
Synthesis of β-cyanoalkylsulfonylated vinyl selenides through a four-component reaction†
Fu-Sheng
He
*a,
Yanfang
Yao
ab,
Zhimei
Tang
a,
Yanjie
Qiu
a,
Wenlin
Xie
b and
Jie
Wu
 *acd
*acd
aSchool of Pharmaceutical and Materials Engineering & Institute for Advanced Studies, Taizhou University, 1139 Shifu Avenue, Taizhou 318000, China. E-mail: jie_wu@fudan.edu.cn; hefs@tzc.edu.cn
bSchool of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
dSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
First published on 28th October 2021
Abstract
A mild copper-catalyzed four-component selenosulfonylation of alkynes, cycloketone oxime esters, DABCO (SO2)2 and diselenides has been developed. This method enables the rapid assembly of β-cyanoalkylsulfonylated vinyl selenides in moderate to good yields. Advantages of this protocol include a broad substrate scope, good functional group tolerance and the late-stage functionalization of complex molecules. Moreover, the potential utility of this methodology is demonstrated through simple oxidation of the products to access synthetically important alkynyl sulfones. Mechanistic studies suggest that a cyanoalkylsulfonyl radical intermediate is involved in this process.
Owing to their widespread synthetic applications and important biological properties, sulfone derivatives have attracted considerable attention from both synthetic and medicinal communities.1 In particular, vinyl sulfones are not only present in many pharmaceuticals serving as neuroprotective agents, nuclear factor erythroid 2-related factor 2 activators and cysteine protease inhibitors, but they are also valuable building blocks in modern organic synthesis.2 Accordingly, various methodologies for the assembly of vinyl sulfones have been developed, such as the Knoevenagel condensation of aldehydes with sulfonylacetic acids, β-elimination of halosulfones and oxidation of vinyl sulfides, which generally require prefunctionalized reagents, harsh reaction conditions or tedious steps.3 On the other hand, vinyl selenides are versatile intermediates and important therapeutic entities, which exhibit a variety of biological activities.4 By combining vinyl sulfone and vinyl selenide units, β-selenovinyl sulfones can be easily transformed into valuable products such as allenes, acetylenes and β-keto sulfones,5 thus rendering the significance of developing efficient approaches towards these compounds (Scheme 1a).6
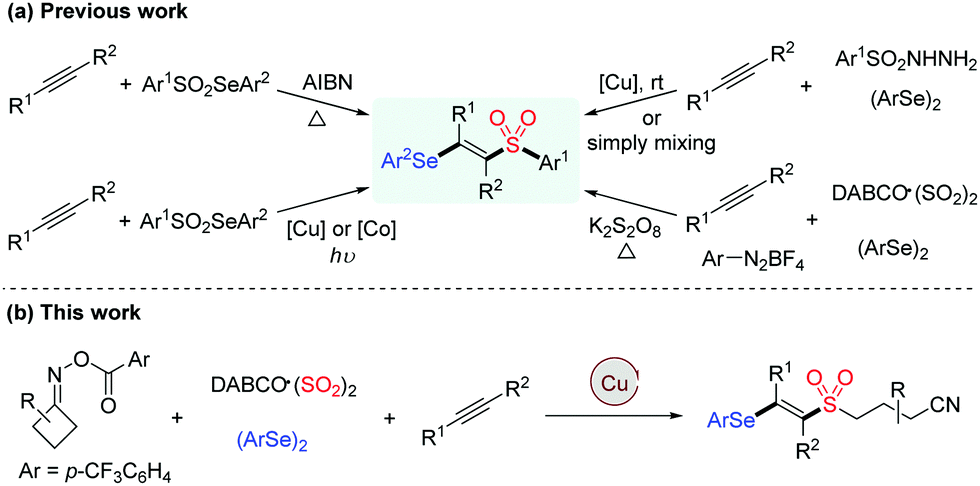 | ||
| Scheme 1 Synthesis of β-selenovinyl sulfones. | ||
Vicinal difunctionalization of alkynes represents an attractive and efficient method for the synthesis of highly functionalized olefins, which results in the simultaneous incorporation of two functional moieties across the triple bond.7 In this context, sulfonyl radical-involved 1,2-difunctionalization of alkynes has emerged as a powerful strategy for accessing structurally diverse vinyl sulfones with high stereoselectivity.8 In recent years, the generation of sulfonyl radicals through the insertion of sulfur dioxide has been extensively employed in a variety of sulfonylation reactions.9 Among these, considerable efforts have been devoted to constructing sulfonyl compounds by utilizing DABCO·((SO2)2),10 inorganic metabisulfite11 or thiourea dioxide12 as the efficient sulfur dioxide surrogates. Moreover, as an alternative approach for the preparation of β-selenovinyl sulfones, a selenosulfonation of alkynes with aryldiazonium tetrafluoroborates, DABCO (SO2)2 and diselenides was reported by Sun et al.13 However, this approach had some limitations, such as the safety and stability of using aryldiazonium tetrafluoroborates and the requirement of using excess K2S2O8 as the oxidant. Therefore, the development of efficient methods for the synthesis of β-selenovinyl sulfones from sulfur dioxide is still highly desirable. Driven by the recent advances in iminyl radical-triggered C–C bond cleavage reactions14 and our continuous interest in the chemistry of sulfur dioxide insertion, herein, we described a facile copper-catalyzed selenosulfonylation of alkynes, cycloketone oxime esters, DABCO (SO2)2 and diselenides, affording β-cyanoalkylsulfonylated vinyl selenides in moderate to good yields under mild conditions (Scheme 1b). Moreover, the synthetic utility of this methodology is illustrated by the late-stage functionalization of complex molecules and the successful application of products to generate alkynyl sulfones.
Initially, cycloketone oxime ester 1a, DABCO·(SO2)2, diphenyl diselenide 2a and phenylacetylene 3a were selected as the model substrates to optimize the reaction conditions (Table 1). To our delight, the desired product 4a was obtained in 53% yield when the reaction occurred in the presence of CuOAc at 80 °C for 12 h in DCE (Table 1, entry 1). A brief screening of other copper salts such as Cu(OTf)2, Cu(TFA)2, CuBr2, CuCl2 and Cu(OAc)2 indicated that Cu(OAc)2 was the best choice, affording to product 4a in 60% yield (Table 1, entries 2–6). Subsequently, the effect of other solvents, including CH3CN, THF and DMSO, was examined (Table 1, entries 7–9). However, better results were not obtained. A survey of temperatures revealed that increasing the reaction temperature to 100 °C would lead to a slightly decreased yield, whereas it increased when the reaction was at 60 °C (Table 1, entries 10 and 11). Gratifyingly, the yield of product 4a could be further improved to 72% when the amounts of cycloketone oxime ester 1a and phenylacetylene 3a were changed (Table 1, entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | Yieldb (%) |
| a Reactions conditions: cycloketone oxime ester 1a (0.4 mmol), DABCO·(SO2)2 (0.2 mmol), diphenyl diselenide 2a (0.2 mmol), phenylacetylene 3a (0.2 mmol), copper catalyst (10 mol%), solvent (2.0 mL), N2, 12 h. b Yield was determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard (isolated yield in parentheses). c Cycloketone oxime ester 1a (0.2 mmol), phenylacetylene 3a (0.3 mmol). | ||||
| 1 | CuOAc | DCE | 80 | 53 |
| 2 | Cu(OTf)2 | DCE | 80 | 52 |
| 3 | Cu(TFA)2 | DCE | 80 | 46 |
| 4 | CuBr2 | DMF | 80 | 52 |
| 5 | CuCl2 | DCE | 80 | 44 |
| 6 | Cu(OAc)2 | DCE | 80 | 60 |
| 7 | Cu(OAc)2 | CH3CN | 60 | 54 |
| 8 | Cu(OAc)2 | THF | 40 | 39 |
| 9 | Cu(OAc)2 | DMSO | 80 | 50 |
| 10 | Cu(OAc)2 | DCE | 100 | 43 |
| 11 | Cu(OAc)2 | DCE | 60 | 64 |
| 12c | Cu(OAc)2 | DCE | 60 | 72 (70) |
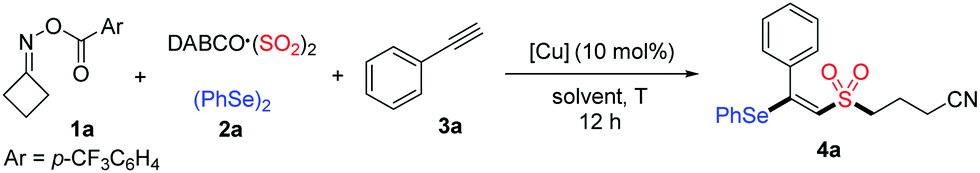
With the optimal conditions established, we began to explore the substrate scope of this copper-catalyzed selenosulfonation of alkynes, cycloketone oxime ester 1a, DABCO (SO2)2 and diphenyl diselenide 2a. The results are summarized in Table 2. In general, a variety of terminal and internal alkynes with electron-neutral, electron-rich, or electron-poor substituents were compatible in this reaction, producing the corresponding products in moderate to good yields. Aryl terminal alkynes bearing different functional groups such as Me, OMe, OH, CHO, F, Cl and Br were well tolerated under the reaction conditions, affording the desired β-cyanoalkylsulfonylated vinyl selenides 4b–4i in 45–80% yields. The structure of product 4i was unambiguously determined by X-ray diffraction analysis.15 Moreover, heteroaryl alkynes were also suitable in this transformation to afford the targeted products 4j–4l in 48–65% yields. The reactions of less reactive aliphatic alkynes, including cyclohexylacetylene, 5-chloro-1-pentyne and 1-hexyne, proceeded smoothly to afford 4m–4o in 31–72% yields. Notably, this protocol was also applicable to internal alkynes. For example, unsymmetrical internal alkynes such as 1-phenyl-1-propyne and methyl phenylpropiolate could convert into the corresponding tetrasubstituted vinyl sulfones 4p and 4r in 72% and 44% yields, respectively.
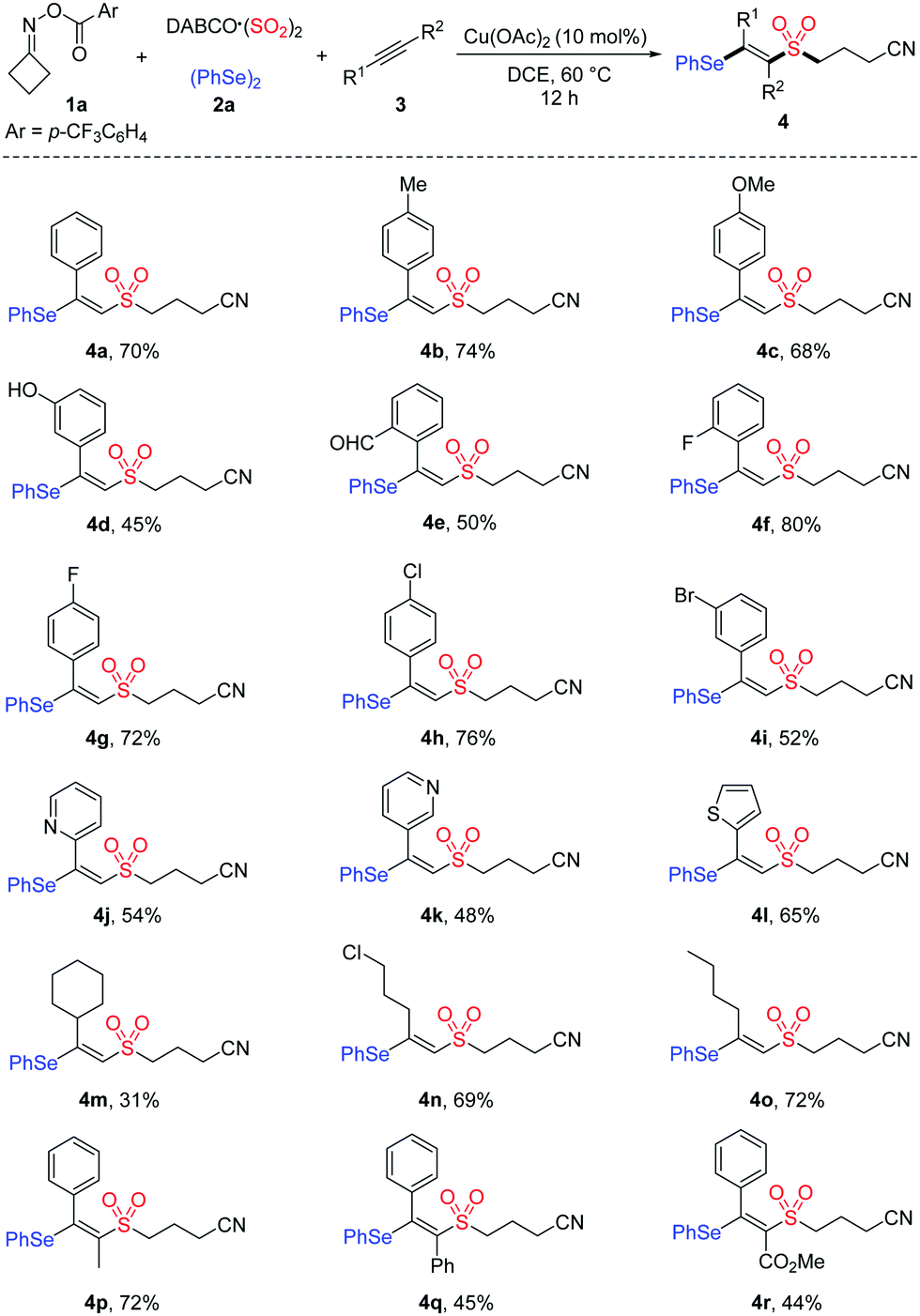
Next, the generality of cycloketone oxime esters and diselenides was evaluated. As shown in Table 3, the mono-substituted cycloketone oxime esters with functionalities including ester, phenyl, ether, and alkyl were well tolerated, affording the desired products 5a–5e with yields from 46% to 72%. In the cases of 3,3-disubstituted cycloketone oxime esters bearing spirocyclic units, the targeted products 5g and 5h were obtained in 55% and 65% yields, respectively. This reaction was also suitable with cycloketone oxime ester containing a Cbz protected-amine, delivering product 5i in 47% yield. Furthermore, diselenides with electron-donating or electron-withdrawing substituents on the aryl ring were all compatible in this transformation (5j–5m).
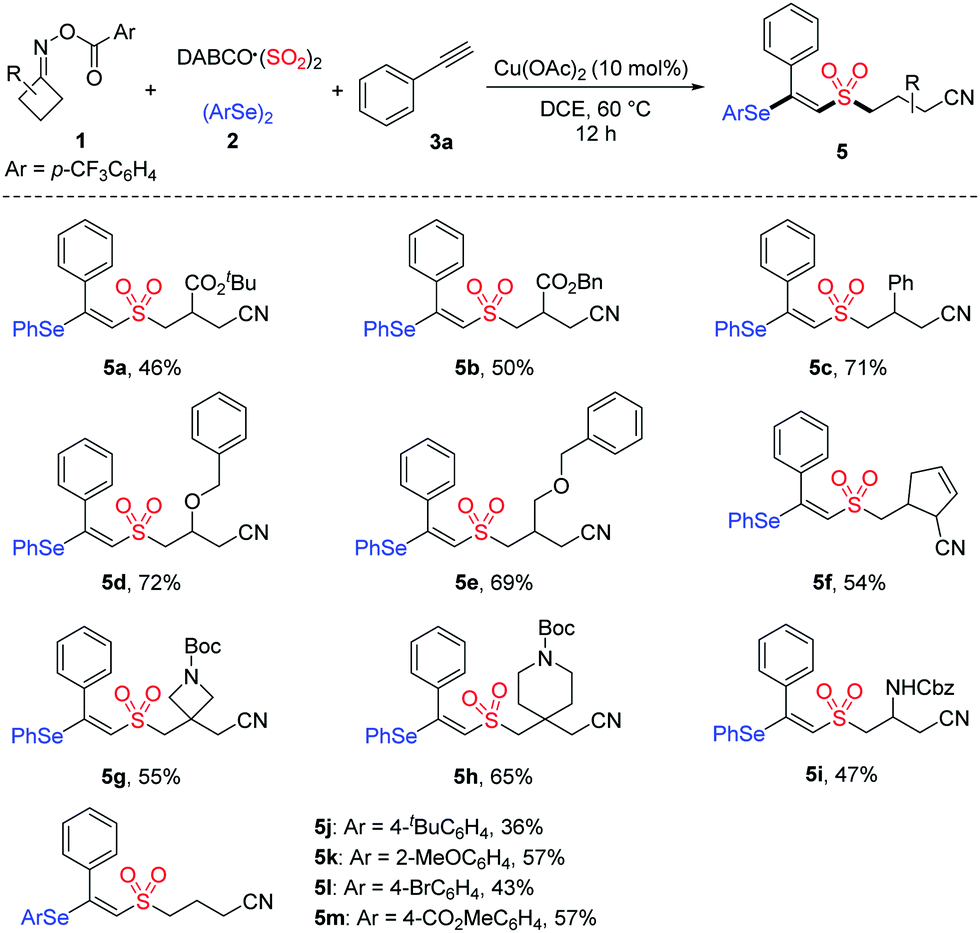
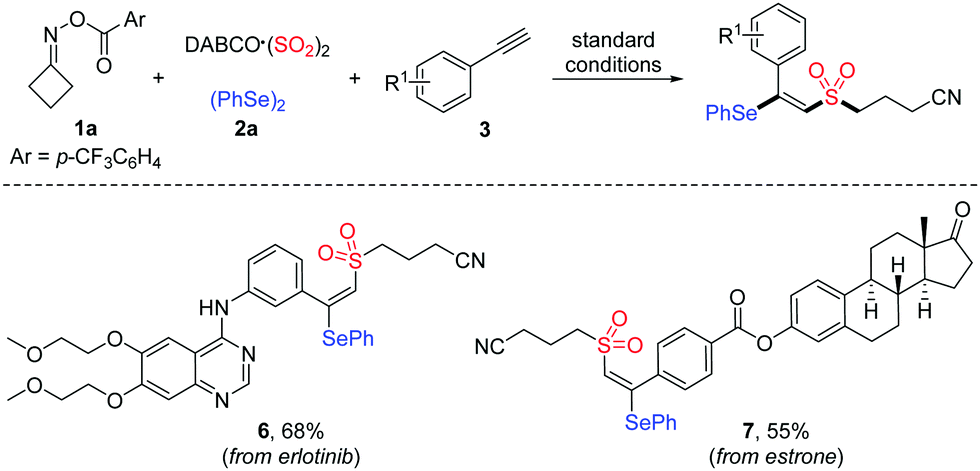
To further demonstrate the applicability of this mild protocol, we applied this copper-catalyzed four-component process in the late-stage functionalization of pharmaceutical derivatives and natural products (Table 4), with an anticipation that these compounds would be promising for several biological evaluations in our laboratory. As expected, erlotinib bearing a terminal alkyne and estrone-derived alkyne reacted smoothly with cycloketone oxime ester 1a, DABCO·(SO2)2 and diphenyl diselenide 2a, affording the corresponding products 6 and 7 in 68% and 55% yields, respectively.
|
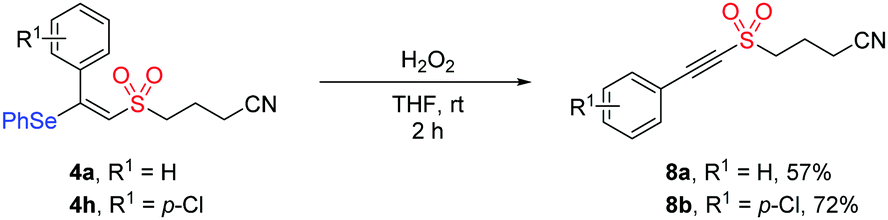
In addition, the synthetic utility of this methodology was investigated through the transformation of the product under oxidation conditions. As illustrated in Scheme 2, the treatment of compound 4a or 4h with hydrogen peroxide in THF afforded alkynyl sulfone product 8a (57%) or 8b (72%). Given that alkynyl sulfone bearing a cyanoalkyl group could not be accessed by the previous methods, this protocol showed potential application in organic synthesis.
 | ||
| Scheme 2 Synthetic application. | ||
To gain mechanistic insight into the current selenosulfonation reaction, several control experiments were performed (Scheme 3). The formation of product 4a was completely hindered when 2.0 equivalents of 2,2,6,6-tetramethylpiperidineoxy (TEMPO) were added to the model reaction under standard conditions, indicating that the reaction proceeded via a radical pathway (Scheme 3, eqn (1)). Additionally, when 2.0 equivalents of butylated hydroxytoluene (BHT) were introduced into the reaction, the trapping-product 9 was isolated in 18% yield. This result suggested that a cyanoalkylsulfonyl radical intermediate was generated in this transformation (Scheme 3, eqn (2)).
 | ||
| Scheme 3 Control experiments. | ||
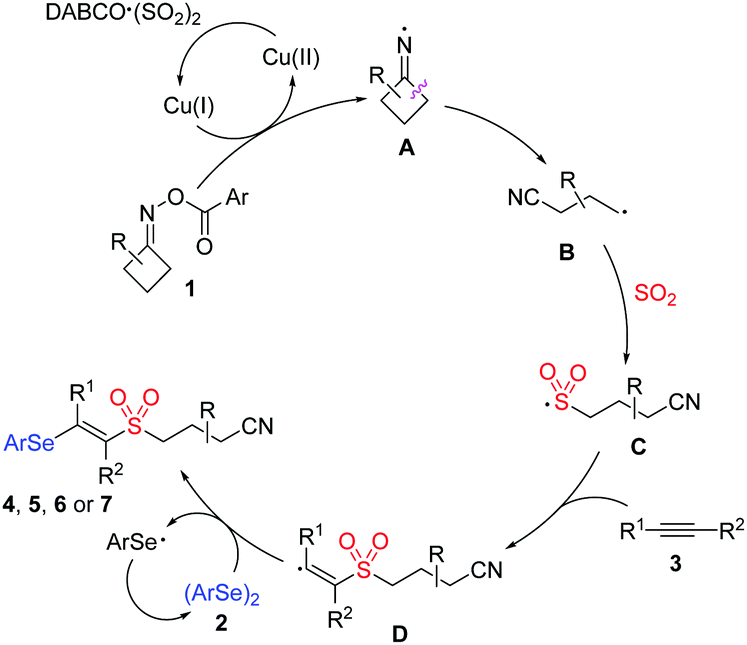
On the basis of the above-mentioned results and literature reports,6,9,13,14 we proposed a plausible mechanism for this copper-catalyzed selenosulfonation of alkynes, cycloketone oxime esters, DABCO (SO2)2 and diselenides (Scheme 4). We postulated that initially iminyl radical A was generated via the Cu(I)-mediated single-electron reduction of cycloketone oxime ester 1, which would undergo ring-opening by the selective β-C–C bond cleavage to produce a highly reactive cyanoalkyl radical B. Then, radical B was captured by sulfur dioxide, leading to the generation of cyanoalkylsulfonyl radical C. Following that, the regiospecific addition with alkyne 3 would produce vinyl radical D, which might react with diphenyl diselenide to afford the targeted product.
 | ||
| Scheme 4 Plausible mechanism. | ||
In conclusion, we have disclosed a copper-catalyzed four-component reaction of cycloketone oxime esters, DABCO (SO2)2, diselenides and alkynes, affording β-cyanoalkylsulfonylated vinyl selenides in moderate to good yields. This vicinal selenosulfonation process proceeded under mild conditions and featured a broad substrate scope and high functional group tolerance. Furthermore, the late-stage functionalization of complex molecules and the successful application of products to generate alkynyl sulfones highlight the potential utility of this methodology. Mechanistic studies indicate that the cyanoalkylsulfonyl radical intermediate has participated.
Financial support from the National Natural Science Foundation of China (No. 21871053, 22101199 and 22171206), the Natural Science Foundation of Zhejiang Province (LQ21B020002), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2019R01005) and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2020ZD04) is gratefully acknowledged.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) T. Zhou, B. Peters, M. F. Maldonado, T. Govender and P. G. Andersson, J. Am. Chem. Soc., 2012, 134, 13592 CrossRef CAS PubMed; (b) J. T. Palmer, D. Rasnick, J. L. Klaus and D. Brömme, J. Med. Chem., 1995, 38, 3193 CrossRef CAS PubMed; (c) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793 CrossRef CAS PubMed.
- (a) S. Y. Woo, J. H. Kim, M. K. Moon, S. H. Han, S. K. Yeon, J. W. Choi, B. K. Jang, H. J. Song, Y. G. Kang, J. W. Kim, J. Lee, D. J. Kim, O. Hwang and K. D. Park, J. Med. Chem., 2014, 57, 1473 CrossRef CAS PubMed; (b) J. W. Choi, S. Kim, J. H. Park, H. J. Kim, S. J. Shin, J. W. Kim, S. Y. Woo, C. Lee, S. M. Han, J. Lee, A. N. Pae, G. Han and K. D. Park, J. Med. Chem., 2019, 62, 811 CrossRef CAS PubMed; (c) M. Uttamchandani, K. Liu, R. C. Panicker and S. Q. Yao, Chem. Commun., 2007, 1518 RSC.
- (a) S. Chodroff and W. F. Whitmore, J. Am. Chem. Soc., 1950, 72, 1073 CrossRef CAS; (b) P. B. Hopkins and P. L. Fuchs, J. Org. Chem., 1978, 43, 1208 CrossRef CAS; (c) L. Wang, H. Yue, D. Yang, H. Cui, M. Zhu, J. Wang, W. Wei and H. Wang, J. Org. Chem., 2017, 82, 6857 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. Perin, E. J. Lenardão, R. G. Jacob and R. B. Panatieri, Chem. Rev., 2009, 109, 1277 CrossRef CAS PubMed; (b) G. Mugesh, W.-W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125 CrossRef CAS PubMed.
- (a) T. G. Back and S. Collins, Tetrahedron Lett., 1981, 22, 5111 CrossRef CAS; (b) T. G. Back, M. V. Krishna and K. R. Muralidharan, J. Org. Chem., 1989, 54, 4146 CrossRef CAS; (c) J. V. Comasseto, L. W. Ling, N. Petragnani and H. A. Stefani, Synthesis, 1997, 373 CrossRef CAS.
- For selected examples, see: (a) T. G. Back, S. Collins and R. G. Kerr, J. Org. Chem., 1983, 48, 3077 CrossRef CAS; (b) Y. Liu, G. Zheng, Q. Zhang, Y. Li and Q. Zhang, J. Org. Chem., 2017, 82, 2269 CrossRef CAS PubMed; (c) K. Sun, X. Wang, F. Fu, C. Zhang, Y. Chen and L. Liu, Green Chem., 2017, 19, 1490 RSC; (d) R. Zhang, P. Xu, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2019, 84, 12324 CrossRef CAS PubMed.
- M.-H. Huang, W.-J. Hao, G.-G. Li, S.-J. Tu and B. Jiang, Chem. Commun., 2018, 54, 10791 RSC.
- For selected examples, see: (a) A. García-Domínguez, S. Müller and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 9949 CrossRef PubMed; (b) Y. Ning, Q. Ji, P. Liao, E. A. Anderson and X. Bi, Angew. Chem., Int. Ed., 2017, 56, 13805 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. Zheng and J. Wu, Sulfur Dioxide Insertion Reactions for Organic Synthesis, Nature Springer, Berlin, 2017 CrossRef; (b) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691 RSC; (c) K. Hofman, N. Liu and G. Manolikakes, Chem. – Eur. J., 2018, 24, 11852 CrossRef CAS PubMed; (d) G. Qiu, L. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405 RSC; (e) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561 RSC; (f) S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013 RSC; (g) S. Ye, M. Yang and J. Wu, Chem. Commun., 2020, 56, 4145 RSC; (h) D. Zeng, M. Wang, W.-P. Deng and X. Jiang, Org. Chem. Front., 2020, 7, 3956 RSC.
- For selected examples, see: (a) D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451 CrossRef CAS PubMed; (b) F. Liu, J.-Y. Wang, P. Zhou, G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570 CrossRef CAS PubMed; (c) J. Zhang, M. Yang, J.-B. Liu, F.-S. He and J. Wu, Chem. Commun., 2020, 56, 3225 RSC; (d) X. Wang, Y. Lin, J.-B. Liu, F.-S. He, Y. Kuang and J. Wu, Chin. J. Chem., 2020, 38, 1098 CrossRef CAS; (e) Y. Yao, Z. Yin, F.-S. He, X. Qin, W. Xie and J. Wu, Chem. Commun., 2021, 57, 2883 RSC; (f) S. Cao, W. Hong, Z. Ye and L. Gong, Nat. Commun., 2021, 12, 2377 CrossRef CAS PubMed; (g) F.-S. He, P. Bao, F. Yu, L.-H. Zeng, W.-P. Deng and J. Wu, Org. Lett., 2021, 23, 7472 CrossRef CAS PubMed; (h) F.-S. He, Y. Yao, Z. Tang, W. Xie and J. Wu, Org. Chem. Front., 2021, 8, 6119 RSC.
- For selected examples, see: (a) F.-S. He, X. Gong, P. Rojsitthisak and J. Wu, J. Org. Chem., 2019, 84, 13159 CrossRef CAS PubMed; (b) X. Gong, M. Yang, J.-B. Liu, F.-S. He, X. Fan and J. Wu, Green Chem., 2020, 22, 1906 RSC; (c) X. Gong, M. Yang, J.-B. Liu, F.-S. He and J. Wu, Org. Chem. Front., 2020, 7, 938 RSC; (d) Y. Liu, Q.-L. Wang, Z. Chen, H. Li, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, Chem. Commun., 2020, 56, 3011 RSC; (e) F.-S. He, Y. Yao, W. Xie and J. Wu, Chem. Commun., 2020, 56, 9469 RSC.
- (a) F.-S. He, M. Yang, S. Ye and J. Wu, Chin. Chem. Lett., 2021, 32, 461 CrossRef CAS; (b) Y. Li, J.-B. Liu, F.-S. He and J. Wu, Chin. J. Chem., 2020, 38, 361 CrossRef CAS; (c) H. Zhang, M. Wang and X. Jiang, Green Chem., 2020, 22, 8238 RSC; (d) J. Chen, N. Liu, Q. Hu, J. Liu, J. Wu, Q. Cai and J. Wu, Org. Chem. Front., 2021, 8, 5316 RSC.
- K. Sun, Z. Shi, Z. Liu, B. Luan, J. Zhu and Y. Xue, Org. Lett., 2018, 20, 6687 CrossRef CAS PubMed.
- For selected reviews and examples, see: (a) T. Xiao, H. Huang, D. Anand and L. Zhou, Synthesis, 2020, 1585 CrossRef CAS; (b) W. Xiao and J. Wu, Chin. Chem. Lett., 2020, 31, 3083 CrossRef CAS; (c) F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943 CrossRef CAS; (d) C. Song, X. Shen, F. Yu, Y. He and S. Yu, Chin. J. Org. Chem., 2020, 40, 3748 CrossRef CAS; (e) F.-S. He, M. Zhang, M. Zhang, X. Luo and J. Wu, Org. Chem. Front., 2021, 8, 3746 RSC; (f) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506 CrossRef CAS PubMed; (g) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, Acc. Chem. Res., 2020, 53, 1066 CrossRef CAS PubMed; (h) F. Xiao, Y. Guo and Y.-F. Zeng, Adv. Synth. Catal., 2021, 363, 120 CrossRef CAS.
- CCDC 2102437 contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data, copies of 1H and 13C NMR spectra. CCDC 2102437. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc05690e |
| This journal is © The Royal Society of Chemistry 2021 |