zhang, Chem. Commun., 2018, 54, 11681 RSC.
- Y.-P. Chang, W. Levason, M. E. Light and G. Reid, Dalton Trans., 2016, 45, 16262 RSC.
- S. L. Benjamin, Y.-P. Chang, C. Gurnani, A. L. Hector, M. Huggon, W. Levason and G. Reid, Dalton Trans., 2014, 43, 16640 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic parameters (Table S1), IR and UV/visible spectra for the new complexes. CCDC 2050667: [{MoOCl2(SeMe2)}(μ-Cl)2], 2050668: [MoOCl3(Me2P(CH2)2PMe2)], 2050669: [MoOCl3(PhS(CH2)2SPh)], 2050670: [MoOCl3(MeSe(CH2)2SeMe)], 2050671: [MoOCl3(iPrS(CH2)2SiPr)], 2050672: [{MoOCl2(SMe2)}(μ-Cl)2], 2050673: [MoCl{Me2P(CH2)2PMe2}2(μ-O)(MoOCl4)], 2050674: [MoOCl3(MeS(CH2)3SMe)] 2050891: [MoCl{o-C6H4(TeMe)2}2(μ-O)MoOCl4]·CH2Cl2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt00038a |
| This journal is © The Royal Society of Chemistry 2021 |