
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive oxygen species, proinflammatory and immunosuppressive mediators induced in COVID-19: overlapping biology with cancer†
Balaraman
Kalyanaraman

Department of Biophysics, Cancer Center, Center for Disease Prevention Research, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI 53226, USA. E-mail: balarama@mcw.edu
First published on 30th June 2021
Abstract
This review analyzes the published literature linking the different mechanisms focused on oxidative stress and inflammation that contribute to COVID-19 disease severity. The objective is to bring together potential proinflammatory mechanisms of COVID-19 pathogenesis and address mitigation strategies using naturally occurring compounds and FDA-approved drugs. Outstanding questions addressed include the following: What is the mechanistic basis for linking enhanced vulnerability in COVID-19 to increased oxidative damage and proinflammatory mediators (e.g., cytokines), especially in high-risk people? Can we repurpose anti-inflammatory and immunomodulatory agents to mitigate inflammation in COVID-19 patients? How does 2-deoxy-D-glucose function as an anti-COVID drug? COVID-19, cancer biology, and immunotherapy share many mechanistic similarities. Repurposing drugs that already have been FDA-approved for mitigating inflammation and immunosuppression in cancer may be a way to counteract disease severity, progression, and chronic inflammation in COVID-19. What are the long-term effects of reactive oxygen species-inducing immune cells and sustained inflammation in so-called long-haulers (long COVID) after recovery from COVID-19? Can we use mitochondria-targeted agents prophylactically to prevent inflammation and boost immunity in long-haulers? Addressing the oxidative chemical biology of COVID-19 and the mechanistic commonalities with cancer may provide new insights potentially leading to appropriate clinical trials and new treatments.
Introduction
Numerous clinical trials are underway to investigate the effects of antioxidants, anti-inflammatory agents, and immunity boosting agents (e.g., vitamin C, vitamin E, N-acetylcysteine, vitamin D, melatonin) and an antioxidant-enriched diet as an adjuvant therapy to standard therapy for coronavirus disease 19 (COVID-19) treatment.1 The goal of these clinical trials is to test the hypothesis that adjuvant therapies with antioxidants and nutritional supplements will decrease the need for mechanical ventilators through decreased oxidative mechanism and inflammation. The role of reactive oxygen species (ROS)-induced oxidative stress and COVID-19 has been addressed in several reviews.2–4 This review discusses the potential mechanisms and therapeutic implications of drugs impacting leukocyte biology—from cancer therapeutics to COVID-19—as follows: nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (Nox2) inhibitors, classical chain-breaking antioxidants, iron chelators, N-acetylcysteine/glutathione (GSH) supplements, nuclear factor erythroid 2-related factor 2 (Nrf2)-activating agents, mitochondria-targeted AMP-activated protein kinase (AMPK) activators, and sirtuin activators. For ease of reference, Table 1 lists the abbreviations used in this review article.| Abbreviation | Term |
|---|---|
| 2-DG | 2-Deoxy-D-glucose |
| 8-OHdG | 8-Hydroxy deoxyguanosine |
| AT-1,7 | Angiotensin 1,7 |
| AT-II | Angiotensin II |
| ACE2 | Angiotensin-converting enzyme 2 |
| AMPK | AMP-activated protein kinase |
| COVID-19 | Coronavirus disease 19 |
| CXCL10 | Chemokines |
| Fe-S | Iron–sulfur |
| GSH | Glutathione |
| GPx | Glutathione peroxidase |
| H2O2 | Hydrogen peroxide |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HOC | Hypochlorous acid |
| IFN-γ | Interferon gamma |
| MDSC | Myeloid-derived suppressor cells |
| Mito-ATO | Mito-atovaquone |
| MPO | Myeloperoxidase |
| mtDNA | Mitochondrial DNA |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NET | Neutrophil extracellular trap |
| NO˙ | Nitric oxide |
| Nox | NADPH oxidase |
| Nox2 | NADPH oxidase 2 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| O2˙− | Superoxide anion |
| ˙OH | Hydroxyl radical |
| ONOO− | Peroxynitrite |
| OXPHOS | Oxidative phosphorylation |
| PAD4 | Protein-arginine deiminase type 4 |
| PD-L1 | Programmed cell death protein 1 |
| PGC-1α | Proliferator-activated receptor-gamma coactivator |
| PHGPx | Phospholipid glutathione peroxidase |
| ROS | Reactive oxygen species |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| STAT | Signal transducer and activator of transcription |
| Teffs | Effector T cells |
| TLR4 | Toll-like receptor 4 |
| TME | Tumor microenvironment |
| TMPRSS2 | Transmembrane serine protease 2 |
| TPP+ | Triphenylphosphonium |
| Tregs | Regulatory T cells |
SARS-CoV-2 and reactive oxygen species
After entering the respiratory tract, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) uses a spike protein to latch onto the angiotensin-converting enzyme 2 (ACE2) receptor present on the surface of the cell membrane, and fuses into the lung cell membrane. Its entry into host cells is facilitated by proteolytic cleaving and activation of glycoproteins by the transmembrane serine protease 2 (TMPRSS2) (Fig. 1).5,6 ACE2, a membrane-bound enzyme responsible for maintenance of blood pressure, converts angiotensin II (AT-II), which is vasoconstrictive, to angiotensin 1,7 (AT-1,7), which exerts vasodilatory effects. Lung inflammation is enhanced by AT-II, which activates superoxide anion (O2˙−), and diminished in the presence of AT-1,7, which decreases O2˙− in vascular cells.7 SARS-CoV-2 decreases ACE2 receptors; consequently, AT-II levels are elevated, resulting in enhanced oxidant formation, oxidative stress, and inflammation.8,9 ACE2 deficiency increases Nox2-mediated oxidant formation.10,11 ACE2 expression in lung tissues is enhanced in smokers and in chronic obstructive pulmonary disease patients. Depending on the type of mutation in the spike protein, its affinity for ACE2 varies.12 For example, the naturally occurring D614G mutation (that is predominantly found in the vast majority of isolates) in the spike protein binds to ACE2 with much more affinity, which enhances its entry into the host cells.12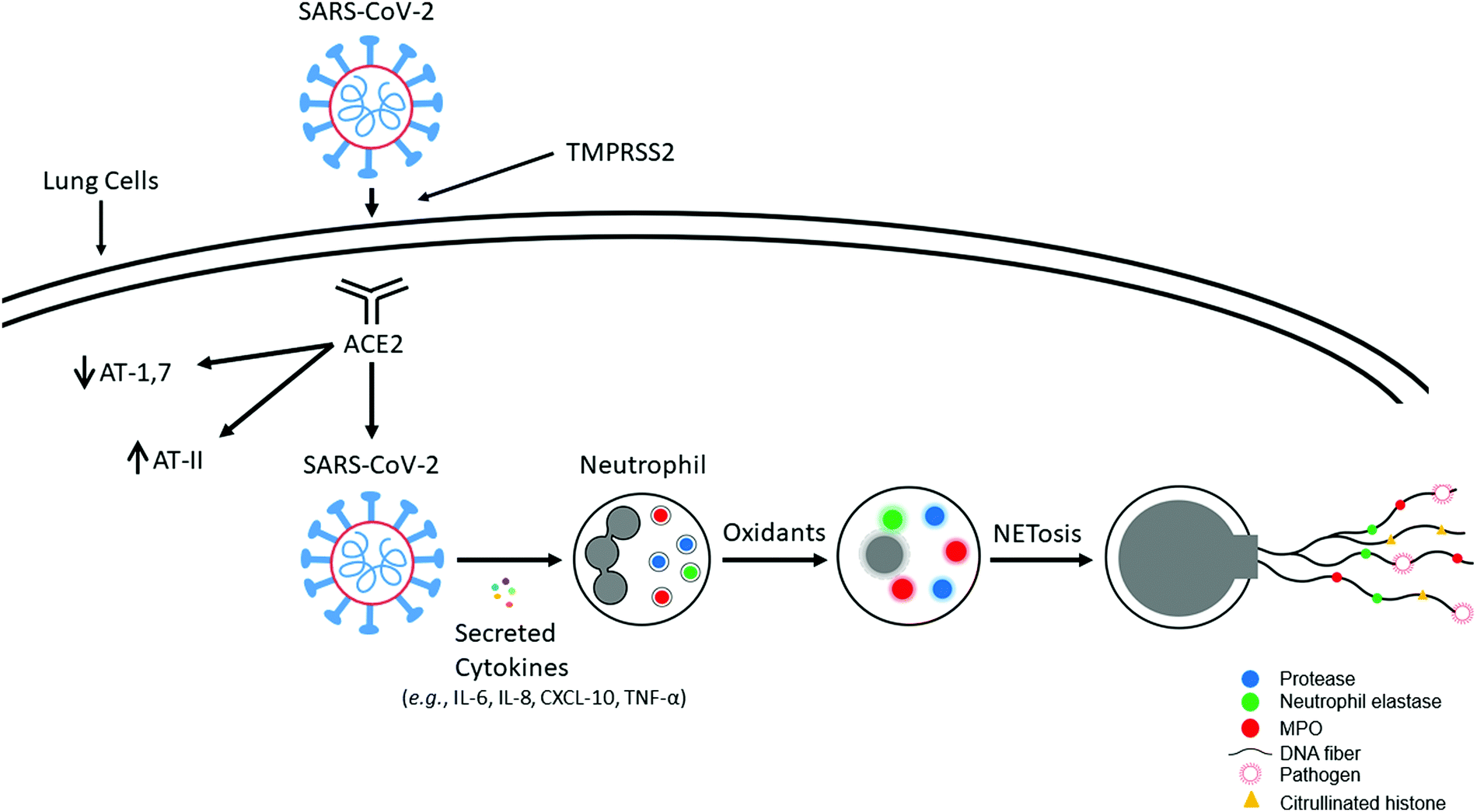 | ||
| Fig. 1 SARS-CoV-2 uptake into the lung cells and induction of NET formation. Modified from Redox Biology, 37, Balaraman Kalyanaraman, Do free radical NETwork and oxidative stress disparities in African Americans enhance their vulnerability to SARS-CoV-2 infection and COVID-19 severity? 101721, Copyright (2020), with permission from Elsevier. | ||
SARS-Cov-2 induces proinflammatory mediators (IL-6, IL-8, CXCL10, TNF-α), and recruitment of neutrophils occurs in response to inflammation (Fig. 1). Baricitinib, an antiviral, an anticytokine, and a Janus kinase/signal transducer and activator of transcription (STAT) inhibitor blocks viral entry of SARS-CoV-2 and prevents the type I, interferon gamma (IFN-γ) mediated increase in expression of ACE2, the receptor for SARS-CoV-2.13 SARS-CoV-2-mediated neutrophil extracellular trap (NET) formation was proposed to be responsible for COVID-19 pathogenesis.14
Neutrophils and NET formation
Neutrophils, belonging to the family of polymorphonuclear leukocytes (white blood cells), are the first immune cell responders for infection. In response to bacterial, fungal, and viral infections, neutrophils activate other immune cells, engulf pathogens by phagocytosis, and destroy the invading pathogens. The respiratory, or oxidative, burst pathway activates Nox2 and generates O2˙−, using NADPH as a cofactor via a one-electron reduction of oxygen as part of their antimicrobial mechanism.15 Dismutation of O2˙− forms additional oxidants (e.g., hydrogen peroxide [H2O2]) that, in the presence of myeloperoxidase (MPO) and the chloride ion, generate hypochlorous acid (HOCl), or bleach, as a potent antibacterial agent. Other potent oxidants such as peroxynitrite (ONOO−) and hydroxyl radicals (˙OH) are formed from the reaction between O2˙− and nitric oxide (NO˙), which is formed from the nitric oxide synthase-catalyzed oxidation of L-arginine (Fig. 2).16 In 2004, it was proposed that in addition to the intracellular antimicrobial killing mechanism, neutrophils trap and kill pathogens by casting a toxic extracellular net.17,18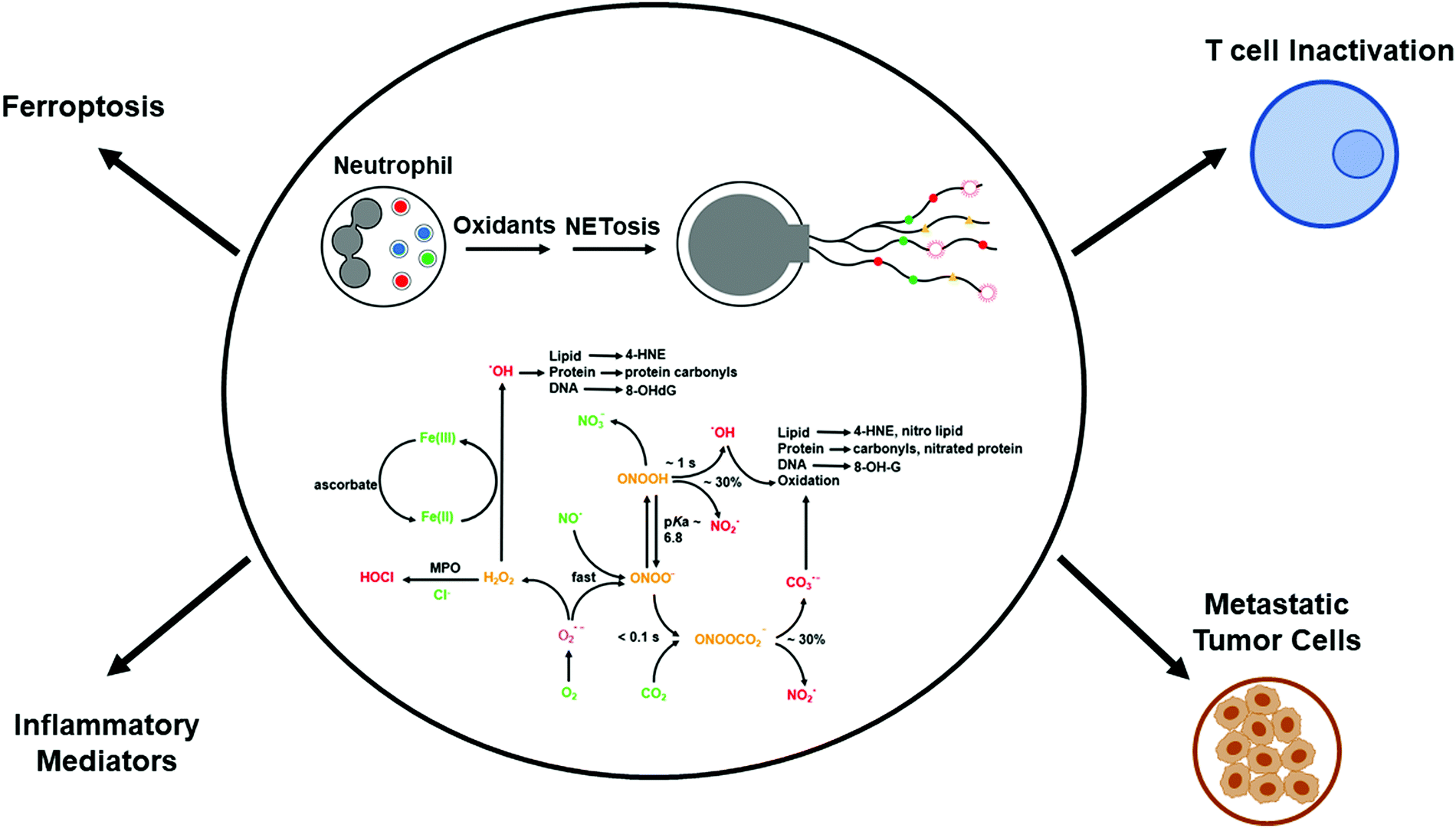 | ||
| Fig. 2 Reactive oxygen/nitrogen species formation and NETosis. Modified from Redox Biology, 37, Balaraman Kalyanaraman, Do free radical NETwork and oxidative stress disparities in African Americans enhance their vulnerability to SARS-CoV-2 infection and COVID-19 severity? 101721, Copyright (2020), with permission from Elsevier. | ||
Neutrophil extracellular traps and NETosis: a double-edged sword
NETosis is a form of cell death that is distinctly different from apoptosis or necrosis.19,20 During NETosis, neutrophils extrude DNA and antimicrobial proteins into the extracellular space, forming a net-like structure, or NETs, to trap and kill invading pathogens (similar to a spider web), including bacteria, fungi, and viruses.19–21 NETs consist of extracellular DNA decorated with histones, cytosolic proteins, and granular proteins; neutrophil elastase; and oxidative enzymes, Nox2, MPO, and nitric oxide synthase (Fig. 2).22,23 Histones are positively charged and form a nucleosome with chromatin. Neutrophil elastase, a serine protease, translocates to the nucleus and instigates NET formation. NET formation is triggered by innate immunity receptors activating Nox2 and/or mitochondria that subsequently activate MPO, neutrophil elastase, and calcium-dependent protein-arginine deiminase type 4 (PAD4). PAD4 citrullinates histones and decreases the net positive charge by converting the positively charged arginine to the neutral citrulline, promoting chromatin decondensation.24 Mutations in Nox2, which do not generate O2˙−, occur in neutrophils derived from patients with chronic granulomatous disease. NET formation is not induced under these conditions.25 Both Nox2 and MPO are implicated in NET formation in plasma isolated from patients with autoimmune conditions.26 The 8-hydroxy deoxyguanosine (8-OHdG)-enriched DNA present in NETs binds to a transmembrane protein (e.g., Cdc25) on tumor cells and facilitates their metastatic potential (Fig. 2).27,28The excessive accumulation of NETs causes damage to the host by inducing proinflammatory mechanisms.29,30 The timely removal of NETs is crucial for preventing self-antigens.31 Degradation of NETs in ischemia/reperfusion-challenged intestinal tissue with deoxyribonuclease 1 is proposed as an effective treatment against intestinal ischemia/reperfusion injury.32 Enhanced NET levels were linked to the onset of acute and chronic inflammation and autoimmune disorders.21 A decreased ability to degrade NETs is pronounced in systemic lupus erythematosus lupus patients with antiphospholipid syndrome33–36 and nephritis,31 and degradation of NETs is inhibited by the presence of autoantibodies, increasing the risk of thrombosis.
Enhanced release of NETs was reported to occur in severe cases of COVID-19.37,38 Sera from COVID-19 patients revealed elevated levels of extracellular DNA (cell-free DNA), and two specific markers of NETs: MPO-DNA and citrullinated histone H3. The levels of cell-free DNA showed a strong correlation with other markers of inflammation (e.g., C-reactive protein, D-dimer). Both cell-free DNA and MPO-DNA were much higher in patients on mechanical ventilation as compared with patients breathing room air.
Another interesting finding of this study21 is that sera from COVID-19 patients triggered NET release from control neutrophils in vitro, indicating that circulating NETs could be used to predict the extent of disease severity. NETs stimulated immunothrombosis in COVID-19 patients and were suggested as suitable therapeutic targets to mitigate prothrombotic complications in COVID-19 patients.39
The interaction of SARS-CoV-2 with hemoglobin causes dysregulated iron metabolism and iron-mediated oxidative stress and hyperinflammation. Ferroptosis—regulated cell death by iron—is initiated by iron- and oxidant-induced lipid peroxidation (Fig. 2).40 Oxidized phospholipid accelerates NET formation.41–45 Deferoxamine, a widely used iron chelator to mitigate iron toxicity, is currently undergoing a clinical trial to determine if it mitigates inflammation in COVID-19 patients.46 Selenium is essential for the effective functioning of glutathione peroxidase (GPx) enzymes, which are critical for detoxifying lipid and phospholipid hydroperoxides.47 Either chelation of redox active iron and/or enhancing the GPx/phospholipid glutathione peroxidase (PHGPx) activity is considered to be therapeutically significant for mitigation of COVID-19-induced inflammation.48 Ebselen, which is a small-molecule therapeutic that has been used as a GPx/PHGPX peroxidase mimetic49 and an inhibitor of Nox enzymes,50 can be used to mitigate NET-induced and hydroperoxide-mediated inflammation. Melatonin, N-acetyl-5-methoxytryptamine, a brain hormone that regulates the circadian rhythm and is activated in the dark (often referred to as an antioxidant), inhibits hemolysis induced by SARS-CoV-2 binding to red blood cells.51 Melatonin was shown to inhibit ferroptosis induced by high glucose.52 Whether melatonin inhibits ferroptosis in COVID-19 remains to be determined. N-Acetylcysteine is undergoing clinical trials for treating COVID-19.53N-Acetylcysteine enhances intracellular GSH levels and its protective mechanism is not due to scavenging of ROS.
In cancer, the opposite scenario occurs. Inhibition of seleno cysteine-containing peroxidase enzymes, which detoxify lipid peroxides and prevent iron-catalyzed reactions, induces ferroptotic cell death.54–56 A characteristic feature in ferroptosis-therapy resistant cancer cells is the downregulated activity of GPx and PHGPx.57,58 Drugs that inhibit GPx expression and activity (e.g., RSL3) potentiate ferroptosis-inducing therapy in resistant cancer cells.57,59
NET, inflammation, and the cytokine storm
During the early stages of the COVID-19 pandemic, many patients on ventilators developed respiratory illness and died. More recently, due to the timely use of anti-inflammatory steroids (e.g., dexamethasone), respiratory illness-related deaths have dramatically decreased. COVID-19 patients admitted into intensive care showed elevated levels of cytokines as compared with those who were not admitted. Dexamethasone, an FDA-approved immunosuppressive steroid that has been used in the clinic for more than 50 years, prevented mortality in seriously ill COVID-19 patients.60 Following SARS-CoV-2 infection, the immune system is activated through recruitment of T cells, macrophages, and dendritic cells. These immune cells secrete molecules, cytokines, and chemokines to combat the viral infection. The “cytokine storm” (also known as the “inflammation storm”) refers to an uncontrolled, hyper-activation of immune cells, proinflammatory cytokines (IL-6, IL-8, IFN-γ, TNF-α), and chemokines (CXCL10). NETs induce proinflammatory cytokines that further stimulate NETs and cause a “feedback” cascading effect.61 Drugs decreasing the NET levels have been shown to prevent COVID-19-associated cytokine release.62However, recent findings suggest that the hyperinflammatory cytokine storm is not solely responsible for the morbidity and mortality in COVID-19 patients.63
Cancer and COVID-19: overlapping biology and potential therapies
A recent report reveals promising therapeutic approaches for treating COVID-19 that originate from advances made in cancer research.64 The mechanistic insights on the leukocyte biology of the tumor microenvironment (TME) gained from decades of cancer research may become very relevant to COVID-19 treatment. Fig. 3 (modified from Bakouny et al.64) illustrates the interconnected biology and ROS involvement between cancer and COVID-19. Recruitment of myeloid-derived suppressor cells (MDSCs) into tumors triggers immunosuppression in cancers. The antitumor immunity was enhanced in lung cancer by targeting myeloid-derived suppressive pathways.65 Inhibiting MDSC trafficking was reported to enhance T cell-based immunotherapeutic efficacy (Fig. 4).66 Drugs (SX-682) suppressing MDSCs66–68 and entinostat, an orally available histone deacetylase inhibitor,69 reenergize T cells in the TME. A similar approach is plausible for reenergizing the host cell response and mitigating proinflammatory mechanisms in COVID-19.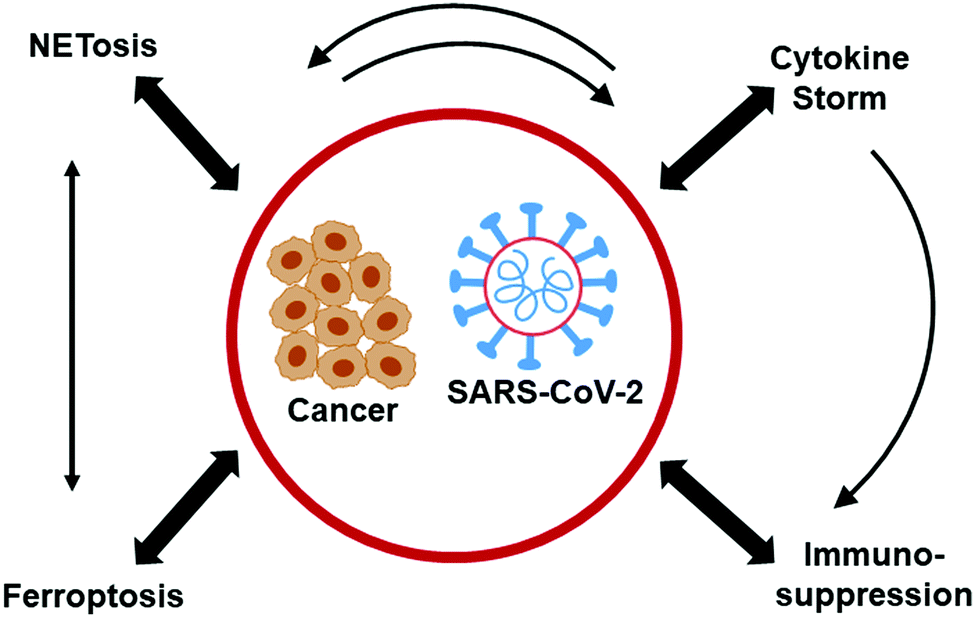 | ||
| Fig. 3 Similarities in proinflammatory mechanisms between cancer and COVID-19. Modified from Cancer Cell, 38, Ziad Bakouny, Jessica Hawley, Toni Choueiri, Solange Peters, Brian Rini, Jermy Warner, Corrie Painter, COVID-19 and Cancer: Current Challenges and Perspectives, 629–646, Copyright (2020), with permission from Elsevier. | ||
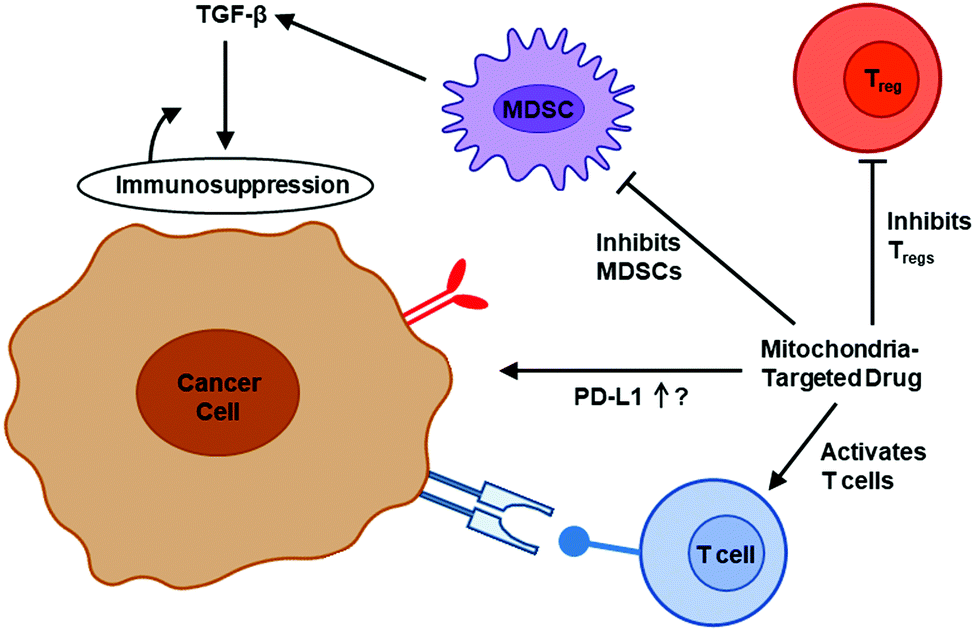 | ||
| Fig. 4 The effect of mitochondria-targeted agents on immune cells in the TME. Modified from an image created by Margaret Gatti-Mays, MD, that appeared in the Dec. 29, 2020, National Cancer Institute's Center for Cancer Research news article, New Clinical Trial Studies Immunotherapy Combination for Metastatic Breast Cancer, with permission from the National Cancer Institute. | ||
Enhanced NET formation compromises the adaptive immune system. T cells necessary for the killing of SARS-CoV-2-infected cells are suppressed by mediators released from activation of macrophages and monocytes. In the TME, the T cell activation is suppressed by MDSCs.70 Thus, agents inhibiting MDSCs could increase the activity of cytotoxic T cells.
There are mechanistic similarities between the mechanisms responsible for toxicity related to cancer immunotherapy and the release of proinflammatory cytokines, TNF-α, IL-1, IL-6, IL-8 (i.e., the COVID-19 cytokine storm) (Fig. 3).71,72 As previously indicated, the proinflammatory cytokine, IL-6, was shown to promote NETosis in the early phase of severe COVID-19.73 Tocilizumab, an anti-IL-6 receptor antibody used to counteract cytokine release during immunotherapy, was tested with little success in COVID-19 patients. Acalabrutinib, a selective Bruton tyrosine kinase, used in the treatment of lymphoid cancers, has been repurposed to treat COVID-19 patients with some success with regard to improvement in oxygen requirements and decrease in inflammatory markers (e.g., IL-6).74
In the RECOVERY trial, dexamethasone was discovered as the drug of choice to treat severely ill COVID-19 patients. Dexamethasone inhibits proinflammatory cytokine production by shifting toll-like receptor 4 (TLR4) signaling away from NF-κB-driven proinflammatory responses. Although the exact mechanism by which dexamethasone mitigates inflammation in severe COVID-19 patients is unknown, results from preclinical disease models suggest that dexamethasone inhibits NET formation and NET-induced TLR4 signalling.75
Recent prostate cancer research has provided additional insights for COVID-19 treatment.64,76 TMPRSS2 is highly expressed in prostate as well as lung tissue. TMPRSS2 expression is modulated by the androgen receptor in normal prostate tissue and in prostate cancer.77 Although male COVID-19 patients have worse outcomes than their female counterparts, COVID-19 prostate cancer patients who underwent androgen deprivation therapy required less hospitalization and oxygen supplementation. Randomized clinical trials are underway using androgen receptor antagonists to treat COVID-19 patients.
A cyclic depsipeptide, plitidepsin (aplidin), approved to treat multiple myeloma in Australia, was shown to be 25-fold more effective than remdesivir in inhibiting replication of SARS-CoV-2.78 This drug exerts a cytostatic not cytotoxic mechanism of action in the in vitro cell culture system.78 Dexamethasone, the anti-inflammatory drug used in COVID-19 patients, also was used to treat multiple myeloma.
NET, cancer metastasis, and immunosuppression
Circulating tumors secrete chemokine receptor antagonists, CXCR1 and CXCR2, inducing formation of NETs that interfere with immune cytotoxicity.79 Inhibition of NETosis sensitized tumors to immunotherapy with checkpoint blockade (programmed cell death protein 1 [PD-L1] and cytotoxic T-lymphocyte-associated protein 4). NETs coat the tumor cells, protecting against tumor cell-targeted cytotoxic lymphocytes and/or natural killer cells.80 Treating with a small molecule inhibitor of NETosis, GSK 484, decreased tumor micrometastases. GSK 484 hydrochloride is a selective and reversible inhibitor of PAD4, and it blocks the citrullination of PAD4 target proteins in human neutrophils. Removal of NETs by treating with deoxyribonuclease 1 or pharmacological inhibitors inhibited tumor-induced inflammation and metastasis. The use of GSK 484 in mitigating NET-mediated inflammation in COVID-19 may be a viable and alternate anti-inflammatory therapeutic approach.Increased levels of NETs were identified in metastatic lesions.27,28 A recent study showed that NETosis and enhanced NET formation in distant organs preceded cancer metastasis. This finding raises the possibility that NETs in blood could be used as a predictive biomarker of metastasis.27 8-OHdG, a characteristic hallmark of NET-DNA, is an extracellular DNA sensor. The interaction between the transmembrane protein Cdc25 and NET-DNA supports the proliferation of metastatic cells.27,28 Other markers of NETosis and serum NET levels (e.g., MPO-DNA) were found to be higher in breast cancer patients with liver metastases.27 Targeting Cdc25 and mitigating NETs in cancer patients may be an effective therapeutic approach to prevent cancer metastasis.27,28 Targeting NETosis in the tumor immune microenvironment was proposed as a promising anti-metastatic strategy.27,28,81 The NET-DNA complex acts as a chemotactic factor and attracts metastatic cells to new sites. The transmembrane protein, Cdc25, acts as a NET-DNA receptor on cancer cells and senses extracellular DNA, activating pathways and enhancing cellular motility27 NET-mediated metastasis was abrogated in Cdc25-knockout cells.
Neutrophils constitute a major fraction of the inflammatory cells in the TME.82 Increased NET formation associated with tumor portends poor prognosis in cancer patients. Increasing evidence suggests that neutrophils promote tumor growth and metastatic progression via the formation of NETs.28,83 Surprisingly, the proposed mechanism involves NET stimulation of mitochondrial biogenesis and bioenergetics in tumor cells through induction and activation of the TLR and peroxisome proliferator-activated receptor-gamma coactivator (PGC-1α), a key regulator of cellular energy metabolism.84 Clearly, suppression of NET is a therapeutic strategy for mitigating tumor growth and metastasis.
Role of mitochondria in NET: repurposing OXPHOS-targeted agents as anti-inflammatory and immunosuppressive drugs in cancer and COVID-19
SARS-CoV-2 enters human cells by interacting with ACE2 receptors and causes a subsequent decrease in ACE2 levels and impaired mitochondrial function. Emerging research connects NETs and mitochondria in cancer and COVID-19;84–86 therefore, a promising approach is to repurpose mitochondrial drugs discovered in cancer to prevent COVID-1987 or mitigate its severity. Mitochondria-targeting drugs exhibit antiproliferative and antiviral effects.88 Recent reports suggest that after entering cells, SARS-CoV-2 RNA and proteins sequester into mitochondria, are able to “hijack” mitochondrial DNA (mtDNA), and are responsible for mitochondrial dysfunction and systemic inflammation by mtDNA.89 SARS-CoV-2, similar to NETosis, induces the release of mtDNA from inside the cell.90Previous reports suggest that inhibitors of mitochondrial respiratory chain complexes decrease neutrophil activation and oxidative burst.91,92 Rotenone and metformin, which are mitochondrial complex I inhibitors, significantly inhibited the recruitment of neutrophils in a lipopolysaccharide-induced lung inflammation mouse model.93 Using a neutrophil-specific knockout zebrafish model, the first in vivo evidence was provided for mitochondrial regulation of neutrophil function.94 Proper functioning of the mitochondrial electron transport chain and maintenance of mitochondrial membrane potential are required for neutrophil motility and chemotaxis to occur.94 Perturbation of mitochondrial function was shown to greatly decrease the antimicrobial potency of inflammatory neutrophils,92 although the role of mitochondria in the neutrophil-induced oxidative burst still remains debatable.95,96
Mitochondria-targeted drugs decreased Nox2 levels, oxidative damage, and inflammation in a mouse model of Parkinson's disease.97 Mitochondria-targeted apocynin, a nonspecific inhibitor of Nox2, attenuated Nox2 and oxidative/nitrative modification of proteins and inflammation in microglia.97 Mito-Q (i.e., co-enzyme Q conjugated to the triphenylphosphonium [TPP+]) moiety, decreased NET formation in a lupus mouse model.98,99 Mito-Q treatment of SARS-CoV-2-infected monocytes from diabetic patients decreased viral replication and cytokine upregulation.100 The exact mechanism by which mitochondria-targeted drugs inhibit NET formation is not known; however, reports suggest that these drugs (e.g., metformin) inhibit protein kinase C beta II translocation to the plasma membrane, preventing the activation of Nox and NET component expulsion (DNA–protein complex) to the extracellular space.101 This is proposed as a plausible mechanism by which metformin inhibits adhesion of cancer cells to NET and metastasis. Selected drugs that target mitochondria and inhibit Nox2 activity and inflammation are shown in Fig. 5. These and related drugs are possible candidates for mitigation of NETs.102 IACS-010759, which was shown to inhibit OXPHOS complex I, also inhibited cancer metastases.103–106 The proposed mechanism involved inhibition of MDSCs in metastatic tumor cells.
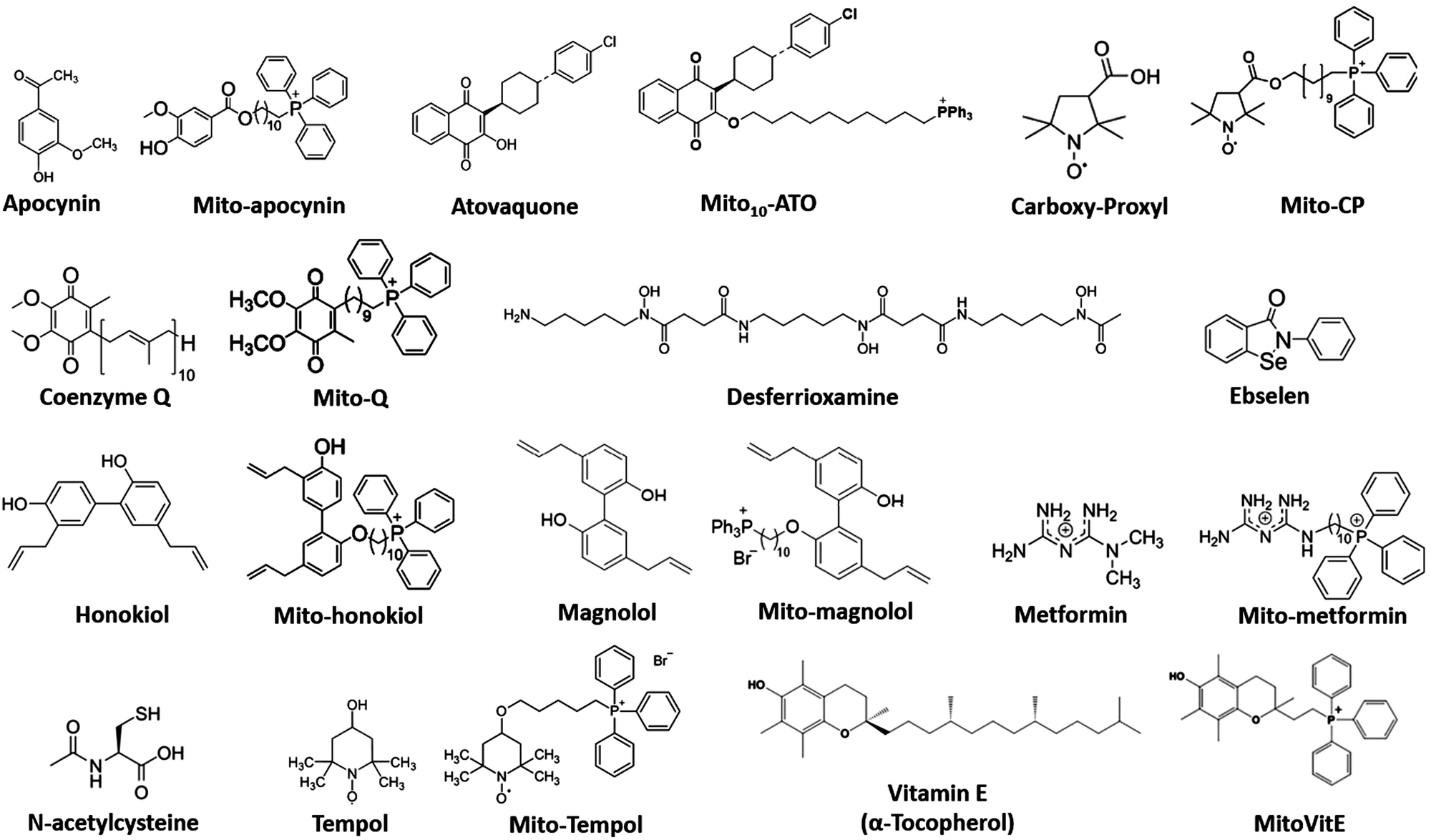 | ||
| Fig. 5 Structures of selected agents used in cancer and COVID-19 research. | ||
Selective inhibition of mitochondrial complex III was reported to activate the function of effector T cells (Teffs) through mitigation of immunosuppression by regulatory T cells (Tregs) (Fig. 4). Tregs are increased in the TME of most cancers.107,108 The survival of cancer patients is lower in part due to increased presence of Tregs, which hamper immunotherapy. Drug therapy targeting Tregs is emerging as a promising antitumor approach.109 Emerging research suggests that augmenting T cell function (and decreasing T cell exhaustion) is effective in inhibiting the COVID-19 variants.110 New findings from melanoma research indicate that mitochondria-targeting drugs can boost T cell activity and enhance immunotherapy.111
Recent research revealed that mitochondrial complex III activity is critical for preserving the suppressive function of Tregs.112 Tregs with decreased complex III activity exhibit decreased immune function. There are only a handful of mitochondrial complex III inhibitors (e.g., antimycin A, atovaquone, arsenic). Atovaquone is an FDA-approved antimalarial drug that is targeted to mitochondrial complex III. Atovaquone is currently undergoing Phase 2 clinical trial for treatment of COVID-19.113 This is a randomized, double-blind investigation of atovaquone in adults hospitalized with COVID-19.
Recently, a new class of mitochondria-targeted complex I and complex III inhibitors was developed through conjugation of the hydroxyl group in atovaquone with TPP+ containing an aliphatic side chain.114 These modified atovaquone derivatives are called Mito-atovaquone (Mito-ATO) (Fig. 5). Using different sidechain lengths, several Mito-ATO derivatives were synthesized (Mito4-ATO, Mito10-ATO, Mito12-ATO, Mito16-ATO, etc.). Mito12-ATO strongly inhibited both complex I- and complex III-stimulated mitochondrial oxygen consumption. The longer-chain Mito-ATO (Mito12-ATO, Mito16-ATO) inhibited only complex I- but not complex III-induced oxygen consumption. Mito10-ATO inhibited Tregs and stimulated Teffs. The longer-chain Mito-ATOs devoid of complex III inhibitory activity did not have any effect on Teffs or Tregs. Mito10-ATO also inhibited the function of MDSCs. Recent research indicates that a mitochondria-targeted polyphenolic compound activated the infiltration of cytotoxic T cells in the TME, stimulating its antitumor function in a cancer mouse xenograft model.115
Mito-honokiol inhibits the brain metastasis of lung cancer in mice xenografts.116 Mito-honokiol downregulates cytoplasmic and mitochondrial STAT3 phosphorylation.116 STAT3 has been identified as a promising immunotherapeutic target for anticancer drugs. Inhibition of STAT3 phosphorylation has been reported to inhibit MDSCs, inflammatory cytokines, immunosuppression, and tumor metastasis.117 Targeting and inhibiting MDSCs with phytochemicals may prevent SARS-CoV-2-induced MDSCs and inhibit immunosuppressive mechanisms in COVID-19.118,119
Use of 2-deoxy-D-glucose as an anti-COVID-19 drug
Recently, in a limited clinical trial consisting of 200–300 moderately and severely ill COVID-19 patients in India, 2-deoxy-D-glucose (2-DG) was found to be effective in reducing the viral load and decreasing the patients’ dependency on supplemental oxygen therapy and the extent of hospitalization. This was attributed to enhanced accumulation of 2-DG in infected lung cells, which resulted in decreased glycolysis and energy production in infected lung cells and inhibition of viral replication. Undoubtedly, this limited success needs to be substantiated with more extensive and rigorous phase 2–3 trials on the therapeutic efficacy of 2-DG as an anti-COVID-19 drug.The aerobic dependency of cancer cells on glycolysis (the Warburg effect) has long stimulated research on the use of 2-DG as an anticancer drug. 2-DG has been used alone and in combination with other standard-of-care cancer therapeutics in numerous clinical trials. Because of neurotoxicity, 2-DG was not approved by FDA in cancer treatment. Like cancer cells, highly proliferating neuronal cells take up 2-DG, resulting in decreased glycolysis and energy production in neuronal cells. Reports also indicate that 2-DG in combination with mitochondria-targeted OXPHOS inhibitors synergistically enhance the antitumor effects of 2-DG.120 This may be significant in COVID-19 research because metformin, a weak mitochondria-targeting drug, has been used in COVID-19 patients. Thus, it is important to understand the antiviral mechanisms and alterations in metabolic reprogramming of immune cells in the presence of 2-DG alone and in combination with metformin and other OXPHOS inhibitors.121 It is also important to fully understand the adverse effects of long-term and short-term administration of 2-DG.122
Recent research showed that elevated glucose and glycolysis enhance SARS-CoV-2 replication and cytokine production in monocytes through a mechanistic pathway involving mitochondrial ROS, hypoxia-inducible factor 1-alpha (HIF-1α) stabilization, stimulation of glycolytic genes, cytokine production, T cell dysfunction, and lung epithelial cell death.123 HIF-1α, a master regulator of glycolysis and glycolytic genes, was induced in SARS-CoV-2–infected monocytes, leading to elevated glycolysis. The virus caused the metabolic remodelling in lung cells to aerobic glycolysis to provide energy and enable its rapid replication.100,101 2-DG inhibits glycolysis in virus-infected cells and induction of proinflammatory cytokines, leading to restoration of T cell function. Pretreatment with Mito-Q or N-acetylcysteine inhibited SARS-CoV-2 replication and cytokine.100 The only limitation of this study is that Mito-SOX oxidation cannot be equated to mitochondrial O2˙−, as O2˙− alone does not react with Mito-SOX to form the red-fluorescent product. Thus, the involvement of mitochondrial O2˙− in the stabilization of HIF-1α has not been demonstrated. The mechanisms by which Mito-Q and N-acetylcysteine inhibit SARS-CoV-2 may involve other mechanisms, including an increase in intracellular GSH. This study is significant in explaining why diabetics with uncontrolled blood glucose levels have a greater risk for developing severe to deadly COVID-19.100,101
Long COVID (or long-hauler syndrome)
The common symptoms of COVID-19 are fever or chills, shortness of breath, fatigue, muscle or body aches, headache, dry cough, loss of taste and smell, sore throat, congestion or runny nose, nausea or vomiting, and diarrhea. Individuals experiencing long COVID, or long-haulers, are increasing in numbers. Nearly 10% of surviving COVID-19 patients experience cardiovascular or neurological complications. It is critically important to develop therapies for persistent symptoms (memory fog, loss of smell, and digestive and heart problems) and to reduce cardiovascular inflammation in long COVID patients. Prophylactic treatment may include supplementation with N-acetylcysteine, GSH, CuraMed (a curcumin supplement), and omega-3. Both weakly and strongly targeted mitochondrial drugs (e.g., metformin, honokiol, Mito-Q, Mito-magnolol, Mito-apocynin) (Fig. 5) have been shown to inhibit immunosuppressive cells and inflammation.97 Because these drugs are relatively nontoxic and do not suppress the immune system like the conventional steroidal drugs, they can be used prophylactically in a preventative setting. Clearly, using mitochondria-targeted drugs and other known immune boosters to enhance mitochondrial fitness and the immune system of host cells infected with SARS-CoV-2124 may provide protection against or lessen the severity of COVID-19.124 Female COVID-19 patients, as compared with male COVID-19 patients, elicit much higher T cell activation.125 Male COVID-19 patients have higher plasma levels of innate immune cytokines (e.g., IL-8).125 Drugs selectively enhancing mitochondrial biogenesis (resveratrol, quercetin) and mitochondrial function should be tested in clinical trials for COVID-19 treatment. Honokiol, an active anti-inflammatory component extracted from the bark of the Magnolia officinalis has been shown to improve mitochondrial function via activation of sirtuin 3.126 Phytotherapy with plant-derived polyphenolics was suggested as a plausible method to mitigate SARS-CoV-2-induced oxidative stress and inflammation.127 Combining mitochondria-targeted drugs with iron chelators128 is another approach to more effectively mitigating inflammatory mechanisms.Tempol, a new oral antiviral drug?
Emerging research from the National Institutes of Health has revealed that a five-membered nitroxide, Tempol, can decrease COVID-19 infections by inhibiting the RNA-dependent RNA polymerase and blocking the replication of the virus inside the host cells.129 Previous reports showed that Tempol was able to oxidize and disassemble the iron–sulfur (Fe–S) cluster of cytosolic aconitase.130 The Fe–S cluster present in the catalytic subunit of the RNA-dependent RNA polymerase is a cofactor of the enzyme that is responsible for the replication of the SARS-CoV-2.129 Fe–S clusters are sensitive to oxidative degradation,131 blocking the viral replication. Tempol did not affect the activity of other Fe-S enzymes, mitochondrial respiratory enzymes, or mitochondrial aconitase at higher concentrations of Tempol. Tempol (the nitroxide moiety) directly reacts with the Fe–S cluster. This is similar to the reaction between nitric oxide, released from the nitric oxide donor molecules, and the Fe–S cluster. Tempol and remdesivir synergistically inhibited the activity of the RNA polymerase and blocked the replication of the SARS-CoV-2.129 Pioneering research from the National Institutes of Health has previously shown that Tempol has low cytotoxicity and high tissue permeability and exerts an anticancer effect.130,132,133A Phase 2/3, adaptive, randomized, placebo-controlled clinical trial (NCT04729595) was started to examine the effects of Tempol (MBM-02) in preventing COVID-19-related hospitalization in subjects with COVID-19 infection. The estimated completion date of the trial is early 2022.
Although the pro-oxidant function of Tempol is responsible for the inhibition of viral replication, the antioxidant, anti-inflammatory, and anti-coagulant mechanisms of Tempol have been shown to decrease pro-inflammatory cytokines (e.g., TNFα, IL-6). Tempol has a superoxide dismutase-mimetic property that decreases intracellular O2˙− levels.134 If the clinical trial is successful, there is a distinct possibility that Tempol may be considered as a therapeutic treatment for COVID-19.
Other antioxidative mechanisms
Other antioxidative mechanisms include the major pathways regulating cellular oxidant balance is the transcription factor, Nrf2.135,136 Nrf2 regulates the expression of antioxidant proteins that protect against oxidative damage induced by inflammation. Several FDA-approved drugs (ursodiol, dimethyl fumarate) and natural compounds (sulforaphane, curcumin, resveratrol, quercetin) are known to activate Nrf2.137 Reports indicate that Nrf2 activation may significantly decrease the intensity of the cytokine storm in COVID-19.138,139 Recent reports suggest that the Nrf2 activators may be a potential therapeutic for COVID-19.138,140Conclusions
In this review, I have attempted to highlight the overlapping oxidative biology (ROS formation, NETosis, ferroptosis, induction of proinflammatory mediators, role of mitochondrial oxidative phosphorylation [OXPHOS], immunosuppression, and immune activation) between cancer and late-stage COVID-19. NET-dependent mechanisms promote the invasive and proliferative properties of tumors. Several drugs targeted to OXPHOS potently inhibit immunosuppressive cells (MDSCs and suppressive neutrophils) and are undergoing clinical trials for treatment of metastatic cancers. Given the overlapping oxidative and proinflammatory and immunosuppressive mechanisms in cancer and COVID-19, promising drugs discovered from decades of clinical cancer research that are already in clinical trials or in consideration for clinical trials (e.g., Nrf2 activators, NF-kappaB inhibitors, iron chelators, classic antioxidants inhibiting NET formation, specific inhibitors of proinflammatory cytokines, mitochondria-targeted AMPK activators) may provide new insights into mitigating inflammation in survivors of COVID-19.Author contributions
B. K. prepared, wrote, and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to thank all my collaborators for insightful applications of mitochondria-targeted drugs in cancer, neurological, and cardiovascular science research. Also, thanks to Lydia Washechek for editorial assistance and for preparing the manuscript including the figures. This work was supported in part by the Harry R. and Angeline E. Quadracci Professor in Parkinson’s Research Endowment.References
- J. Beltrán-García, R. Osca-Verdegal, F. V. Pallardó, J. Ferreres, M. Rodríguez, S. Mulet, F. Sanchis-Gomar, N. Carbonell and J. L. García-Giménez, Antioxidants, 2020, 9 Search PubMed.
- M. Laforge, C. Elbim, C. Frère, M. Hémadi, C. Massaad, P. Nuss, J. J. Benoliel and C. Becker, Nat. Rev. Immunol., 2020, 20, 515–516 CrossRef CAS PubMed.
- R. Cecchini and A. L. Cecchini, Med. Hypotheses, 2020, 143, 110102 CrossRef CAS PubMed.
- B. Kalyanaraman, Redox Biol., 2020, 37, 101721 CrossRef CAS PubMed.
- I. Hamming, W. Timens, M. L. Bulthuis, A. T. Lely, G. Navis and H. van Goor, J. Pathol., 2004, 203, 631–637 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280 CrossRef CAS PubMed.
- A. M. Papinska, M. Soto, C. J. Meeks and K. E. Rodgers, Pharmacol. Res., 2016, 107, 372–380 CrossRef CAS PubMed.
- S. Derouiche, J. Infect. Dis. Epidemiol., 2020, 6, 121 Search PubMed.
- M. Gheblawi, K. Wang, A. Viveiros, Q. Nguyen, J. C. Zhong, A. J. Turner, M. K. Raizada, M. B. Grant and G. Y. Oudit, Circ. Res., 2020, 126, 1456–1474 CrossRef CAS PubMed.
- J. Wysocki, D. I. Ortiz-Melo, N. K. Mattocks, K. Xu, J. Prescott, K. Evora, M. Ye, M. A. Sparks, S. K. Haque, D. Batlle and S. B. Gurley, Physiol. Rep., 2014, 2, e00264 CrossRef PubMed.
- L. A. Rabelo, N. Alenina and M. Bader, Hypertens. Res., 2011, 34, 154–160 CrossRef CAS PubMed.
- S. Ozono, Y. Zhang, H. Ode, K. Sano, T. S. Tan, K. Imai, K. Miyoshi, S. Kishigami, T. Ueno, Y. Iwatani, T. Suzuki and K. Tokunaga, Nat. Commun., 2021, 12, 848 CrossRef CAS PubMed.
- J. Stebbing, G. Sánchez Nievas, M. Falcone, S. Youhanna, P. Richardson, S. Ottaviani, J. X. Shen, C. Sommerauer, G. Tiseo, L. Ghiadoni, A. Virdis, F. Monzani, L. R. Rizos, F. Forfori, A. Avendaño-Céspedes, S. De Marco, L. Carrozzi, F. Lena, P. M. Sánchez-Jurado, L. G. Lacerenza, N. Cesira, D. Caldevilla-Bernardo, A. Perrella, L. Niccoli, L. S. Méndez, D. Matarrese, D. Goletti, Y. J. Tan, V. Monteil, G. Dranitsaris, F. Cantini, A. Farcomeni, S. Dutta, S. K. Burley, H. Zhang, M. Pistello, W. Li, M. M. Romero, F. Andrés Pretel, R. S. Simón-Talero, R. García-Molina, C. Kutter, J. H. Felce, Z. F. Nizami, A. G. Miklosi, J. M. Penninger, F. Menichetti, A. Mirazimi, P. Abizanda and V. M. Lauschke, Sci. Adv., 2021, 7(1), eabe4724 CrossRef CAS PubMed.
- F. P. Veras, M. C. Pontelli, C. M. Silva, J. E. Toller-Kawahisa, M. de Lima, D. C. Nascimento, A. H. Schneider, D. Caetité, L. A. Tavares, I. M. Paiva, R. Rosales, D. Colón, R. Martins, I. A. Castro, G. M. Almeida, M. I. F. Lopes, M. N. Benatti, L. P. Bonjorno, M. C. Giannini, R. Luppino-Assad, S. L. Almeida, F. Vilar, R. Santana, V. R. Bollela, M. Auxiliadora-Martins, M. Borges, C. H. Miranda, A. Pazin-Filho, L. L. P. da Silva, L. D. Cunha, D. S. Zamboni, F. Dal-Pizzol, L. O. Leiria, L. Siyuan, S. Batah, A. Fabro, T. Mauad, M. Dolhnikoff, A. Duarte-Neto, P. Saldiva, T. M. Cunha, J. C. Alves-Filho, E. Arruda, P. Louzada-Junior, R. D. Oliveira and F. Q. Cunha, J. Exp. Med., 2020, 217(12), e20201129 CrossRef CAS PubMed.
- B. M. Babior, N. Engl. J. Med., 1978, 298, 659–668 CrossRef CAS PubMed.
- J. S. Beckman, T. W. Beckman, J. Chen, P. A. Marshall and B. A. Freeman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1620–1624 CrossRef CAS.
- V. Brinkmann, U. Reichard, C. Goosmann, B. Fauler, Y. Uhlemann, D. S. Weiss, Y. Weinrauch and A. Zychlinsky, Science, 2004, 303, 1532–1535 CrossRef CAS PubMed.
- V. Brinkmann and A. Zychlinsky, Nat. Rev. Microbiol., 2007, 5, 577–582 CrossRef CAS PubMed.
- B. G. Yipp and P. Kubes, Blood, 2013, 122, 2784–2794 CrossRef CAS PubMed.
- N. Sorvillo, D. Cherpokova, K. Martinod and D. D. Wagner, Circ. Res., 2019, 125, 470–488 CrossRef CAS PubMed.
- V. Delgado-Rizo, M. A. Martínez-Guzmán, L. Iñiguez-Gutierrez, A. García-Orozco, A. Alvarado-Navarro and M. Fafutis-Morris, Front. Immunol., 2017, 8, 81 Search PubMed.
- V. Papayannopoulos, K. D. Metzler, A. Hakkim and A. Zychlinsky, J. Cell Biol., 2010, 191, 677–691 CrossRef CAS PubMed.
- D. Dąbrowska, E. Jabłońska, M. Garley, W. Ratajczak-Wrona and A. Iwaniuk, Scand. J. Immunol., 2016, 84, 317–322 CrossRef.
- I. Neeli, S. N. Khan and M. Radic, J. Immunol., 2008, 180, 1895–1902 CrossRef CAS PubMed.
- G. P. Hule, U. A. Bargir, M. Kulkarni, P. Kambli, P. Taur, M. Desai and M. R. Madkaikar, Front. Immunol., 2019, 10, 1739 CrossRef CAS PubMed.
- J. McHugh, Nat. Rev. Rheumatol., 2018, 14, 560 CrossRef PubMed.
- L. Yang, Q. Liu, X. Zhang, X. Liu, B. Zhou, J. Chen, D. Huang, J. Li, H. Li, F. Chen, J. Liu, Y. Xing, X. Chen, S. Su and E. Song, Nature, 2020, 583, 133–138 CrossRef CAS PubMed.
- R. F. Rayes, J. G. Mouhanna, I. Nicolau, F. Bourdeau, B. Giannias, S. Rousseau, D. Quail, L. Walsh, V. Sangwan, N. Bertos, J. Cools-Lartigue, L. E. Ferri and J. D. Spicer, JCI Insight, 2019, 5, e128008 CrossRef PubMed.
- V. Papayannopoulos, Nat. Rev. Immunol., 2018, 18, 134–147 CrossRef CAS PubMed.
- M. J. Kaplan and M. Radic, J. Immunol., 2012, 189, 2689–2695 CrossRef CAS PubMed.
- A. Hakkim, B. G. Fürnrohr, K. Amann, B. Laube, U. A. Abed, V. Brinkmann, M. Herrmann, R. E. Voll and A. Zychlinsky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9813–9818 CrossRef CAS PubMed.
- S. Wang, T. Xie, S. Sun, K. Wang, B. Liu, X. Wu and W. Ding, Sci. Rep., 2018, 8, 17788 CrossRef CAS PubMed.
- J. Leffler, L. Stojanovich, Y. Shoenfeld, G. Bogdanovic, R. Hesselstrand and A. M. Blom, Clin. Exp. Rheumatol., 2014, 32, 66–70 Search PubMed.
- M. Bruschi, A. Bonanni, A. Petretto, A. Vaglio, F. Pratesi, L. Santucci, P. Migliorini, R. Bertelli, M. Galetti, S. Belletti, L. Cavagna, G. Moroni, F. Franceschini, M. Fredi, G. Pazzola, L. Allegri, R. A. Sinico, G. Pesce, M. Bagnasco, A. Manfredi, G. A. Ramirez, P. Ramoino, P. Bianchini, F. Puppo, F. Pupo, S. Negrini, F. Mattana, G. Emmi, G. Garibotto, D. Santoro, F. Scolari, A. Ravelli, A. Tincani, P. Cravedi, S. Volpi, G. Candiano and G. M. Ghiggeri, J. Rheumatol., 2020, 47, 377–386 CrossRef CAS PubMed.
- R. Salemme, L. N. Peralta, S. H. Meka, N. Pushpanathan and J. J. Alexander, J. Cell Immunol., 2019, 1, 33–42 Search PubMed.
- J. McHugh, Nat. Rev. Rheumatol., 2019, 15, 512 Search PubMed.
- Y. Zuo, S. Yalavarthi, H. Shi, K. Gockman, M. Zuo, J. A. Madison, C. Blair, A. Weber, B. J. Barnes, M. Egeblad, R. J. Woods, Y. Kanthi and J. S. Knight, JCI Insight, 2020, 5, e138999 Search PubMed.
- C. Mozzini and D. Girelli, Thromb. Res., 2020, 191, 26–27 CrossRef CAS.
- E. A. Middleton, X. Y. He, F. Denorme, R. A. Campbell, D. Ng, S. P. Salvatore, M. Mostyka, A. Baxter-Stoltzfus, A. C. Borczuk, M. Loda, M. J. Cody, B. K. Manne, I. Portier, E. Harris, A. C. Petrey, E. J. Beswick, A. F. Caulin, A. Iovino, L. M. Abegglen, A. S. Weyrich, M. T. Rondina, M. Egeblad, J. D. Schiffman and C. C. Yost, Blood, 2020, 136(10), 1169–1179 CrossRef CAS PubMed.
- H. Bayır, T. S. Anthonymuthu, Y. Y. Tyurina, S. J. Patel, A. A. Amoscato, A. M. Lamade, Q. Yang, G. K. Vladimirov, C. C. Philpott and V. E. Kagan, Cell Chem. Biol., 2020, 27, 387–408 CrossRef.
- S. Yotsumoto, Y. Muroi, T. Chiba, R. Ohmura, M. Yoneyama, M. Magarisawa, K. Dodo, N. Terayama, M. Sodeoka, R. Aoyagi, M. Arita, S. Arakawa, S. Shimizu and M. Tanaka, Sci. Rep., 2017, 7, 16026 CrossRef.
- M. Sato, R. Kusumi, S. Hamashima, S. Kobayashi, S. Sasaki, Y. Komiyama, T. Izumikawa, M. Conrad, S. Bannai and H. Sato, Sci. Rep., 2018, 8, 968 CrossRef PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison, 3rd and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- K. Hadian and B. R. Stockwell, Cell, 2020, 181, 1188 CrossRef CAS PubMed.
- J. Cao, X. Chen, L. Jiang, B. Lu, M. Yuan, D. Zhu, H. Zhu, Q. He, B. Yang and M. Ying, Nat. Commun., 2020, 11, 1251 CrossRef CAS PubMed.
- Y. Shakiba, Kermanshah University of Medical Sciences, Application of Desferal to Treat COVID-19, 2020, Identifier NCT04333550, https://clinicaltrials.gov/ct2/show/NCT04333550.
- F. Ursini and M. Maiorino, in Encyclopedia of Biological Chemistry, ed. W. J. Lennarz and M. D. Lane, Elsevier, New York, 2004, pp. 224–228 Search PubMed.
- B. B. Muhoberac, Front. Cell. Infect. Microbiol., 2020, 10, 569709 CrossRef.
- H. Sies and M. J. Parnham, Free Radical Biol. Med., 2020, 156, 107–112 CrossRef CAS.
- S. M. Smith, J. Min, T. Ganesh, B. Diebold, T. Kawahara, Y. Zhu, J. McCoy, A. Sun, J. P. Snyder, H. Fu, Y. Du, I. Lewis and J. D. Lambeth, Chem. Biol., 2012, 19, 752–763 CrossRef CAS.
- D. Loh, Melatonin Res., 2020, 3, 380–416 CrossRef.
- H. Ma, X. Wang, W. Zhang, H. Li, W. Zhao, J. Sun and M. Yang, Oxid. Med. Cell. Longevity, 2020, 2020, 9067610 Search PubMed.
- S. De Flora, R. Balansky and S. La Maestra, FASEB J., 2020, 34, 13185–13193 CrossRef CAS PubMed.
- S. J. Dixon and B. R. Stockwell, Annu. Rev. Cancer Biol., 2019, 3, 35–54 CrossRef.
- C. M. Bebber, F. Müller, L. Prieto Clemente, J. Weber and S. von Karstedt, Cancers, 2020, 12 Search PubMed.
- B. R. Stockwell and X. Jiang, Cell Chem. Biol., 2020, 27, 365–375 CrossRef CAS PubMed.
- V. S. Viswanathan, M. J. Ryan, H. D. Dhruv, S. Gill, O. M. Eichhoff, B. Seashore-Ludlow, S. D. Kaffenberger, J. K. Eaton, K. Shimada, A. J. Aguirre, S. R. Viswanathan, S. Chattopadhyay, P. Tamayo, W. S. Yang, M. G. Rees, S. Chen, Z. V. Boskovic, S. Javaid, C. Huang, X. Wu, Y. Y. Tseng, E. M. Roider, D. Gao, J. M. Cleary, B. M. Wolpin, J. P. Mesirov, D. A. Haber, J. A. Engelman, J. S. Boehm, J. D. Kotz, C. S. Hon, Y. Chen, W. C. Hahn, M. P. Levesque, J. G. Doench, M. E. Berens, A. F. Shamji, P. A. Clemons, B. R. Stockwell and S. L. Schreiber, Nature, 2017, 547, 453–457 CrossRef CAS.
- J. H. Woo, Y. Shimoni, W. S. Yang, P. Subramaniam, A. Iyer, P. Nicoletti, M. Rodríguez Martínez, G. López, M. Mattioli, R. Realubit, C. Karan, B. R. Stockwell, M. Bansal and A. Califano, Cell, 2015, 162, 441–451 CrossRef CAS PubMed.
- J. P. Friedmann Angeli, D. V. Krysko and M. Conrad, Nat. Rev. Cancer, 2019, 19, 405–414 CrossRef CAS.
- H. Ledford, Nature, 2020, 582, 469 CrossRef CAS PubMed.
- F. Pedersen, B. Waschki, S. Marwitz, T. Goldmann, A. Kirsten, A. Malmgren, K. F. Rabe, M. Uddin and H. Watz, Eur. Respir. J., 2018, 51, 1700970 CrossRef PubMed.
- S. F. Assimakopoulos and M. Marangos, Med. Hypotheses, 2020, 140, 109778 CrossRef CAS PubMed.
- K. E. Remy, M. Mazer, D. A. Striker, A. H. Ellebedy, A. H. Walton, J. Unsinger, T. M. Blood, P. A. Mudd, D. J. Yi, D. A. Mannion, D. F. Osborne, R. S. Martin, N. J. Anand, J. P. Bosanquet, J. Blood, A. M. Drewry, C. C. Caldwell, I. R. Turnbull, S. C. Brakenridge, L. L. Moldwawer and R. S. Hotchkiss, JCI Insight, 2020, 5(17), e140329 CrossRef PubMed.
- Z. Bakouny, J. E. Hawley, T. K. Choueiri, S. Peters, B. I. Rini, J. L. Warner and C. A. Painter, Cancer Cell, 2020, 38, 629–646 CrossRef CAS PubMed.
- A. Sawant, C. C. Schafer, T. H. Jin, J. Zmijewski, H. M. Tse, J. Roth, Z. Sun, G. P. Siegal, V. J. Thannickal, S. C. Grant, S. Ponnazhagan and J. S. Deshane, Cancer Res., 2013, 73, 6609–6620 CrossRef CAS PubMed.
- L. Sun, P. E. Clavijo, Y. Robbins, P. Patel, J. Friedman, S. Greene, R. Das, C. Silvin, C. Van Waes, L. A. Horn, J. Schlom, C. Palena, D. Maeda, J. Zebala and C. T. Allen, JCI Insight, 2019, 4(7), e126853 CrossRef PubMed.
- S. Greene, Y. Robbins, W. K. Mydlarz, A. P. Huynh, N. C. Schmitt, J. Friedman, L. A. Horn, C. Palena, J. Schlom, D. Y. Maeda, J. A. Zebala, P. E. Clavijo and C. Allen, Clin. Cancer Res., 2020, 26, 1420–1431 CrossRef CAS.
- C. Kang, S. Y. Jeong, S. Y. Song and E. K. Choi, Radiat. Oncol. J., 2020, 38, 1–10 CrossRef.
- A. Orillion, A. Hashimoto, N. Damayanti, L. Shen, R. Adelaiye-Ogala, S. Arisa, S. Chintala, P. Ordentlich, C. Kao, B. Elzey, D. Gabrilovich and R. Pili, Clin. Cancer Res., 2017, 23, 5187–5201 CrossRef CAS PubMed.
- C. Groth, X. Hu, R. Weber, V. Fleming, P. Altevogt, J. Utikal and V. Umansky, Br. J. Cancer, 2019, 120, 16–25 CrossRef CAS PubMed.
- C. Turnquist, B. M. Ryan, I. Horikawa, B. T. Harris and C. C. Harris, Cancer Cell, 2020, 38, 598–601 CrossRef CAS.
- M. Merad and J. C. Martin, Nat. Rev. Immunol., 2020, 20, 355–362 CrossRef CAS.
- S. Mukhopadhyay, S. Sinha and S. K. Mohapatra, 2020, medRxiv, [Preprint], 2020.2010.2013.20211425.
- M. Roschewski, M. S. Lionakis, J. P. Sharman, J. Roswarski, A. Goy, M. A. Monticelli, M. Roshon, S. H. Wrzesinski, J. V. Desai, M. A. Zarakas, J. Collen, K. Rose, A. Hamdy, R. Izumi, G. W. Wright, K. K. Chung, J. Baselga, L. M. Staudt and W. H. Wilson, Sci. Immunol., 2020, 5(48), eabd0110 CrossRef CAS PubMed.
- T. Wan, Y. Zhao, F. Fan, R. Hu and X. Jin, Front. Immunol., 2017, 8, 60 Search PubMed.
- V. Mollica, A. Rizzo and F. Massari, Future Oncol., 2020, 16, 2029–2033 CrossRef CAS.
- K. H. Stopsack, L. A. Mucci, E. S. Antonarakis, P. S. Nelson and P. W. Kantoff, Cancer Discovery, 2020, 10, 779–782 CrossRef CAS PubMed.
- K. M. White, R. Rosales, S. Yildiz, T. Kehrer, L. Miorin, E. Moreno, S. Jangra, M. B. Uccellini, R. Rathnasinghe, L. Coughlan, C. Martinez-Romero, J. Batra, A. Rojc, M. Bouhaddou, J. M. Fabius, K. Obernier, M. Dejosez, M. J. Guillén, A. Losada, P. Avilés, M. Schotsaert, T. Zwaka, M. Vignuzzi, K. M. Shokat, N. J. Krogan and A. García-Sastre, Science, 2021, 371(6532), 926–931 CrossRef CAS.
- Á. Teijeira, S. Garasa, M. Gato, C. Alfaro, I. Migueliz, A. Cirella, C. de Andrea, M. C. Ochoa, I. Otano, I. Etxeberria, M. P. Andueza, C. P. Nieto, L. Resano, A. Azpilikueta, M. Allegretti, M. de Pizzol, M. Ponz-Sarvisé, A. Rouzaut, M. F. Sanmamed, K. Schalper, M. Carleton, M. Mellado, M. E. Rodriguez-Ruiz, P. Berraondo, J. L. Perez-Gracia and I. Melero, Immunity, 2020, 52, 856–871 CrossRef.
- A. Flemming, Nat. Rev. Immunol., 2020, 20, 352–353 CrossRef CAS PubMed.
- M. Binnewies, E. W. Roberts, K. Kersten, V. Chan, D. F. Fearon, M. Merad, L. M. Coussens, D. I. Gabrilovich, S. Ostrand-Rosenberg, C. C. Hedrick, R. H. Vonderheide, M. J. Pittet, R. K. Jain, W. Zou, T. K. Howcroft, E. C. Woodhouse, R. A. Weinberg and M. F. Krummel, Nat. Med., 2018, 24, 541–550 CrossRef CAS PubMed.
- L. Wu, S. Saxena, M. Awaji and R. K. Singh, Cancers, 2019, 11 Search PubMed.
- W. Wang, J. Zhang, N. Zheng, L. Li, X. Wang and Y. Zeng, Clin. Transl. Med., 2020, 10, e126 Search PubMed.
- H. O. Yazdani, E. Roy, A. J. Comerci, D. J. van der Windt, H. Zhang, H. Huang, P. Loughran, S. Shiva, D. A. Geller, D. L. Bartlett, A. Tsung, T. Sheng, R. L. Simmons and S. Tohme, Cancer Res., 2019, 79, 5626–5639 CrossRef CAS PubMed.
- D. C. Wallace, Nat. Rev. Cancer, 2012, 12, 685–698 CrossRef CAS PubMed.
- R. Ganji and P. H. Reddy, Front. Aging Neurosci., 2020, 12, 614650 CrossRef CAS.
- L. Ouyang and J. Gong, Med. Hypotheses, 2020, 144, 110161 CrossRef CAS PubMed.
- Z. Cheng, D. Zhang, J. Chen, Y. Wu, X. Liu, L. Si, Z. Zhang, N. Zhang, Z. Zhang, W. Liu, H. Liu, L. Zhang, L. Song, L. S. C. Dunmall, J. Dong, N. R. Lemoine and Y. Wang, 2020, bioRxiv, [Preprint], 2020.2010.2019.344713.
- K. K. Singh, G. Chaubey, J. Y. Chen and P. Suravajhala, Am. J. Physiol.: Cell Physiol., 2020, 319, C258–C267 CrossRef PubMed.
- D. Scozzi, M. Cano, L. Ma, D. Zhou, J. H. Zhu, J. A. O’Halloran, C. Goss, A. M. Rauseo, Z. Liu, S. K. Sahu, V. Peritore, M. Rocco, A. Ricci, R. Amodeo, L. Aimati, M. Ibrahim, R. R. Hachem, D. Kreisel, P. A. Mudd, H. S. Kulkarni and A. E. Gelman, JCI Insight, 2021, 6(4), e143299 Search PubMed.
- Y. Bao, C. Ledderose, T. Seier, A. F. Graf, B. Brix, E. Chong and W. G. Junger, J. Biol. Chem., 2014, 289, 26794–26803 CrossRef CAS PubMed.
- G. Fossati, D. A. Moulding, D. G. Spiller, R. J. Moots, M. R. White and S. W. Edwards, J. Immunol., 2003, 170, 1964–1972 CrossRef CAS PubMed.
- J. W. Zmijewski, E. Lorne, X. Zhao, Y. Tsuruta, Y. Sha, G. Liu, G. P. Siegal and E. Abraham, Am. J. Respir. Crit. Care Med., 2008, 178, 168–179 CrossRef CAS PubMed.
- W. Zhou, L. Cao, J. Jeffries, X. Zhu, C. J. Staiger and Q. Deng, Dis. Models Mech., 2018, 11, dmm033027 CrossRef PubMed.
- A. J. Sbarra and M. L. Karnovsky, J. Biol. Chem., 1959, 234, 1355–1362 CrossRef CAS.
- J. V. Bannister and W. H. Bannister, Environ. Health Perspect., 1985, 64, 37–43 CrossRef CAS PubMed.
- M. Langley, A. Ghosh, A. Charli, S. Sarkar, M. Ay, J. Luo, J. Zielonka, T. Brenza, B. Bennett, H. Jin, S. Ghaisas, B. Schlichtmann, D. Kim, V. Anantharam, A. Kanthasamy, B. Narasimhan, B. Kalyanaraman and A. G. Kanthasamy, Antioxid. Redox Signaling, 2017, 27, 1048–1066 CrossRef CAS.
- K. A. Fortner, L. P. Blanco, I. Buskiewicz, N. Huang, P. C. Gibson, D. L. Cook, H. L. Pedersen, P. S. T. Yuen, M. P. Murphy, A. Perl, M. J. Kaplan and R. C. Budd, Lupus Sci. Med., 2020, 7, e000387 CrossRef PubMed.
- M. P. Murphy and R. C. Hartley, Nat. Rev. Drug Discovery, 2018, 17, 865–886 CrossRef CAS PubMed.
- A. C. Codo, G. G. Davanzo, L. B. Monteiro, G. F. de Souza, S. P. Muraro, J. V. Virgilio-da-Silva, J. S. Prodonoff, V. C. Carregari, C. A. O. de Biagi Jr, F. Crunfli, J. L. Jimenez Restrepo, P. H. Vendramini, G. Reis-de-Oliveira, K. Bispo Dos Santos, D. A. Toledo-Teixeira, P. L. Parise, M. C. Martini, R. E. Marques, H. R. Carmo, A. Borin, L. D. Coimbra, V. O. Boldrini, N. S. Brunetti, A. S. Vieira, E. Mansour, R. G. Ulaf, A. F. Bernardes, T. A. Nunes, L. C. Ribeiro, A. C. Palma, M. V. Agrela, M. L. Moretti, A. C. Sposito, F. B. Pereira, L. A. Velloso, M. A. R. Vinolo, A. Damasio, J. L. Proença-Módena, R. F. Carvalho, M. A. Mori, D. Martins-de-Souza, H. I. Nakaya, A. S. Farias and P. M. Moraes-Vieira, Cell Metab., 2020, 32, 437–446 CrossRef CAS PubMed.
- F. K. Tsogas, D. Majerczyk and P. C. Hart, Int. J. Mol. Sci., 2021, 22(2), 867 CrossRef CAS.
- G. Schönrich, M. J. Raftery and Y. Samstag, Adv. Biol. Regul., 2020, 77, 100741 CrossRef PubMed.
- J. R. Molina, Y. Sun, M. Protopopova, S. Gera, M. Bandi, C. Bristow, T. McAfoos, P. Morlacchi, J. Ackroyd, A. A. Agip, G. Al-Atrash, J. Asara, J. Bardenhagen, C. C. Carrillo, C. Carroll, E. Chang, S. Ciurea, J. B. Cross, B. Czako, A. Deem, N. Daver, J. F. de Groot, J. W. Dong, N. Feng, G. Gao, J. Gay, M. G. Do, J. Greer, V. Giuliani, J. Han, L. Han, V. K. Henry, J. Hirst, S. Huang, Y. Jiang, Z. Kang, T. Khor, S. Konoplev, Y. H. Lin, G. Liu, A. Lodi, T. Lofton, H. Ma, M. Mahendra, P. Matre, R. Mullinax, M. Peoples, A. Petrocchi, J. Rodriguez-Canale, R. Serreli, T. Shi, M. Smith, Y. Tabe, J. Theroff, S. Tiziani, Q. Xu, Q. Zhang, F. Muller, R. A. DePinho, C. Toniatti, G. F. Draetta, T. P. Heffernan, M. Konopleva, P. Jones, M. E. DiFrancesco and J. R. Marszalek, Nat. Med., 2018, 24, 1036–1046 CrossRef CAS PubMed.
- G. M. Fischer, A. Jalali, D. A. Kircher, W. C. Lee, J. L. McQuade, L. E. Haydu, A. Y. Joon, A. Reuben, M. P. de Macedo, F. C. L. Carapeto, C. Yang, A. Srivastava, C. R. Ambati, A. Sreekumar, C. W. Hudgens, B. Knighton, W. Deng, S. D. Ferguson, H. A. Tawbi, I. C. Glitza, J. E. Gershenwald, Y. N. Vashisht Gopal, P. Hwu, J. T. Huse, J. A. Wargo, P. A. Futreal, N. Putluri, A. J. Lazar, R. J. DeBerardinis, J. R. Marszalek, J. Zhang, S. L. Holmen, M. T. Tetzlaff and M. A. Davies, Cancer Discovery, 2019, 9, 628–645 CrossRef CAS PubMed.
- American Association for Cancer Research, Cancer Discovery, 2019, 9, Of10 Search PubMed.
- L. Han, A. Cavazos, N. Baran, Q. Zhang, V. M. Kuruvilla, J. P. Gay, N. Feng, V. L. Battula, H. M. Kantarjian, N. G. Daver, J. R. Marszalek, M. Andreeff and M. Y. Konopleva, Blood, 2019, 134, 5161 CrossRef.
- X. Shen, N. Li, H. Li, T. Zhang, F. Wang and Q. Li, J. Cancer Res. Clin. Oncol., 2010, 136, 1745–1754 CrossRef CAS.
- B. Ondondo, E. Jones, A. Godkin and A. Gallimore, Front. Immunol., 2013, 4, 197 CAS.
- M. Beyer and J. L. Schultze, Curr. Pharm. Des., 2009, 15, 1879–1892 CrossRef CAS PubMed.
- H. Ledford, Nature, 2021, 590, 374–375 CrossRef CAS PubMed.
- Y. G. Najjar, A. V. Menk, C. Sander, U. Rao, A. Karunamurthy, R. Bhatia, S. Zhai, J. M. Kirkwood and G. M. Delgoffe, JCI Insight, 2019, 4(5), e124989 CrossRef PubMed.
- S. E. Weinberg, B. D. Singer, E. M. Steinert, C. A. Martinez, M. M. Mehta, I. Martinez-Reyes, P. Gao, K. A. Helmin, H. Abdala-Valencia, L. A. Sena, P. T. Schumacker, L. A. Turka and N. S. Chandel, Nature, 2019, 565, 495–499 CrossRef CAS PubMed.
- M. Jain, University of Texas Southwestern Medical Center, Atovaquone for Treatment of COVID-19, 2020, Identifier NCT04456153, https://clinicaltrials.gov/ct2/show/NCT04456153.
- G. Cheng, M. Hardy, P. Topchyan, R. Zander, P. Volberding, W. Cui and B. Kalyanaraman, Sci. Rep., 2020, 10, 17872 CrossRef CAS PubMed.
- G. Cheng, M. Hardy, P. Topchyan, R. Zander, P. Volberding, W. Cui and B. Kalyanaraman, iScience, 2021, 24, 102673 Search PubMed.
- J. Pan, Y. Lee, G. Cheng, J. Zielonka, Q. Zhang, M. Bajzikova, D. Xiong, S. W. Tsaih, M. Hardy, M. Flister, C. M. Olsen, Y. Wang, O. Vang, J. Neuzil, C. R. Myers, B. Kalyanaraman and M. You, iScience, 2018, 3, 192–207 CrossRef CAS.
- D. Vasquez-Dunddel, F. Pan, Q. Zeng, M. Gorbounov, E. Albesiano, J. Fu, R. L. Blosser, A. J. Tam, T. Bruno, H. Zhang, D. Pardoll and Y. Kim, J. Clin. Invest., 2013, 123, 1580–1589 CrossRef CAS PubMed.
- J. Schulte-Schrepping, N. Reusch, D. Paclik, K. Baßler, S. Schlickeiser, B. Zhang, B. Krämer, T. Krammer, S. Brumhard, L. Bonaguro, E. De Domenico, D. Wendisch, M. Grasshoff, T. S. Kapellos, M. Beckstette, T. Pecht, A. Saglam, O. Dietrich, H. E. Mei, A. R. Schulz, C. Conrad, D. Kunkel, E. Vafadarnejad, C. J. Xu, A. Horne, M. Herbert, A. Drews, C. Thibeault, M. Pfeiffer, S. Hippenstiel, A. Hocke, H. Müller-Redetzky, K. M. Heim, F. Machleidt, A. Uhrig, L. Bosquillon de Jarcy, L. Jürgens, M. Stegemann, C. R. Glösenkamp, H. D. Volk, C. Goffinet, M. Landthaler, E. Wyler, P. Georg, M. Schneider, C. Dang-Heine, N. Neuwinger, K. Kappert, R. Tauber, V. Corman, J. Raabe, K. M. Kaiser, M. T. Vinh, G. Rieke, C. Meisel, T. Ulas, M. Becker, R. Geffers, M. Witzenrath, C. Drosten, N. Suttorp, C. von Kalle, F. Kurth, K. Händler, J. L. Schultze, A. C. Aschenbrenner, Y. Li, J. Nattermann, B. Sawitzki, A. E. Saliba and L. E. Sander, Cell, 2020, 182, 1419–1440 CrossRef CAS.
- A. Salminen, K. Kaarniranta and A. Kauppinen, Int. Immunopharmacol., 2018, 61, 231–240 CrossRef CAS PubMed.
- G. Cheng, J. Zielonka, B. P. Dranka, D. McAllister, A. C. Mackinnon, Jr., J. Joseph and B. Kalyanaraman, Cancer Res., 2012, 72, 2634–2644 CrossRef CAS.
- S. Y. Tan, Y. Kelkar, A. Hadjipanayis, A. Shipstone, T. A. Wynn and J. P. Hall, J. Immunol., 2020, 205, 957–967 CrossRef CAS PubMed.
- B. Dwarakanath and V. Jain, Future Oncol., 2009, 5, 581–585 CrossRef CAS.
- C. Wang, Z. Wang, G. Wang, J. Y. Lau, K. Zhang and W. Li, Signal Transduct. Target. Ther., 2021, 6, 114 CrossRef CAS.
- J. Burtscher, G. Cappellano, A. Omori, T. Koshiba and G. P. Millet, iScience, 2020, 23, 101631 CrossRef CAS.
- T. Takahashi, M. K. Ellingson, P. Wong, B. Israelow, C. Lucas, J. Klein, J. Silva, T. Mao, J. E. Oh, M. Tokuyama, P. Lu, A. Venkataraman, A. Park, F. Liu, A. Meir, J. Sun, E. Y. Wang, A. Casanovas-Massana, A. L. Wyllie, C. B. F. Vogels, R. Earnest, S. Lapidus, I. M. Ott, A. J. Moore, A. Shaw, J. B. Fournier, C. D. Odio, S. Farhadian, C. Dela Cruz, N. D. Grubaugh, W. L. Schulz, A. M. Ring, A. I. Ko, S. B. Omer and A. Iwasaki, Nature, 2020, 588, 315–320 CrossRef CAS PubMed.
- V. B. Pillai, A. Kanwal, Y. H. Fang, W. W. Sharp, S. Samant, J. Arbiser and M. P. Gupta, Oncotarget, 2017, 8, 34082–34098 CrossRef PubMed.
- E. Levy, E. Delvin, V. Marcil and S. Spahis, Am. J. Physiol.: Endocrinol. Metab., 2020, 319, E689–E708 CrossRef CAS.
- G. Cheng, J. Zielonka, M. Hardy, O. Ouari, C. R. Chitambar, M. B. Dwinell and B. Kalyanaraman, Oncotarget, 2019, 10, 3518–3532 CrossRef.
- N. Maio, B. A. P. Lafont, D. Sil, Y. Li, J. M. Bollinger, C. Krebs, T. C. Pierson, W. M. Linehan and T. A. Rouault, Science, 2021, eabi5224 Search PubMed.
- M. C. Ghosh, W. H. Tong, D. Zhang, H. Ollivierre-Wilson, A. Singh, M. C. Krishna, J. B. Mitchell and T. A. Rouault, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12028–12033 CrossRef CAS PubMed.
- T. A. Rouault and N. Maio, Mol. Cell, 2020, 78, 1–3 CrossRef CAS.
- A. P. Cotrim, F. Hyodo, K. Matsumoto, A. L. Sowers, J. A. Cook, B. J. Baum, M. C. Krishna and J. B. Mitchell, Clin. Cancer Res., 2007, 13, 4928–4933 CrossRef CAS.
- J. B. Mitchell, M. R. Anver, A. L. Sowers, P. S. Rosenberg, M. Figueroa, A. Thetford, M. C. Krishna, P. S. Albert and J. A. Cook, Cancer Res., 2012, 72, 4846–4855 CrossRef CAS PubMed.
- M. C. Krishna, A. Russo, J. B. Mitchell, S. Goldstein, H. Dafni and A. Samuni, J. Biol. Chem., 1996, 271, 26026–26031 CrossRef CAS PubMed.
- Q. Ma, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 401–426 CrossRef CAS.
- R. M. da Costa, D. Rodrigues, C. A. Pereira, J. F. Silva, J. V. Alves, N. S. Lobato and R. C. Tostes, Front. Pharmacol., 2019, 10, 382 CrossRef CAS PubMed.
- N. Robledinos-Antón, R. Fernández-Ginés, G. Manda and A. Cuadrado, Oxid. Med. Cell. Longevity, 2019, 2019, 9372182 Search PubMed.
- J. M. McCord, B. M. Hybertson, A. Cota-Gomez, K. P. Geraci and B. Gao, Antioxidants, 2020, 9 Search PubMed.
- R. A. Zinovkin and O. A. Grebenchikov, Biochemistry, 2020, 85, 833–837 CAS.
- A. Cuadrado, M. Pajares, C. Benito, J. Jiménez-Villegas, M. Escoll, R. Fernández-Ginés, A. J. Garcia Yagüe, D. Lastra, G. Manda, A. I. Rojo and A. T. Dinkova-Kostova, Trends Pharmacol. Sci., 2020, 41, 598–610 CrossRef CAS PubMed.
Footnote |
| † Dedicated to my longtime friend and mentor, Sampath (Sam) Parthasarathy, who recently passed away due to COVID-19. |
| This journal is © The Royal Society of Chemistry 2021 |