
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron transfer within β-diketiminato nickel bromide and cobaltocene redox couples activating CO2†‡
Philipp
Zimmermann
a,
Alexander F. R.
Kilpatrick
 a,
Deniz
Ar
a,
Serhiy
Demeshko
b,
Beatrice
Cula
a and
Christian
Limberg
*a
a,
Deniz
Ar
a,
Serhiy
Demeshko
b,
Beatrice
Cula
a and
Christian
Limberg
*a
aHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: christian.limberg@chemie.hu-berlin.de
bUniversität Göttingen, Institut für Anorganische Chemie, Tammannstr. 4, D-37077 Göttingen, Germany
First published on 16th December 2020
Abstract
Reduction of β-diketiminato nickel(II) complexes (LtBuNiII) to the corresponding nickel(I) compounds does not require alkali metal compounds but can also be performed with the milder cobaltocenes. LtBuNiBr and Cp2Co have rather similar redox potentials, so that the equilibrium with the corresponding electron transfer compound [LtBuNiIBr][Cp2CoIII] (ETC) clearly lies on the side of the starting materials. Still, the ETC portion can be used to activate CO2 yielding a mononuclear nickel(II) carbonate complex and ETC can be isolated almost quantitatively from the solutions through crystallisation. The more negative reduction potential of Cp*2Co shifts the equilibrium formed with LtBuNiBr strongly towards the ETC and accordingly the reaction of such solutions with CO2 is much faster.
Monovalent nickel has the reductive power to activate substrates by injecting an electron or by transferring electron density.1 A couple of nickel enzymes are known or proposed to involve nickel(I) intermediates in developing functions like the conversion of carbon oxides.1 Consistently, in the last decade the β-diketiminato nickel(I) moiety has demonstrated its potential to activate small molecules, such as CO2 and CO, but also N2, H2, P4, SF6, O2, formate, NO, Azides and organonitro and -nitroso compounds.1–16 Typically, it is generated by the reaction of a β-diketiminato nickel(II) bromido precursor with elemental sodium or potassium, KC8 or Na/Hg, and subsequently it has three possibilities to reach a stable coordinative saturation: (i) it binds a solvent donor molecule or a donor from the gas phase, like N2; (ii) the alkali metal halide generated concomitantly remains in the coordination sphere; (iii) the aryl ring of a second molecule is coordinated so that a dimerization occurs.4,9,17 A further option becomes conceivable, if reductants are used that produce a weakly coordinating cation, namely, the preservation of the β-diketiminato nickel bromide core. We were interested to examine the behaviour of such a system, especially in a redox regime that is not as harsh as under conditions where alkali metals are used as reductants, bearing in mind that nickel enzymes, which reductively activate CO or CO2, also work at comparatively mild potentials.
For the selection of suitable reductants, the half-wave potentials had to be considered. In THF solution LtBuNiBr (LtBu = [HC(C(CMe3)NC6H3(iPr)2)2]−) exhibits a reversible redox event at −1.37 V (vs. Fc/Fc+) that corresponds to the NiII/NiI redox couple (see Fig. S3, ESI‡). Contemplating the chemical conversion of the nickel(II) complex to a reduced product, the Nernst equation for reversible electron-transfer processes requires the reductant to have a redox potential that is 0.118 V more negative to achieve a yield of 91%. The formal potentials of Cp2Co and Cp*2Co in THF are −1.3318 V and −1.8519 V (vs. Fc/Fc+), respectively. Hence, in the case of Cp*2Co an almost quantitative electron transfer was to be expected. However, Cp2Co was anticipated to represent an interesting case, too, as the formal potentials of both reactants are almost at the same level, so that the corresponding redox equilibrium should be subject to subtle influences.
Combining toluene solutions of LtBuNiBr and Cp*2Co led to red solutions, from which upon standing for 1 h a brown crystalline solid precipitated. Single crystal X-ray analysis combined with spectroscopic data revealed that it corresponded to the expected product [LtBuNiIBr][Cp*2CoIII], 1. The determined structure (Fig. 1) features a LtBuNiBr beside a Cp*2Co entity, and the relevant bond lengths and angles indicate that the electron has been transferred from Co to Ni, i.e. that 1 corresponds to an ionic compound: The bond distances around the nickel centre of the [LtBuNiBr]− anion in 1 (d(Ni–N) = 1.895(4), 1.904(4) Å; d(Ni–Br) = 2.3447(8) Å) are significantly longer than those found in the neutral complex LtBuNiBr (see Fig. S16 (ESI‡), d(Ni–N) = 1.8112(16), Å; d(Ni–Br) = 2.2781(5) Å) and fall within the range found for corresponding bonds in other NiI complexes containing that β-diketiminato ligand.4,9 The bond distances involving the cobalt atom in 1 (ave. Co–CCp = 2.051(6) Å; Co–Cpcent = 1.655(3) Å) are typical of those found in a decamethylcobaltocenium cation (e.g. [Cp*2Co][PF6]; ave. Co–CCp = 2.053(3) Å; Co–Cpcent = 1.655(2) Å).20 Consistently, the solid showed an EPR signal that matches well previous signals observed for LtBuNiI complexes.
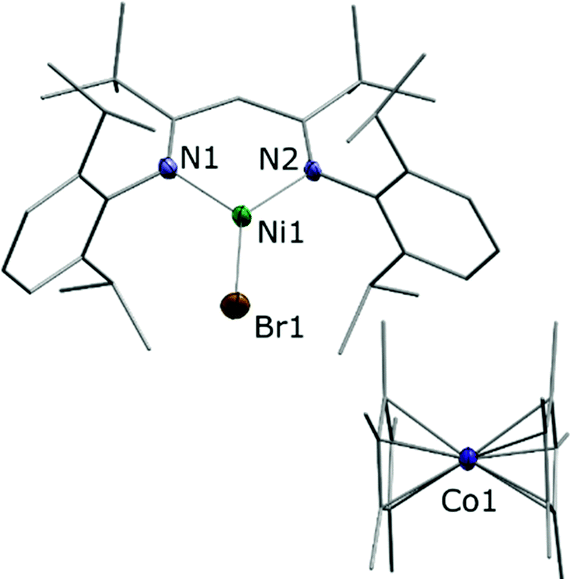 | ||
| Fig. 1 Molecular structure of [LtBuNiBr][Cp*2Co]·3C7H8. Hydrogen atoms are omitted for clarity. Selected bond lengths/Å and angles/°: N1–Ni1 1.895(4), N2–Ni1 1.904(4), Ni1–Br1 2.3447(8), N2–Ni1–N1 98.67(16), N1–Ni1–Br1 134.78(12), N2–Ni1–Br1 126.53(12), ave. Co–CCp = 2.051(6), ave. Co–Cpcent = 1.655(3). | ||
Astonishingly, the reaction between LtBuNiBr and Cp2Co proceeded similarly, despite the much milder reduction potential of Cp2Co. As before a brown solid crystallised directly from the reaction solution when the latter was sufficiently concentrated, and a single crystal X-ray analysis revealed a product constitution consisting of a LtBuNiBr and a Cp2Co entity (Fig. 2). The Cp–Co distances (ave. Co–CCp = 2.026(5) Å; Co–Cpcent = 1.629(3) Å) are consistent with those of a cobaltocenium cation ([Cp2Co][PF6]; ave. Co–CCp = 2.026(4) Å; Co–Cpcent = 1.637(5) Å),21 and for the same arguments as before again the structural data of the LtBuNiBr unit point to an ionic compound, i.e. [LtBuNiIBr][Cp2CoIII], 2.
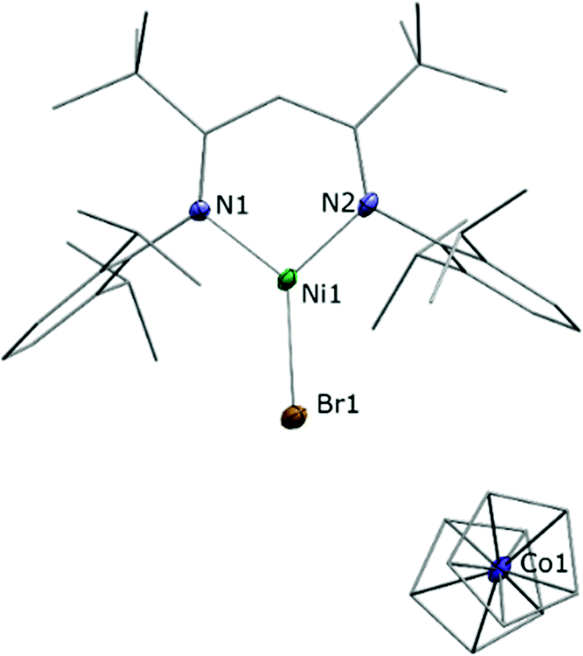 | ||
| Fig. 2 Molecular structure of [LtBuNiBr][Cp2Co]. Hydrogen atoms are omitted for clarity. Selected bond lengths/Å and angles/°: N1–Ni1 1.898(4), N2–Ni1 1.891(4), Ni1–Br1 2.3443(8), N2–Ni1–N1 99.18(17), N1–Ni1–Br1 130.55(12), N2–Ni1–Br1 130.26(12), ave. Co–CCp = 2.026(5), ave. Co–Cpcent = 1.629(3). | ||
Indeed SQUID magnetometry measurements are consistent with an S = 1/2 system over the entire temperature range (between 2 and 295 K, see Fig. S15, ESI‡), and EPR spectroscopy of polycrystalline 2 shows a signal typical for LtBuNiI compounds.
Consequently, the electron transfer compound [LtBuNiIBr][Cp2CoIII] had precipitated in a clean manner from the reaction mixture. However, when the same red brown solid was dissolved, investigations performed with the solution revealed a rather different situation. Unlike solutions of 1 in toluene, which are red and lead to an intense EPR signal, toluene solutions of 2 are green brown at RT (Fig. S1, ESI‡) and in the EPR spectrum no signal could be observed, which supports the case for a reverse of electron transfer: Resulting LtBuNiIIBr is an even-spin system and does not show an EPR signal in the perpendicular mode, while for Cp2Co an EPR spectrum can only be expected at rather low temperatures, due to the fast relaxation of its excited state.22,23 Upon cooling toluene solutions of 2, however, the colour brightens and eventually becomes comparable to that of 1 in toluene (Fig. S1, ESI‡). At the same time a characteristic NiI signal starts to emerge with decreasing temperature (Fig. 3), pointing to an equilibrium as depicted in eqn (1) that can be influenced by the temperature.
| LtBuNiIIBr + Cp2CoII ⇌ [LtBuNiIBr][Cp2CoIII] | (1) |
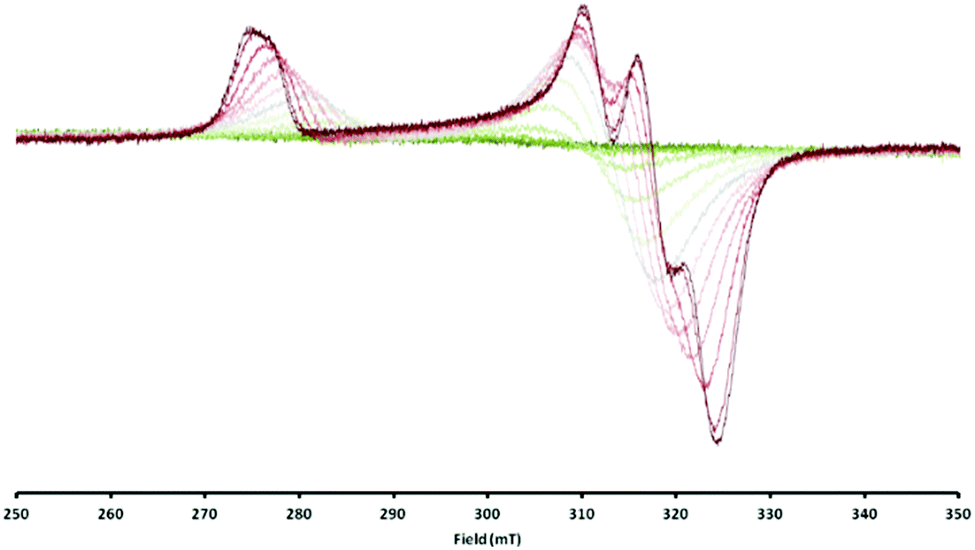 | ||
| Fig. 3 Overlaid EPR spectra of [LtBuNiBr][Cp2Co] in toluene measured between 20 °C (dark green, silent) and −95 °C (dark red, up to ∼60%). | ||
Integration of the EPR signals (in comparison to a standard) allowed the determination of the equilibrium constants at different temperatures (Fig. S14, ESI‡) and via the van’t Hoff equation a 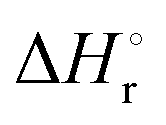 of −20.3 kJ mol−1 as well as a
of −20.3 kJ mol−1 as well as a 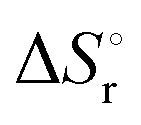 of −102.1 J mol−1 K−1 was determined, giving a
of −102.1 J mol−1 K−1 was determined, giving a 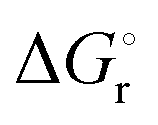 of 9.8 kJ mol−1.
of 9.8 kJ mol−1.
Hence, at room temperature this equilibrium lies far on the left-hand side and the two components are largely uncoupled with less than 2% of electron transfer complex [LtBuNiIBr][Cp2CoIII], 2, formed. However, apparently despite this small fraction the low solubility of 2 leads to its crystallisation and thus to the continuous shifting of the equilibrium to the right-hand side. In the solid state – removed from the equilibrium – [LtBuNiIBr][Cp2CoIII] is stable as an ion-pair.
Naturally, it was of interest how such a solution behaves electrochemically in comparison to solutions of [LtBuNiBr] and 1 (Fig. 4). While LtBuNiIIBr shows the abovementioned single NiII/NiI redox event at −1.37 V (process I), CV scans of 1 as expected show two further reversible events at more negative potentials. Process II is assigned to the CoIII/CoII couple, while process III at −3.16 V is typical for CoII/CoI of Cp*2Co. Consequently, three events should be expected for 2, too, but the lower electron donating effect of Cp with respect to Cp* for the cobaltocene means here the NiII/NiI and CoIII/CoII events coincidentally overlap (E(I)1/2 = −1.37 V), and the CoII/I event occurs at a more positive potential (E(II)1/2 = −2.55 V).
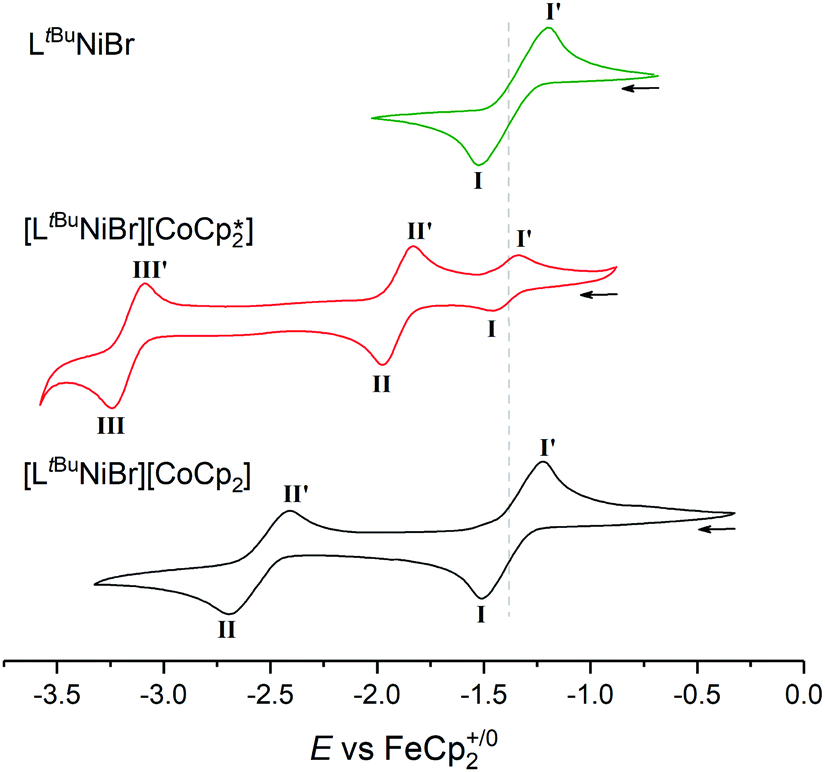 | ||
| Fig. 4 Offset cyclic voltammograms of LtBuNiBr (green), [LtBuNiBr][Cp*2Co] (red) and [LtBuNiBr][Cp2Co] (black) in THF/0.1 M [nBu4N][PF6], scan rate 100 mV s−1. Dotted line denotes NiII/I couple. | ||
The increased current response of process I with respect to process II (average i(I)p/i(II)p = 1.2) is consistent with this interpretation.
Finally, bearing in mind the background outlined in the introduction, the potential of 1 and 2 to activate CO2 was investigated. The nickel(I) complex [LtBuNiI–N2–NiILtBu] containing a labile dinitrogen ligand had been found to reductively couple CO2 to give oxalate.2 After double reduction of [LtBuNiI–N2–NiILtBu] with two equivalents of potassium the resulting [LtBuNiI–N2–NiILtBu]K2 had proved capable of cleaving CO2 to give nickel-bound CO and a carbonate complex [LtBuNiII–CO3]K, which aggregates through interactions involving the potassium ions to yield a hexamer.2 As in 1 and 2 the nickel complex anion contains only one reducing equivalent, a reaction analogous to [LtBuNiI–N2–NiILtBu] with formation of oxalate appeared intuitively more likely, but instead of the readily dissociating neutral N2 ligand more strongly bound anionic bromido ligands are found in 1 and 2, so that the nickel centres are not similarly accessible. Indeed, this significantly altered the course of the reaction. Upon addition of CO2 to solutions of 1 in C6D6 or toluene, within a minute the mixture became cloudy and a microcrystalline solid precipitated. This was analyzed by means of ATR-IR spectroscopy and while no Ni–CO3 or oxalate2 species were detected, bands between 1550 and 1650 cm−1 were detected, indicating the formation of a carbonate product (further assignment is difficult due to band overlap). In contrast, reaction of 2 with CO2 took much longer and thus only after 30 minutes the solution started to become cloudy, which is reasonable, as 2 can only react out of an equilibrium, in which it exists as a minor component. However, the slow reaction rate led to the formation of a crystalline solid after storage for one week and a crystal structure analysis indeed revealed the formation of the mononuclear carbonate complex [LtBuNiCO3][Cp2Co], 3 (Fig. 5). Extensive efforts to grow high quality crystals remained without avail, so that the data quality does not permit a discussion of metric parameters, but the molecular structure determined and the constitution are without doubt. A carbonate anion is symmetrically coordinating the nickel centre in a square planar fashion, so that the structure of 3 is rather different as compared to the one of {[LtBuNiII–CO3]K}6, where the K+ counterions actively participate in the structure construction and lead to aggregation to a hexanuclear complex.2,24,25 A similar outcome as in case of the reaction between 1 and CO2 was also revealed for 2 by ATR-IR spectroscopy (bands between 1550 and 1650 cm−1) and also in this case no Ni–CO3 or oxalate2 complexes were detected.
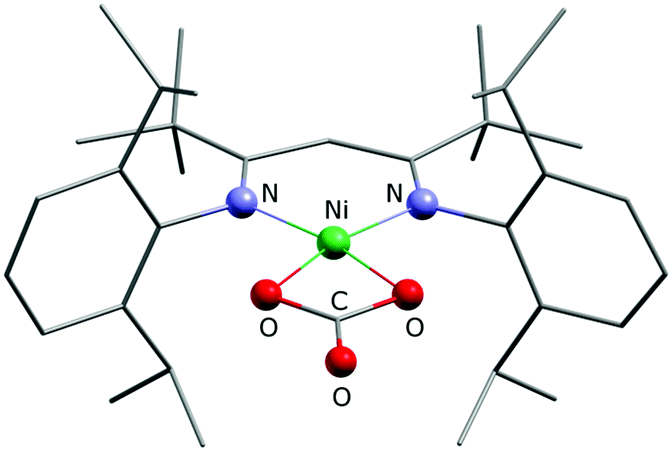 | ||
| Fig. 5 Ball and stick drawing of the solid state structure of the [LtBuNiCO3]− anion in 3. | ||
Formation of 3 requires cooperation of two equivalents of 2, which activate one CO2 molecule, likely yielding in a NiII–CO22− intermediate, which can react with a second molecule of CO2 to yield CO and carbonate (Scheme 1).26 Hence, concomitantly one equivalent of [LtBuNiBr] should be produced as well as one equivalent of [Cp2Co]Br, which, however, was not isolated. Similar cooperativity of two NiI centres in CO2 activation has been reported utilising different PNP-pincer ligands (LPNP). While in case of a rigid ligand the resulting LPNPNiII–CO22−–NiIILPNP appeared stable in contact with excessive CO2,27 utilising a more flexible ligand did also lead to concomitant reaction with an additional molecule of CO2, forming a dinuclear LPNPNiII–CO3–NiIILPNP unit and CO.28 However, no examples have been reported so far where a cooperative activation of CO2 leads to a mononuclear nickel complex, and thus our system is closing this gap. We hypothesise that the presence of the bromido ligand, which remained in the coordination sphere of the reduced nickel centre due to the employment of cobaltocenes as reducing agents (converting into non-coordinating cations), is decisive for the course of the reaction. If it is not there, as in case of [LtBuNiI–N2–NiILtBu], which in solution exists in a mononuclear form [LtBuNiI–N2]17 comparable to 2 but with a rather labile co-ligand, oxalate is formed. The differing findings made for 2, where one coordination site is blocked by the bromido ligand, suggests that formation of oxalate does not occur when only a single coordination site is available for CO2 activation at a nickel(I) centre, presumably as this hinders the fast dimerization of the transient CO2˙− species, which thus accepts a further electron instead.
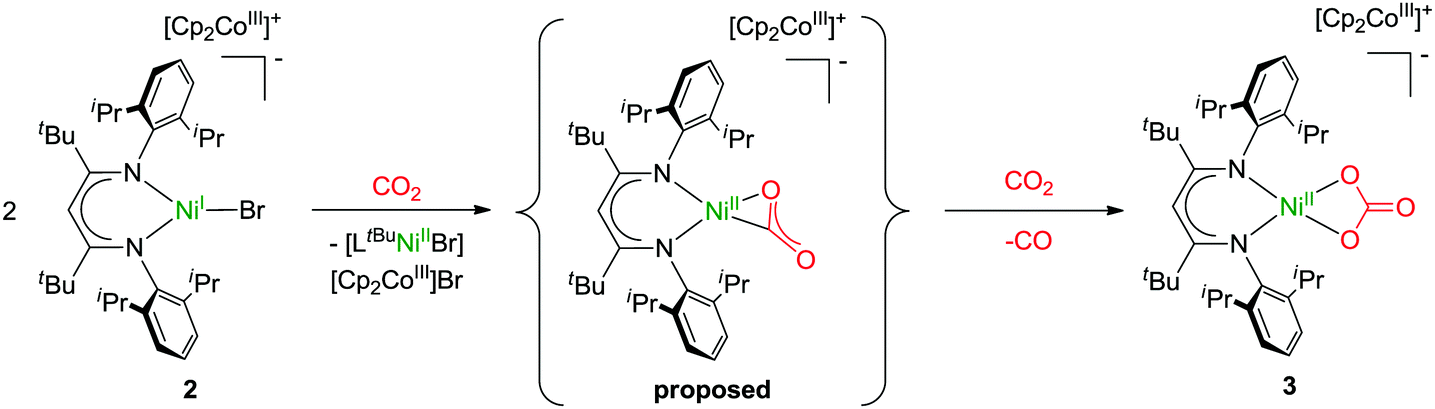 | ||
| Scheme 1 Reaction of 2 with CO2 leads to the formation of 3via a possible NiII–CO22− intermediate. | ||
Consequently, carbonate is formed from solutions of both 1 and 2 in contact with CO2 gas, which is notable as solutions of 2 at ambient temperature – unlike those of 1 – do not even show an EPR signal indicative for nickel(I) species (Cp2Co alone does not react with CO2). Thus, small molecule activation at reduced metal centres does not necessarily require quantitative electron transfer from the reductant, which should also be considered when discussing oxidation states in biological systems.
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038. We are grateful to Professor Dr Rainer Winter for helpful discussions.
Conflicts of interest
There are no conflicts to declare.References
- P. Zimmermann and C. Limberg, J. Am. Chem. Soc., 2017, 139, 4233 CrossRef CAS.
- B. Horn, C. Limberg, C. Herwig and B. Braun, Chem. Commun., 2013, 49, 10923 RSC.
- B. Horn, S. Pfirrmann, C. Limberg, C. Herwig, B. Braun, S. Mebs and R. Metzinger, Z. Anorg. Allg. Chem., 2011, 637, 1169 CrossRef CAS.
- S. Pfirrmann, C. Limberg, C. Herwig, R. Stößer and B. Ziemer, Angew. Chem., Int. Ed., 2009, 48, 3357 CrossRef CAS.
- S. Yao, Y. Xiong, C. Milsmann, E. Bill, S. Pfirrmann, C. Limberg and M. Driess, Chem. – Eur. J., 2010, 16, 436 CrossRef CAS.
- P. Holze, B. Horn, C. Limberg, C. Matlachowski and S. Mebs, Angew. Chem., Int. Ed., 2014, 53, 2750 CrossRef CAS.
- S. Yao, E. Bill, C. Milsmann, K. Wieghardt and M. Driess, Angew. Chem., Int. Ed., 2008, 47, 7110 CrossRef CAS.
- S. Yao and M. Driess, Acc. Chem. Res., 2012, 45, 276 CrossRef CAS.
- S. Pfirrmann, S. Yao, B. Ziemer, R. Stösser, M. Driess and C. Limberg, Organometallics, 2009, 28, 6855 CrossRef CAS.
- S. C. Puiu and T. H. Warren, Organometallics, 2003, 22, 3974 CrossRef CAS.
- E. Kogut, H. L. Wiencko, L. Zhang, D. E. Cordeau and T. H. Warren, J. Am. Chem. Soc., 2005, 127, 11248 CrossRef CAS.
- M. M. Melzer, S. Jarchow-Choy, E. Kogut and T. H. Warren, Inorg. Chem., 2008, 47, 10187 CrossRef CAS.
- S. Wiese, P. Kapoor, K. D. Williams and T. H. Warren, J. Am. Chem. Soc., 2009, 131, 18105 CrossRef CAS.
- P. Zimmermann, D. Ar, M. Rößler, P. Holze, B. Braun, C. Herwig and C. Limberg, Angew. Chem., Int. Ed. DOI:10.1002/anie.202010180.
- T. Kothe, U. H. Kim, S. Dechert and F. Meyer, Inorg. Chem., 2020, 59, 14207 CrossRef CAS.
- H. Stevens, P.-C. Duan, S. Dechert and F. Meyer, J. Am. Chem. Soc., 2020, 142, 6717 CrossRef CAS.
- P. Holze, B. Braun-Cula, S. Mebs and C. Limberg, Z. Anorg. Allg. Chem., 2018, 644, 973 CrossRef CAS.
- D. V. Yandulov and R. R. Schrock, Inorg. Chem., 2005, 44, 1103 CrossRef CAS.
- D. Nied, E. Matern, H. Berberich, M. Neumaier and F. Breher, Organometallics, 2010, 29, 6028 CrossRef CAS.
- D. Braga, O. Benedi, L. Maini and F. Grepioni, J. Chem. Soc., Dalton Trans., 1999, 2611 RSC.
- T. Zhu, Y. Sha, H. A. Firouzjaie, X. Peng, Y. Cha, D. M. M. M. Dissanayake, M. D. Smith, A. K. Vannucci, W. E. Mustain and C. Tang, J. Am. Chem. Soc., 2020, 142, 1083 CrossRef CAS.
- J. H. Ammeter and J. M. Brom, Chem. Phys. Lett., 1974, 27, 380 CrossRef CAS.
- J. H. Ammeter and J. D. Swalen, J. Chem. Phys., 1972, 57, 678 CrossRef CAS.
- B. Horn, C. Limberg, C. Herwig, M. Feist and S. Mebs, Chem. Commun., 2012, 48, 8243 RSC.
- N. J. Hartmann, G. Wu and T. W. Hayton, Chem. Sci., 2018, 9, 6580 RSC.
- P. Zimmermann, S. Hoof, B. Braun-Cula, C. Herwig and C. Limberg, Angew. Chem., Int. Ed., 2018, 57, 7230 CrossRef CAS.
- C. Yoo and Y. Lee, Angew. Chem., Int. Ed., 2017, 56, 9502 CrossRef CAS.
- F. Schneck, F. Schendzielorz, N. Hatami, M. Finger, C. Würtele and S. Schneider, Angew. Chem., Int. Ed., 2018, 57, 14482 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Dr Wolfgang Kaim on occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental section, additional figures and X-Ray crystallographic data. CCDC 2023970, 2023971, 2024643. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06983c |
| This journal is © The Royal Society of Chemistry 2021 |