
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies to promote permeation and vectorization, and reduce cytotoxicity of metal complex luminophores for bioimaging and intracellular sensing
Lorcan
Holden
,
Christopher S.
Burke
,
David
Cullinane
and
Tia E.
Keyes
 *
*
School of Chemical Sciences, and National Centre for Sensor Research Dublin City University, Dublin 9, Ireland. E-mail: holden.lorcan2@mail.dcu.ie; christopher.burke42@mail.dcu.ie; david.cullinane5@mail.dcu.ie; tia.keyes@dcu.ie
First published on 5th May 2021
Abstract
Transition metal luminophores are emerging as important tools for intracellular imaging and sensing. Their putative suitability for such applications has long been recognised but poor membrane permeability and cytotoxicity were significant barriers that impeded early progress. In recent years, numerous effective routes to overcoming these issues have been reported, inspired in part, by advances and insights from the pharmaceutical and drug delivery domains. In particular, the conjugation of biomolecules but also other less natural synthetic species, from a repertoire of functional motifs have granted membrane permeability and cellular targeting. Such motifs can also reduce cytotoxicity of transition metal complexes and offer a valuable avenue to circumvent such problems leading to promising metal complex candidates for application in bioimaging, sensing and diagnostics. The advances in metal complex probes permeability/targeting are timely, as, in parallel, over the past two decades significant technological advances in luminescence imaging have occurred. In particular, super-resolution imaging is enormously powerful but makes substantial demands of its imaging contrast agents and metal complex luminophores frequently possess the photophysical characteristics to meet these demands. Here, we review some of the key vectors that have been conjugated to transition metal complex luminophores to promote their use in intra-cellular imaging applications. We evaluate some of the most effective strategies in terms of membrane permeability, intracellular targeting and what impact these approaches have on toxicity and phototoxicity which are important considerations in a luminescent contrast or sensing agent.
Introduction
Fluorescence microscopy is one of the key underpinning research tools in cell biology and is a powerful method for visualising the cellular environment and that of its organelles. In live cells, in particular, fluorescence microscopy is used to understand and interrogate dynamic changes to subcellular molecular constituents such as nucleic acid, enzymes and protein–receptor interactions and membranes. The most widely applied imaging method is probably confocal fluorescence microscopy as it provides high, but diffraction limited, resolution in imaging and offers improved axial and lateral resolution as well as background elimination over wide field methods. However, over the past 20 years optical imaging technology has advanced dramatically, overcoming the notable hurdle of Abbe's diffraction limit in fluorescence based super-resolution microscopy techniques such as stimulated emission depletion (STED) and Stochastic Optical Reconstruction Microscopy (STORM).1,2Fluorescence microscopy, in all its forms, including related methods such as flow cytometry, rely on fluorescence contrast agents. Small molecule, organic fluorophores are the most widely applied contrast agents in fluorescence imaging because they frequently have molecular weights below 500 Da and often show good permeability to the cell membrane. Frequently, those with superior permeability are very lipophilic compounds that may require dissolution in organic solvent. Although this can promote permeabilization it can also damage cell membrane and organic solvent above about 4 or 5% v/v in cell media can induce cytotoxicity. A key advantage of organic fluorophores is that they can have outstanding molecular brightness with very high extinction coefficients at their excitation wavelength (>50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and quantum yields in the range 0.3 to 0.6 or higher. Conversely, they also typically have short emission lifetimes (approx. <10 ns), tendency to photobleach and small Stokes shifts (<50 nm). The latter can lead to issues of emission artifacts from inner-filter effects and self-quenching where local concentrations are high, e.g. when targeted in imaging applications. These issues have led to demand for more specialised dyes that fulfil specific photophysical requirements of the given imaging application or technique and in many cases these demands, can potentially be fulfilled by transition metal complexes.3
000 M−1 cm−1) and quantum yields in the range 0.3 to 0.6 or higher. Conversely, they also typically have short emission lifetimes (approx. <10 ns), tendency to photobleach and small Stokes shifts (<50 nm). The latter can lead to issues of emission artifacts from inner-filter effects and self-quenching where local concentrations are high, e.g. when targeted in imaging applications. These issues have led to demand for more specialised dyes that fulfil specific photophysical requirements of the given imaging application or technique and in many cases these demands, can potentially be fulfilled by transition metal complexes.3
Correspondingly, over the past 15 years the application of transition metal complexes luminophores to cell imaging has expanded dramatically.4 This has been driven in large part by creative solutions to the longstanding problems of poor permeation of coordination compounds through the cell membrane.5–9 In many cases these solutions are inspired by strategies employed in the pharmaceutical industry to promote membrane permeability of therapeutics and their delivery to intracellular targets. In eukaryotic organisms, cell permeation of exogenous species can occur via passive and/or active transport or through vesicle mediated mechanisms, the latter are also activated mechanisms. Mode of cell uptake, where it occurs, is dictated by the nature of the molecular or ionic species to cross the cell membrane. Passive transport is the simple diffusion of a molecular species across the membrane along a concentration gradient without energy input. Active molecular transport can be primary active transport where movement is mediated by a transport protein (of which there are numerous examples), powered by ATP hydrolysis or through a secondary active transport mechanism that is facilitated by a redox gradient, often occurring for ions. Endocytosis is an active bulk transport mechanism, that mediates movement of particles and large molecular species across the membrane via membrane invagination and encapsulation of the species into a membrane bound vesicle for transport. There are multiple mechanisms behind endocytosis, the prevalent one depends on the nature and notably the size of the species taken up. The topic is reviewed in detail elsewhere.10,11
Obviously, for drugs with intracellular targets, cell membrane permeability is a key determinant of efficacy and many conventional small molecule therapeutics are passively membrane permeable. Traditionally, the Lipinski rule of 5 has been widely used to predict oral availability of a drug and correlates strongly with passive membrane permeability of a molecule, defining parameters; logP (<5), molecular weight (less than 500 Da) and number of H-bond donors and acceptors as multiples of 5 that a molecule must possess to have drug-like behaviour.12 Although, in practise, passively permeable small molecules frequently deviate from these parameters, they remain a useful starting point to anticipate passive permeability and physicochemical properties of transition metal complex luminophores usually deviate significantly from the rule parameters. But so also do many non-traditional therapeutics, most notably biologics, such as proteins and nucleic acids. Consequently, a wide variety of methods have been exploited to drive such species across the membrane in therapeutic applications, in particular, as discussed below, the use of delivery vectors or signal or cell penetrating peptides (CPPs) to drive membrane permeation and targeting.13
So, instead of just relying on means to promote passive permeation, means to promote active processes that drive impermeable species across the membrane and that can even target them to specific receptors or destinations have been widely studied and exploited in therapeutics. As discussed herein, these methods can be effectively adapted to transition metal complex luminophores. Many of the strategies discussed, of course also apply to metal complex therapeutics but in this short review, we focus on only metal complexes as imaging agents and some instance theranostic agents, rather than metallodrugs and direct the reader to other reviews for this focus.14
Transition metal luminophores for cellular imaging
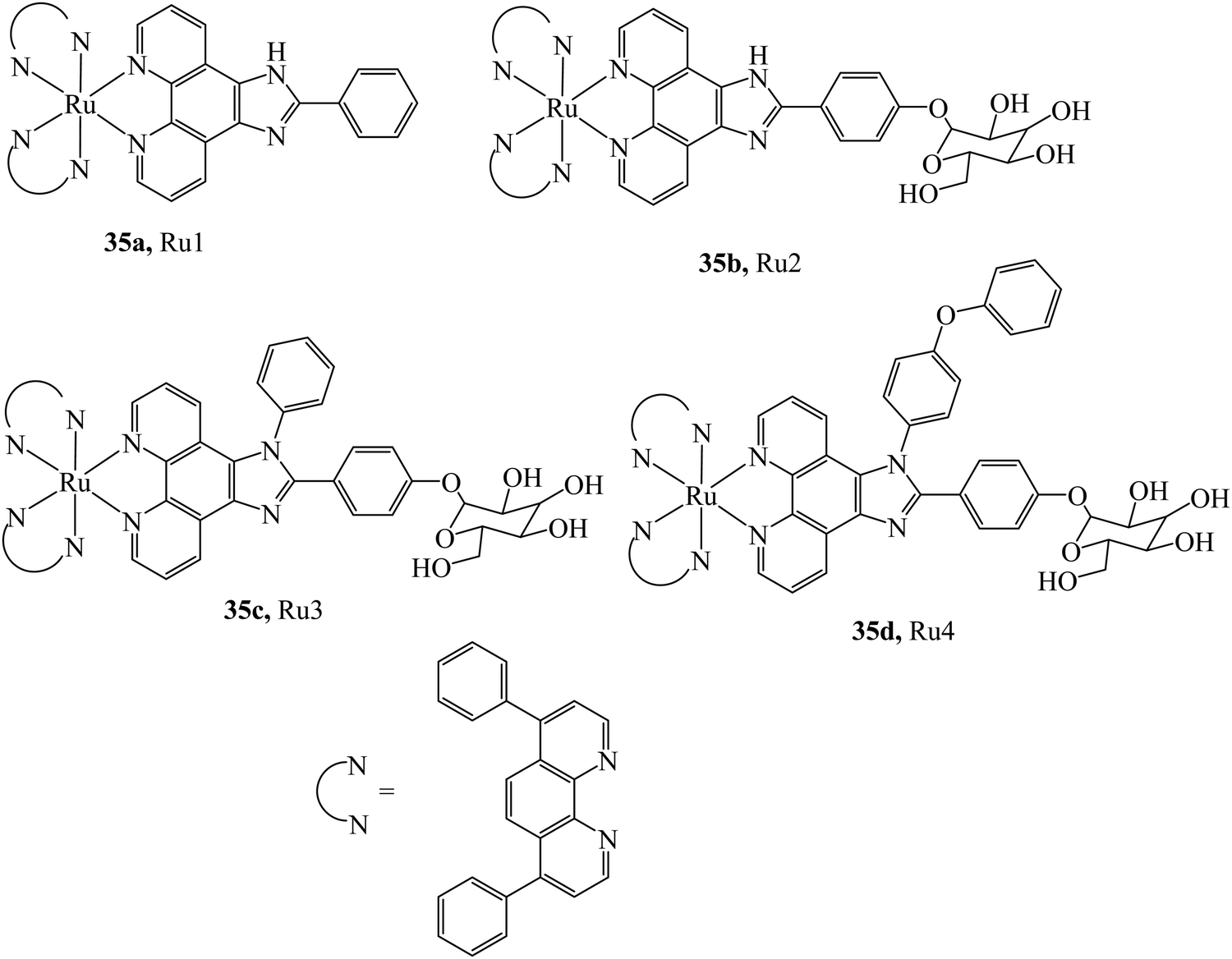
Transition metal complexes luminophores typically exhibit triplet charge transfer excited states that confer emission characteristics very different to those of organic fluorophores. Not all are advantageous for every application, for example in single photon counting methods, used for example in Fluorescence (or phosphorescence lifetime imaging) or in fluorescence lifetime correlation the long lived excited states of metal complexes can dramatically extend the time taken to acquire data. Nonetheless there are many photophysical characteristics that make metal complexes potentially very useful as an imaging probe. Such properties include; long luminescent lifetimes, often associated with long-lived triplet metal–ligand charge transfer (3MLCT) excited states that can confer environmental sensitivity on the probe; excellent photostability, that can be tuned by ligand modification; and large Stoke shifts, that can help avoid artifactual effects caused by high local probe concentration. The Stokes shift in emission also permit multimodal methods to be applied such as resonance Raman with emission imaging, this is usually rendered impossible by the large fluorescence background on the weaker Raman signal observed when the emission is not Stokes shifted.15Another key advantage that the chemist can draw upon in designing transition metal luminophores for cell imaging are the well-established synthetic methods and the vast library of reported structures bearing different metal and ligand combinations that can be applied to tune the physico-chemistry of metal complex luminophores. Metal ion identity and the σ–donor and π*–acceptor abilities of the ligands can be readily altered to tune excited state properties. And, in 6 coordinate complexes like Ru(II) or Os(II) mixed ligands can be applied to the metal centre to achieve a specific physiochemical goal. For example, solvent dependant emission switching can be exploited using phenazine ligands like dipyridophenazine (dppz) that render their complexes non-emissive in water, thus providing a contrast probe with effectively zero background.16,17 Photoactive excited states can be manipulated to stimulate photo-induced redox reactions with biomolecules, ligand ejection or ROS generation, all of which can be exploited for phototherapeutic applications of metal complexes but such approaches can also offer selective imaging advantages.18–20 Cyclometallation, particularly for Ir(III) complexes, enhances lipophilicity and offers outstanding tuneability, because of the mixed 3MLCT and 3IL nature of the excited states, of emission wavelength and lifetime.21 Complexes bearing reactive functional groups are also often included in probe design to permit facile follow-on conjugation or click chemistries for vectorisation or for targeted cellular or bio-orthogonal reactions.22–26
Lastly, the long-lived and triplet nature of metal complex excited states can be harnessed for intracellular sensing, most notably through quenching by oxygen (O2) and detection of reactive oxygen species (ROS).27,28
The most widely reported metal luminophores applied to cellular imaging to date are those of Ru(II), Ir(III), Re(I) and Pt(II) coordination compounds, but there are exciting recent examples that have focussed on Os(II) and Zn(II), as outlined below. Ru(II) complexes used in bioimaging typically bear polypyridine ligands (N^N) and are characterised by their blue excitation into 1MLCT excited states to yield red-orange 3MLCT emission. Although less uncommon, NIR emission is accessible in such complexes using ligand systems like 2,2′-biquinoline (biq) or 2,6-bis(8′-quinolinyl)pyridine (bqp).29–31 The majority of ligands applied to Ru(II) (and Os(II)) luminophores are bidentate as quantum yields and lifetimes are typically shorter in tridentate terpyridyl systems. Although there are synthetic strategies to enhance photophysical properties in such complexes, there have been few reports to date on their application as imaging probe.32–34 A key drawback with Ru(II) probes is that, with the exception of more lipophilic complexes (e.g. bearing 4,7-diphenyl-1,10-phenanthroline (dpp) or cyclometalated ligands), Ru(II) complexes tend to be poorly membrane permeable.35,36 In contrast, Ir(III) complexes appear to have much higher cellular permeability, due to their greater lipophilicity particularly with cyclometalated ligand systems based on phenyl-pyridines (C^N), typically as; [Ir(C^N)2(N^N)]+.37 Such ligands also confer opportunity for substantial photophysical tuning of long-lived 3IL and 3MLCT states. Although, Ir(III) probes are often limited in the imaging context by higher cytotoxicity. Research into Re(I) complexes for bioimaging has long been driven by the use of Re(I) as a cold and luminescent analogue for 99mTc radiopharmaceuticals. Most Re(I) complexes designed for luminescence imaging are comprised of a tricarbonyl and N3-polypyridine ligand core, [Re(N^N^N)(CO)3]+, and such Re(I) probes exhibit longer-lived 3MLCT excited states and relatively higher quantum yields than ruthenium complexes. They are limited, however, in the context of bioimaging by their blue/UV excitation wavelengths, which is a poorly transmissive through biological media and typically generates interfering autofluoresence.38 Interestingly, though, the carbonyl ligand, because of its distinctive vibrational frequency, offers the opportunity to map the probe distribution by vibrational microscopy where UV/blue excitation is not required, some examples are discussed below.39 The coordination geometry of Pt(II) complexes tends to be square-planar, offering distinct physicochemical and thus biochemical impact as well as distinctive photophysical properties compared to the more widely studied 6-coordinate metal systems. For example, aggregation and disaggregation of Pt(II) planar units can induce emission switching.40 Photophysical tuning of their 3ILCT (intraligand charge transfer) states is readily achieved within the common design of using tridentate ligands, particularly via cyclometalated ligands, which can confer long-lived excited states suited to two-photon and time-gated imaging.41
With growing interest in metallo-probes, focus is also moving toward less-explored metal systems such as the NIR imaging capabilities of Os(II), and also toward, the use of Zn(II) as a first-row transition metal with potential for super-resolution imaging.42–44
Strategies to promote cellular uptake of metal complex luminophores
The application of transition metal luminophores to cell imaging has, in the past, been limited by generally poor or unpredictable uptake, poor intracellular targeting and high cytotoxicity of such metal complexes. As described, few metal complex luminophores conform well to the rule of 5, and although there are some examples where very lipophilic metal complexes appear to be capable of passive uptake,35,45 it has been shown that judicious complex conjugation to uptake vectors and targeting agents can promote reliable permeation and targeting. This has led over the past decade to increased interest in the application of metal complexes luminophores for imaging. Furthermore, precision targeting is often accompanied by reduced toxicity, for example, by limiting mitochondrial access if this is not the imaging target.A disadvantage to applying drug delivery approaches to metal complexes though, may be inefficient endosome escape, which is avoided in passive diffusion. Endosomal entrapment can often be identified as punctate staining patterns in cell images coincident with the late endosomes and/or lysosomes. Fortunately, there are many reports of successful subcellular targeting by vectorisation and in the following sections, we outline some of the key metal complex vectorisation strategies employed to date for cellular imaging and their relative successes and failures.
Promoting cell permeation of metal complexes by modulating lipophilicity using organic pendants
Lipophilicity is a key physicochemical property that can influence a species’ cell membrane permeability, and the mechanism of uptake, i.e. through active mechanism such as endocytosis or passive permeation. Though for many species a mixture of uptake mechanisms apply. Small changes in lipophilicity between probe derivatives can drastically alter cellular uptake and distribution. The lipophilic/cationic balance required to induce uptake and influence localisation of metal complexes remains poorly defined. Some commonly explored modifications made to metal complexes to alter lipophilicity include conjugation to alkyl chains, poly(ethylene glycol) (PEG), triphenylphosphonium (TPP) and lipophilic organic moieties, or alternatively through direct tuning of ligand lipophilicity.Numerous studies have explored the lipophilic/cationic balance required to achieve uptake of metal complexes by vectorisation with alkyl and alkane thiols chains of varying length. Some of the earliest work investigating metal–alkane conjugates were centred on rhenium complexes. It was demonstrated that alkyl ester derivatives (n = 6 to 16) of complexes 1a–c, (Fig. 1) were permeable to Spironucleus vortens, indicating the importance of strong lipophilic character.46 This report was followed by a more detailed investigation into these complexes (Fig. 1) where uptake into yeast cells was poor but uptake into MCF-7 human cells, was greatly enhanced. The complexes showed generally low toxicity, although toxicity increased with increasing lipophilicity of the complex in those with longer alkyl chains. Intracellular localiseation was also shown to be dependent on lipophilicty where membrane, nucleolus and mitochondrial membrane localisation was evident from confocal microscopy for lipophilic derivatives whereas mitochondrial uptake was more prominent for more polar complexes. Incubation of the complexes at 4 °C was investigated to understand if the uptake mechanism was energy independent. Under these conditions, the cationic species permeated the cell membrane, suggesting passive diffusion.47 Other reported rhenium alkane conjugates include the series 3a–c (Fig. 1). The NCS-free derivatives localised within the mitochondria of HeLa cells and showed very high toxicity. Incubation at 4 °C greatly reduced cellular uptake indicating an energy-dependent uptake pathway such as endocytosis.48
 | ||
| Fig. 1 Rhenium carbonyl complexes vectorised with differing alkyl chains. | ||
Massi et al. have shown that by modulating charge on Ir(III)tetrazolato complexes the uptake and toxicity to bacteria could be controlled leading to effective strategies for imaging bacteria with these compounds.49,50 Lincoln et al. reported the complex series 4a–c (Fig. 2) with alkyl chains of varying length that were investigated for cellular uptake and localization.51 They observed that the least lipophilic complex, 4a, stained the nucleus whereas the more lipophilic species, 4c, localised at the plasma membrane and endoplasmic reticulum. Lipophilicity of the complexes was inferred from length of alkyl chain, but a quantitative value such as logP, was not reported. Interestingly, the membrane staining complexes induced membrane permeabilization when photoactivated. This was attributed to damage caused by singlet oxygen generated locally by the excited state complex. The series was further investigated to achieve a greater understanding of their biomolecular binding properties in cells. Varying the alkyl chain length altered staining patterns in confocal microscopy reflecting altered recognition of RNA, DNA and phospholipid membranes reflecting the effect of lipophilicity on cellular localization.52 Lincoln et al. reported 5a–c (Fig. 2), developed for the purpose of two-photon photosensitizer for PDT targeting lysosomes. The two-photon absorbance cross-section of the complexes was found to be between 700 nm to 900 nm, coinciding well with the biological window. Lysosome targeting was confirmed through colocalization with LysoTrackerGreen where a correlation value 0.85 was measured, whereas a value of 0.13 and 0.08 were reported for MitoTrackerGreen and Hoechst 33342 respectively. And, importantly, the complexes showed low dark cytotoxicity but induced necrocytosis in the cells under two photon irradiation.53
 | ||
| Fig. 2 Ru(II) polypyridyl structures with alkyl chains reported by Lincoln et al.35,36 | ||
Massi et al. reported that fatty acid pendants were effective in driving Iridium Tetrazolato complexes 7a–e into live cells mainly to the endoplasmic reticulum in and importantly, in spite of the lipophilicty the complexes showed low cytotoxicity. In general, increasing the lipophilicity of the metal through modification of the ligand or applying a lipophilic pendant, has been successful approach to promoting metal complex uptake, drawbacks of this approach are that such complexes can have low water solubility and so require organic solvent for dissolution.54 As commonly used solvents in imaging, DMSO and ethanol increase membrane permeability, it can be difficult to assess the true permeability of probes that are pre-dissolved in such solvent. Furthermore, although by no means a universal effect, increasing lipophilicity can promote dark cytotoxicity, often because the mitochondria is targeted. An example that bucked the trend, was reported by Glazer et al., where uptake, localisation and toxicity of tris homoleptic complexes 8 and 9 were compared. Whereas the more lipophilic dpp complex showed strong uptake, mitochondrial localisation and cytotoxicity, surprisingly, given its charge, the anionic complex showed fair uptake into cells. Because it did not target the mitochondria it showed much lower cytotoxicity in the dark then the lipophilic analogue but did show photocytotoxicity, this is an advantage in terms of phototherapy but may also be a useful approach in imaging whereby balancing charge and lipophilicity may permit control of localisation and reduce cytotoxicity.55
Appending the polymer poly(ethylene glycol) (PEG) to ligands can overcome some of the less desirable characteristics sometimes associated with alkyl chain conjugation to metal complexes. Notably, PEGylation can promote water solubility/hydrophilicity and reduce cytotoxicity. It has been hypothesised that the reduction in cytotoxicity is a result of the PEG pendant impeding the interaction of metal complexes with organelles and biomolecules.56 In addition to using PEG as a stand-alone pendant, studies have also reported using PEG as a linker to other functional moieties to promote aqueous solubility, this has been effectively utilised to link a luminescent metal core to bioorthogonal compounds.57
Li et al. synthesised a series of PEGylated Ir(III) complexes and studied their cellular uptake and distribution. These Ir(III) complexes had high water solubility and excellent biocompatibility, i.e. low cytotoxicity.58 The group further investigated Ir(III)-PEG series 10–12 (Fig. 3) (Fig. 3) where C^N is ppy, pq, pba,with a focus on studying biological applications. These PEG vectorised complexes exhibited minimal dark cytotoxicity but substantial light-induced toxicity, in contrast to the free complexes that were dark toxic. The [Ir(pq)2(bpy-CONH-PEG)]+ complex was visualised in a zebrafish model 1 h and 24 h after injection. The complex was distributed throughout the zebrafish in the head space, around the yolk sac and cardiac cavity after 24 h with intense emission suggesting slow clearance of the complex.56
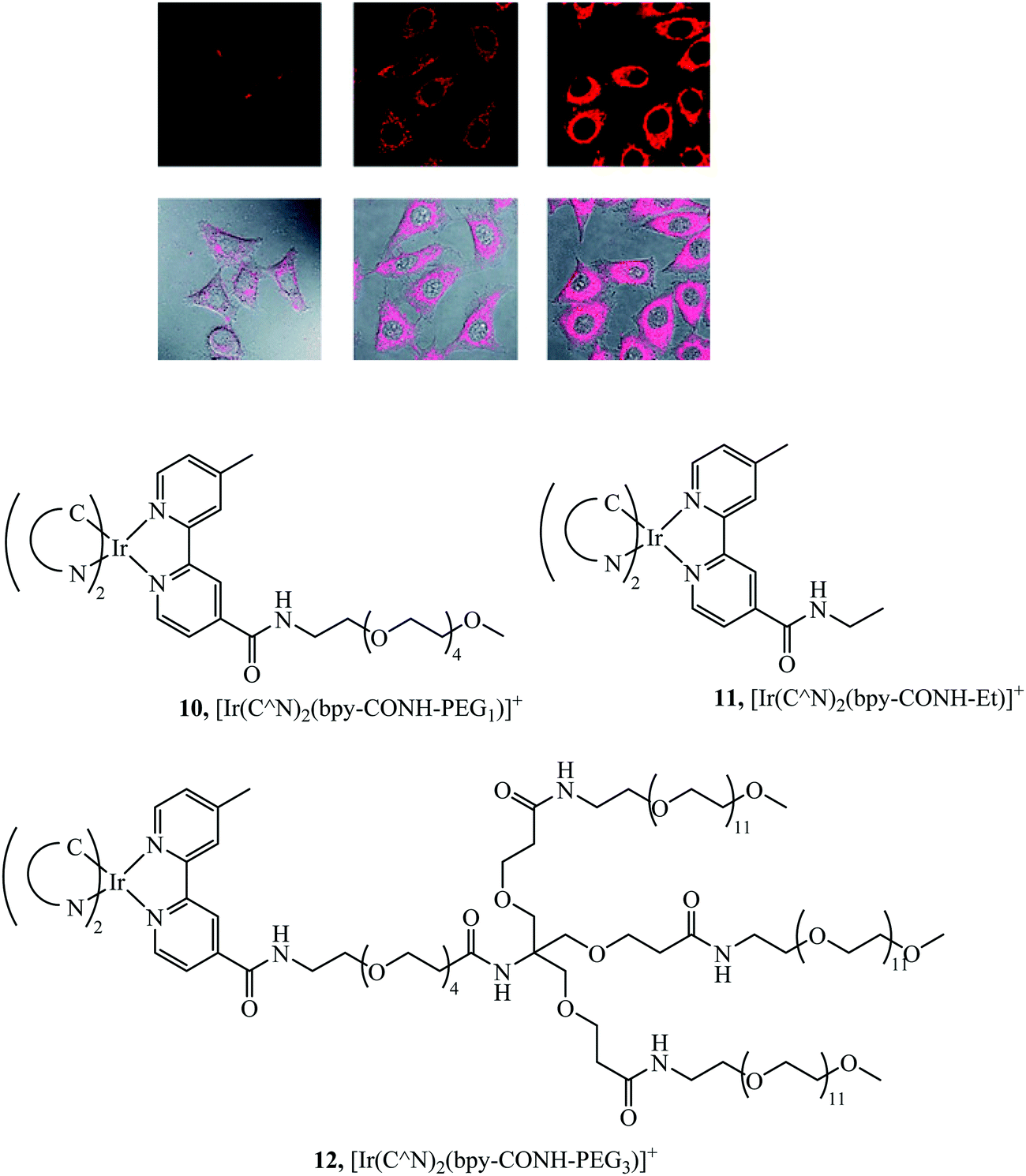 | ||
| Fig. 3 Pegylated IR complexes visualised with HeLa cells with complex 10 at 10 mm for 1 h (left), 100 mm for 1 h (middle), and 200 mm for 2 h (right) at 378 C. Reproduced from ref. 58, S. P.-Y. Li, H.-W. Liu, K. Y. Zhang and K. K.-W. Lo, Chem. – Eur. J., 2010, 16, 8329–8339, Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Triphenylphosphonium (TPP) has been quite widely reported as a lipophilic cationic moiety, that when appended to a ligand, is effective in promoting cellular uptake and in particular in promoting mitochondrial targeting of complex luminophores. The mitochondria are a dual membrane bound structure, the outer membrane is similar to plasma membrane in terms of permeability but the inner mitochondrial membrane which is the barrier to the mitochondrial matrix, exhibits a unique transmembrane potential (Δψm). This is due to electrochemical gradient formed from the respiratory chain complexes of the mitochondria, the value varies with cell type and disease state but is roughly −180 mV. This anionic potential can be exploited to promote mitochondrial targeting and membrane permeation. Mitochondrial dysfunction is implicated in cardiovascular and neurological diseases, so diagnostic/sensor probes particularly those focussing on oxygen or ROS and capable of targeting the mitochondria and reporting on mitochondrial health are important.59 In drug delivery delocalized lipophilic cations (DLCs) have been an important tool for mitochondrial delivery promoted by their dual lipophilic and cationic characteristics that attract them into the negatively charged mitochondrial matrix permitting their accumulation in the mitochondria.60
TPP is one of the most prevalent DLCs used in drug delivery with a cationic charge/lipophilicity ratio that readily promotes membrane targeting and thus it has also become a key moiety applied to metal complexes. Zielonka et al. have published a comprehensive review of TPP as a mitochondrial target which concentrates on organic conjugates and also reviews some transition metal conjugates.61
Murase et al. reported the iridium complexes, 12 and 13 (Fig. 4), as oxygen sensors that localise in the endoplasmic reticulum and mitochondria of HeLa cells respectively. The luminescence of the complex was quenched under normoxic conditions (20% O2) whereas in hypoxic conditions (2.5% O2) the complex was brightly phosphorescent. Imaging was performed using luminescence lifetime imaging, with an incubation concentration of just 500 nM.62 Zhou et al. investigated two iridium complexes, 14 and 15, (Fig. 4) as green and NIR emitters, both complexes modified with TPP and effectively targeting mitochondria. The dyes showed excellent anti-photobleaching capability in continuous live cell imaging whilst maintaining relatively low photocytotoxicity.63 TPP has also been effective at mitochondrial targeting of large macromolecules, Rau et al. synthesised the [Ru(bpy)2(pic)]2+–protein (HSA)–PEO–tpp complex which contained a ruthenium photosensitiser, PEO chain and the HSA protein which was highly photocytotoxic.64
 | ||
| Fig. 4 Cyclometalated Iridium complexes with triphenylphosphine pendants. | ||
In contrast, TPP vectorisation did not redirect the platinum(II) luminescent complex 16 (Fig. 5) to the mitochondria. The complex was water soluble and localised in the plasma membrane of HeLa cells. Bioimaging was achieved through two-photon induced luminescence with low cytotoxicity. Cell viability was reported as 90% in HeLa cells after 25 hours of exposure at a concentration of 10 μg ml−1.65
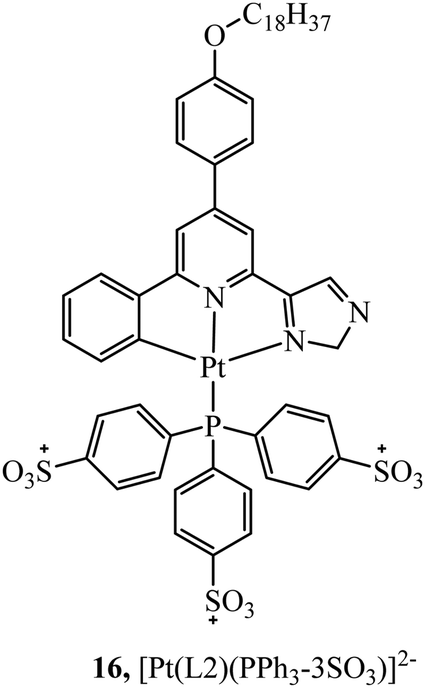 | ||
| Fig. 5 Platinum complexes containing triphenylphosphine. | ||
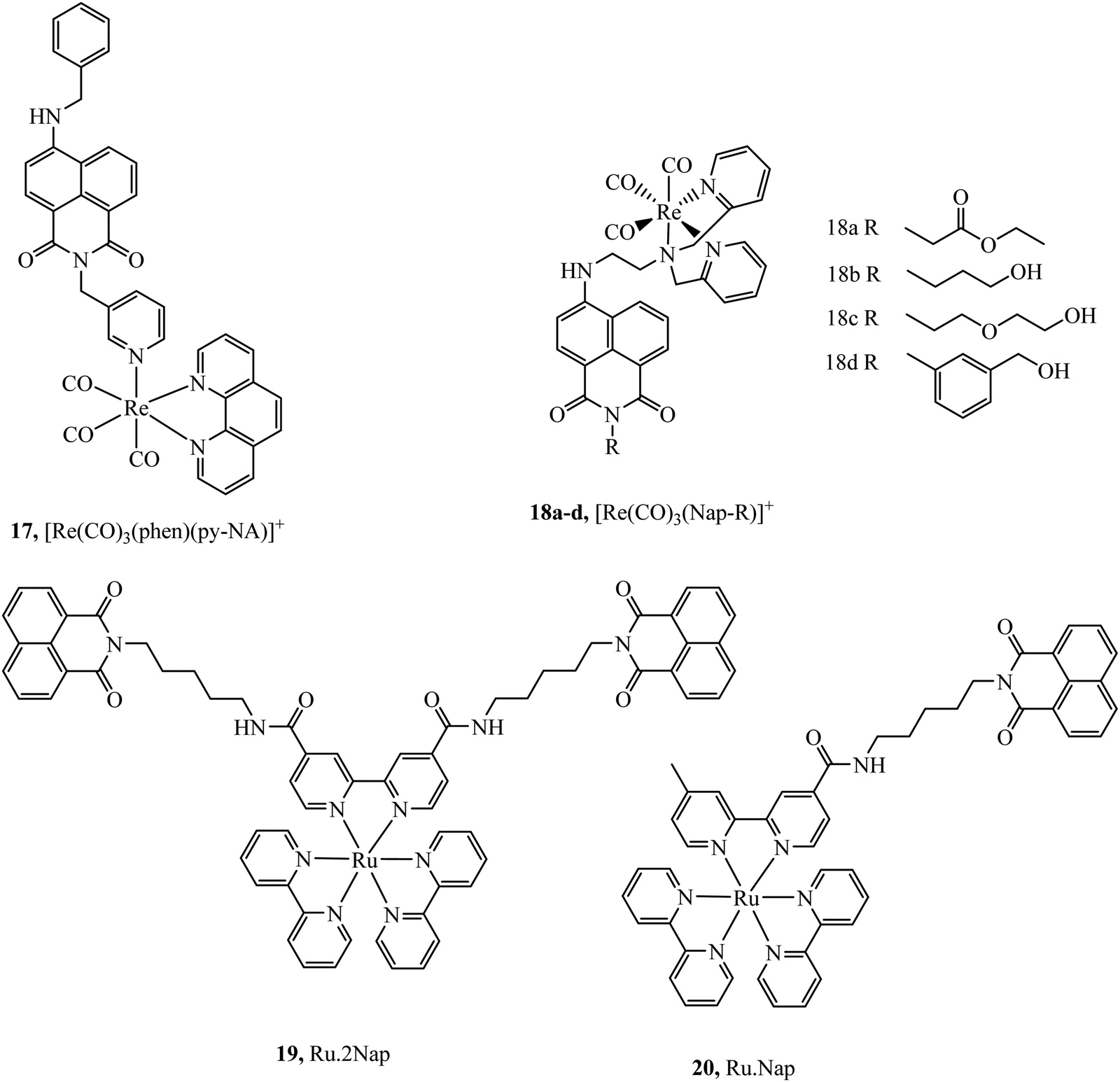
Organic fluorophores have also been applied as lipophilic pendants to promote cellular uptake and confer additional photophysical properties on the metal complex. Notably 1,8-naphthalimide and CY5 have been conjugated to metal complex luminophores and shown to enhance cellular uptake. Complex 17 (Fig. 6) exhibited uptake into yeast cells attributed to the lipophilicity conferred by the naphthalimide ligand. The most lipophilic of the derivatives studied entered nuclei, particularly in dividing cells.66 Prior to this report, naphthalimide modified complexes had been reported for a different series of rhenium–naphthalimide complexes, 18a–d (Fig. 6) and showed variable cell uptake characteristics in human osteoarthritic cells that depended on functionalisation. 18b showed the most promise, exhibiting uptake and bright staining of the ER membranes and Golgi. Higher probe concentration led to apoptosis. In anaerobically grown aerotolerant protistan fish parasite Spironucleus vortens, uptake to hydrogenosomes was observed with low toxicity.67
 | ||
| Fig. 6 1,8 Naphthalimide–metal conjugates. | ||
Ryan et al. investigated the effects of mono bis-1,8-naphthalimide conjugation on DNA binding properties of a ruthenium complex, 20 (Fig. 6). The complex showed the ability to cleave plasmid DNA upon light activation whereas 19 with dual substituted bis-1,8-naphthalimides (Fig. 6) showed poor cleavage ability. These results were reinforced when the complexes were incubated in HeLa cells, the mono complex reduced their viability when photoactivated and the disubstituted complex was nontoxic under the same conditions.68
At elevated levels, peroxynitrite can oxidise proteins, nucleic acids and lipids leading to the apoptosis of cells. It is one of a number of reactive nitrogen species that are of interest for intracellular sensor development. A Ruthenium-CY5 FRET probe, 21 (Fig. 7), has been reported to localise in the mitochondria and ratiometrically sense peroxynitrite. The presence of peroxynitrite cleaves the CY5 polymethine bridge of the FRET probe resulting in an interruption of energy transfer from the ruthenium complex to CY5 that leads to a reduced emission from Cy5 and an increase from the ruthenium complex. The probe showed low cytotoxicity with cell viability of 84% after incubation of the complex after 24 hours at 50 μ M, and was capable of peroxynitrite ratiometric sensing using confocal microscopy.69
 | ||
| Fig. 7 Ruthenium-Cy5 FRET probe. | ||
The simplest approach to altering lipophilicity that does not require bulky vectorisation is through the modification of ancillary ligands coordinated directly to the metal centre. 4,7-Diphenyl-1,10-phenanthroline (dpp) has been reported to greatly increase cellular uptake when compared to bpy or phen analogues. For example, uptake of 22 (Fig. 8) in HeLa cells occurs through passive diffusion whereas bpy and phen analogues have not been reported to permeate through live cell membranes.35 Similar studies have shown increased cellular uptake is observed on increasing the aromatic surface area of the heteroligand in 23a–e (Fig. 8). However, this also served to increase complex cytotoxicity.70
 | ||
| Fig. 8 Ruthenium polypyridyl complexes with differing aromatic areas to tune lipophilicity. | ||
Multi-metallic systems have also proven effective as imaging dyes with numerous examples including both dinuclear ruthenium complexes and heterodinuclear complexes containing ruthenium and iridium.71–73 The Thomas group have performed extensive investigation into dinuclear complexes for imaging utilising the tetrapyridophenazine (tpphz) bridging ligand to prepare ruthenium polypyridyl species in the form [(N^N)2Ru(tpphz)Ru(N^N)2]4+. Similar to tuning mononuclear ruthenium ancillary ligands the tuning of ancillary ligands in dinuclear species has a large impact on uptake and toxicity. This is evident on comparing the Ru(II) tpphz dinuclear analogues using bpy, phen and dpp as ancillary ligands. The bpy complex has poor cellular uptake in live cells but on increasing lipophilicity by using phen ligands excellent cellular uptake was observed with concentration dependant localisation to mitochondria or nucleus where high resolution DNA imaging was accomplished.71,74 A further increase in lipophilicity by coordinating the dpp ligand maintains high uptake with localisation to the endoplasmic reticulum but also greatly increases cytotoxicity.75 The hetero-bimetallic Ir(III)–Ru(II) complex 24 (Fig. 9) was reported to accumulate in the mitochondria after entering through active transport. This complex was designed to provide a dual therapeutic effect where, upon irradiation, photodissociation occurs producing a photoactivated chemotherapeutic aqua ruthenium moiety and a photodynamic therapeutic iridium moiety. 24 was imaged in A549R cells where apoptosis was observed from mtDNA damage and mitochondrial dysfunction after irradiation at 405 nm.76 A series of luminescent Re(I)–Au(I) complexes, 25a–d (Fig. 9), have been recently reported with the aim of developing an anticancer theranostic that detects the moment of interaction with their biological targets. The gold fragment aided biodistribution of the bimetallic series in HeLa cells with membrane localisation observed whereas in A459 cells no internalisation was evident.77
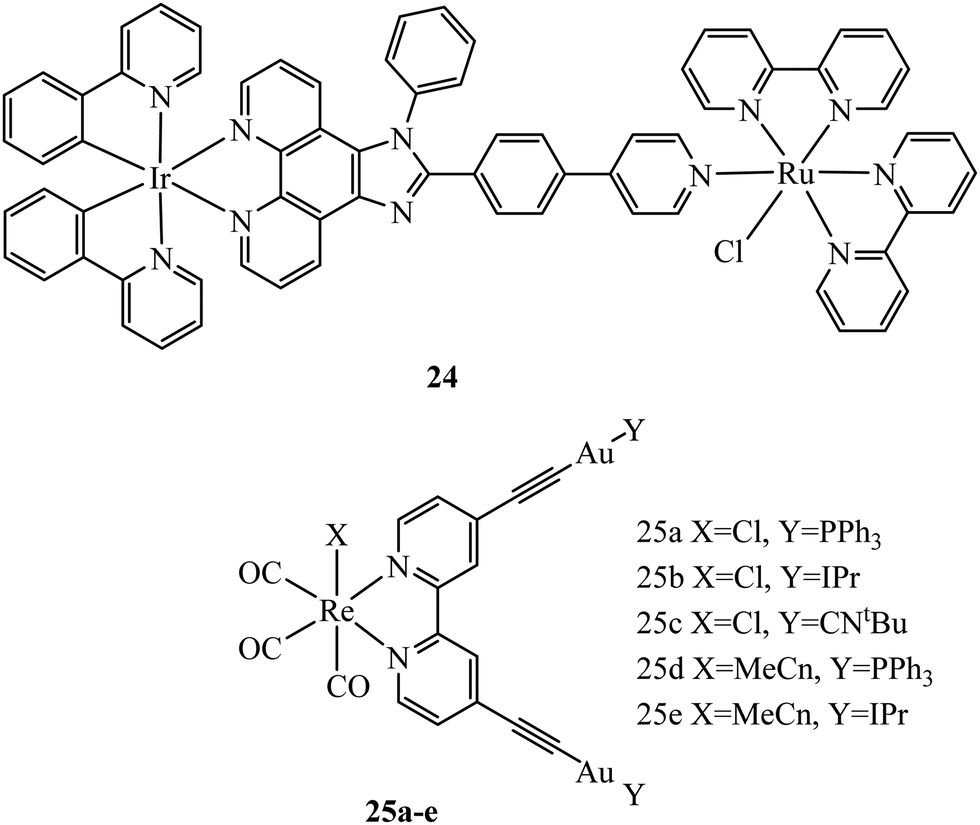 | ||
| Fig. 9 Structures of Compounds 24 and 25. | ||
Cyclometalation reduces the charge of the cationic metal and increases lipophilicity and is a strategy employed increasingly to promote permeation. It has proven particularly effective for ruthenium complexes but has the problematic drawback of increased dark cytotoxicity and reduced luminescence compared to non-cyclometalated analogues. Chao et al. compared the uptake and biological activity of two ruthenium complexes, [Ru(bpy)2(dppz)]2+ and a cyclometalated derivative [Ru(bpy)(ppy)(N^N)]+ where ppy is 2-phenylpyridine.36 This study prompted the synthesis of additional cyclometalated derivatives of ruthenium complexes in the form [Ru(bpy)(ppy)(N^N)]2+ where N^N is pip, dqp, dppn and IP (imidazo[4,5-f] [1,10]phenanthroline). The cyclometalated complexes were compared to noncyclometalated analogues investigating the difference in 3D multicellular tumour spheroids cytotoxicity. The cyclometalated series showed increased cellular uptake and higher dark cytotoxicity compared their polypyridyl analogues.78
McFarland et al., reported the series 24a–d of ruthenium(II) complexes coordinated to π-extended benzo[h]imidazo[4,5-f]quinoline (IBQ) cyclometalating ligands (C^N) appended to thienyl rings (n) as shown in Fig. 10.79 The complexes showed interesting photophysical properties including dual emission originating from a singlet interligand fluorescence and 3MLCT phosphorescence that could be selectively excited. The Log P (octane/water) were measured for these complexes and this was observed to increase with number of thienyl rings and all four complexes showed cell uptake but distribution varied and compounds 1 and 2 showed high dark cytotoxicity attributed to their entry to the nucleus and DNA aggregation whereas compound 3 and particularly 4 showed photocytotoxicity along with reduced dark cytotoxicity at melanoma cells, offering the prospect of a useful phototherapeutic.
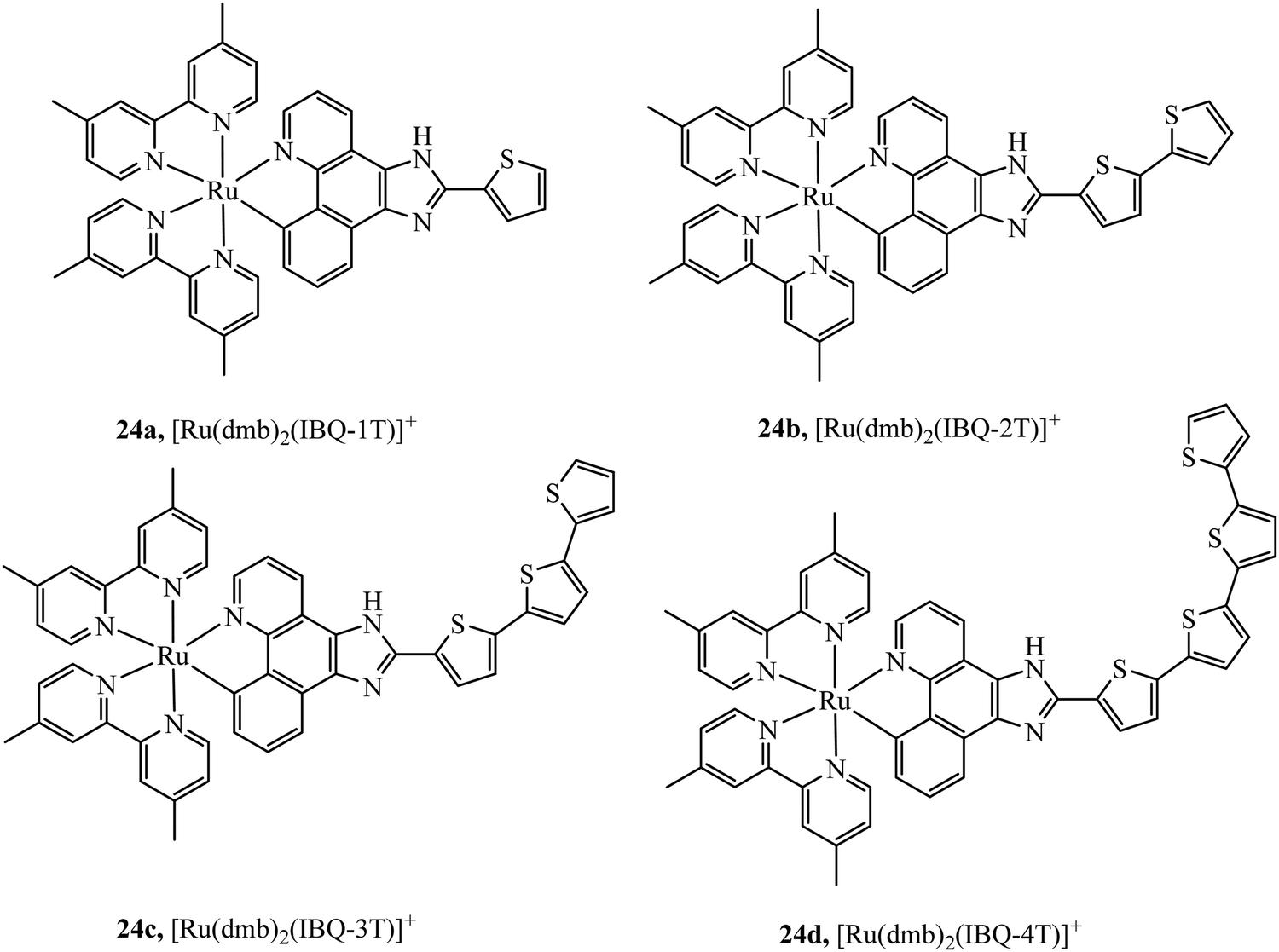 | ||
| Fig. 10 Ruthenium(II) complexes coordinated to π-extended benzo[h]imidazo[4,5-f]quinoline (IBQ) cyclometalating ligands (C^N) appended to thienyl rings. | ||
Complex 25 (Fig. 11) was investigated as a potential dye for STORM, the complex undergoes photoactivation dependant on photooxidation of thioesters to sulfoxides.3925 was imaged in both HeLa and COS7 cells, where mitochondrial localisation was evident (Pearson value of 0.79). STORM images were collected from incubation of 25 with COS7 where localisation precision was reported to be ca. 12 nm. The zinc terpyridine complex 27 (Fig. 11) was imaged in HepG2 cells using STED microscopy.80 Thiophene modification of terpyridine provides an electron rich bridge, allowing extended electronic delocalisation to improve performance as an imaging agent. 27 was found to bind with preference for mtDNA providing super resolution micrographs of mitochondria. The osmium series 26a–c (Fig. 11) were prepared and isolated as fac and mer isomers.81 Both isomers of 26a were imaged in S. aureus using structured illumination microscopy. The mer26a complex showed greater antimicrobial activity and brighter cell staining properties which were attributed to higher cellular uptake.
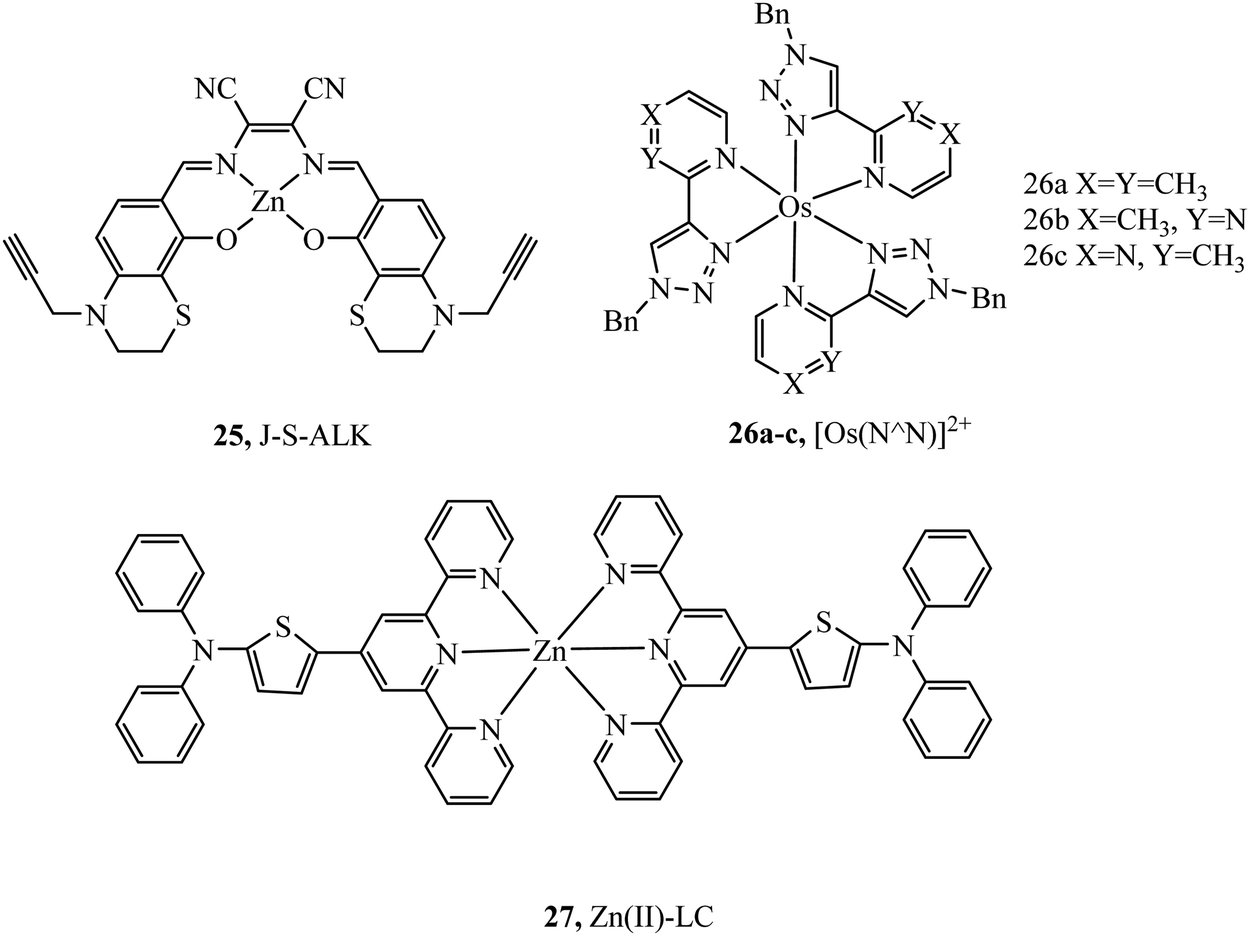 | ||
| Fig. 11 Zinc and osmium dyes reported for use with super-resolution microscopy. | ||
Receptor targeted uptake of metal complex luminophores
Receptor targeting can promote active transport of compounds into cells achieved through conjugation of ligands that bind specific surface receptors. Overexpressed membrane-based receptors can be a common feature of cancer cell lines and targeting these receptors with their specific ligands can lead to cell surface labelling and sometimes selective uptake. This can be a helpful approach in design of imaging probes to distinguish healthy cells and offers an obvious benefit in terms of targeted therapy.82The biomolecule biotin, also known as vitamin H, is transported into cells through a sodium-dependent multivitamin transporter and a high-affinity biotin transporter which is overexpressed in certain cancer cell lines (ovarian, colon and breast). Overexpression of biotin receptors offers the prospect of selective targeting of certain cancer cell lines using biotin as a conjugation vector creating desirable theranostic agents.83
Biotinylated probes have also been reported as diagnostic tools to investigate avidin binding. For example, a series of [Ru(dpp/phen/bpy)2(N^N-biotin)]2+ (Fig. 12) complexes with spacers of varying lengths were studied against avidin binding and luminescent response. Increases in lifetime and emission intensity were reported with the probe undergoing visualisation through immobilisation on the surface of microspheres.84
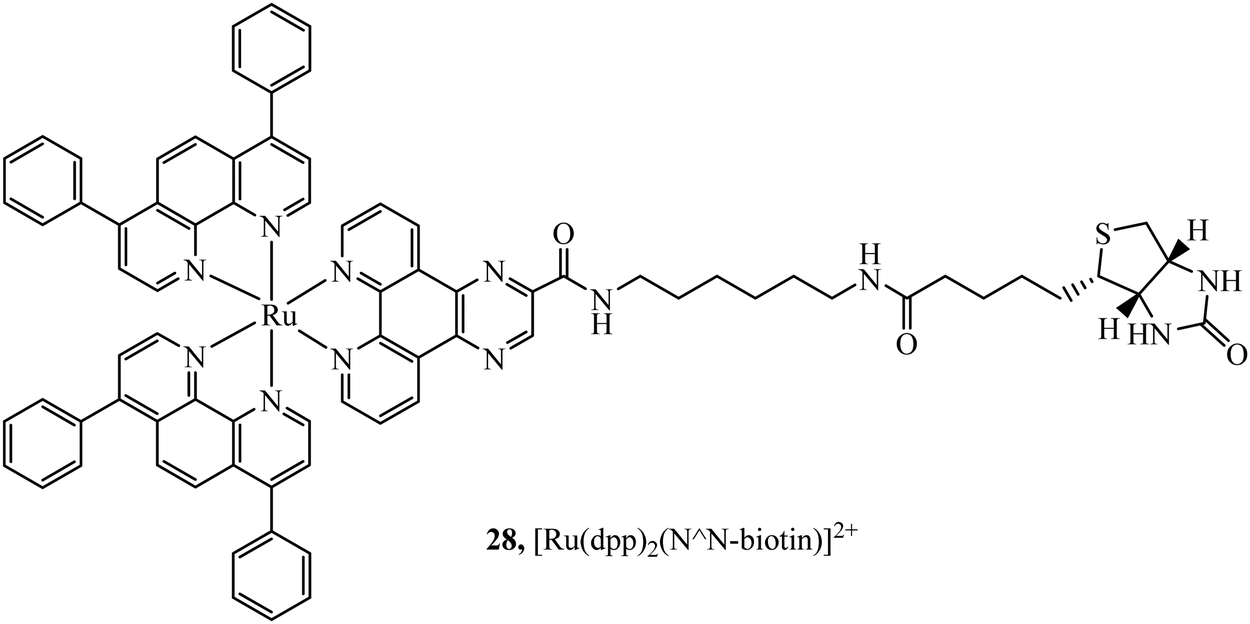 | ||
| Fig. 12 [Ru(dpp)2(N^N-biotin)]2+ diagnostic tool for avidin binding. | ||
A dual emissive iridium–biotin conjugate series 29 (Fig. 13) was developed as a ratiometric sensors for avidin binding. The conjugate series was studied in live HeLa cells where the biotin conjugate rapidly underwent cell uptake and localised in the cell membrane and mitochondria. Complexes with a secondary amine on the cyclometalated ligands had dual emission.85 Early rhenium tricarbonyl conjugates in the form [Re(N^N)(py)(CO)3]+ where N^N is 4,4′-bis{[2-(biotinamido)ethyl]aminocarbonyl}-2,2′-bipyridine, showed similar effects on Era protein binding, where emission enhancement, hypsochromic shifts in emission maxima and longer fluorescent lifetimes were evident. Cytotoxicity was examined for these rhenium complexes in HeLa cells where the biotin conjugated complexes remain noncytoxic whereas the parent rhenium complex is cytotoxic. Cell uptake in HeLa cells was determined through confocal microscopy.86
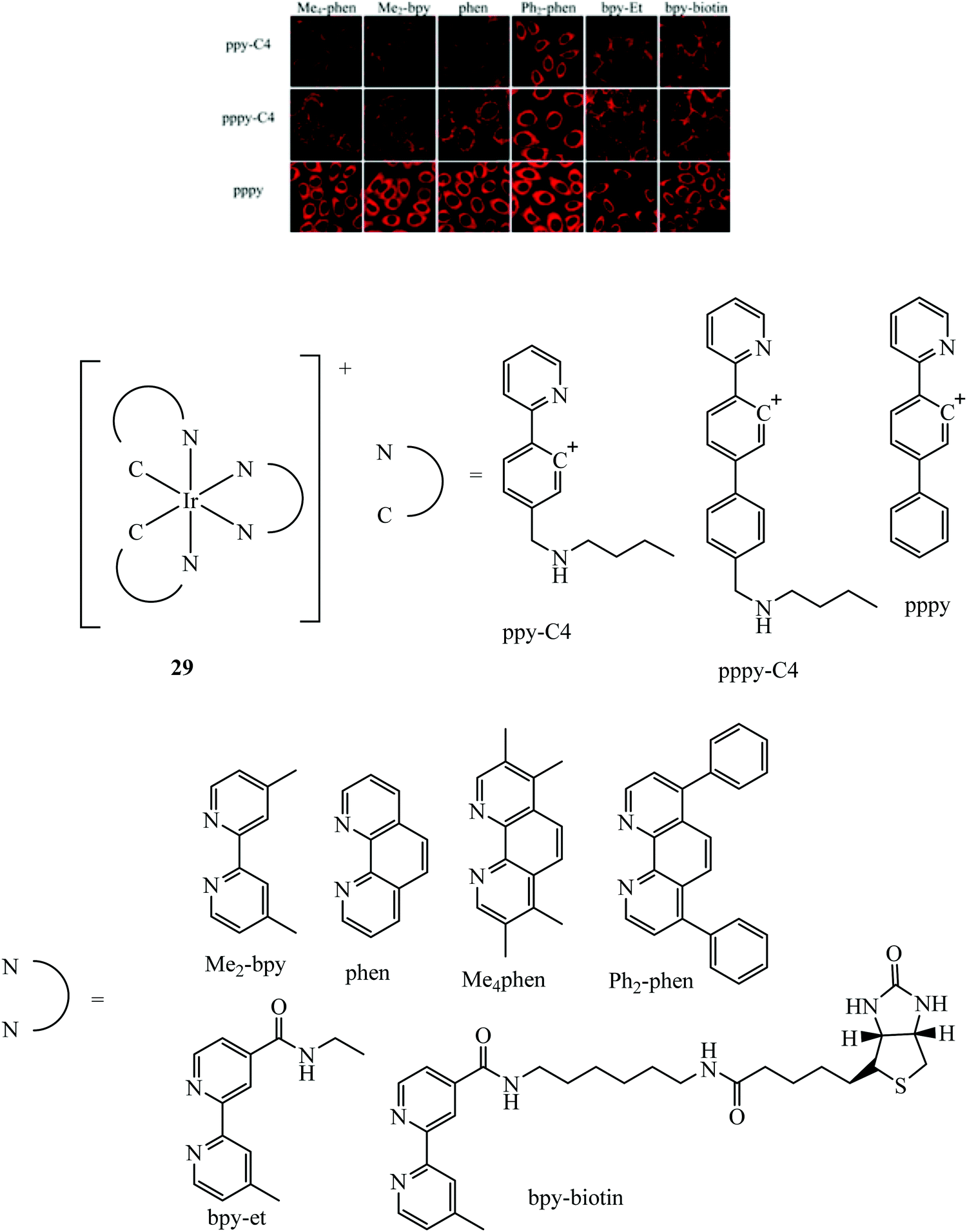 | ||
| Fig. 13 Iridium biotin conjugates imaged with confocal microscopy in HeLa cells. Reproduced from ref. 85. Copyright 2015 American Chemical Society. | ||
Selective cell uptake in cell lines with overexpressed biotin receptors has been reported for several ruthenium biotin conjugates with the aim of producing anticancer phototherapeutics. The first, the Ru–biotin complex 31 (Fig. 14), capable of excitation via a two-photon effect was investigated as an anti-cancer agent against the A549R cell line which overexpressed biotin receptors. High affinity for biotin receptors was observed and cells were imaged through confocal microscopy where the complex was observed to induce apoptosis through photodamage and activation of caspase3/7.87 In another study, PEGylated-BODIPY was exploited for NIR sensitisation in combination with a ruthenium polypyridyl biotinylated complex to target A549R cells. The conjugate displayed no dark toxicity but excellent photodynamic activity. Biotinylation of anthracene modified Ru(II) complexes was also shown to be effective at promoting uptake in cancerous cell lines.88
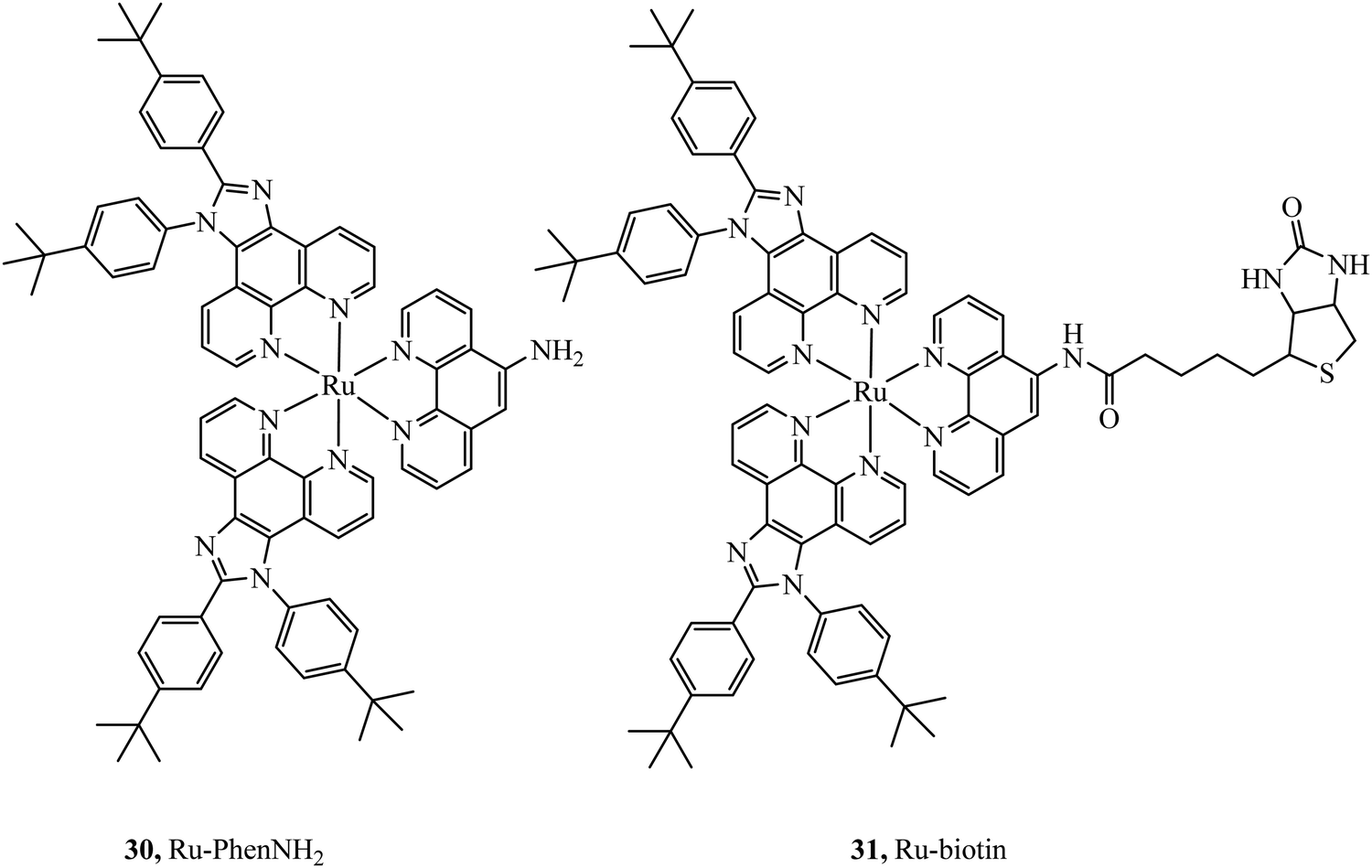 | ||
| Fig. 14 Ru–biotin conjugate designed to achieve secretive cell uptake. | ||
Steroid hormones, such as estrogen, progesterone and testosterone are readily permeable to the plasma membrane as following their secretion in vivo they must passively permeate cell membranes to reach their intracellular targets. Thus, steroids are a clear contender for bioconjugation to metal complexes to promote cell permeation. Hormone receptors can also be over-expressed in certain disease states and so offer prospects in diagnostic imaging. In practise, it is mainly estrogen that has been applied in this regard to date for metal complex luminophores. Estrogens, are responsible for female secondary sexual characteristics, regulating the differentiation and maintenance of neural, skeletal, cardiovascular, and reproductive tissues. In addition, they participate in the development of estrogen-dependent cancer such as breast, ovarian, colon, prostate, and endometrial cancers.89,90 Receptor content can be used to gather an accurate index of cancer. As such, developing a probe that binds to receptor eliciting a strong change in photophysical properties can be a useful diagnostic and prognostic tool.
Lo and co-workers have reported rhenium, iridium and ruthenium–estradiol conjugates and investigated their interaction with estrogen receptors (ERs). The group first reported synthesis of a series of Ir(III)-estradiol of the form [Ir(N^C)2(bpy-ph-est)]+ where C^N is 2-phenylpyridine, 1-phenylpyrazole, 7,8-benzoquinoline, or 2-phenylquinoline. These iridium complexes showed increased emission intensity and lifetime on specific binding to the estrogen receptor α. This series pointed toward the potential of estradiol–metal conjugates as a diagnostic tool, although, the series was not studied in cells.91 This was followed by the investigation into two complexes 32 and 33+ (Fig. 15) where the Ru(II) dpp complexes showed extended lifetimes and enhanced emissions when bound to an estradiol receptor. Uptake and Imaging was performed using confocal microscopy at HeLa cells and the dpp complex, similar to many series containing this ligand showed superior uptake to its bpy counterparts. The complex was observed to disperse in the cytoplasm and particularly in the perinuclear region of the cell. The complexes showed low cytotoxicity when compared to the anticancer agent cisplatin, a benefit where the complexes are intended as probes or diagnostic agents.92
 | ||
| Fig. 15 Ruthenium estradiol complexes. | ||
Sugar/carbohydrate conjugates have been employed due to their biocompatibility and capacity for increased cellular uptake. As key functional, structural, and nutritional components of cells, sugars have their own transport mechanisms across the cell membrane, mainly through protein mediated process that can be hitched onto by glycoconjugates of metal complexes. The ruthenium sugar series 34a–c, (Fig. 16), was synthesised to achieve cellular uptake in HepG2 cells and retain their photophysical properties. The complexes were generated using s-glycosylated bonds to resist endogenous hydrolysis. To reduce surface lectin interaction the carbohydrates were bound with small linkers to bipyridine. The complexes were capable of slow cellular uptake after 24 h with the D-glucose complex having the clearest cellular accumulation.93 Sugar conjugates have also been explored as possible avenues for cell uptake selectivity as certain cancer cell lines can overexpress certain sugar transporters such as GLUT and SGLT.9435a–d (Fig. 17) were synthesised to investigate their potential as PDT agents.95 The complexes localised within the mitochondria and had excellent levels of singlet oxygen generation. Further, the complexes were tested in vivo by testing against tumours located in mice. The complexes produced a decrease in tumour volume when implemented using two-photon activation.
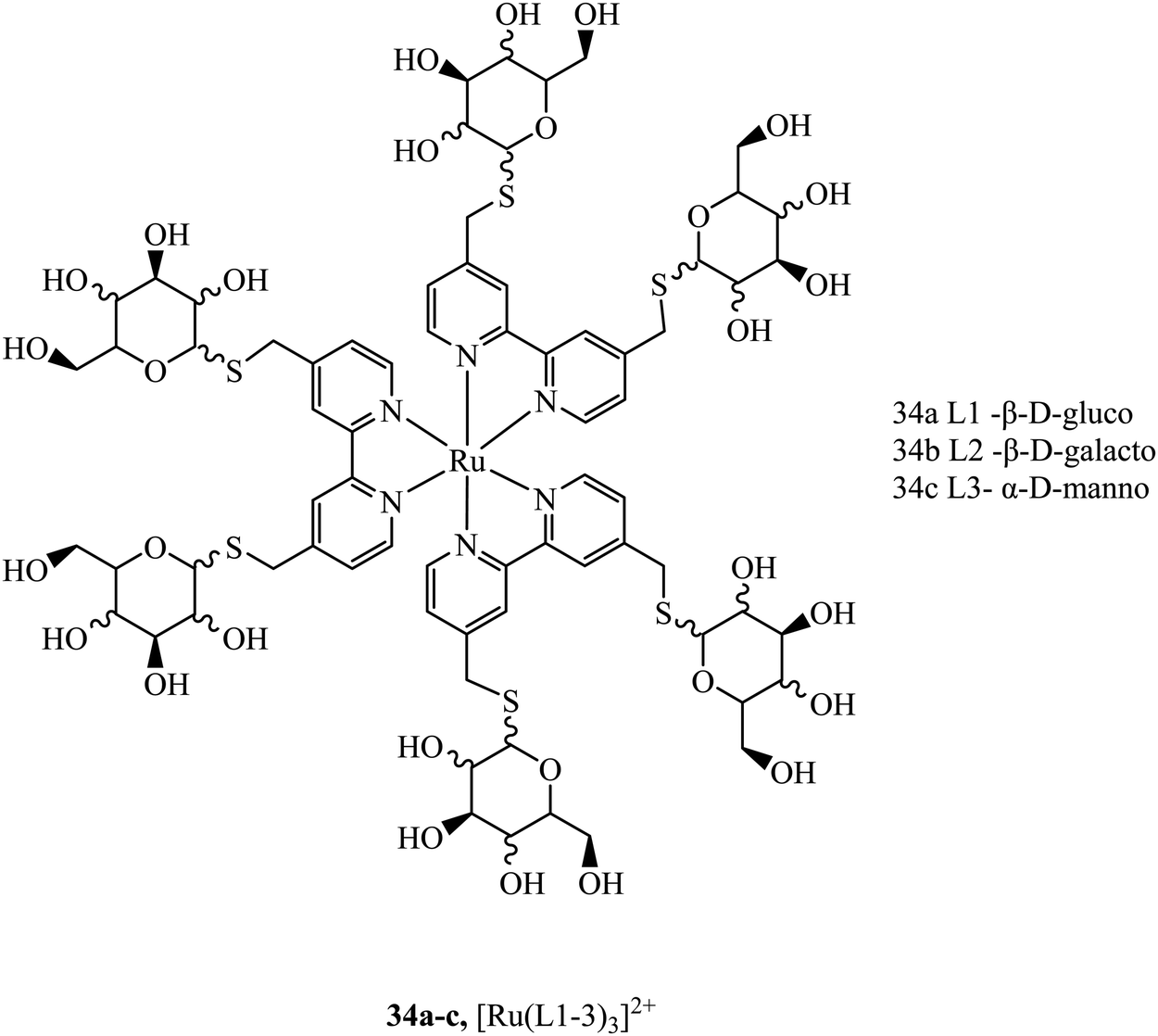 | ||
| Fig. 16 Ruthenium polypyridyl s-glycosylated sugar conjugates. | ||
 | ||
| Fig. 17 [Ru(dpp)2(pic-glucose)]2+.73 | ||
Hua-Wei Liu et al. have explored sugar conjugation to luminescent Ru(II), Ir(III) and Re(I) complexes. Initially, a series of Iridium(III) complexes in the form [(Ir(hqp)2)mbpy-TEG-sugar]+ (37Fig. 18) was synthesised where the sugar analogues were was glucose, galactose, lactose and maltose. Glucose conjugates were found to be the most cytotoxic which is attributed to their more effective cell uptake compared to other sugar conjugates.96 However, following this study, the group investigated 36a–c (Fig. 18) with and without a glucose pendant. Interestingly, in that case, glucose conjugation resulted in lower cytotoxicity across HeLa, MCF-7, HEK293T and NIH/3T3 cell lines relative to the free complexes.97 The group further explored rhenium–fructose conjugates and their relative cytotoxicity across a series of cell lines. The complexes were highly cytotoxic and photocytotoxic but demonstrated selective uptake for breast cancer cells.97 Ruthenium–fructose conjugates have similarly been investigated and found to stain the mitochondria. They did not induce significant cell death in MCF-7 cells up to a concentration of 200 μM.98
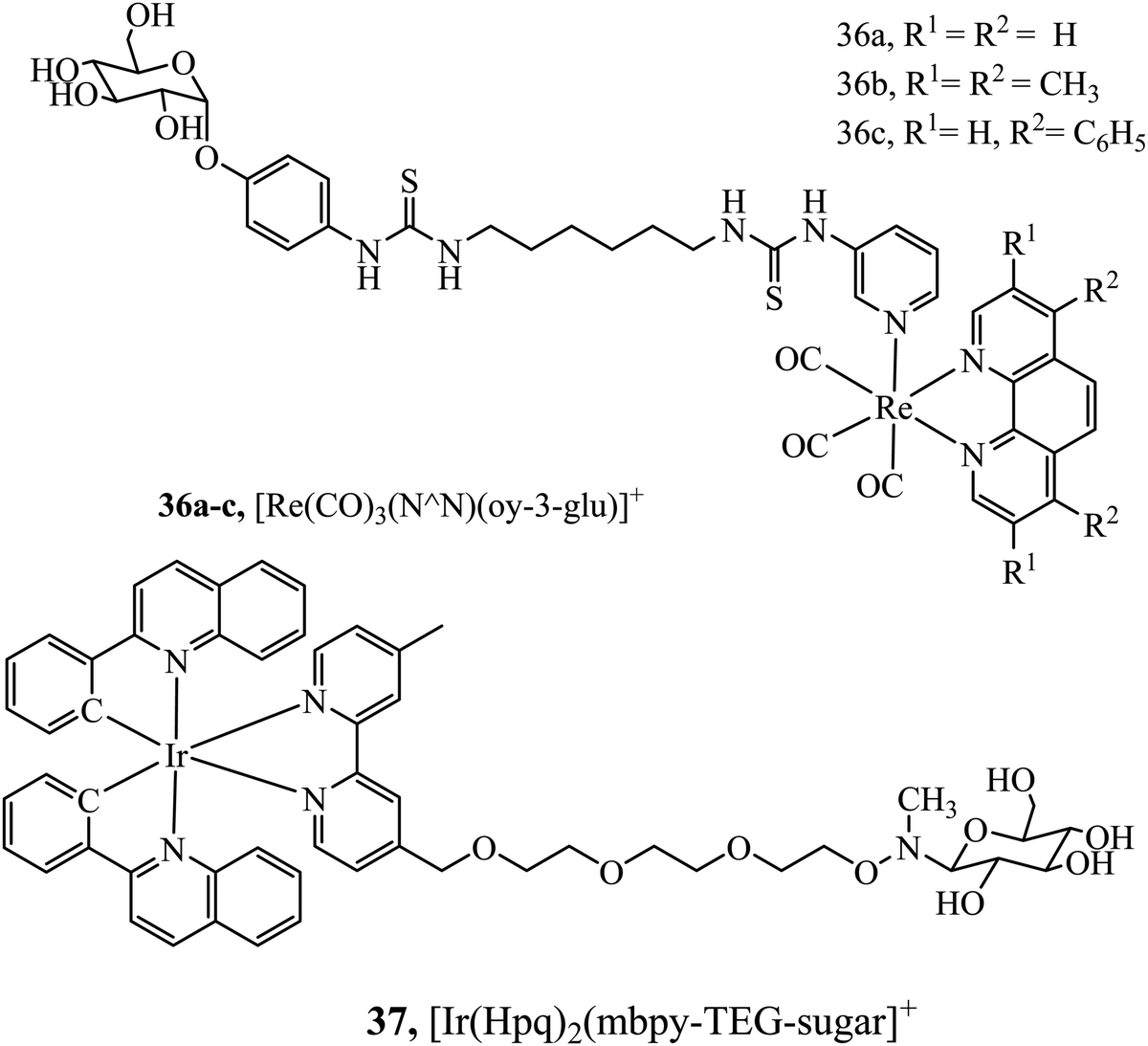 | ||
| Fig. 18 Rhenium and iridium conjugates bearing a single sugar pendant. | ||
The folate receptor is a glycoprotein that is overexpressed in a variety of tumour types including ovarian, lung and brain, offering an avenue to achieve selective cell uptake of drug-folate conjugates. A rhenium bisquinoline complex, 38, (Fig. 19) conjugated to folate via a PEG linker was investigated a theranostic tool for FR-overexpressing cancer cells. The conjugate showed selective uptake in A2780/AD cells with no uptake in CHO cells after 24 h. The folate conjugated complex showed higher and accelerated cell uptake when compared to the Re-PEG parent complex which only achieved permeation through passive diffusion.99
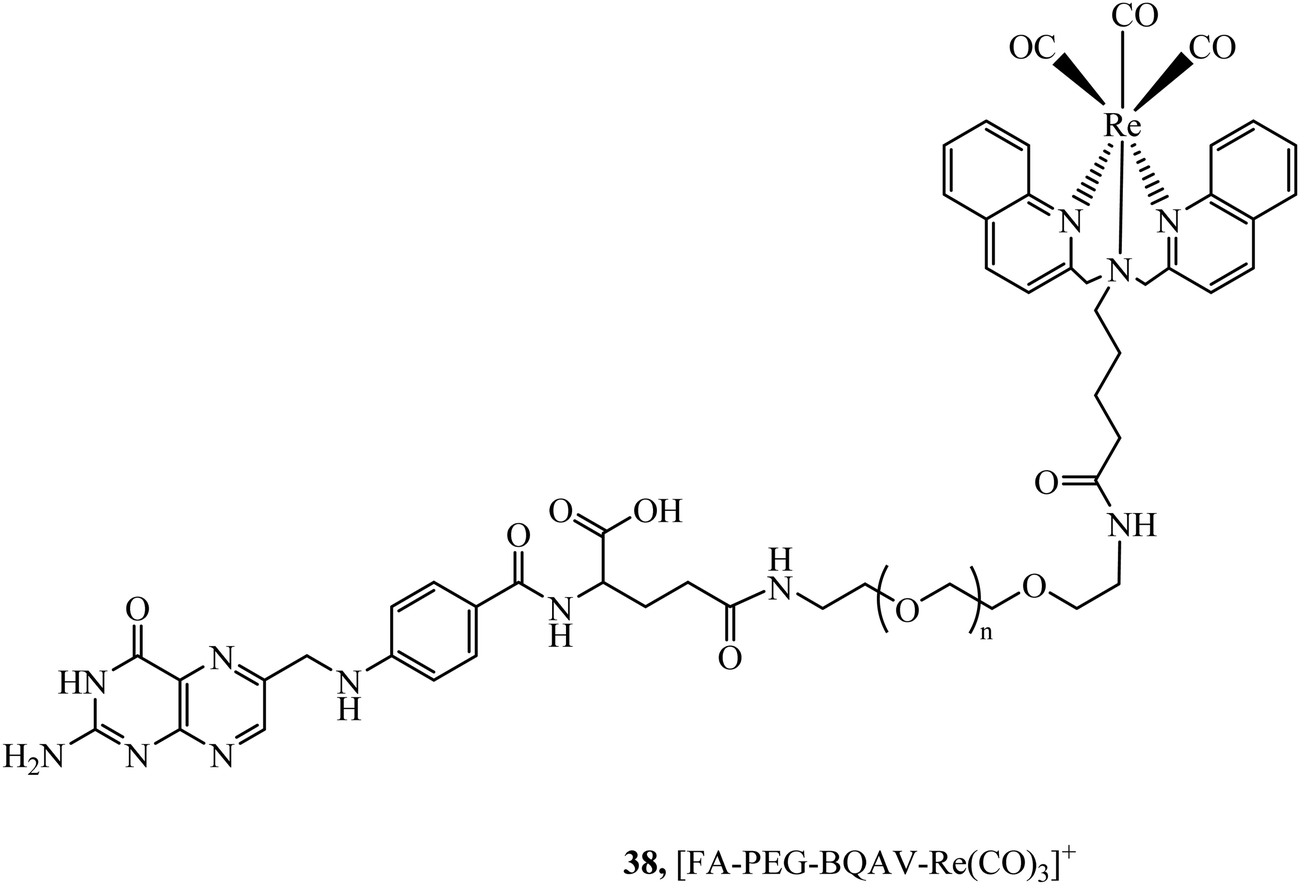 | ||
| Fig. 19 Rhenium–folate complex. | ||
The cubilin receptor is a transmembrane protein of particular interest due to the mutations in the CUBN gene which encodes it. These mutations of CUBN have been associated with megaloblastic anaemia, creating a desire for cubilin diagnostic agents.100 Uptake of vitamin B12 is dependent on an Intrinsic Factor protein (IF)-cubilin mediated pathway to form the cubilin-IF-B12 complex resulting in the potential use of B12 as a vector to synthesise cubilin diagnostic agents. The rhenium-vitamin B12 complex, 39a (Fig. 20) first demonstrated uptake via an IF-cubilin mediated pathway into placental BeWo cells where it distributed throughout cytoplasm and nuclear regions.101 Limitations of this complex include water solubility, weak fluorescence and poor distinguishability from background fluorescence. Building on the previous study the rhenium-cubilin complex, 39b (Fig. 20), was reported. 39b contained a thiazolate chelating ligand to rhenium that improved water solubility.102 IF binding could be monitored with ease; upon conjugation to B12 the fluorescence of Re complex is quenched but when the conjugate binds the intrinsic factor, a 15-fold increase in emission intensity occurs with concomitant blue-shift. To overcome autofluorescence, dual excitation was employed. Cubilin receptor mediated uptake of the conjugate into A549 cells was confirmed by confocal microscopy, thus revealing the presence of the cubilin in lung cancer cells.
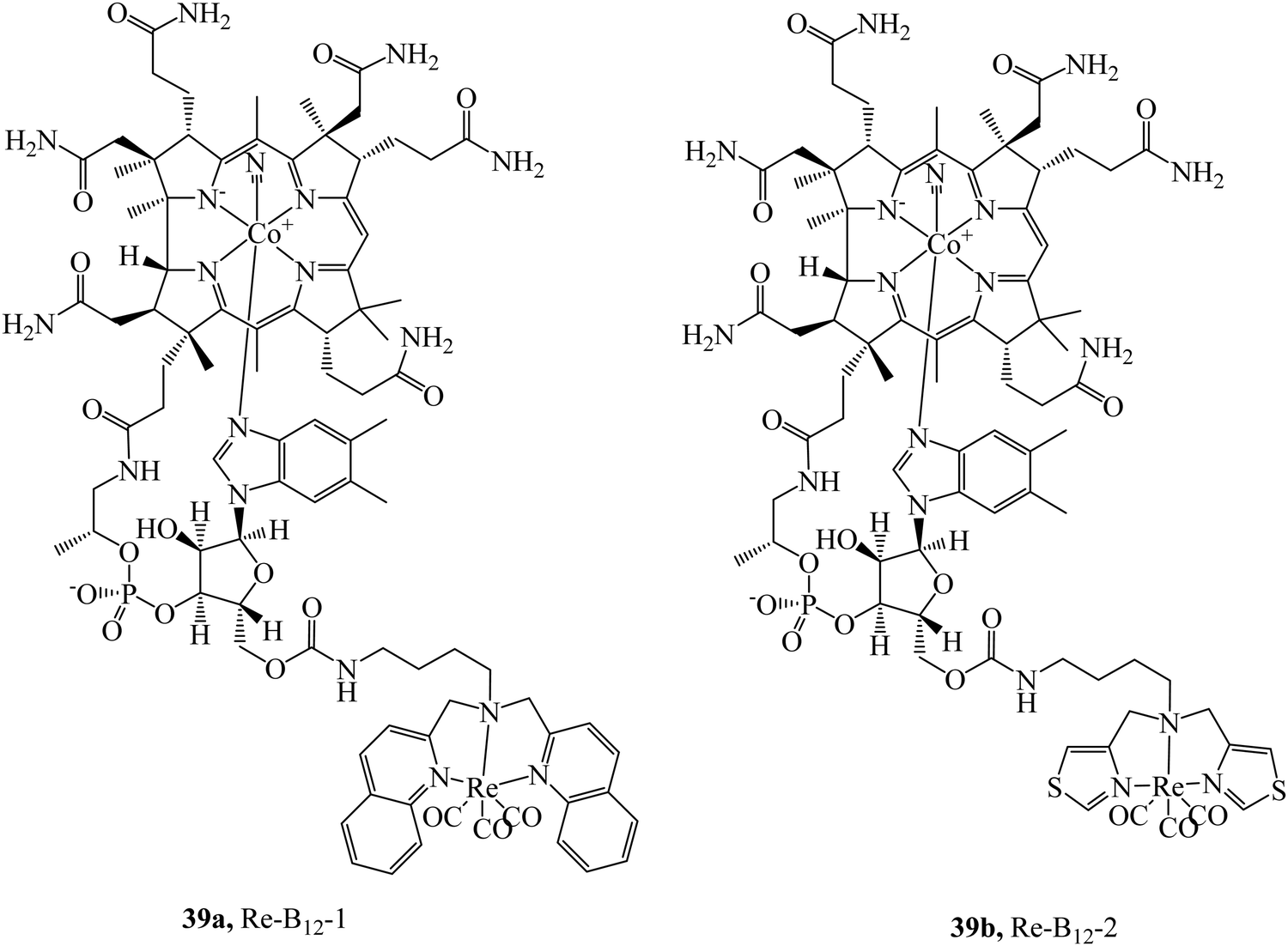 | ||
| Fig. 20 Rhenium-B12 conjugates designed target the cubilin receptor. | ||
Uptake strategies based on supramolecular or nanoparticle vectors
Nanoparticles offer several unique advantages as delivery vectors for sensing and imaging agents, including tailored sizing and surface functionality, broad biocompatibility and stability as well as distinctive optical properties either inherent to their electroclinic structure in the case of metal or quantum dot structures or arising from modification of photophysically innocent particles (such as polymer or silicate particles or liposomes or lipoplexes) with high concentrations of highly localised luminophore. There are also increasingly diverse ways of controlling distribution of probe in nanoparticle structures such as multilayering of cargo within a core or as part of the shell or at the surface of a core–shell structure.Nanoparticle uptake across the cell membrane occurs almost invariably by endocytosis but the specific endocytosis mechanism, e.g. clathrin–caveolin independent or caveolae-mediated endocytosis macropinocytosis or phagocytosis, is dictated by the physicochemical properties of the particle, including surface chemistry, charge/zeta potential, shape, and crucially; size.103
Although the physicochemical properties that influence uptake are aggregate, a general rule of thumb that has emerged is that particles of roughly 50 nm diameter are optimally mobile across the bilayer.104 In some applications the particle is merely a delivery vector with, much like drug delivery, the release of the probe required on arrival at its cellular destination. In other cases, the particle itself is the imaging probe. In the latter case in particular, the toxicity of particle may be an issue and metal complex doped nanoparticles vary widely in their toxicology, but like permeability, this tends to correlate with scale, smaller particles tend to be more toxic, likely because of their greater mobility within the cell.105 For metal complex luminophore modified particles surface doped nanoparticles have been reported as being highly cytotoxic whereas nanoparticles that encapsulate the metal complexes can prevent interaction of the transition metals with biomolecules or organelles in the cell limiting their toxicity which presents a key opportunity to use metal complexes that are otherwise unsuited to imaging.106
Gold nanoparticles are attractive candidates as uptake vehicles for surface appended metal complexes because they are commercially available or can be prepared with low dispersity indices and they are easily functionalised with a wide variety of well characterised surface chemistries available to gold. Furthermore, gold particles tend to be stable and biocompatible with many studies reported on their uptake bey cells.107 With appropriate termini, such as thiol or pyridine functionality, metal complexes can be readily tethered to gold particles to create luminescent or surface enhanced Raman active probes. Tendency for quenching of the excited state by gold can be reduced by using relatively long linkers that create some distance between luminophore and particle, indeed if the balance is correct metal enhanced emission can facilitate bright emission.108
For example, Elmes et al. reported a series of luminescent ruthenium gold nanoparticles with the complexes [Ru(N^N)2(phen-R-SH)]2+ (where (N^N) is bpy, phen or tap), the functionalisation of the ruthenium complexes with an alky thiol chain facilitates the formation of the nanoparticle. The complexes underwent cell uptake efficiently with imaging from confocal microscopy and TEM showing localisation in the cytoplasm and nucleus. DNA binding was reported with the complexes remaining nontoxic.109 Similarly, Pikramenou et al. investigated 40 (Fig. 21) s bound to gold nanoparticles through use of a thiol linker. These particles were reported for their intense luminescent properties allowing imaging at a single nanoparticle level. The nanoparticles were not significantly cytotoxic and localised in the nuclei of A549 cells.110 Recently, this approach was extended to a Raman imaging sensor application where 5-phenylethynyl-2,1,3-benzothiadiazole(PTDB)/ruthenium(II) complex was immobilised on a gold-silver alloy nanoparticles used as ratiometric surface enhanced Raman spectroscopy (SERS) nanoprobe to monitor CO in hepG2 cells and in ex vivo liver tissue from mice. The nanoparticle bound ruthenium complex in this case was not luminescent, but coordinated to a ligand containing an alkyne group that was displaced by CO within the cell leading to loss of the alkyne vibration (∼2206 cm−1) and the appearance of metal carbonyl Raman signature at approximately 2100 cm−1. The nanoprobe thus reported ratiometrically on CO and was permeable to both cells and tissues and appeared to be non-toxic at detection level concentrations.111
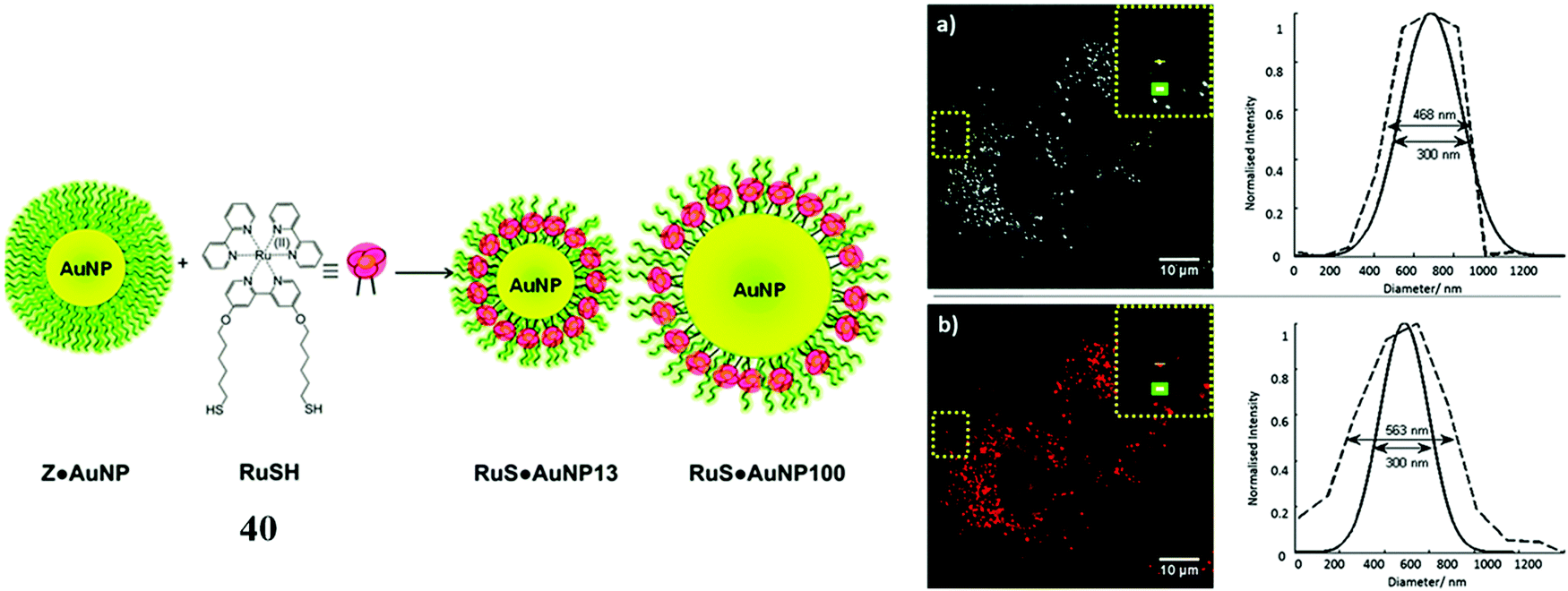 | ||
| Fig. 21 RuSH gold nanoparticles imaged in A548 cells. Reproduced from ref. 110 with permission from the Royal Society of Chemistry, copyright 2013. | ||
Mesoporous silica nanoparticles offer similar advantages to gold nanoparticles in terms of functionalisation and size tuning, through the reliable Stöber method.112 And, while they do not offer the advantages of plasmonic interactions, they are optically transparent can often show excellent biocompatibility. Silica nanoparticle mesoporosity is particularly useful for sensors applications allowing free diffusion of e.g. oxygen through the particle. A key advantage of many metal complex luminophores is their very large Stokes shift in their emission spectra, this enables their localisation at high concentration, inside or at the surface of nanostructures without artefactual effects on their photophysics such as inner filter or self-quenching effects.113 Metal complex luminophores, particularly Ruthenium(II) polypyridyl complexes have been widely studied encapsulated in such mesoporous silica nanoparticles, particularly for their oxygen sensing capabilities.114 The first example of mesoporous silicate nanoparticles modified with metal complex luminophore applied to cell imaging was reported by Philbert et al., in 2001. Their particles were relatively large so they were inserted into cells with a gene insertion gun and they reported a ratiometric oxygen response within living rat C6 glioma cells for the first time. There have been a number of studies exploiting mesoporous silicate particles with ruthenium polypyridyl sensors applied to cell imaging since and reports beyond oxygen.115–118 A drawback of oxygen sensing through triplet energy transfer quenching is generation of high local concentrations of toxic singlet oxygen. Tanabe et al. took advantage of trapping a Ru(II) oxygen sensor complex within mesoporous silica to confine ROS generated while sensing to the particle, thus reducing toxic effects. Critically, the particles were less toxic compared to the free complex which demonstrates another benefit of using particle vehicles.119
More recently Wang et al. reported silicate particles of 52 nm diameter encapsulating Ru(II)tris-phenanthroline complex and surface modified using silane chemistry, with N,N′-bis(carboxypropyl)-9,9′-biacridine (BAC), a Cl− sensor. In this case the metal complex provided the reference signal and ratiometric emission sensing of chloride at the particles was investigated in cells. The particles showed low cross sensitively to pH and excellent photostability but the impact of oxygen on the ratiometric signal was not explored. Although useful in the context of Cl− sensing because of local chloride concentration, the particles were found to localise in the lysosome, a common consequence of low endosome escape in nanoparticle uptake.120
Frasconi et al. immobilised the complex [Ru(terpy)(dppz)H2O]2+, where terpy is 2,2′:6′,2′′-terpyridine, at silica nanoparticles by coordination of a monodentate ligand with nitrile group which anchors the complex to the nanoparticles. The Ru-nanoparticles exhibited low dark cytotoxicity until photoactivation triggered nitrile ligand cleavage and ejection of a highly cytotoxic Ru-aquo solvate complex and separately, paclitaxel drug loaded within the silica nanoparticle. Uptake of the particles was rapid, with distribution observed throughout the cytoplasm of MDA-MB breast cancer cells.121
A ruthenium polypyridyl complex has been vectorised onto silica particles through coupling of amide and carboxylic acid functionalities. The nanoparticle developed was a two-photon-excitable red fluorescent probe capable of imaging HeLa cells, zebrafish and nude mice. The nanoparticles showed low levels of cytotoxicity in HeLa cells with viability over 80% after 24 h incubation at 200 μg mL−1.122
A possible drawback of silicate nanoparticles for imaging purposes is that they have been shown to be vulnerable to erosion by the cellular medium and given many such species route to the lysosome on uptake this may be exacerbated in it's acidic environment.123
Core–shell nanoparticles permit the inclusion of probes at the core or at the surface of the particle depending on their amphiphilicity. The water insoluble luminescent complexes 41a and 41b (Fig. 22) were encapsulated in a core–shell particles of diameter between 30 and 50 nm. The Re particles successfully entered 293T cells and demonstrated good DNA transfection capability.124 Our group reported a core shell ratiometric oxygen sensing nanoparticle where a Ru oxygen probe, 42+, was present on the shell of the particle through conjugation with poly(L-lysine) and a reference BODIPY dye was confined to the polystyrene core. Co-Excitation of both the Ru(II) outer shell and BODIPY core was achieved without cross talk between probes. The nanoparticle bound strongly to the cell membrane requiring CTAB to stimulate uptake. Ratiometric oxygen sensing at the lysosomes was possible once the particle was internalised by the cell.125
 | ||
| Fig. 22 Rhenium parent complexes prior to nanoparticle vectorisation. | ||
A semiconducting polymer containing BODIPY-iridium FRET pair, 43, (Fig. 23) was reported to assemble into nanoparticles. Designed as a photosensitiser for photodynamic therapy, achieving a high singlet oxygen quantum yield (0.97), the photosensitiser was imaged in HeLa cells and xenograft zebrafish with CLSM which showed cancer inhibition upon singlet oxygen sensitization. Anticancer studies were performed with tumour bearing mice indicating good anticancer ability and biocompatibility.126 Two lanthanide doped up-conversion nanoparticles had their surface decorated covalently with iridium complexes to achieve NIR PDT. The nanoparticles maintained efficient singlet oxygen generation at low O2 levels due to the long lifetime of the iridium complex resulting in good PDT efficiency even in hypoxic conditions.127 Similarly, lanthanide doped up-conversion particles were functionalised with [Re(CO)3(dpp)(cpy)]+ complex permitting their endocytic uptake into A2780cis cells and the ability to trigger PDT activity using NIR sensitisation which conveniently overcomes the need for blue/UV direct excitation using Re(I) complexes.128
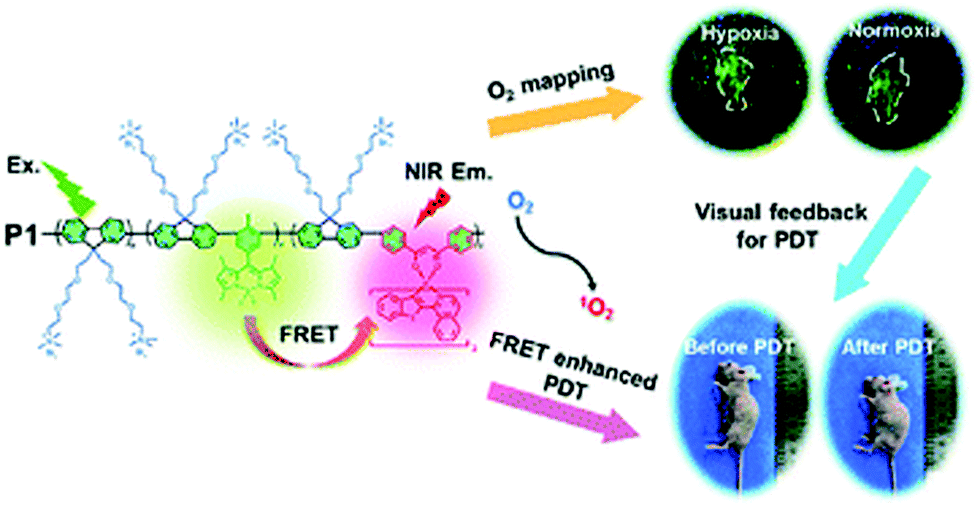 | ||
| Fig. 23 Ir-BODIPY FRET pair imaged in HeLa cells. Reproduced from ref. 126 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Aside from modified nanoparticles, self-assembled nano-dimensioned structures that comprise of the metal complex luminophore are an alternative and very useful approach to driving metal complexes across the cell membrane, as again, such structures, if they are of the right dimensions, tend to be endocytosed. Although not a sugar occurring naturally in mammalian cells, cyclodextrins are oligosaccharide rings structures that have found wide application in supramolecular chemistry due to their excellent water solubility and capability as a molecule host.129 Because of their hydrophilicity and high number of H-donors, they are poorly membrane permeable. But interestingly, 44 (Fig. 24) was reported to induce the aggregation of DNA within the hydrophobic interior of cyclodextrin. The thus formed DNA/CD-Ru aggregate was found to translocate across the membrane of 293T cells where the ruthenium centre permitted tracking of the DNA. Such an approach holds potential for trackable drug carriers of hydrophobic molecules or as a conduit for oligonucleotides for use in anti-sense therapy.130
 | ||
| Fig. 24 Imaging of 293T cells with a Ru–cyclodextrin conjugate. Reproduced from ref. 130. Copyright 2007 American Chemical Society. | ||
A similar effect was reported by Mao et al., where, nanoaggregate assembly of a luminescent dual cyclodextrin-functionalized Ru(II) complex on host recognition of an adamantane-appended tumor-targeting peptide, rendered the complex permeable to integrin αvβ3 rich tumour cells U87MG and MCF-7 cells. The Ru-CD-RGD assembly was observed to localizes specifically to lysosomes shown through co-localisation with LysoTracker. Again, a common destination for endocytosed particles and apoptotic cell death was caused through lysosomal damage, ROS species and caspase activation.131
A similar strategy employing a Ru-cyclodextrin to capture a conjugated adamantane–anthracene dyad was also reported. Anthracene acts as a photoactivatable cleavage agent by generating singlet oxygen and endoperoxides. The complex accumulated in the nuclei of cancer cells and showed excellent phototoxicity and anticancer activity.132 Although these examples describe cytotoxic applications, self-assembled cyclodextrin aggregates could yet offer an untapped uptake strategy for metal complex imaging probes, particularly if combined with additional targeting agents.
Self-aggregation of metal complex luminophores with pendant alkyl, lipid tails, or amphiphiles has been demonstrated for a number of metal complexes and is an effective way of preparing luminescent nanoparticles.133,134 But, until recently, there were few examples of such an approach to promote cell uptake. In one such report, bioconjugation with the biomolecule squalene was reported to promote cellular uptake where the Ru-BPPBI-hx-SQ complex was shown to self-assemble into nanoparticles in aqueous media. The ‘”Ru-BPPBI-hx-SQ” complex was observed to concentrate within cells and was also found to permeate tumour spheroids, where it was observed to diffuse throughout an organised multicellular spheroid without significant toxicity.45 We recently reported on self-aggregation, in aqueous media, of a family of lipophilic Ru complexes, [Ru(dpp)2(x-ATAP)]2+, where dpp is 4,7-diphenyl-1,10-phenanthroline and x-ATAP is 5-amino-1,10-phenanthroline with pendant alkyl-acetylthio chains of length x = 6, 8, 11, and 16. The nanoaggregates were characterised by dynamic light scattering and found to be between 140 and 180 nm depending on alkyl chain length. The aggregates readily permeated HeLa and CHO cells in a temperature dependent manner, attributed to endocytosis. And interestingly, organelle localization varied with chain length and cell line. In HeLa cells, [Ru(dpp)2(11-ATAP)]2+ localized highly specifically to the Golgi apparatus but to endosomes in CHO. Whereas the [Ru(dpp)2(16-ATAP)]2+ localised to the mitochondria in HeLa and to but to lipid rich lipid droplets in CHO.135
Liposomes as nanocarriers of cargo into cells and tissues have been well studied in the context of drug delivery due to their ability to entrap both lipophilic and hydrophilic species within the lipid bilayer or the discrete aqueous space they enclose.136 While there have been several reports on metal complex luminophore reconstitution into liposomes, their application in complex delivery for imaging has had surprisingly little attention to date.137–143 One pertinent study created liposomes comprised of dipalmitoylphosphatidylcholine (DPPC) and cholesterol functionalised with PEG to increase hydrophilicity and decrease immunological recognition. The lipid layer was loaded with [Ru(phen)2(dppz)][(ClO4)2], where luminescence from the metal complex allowed tracking of uptake and nuclear DNA-targeting in MDA-MB-231 breast cancers cells.144 Furthermore, the liposome vehicle permitted permeation into a mouse model of triple negative breast cancer leading to reduced tumour growth. The Iridium polypyridyl complexes, [Ir(ppy)2(HPIP)](PF6) (Ir-1), [Ir(ppy)2(BHPIP)](PF6), where ppy is 2-phenylpyridine and BHPIP is 2-(3-bromo-4-hydroxy)phenylimidazo, have also been introduced to a range of cancer cells by liposomal encapsulation as a drug delivery strategy. Toxicity of the complexes in liposomes was greatly increased when compared to free complexes demonstrating enhanced cellular uptake.145 Tunik, Urtti et al. reported on preparation of PEGylated DPPC liposomes labelled with red emitting (∼650 nm) cyclometallated Ir complexes with long alkyl tails and demonstrated that the liposome formulation were effective as imaging probes, promoting probe upake in Retinal pigment epithelium cell line (ARPE-19) and indeed in vivo in ocular imaging where their red emission made imaging possible without contributing autofluorescence and the probes could be used for in vivo fundoscopy and OCT as contrast agents.146
Overall, although nanoparticles are a highly effective means of promoting probe delivery, there are two key limitations to their use in imaging. The first an inherent drawback associated with their endocytotic uptake mechanism, is that they can become trapped in the endosome where they then go on to terminate at the lysosome, often resulting in particle degradation in its acidic, enzyme rich environment. This is obviously a significant drawback if the purpose is to direct the particle or the probe it is carrying to a given organelle or structure within the cell. The second issue is toxicity, as described, nanoparticles can show significant cell toxicity through a number of mechanisms often showing membrane toxicity in particular causing loss of membrane integrity. Modification of the physicochemical properties of the particle make the latter problem more tractable but the issue of endosome escape is and is an ongoing issue in drug delivery and applies to molecular as well as, in particular nanoparticle species, a number of methods to overcome it are emerging that should aid in delivery of complex luminophores in the future.147
Cell penetrating peptides enhance uptake, mediate toxicity and are capable of discrete subcellular targeting
Peptides have emerged over the past 20 years as potent tools in drug delivery. Natural sequences, in the form of signal peptides have evolved to regulate biomolecule transport and targeting expressed within the transduction domains of proteins. And, for many viruses, cell-membrane permeation of the host is peptide-mediated. These latter sequences in particular, both natural and related synthetic sequences have formed the basis of cell penetrating peptides (CPPs) that are widely used in drug delivery. The development of CPPs has centred fundamentally on the uptake of short cationic sequences, typically less than 30 amino acids long containing arginine or lysine residues, driven by electrostatic interaction with the cell membrane that bears a negative surface charge.148 The most common CPP originates from investigations into the action of the HIV transactivator protein and its constituent Tat signal peptide sequences.149,150 But there are now many naturally derived CPPs investigated for their vectorisation efficiency and there is a growing library of synthetically derived CPPs that offer bespoke designs, often using non-natural amino acids. The precise uptake mechanisms of CPP conjugates are still the subject of much debate and likely varies dependant on the nature of the CPP which may promote direct penetration or activated uptake through endocytosis.151 Key advantages of CPPs and peptides for delivery in general, are their ease of synthesis using solid state methods that are often automated and the ease of bioconjugation to their cargo. Drawbacks, which have limited their success in some clinical trials, relates to issue of longer-term biostability as they are prone to proteolysis in vivo, and similarly to nanoparticles because they are permeant through endocytosis, they are prone to endosomal entrapment and cell selectivity. Nonetheless the repertoire of CPPS and targeting sequences is growing and, CPPs are being finding wide application in many medical research domains including in delivery and targeting of metal complexes in vitro and vivo.152,153Early applications of CPPs to metal complex probes exploited polyarginine sequences with broad success as long as the arginine chain was sufficiently long (typically (Arg)6 or higher). The length dependence on uptake is consistent with studies of polyarginine conjugates of organic fluorophores.154 In general, polyarginine conjugation is effective at translocating the cellular membrane but has little predictable targeting capability thereafter. The Barton group reported octaarginine conjugates with [Ru(bpy-R)(phen)(dppz)]2+ and [Rh(chrysi)(phen)(bpy-R)]3+ complexes where bipyridine was conjugated to octaarginine achieving cytoplasmic localisation.14,15 Interestingly, the group demonstrated that nuclear penetration was achieved with the appendage of fluorescein to the polyarginine tail, permitting DNA mismatch targeting by Rh(III) complexes, but is still unknown if this is a general nucleus targeting strategy. Early studies by our group similarly revealed that [Ru(bpy)2(pip-(Arg)8)]10+ could traverse the membrane of myeloma cells for cytoplasmic staining but the penta-arginine conjugate was impermeable.155 Since then, we have demonstrated that octa-arginine is effective at promoting cellular uptake and cytosolic distribution of a range of other complexes including [Os(bpy)2(pip-(Arg)8)]10+ (46Fig. 25), [Ir(F2ppy)2(pip-(Arg)8)]9+, [Ru(dppz)2(pic-(Arg)8)]10+, [Ru(bpy)2(phen-Ar-(Arg)8)]10+ (45Fig. 25), [Ru(tap)2(bpy-Ar-(Arg)8)]10+, [Ru(dppz)(bpy)(bpy-Ar-(Arg)8)]10+ and [Ru(biq)2(trzbenz-(Arg)8)]9+ (47Fig. 25).138,156–161 Notably, in our hands, the parent complexes all failed to permeate the cell membrane under the same conditions. Nona-arginine appears to be similarly effective for vectorisation of metal complexes as reported by the Policar group who studied Re(I) derivatives including [ReCl(CO)3(phen-(Arg)9)]9+ and [Re(CO)3(Py)(phen-(Arg)9)]10+ for membrane labelling. These Re(I) derivatives which bear carbonyl ligands can also be exploited for multimodal imaging using IR synchrotron and Raman imaging in CHO cells and skin biopsies.162,163 The evident diversity of metal complex cargo that can be transported using polyarginines is a significant step towards achieving predictable uptake properties for metal-based imaging probes. However, beyond uptake, polyarginines alone tend not to target the complex to specific organelles, although in some examples punctate appearance of images suggest endosomal entrapment. But lack of targeting, may lead to higher dark toxicities compared to other CPP metal complex conjugates.
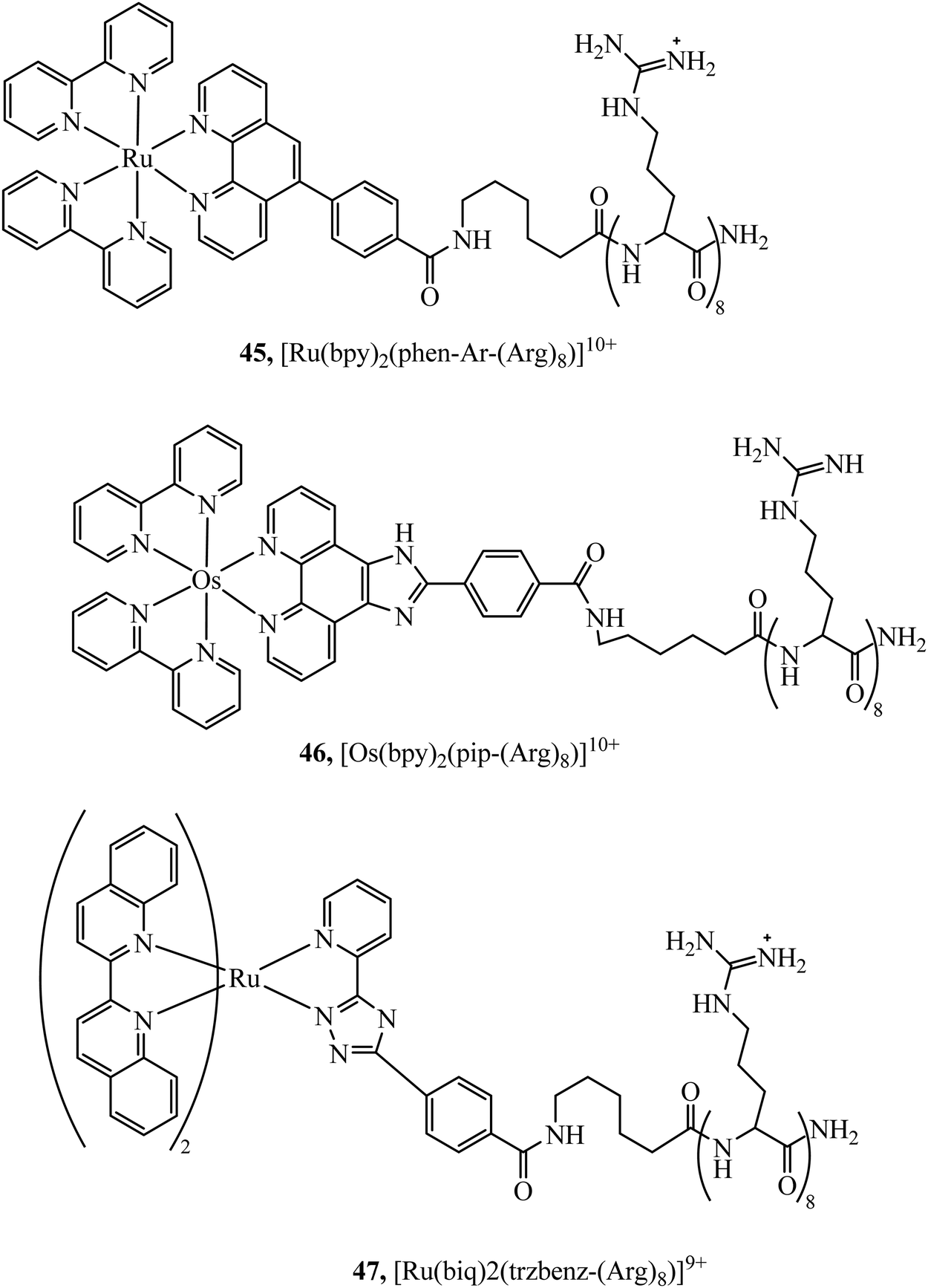 | ||
| Fig. 25 Ruthenium/osmium octaarginine complexes. | ||
Mitochondrial penetrating peptides (MPPs) have been specifically designed to exhibit the necessary balance of charge and lipophilicity to overcome initial cellular penetration and also the barrier of the double-membrane of the mitochondria and its potential gradient. The Kelley group has engineered several iterations of MPP that enable the transport of various cargo into the mitochondria that exhibit these characteristics.164,165 Our group adopted one of their sequences, FrFKFrFK (where r is D-arginine) to prepare a peptide-bridged dinuclear Ru(II) conjugate that localised precisely and rapidly to the interior of the mitochondria of live HeLa cells.166 Once localised, the Ru(II) probe was capable of dynamic luminescence lifetime response to changing oxygen and ROS concentrations. Next, we tagged a DNA-light switch probe with this sequence to create a Ru(II) conjugate, 48a (Fig. 26), that efficiently localised to mitochondria in live HeLa cells to enable the high contrast visualisation of mitochondrial nucleoids.160 Targeting could be confirmed using confocal microscopy, lifetime imaging and resonance Raman microscopy. More recently, we doubly labelled a NIR emissive Os(II) terpyridine complex with this sequence and discovered that although mitochondrial targeting in HeLa and MCF-7 cells was retained at low concentrations (below 30 μM), higher concentrations led to non-specific distribution, and high toxicity was observed in either scenario via different mechanisms.167 Interestingly, we also found that conjugation to the more lipophilic [Ru(biq)2(trzbenz)]+ complex also resulted in higher dark toxicity than expected.161 Combined, these recent examples of probes bearing higher charge (Os(II) case) and higher lipophilicity (Ru-biq case) indicates that there is a limit to the robustness of MPP vectorisation, but it is nonetheless a powerful method to target the mitochondria with metal complex imaging probes where the balance is met.
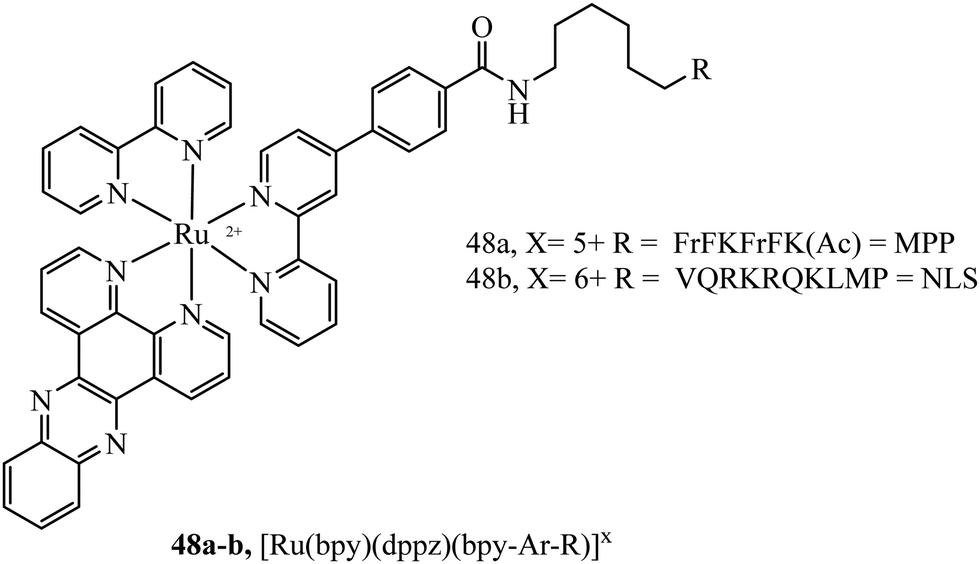 | ||
| Fig. 26 Trisheteroleptic ruthenium probe with MPP and NLS peptide bioconjugates. | ||
A Nuclear Localisation Signal (NLS) peptide is tagged onto various proteins to permit their delivery to the cell nucleus, exemplified from the initial discovery of PKKKRKV as the NLS of SV40 large T-antigen.168 Based on work by Ragin et al.,169 we explored the use of VQRKRQKLMP derived from transcription factor NF-κB as an NLS, through its conjugation to two Ru(II) complexes, [Ru(dpp)2(pic-R)]2+ and [Ru(bpy)2(pic-R)]2+. While the parent complexes did not exhibit uptake, the NLS conjugates penetrated the nuclear envelope with high selectivity, the more lipophilic derivative concentrating in the nucleolus of CHO cells.170 Using the same NLS sequence, we have since demonstrated the successful nuclear delivery of 48b for DNA targeted light-switch imaging, permitting mitosis visualisation in remarkable contrast and resolution using STED microscopy (Fig. 27).158 Also, [Ru(tap)2(bpy-Ar-NLS)]6+, was delivered with high selectivity to the nucleus of HeLa cells where its emission switch-off signified its DNA binding so that it could be triggered to photo-induce DNA oxidation and damage with spatiotemporal control.159 Recently, PAAKRVKLD, a NLS derived from the human c-Myc regulator gene, was conjugated to a cyclometalated Ir(III) complex leading to NLS driven nuclear uptake of the peptide conjugate with low toxicity compared to nuclear exclusion and high toxicity of the free complex.171 Re(I) quinolyl derivatives have also been investigated in combination with NLS peptides – a study reported by Gasser et al. demonstrated successful nuclear delivery using CRRRK in HeLa cells but with potent toxicity.172 Consistent nuclear uptake remains a difficult challenge to overcome for the majority of metal complexes but the successes demonstrated by NLS targeting potentially offers a robust strategy for future progress.
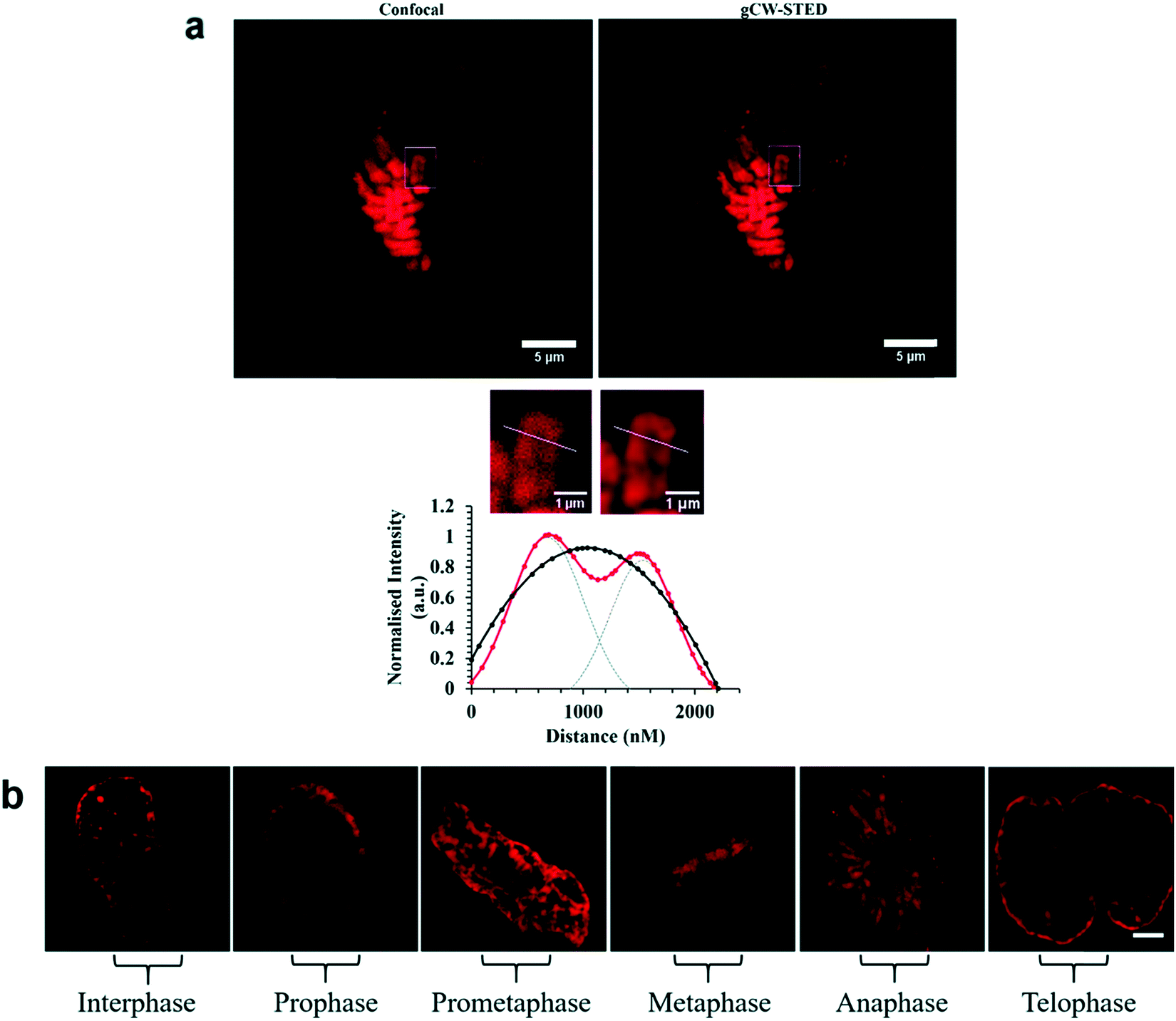 | ||
| Fig. 27 STED capability of imaging [Ru(dppz)(bpy)(bpy-Ar-NLS)]6+. Reproduced from ref. 158 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Recognition peptides that are specific receptors for protein targets have also been applied to metal complex luminophores for imaging intracellularly or at the cell membrane. The peptide sequence arginine–glycine–aspartate (RGD) is a particularly important motif that is recognised by integrins. Integrins are a class of membrane glycoprotein involved in adhesion for example of blood platelets and involved in tumour angiogenesis which is an essential mechanism for the growth of new tumours. Integrin α2Bβ3 is an important integrin involved in activation and aggregation of blood platelets 49a and 49b (Fig. 28) have been reported to target platelet integrin with an eight-fold luminescence increase and could distinguish, different conformational states associated with activation of integrin allowing dynamic monitoring of integrin activation status.173 The integrin αvβ3 in particular, plays an important role in cancer, where it is highly expressed on activated endothelial cells and some cancer cells but is not present on resting endothelial cells leading to potential cell selectivity when targeted.174 An Ir(III) histidine complex with a cyclic RGD peptide has also been reported to show cell membrane permeability and good luminescent properties for tracking.175 The peptide agonist WKYMVm was conjugated to [Ir(ppy)2(phen-R)]+ to produce a conjugate with low toxicity that was demonstrated to successfully target and inhibit formyl peptide receptor 2 (FPR2) in live HUVEC cells.176 Similarly, focussed on cancer, Kühn et al. reported terpyridine-based ruthenium(II) complexes conjugated to a cyclic RGD to target αvβ3. The report focussed on the antiproliferative properties of the complex, rather than emission properties which are often relatively weak for Ru(II)terpyridine complexes and they were found to be non-toxic. This may be because the complex is bound by integrin at the cell surface rather than permeating to the cytoplasm, but such an approach is nonetheless potentially very useful in cancer imaging.177
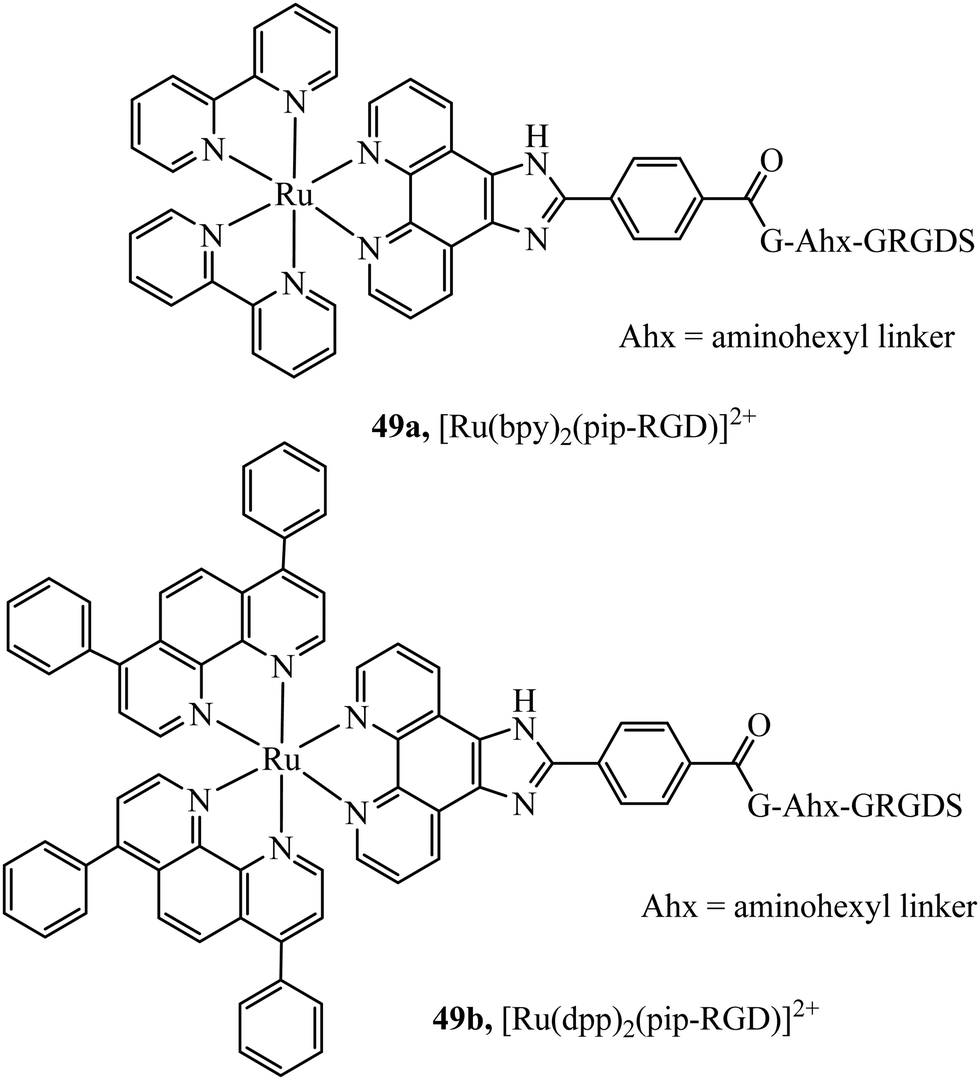 | ||
| Fig. 28 Ruthenium polypyridyl complex vectorised with the RGD peptide through the use of an aminohexyl linker. | ||
Fernández-Moreira et al. have contributed recent work in this field, synthesising the peptide linked heterobimetallic Ir(III)/Au(I) complexes 50 and 51 (Fig. 29).178 Designed as potential theranostic agents, the Ir(III) fragment provides emissive properties and the Au(I) fragment can provide bioactivity. Use of a peptide linker was rationalised to overcome potential aqueous solubility issues surrounding the Ir(III) fragment. Imaging of 50 and 51 in A549 cells was completed using confocal microscopy where lysosome localisation was determined from co-staining with LysoTracker. The authors hypothesise 50 and 51 did not provide the expected anti-cancer properties due to lysosomal localisation, which was still observable after 24 h incubation indicating slow lysosomal escape.
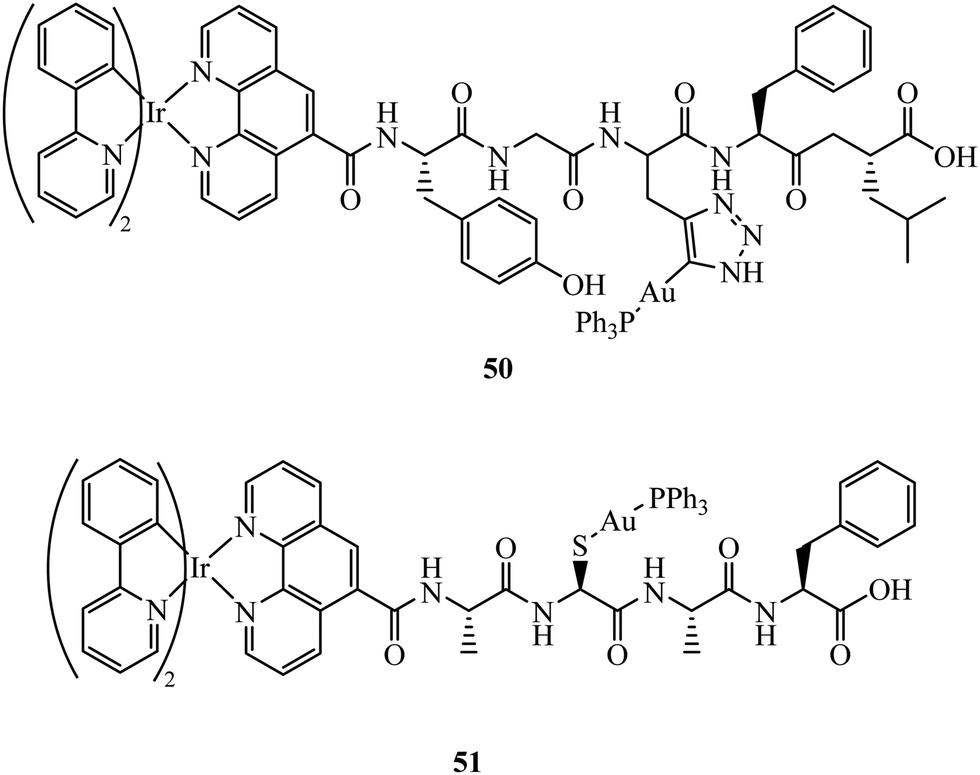 | ||
| Fig. 29 Ir(III)/Au(I) bimetallic peptide linked theranostic agents. | ||
Peptide nucleic acids (PNAs) are a synthetic DNA mimic where N-(2-aminethylglycine) units form a chain with nucleobases. The neutral chains of PNAs allow stronger and more selective binding activities to complementary nucleic acid sequences, in addition to enhanced resistance to enzyme cleavage. However, this is balanced against typically low aqueous solubility and poor cellular uptake which has limited their widespread implementation. PNA conjugation offers a unique uptake and nuclear vectorisation potential for metal complex imaging probes. Although DMSO was required for cell uptake, a preliminary study by Licandro et al. using dinuclear diazine Re(I) carbonyl complexes functionalised with PNA demonstrated that rapid nuclear staining could be achieved in HEK-293 cells with imaging by two-photon excitation.179 Later, Mari et al. reported similar 52 (Fig. 30) conjugates where nuclear staining was observed in HEK-293 cells for a neutral PNA whereas the cationic derivative appeared to be confined to endosomes.180 More recently, these conjugates have been investigated with varying linker length between complex and PNA. While longer linkers led to detrimental aggregation within DU145 cells, a short linker appeared to suggest potential for the Re-PNA conjugate to act as a luminescent miRNA inhibitor.181
 | ||
| Fig. 30 Rhenium–peptide nucleic acid. | ||
Conclusions and future perspectives
The design and optimisation of transition metal complexes for application as emission contrast agents for bioimaging has dramatically expanded over recent years with increasing focus on the development of specialised sensing probes and imaging pro-drugs for theranostics. The exploitation of motifs and vectors for promoting cell membrane permeability a key barrier to metal complex luminophore application in these domains has established a range of transition metal probes with impressive capacities for targeting that offer opportunities across numerous biological applications such as super-resolution microscopy, DNA mismatch sensing and anti-cancer therapies. Although we have focussed in this short review on imaging applications many of the approaches for permeation described here also apply and have been applied to metal complexes applied to therapeutic applications.Means to promote metal complex permeation, vary from direct approaches that increase the lipophilicity of the complex, including modifications to the ligand lipophilicity directly or by appending pendant lipophilic moieties to the ligands to bioconjugation to sugars, peptides or receptors such as vitamins. Another less direct but very effective means to promote permeation is to use nanoparticles as vectors. Nanoparticles in the range of 20 to 100 nm diameter tend to effectively permeate the plasma membrane, depending on the shape and charge of the particle. And metal complex can be introduced through such vectors either by encapsulating into or adsorption onto pre-assembled particles such as metal and polymer particles. Or, by using complexes that self-assemble into particles for example by appending extended lipophilic such as alkyl or fatty acid tails. Within this approach such complexes can also be assembled into liposomes with other amphiphilic constituents. Examples of all of these approaches have been described herein that achieve good cell permeability and in some cases vectorisation. A key limitation of nanoparticle vectorisation is the tendency for uptake to occur by endocytosis and this can lead to endosomal trapping or direction to the lysosome. Vectorisation of metal complexes is harder to achieve reliably than permeation. And indeed, in general is best achieved to date by using bioconjugation, exploiting receptors or making use of species such as signal peptides that naturally act as vectors in vivo.
In recent years bio-orthogonal click chemistry has emerged as a powerful alternative to conventional bioconjugation methods. It offers the prospect of highly selective in-cellulo chemistry with limited cross-reaction leading to off-target conjugation or side reactions that produce cytotoxic products. The application of these methods to transition metal complex luminophores is a growing application area that has evolved strongly to date on radiolabels.23 However, there is plenty of opportunity to use these synthetic approaches with luminescent metal complex and related pro-drugs and this is likely to see significant breakthroughs in the coming years.
Non-viral drug delivery vectors have emerged very strongly in the pharmaceutical world as means of delivering biologics particularly genes and mRNA to cells and given the wide breadth of research on metal complex luminophore interaction with nucleic acids, these routes offer very useful opportunities to introduce metal complex linked nucleic acid materials to cells and organelles. Opening up exciting prospects in theranostics. Also, approaches are being found to avoid or release from the endosomal trap of endocytosis in non-viral drug delivery and no doubt these approaches will also make their way into the field of metal complex luminophores.
Although the majority of imaging applications to date for metal complex luminophores have focussed on conventional confocal or wide-field luminescence imaging methods. An area of significant application growth for metal complex luminophores is likely to be in super-resolution microscopy. Super-resolution methods span a range of different methods, including photoactivation localization microscopy (PALM), stimulated emission depletion (STED) microscopy, stochastic optical reconstruction microscopy (STORM), super-resolution structured illumination microscopy (SR-SIM).182 All offer outstanding, sub-diffraction resolved structural and potentially dynamic insights into the cell but make unique demands of the luminescent agents used for contrast. The photophysical characteristics of metal complex luminophores, in particular their Stokes shift and robust photostability, make them particularly amenable to many of these approaches and examples have demonstrated suitability of Ruthenium(II) complexes to such methods.74,158 In addition reports of less commonly studied metal luminophores such as Zn(II) and Os(II) have also been demonstrated as effective super-resolution probes. For optimal super-resolution imaging, obviously probe permeability to live cells, and precision targeting of probe to the region or species of interest is critically important. As outlined in this review, the capacity to achieve these goals with is now well within reach for metal complex luminophores and they are thus likely to see expanded application at the forefront of cell imaging.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is also based upon work supported by Science Foundation Ireland under Grant No 19/FFP/6428. LH, CB and TEK gratefully acknowledge the Irish Research Council for funding under Government of Ireland Postgraduate Scholarship GOIPG/2019/2426 and Government of Ireland Postdoctoral Fellowship, GOIPD/2018/495.References
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 Search PubMed.
- F. Göttfert, C. A. Wurm, V. Mueller, S. Berning, V. C. Cordes, A. Honigmann and S. W. Hell, Biophys. J., 2013, 105, L01–L03 Search PubMed.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 Search PubMed.
- M. P. Coogan and V. Fernández-Moreira, Chem. Commun., 2014, 50, 384–399 Search PubMed.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159–1170 Search PubMed.
- M. J. S. A. Silva, P. M. P. Gois and G. Gasser, ChemBioChem, 2021 DOI:10.1002/cbic.202100015.
- M. Martínez-Alonso and G. Gasser, Coord. Chem. Rev., 2021, 434, 213736 Search PubMed.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 Search PubMed.
- X. Zhen, R. Qu, W. Chen, W. Wu and X. Jiang, Biomater. Sci., 2021, 9, 285–300 Search PubMed.
- J. G. Joseph and A. P. Liu, Adv. Biosyst., 2020, 4, 1900278 Search PubMed.
- S. Kumari, S. Mg and S. Mayor, Cell Res., 2010, 20, 256–275 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 Search PubMed.
- P. D. Dobson and D. B. Kell, Nat. Rev. Drug Discovery, 2008, 7, 205–220 Search PubMed.
- M. Hu, X. Ai, Z. Wang, Z. Zhang, H. Cheong, W. Zhang, J. Lin, J. Li, H. Yang and B. Xing, Nano Res., 2018, 11, 5474–5498 Search PubMed.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer, Berlin, Heidelberg, 2007, pp. 117–214 Search PubMed.
- F. E. Poynton, J. P. Hall, P. M. Keane, C. Schwarz, I. V. Sazanovich, M. Towrie, T. Gunnlaugsson, C. J. Cardin, D. J. Cardin, S. J. Quinn, C. Long and J. M. Kelly, Chem. Sci., 2016, 7, 3075–3084 Search PubMed.
- G. Li, L. Sun, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13261–13276 Search PubMed.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126 Search PubMed.
- C. Moucheron, A. Kirsch-De Mesmaeker and J. M. Kelly, J. Photochem. Photobiol., B, 1997, 40, 91–106 Search PubMed.
- S. L. Hopkins and S. Bonnet, Ruthenium Complexes, John Wiley & Sons, Ltd, 2018, pp. 89–116 Search PubMed.
- C.-J. Li, S.-Y. Yin, H.-P. Wang, Z.-W. Wei and M. Pan, J. Photochem. Photobiol., A, 2019, 379, 99–104 Search PubMed.
- T. Mede, M. Jäger and U. S. Schubert, Chem. Soc. Rev., 2018, 47, 7577–7627 Search PubMed.
- K. K.-W. Lo, Acc. Chem. Res., 2020, 53, 32–44 Search PubMed.
- W. Wang, L. Lu, K.-J. Wu, J. Liu, C.-H. Leung, C.-Y. Wong and D.-L. Ma, Sens. Actuators, B, 2019, 288, 392–398 Search PubMed.
- W. Wang, Z.-Z. Dong, C. Yang, G. Li, Y.-C. Tse, C.-H. Leung and D.-L. Ma, Sens. Actuators, B, 2017, 251, 374–379 Search PubMed.
- W. Wang, K. Vellaisamy, G. Li, C. Wu, C.-N. Ko, C.-H. Leung and D.-L. Ma, Anal. Chem., 2017, 89, 11679–11684 Search PubMed.
- V. Mirabello, F. Cortezon-Tamarit and S. I. Pascu, Front. Chem., 2018, 6, 27 Search PubMed.
- T. Yoshihara, Y. Hirakawa, M. Hosaka, M. Nangaku and S. Tobita, J. Photochem. Photobiol., C, 2017, 30, 71–95 Search PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. Clive Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 Search PubMed.
- M. Abrahamsson, M. Jäger, T. Österman, L. Eriksson, P. Persson, H.-C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2006, 128, 12616–12617 Search PubMed.
- T. E. Keyes, J. G. Vos, J. A. Kolnaar, J. G. Haasnoot, J. Reedijk and R. Hage, Inorg. Chim. Acta, 1996, 245, 237–242 Search PubMed.
- A. K. Pal, S. Serroni, N. Zaccheroni, S. Campagna and G. S. Hanan, Chem. Sci., 2014, 5, 4800–4811 Search PubMed.
- F. Schramm, V. Meded, H. Fliegl, K. Fink, O. Fuhr, Z. Qu, W. Klopper, S. Finn, T. E. Keyes and M. Ruben, Inorg. Chem., 2009, 48, 5677–5684 Search PubMed.
- M. Abrahamsson, M. Jäger, R. J. Kumar, T. Österman, P. Persson, H.-C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2008, 130, 15533–15542 Search PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 Search PubMed.
- H. Huang, P. Zhang, B. Yu, Y. Chen, J. Wang, L. Ji and H. Chao, J. Med. Chem., 2014, 57, 8971–8983 Search PubMed.
- K. K.-S. Tso and K. K.-W. Lo, Iridium(III) in Optoelectronic and Photonics Applications, John Wiley & Sons, Ltd, 2017, pp. 415–477 Search PubMed.
- L. C.-C. Lee, K.-K. Leung and K. K.-W. Lo, Dalton Trans., 2017, 46, 16357–16380 Search PubMed.
- S. Clède, F. Lambert, C. Sandt, Z. Gueroui, M. Réfrégiers, M.-A. Plamont, P. Dumas, A. Vessières and C. Policar, Chem. Commun., 2012, 48, 7729–7731 Search PubMed.
- C. Y.-S. Chung, S. P.-Y. Li, M.-W. Louie, K. K.-W. Lo and V. W.-W. Yam, Chem. Sci., 2013, 4, 2453–2462 Search PubMed.
- S. W. Botchway, M. Charnley, J. W. Haycock, A. W. Parker, D. L. Rochester, J. A. Weinstein and J. A. G. Williams, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16071–16076 Search PubMed.
- S. A. E. Omar, P. A. Scattergood, L. K. McKenzie, H. E. Bryant, J. A. Weinstein and P. I. P. Elliott, Molecules, 2016, 21, 1382 Search PubMed.
- C. Ge, H. Huang, Y. Wang, H. Zhao, P. Zhang and Q. Zhang, ACS Appl. Bio Mater., 2018, 1, 1587–1593 Search PubMed.
- J. Tang, M. Zhang, H.-Y. Yin, J. Jing, D. Xie, P. Xu and J.-L. Zhang, Chem. Commun., 2016, 52, 11583–11586 Search PubMed.
- F. Dosio, B. Stella, A. Ferrero, C. Garino, D. Zonari, S. Arpicco, L. Cattel, S. Giordano and R. Gobetto, Int. J. Pharm., 2013, 440, 221–228 Search PubMed.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernández-Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066–3068 Search PubMed.
- V. Fernández-Moreira, F. L. Thorp-Greenwood, A. J. Amoroso, J. Cable, J. B. Court, V. Gray, A. J. Hayes, R. L. Jenkins, B. M. Kariuki, D. Lloyd, C. O. Millet, C. F. Williams and M. P. Coogan, Org. Biomol. Chem., 2010, 8, 3888–3901 Search PubMed.
- M.-W. Louie, T. T.-H. Fong and K. K.-W. Lo, Inorg. Chem., 2011, 50, 9465–9471 Search PubMed.
- V. Fiorini, I. Zanoni, S. Zacchini, A. L. Costa, A. Hochkoeppler, V. Zanotti, A. M. Ranieri, M. Massi, A. Stefan and S. Stagni, Dalton Trans., 2017, 46, 12328–12338 Search PubMed.
- A. M. Ranieri, C. Caporale, V. Fiorini, A. Hubbard, P. Rigby, S. Stagni, E. Watkin, M. I. Ogden, M. J. Hackett and M. Massi, Chem. – Eur. J., 2019, 25, 10566–10570 Search PubMed.
- F. R. Svensson, M. Matson, M. Li and P. Lincoln, Biophys. Chem., 2010, 149, 102–106 Search PubMed.
- M. Matson, F. R. Svensson, B. Nordén and P. Lincoln, J. Phys. Chem. B, 2011, 115, 1706–1711 Search PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 Search PubMed.
- C. Caporale, A. M. Ranieri, S. Paternoster, C. A. Bader, M. Falasca, S. E. Plush, D. A. Brooks, S. Stagni and M. Massi, Inorganics, 2020, 8, 23 Search PubMed.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 Search PubMed.
- S. P.-Y. Li, C. T.-S. Lau, M.-W. Louie, Y.-W. Lam, S. H. Cheng and K. K.-W. Lo, Biomaterials, 2013, 34, 7519–7532 Search PubMed.
- A. M.-H. Yip and K. K.-W. Lo, Coord. Chem. Rev., 2018, 361, 138–163 Search PubMed.
- S. P.-Y. Li, H.-W. Liu, K. Y. Zhang and K. K.-W. Lo, Chem. – Eur. J., 2010, 16, 8329–8339 Search PubMed.
- G. S. Gorman, P. F. Chinnery, S. DiMauro, M. Hirano, Y. Koga, R. McFarland, A. Suomalainen, D. R. Thorburn, M. Zeviani and D. M. Turnbull, Nat. Rev. Disease Primers, 2016, 2, 1–22 Search PubMed.
- J. S. Modica-Napolitano and J. R. Aprille, Adv. Drug Delivery Rev., 2001, 49, 63–70 Search PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 Search PubMed.
- T. Murase, T. Yoshihara and S. Tobita, Chem. Lett., 2012, 41, 262–263 Search PubMed.
- Q. Zhang, R. Cao, H. Fei and M. Zhou, Dalton Trans., 2014, 43, 16872–16879 Search PubMed.
- S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau and T. Weil, J. Am. Chem. Soc., 2017, 139, 2512–2519 Search PubMed.
- C.-K. Koo, K.-L. Wong, C. W.-Y. Man, H.-L. Tam, S.-W. Tsao, K.-W. Cheah and M. H.-W. Lam, Inorg. Chem., 2009, 48, 7501–7503 Search PubMed.
- E. E. Langdon-Jones, C. F. Williams, A. J. Hayes, D. Lloyd, S. J. Coles, P. N. Horton, L. M. Groves and S. J. A. Pope, Eur. J. Inorg. Chem., 2017, 5279–5287 Search PubMed.
- E. E. Langdon-Jones, N. O. Symonds, S. E. Yates, A. J. Hayes, D. Lloyd, R. Williams, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2014, 53, 3788–3797 Search PubMed.
- G. J. Ryan, F. E. Poynton, R. B. P. Elmes, M. Erby, D. C. Williams, S. J. Quinn and T. Gunnlaugsson, Dalton Trans., 2015, 44, 16332–16344 Search PubMed.
- W. Zhang, Y. Liu, Q. Gao, C. Liu, B. Song, R. Zhang and J. Yuan, Chem. Commun., 2018, 54, 13698–13701 Search PubMed.
- U. Schatzschneider, J. Niesel, I. Ott, R. Gust, H. Alborzinia and S. Wölfl, ChemMedChem, 2008, 3, 1104–1109 Search PubMed.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 Search PubMed.
- A. Wragg, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem. – Eur. J., 2014, 20, 14004–14011 Search PubMed.
- A. Wragg, M. R. Gill, L. McKenzie, C. Glover, R. Mowll, J. A. Weinstein, X. Su, C. Smythe and J. A. Thomas, Chem. – Eur. J., 2015, 21, 11865–11871 Search PubMed.
- S. Sreedharan, M. R. Gill, E. Garcia, H. K. Saeed, D. Robinson, A. Byrne, A. Cadby, T. E. Keyes, C. Smythe, P. Pellett, J. Bernardino de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2017, 139, 15907–15913 Search PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 Search PubMed.
- C. Zhang, R. Guan, X. Liao, C. Ouyang, T. W. Rees, J. Liu, Y. Chen, L. Ji and H. Chao, Chem. Commun., 2019, 55, 12547–12550 Search PubMed.
- A. Luengo, M. Redrado, I. Marzo, V. Fernández-Moreira and M. C. Gimeno, Inorg. Chem., 2020, 59, 8960–8970 Search PubMed.
- H. Huang, P. Zhang, H. Chen, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 715–725 Search PubMed.
- G. Ghosh, K. L. Colón, A. Fuller, T. Sainuddin, E. Bradner, J. McCain, S. M. A. Monro, H. Yin, M. W. Hetu, C. G. Cameron and S. A. McFarland, Inorg. Chem., 2018, 57, 7694–7712 Search PubMed.
- Y. Shen, T. Shao, B. Fang, W. Du, M. Zhang, J. Liu, T. Liu, X. Tian, Q. Zhang, A. Wang, J. Yang, J. Wu and Y. Tian, Chem. Commun., 2018, 54, 11288–11291 Search PubMed.
- K. L. Smitten, P. A. Scattergood, C. Kiker, J. A. Thomas and P. I. P. Elliott, Chem. Sci., 2020, 11, 8928–8935 Search PubMed.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 Search PubMed.
- W. X. Ren, J. Han, S. Uhm, Y. J. Jang, C. Kang, J.-H. Kim and J. S. Kim, Chem. Commun., 2015, 51, 10403–10418 Search PubMed.
- K. K.-W. Lo and T. K.-M. Lee, Inorg. Chim. Acta, 2007, 360, 293–302 Search PubMed.
- K. Y. Zhang, H.-W. Liu, M.-C. Tang, A. W.-T. Choi, N. Zhu, X.-G. Wei, K.-C. Lau and K. K.-W. Lo, Inorg. Chem., 2015, 54, 6582–6593 Search PubMed.
- M.-W. Louie, M. H.-C. Lam and K. K.-W. Lo, Eur. J. Inorg. Chem., 2009, 4265–4273 Search PubMed.
- J. Li, L. Zeng, K. Xiong, T. W. Rees, C. Jin, W. Wu, Y. Chen, L. Ji and H. Chao, Chem. Commun., 2019, 55, 10972–10975 Search PubMed.
- S. Paul, P. Kundu, U. Bhattacharyya, A. Garai, R. C. Maji, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2020, 59, 913–924 Search PubMed.
- L. L. Hart and J. R. Davie, Biochem. Cell Biol., 2002, 80, 335–341 Search PubMed.
- J. Liang and Y. Shang, Annu. Rev. Physiol., 2013, 75, 225–240 Search PubMed.
- K. K.-W. Lo, K. Y. Zhang, C.-K. Chung and K. Y. Kwok, Chem. – Eur. J., 2007, 13, 7110–7120 Search PubMed.
- K. K.-W. Lo, T. K.-M. Lee, J. S.-Y. Lau, W.-L. Poon and S.-H. Cheng, Inorg. Chem., 2008, 47, 200–208 Search PubMed.
- M. Gottschaldt, U. S. Schubert, S. Rau, S. Yano, J. G. Vos, T. Kroll, J. Clement and I. Hilger, ChemBioChem, 2010, 11, 649–652 Search PubMed.
- M. B. Calvo, A. Figueroa, E. G. P ulido, R. G. Campelo and L. A. Aparicio, Int. J. Endocrinol., 2010, 2010, 205357 Search PubMed.
- J. Liu, X. Liao, K. Xiong, S. Kuang, C. Jin, L. Ji and H. Chao, Chem. Commun., 2020, 56, 5839–5842 Search PubMed.
- H.-W. Liu, K. Y. Zhang, W. H.-T. Law and K. K.-W. Lo, Organometallics, 2010, 29, 3474–3476 Search PubMed.
- M.-W. Louie, H.-W. Liu, M. H.-C. Lam, Y.-W. Lam and K. K.-W. Lo, Chem. – Eur. J., 2011, 17, 8304–8308 Search PubMed.
- C. T.-S. Lau, C. Chan, K. Y. Zhang, V. A. L. Roy and K. K.-W. Lo, Eur. J. Inorg. Chem., 2017, 5288–5294 Search PubMed.
- N. Viola-Villegas, A. E. Rabideau, J. Cesnavicious, J. Zubieta and R. P. Doyle, ChemMedChem, 2008, 3, 1387–1394 Search PubMed.
- M. Aminoff, J. E. Carter, R. B. Chadwick, C. Johnson, R. Gräsbeck, M. A. Abdelaal, H. Broch, L. B. Jenner, P. J. Verroust, S. K. Moestrup, A. de la Chapelle and R. Krahe, Nat. Genet., 1999, 21, 309–313 Search PubMed.
- N. Viola-Villegas, A. E. Rabideau, M. Bartholomä, J. Zubieta and R. P. Doyle, J. Med. Chem., 2009, 52, 5253–5261 Search PubMed.
- A. R. Vortherms, A. R. Kahkoska, A. E. Rabideau, J. Zubieta, L. L. Andersen, M. Madsen and R. P. Doyle, Chem. Commun., 2011, 47, 9792–9794 Search PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13, 339 Search PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 Search PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 Search PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 Search PubMed.
- X. Hu, Y. Zhang, T. Ding, J. Liu and H. Zhao, Front. Bioeng. Biotechnol., 2020, 8, 990 Search PubMed.
- R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2009, 253, 1833–1853 Search PubMed.
- R. B. P. Elmes, K. N. Orange, S. M. Cloonan, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2011, 133, 15862–15865 Search PubMed.
- N. J. Rogers, S. Claire, R. M. Harris, S. Farabi, G. Zikeli, I. B. Styles, N. J. Hodges and Z. Pikramenou, Chem. Commun., 2013, 50, 617–619 Search PubMed.
- X. Qin, Y. Si, Z. Wu, K. Zhang, J. Li and Y. Yin, Anal. Chem., 2020, 92, 924–931 Search PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 Search PubMed.
- D. Wencel, C. Dolan, M. Barczak, T. E. Keyes and C. McDonagh, Nanotechnology, 2013, 24, 365705 Search PubMed.
- L. M. Rossi, L. Shi, F. H. Quina and Z. Rosenzweig, Langmuir, 2005, 21, 4277–4280 Search PubMed.
- S. Lechevallier, R. Mauricot, H. Gros-Dagnac, S. Chevreux, G. Lemercier, E. Phonesouk, M. Golzio and M. Verelst, ChemPlusChem, 2017, 82, 770–777 Search PubMed.
- H. Xu, J. W. Aylott, R. Kopelman, T. J. Miller and M. A. Philbert, Anal. Chem., 2001, 73, 4124–4133 Search PubMed.
- Y. Ellahioui, M. Patra, C. Mari, R. Kaabi, J. Karges, G. Gasser and S. Gómez-Ruiz, Dalton Trans., 2019, 48, 5940–5951 Search PubMed.
- L. He, Y. Huang, H. Zhu, G. Pang, W. Zheng, Y.-S. Wong and T. Chen, Adv. Funct. Mater., 2014, 24, 2754–2763 Search PubMed.
- N. Kitajima, Y. Umehara, A. Son, T. Kondo and K. Tanabe, Bioconjugate Chem., 2018, 29, 4168–4175 Search PubMed.
- L. Ding, Y. Lian, Z. Lin, Z. Zhang and X. Wang, ACS Sens., 2020, 5, 3971–3978 Search PubMed.
- M. Frasconi, Z. Liu, J. Lei, Y. Wu, E. Strekalova, D. Malin, M. W. Ambrogio, X. Chen, Y. Y. Botros, V. L. Cryns, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 11603–11613 Search PubMed.
- Y.-K. Dou, Y. Shang, X.-W. He, W.-Y. Li, Y.-H. Li and Y.-K. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 13954–13963 Search PubMed.
- S.-A. Yang, S. Choi, S. M. Jeon and J. Yu, Sci. Rep., 2018, 8, 185 Search PubMed.
- L. Liu, X. Li, S. Hou, Y. Xue, Y. Yao, Y. Ma, X. Feng, S. He, Y. Lu, Y. Wang and X. Zeng, Chem. Commun., 2009, 6759–6761 Search PubMed.
- A. Byrne, J. Jacobs, C. S. Burke, A. Martin, A. Heise and T. E. Keyes, Analyst, 2017, 142, 3400–3406 Search PubMed.
- J. Jiang, Y. Qian, Z. Xu, Z. Lv, P. Tao, M. Xie, S. Liu, W. Huang and Q. Zhao, Chem. Sci., 2019, 10, 5085–5094 Search PubMed.
- J. Zhao, S. Sun, X. Li, W. Zhang and S. Gou, ACS Appl. Bio Mater., 2020, 3, 252–262 Search PubMed.
- M. Hu, J. Zhao, X. Ai, M. Budanovic, J. Mu, R. D. Webster, Q. Cao, Z. Mao and B. Xing, Dalton Trans., 2016, 45, 14101–14108 Search PubMed.
- Y. Pellegrin, R. J. Forster and T. E. Keyes, Inorg. Chim. Acta, 2008, 361, 2683–2691 Search PubMed.
- Y. Liu, Y. Chen, Z.-Y. Duan, X.-Z. Feng, S. Hou, C. Wang and R. Wang, ACS Nano, 2007, 1, 313–318 Search PubMed.
- S.-S. Xue, C.-P. Tan, M.-H. Chen, J.-J. Cao, D.-Y. Zhang, R.-R. Ye, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2017, 53, 842–845 Search PubMed.
- N. Cheng, Y. Chen, J. Yu, J. Li and Y. Liu, Bioconjugate Chem., 2018, 29, 1829–1833 Search PubMed.
- C. Draeger, C. Böttcher, C. Messerschmidt, A. Schulz, L. Ruhlmann, U. Siggel, L. Hammarström, H. Berglund-Baudin and J.-H. Fuhrhop, Langmuir, 2000, 16, 2068–2077 Search PubMed.
- A. Guerrero-Martínez, Y. Vida, D. Domínguez-Gutiérrez, R. Q. Albuquerque and L. De Cola, Inorg. Chem., 2008, 47, 9131–9133 Search PubMed.
- S. Finn, A. Byrne, K. S. Gkika and T. E. Keyes, Front. Chem., 2020, 8, 638 Search PubMed.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Front. Pharmacol., 2015, 6, 286 Search PubMed.
- K. P. McNamara, N. Rosenzweig and Z. Rosenzweig, Mikrochim. Acta, 1999, 131, 57–64 Search PubMed.
- L. Cosgrave, M. Devocelle, R. J. Forster and T. E. Keyes, Chem. Commun., 2010, 46, 103–105 Search PubMed.
- Y. Hisamatsu, A. Shibuya, N. Suzuki, T. Suzuki, R. Abe and S. Aoki, Bioconjugate Chem., 2015, 26, 857–879 Search PubMed.
- W. Jiang, Y. Gao, Y. Sun, F. Ding, Y. Xu, Z. Bian, F. Li, J. Bian and C. Huang, Inorg. Chem., 2010, 49, 3252–3260 Search PubMed.
- K. K.-W. Lo, P.-K. Lee and J. S.-Y. Lau, Organometallics, 2008, 27, 2998–3006 Search PubMed.
- S. Bonnet, B. Limburg, J. D. Meeldijk, R. J. M. Klein Gebbink and J. A. Killian, J. Am. Chem. Soc., 2011, 133, 252–261 Search PubMed.
- A. Mechler, B. D. Stringer, M. S. H. Mubin, E. H. Doeven, N. W. Phillips, J. Rudd-Schmidt and C. F. Hogan, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 2939–2946 Search PubMed.
- J. Shen, H.-C. Kim, J. Wolfram, C. Mu, W. Zhang, H. Liu, Y. Xie, J. Mai, H. Zhang, Z. Li, M. Guevara, Z.-W. Mao and H. Shen, Nano Lett., 2017, 17, 2913–2920 Search PubMed.
- W.-Y. Zhang, F. Du, M. He, L. Bai, Y.-Y. Gu, L.-L. Yang and Y.-J. Liu, Eur. J. Med. Chem., 2019, 178, 390–400 Search PubMed.
- J. R. Shakirova, A. Sadeghi, A. A. Koblova, P. S. Chelushkin, E. Toropainen, S. Tavakoli, L.-S. Kontturi, T. Lajunen, S. P. Tunik and A. Urtti, RSC Adv., 2020, 10, 14431–14440 Search PubMed.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 Search PubMed.
- K. M. Stewart, K. L. Horton and S. O. Kelley, Org. Biomol. Chem., 2008, 6, 2242–2255 Search PubMed.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 Search PubMed.
- M. Green and P. M. Loewenstein, Cell, 1988, 55, 1179–1188 Search PubMed.
- S. Futaki and I. Nakase, Acc. Chem. Res., 2017, 50, 2449–2456 Search PubMed.
- K. Kardani, A. Milani, S. H. Shabani and A. Bolhassani, Expert Opin. Drug Delivery, 2019, 16, 1227–1258 Search PubMed.
- J. Xie, Y. Bi, H. Zhang, S. Dong, L. Teng, R. J. Lee and Z. Yang, Front. Pharmacol., 2020, 11, 697 Search PubMed.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 Search PubMed.
- U. Neugebauer, Y. Pellegrin, M. Devocelle, R. J. Forster, W. Signac, N. Moran and T. E. Keyes, Chem. Commun., 2008, 5307–5309 Search PubMed.
- A. Byrne, C. Dolan, R. D. Moriarty, A. Martin, U. Neugebauer, R. J. Forster, A. Davies, Y. Volkov and T. E. Keyes, Dalton Trans., 2015, 44, 14323–14332 Search PubMed.
- C. Dolan, R. D. Moriarty, E. Lestini, M. Devocelle, R. J. Forster and T. E. Keyes, J. Inorg. Biochem., 2013, 119, 65–74 Search PubMed.
- A. Byrne, C. S. Burke and T. E. Keyes, Chem. Sci., 2016, 7, 6551–6562 Search PubMed.
- C. S. Burke, A. Byrne and T. E. Keyes, J. Am. Chem. Soc., 2018, 140, 6945–6955 Search PubMed.
- C. S. Burke, A. Byrne and T. E. Keyes, Angew. Chem., Int. Ed., 2018, 57, 12420–12424 Search PubMed.
- D. Cullinane, K. S. Gkika, A. Byrne and T. E. Keyes, J. Inorg. Biochem., 2020, 111032 Search PubMed.
- S. Hostachy, J.-M. Swiecicki, C. Sandt, N. Delsuc and C. Policar, Dalton Trans., 2016, 45, 2791–2795 Search PubMed.
- S. Clède, N. Delsuc, C. Laugel, F. Lambert, C. Sandt, A. Baillet-Guffroy and C. Policar, Chem. Commun., 2015, 51, 2687–2689 Search PubMed.
- K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Chem. Biol., 2008, 15, 375–382 Search PubMed.
- S. R. Jean, M. Ahmed, E. K. Lei, S. P. Wisnovsky and S. O. Kelley, Acc. Chem. Res., 2016, 49, 1893–1902 Search PubMed.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, J. Am. Chem. Soc., 2014, 136, 15300–15309 Search PubMed.
- K. S. Gkika, A. Byrne and T. E. Keyes, Dalton Trans., 2019, 48, 17461–17471 Search PubMed.
- D. Kalderon, B. L. Roberts, W. D. Richardson and A. E. Smith, Cell, 1984, 39, 499–509 Search PubMed.
- A. D. Ragin, R. A. Morgan and J. Chmielewski, Chem. Biol., 2002, 9, 943–948 Search PubMed.
- L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Forster, M. Devocelle and T. E. Keyes, Chem. Commun., 2013, 49, 2658–2660 Search PubMed.
- A. H. Day, M. H. Übler, H. L. Best, E. Lloyd-Evans, R. J. Mart, I. A. Fallis, R. K. Allemann, E. A. H. Al-Wattar, N. I. Keymer, N. J. Buurma and S. J. A. Pope, Chem. Sci., 2020, 11, 1599–1606 Search PubMed.
- A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044–4056 Search PubMed.
- K. Adamson, C. Dolan, N. Moran, R. J. Forster and T. E. Keyes, Bioconjugate Chem., 2014, 25, 928–944 Search PubMed.
- Z. Liu, F. Wang and X. Chen, Drug Dev. Res., 2008, 69, 329–339 Search PubMed.
- X. Ma, J. Jia, R. Cao, X. Wang and H. Fei, J. Am. Chem. Soc., 2014, 136, 17734–17737 Search PubMed.
- K. Vellaisamy, G. Li, W. Wang, C.-H. Leung and D.-L. Ma, Chem. Sci., 2018, 9, 8171–8177 Search PubMed.
- E. M. Hahn, N. Estrada-Ortiz, J. Han, V. F. C. Ferreira, T. G. Kapp, J. D. G. Correia, A. Casini and F. E. Kühn, Eur. J. Inorg. Chem., 2017, 1667–1672 Search PubMed.
- A. Luengo, I. Marzo, M. Reback, I. M. Daubit, V. Fernández-Moreira, N. Metzler-Nolte and M. C. Gimeno, Chem. – Eur. J., 2020, 26, 12158–12167 Search PubMed.
- E. Ferri, D. Donghi, M. Panigati, G. Prencipe, L. D’Alfonso, I. Zanoni, C. Baldoli, S. Maiorana, G. D’Alfonso and E. Licandro, Chem. Commun., 2010, 46, 6255–6257 Search PubMed.
- C. Mari, M. Panigati, L. D’Alfonso, I. Zanoni, D. Donghi, L. Sironi, M. Collini, S. Maiorana, C. Baldoli, G. D’Alfonso and E. Licandro, Organometallics, 2012, 31, 5918–5928 Search PubMed.
- S. Cauteruccio, M. Panigati, L. Veronese, N. Zaffaroni, M. Folini and E. Licandro, J. Organomet. Chem., 2019, 887, 32–39 Search PubMed.
- L. Schermelleh, A. Ferrand, T. Huser, C. Eggeling, M. Sauer, O. Biehlmaier and G. P. C. Drummen, Nat. Cell Biol., 2019, 21, 72–84 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |