
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bioinspired oxoiron(IV) motif supported on a N2S2 macrocyclic ligand†
Jennifer
Deutscher
 a,
Philipp
Gerschel
b,
Katrin
Warm
a,
Uwe
Kuhlmann
c,
Stefan
Mebs
d,
Michael
Haumann
d,
Holger
Dau
d,
Peter
Hildebrandt
c,
Ulf-Peter
Apfel
*be and
Kallol
Ray
*a
a,
Philipp
Gerschel
b,
Katrin
Warm
a,
Uwe
Kuhlmann
c,
Stefan
Mebs
d,
Michael
Haumann
d,
Holger
Dau
d,
Peter
Hildebrandt
c,
Ulf-Peter
Apfel
*be and
Kallol
Ray
*a
aInstitut für Chemie Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489, Berlin, Germany. E-mail: kallol.ray@hu-berlin.de
bAnorganische Chemie 1 Ruhr-Universität Bochum, Universitätsstraße 150, 44780, Bochum, Germany
cInstitut für Chemie Technische, Universität Berlin, Fakultät II Straße des 17, Juni 135, 10623, Berlin, Germany
dInstitut für Physik Freie, Universität Berlin, Arnimallee 14, 14195 Berlin, Germany
eDepartment of Electrosynthesis, Fraunhofer UMSICHT, Osterfelder Str. 3, 46047 Oberhausen, Germany
First published on 17th February 2021
Abstract
A mononuclear oxoiron(IV) complex 1-trans bearing two equatorial sulfur ligations is synthesized and characterized as an active-site model of the elusive sulfur-ligated FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediates in non-heme iron oxygenases. The introduction of sulfur ligands weakens the FeO bond and enhances the oxidative reactivity of the FeIVO unit with a diminished deuterium kinetic isotope effect, thereby providing a compelling rationale for nature's use of the cis-thiolate ligated oxoiron(IV) motif in key metabolic transformations.
O intermediates in non-heme iron oxygenases. The introduction of sulfur ligands weakens the FeO bond and enhances the oxidative reactivity of the FeIVO unit with a diminished deuterium kinetic isotope effect, thereby providing a compelling rationale for nature's use of the cis-thiolate ligated oxoiron(IV) motif in key metabolic transformations.
Sulfur-ligated oxoiron(IV) centers are proposed as key oxidants in the catalytic cycles of various heme and non-heme iron oxygenases (Scheme 1).1–3 Iron(IV)–oxo porphyrin π-cation radical (Cpd I) intermediates containing a thiolate ligand trans to the oxo group have been isolated and spectroscopically characterized in a number of heme enzymes.4–6 The increased basicity of the oxoiron(IV) core caused by the strong electron donation from the trans-sulfur ligand, is discussed as a strategy to perform hydrogen atom abstraction by Cpd I at a lower redox potential without performing oxidative destruction of the surrounding enzyme environment.5,7–10 However, similar knowledge on the effect of cis-sulfur ligands on the reactivity of oxoiron(IV) cores in non-heme enzymes is lacking. Notably, identification of cis thiolate-ligated oxoiron(IV) species remained elusive in biology, although they are suggested to be reactive intermediates for a wide range of chemical transformations, including sulfur-oxygenation, hydrogen-atom abstraction, and C–S bond formation reactions in non-heme enzymes.
 | ||
| Scheme 1 Top: Proposed structures of the thiolate-ligated oxoiron(IV) reactive intermediates in biology; bottom: structures of [(TMCS)FeIV(O)]+, 1-trans and 2-trans. | ||
For over 40 years, small-molecule complexes synthesized as active-site models of the high-valent intermediates in heme and non-heme oxygenases have advanced our understanding of the catalytic cycles.3,11–20 Despite these efforts, the synthesis of an oxoiron(IV) porphyrin complex with a thiolate ligand has not yet been achieved. Furthermore, [(TMCS)FeIV(O)]+ (TMCS = 1-mercaptoethyl-4,8,11-trimethyl-1,4,8,11-tetraazacyclotetra-decane; (Scheme 1)), represents the only synthetic complex21 thus far to model the RS-FeIVO unit associated with the active oxidants of cytochrome P4502,6 and chloroperoxidase.4,5,10 Similarly, a recently reported [(Me3TACN)FeIV(O)(S2Si(CH3)2)] (Me3TACN = 1,4,7-trimethyl-1,4,7-triazacyclononane)13 complex represents the only model complex for the postulated oxoiron(IV) core containing a sulfur ligation cis to the oxo group in non-heme oxygenases.22,23 However, the thermal instability of the compound has prevented any reactivity studies. Herein we report the synthesis and characterization of the S = 1 FeIVO complex [(dithiacyclam)FeIV(O)(CH3CN)]2+ (1-trans, dithiacyclam24,25 = 1,8-dithia-4,11-diazacyclotetra-decane), which contains two thioether sulfur coordination sites poised cis to the oxo group. A comparative study between 1-trans, containing a N2S2 macrocyclic ligand, and [FeIV(O)(cyclam)(CH3CN)]2+ (2-trans; cyclam = 1,4,8,11-tetraazacyclotetradecane; (Scheme 1)),26 based on the popular N4-donor cyclam ligand, provides some insight how cis-sulfur coordination influences the reactivity and spectroscopic properties of the oxoiron(IV) unit.
Combining the tetradentate dithiacyclam ligand with Fe(OTf)2(CH3CN)2 in acetonitrile yielded the iron(II) complex in two isomeric forms [FeII(dithiacyclam)(CH3CN)2](OTf)2 (1a-trans) and [FeII(dithiacyclam)(OTf)2] (1a-cis). Crystals suitable for X-ray diffraction analysis were grown by vapour diffusion of diethyl ether into an acetonitrile solution of the iron(II) complex at −15 °C for 1a-cis or at −40 °C for 1a-trans. The X-ray structure of 1a-trans displays a six-coordinate geometry with axially bound CH3CN ligands (Fig. 1; Table S2, ESI†). The N2S2 donor atoms of dithiacyclam occupy the equatorial coordination sites and show average Fe–S and Fe–N distances of 2.252(6) Å and 1.985(18) Å, respectively. In contrast, in 1a-cis the sulfur donor atoms occupy the axial coordination sites and show average Fe–S distances of 2.466(14) Å (Table S1, ESI†); the nitrogen atoms of the dithiacyclam and the oxygen atoms of the triflate (OTf) anions occupy equatorial positions with average Fe–N and Fe–O distances of 2.2085(4) Å and 2.143(3) Å, respectively. The zero-field Mößbauer spectrum of 1a-cis in acetone (Fig. S1, ESI†) at 15 K reveals a single quadrupole doublet with an isomer shift of δ = 1.10 mm s−1 and a large quadrupole splitting of (ΔEQ) = 3.26 mm s−1, demonstrating that the iron(II) center remains in the high-spin configuration (S = 2). In addition, the 1H- and 19F-NMR spectra (Fig. S2, ESI†) of 1a-cis in d6-acetone at −85 °C display paramagnetically shifted peaks, indicative of the coordination of both OTf anions and further supporting the high-spin iron(II) assignment. In CH2Cl2/CH3CN solution, CH3CN gradually replaces the bound triflates of 1a-cis to form 1a-trans with an S = 0 FeII ground-state, as evident from 1H-NMR (at −85 °C), which reveals peaks between 0 and 4 ppm (Fig. S3, ESI,† left), and 19F-NMR (at −85 °C; Fig. S3, ESI,† right), which shows a singlet at −79.0 ppm corresponding to free OTf anions. Freezing the solution leads to the partial re-binding of OTf anion to FeII; zero-field Mößbauer measurement (Fig. S4, ESI†) at 15 K shows a major quadrupole doublet with a new high-spin FeII signal (δ = 1.15 mm s−1 and ΔEQ = 2.31 mm s−1; 73%) with a significantly reduced ΔEQ relative to 1a-cis, presumably corresponding to the trans-[(dithiacyclam)FeII(OTf)(CH3CN)]+ complex. An additional doublet with δ = 0.52 mm s−1 and ΔEQ = 0.26 mm s−1 corresponds to the low-spin S = 0 FeII center in 1a-trans (27%). Thus, CH3CN-binding favours the trans configuration and the coordination of both CH3CN is necessary for stabilizing the low-spin FeII state in 1a.
 | ||
| Fig. 1 Molecular structures of 1a-cis and 1a-trans obtained by XRD. Atoms are displayed as thermal ellipsoids at 50% probability level; triflate counter ions for 1a-trans are omitted for clarity. Atom types: Fe: orange; N: blue; C: grey; O: red; S: yellow; F: green; H: white. | ||
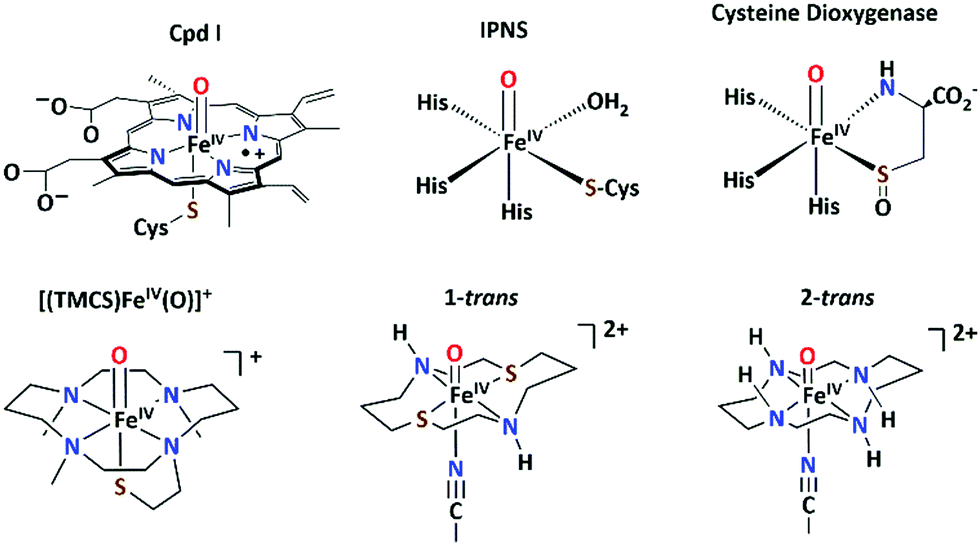
A solution of 1a-trans in a CH2Cl2/CH3CN solvent mixture (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) at −85 °C with 4 equiv. of 2-(tert-butylsulfonyl)iodosylbenzene (tBuSO2C6H4IO, sPhIO)27 led to the formation of a pale green intermediate 1-trans (t1/2 = 10000 s at −65 °C) with absorption maxima at 596 nm (εmax = 226 M−1 cm−1) and 815 nm (εmax = 549 M−1 cm−1), which are typical of S = 1 oxoiron(IV) cores (Fig. 2; left).11 The characteristic near-infrared band in 1-trans @ 815 nm is significantly red-shifted relative to that in 2-trans,26 which is consistent with a weakened equatorial field in 1-trans. Notably, in the absence of CH3CN, 1-trans was not generated.28 An electron spray ionization mass spectrum (Fig. S5, ESI†) of 1-trans exhibited a signal at m/z = 794.96, consistent with its formulation as {[(dithiacyclam)FeIV(O)(OTf)](SPhIO)}+ (m/z calc = 794.97), which is shifted by 4 units to m/z = 798.97, when sPhI18O was used to generate 1-trans. 19F-NMR (at −85 °C in a 95:5 mixture of CD2Cl2 and CD3CN) shows a singlet at −77.0 ppm corresponding to the free OTf anions in 1-trans (Fig. S6, ESI†). The zero-field Mößbauer spectrum of 1-trans in frozen acetone/CH2Cl2/CH3CN solution and recorded at 15 K exhibits a doublet representing about 84% of the iron with ΔEQ = 1.21 mm s−1 and δ = 0.13 mm s−1 corresponding to the presence of an FeIV center (Fig. 2; right); the remaining 16% of the signals with ΔEQ = 1.57 mm s−1 and δ = 0.55 mm s−1 correspond to a high-spin FeIII product, arising from the decay of 1-trans.
:5) at −85 °C with 4 equiv. of 2-(tert-butylsulfonyl)iodosylbenzene (tBuSO2C6H4IO, sPhIO)27 led to the formation of a pale green intermediate 1-trans (t1/2 = 10000 s at −65 °C) with absorption maxima at 596 nm (εmax = 226 M−1 cm−1) and 815 nm (εmax = 549 M−1 cm−1), which are typical of S = 1 oxoiron(IV) cores (Fig. 2; left).11 The characteristic near-infrared band in 1-trans @ 815 nm is significantly red-shifted relative to that in 2-trans,26 which is consistent with a weakened equatorial field in 1-trans. Notably, in the absence of CH3CN, 1-trans was not generated.28 An electron spray ionization mass spectrum (Fig. S5, ESI†) of 1-trans exhibited a signal at m/z = 794.96, consistent with its formulation as {[(dithiacyclam)FeIV(O)(OTf)](SPhIO)}+ (m/z calc = 794.97), which is shifted by 4 units to m/z = 798.97, when sPhI18O was used to generate 1-trans. 19F-NMR (at −85 °C in a 95:5 mixture of CD2Cl2 and CD3CN) shows a singlet at −77.0 ppm corresponding to the free OTf anions in 1-trans (Fig. S6, ESI†). The zero-field Mößbauer spectrum of 1-trans in frozen acetone/CH2Cl2/CH3CN solution and recorded at 15 K exhibits a doublet representing about 84% of the iron with ΔEQ = 1.21 mm s−1 and δ = 0.13 mm s−1 corresponding to the presence of an FeIV center (Fig. 2; right); the remaining 16% of the signals with ΔEQ = 1.57 mm s−1 and δ = 0.55 mm s−1 correspond to a high-spin FeIII product, arising from the decay of 1-trans.
 | ||
| Fig. 2 Left: UV-Vis-spectrum of 1a-trans (black) and 1-trans (blue) in a 95:5 mixture of CH2Cl2 and CH3CN at −90 °C; the inset shows the resonance Raman spectra of 16O- (black) and 18O-labelled (red) 1-trans (4 mM) upon 413 nm irradiation at −90 °C. Solvent signal is indicated by an asterisk. Right: Zero-field Mößbauer spectrum (black) of a frozen sample of 1-trans in a solvent mixture of acetone/CH2Cl2/CH3CN (10:0.95:0.05) and simulation (red) with δ = 0.13 mm s−1 and ΔEQ = 1.21 mm s−1 for the main species (blue, 84%). The minor species with δ = 0.55 mm s−1 and ΔEQ = 1.57 mm s−1 corresponds to the decay of 1-trans (brown, 16%). | ||
The Fe K-edge X-ray absorption (Fig. S7 and Table S4, ESI†) spectrum of 1-trans reveals a K-edge energy of 7122.7 eV, which is lower relative to 2-trans (7123.9 eV). Furthermore, the pre-edge transition in 1-trans is less intense, which may reflect a less covalent FeO bond in 1-trans relative to 2-trans. The resonance Raman (rR) spectrum of 1-trans exhibits a ν(FeO) stretching mode at 793 cm−1 (Fig. 2, inset), which is red-shifted by 49 cm−1 relative to that in 2-trans (ν(FeO) = 842 cm−1),26 thereby demonstrating an elongation of the FeO bond by ∼0.02 Å in 1-trans relative to 2-trans.29 However, within the error of the extended X-ray absorption fine structure (EXAFS) analysis, the FeO distances in 1-trans and 2-trans are not discernible; in both cases a distance of 1.67 ± 0.02 Å has been obtained.26 The DFT optimized geometry of 1-trans in the S = 1 state slightly underestimates the Fe–O bond (calculated @ 1.655 Å) from the EXAFS data (Tables S5 and S6, ESI†), and as a result the calculated ν(Fe–O) is overestimated. This is a common problem that is encountered in the oxoiron(IV) chemistry.8,13 However, a geometry scan of the Fe–O bond length (Table S6, ESI†) reveals a relatively flat surface potential, with the structures exhibiting Fe–O bond lengths of 1.66–1.70 Å being within 1 kcal mol−1 in energy from the lowest energy structure. In particular, a constrained optimization with a fixed Fe–O distance @ 1.68 Å for 1-trans gives a calculated ν(Fe–O) of 794 cm−1, in good agreement with the experiment. Similarly, a constrained geometry with an Fe–O distance of 1.66 Å can account for the experimental ν(Fe–O) of 842 cm−1 for 2-trans. In summary, a 49 cm−1 red-shift in the ν(Fe–O) of 1-trans relative to 2-trans, translates to an FeO elongation of only 0.02 Å (based on both constrained DFT optimization and Badger's rule29), which is not clearly discernible within the error of the EXAFS analysis, but is reflected in a less-intense pre-edge transition at the Fe K-edge.
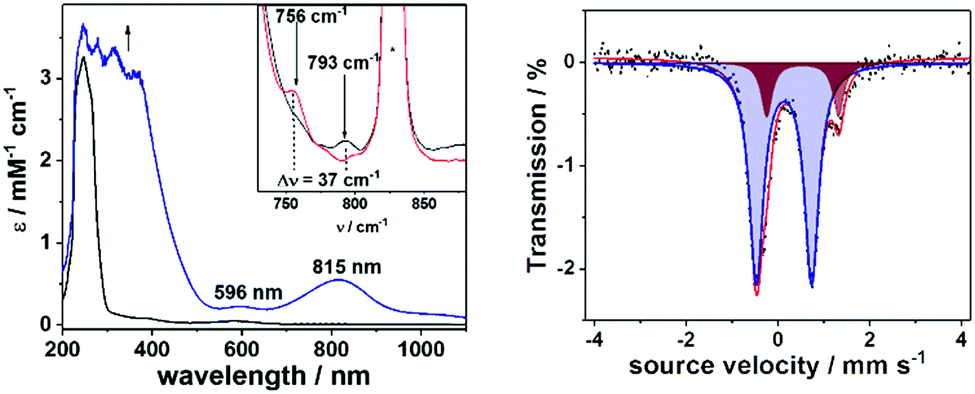
The introduction of the equatorial sulfur ligands also exhibits a significant effect on the reactivity of the oxoiron(IV) unit (Fig. 3 and Fig.s S8–S12, ESI†). In reactions with substrates containing C-H bonds like xanthene, 1,4-cyclohexadiene (CHD), dihydroanthracene (DHA), fluorene and indene, 1-trans reacts at least 3–4 orders of magnitude faster than 2-trans (Table 1). Furthermore, low kinetic isotope effects (KIEs) of 1.47 and 2.82 were determined in the reaction of 1-trans with xanthene (Fig. 3, top right) and DHA (Fig. 3, bottom left), respectively, which are in contrast to the previously reported value of 20.0 for reaction of xanthene with 2-trans.26 Nevertheless, when the logarithms of the second order rate constants (k2′) were plotted vs. the BDE C–H values of the substrates, the linear correlation previously reported for 2-trans were found to be also valid for 1-trans (Fig. 3, bottom right). Thus, although proton-transfer is involved in the rate-determining step of the oxidation of C-H bonds by 1-trans, the large tunneling contribution in hydrogen atom abstraction (HAA), which is observed in 2-trans and in most high-valent metal-oxo mediated HAA reactions,11,12,30 is not applicable for 1-trans. In particular, in a previous study the axial thiolate ligand of [FeIV(O)(TMCS)]+ has been suggested to play a unique role in facilitating tunnelling, thereby resulting in a large KIE of 80 for DHA oxidation.31 A contrasting effect is now demonstrated for the equatorial sulphur ligation, which reduces the tunnelling contribution to a minimum. The effect of N versus S donors in an otherwise identical ligand environment is also reflected in the higher oxygen atom transfer (OAT) ability of 1-trans relative to 2-trans (Table 1).
 | ||
| Fig. 3 Top left: Changes in the UV-Vis-spectra of a 1 mM solution of 1-trans in CH2Cl2/CH3CN (95:5) at −80 °C upon addition of 100 eq xanthene; the inset shows the time trace for the decay of the 815 nm band and its pseudo-first order fit; top right: plots of the pseudo-first order rate constants kobsvs. the substrate concentrations for xanthene and d2-xanthene in order to determine the kinetic isotopic effect (KIE) for the reaction of 1-trans with xanthene; bottom left: plots of the pseudo-first order rate constants kobsvs. the substrate concentrations for 9,10-dihydroanthracene (DHA) and d4-DHA in order to determine the kinetic isotopic effect (KIE) for the reaction of 1-trans with DHA; bottom right: plot of the logarithms of the second-order rate constants k2′ vs. the C-H BDEs of the substrates with 1-trans in CH2Cl2/CH3CN (95:5). | ||
| Substrate | BDEa [kcal mol−1] | k 2 [M−1s−1] 1-trans | k 2[M−1s−1] 2-trans | Product (yield)d |
|---|---|---|---|---|
| a Values taken from ref. 32. b Values calculated for 20 °C from experimental values determined at −80 °C using van’t Hoff equation. c Values calculated for 15 °C from experimental values determined at −65 °C using van’t Hoff equation. d Specified yields correspond to the reactivity of 1-trans. n.d: the reaction was too fast for kinetic studies. | ||||
| Xanthene | 75.2 | 205b | 1.1 × 10−1 | Xanthone (36%) |
| 1,4-CHD | 76 | 370b | 9.7 × 10−2 | |
| DHA | 76.3 | 355b | 4.9 × 10−2 | Anthracene (48%) |
| Fluorene | 82.2 | 18.2c | 7.1 × 10−3 | Fluorenone (11%) |
| Indene | 83 | 17.6c | 5.8 × 10−3 | Indenone |
| Thioanisol | — | 87c | — | |
| PPh3 | — | n.d | 5.9 | OPPh3 (28%) |
In summary, a minor 0.02 Å elongation of the FeO bond upon introduction of the equatorial sulfur ligands is shown to have a dramatic influence on the spectroscopic (Table S7, ESI†) and oxidative reactivity properties of the FeIVO unit. Notably, 1-trans, similar to the previously reported13 [(Me3TACN)FeIV(O)(S2Si(CH3)2)] complex features a very low ν(Fe–O), which establishes a general trend of the activation of the Fe–O bond in oxoiron(IV) complexes involving cis-sulphur ligands. The enhanced reactivity of 1-trans relative to 2-trans, can presumably be attributed to the positive shift in the redox potential upon sulphur ligation, as evident from cyclic voltammetry experiments, which shows a 170 mV positive shift in the Fe2+/3+ potential (Fig. S13, ESI†) in 1a-trans relative to 2a-trans. In addition, a change in mechanism in the C–H bond oxidation reactions from HAA to proton coupled electron transfer (PCET)33 is evident from the reduction of KIE from a value of 20 in 2-trans to 1.47 in 1-trans (using xanthene as a substrate). This drastic downshift in KIE is unique for sulfur substitution and is not observed in the oxygen substituted oxoiron(IV) center.34 Understanding this lowering of KIE will require further experimental and computational work. However the significant effect of the equatorial sulfur ligation on the physical and chemical properties of oxoiron(IV) cores may provide a compelling rationale for nature's use of the cis-thiolate ligated oxoiron(IV) motif in key metabolic transformations that involve the activation of strong C–H bonds.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat to K. R., P. H., and H. D., Excellence Strategy – EXC-2033 – Project number 390677874 to U.-P. A. and P. G., AP242/5-1 to U.-P. A. and the Heisenberg-Professorship and RA/2409/8-1 to K. R. We also thank the Fraunhofer Internal Programs under Grant No. Attract 097-602175 (U.-P. A.) and the Bundesministerium für Bildung und Forschung (BMBF, Operando-XAS project, 05K19KE1). The Helmholtz-Zentrum Berlin (HZB) is thanked for enabling the XAS experiments at KMC-3 of the BESSY synchrotron.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS.
- J. B. Gordon and D. P. Goldberg, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2020 DOI:10.1016/B978-0-12-409547-2.14906-6.
- M. T. Green, J. H. Dawson and H. B. Gray, Science, 2004, 304, 1653–1656 CrossRef CAS.
- K. L. Stone, R. K. Behan and M. T. Green, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16563–16565 CrossRef CAS.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS.
- F. Ogliaro, S. P. de Visser and S. Shaik, J. Inorg. Biochem., 2002, 91, 554–567 CrossRef CAS.
- M. T. Green, J. Am. Chem. Soc., 2006, 128, 1902–1906 CrossRef CAS.
- R. K. Behan, L. M. Hoffart, K. L. Stone, C. Krebs and M. T. Green, J. Am. Chem. Soc., 2006, 128, 11471–11474 CrossRef CAS.
- K. L. Stone, R. K. Behan and M. T. Green, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12307–12310 CrossRef CAS.
- X. Engelmann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef CAS.
- R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS.
- J. B. Gordon, A. C. Vilbert, I. M. DiMucci, S. N. MacMillan, K. M. Lancaster, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2019, 141, 17533–17547 Search PubMed.
- L. Vicens, G. Olivo and M. Costas, ACS Catal., 2020, 10, 8611–8631 CrossRef CAS.
- J. Chen, Z. Jiang, S. Fukuzumi, W. Nam and B. Wang, Coord. Chem. Rev., 2020, 421, 213443 Search PubMed.
- T. Devi, Y.-M. Lee, W. Nam and S. Fukuzumi, Coord. Chem. Rev., 2020, 410, 213219 Search PubMed.
- S. Fukuzumi, K.-B. Cho, Y.-M. Lee, S. Hong and W. Nam, Chem. Soc. Rev., 2020, 49, 8988–9027 RSC.
- S. Hong, Y.-M. Lee, K. Ray and W. Nam, Coord. Chem. Rev., 2017, 334, 25–42 Search PubMed.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2018, 51, 2014–2022 CrossRef CAS.
- M. Sankaralingam, Y.-M. Lee, W. Nam and S. Fukuzumi, Coord. Chem. Rev., 2018, 365, 41–59 Search PubMed.
- M. R. Bukowski, K. D. Koehntop, A. Stubna, E. L. Bominaar, J. A. Halfen, E. Münck, W. Nam and L. Que, Science, 2005, 310, 1000–1002 CrossRef CAS.
- D. Kumar, W. Thiel and S. P. de Visser, J. Am. Chem. Soc., 2011, 133, 3869–3882 CrossRef CAS.
- S. Aluri and S. P. de Visser, J. Am. Chem. Soc., 2007, 129, 14846–14847 CrossRef CAS.
- P. Gerschel, B. Battistella, D. Siegmund, K. Ray and U.-P. Apfel, Organometallics, 2020, 39, 1497–1510 CrossRef CAS.
- P. Gerschel, K. Warm, E. Farquhar, U. Englert, M. Reback, D. Siegmund, K. Ray and U.-P. Apfel, Dalton Trans., 2019, 48, 5923–5932 RSC.
- D. Kass, T. Corona, K. Warm, B. Braun-Cula, U. Kuhlmann, E. Bill, S. Mebs, M. Swart, H. Dau and M. Haumann, J. Am. Chem. Soc., 2020, 142, 5924–5928 CrossRef CAS.
- D. Macikenas, E. Skrzypczak-Jankun and J. D. Protasiewicz, J. Am. Chem. Soc., 1999, 121, 7164–7165 CrossRef CAS.
- Since we start with a pure solution of 1a-trans, the sole product that is formed is 1-trans (see also ref. 33). Oxidation of a solution of 1a-cis leads to only iron(III)products; so we presume that 1-cis is too unstable to be trapped in high-yields.
- Acoording to Badger's rule ( R. M. Badger, J. Chem. Phys., 1935, 3, 710–714 CrossRef CAS ), which is previously shown to be valid for oxoiron(IV) complexes (see ref. 8).
- (a) D. Mandal and S. Shaik, J. Am. Chem. Soc., 2016, 138, 2094–2097 CrossRef CAS; (b) D. Mandal, D. Mallick and S. Shaik, Acc. Chem. Res., 2018, 51, 107–117 CrossRef CAS; (c) E. J. Klinker, S. Shaik, H. Hirao and L. Que Jr., Angew. Chem., Int. Ed., 2009, 48, 1291–1295 CrossRef CAS.
- J. E. M. N. Klein, D. Mandal, W.-M. Ching, D. Mallick, L. Que, Jr. and S. Shaik, J. Am. Chem. Soc., 2017, 139, 18705–18713 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007, DOI:10.1201/9781420007282.
- J. M. Meyer, Acc. Chem. Res., 2010, 44, 36–46 CrossRef.
- I. Monte-Pérez, X. Engelmann, Y.-M. Lee, M. Yoo, E. Kumaran, E. R. Farquhar, E. Bill, J. England, W. Nam, M. Swart and K. Ray, Angew. Chem., Int. Ed., 2017, 56, 14384–14388 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2054129 and 2054130. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc00250c |
| This journal is © The Royal Society of Chemistry 2021 |