
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determining the photostability of avobenzone in sunscreen formulation models using ultrafast spectroscopy†
Emily L.
Holt
 ab,
Natércia d. N.
Rodrigues
ac,
Juan
Cebrián
c and
Vasilios G.
Stavros
*a
ab,
Natércia d. N.
Rodrigues
ac,
Juan
Cebrián
c and
Vasilios G.
Stavros
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk
bMolecular Analytical Science Centre for Doctoral Training, Senate House, University of Warwick, Coventry, CV4 7AL, UK
cLubrizol Life Science Beauty, Calle Isaac Peral, 17 Pol. Ind. Camí Ral, 08850 Barcelona, Spain
First published on 25th October 2021
Abstract
Avobenzone is an ultraviolet (UV) filter that is often included in sunscreen formulations despite its lack of photostability. Its inclusion is necessary due to few existing alternatives for photoprotection in the UVA region (320–400 nm). To better understand and predict the photostability of avobenzone, ultrafast transient electronic absorption spectroscopy (TEAS) has been used to study the effects of solvent (including emollients), concentration and skin surface temperature on its excited-state relaxation mechanism, following photoexcitation with UVA radiation (∼350 nm). Subtle differences between the excited-state lifetimes were found between the systems, but the TEAS spectral features were qualitatively the same for all solution and temperature combinations. Alongside TEAS measurements, UV filter/emollient blends containing avobenzone were irradiated using simulated solar light and their degradation tracked using steady-state UV-visible spectroscopy. Sun protection factor (SPF) and UVA protection factor (UVA-PF) assessments were also carried out on representative oil phases (higher concentration blends), which could be used to formulate oil-in-water sunscreens. It was found that there was an apparent concentration dependence on the long-term photoprotective efficacy of these mixtures, which could be linked to the ultrafast photodynamics by the presence of a ground-state bleach offset. This combination of techniques shows potential for correlating long-term behaviours (minutes to hours) of avobenzone with its ultrafast photophysics (femtoseconds to nanoseconds), bridging the gap between fundamental photophysics/photochemistry and commercial sunscreen design.
Introduction
Avobenzone (also known as butyl methoxydibenzoylmethane) is a UVA (320–400 nm) filter that is commonly included in sunscreen formulations to protect the skin from the detrimental effects of solar UV radiation.1 Despite its complex photochemistry and reported lack of photostability upon UV light exposure,2,3 its inclusion in formulations is often a necessity due to a lack of approved UVA filters, particularly in the United States.4Avobenzone exists predominantly in a chelated enol form in its ground electronic state, however, photoproducts can form via enol–keto tautomerization, following photoexcitation induced by incident solar rays. The diketo tautomer photoproduct can then undergo further photochemistry that introduces reactive triplet states; these, in turn, can cause indirect degradation of formulations and induce damage on the skin via oxidative stress.3,5,6 Furthermore, the diketo form absorbs UVA radiation much less efficiently than the enol form, thus the level of protection offered by avobenzone-containing formulations after irradiation is decreased.7 The structures of the enol and diketo tautomers, along with their absorption profiles in ethanol before and after irradiation, are shown in Fig. 1. The tautomerisation and subsequent decrease in efficacy of irradiated avobenzone has also been demonstrated using several different comparable methods in earlier publications.7–14
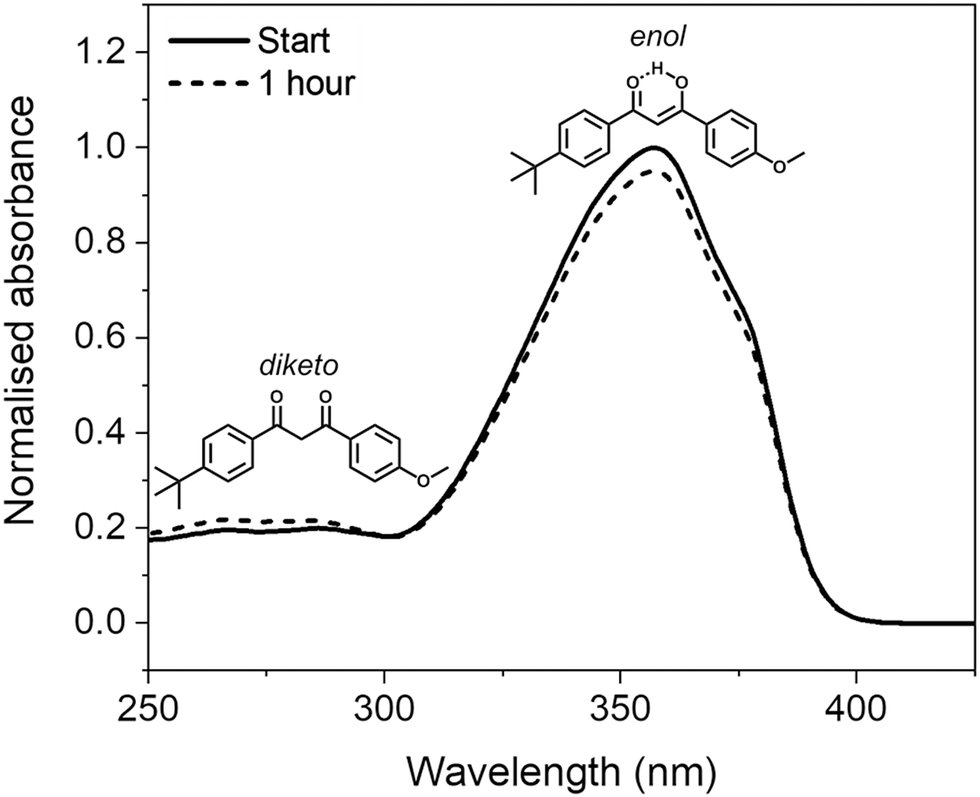 | ||
| Fig. 1 Normalised UV-visible spectra of 5 μg mL−1 (∼16 μM) solution of avobenzone in ethanol taken before and after one hour of irradiation (solid and dotted lines, respectively), using a xenon lamp at solar irradiance (∼1000 W m−2). Inset are the molecular structures of the chelated enol and diketo tautomers of avobenzone that give rise to the spectral peaks at ∼350 nm and ∼270 nm, respectively. | ||
Previous studies have sought to identify ingredients that stabilise the more effective chelated enol form of avobenzone, such as compatible UV filters or antioxidants.15–17 Micellar and lipid microparticle encapsulation of avobenzone have also been shown to have a role in enhancing its photostability.9,18 Deuteration of the diketo methylene/enol moiety affects the tautomerisation equilibrium, by shifting it towards the ketone form.19 Many photofragments of avobenzone have also been identified, which in turn may induce cytotoxic and skin sensitization effects.20–22
Transient electronic absorption spectroscopy (herein abbreviated as TEAS), is an ultrafast pump–probe technique that has been utilised previously to explore the photoprotection mechanisms of many individual UV filters in solution, including avobenzone; additional examples include benzophenone-3, ethylhexyl methoxycinnamate, homosalate, ethylhexyl salicylate, octocrylene and diethylamino hydroxybenzoyl hexyl benzoate.23–28 An ideal sunscreen filter should be able to dissipate excited state energy safely (ideally non-radiatively) and rapidly (within femtoseconds, 1 fs = 10−15 s, to picoseconds, 1 ps = 10−12 s). This reduces the likelihood of harmful side reactions such as forming triplet states, or fragmentation of the molecule.29,30 Furthermore, a rapid return to the ground state after photoexcitation implies that the molecule quickly becomes available to absorb UV radiation again. To investigate properties such as these, the femtosecond resolution of TEAS can be used to full advantage to determine the excited-state relaxation mechanisms of UV filters in real-time.31
A previous TEAS study of avobenzone in cyclohexane, methanol and acetonitrile by Dunkelberger et al. identified changes to its electronic excited-state lifetime that were dependent on solvent polarity.23 Solvent-dependent effects have also been observed by chromatography, NMR and UV-visible spectroscopy;7–10,32,33 these techniques (amongst others) are often used in conjunction with TEAS to correlate the long-term behaviours (minutes to hours) of molecules such as avobenzone, with their ultrafast photophysical processes. Computational methods such as density functional theory (DFT), including time-dependent DFT (TD-DFT), can also be used to guide the assessment of a molecule's suitability for sunscreen use.28,34,35 As an example, Wang et al. include both experimental (including ultrafast) data and computational methods in their study of UV-induced photochemical reactions in diketo avobenzone derivatives.36
However, TEAS studies have not been widely used in conjunction with industry-standard efficacy tests that sunscreen formulations are subject to, such as sun protection factor (SPF) and UVA protection factor (UVA-PF) determination. These tests are ultimately conducted in vivo on human volunteers, but due to practical and ethical considerations are initially performed using analogous in vitro techniques.37,38In vitro testing involves applying a thin layer (up to 2.5 mg cm−2) of a sunscreen to a synthetic skin substrate and determining the SPF/UVA-PF via spectrophotometric absorbance and/or transmittance measurements.31,39In vitro SPF values are both measured and predicted upon solar irradiation by Binks et al., in their study of mixtures (solutions and emulsions) of UV filters including avobenzone, using squalane and propane-1,2-diol as solvent.13
A fundamental understanding of the underlying photochemical processes that UV filters (such as avobenzone) undergo is crucial for improving overall sunscreen performance. However, if these studies could mimic the final sunscreen and conditions of use more closely, they may better inform formulation design. Therefore, the present study aims to enhance the understanding of what causes instability of avobenzone on a molecular level and how this may affect long-term sunscreen performance. Importantly, the TEAS methods used in this study have been extended to more closely mimic real-life conditions. We demonstrate, to the best of our knowledge, the first examples of TEAS measurements targeting the effects of skin surface temperature on avobenzone photostability in industry-relevant emollients, compared to ethanol and cyclohexane. Temperature effects in avobenzone and related enol benzoylacetones observed previously include differing decay rates of avobenzone photoproducts and changes to the intramolecular H-bond strength, respectively.3,40 In addition to TEAS, steady-state UV-visible spectroscopy and SPF/UVA-PF tests were used to correlate how the ultrafast dynamics could be influencing the long-term absorption profile of avobenzone upon prolonged irradiation, in combination with additional UV filters.
Alongside recent spectroscopic advances examining the photodynamics of UV sunscreen filter candidates on skin mimics at room temperature,41–43 our temperature-dependent TEAS setup could be incorporated into studies of this type in the future. These latest developments further increase the applicability of laser spectroscopy methods to commercial sunscreen design.
Experimental
Sample preparation
The avobenzone (Eusolex® 9020 by Merck) studied in solution and emollient mixtures was used as purchased without further purification. Solutions of avobenzone for ultrafast TEAS measurements were prepared to a concentration of ∼10 mM (equivalent to ∼3 mg mL−1) and ∼1 mM (equivalent to ∼0.3 mg mL−1) in ethanol (absolute, Fisher Scientific) and cyclohexane (>99.9%, VWR Chemicals), respectively, and ∼10 mM in the cosmetic grade emollients diisopropyl adipate (Schercemol™ DIA ester, referred to as DIA) and lauryl lactate (Schercemol™ LL ester, herein LL), both provided by Lubrizol. All solvents and emollients were used as acquired.For the data presented in Fig. 1, a solution of avobenzone in ethanol was prepared to an exact concentration of 5 μg mL−1 (∼16 μM). For the steady-state UV-visible spectroscopy measurements shown in Fig. 3, mixtures of avobenzone and additional UV filters in DIA and LL, were prepared by combining ∼4 μg mL−1 of avobenzone, ∼10 μg mL−1 ethylhexyl methoxycinnamate (EHMC, Eusolex® 2292, Merck) and ∼13 μg mL−1 octocrylene (Eusolex® OCR, Merck) in each emollient and stirred until the filters were dissolved. These masses correspond to a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7.5:10 (avobenzone:EHMC:octocrylene), mimicking the ratios of the maximum permitted w/w% in a completed sunscreen blend in the United States.
:7.5:10 (avobenzone:EHMC:octocrylene), mimicking the ratios of the maximum permitted w/w% in a completed sunscreen blend in the United States.
For sun protection factor (SPF) and UVA-PF testing (more details on the exact methodologies for these tests are given below), five oil phase samples containing varying amounts of avobenzone, EHMC and octocrylene in emollient were made. These mixtures were prepared in batches of 25 g; Table S1 (ESI†) shows the mass of each ingredient included in each batch. The oil phases were made by combining the ingredients in a beaker and mixing using an overhead mechanical stirrer until a homogeneous mixture was achieved. The mixture was gently heated on a hot plate (∼30 °C) as it was mixed to allow for the solvation of avobenzone. Once the oil phase mixture was clear, stirring and heating were stopped, and the mixture was left to cool to room temperature. Samples 1 and 2 contain a mixture of the three UV filters (avobenzone, EHMC and octocrylene) in the same proportion as the samples for steady-state measurements, i.e., 3:7.5:10, respectively, differing only in the emollient used: DIA was used for Sample 1 and LL for Sample 2. Samples 3 and 4 constitute a 5-fold and 10-fold decrease in the w/w% of UV filters respectively, compared to Sample 1. Finally, Sample 5 reverses the w/w% of EHMC and octocrylene included in the blend, compared to Sample 1. For comparison to the concentrations used for TEAS measurements, the 10 mM solution in ethanol is approximately equal to a concentration of 0.4% w/w.
Instrumentation
:1 ratio to create the pump and probe beams needed for TEAS experiments. The 2.5 W portion of the fundamental 800 nm beam is converted into the 350 nm required for photoexcitation using an optical parametric amplifier (TOPAS Prime with UV extension, Light Conversion). The power of the pump beam was recorded to be approximately 0.5 mW at the sample, which corresponds to a fluence of 0.65 mJ cm−2. This was kept consistent except for the ∼1 mM solutions in ethanol and cyclohexane, where the fluence was increased, up to ∼1.1 mJ cm−2, to increase signal. Only ∼5% power of the remaining 1 W fundamental beam is required to generate the supercontinuum white-light (320–720 nm) probe pulses; this is achieved by focusing the fundamental 800 nm beam (< 50 mW) onto a vertically translated CaF2 crystal (2.5 cm diameter, 2 mm thick). The relative polarization between the pump and probe is maintained at magic angle (54.7°). The different time delays are created by a gold retroreflector, mounted on to an automatic motorised delay stage in the probe beam line, which lengthens or shortens this beam path according to the requirements of the experiment. An optical chopper blocks every other excitation pulse to attain the reported difference spectra between “pump on” and “pump off”. All reported TEAS datasets were the average of at least 5 scans, with a scan comprising a data matrix of all wavelengths and time delays. Each time delay in turn averages a total of 2500 “shots”, i.e. pump-on, pump off pairs. Each set of ≥ 5 scans were repeated twice, using freshly made samples on separate days, with similar results both times.
All transients were analysed with the software package Glotaran45 using global kinetic analysis with a parallel model. A parallel model sums exponential functions that decay independently of one another.46 The implementation of this method and its assumptions have been excellently summarised in the review by Beckwith et al.,47 briefly, a parallel model assumes that all dynamic processes occur instantaneously and concurrently following excitation. For presentation purposes, all transient absorption spectra were chirp-corrected using the KOALA package detailed in the publication by Grubb et al.48 Characterisation of the instrument response is given in the ESI† (Supplementary Methods and Fig. S1).
Increasing the sample temperature to ∼35 °C in the interaction region, to mimic the surface temperature of the skin, was attained by using a temperature-controllable flow-through cell (Harrick Scientific), which was connected via a K-type thermocouple to a home-built controller enclosing Panasonic KT4 electronics. The path length of the sample was 25 μm as set by PTFE spacers placed between two CaF2 windows. The CaF2 windows were 2.5 cm in diameter and 2 mm thick. The sample was photoexcited at ∼350 nm and replenished between each pair of pulses using a diaphragm pump (Simdos 02). For measurements at room temperature (recorded as being between ∼21–23 °C), all apparatus and procedures remained the same, except that the heater was switched off for the duration of these measurements.
Results and discussion
The photostability and photoprotection afforded by avobenzone were assessed using TEAS at skin surface temperature to reflect real-world conditions of use, by heating the sample at the interaction region. A review of the literature, which is included in the Supplementary Discussion in the ESI,† concludes that any temperature within the range 32–38 °C is justifiable as a skin surface temperature for investigations relating to sunscreens; therefore, the experiments reported here were performed at 35 °C as the midpoint of this range. The results for the ∼10 mM ethanol and cyclohexane solutions at skin surface temperature are shown in the top two rows of Fig. 2, with equivalent measurements for solutions at room temperature and with a 10-fold reduction in concentration to ∼1 mM, shown in Fig. S2 and S3 (ESI†), respectively. The TEAS results for the ∼10 mM avobenzone/emollient solutions, diisopropyl adipate (DIA) and lauryl lactate (LL), also shown in Fig. 2 will be returned to below, once the dynamics in the volatile solvents have been discussed.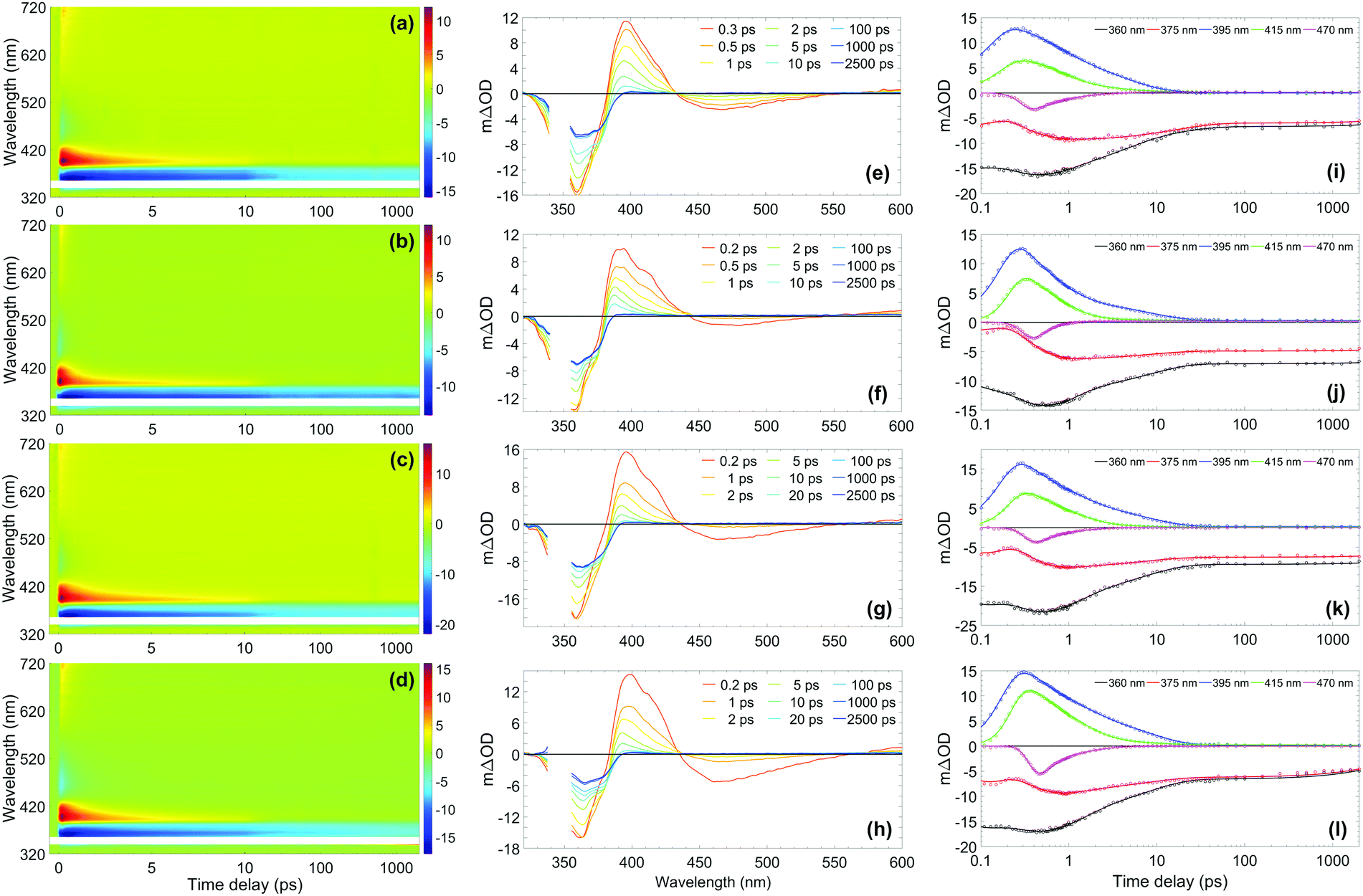 | ||
| Fig. 2 (Left-hand column) Two-dimensional colour maps showing all transient absorption spectra (TAS) of ∼10 mM avobenzone photoexcited at ∼350 nm, with sample cell temperature set to 35 °C, in (a) ethanol, (b) cyclohexane, (c) diisopropyl adipate (DIA) and (d) lauryl lactate (LL). The region between 340–355 nm has been omitted due to oversaturation from the pump pulse. Zoomed-in false colour maps of the wavelength region > 400 nm can be found in the ESI† (Fig. S9). (Middle column) TAS at selected pump–probe time delays for avobenzone in (e) ethanol, (f) cyclohexane, (g) DIA and (h) LL. These plots are attained by taking vertical slices through the colour maps at the given time delay and are presented using the same mΔOD scale as the corresponding colour map, with the same wavelength region omitted. (Right-hand column) Lineouts to show spectral features at specific probe wavelengths in (i) ethanol, (j) cyclohexane, (k) DIA and (l) LL. The features at 360 nm and 375 nm are ground-state bleach features, 395 nm and 415 nm are excited-state absorption features, and 470 nm is stimulated emission. The circles denote the raw datapoints and the solid line in each case is the fit attained using a parallel model (time constants from fitting are presented in Table 1). The time delay axis is presented on a logarithmic scale. Time delays prior to 0.1 ps are presented on a linear scale in the ESI† (Fig. S6 for ethanol and cyclohexane, Fig. S7 for DIA and LL). | ||
From the results in Fig. 2, the spectral features (which appear instantaneously following photoexcitation) are comparable between the ethanol and cyclohexane datasets, as such the following description applies to both solute/solvent combinations. The overlap of probe wavelengths with the ground-state absorption region of avobenzone gives rise to the intense ground-state bleach (GSB) feature between 330–380 nm; a more detailed exploration into the implications of the incomplete GSB recovery observed will follow. It is noted that the GSB region is partially obscured due to pump bleaching artefacts. Other significant spectral characteristics include an excited-state absorption (ESA) between 380 and 430 nm, and a short-lived stimulated emission (SE) between 450 and 550 nm. These ESA and SE features have been observed previously by Dunkelberger et al.23 in TEAS measurements of avobenzone in methanol and cyclohexane, and also in the work of Verma et al., who performed TEAS on avobenzone precursors: dibenzoylmethane (DBM) and benzoylacetone (BZA) in acetonitrile and hexane at room temperature.50,51 Combining the insights from these room-temperature studies23,50,51 with the results of our quantitative global kinetic fits using a parallel model, presented in Table 1 at skin surface temperature and in Table S3 (ESI†) at room temperature, informed our assignment of: spectral features (Fig. 2 and Fig. S2, ESI†), associated lifetimes τ1–τ4 (Table 1), and the potential effect of the increased temperature. Also, for reference, the decay associated spectra (DAS) obtained from the global fits of avobenzone (in all four solvents) are presented in Fig. S4 of the ESI.† A sequential model, of the general form  was also considered for the extraction of spectral features, where n is the number of lifetimes required to describe the data. However, a parallel model was deemed appropriate considering the quality of the fits, reflected in the lineouts (Fig. 2) and the residuals (Fig. S10, ESI†). A parallel model could also prove more suitable if mixtures of UV filters were to be investigated using TEAS, as multiple molecular processes arising from different molecules would be induced simultaneously, at time zero (Δt = 0).
was also considered for the extraction of spectral features, where n is the number of lifetimes required to describe the data. However, a parallel model was deemed appropriate considering the quality of the fits, reflected in the lineouts (Fig. 2) and the residuals (Fig. S10, ESI†). A parallel model could also prove more suitable if mixtures of UV filters were to be investigated using TEAS, as multiple molecular processes arising from different molecules would be induced simultaneously, at time zero (Δt = 0).
| Time constant | Ethanol | Cyclohexane | Diisopropyl adipate (DIA) | Lauryl lactate (LL) |
|---|---|---|---|---|
| a Outside the time-window of the instrument. | ||||
| τ 1 (fs) | 150 ± 140 | 235 ± 80 | 165 ± 140 | 195 ± 140 |
| τ 2 (ps) | 1.2 ± 0.1 | 0.70 ± 0.08 | 1.0 ± 0.1 | 1.2 ± 0.1 |
| τ 3 (ps) | 8.2 ± 0.1 | 8.4 ± 0.1 | 7.7 ± 0.1 | 8.2 ± 0.1 |
| τ 4 (ns) | >2.5a | >2.5a | >2.5a | >2.5a |
In all cases following photoexcitation, τ1 represents the evolution of the chelated enol form of avobenzone from the Franck–Condon region. It is possible that there are additional ultrafast processes occurring within this time on a proportion of the excited molecules, such as excited-state intramolecular proton transfer, or rotation around the C–O single bond of the hydroxyl group to form non-chelated avobenzone species. These branching pathways have both been suggested in previous studies,12,51,52 although they cannot be confirmed from the evidence presented in this study as τ1 is close to the instrument response timescale.
The lifetime assigned to τ2 relates to the decay of the initially excited S1 state of the chelated enol form of avobenzone via SE, with the faster decay occurring in non-polar cyclohexane. The lineouts at 470 nm in Fig. 2(i) and (j) confirm that the SE does not persist much beyond 1–2 ps in either solvent. This was also observed both in the room temperature lineouts presented in Fig. S5 (ESI†) and also by Dunkelberger et al.23 in their room temperature study of avobenzone, which is the first indication that the temperature increase has had little effect on photodynamics. The timescale of the SE is very short compared to the much longer timescales that would be expected for fluorescence. This indicates that long-lived radiative decay (> ns), which would be of concern for sunscreen applications, does not occur in avobenzone. However, a low intensity positive offset is present in the lineouts at 550 nm at both temperatures (Fig. S8, ESI†), which persists beyond the 2.5 ns timescale of our experiment. At 600 nm, following the decay of the initial ESA as observed in Fig. S8 and S9 (ESI†), a similar offset to that at 550 nm is present. These offsets are akin to similar data presented by Verma et al.50 in DBM at 538 nm and could be attributed to the formation of a triplet state of enolic avobenzone. Such states have previously been proposed as an additional route to the formation of the diketo photoproduct of avobenzone and photodegradation of dibenzoylmethane compounds.50,53
To investigate this possibility further, TD-DFT was used to calculate the energies of the singlet and triplet states of the chelated enol and diketo forms of avobenzone. Table S4 (ESI†) shows the optimised ground-state structures attained using DFT, alongside the calculated vertical excitation energies and state characters of the first five singlet and triplet states using TD-DFT. Table S5 shows the calculated triplet state energies using the ΔSCF method, which is generally considered to be more accurate for T1 energy determination.54 These calculations have returned values of between 460 and 500 nm for the absorption of the T1 relative to the ground state. Furthermore, the T4(3nπ*) state is close in energy to the S1(1ππ*) state, further supporting the observation of fast intersystem crossing from the chelated enol form of avobenzone, in accordance with El Sayed's rule. The singlet-state energies calculated for the chelated enol form align well with experimental values (e.g.Fig. 1); moreover both the singlet and triplet state energies attained are comparable to those in previous computational studies.55,56
To complete the assignment of the time constants given in Table 1, τ3 is assigned to the lifetime of the vibrationally hot chelated enol form of avobenzone in the ground electronic state (S0). In Fig. 2(e) and (f), a blue-shifting ESA peak with concurrent narrowing, a characteristic manifestation of vibrational cooling,57 can be seen in the transients up to and including 10 ps. From the lineouts at 395 nm and 415 nm, the ESA has completely decayed after around 25 ps. The ESA decay coincides almost exactly with the maximum GSB recovery within the time delay limits of our experiments, in both the heated and room temperature models. Both in the case of avobenzone and DBM, these coincident recovery and decay rates are indicative of an intermediate species returning to its original, ground state form. Dunkelberger et al.23 also suggest that, after UVA photoexcitation, the vibrationally excited S0 chelated enol form of avobenzone relaxes in the same timeframe as the GSB recovers. Verma et al.50 also observed a concurrence between the decay of the GSB and the ESA for DBM in acetonitrile, where a complete recovery of the positive ESA feature at 385 nm is observed within 14 ps, leaving behind a constant (negative) GSB offset. The excess energy inducing vibrational motion in the ground electronic state, can then dissipate safely as heat into the surrounding environment, in this case the solvent. Such formation of a vibrationally excited species is considered a favourable relaxation pathway. The main difference, albeit relatively minor, between the two sample temperatures was the lifetime of τ3 in ethanol, where a ∼1 ps decrease in lifetime was observed under heating to skin surface temperature.
The negative offset at 360 and 375 nm, which remains constant from 25 ps up to the remaining time window of our experiments, reveals an incomplete GSB recovery, denoted by τ4 (> 2.5 ns). The extent of GSB recovery is of significant interest for sunscreen applications. Ideally, a 100% GSB recovery would be observed within the TEAS time delay window, which would indicate that all excited-state molecules have recovered to their initial ground-state form. Thereafter, the molecule can reabsorb UV radiation and perform its role as a sunscreen filter. A close to full recovery (i.e. return to zero signal) is observed in UV filters such as octocrylene,27 however, this is not the case with avobenzone, as previously evidenced. The most likely explanation for the incomplete GSB recovery in this instance is the formation of one or more photoproducts, such as long-lived photoisomers. Such isomers have been suggested in earlier laser flash photolysis experiments, with lifetimes on the order of milliseconds.3,12 A photoproduct cannot always be detected using TEAS as it may absorb outside the spectral window of the probe, or its appearance may manifest as a convolution of spectral features. For example, for the asymmetric molecule BZA, there was an observed increase in the ESA region with respect to time,51 which was assigned to the formation of further non-chelated enol species separate to that caused by hydroxyl bond torsion; a scenario that has also been suggested for avobenzone.23 As such, in this instance, it is challenging to quantitatively determine GSB recovery percentage for avobenzone in our experiments, as there may be a convolution of signals from the formation of photoproducts in the GSB region. Furthermore, although not possible to determine an exact quantum yield, a small percentage of the photoexcited avobenzone molecules are likely to undergo intersystem crossing, given the proximity of 1ππ* states to 3nπ* states, as discussed earlier. It follows, therefore, that formation of triplet states is an additional contributing factor to the incomplete GSB recovery.
The effect of reducing the concentration of avobenzone 10-fold from 10 mM to 1 mM was also investigated at both temperatures (Fig. S3, ESI†). This concentration reduction had no qualitative effect on the dynamics observed for avobenzone, nor was there any significant effect observed when the temperature was increased at the lower concentration. It was not possible to test concentrations > 10 mM, as the optical density of the sample was too high at the 25 μm path length. In addition, owing to the similarities of the TEAS spectra at both room (Fig. S2, ESI†) and skin surface temperature (Fig. 2), it can be confidently stated that the increase in temperature does not appear to have significant impact on the photoprotection mechanism of avobenzone, under these experimental conditions.
The knowledge gained on avobenzone (and comparable molecules) in ethanol and cyclohexane can now be used to interpret its behaviours in two emollients at skin surface temperature, representing a cosmetically more relevant system. The effect that emollients can have on sunscreen performance has been of recent experimental interest.58 TEAS measurements at ∼35 °C were performed in two emollients, DIA and LL. The measurements in emollient were conducted at surface skin temperature only, to determine any differences between the solvents in this closer to real-life model. The results are shown in the bottom two rows of Fig. 2, and the global fitting analysis results are shown alongside those for ethanol and cyclohexane in Table 1, to aid comparison. Both datasets reveal similar spectral features; these features, in turn, are also qualitatively comparable to those observed in ethanol and cyclohexane as discussed above. The slight quantitative differences in the time constants, are most likely due to minor variations in emollient polarity. Upon closer inspection of Fig. 2(l), showing the lineouts in LL, there appears to be the beginnings of a GSB recovery in the lineouts at 360 and 375 nm within the time window of our TEAS setup, in contrast to the three other solvents. This could indicate that, the relaxation of an intermediate species can occur faster in LL compared to the other solvents. Overall, the parallels between the TEAS measurements in Fig. 2 indicate that the way avobenzone dissipates excess UV energy on ultrafast timescales is mostly unaffected by the solvent environment, in agreement with the findings of Dunkelberger et al.23 Furthermore, it can be deduced that the concentration of avobenzone does not appear to have a noticeable impact upon its ultrafast photodynamics within the 1–10 mM range.
To investigate the properties of avobenzone in emollients further, in keeping with a bottom-up approach, mixtures of UV filters including avobenzone were made and exposed to solar-simulated radiation for two hours. The results of the steady-state UV-visible spectroscopy used to track photodegradation (Fig. 3) suggest that ∼4 μg mL−1 avobenzone is photostable in DIA and LL, in mixtures where ethylhexyl methoxycinnamate (EHMC) and octocrylene are also present in low concentrations (∼10 μg mL−1 and ∼13 μg mL−1 respectively). This can be seen as the absorbance in the UVA region has remained constant for the duration of the irradiation time. It was not possible to observe whether the diketo photoproduct of avobenzone was being formed, due to saturation of the detector in the UV-visible spectrometer at wavelengths < 290 nm caused by emollient (solvent) absorption. The reduction in the absorption of these mixtures in the region of 290 – 320 nm is assigned to the trans–cis isomerization that is known to occur in EHMC.59–61 Although absorption of the UVB wavelengths is not affected by this isomerisation (i.e. there is no observed red or blue shift), the extinction coefficient of the cis species is markedly lower than the trans species, thus a reduction in absorption in the UVB region is observed.61 The concentrations used for the irradiation studies in Fig. 3 were markedly lower compared to a completed sunscreen formulation (around 104 times more dilute), however the concentrations were maximised to be suitable for this type of analysis.
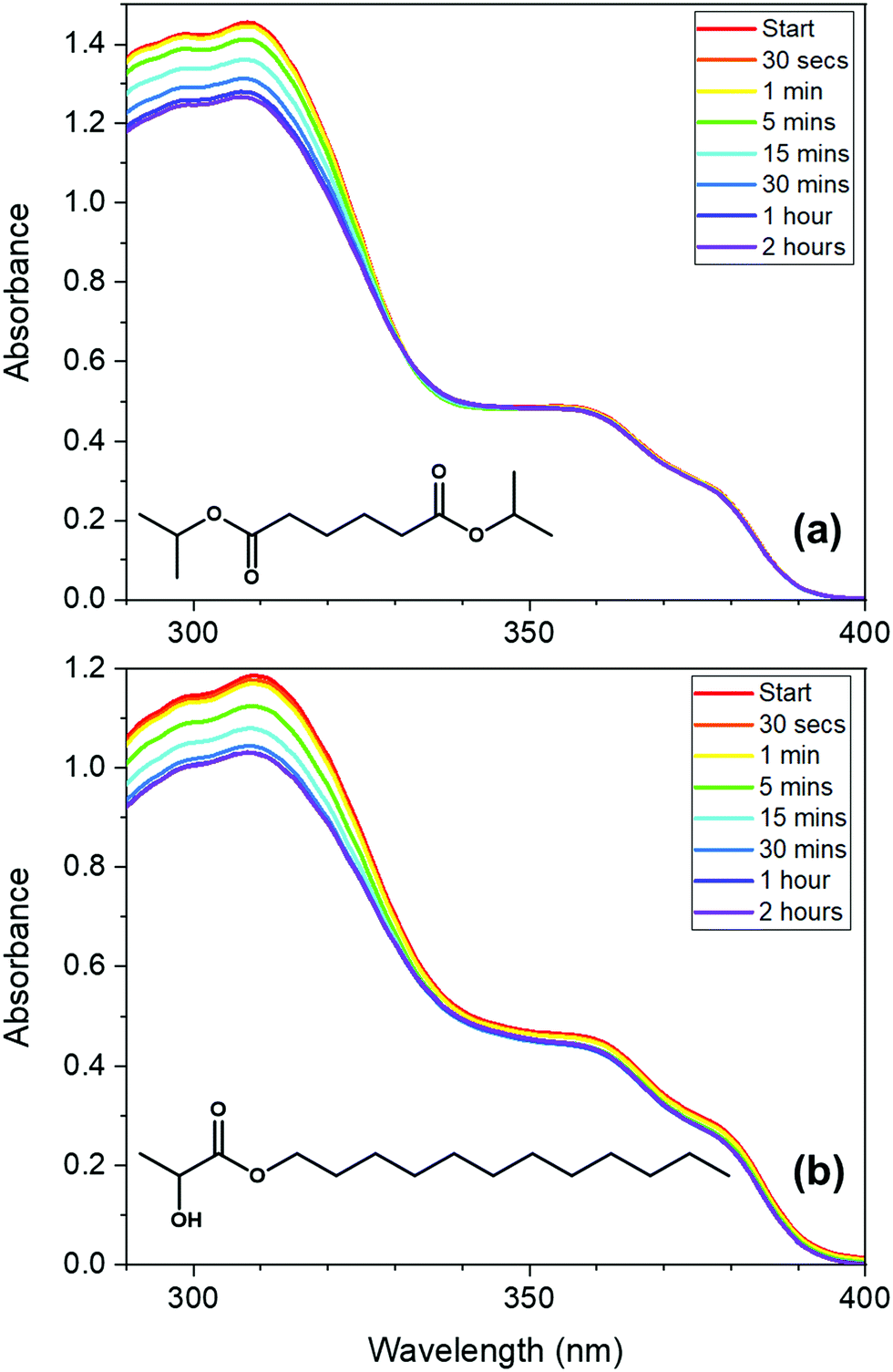 | ||
| Fig. 3 UV-visible absorption spectra of low-concentration avobenzone, EHMC and octocrylene mixtures (∼4 μg mL−1 of avobenzone, ∼10 μg mL−1 EHMC and ∼13 μg mL−1 octocrylene; 3:7.5:10 ratio), taken following irradiation using a solar simulator at regular intervals between 0 and 2 hours, in the following emollients: (a) diisopropyl adipate (DIA) and (b) lauryl lactate (LL). The molecular structure of the emollients is inset on the respective graphs. The spectra at each time point were measured three times consecutively, then averaged to attain the presented data. This was repeated twice on different days with new sample made with the same bulk ingredients, with similar results. | ||
Next, in vitro SPF and UVA-PF values were measured for the five oil phase samples (components detailed in Table S1, ESI†); the results are shown in Table 2. Samples 1–4 contain the same proportion of the three UV filters as the samples used to attain the results in Fig. 3, however SPF and UVA-PF protocols are designed for concentrations in line with those in a completed sunscreen formulation. By using these protocols, the effect of including additional UV filters in an avobenzone/emollient solution could be investigated at clinically relevant concentrations. The most concentrated samples (1 and 2) based upon DIA and LL respectively, demonstrate a similar performance overall, with the percentage decrease in SPF and UVA-PF following irradiation being slightly more favourable in Sample 2. However, upon comparison of Samples 1, 3 and 4, all of which have DIA as the base emollient, there appears to be some variation in performance when the concentrations of the UV filters are changed. Sample 1 has the active ingredients in the highest concentration, followed by Sample 3 (5-fold dilution compared to Sample 1), with Sample 4 having the lowest concentration (a 10-fold reduction compared to Sample 1). From the results in Table 2, it appears that the extent of degradation decreases as the concentration of filters is reduced. It is possible that the emollients are effective in dissipating excited-state energy from the UV filters via energy transfer (heating) from the filter to the solvent, thus minimising degradation. However, the more likely scenario is that there is added protection from a higher proportion of emollient due to spectral overlap, with the absorbance region of the emollients beginning at around 400 nm. This dependence may also explain why almost no degradation in the UVA region is observed in the UV-Vis spectra in Fig. 3, which measured the same ratio of filters in the lowest concentration.
| Test | Sample | % Change SPF | % Change UVA-PF |
|---|---|---|---|
| Different emollients | 1 | −15.8 ± 2.2 | −21.4 ± 1.6 |
| 2 | −14.9 ± 0.9 | −20.3 ± 1.1 | |
| Different concentration of UV filters | 3 | −13.6 ± 1.6 | −9.3 ± 1.2 |
| 4 | −9.7 ± 1.9 | −3.7 ± 1.6 | |
| Different proportions of UV filters | 5 | −21.3 ± 1.3 | −28.0 ± 1.4 |
To determine whether intermolecular interactions that can drive photodegradation occur in the bulk oil phase, Sample 1 and Sample 5 were compared. The amount of avobenzone is the same between each of these samples (equivalent to 2.2% w/w in an oil-in-water formulation, assuming 25% oil), but the proportions of EHMC and octocrylene have been reversed. It is known that EHMC adversely affects the photostability of avobenzone;62–64 conversely, octocrylene improves avobenzone's photostability via triplet–triplet energy state quenching.65–67 This is in line with observations between Sample 1 and Sample 5 (Table 2), whereby the percentage decrease in SPF and UVA-PF is larger for Sample 5 in both cases, which contains the greater proportion of EHMC. The study by Kikuchi et al.62 also concluded that the addition of EHMC to a mixture of avobenzone and octocrylene harms photostability. The results in Table 2 suggest that diffusion-limited energy transfer mechanisms between the UV filters in the mixture are likely have a role to play in the photostability of avobenzone, as previously investigated in the work of Yagi and co-workers,62,68–70 alongside the reduction in photodegradation due to spectral protection (solvent absorbing UV radiation rather than the UV filter).67
It is noted that we opted not to study blends using TEAS for the present work, as a convolution of TEAS signals of all three UV filters may have masked avobenzone signals of interest. We aim, however, to further develop our TEAS setup in future experiments to assess the effect of these intermolecular energy transfer processes by: (1) extending the time-window of our measurements from fs through to ms; and (2) investigating higher concentration effects by minimising path length through the sample via model skin surface studies. The former could also be of great benefit for a more detailed assessment of the effect of GSB recovery on the long-term SPF and UVA-PF metrics.
This section is concluded by considering the TEAS measurements alongside the steady-state SPF and UVA-PF measurements. It is unsurprising that there is a notable reduction in SPF and UVA-PF of Samples 1–5, when one considers the incomplete GSB recovery seen in Fig. 2. Long-lived photoproducts of avobenzone (the presence of which is indicated by the incomplete recovery) not only reduce the UVA protection it provides; they also have the potential to disrupt the efficacy of sunscreen blends as photoproducts can undergo further light-induced processes. TEAS detects the early onset of photoproducts and the routes by which they are formed, which we suggest may offer an alternative tool for increasing emulsion photostability. For example, improvement could be realised by adding or changing functional groups within UV filters such as avobenzone71 towards increasing GSB recovery. Assuming there is no convolution of positive signals in the GSB region, this will indicate that the proportion of UV filter that returns to its electronic ground state has increased, thus reducing the drop in SPF and UVA-PF values. Encouraging evidence that this approach could be successful is indicated in the TEAS data and SPF/UVA-PF metrics for avobenzone in LL, whereby a marginal GSB recovery was observed towards the end of the TEAS spectral window, which is reflected in the slightly improved performance of LL in the SPF and UVA-PF tests compared to DIA.
Conclusions
In this work, the photoprotection mechanism of the UVA filter avobenzone has been studied using transient electronic absorption spectroscopy (TEAS), building upon prior research.23 Specifically, we have tracked the excited state dynamics of avobenzone solutions at elevated temperatures, by heating the sample interaction region to skin surface temperature, thus aligning with closer-to-real-world sunscreen usage. The TEAS results have shown that upon UVA excitation, the excited-state relaxation mechanisms of avobenzone in ethanol, cyclohexane, diisopropyl adipate and lauryl lactate (the latter two being commercial emollients) are very similar. This observation holds whether these TEAS data are taken at skin surface temperature, room temperature and/or with a 10-fold reduction in concentration. In the future, it may be of interest to investigate whether larger deviations in temperature result in directly observable changes in intramolecular H-bond strength, or the decay rate of non-chelated enol photoproducts of avobenzone, as suggested in earlier publications.3,40The present study demonstrates that TEAS, supported here by DFT and steady-state irradiation methodologies, can be used as a complementary analysis tool alongside industry-standard techniques for evaluating sunscreen performance (SPF and UVA-PF testing). By garnering fundamental insight into the ultrafast photodynamics of sunscreen molecules, molecular design of optimal UV filters can be predicted; for example, maximising ground state bleach (GSB) recovery in TEAS measurements limits the potential photodegradation of the UV filter. It would be beneficial for future studies to monitor the evolution of the GSB beyond the nanosecond timescales possible here, to further affirm the link between ultrafast observations and long-term photostability. This, in turn, may lead to improved suncare formulations.
Author contributions
E. L. H. was responsible for preparing the manuscript, data curation, validation, formal analysis, investigation and visualization of the TEAS, steady-state and computational data. N. d. N. R. was responsible for data curation, investigation, formal analysis and validation of the SPF and UVA-PF data. J. C. provided resources, V. G. S. provided supervision and acquired funding. All authors contributed to the conceptualization of the study and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. L. H. thanks the Engineering and Physical Sciences Research Council (EPSRC) for a PhD studentship through the EPSRC Centre for Doctoral Training in Molecular Analytical Science, grant number EP/L015307/1; also, the Warwick Research Technology Platform for Scientific Computing and the HPC Midlands+ consortium (EPSRC grant number EP/P020232) for computational resources. N. d. N. R. acknowledges support and funding from the European Union's Horizon 2020 research and innovation programme under grant agreement number 844177—SUNNRL. V. G. S. thanks the Royal Society for a Royal Society Industry Fellowship and the EPSRC under Grant EP/N010825 for equipment funds (WCUS). The authors would also like to thank Dr Laurent Blasco for the expertise and enthusiasm he provided during helpful discussions, and Dr Claudio Ribeiro (Lubrizol) for conducting preliminary skin surface temperature measurements in São Paulo, Brazil. Finally, the authors thank Rod Wesson (University of Warwick) for building the temperature controller used in this work.References
- S. Q. Wang, P. R. Tanner, H. W. Lim and J. F. Nash, Photochem. Photobiol. Sci., 2013, 12, 197–202 CrossRef CAS PubMed.
- I. Andrae, A. Bringhen, F. Böhm, H. Gonzenbach, T. Hill, L. Mulroy and T. G. Truscott, J. Photochem. Photobiol., B, 1997, 37, 147–150 CrossRef CAS.
- A. Cantrell and D. J. McGarvey, J. Photochem. Photobiol., B, 2001, 64, 117–122 CrossRef CAS.
- J. B. Mancuso, R. Maruthi, S. Q. Wang and H. W. Lim, Am. J. Clin. Dermatol., 2017, 18, 643–650 CrossRef PubMed.
- C. Paris, V. Lhiaubet-Vallet, O. Jimenez, C. Trullas and M. A. Miranda, Photochem. Photobiol., 2009, 85, 178–184 CrossRef CAS PubMed.
- S. Osseiran, E. M. Roider, H. Wang, Y. Suita, M. Murphy, D. E. Fisher and C. L. Evans, J. Biomed. Opt., 2017, 22, 125004 Search PubMed.
- S. Afonso, K. Horita, J. P. S. eSilva, I. F. Almeida, M. H. Amaral, P. A. Lobão, P. C. Costa, M. S. Miranda, J. C. G. E. da Silva and J. M. S. Lobo, J. Photochem. Photobiol., B, 2014, 140, 36–40 CrossRef CAS PubMed.
- G. J. Mturi and B. S. Martincigh, J. Photochem. Photobiol., A, 2008, 200, 410–420 CrossRef CAS.
- K. M. Hanson, M. Cutuli, T. Rivas, M. Antuna, J. Saoub, N. T. Tierce and C. J. Bardeen, Photochem. Photobiol. Sci., 2020, 19, 390–398 CrossRef CAS PubMed.
- J. J. Vallejo, M. Mesa and C. Gallardo, Vitae, 2011, 18, 63–71 CAS.
- A. R. Abid, B. Marciniak, T. Pędziński and M. Shahid, J. Photochem. Photobiol., A, 2017, 332, 241–250 CrossRef CAS.
- M. Yamaji and M. Kida, J. Phys. Chem. A, 2013, 117, 1946–1951 CrossRef CAS PubMed.
- B. P. Binks, P. D. Fletcher, A. J. Johnson, I. Marinopoulos, J. Crowther and M. A. Thompson, J. Photochem. Photobiol., A, 2017, 333, 186–199 CrossRef CAS.
- S. Moi, B. Hosamani, K. Kumar, S. Gunaga, S. Raghothama and K. H. Gowd, J. Photochem. Photobiol., A, 2021, 420, 113488 CrossRef CAS.
- N. A. Shaath, Photochem. Photobiol. Sci., 2010, 9, 464–469 CrossRef CAS PubMed.
- P. Govindu, B. Hosamani, S. Moi, D. Venkatachalam, S. Asha, V. John, V. Sandeep and K. Gowd, Photochem. Photobiol. Sci., 2019, 18, 198–207 CrossRef CAS PubMed.
- J. Kockler, S. Robertson, M. Oelgemöller, M. Davies, B. Bowden, H. G. Brittain and B. D. Glass, Profiles of Drug Substances, Excipients and Related Methodology, Elsevier, 2013, vol. 38, pp. 87–111 Search PubMed.
- S. Scalia and M. Mezzena, AAPS PharmSciTech, 2009, 10, 384–390 CrossRef CAS PubMed.
- R. B. Murphy, J. Staton, A. Rawal and T. A. Darwish, Photochem. Photobiol. Sci., 2020, 19, 1410–1422 CrossRef CAS PubMed.
- I. Karlsson, L. Hillerström, A.-L. Stenfeldt, J. Mårtensson and A. Börje, Chem. Res. Toxicol., 2009, 22, 1881–1892 Search PubMed.
- W. Schwack and T. Rudolph, J. Photochem. Photobiol., B, 1995, 28, 229–234 CrossRef CAS.
- J. A. Berenbeim, N. G. K. Wong, M. C. R. Cockett, G. Berden, J. Oomens, A. M. Rijs and C. E. H. Dessent, J. Phys. Chem. A, 2020, 124, 2919–2930 CrossRef CAS PubMed.
- A. D. Dunkelberger, R. D. Kieda, B. M. Marsh and F. F. Crim, J. Phys. Chem. A, 2015, 119, 6155–6161 CrossRef CAS PubMed.
- L. A. Baker, M. D. Horbury, S. E. Greenough, P. M. Coulter, T. N. V. Karsili, G. M. Roberts, A. J. Orr-Ewing, M. N. R. Ashfold and V. G. Stavros, J. Phys. Chem. Lett., 2015, 6, 1363–1368 CrossRef CAS PubMed.
- Y. Peperstraete, M. Staniforth, L. A. Baker, N. D. Rodrigues, N. C. Cole-Filipiak, W.-D. Quan and V. G. Stavros, Phys. Chem. Chem. Phys., 2016, 18, 28140–28149 RSC.
- E. L. Holt, K. M. Krokidi, M. A. P. Turner, P. Mishra, T. S. Zwier, N. d. N. Rodrigues and V. G. Stavros, Phys. Chem. Chem. Phys., 2020, 22, 15509–15519 RSC.
- L. A. Baker, M. D. Horbury and V. G. Stavros, Opt. Express, 2016, 24, 10700–10709 CrossRef CAS PubMed.
- M.-H. Kao, R. K. Venkatraman, M. Sneha, M. Wilton and A. J. Orr-Ewing, J. Phys. Chem. A, 2021, 125, 636–645 CrossRef CAS PubMed.
- N. d. N. Rodrigues, N. C. Cole-Filipiak, M. A. P. Turner, K. Krokidi, G. L. Thornton, G. W. Richings, N. D. M. Hine and V. G. Stavros, Chem. Phys., 2018, 515, 596–602 CrossRef CAS.
- L. A. Baker and V. G. Stavros, Sci. Prog., 2016, 99, 282–311 CrossRef CAS PubMed.
- E. L. Holt and V. G. Stavros, Int. Rev. Phys. Chem., 2019, 38, 243–285 Search PubMed.
- S. P. Huong, E. Rocher, J.-D. Fourneron, L. Charles, V. Monnier, H. Bun and V. Andrieu, J. Photochem. Photobiol., A, 2008, 196, 106–112 CrossRef CAS.
- P. Trebse, O. Polyakova, M. Baranova, M. Kralj, D. Dolenc, M. Sarakha, A. Kutin and A. Lebedev, Water Res., 2016, 101, 95–102 CrossRef CAS PubMed.
- J. V. T. Gomes, A. C. P. da Silva, M. L. Bello, C. R. Rodrigues and B. A. M. C. Santos, J. Mol. Model., 2019, 25, 1–11 CrossRef PubMed.
- C. T.-L. Chan, C. Ma, R. C.-T. Chan, H.-M. Ou, H.-X. Xie, A. K.-W. Wong, M.-L. Wang and W.-M. Kwok, Phys. Chem. Chem. Phys., 2020, 22, 8006–8020 RSC.
- J. Wang, Y. Lei, Y. Guo, J. Wang and J. Ma, Photochem. Photobiol. Sci., 2019, 18, 3000–3007 CrossRef CAS PubMed.
- L. Ferrero, M. Pissavini and O. Doucet, Photochem. Photobiol. Sci., 2010, 9, 540–551 CrossRef CAS PubMed.
- J. Stanfield, U. Osterwalder and B. Herzog, Photochem. Photobiol. Sci., 2010, 9, 489–494 CrossRef CAS PubMed.
- A. Springsteen, R. Yurek, M. Frazier and K. F. Carr, Anal. Chim. Acta, 1999, 380, 155–164 CrossRef CAS.
- E. V. Borisov, E. V. Skorodumov, V. M. Pachevskaya and P. E. Hansen, Magn. Reson. Chem., 2005, 43, 992–998 CrossRef CAS PubMed.
- M. D. Horbury, E. L. Holt, L. M. M. Mouterde, P. Balaguer, J. Cebrián, L. Blasco, F. Allais and V. G. Stavros, Nat. Commun., 2019, 10, 4748 CrossRef PubMed.
- Y. Liu, X. Zhao, J. Luo and S. Yang, J. Lumin., 2019, 206, 469–473 CrossRef CAS.
- T. T. Abiola, N. d. N. Rodrigues, C. Ho, D. J. Coxon, M. D. Horbury, J. M. Toldo, M. T. do Casal, B. Rioux, C. Peyrot and M. M. Mention, J. Phys. Chem. Lett., 2020, 12, 337–344 CrossRef PubMed.
- J. M. Woolley, M. Staniforth, M. D. Horbury, G. W. Richings, M. Wills and V. G. Stavros, J. Phys. Chem. Lett., 2018, 9, 3043–3048 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- L. J. G. W. van Wilderen, C. N. Lincoln and J. J. van Thor, PLoS One, 2011, 6, e17373 CrossRef CAS PubMed.
- J. Beckwith, C. Rumble and E. Vauthey, Int. Rev. Phys. Chem., 2020, 39, 135–216 Search PubMed.
- M. P. Grubb, A. J. Orr-Ewing and M. N. R. Ashfold, Rev. Sci. Instrum., 2014, 85, 064104 CrossRef PubMed.
- G. Ni, G. Li, S. V. Boriskina, H. Li, W. Yang, T. Zhang and G. Chen, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- P. K. Verma, F. Koch, A. Steinbacher, P. Nuernberger and T. Brixner, J. Am. Chem. Soc., 2014, 136, 14981–14989 CrossRef CAS PubMed.
- P. K. Verma, A. Steinbacher, F. Koch, P. Nuernberger and T. Brixner, Phys. Chem. Chem. Phys., 2015, 17, 8459–8466 RSC.
- S. Tobita, J. Ohba, K. Nakagawa and H. Shizuka, J. Photochem. Photobiol., A, 1995, 92, 61–67 CrossRef CAS.
- A. Aspée, C. Aliaga and J. Scaiano, Photochem. Photobiol., 2007, 83, 481–485 CrossRef PubMed.
- R. J. Charlton, R. M. Fogarty, S. Bogatko, T. J. Zuehlsdorff, N. D. M. Hine, M. Heeney, A. P. Horsfield and P. D. Haynes, J. Chem. Phys., 2018, 148, 104108 CrossRef CAS PubMed.
- M. Kojić, M. Petković and M. Etinski, Phys. Chem. Chem. Phys., 2016, 18, 22168–22178 RSC.
- G. H. G. Trossini, V. G. Maltarollo, R. D. A. Garcia, C. A. S. O. Pinto, M. V. R. Velasco, K. M. Honorio and A. R. Baby, J. Mol. Model., 2015, 21, 319 CrossRef PubMed.
- M. D. Horbury, W.-D. Quan, A. Flourat, F. Allais and V. G. Stavros, Phys. Chem. Chem. Phys., 2017, 19, 21127–21131 RSC.
- M. Sohn, L. Amorós-Galicia, S. Krus, K. Martin and B. Herzog, J. Photochem. Photobiol., B, 2020, 205, 111818 CrossRef CAS PubMed.
- L. R. Gaspar and P. M. Campos, Int. J. Pharm., 2006, 307, 123–128 CrossRef CAS PubMed.
- K. M. Hanson, S. Narayanan, V. M. Nichols and C. J. Bardeen, Photochem. Photobiol. Sci., 2015, 14, 1607–1616 CrossRef CAS PubMed.
- S. Pattanaargson, T. Munhapol, N. Hirunsupachot and P. Luangthongaram, J. Photochem. Photobiol., A, 2004, 161, 269–274 CrossRef CAS.
- A. Kikuchi, Y. Nakabai, N. Oguchi-Fujiyama, K. Miyazawa and M. Yagi, J. Lumin., 2015, 166, 203–208 CrossRef CAS.
- R. M. Sayre, J. C. Dowdy, A. J. Gerwig, W. J. Shlelds and R. V. Lioyd, Photochem. Photobiol., 2005, 81, 452–456 CrossRef CAS PubMed.
- D. Dondi, A. Albini and N. Serpone, Photochem. Photobiol. Sci., 2006, 5, 835–843 CrossRef CAS PubMed.
- V. Lhiaubet-Vallet, M. Marin, O. Jimenez, O. Gorchs, C. Trullas and M. A. Miranda, Photochem. Photobiol. Sci., 2010, 9, 552–558 CrossRef CAS PubMed.
- D. G. Beasley and T. A. Meyer, Am. J. Clin. Dermatol., 2010, 11, 413–421 CrossRef PubMed.
- B. Herzog, J. Giesinger and V. Settels, Photochem. Photobiol. Sci., 2020, 19, 1636–1649 CrossRef CAS PubMed.
- S. Matsumoto, R. Kumasaka, M. Yagi and A. Kikuchi, J. Photochem. Photobiol., A, 2017, 346, 396–400 CrossRef CAS.
- R. Kumasaka, A. Kikuchi and M. Yagi, Photochem. Photobiol., 2014, 90, 727–733 CAS.
- A. Kikuchi and M. Yagi, Chem. Phys. Lett., 2011, 513, 63–66 CrossRef CAS.
- J. Zawadiak and M. Mrzyczek, Spectrochim. Acta, Part A, 2012, 96, 815–819 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/d1cp03610f. The underlying data of this publication can be accessed via the Zenodo archive at DOI: 10.5281/zenodo.5531347 |
| This journal is © the Owner Societies 2021 |