Short oligoalanine helical peptides for supramolecular nanopore assembly and protein cytosolic delivery†
Received
19th June 2020
, Accepted 7th November 2020
First published on 8th December 2020
Abstract
In this work we report a rational design strategy for the identification of new peptide prototypes for the non-disruptive supramolecular permeation of membranes and the transport of different macromolecular giant cargos. The approach targets a maximal enhancement of helicity in the presence of membranes with sequences bearing the minimal number of cationic and hydrophobic moieties. The here reported folding enhancement in membranes allowed the selective non-lytic translocation of different macromolecular cargos including giant proteins. The transport of different high molecular weight polymers and functional proteins was demonstrated in vesicles and in cells with excellent efficiency and optimal viability. As a proof of concept, functional monoclonal antibodies were transported for the first time into different cell lines and cornea tissues by exploiting the helical control of a short peptide sequence. This work introduces a rational design strategy that can be employed to minimize the number of charges and hydrophobic residues of short peptide carriers to achieve non-destructive transient membrane permeation and transport of different macromolecules.
Introduction
The selective non-destructive and transient disruption of membranes constitutes one of the most important challenges for chemistry and macromolecular transport.1–4 Cationic natural or synthetic amphiphiles have been widely employed for membrane perturbation due to the presence of anionic lipids or the abundant anionic glycosaminoglycans at the plasma membrane, which are generally involved in the initial interactions between biological membranes and cell penetrating peptides. Therefore, different cationic lipids, biopolymers and synthetic analogues have been employed for the membrane translocation of hydrophilic macromolecules.1–4 However, strongly cationic molecules and surfactants suffer from non-specific binding to membranes and detergent behavior.1,5,6 Over the last years, different strategies employing cationic synthetic materials3,7,8 have been developed to transport bioactive macromolecules,9 which include peptides,10–21 chimeric proteins,22 polymers,23–25 dynamic covalent bonds,23–26 supramolecular caging,27 transient charge masking,28 lipid particles,29,30 polymersomes,31,32 and highly charged nanoparticles.33–35 Importantly, the presence of anionic lipids, especially in the endosomal compartments, has been proved critical for the correct function of several natural36,37 and artificial cationic macromolecular carriers.12,38 In general, cationic and amphiphilic membrane targeted molecules mainly operate by unspecific disruption of membranes and/or by the direct endosomal annihilation due to osmotic pressure unbalance.5,39 Nevertheless, most of the recently developed synthetic transporters incorporate a high number of cationic residues and/or show strong amphiphilic character, which can easily destroy membranes and cause toxicity.6,40 Despite a high number of charges could interfere with anionic biomolecules and cause toxicity, certain reported peptides reduce these limitations by controlling their membranolytic activity, such as RALA,41 which requires endosomal pH acidification. Despite these clear precedents, little attention has been focused on the development of rational strategies for the design of membrane targeted molecules with selective and transient non-lytic membrane disruption and with a minimum number of cationic and hydrophobic residues.6 Herein, we report a rational strategy for non-disruptive membrane permeation and macromolecular transport, which is based on the maximization of the helicity of a short, simple peptide as it transits from the aqueous to the membrane environment. Experiments in vesicles and dynamic simulations confirmed the targeted peptide helical enhancement and the non-lytic membrane permeation, which triggered the non-covalent transport of different cargos through the membranes of vesicles and cells. This strategy allowed the discovery of a peptide sequence, with only three arginine residues, which transported, with high efficiency and low toxicity, functional proteins and monoclonal antibodies across membranes of cells and tissues.
Results
Design and initial screening
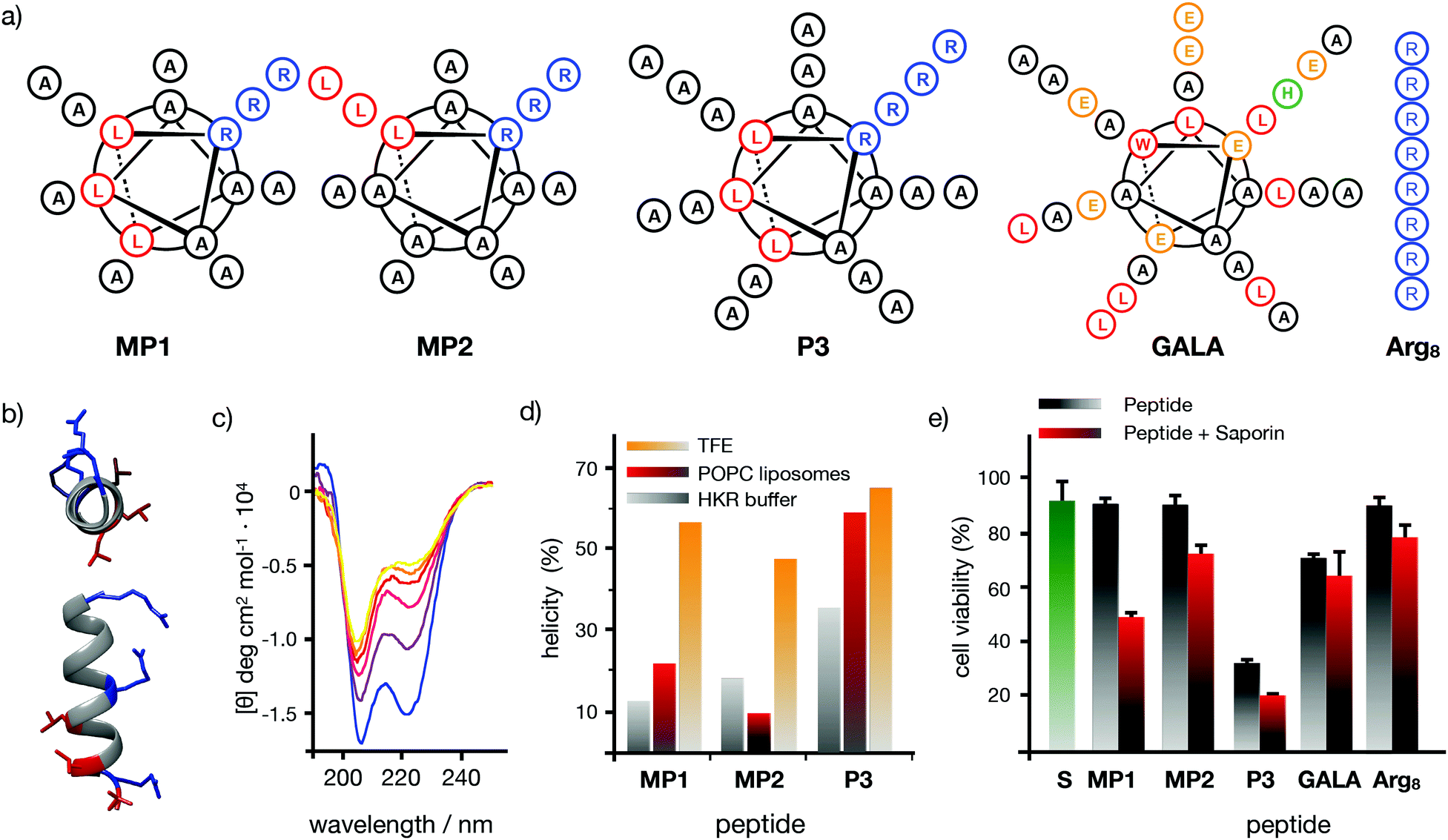
Seminal work from Baldwin and co-workers highlighted the importance of alanine residues to promote helical folding in short peptides in the absence of side chain stabilizing interactions.43,44 In addition, inspiring cationic penetrating oligoalanine scaffolds, originally reported by Steven Dowdy,45 had been shown to efficiently internalize inside cells.46 In these scaffolds, four helical turns are required to quantify helicity by a measurable Cotton effect in circular dichroism (CD).45,46 Therefore, sequences of 16 amino acids were selected to study and optimize peptide helical folding in different environments.42 We started from short peptide scaffolds of alanine, the amino acid with the strongest alpha helical stabilizing character, to adjust the minimal number of substitutions by cationic (Arg) and hydrophobic (Leu) aminoacids that would endow the peptide with penetrating properties. Minimization of cationic residues was targeted in order to reduce the potential complexation with anionic biopolymers47 and three leucines were previously found to maximize peptide uptake.46 Thus, in this work, three oligoalanine peptides46 with different and representative helical behavior43 (MP1, MP2 and P3) were designed and synthesized together with the octaarginine Arg8 and the pore-forming GALA, as peptide controls with higher cationic and hydrophobic character respectively (Fig. 1a). The CD spectra indicated that the minimal peptide MP1, with three aligned arginines and a radial distribution of three leucines close to the N-terminus (Fig. 1a–c), showed low helicity in aqueous buffer that was increased (to 20%) in the presence of zwitterionic neutral liposomes (POPC: 1-palmitoyl-2-oleoylphosphatidylcholine) (Fig. 1d and Fig. S1, ESI†). In contrast, peptide MP2 with three aligned arginines and three aligned leucines, on the same side of the putative alpha helix, showed stronger helical behaviour in buffer (∼20%) that was reduced (∼10%) in neutral lipid membranes (Fig. 1d and Fig. S1, ESI†). Peptide P3, analogous to MP1 but with one extra helical turn (Fig. 1a), showed higher helicity in buffer (∼35%) that was also enhanced in liposomes (∼50%), close to the maximum helicity that can be detected with the helix-stabilizing solvent TFE.48 With these three representative examples of our previously reported library of penetrating oligoalanines,46 a preliminary screening of the uptake and cytosolic release of a cell-impermeable toxin was carried out by the MTT assay (Fig. 1e). Incubation of all peptides with cells, in the absence or in the presence of saporin, indicated optimal transport of the toxin for MP1, lower efficiency for MP2 and strong toxicity of the longer most helical oligoalanine P3 (Fig. 1e). The corresponding control experiments with GALA49 and Arg85 showed lower levels of saporin uptake and release (Fig. 1e).
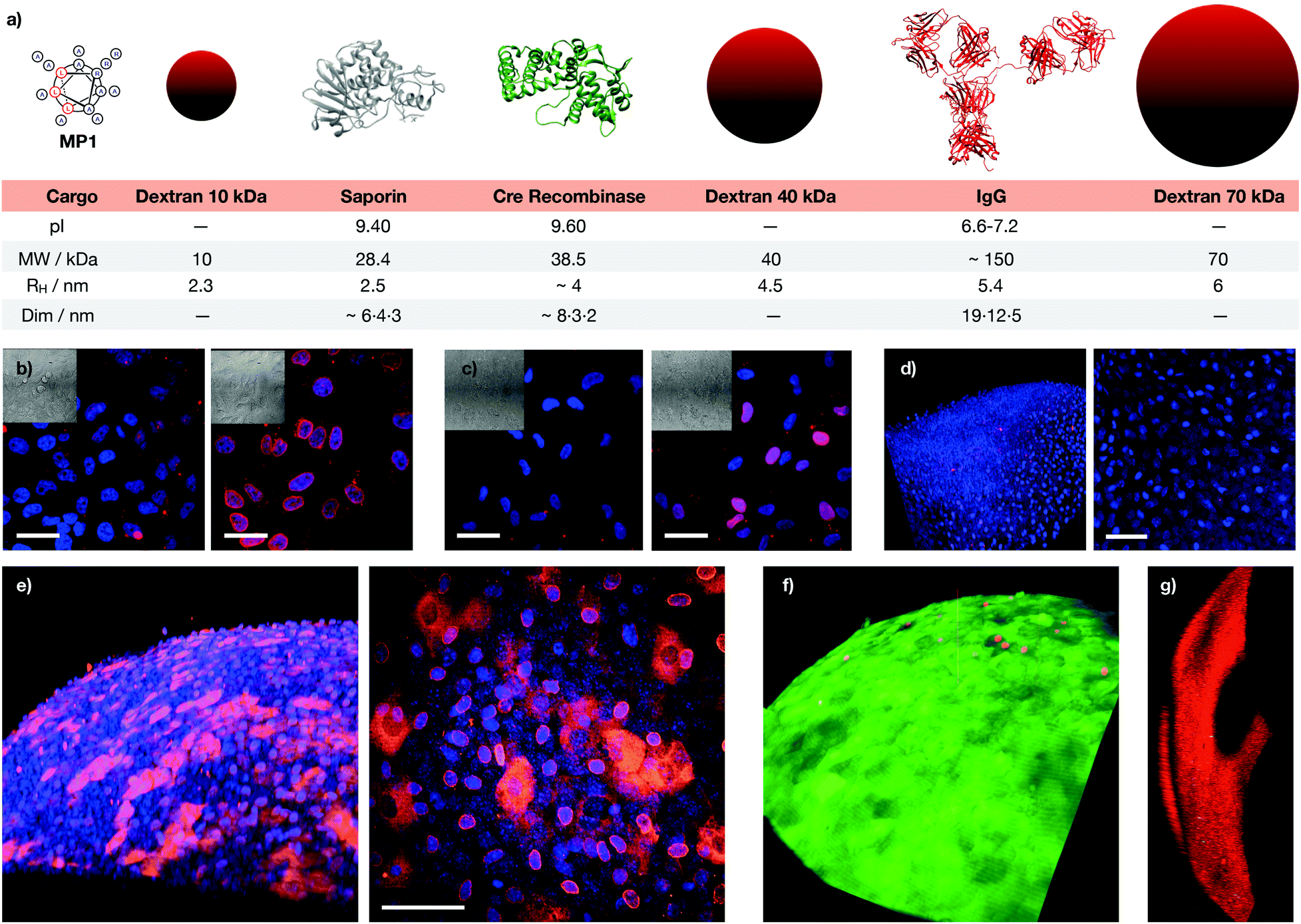
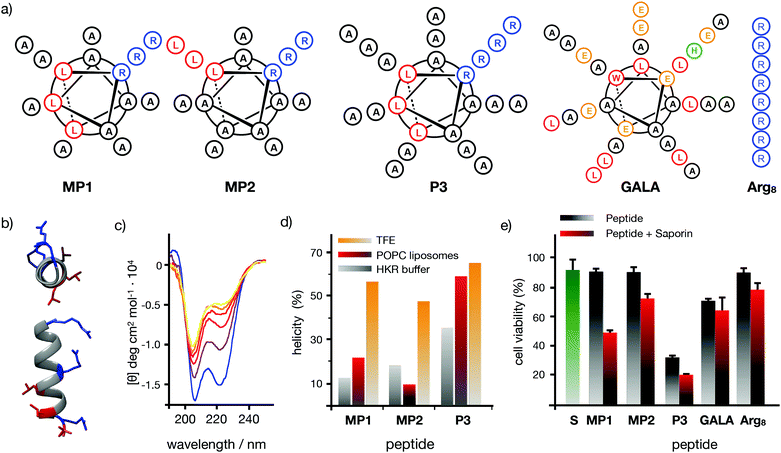 |
| | Fig. 1 Peptide structures, helical content quantification by CD and initial screening. (a) Peptides’ structures. Sequences: MP1 (Ac-LRALAALARAAAAAAR-NH2); MP2 (Ac-LRAAAAALRAAAAALR-NH2); P3 (Ac-LRALAALARAAAAAARAAAAAAR-NH2); GALA (Ac-WEAALAEALAEALAEHLAEALAEALEALAA-NH2); Arg8 (Ac-RRRRRRRR-NH2), (b) 3D prediction of MP1 using PEP-FOLD 3.5.42 Arginines in blue, leucines in red. (c) Circular dichroism spectra of MP1 (100 μM) at different temperatures (10, 20, 30, 40, 50, 60 °C) in HKR buffer. (d) % helicity of MP1, MP2, and P3 in HKR, liposomes (POPC) or TFE at 40 °C (see ESI†). (e) Cell viability in HeLa cells without (grey) or with (red) 10 μg mL−1 of saporin and 30 μM of the indicated peptides for 1 h and further incubation for 6 h before measuring viability by MTT assay. In green the control with saporin alone. Error bars represent standard deviation (SD) of 3 replicates. | |
Selective membrane disruption by MP1
This interesting toxin transport profile and the previously confirmed maximal uptake of MP1 in cells46 prompted a detailed characterization of the interactions of this peptide in different lipid bilayer membranes (Fig. 2). To study the impact of anionic lipids,37,50 the CD of MP1 was measured in anionic vesicles (POPC/POPG 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, POPG: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol, Fig. 2a). A remarkable four-fold helicity enhancement was observed in the transition of MP1 from aqueous to anionic lipid environments (Fig. 2a).37 Fluorescence resonance energy transfer (FRET) between the peptide labelled with a carboxyfluorescein probe (CF-MP1, donor) and the peptide labelled with TAMRA (TM-MP1, acceptor), indicated a much higher self-assembly of MP1 in the presence of anionic membranes compared to that of the aqueous buffer (Fig. 2b). Self-assembly of MP1 in the absence of membranes is supported by the quenching and red shift of the emission of a carboxyfluorescein labelled MP1 at increasing concentrations (Fig. S2, ESI†). FRET experiments with the donor partner (nitrobenzoxadiazole, NBD) installed in a phospholipid counterpart confirmed a higher FRET, and thus membrane affinity, between the TAMRA-labelled peptide (TM-MP1) with anionic vesicles in comparison to neutral vesicles (Fig. 2c).
:1, POPG: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol, Fig. 2a). A remarkable four-fold helicity enhancement was observed in the transition of MP1 from aqueous to anionic lipid environments (Fig. 2a).37 Fluorescence resonance energy transfer (FRET) between the peptide labelled with a carboxyfluorescein probe (CF-MP1, donor) and the peptide labelled with TAMRA (TM-MP1, acceptor), indicated a much higher self-assembly of MP1 in the presence of anionic membranes compared to that of the aqueous buffer (Fig. 2b). Self-assembly of MP1 in the absence of membranes is supported by the quenching and red shift of the emission of a carboxyfluorescein labelled MP1 at increasing concentrations (Fig. S2, ESI†). FRET experiments with the donor partner (nitrobenzoxadiazole, NBD) installed in a phospholipid counterpart confirmed a higher FRET, and thus membrane affinity, between the TAMRA-labelled peptide (TM-MP1) with anionic vesicles in comparison to neutral vesicles (Fig. 2c).
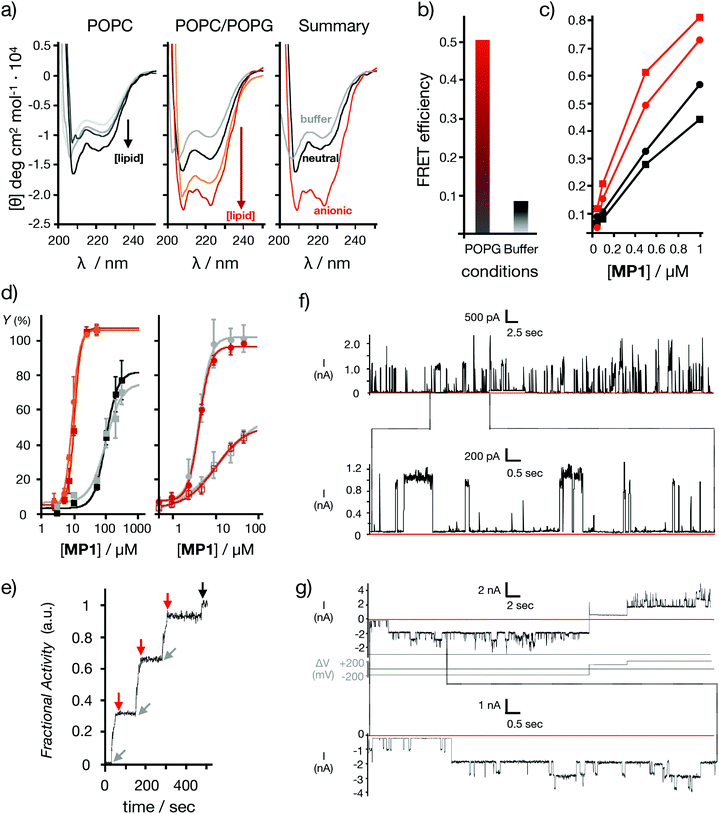 |
| | Fig. 2 Model membranes. (a) Peptide helicity at pH 7.5 (20 °C) in the presence of neutral vesicles (left) and anionic vesicles (middle) with different lipid/peptide ratios (L/P: 1.25, 5, 12, 50). On the right a summary of the CD spectra with buffer (grey), neutral (black) and anionic (red) vesicles at L/P 50 is shown. (b) FRET efficiency between the labelled peptides: CF-MP1 (donor) and TM-MP1 (acceptor), in anionic vesicles (red) and aqueous buffer (grey). [Peptides] = 5 μM each, [vesicles] = 125 μM. (c) FRET efficiency between the NBD labelled phospholipid (donor) and the labelled peptide TM-MP1 (acceptor) in neutral (black) or anionic (red) vesicles. Circles are for head- and squares for the tail-labelled NBD-lipid. [Vesicles] = 125 μM. (d) Left: Dose response curves for LUVs⊂ANTS/DPX release experiments at different MP1 concentrations. Anionic vesicles are in red (pH 7.5) and in orange (pH 5.5) (EC50 = 10.7 and 9.0 μM respectively). Neutral vesicles are in black (pH 7.5) and in grey (pH 5.5) (EC50 = 98.2 and 97.2 μM respectively). Right: Dose response curves for dextran release in anionic vesicles LUVs⊂Dextran at different MP1 concentrations. The 10 kDa FITC-Dex (filled circles) and 40 kDa TM-Dex (empty squares) are shown in red (pH 7.5) and in grey (pH 5.5) respectively. Fractional activity Y against peptide concentration (n = 3). (e) BLM electrical current recordings in DPhPC membrane at 25 μM of peptide in each well (potential in first recording +200 mV; in second recording, +200 mV or −200 mV as indicated). Conditions: 0.5 M KCl, 10 mM MOPS, pH 7.4. The current signals were aquired at 1 kHz and sampled at 5 kHz, resulting data were filtered using low pass Bessel (8-pole). Kinetic fluorescent trace of LUVs⊂ANTS/DPX showing cycles of pore gating at pH 7.5 after sequential additions of MP1 (gray arrows, 10 μM at t = 25, 150, and 275 s) and POPG (red arrows, 50 μM at t = 50, 175, and 300 s). At t = 475 s (black arrow) Triton X-100 was added for complete vesicle lysis (see ESI†). | |
Analogous FRET experiments but with the NBD donor installed in the alkyl chain of the lipid counterpart indicated a preferential orientation of the MP1 peptide with the hydrophobic leucine N-terminus deeply inserted into the non-polar region of the bilayer (Fig. 2c). Release experiments were then performed in large unilamellar vesicles with an encapsulated fluorescent probe and a quencher (LUVs⊂ANTS/DPX, ∼120 nm).38 Dose–response experiments with MP1 showed a strong selectivity for negatively charged membranes, as indicated by a ten times lower EC50 for probe release in anionic vesicles (POPC/POPG 3:1) compared to neutral liposomes (POPC) (Fig. 2d (left) and Fig. S3a, ESI†). High molecular weight dextran (i.e. 10–40 kDa) in vitro release experiments in vesicles revealed marginal dextran release from neutral vesicles but efficient and size-selective leakage of dextran polymers for anionic vesicles (Fig. 2d (right) and Fig. S3b, ESI†).2 Importantly, dynamic light scattering (DLS) measurements of the vesicles in the presence of MP1 confirmed the homogenous size and structural integrity of the vesicles even at very high peptide concentrations (i.e. 800 μM, Fig. S3c, ESI†). These results confirmed a non-destructive membrane disruption mechanism enhanced by the specific folding and the supramolecular assembly of MP1.
Computational modelling
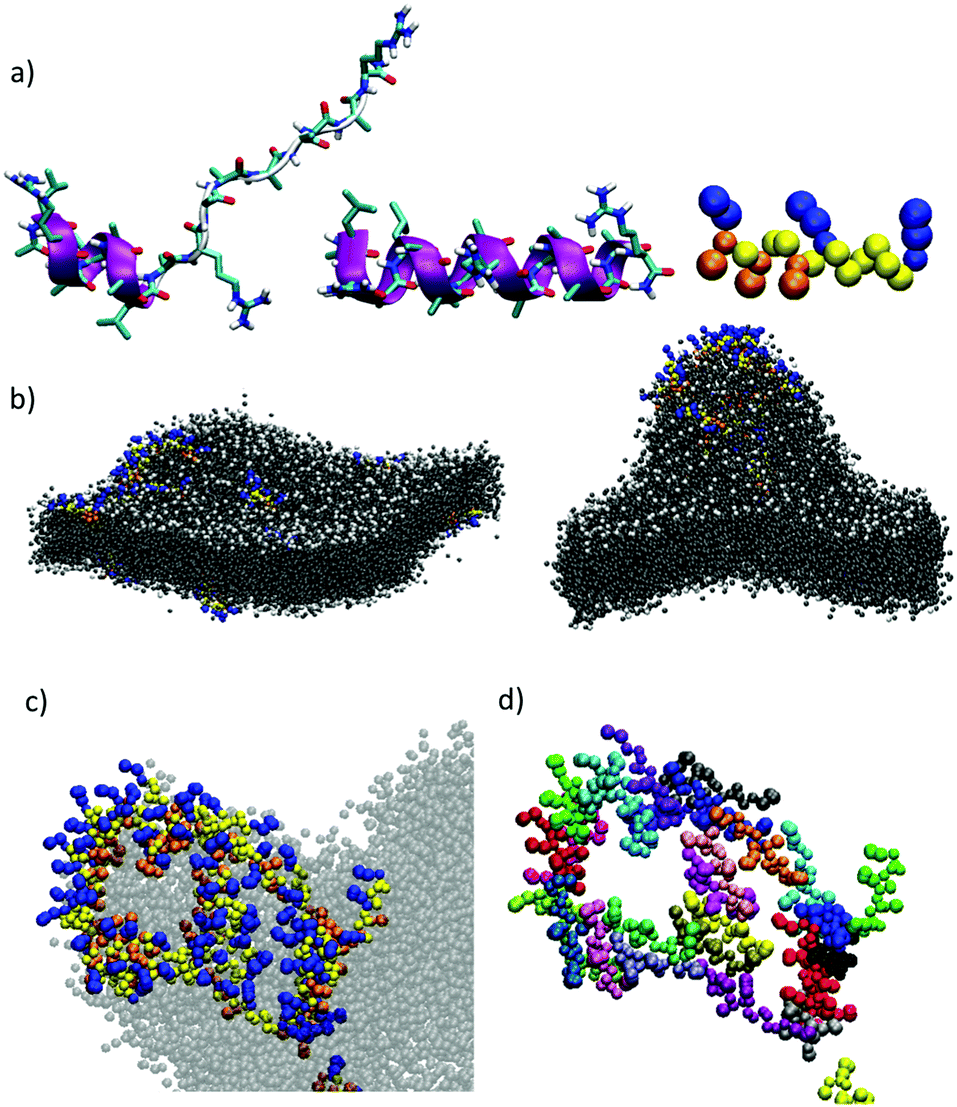
Computational simulations were performed to study MP1 in the highly dynamic environment of the early endosomal membrane (see ESI†). As experimentally confirmed by CD, the atomistic Molecular Dynamics (AT-MD) simulations of MP1 revealed peptide unfolding in aqueous solution and stable helical arrangement in TFE (Fig. 3a). AT-MD simulations are restricted to short timescales, which hinder the observation of multiple peptide folding, aggregation and membrane insertion events.51 Alternatively, coarse-grained (CG-MD) simulations largely increase the computational power and timescales, allowing the simulation of complex dynamic systems. Accordingly, CG-MD simulations using the MARTINI force-field52 were performed starting from different folded units of MP1 in an early endosome anionic model membrane in the presence of an external electric field simulating the endosomal membrane potential (Fig. 3b–d and Fig. S4, ESI†).53 31 units of MP1 in the presence of 1309 lipids, L/P ∼ 42, were used to achieve a L/P ratio corresponding to the lowest active peptide concentration in the vesicle leakage experiments (∼3 μM, Fig. 2d). These experiments confirmed the initial interaction of MP1 with membranes by anchoring and orienting the Leu enriched hydrophobic N-terminus towards the hydrophobic core of the lipid bilayer, and the arginine cationic residues exposed to the solvent environment (Video S1, ESI†), as experimentally supported by FRET (Fig. 2c). Interestingly, beyond the strong interactions with the membrane, adaptive peptide MP1 showed a self-assembly pattern with the Ala residues interacting between peptide monomers and mimicking prefibrillar peptide assemblies in the lipid bilayer (Fig. 3b–d). The self-assembly of the peptides, in a model membrane with external applied electric field, showed the inducement of local membrane curvature and self-assembly of MP1 at the apical regions of the membrane invaginations (Fig. 3b–d). As the simulation evolved with time, a transitory membrane disruption was observed (Fig. 3c, d and Fig. S4, ESI†).
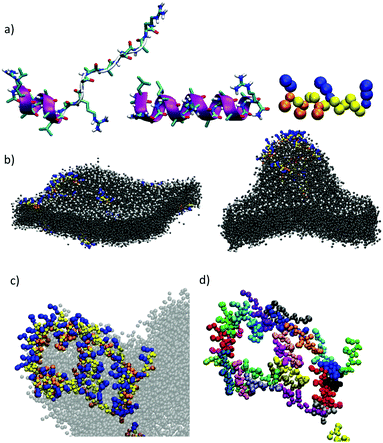 |
| | Fig. 3 (a) Snapshot (t = 40 ns) from AT-MD simulations of peptide MP1 in water (left), TFE (middle) and mapping to CG resolution (right). (b) Snapshots from CG-MD simulations [t = 10 ns, (left) and t = 300 ns (right) respectively] showing the interaction of peptides and an asymmetric bilayer composed by DPPC:DPPE (9:1) and DPPC:DPPE:DPPS (3:5:2), subjected to an external electric field of 30 mV applied across the membrane. A detail of the disposition of the peptides (c and d) is shown. CG residues in (b and c) are represented in yellow (Ala), orange (Leu) and blue (Arg). A different color is used for each peptide in (d) to highlight the independent units of MP1. (DPPC: dipalmitoyl phosphatidylcholine, DPPE: dipalmitoyl phosphatidylethanolamine, DPPS: dipalmitoyl phosphatidylserine). | |
To investigate the potential formation of membrane pores by MP1, gating experiments were carried out by sequential addition to vesicles of MP1 followed by an anionic binder competitor (POPG). Pore gating (opening/closing) was confirmed by three cycles of activation and inhibition of the transport in a single transport kinetics experiment (Fig. 2f). Finally, to unambiguously confirm the potential assembly of supramolecular nanopores from MP1, ion channel conductance recordings were performed in planar bilayer membranes (black lipid membranes, BLM).54 The typical BLM current recordings reported for oligocationic penetrating peptides (i.e. nonarginine) have shown random spikes of current, whose noise fluctuations increased with time.55 In contrast, recordings of MP1 showed individual stochastic gating events of high current, which were indicative of the formation of individual giant channels assembled from MP1 (see ESI† and Fig. 2e). Not surprisingly, as previously shown for other membrane ion channels,56 the formation of MP1 channels was dependent on the applied voltage. However, the remarkably high and identical steady state current intensity (∼1100 pA, 200 mV) of the series of stochastic events, observed along different BLM recordings, demonstrated the assembly of a hitherto not described giant supramolecular membrane pore assembled from a peptide sequence shorter than a membrane leaflet. Intriguingly, the integrity of the planar bilayer was maintained even at long times with MP1 (Fig. 2e) and current recordings eventually showed long-lasting opened states (at 1900 pA) with additional individual stochastic gating events of identical current intensity (1100 pA) on top of the opened state (Fig. 2e).
Dextran transport in cells
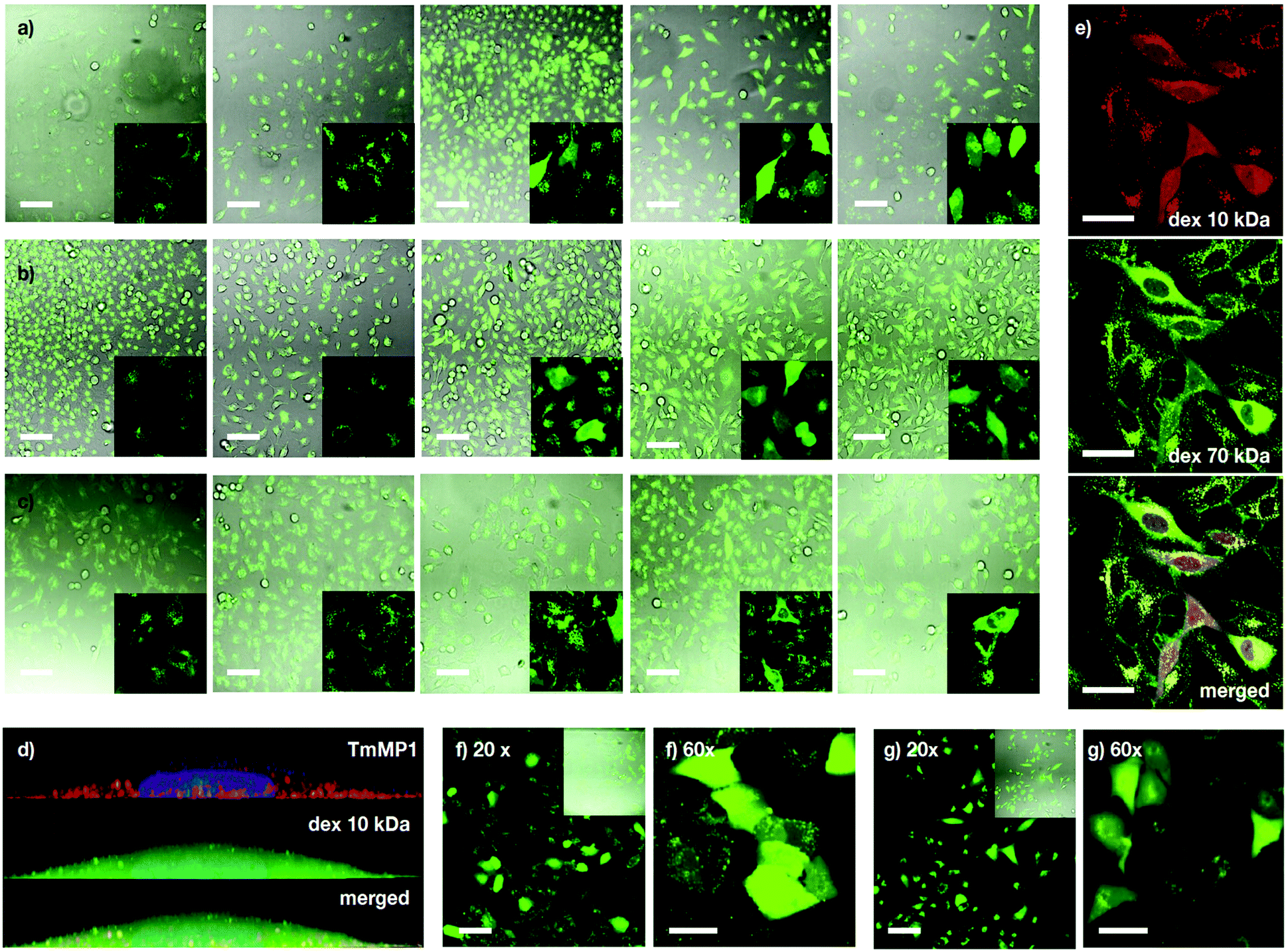
Following the preliminary transport results and the excellent properties of MP1 in model membranes, transport experiments in cells were investigated first with dextran polymers, which are markers of endocytic routes, and are unable to escape from the endosome on their own.9,57 Kinetic experiments in HeLa cells showed mainly punctate fluorescence at short incubation times (i.e. 1 hour) and LysoTracker staining indicated that TM-MP1 and dextran remained trapped together in the endosomes (punctated pattern, Fig. 4d and Fig. S5, ESI†). Despite the peptide did not leave the endosomes, at longer incubation times (3 hours) stronger diffuse cytosolic dextran fluorescence was detected inside the cells, which suggested an initial endocytic uptake followed by dextran release (Fig. S5, ESI†).
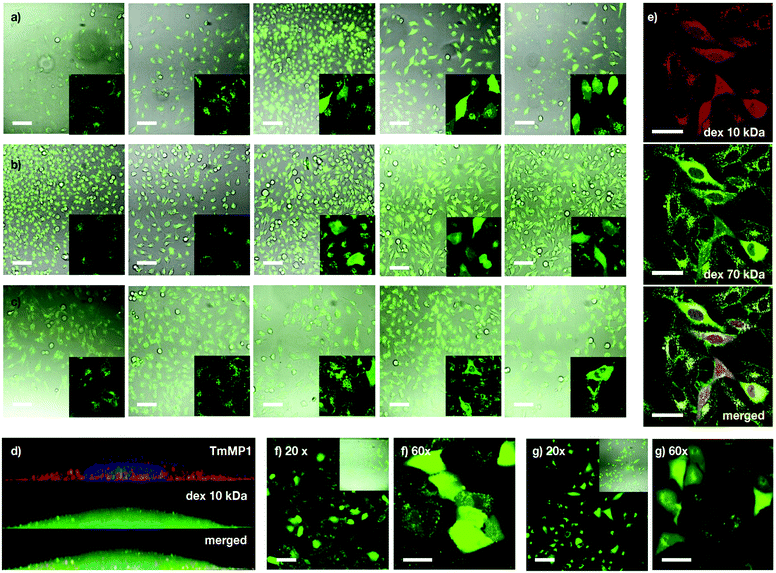 |
| | Fig. 4 Dextran (Dex) transport. (a–c) HeLa cells incubated with MP1 (0, 10, 20, 30, 50 μM) and fluorescently labelled dextran: 10 kDa Alexa488-Dex (a), 40 kDa FITC-Dex (b) and 70 kDa FITC-Dex (c). In the absence of MP1, punctate fluorescence signal was observed, which is indicative of endosomes or lysosomes. Peptide-triggered cytosolic release of the dextran affords a homogeneous staining of the cell. Percentage of cells with intracellular release at 30 μM was 42, 30 and 10% for (a, b and c). (d) 3D view of a HeLa cell with 5 μM TM-MP1 (orange) and 10 kDa Alexa488-Dex (green), showing dextran (cytosol) and peptide (endosomes). Nucleus stained with Hoechst. (e) HeLa cells co-incubated with 10 kDa TM-Dex (red), 70 kDa FITC-Dex (green) and MP1 (30 μM). (f) Vero cells with 10 kDa Alexa488-Dex and 20 μM MP1 (30% release). (g) A549 cells with 10 kDa Alexa488-Dex and 50 μM of MP1 (30% release). Scale bars are 100 μm for (a–c and f) (20×), (g) (20×) and 50 μm for (e and f) (60×) and (g) (60×). | |
Experiments with dextran polymers of different molecular weights (10, 40 and 70 kDa) showed excellent, yet size-selective cytosolic transport of the different dextrans for MP1 at 20 and 30 μM concentration (Fig. 4a–c).58 Mixed solutions of different dextrans again confirmed simultaneous and size selective polymer transport (10 > 70 kDa, Fig. 4e). Importantly, polymer transport was insensitive to the presence of serum proteins, as the same level of dextran uptake and release, about 45%, was confirmed in the presence of 10% FBS (Fig. S6a, ESI†). In addition, we had previously determined that TM-MP1 half-life in 100% FBS was ∼2.2 h.46 Vero (monkey kidney) and the difficult-to-transfect A549 (human lung cancer) cells further confirmed the excellent scope of MP1 (Fig. 4f and g). Studies in the presence of endocytic inhibitors showed only partial inhibition of the uptake and intracellular release, which requires the uptake of both peptide and dextran, by dynasore and EIPA, which block dynamin-dependent endocytosis and macropinocytosis respectively (Fig. S6b, ESI†). Therefore, as these routes are well known to be responsible for the dextran polymer uptake,57 these results suggested a mixed uptake for MP1 and a general mechanism for the transport of the polymers, which was mainly dependent on the uptake of the dextran cargo.
Functional protein transport in cells
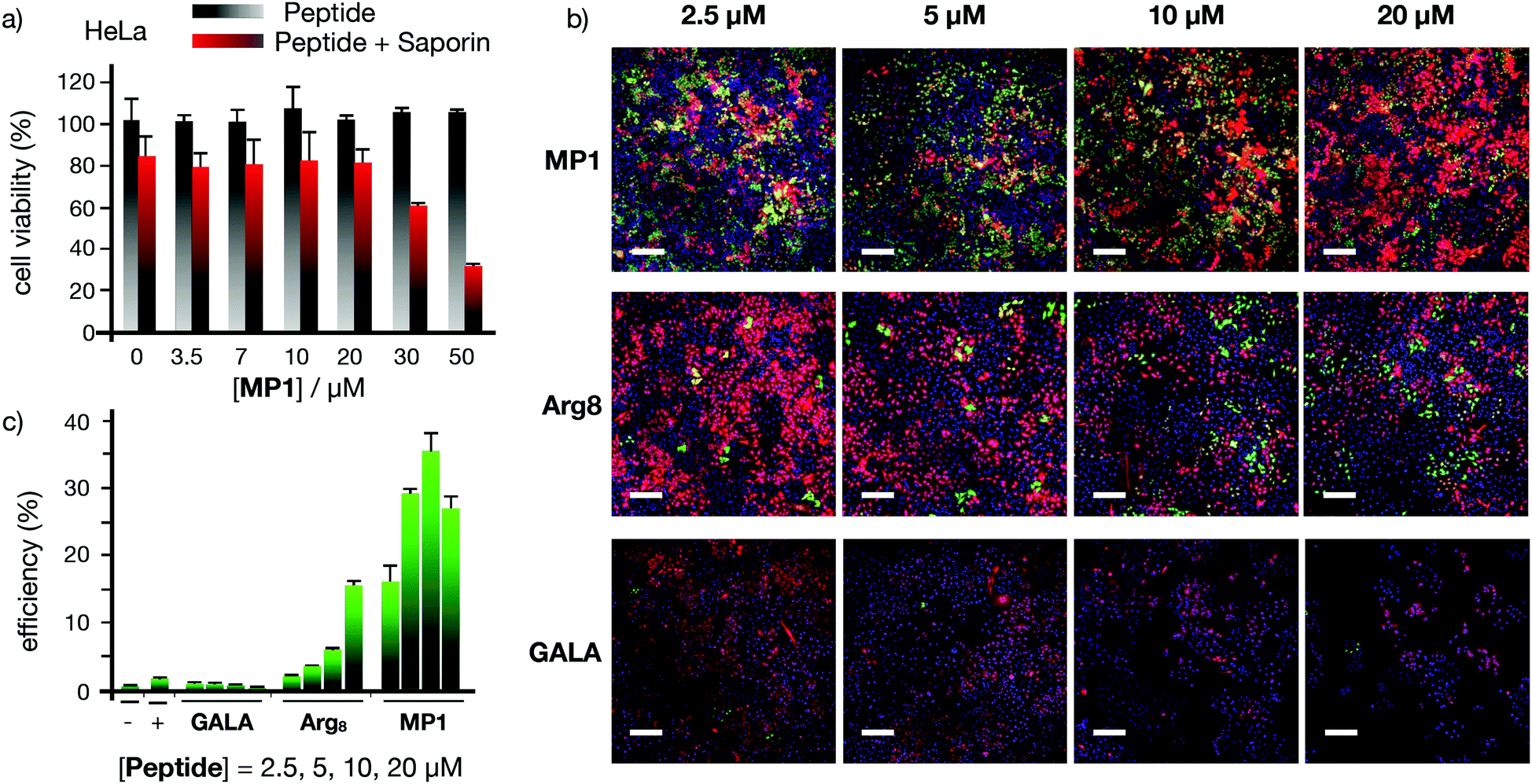
The excellent profile for the membrane transport of MP1 with model hydrophilic polymers prompted the study of functional protein cargos (Fig. 5 and Fig. S7, ESI†). Dose response transport experiments with MP1 (0–50 μM) in HeLa cells in the presence or absence of saporin (10 μg mL−1) showed a respectable 70% toxicity enhancement for MP1 and perfect cell viability for the peptide alone in the absence of the toxin (Fig. 5a). Control experiments showed minor transport for the GALA peptide and no activity for Arg8 after 6 h incubation (Fig. S7a, ESI†). Further control experiments, at longer recovery times (24 hours), showed the enhanced saporin cytosolic transport in HeLa (Fig. S7b, ESI†) and A549 cells (Fig. S7c, ESI†).
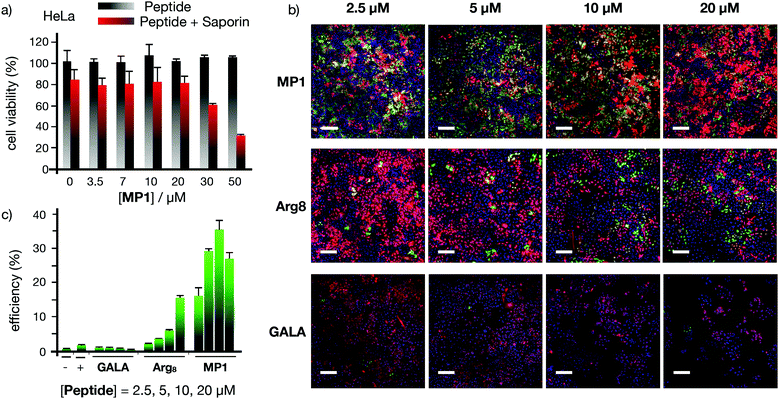 |
| | Fig. 5 Functional protein membrane transport. (a) HeLa cells incubated with 0 (grey bars) or 10 μg mL−1 (red bars) saporin in the presence of increasing concentrations of MP1 for 1 h, medium was then replaced and cells further incubated for 6 h before measuring viability by MTT assay. (b) Cre reporter-HeLa cells 3 days after incubation with the indicated peptides (MP1, Arg8, or GALA) and Cre recombinase. Cre recombination turned the dsRED (red) to EGFP (green) expression. Nuclei stained with Hoechst (blue). Scale bars are 200 μm. (c) Flow cytometry quantification of the fraction of EGFP expressing Cre reporter-HeLa cells. Error bars represent SD of three replicates. | |
The Cre recombinase enzyme catalyses loxP sites recombination followed by gene expression switch from a red (dsRED) to a green (EGFP) fluorescent protein in a reporter cell line (Fig. 5b, c and Fig. S8, ESI†).59 The corresponding microscopy and flow cytometry quantification of the Cre transport in HeLa cells confirmed an excellent level of recombination at low concentrations of MP1, optimal at 10 μM and superior to the controls with Arg8 and GALA (Fig. 5b and c). Finally, supramolecular transport experiments with green fluorescent protein (GFP) equipped with a nuclear localization signal (NLS) also confirmed the transport of this functional cargo by cell's nuclei staining due to GST-NLS-GFP accumulation (Fig. S9a, ESI†). Importantly, independent MTT toxicity assays confirmed that the IC50 toxicity values obtained for MP1 were between four and ten-fold higher than the optimal MP1 release concentration (30 μM and 10 μM for dextran and Cre recombinase respectively, Fig. S10–S12, ESI†). This remarkable ratio in the active/toxic concentration of MP1 confirmed the integrity of the plasma membrane and the non-lytic efficient transport of large macromolecules in live cells.
Transport of antibodies
These excellent results encouraged us to test the potential of the helical responsive MP1 for the transport of antibodies,5–7,40,60,61 which was evaluated with mouse monoclonal Mab414 against the nuclear pore complex proteins (Fig. 6 and Fig. S14, see ESI†). Transport of Mab414 and its functional binding to the nuclear membrane of the cells were confirmed by confocal microscopy (70% of nuclei labelling, Fig. 6b and c). Efficient Mab414 transport in ARPE-19 cells (human retinal pigmentary epithelium) was validated by a 57% nuclear membrane staining in these cells (Fig. 6c). Additionally, a fluorescently labelled immunoglobulin (IgG-CF) was further transported across cell membranes (Fig. S9b, ESI†). Importantly, all controls in the absence of MP1 resulted in endosomal entrapment and weaker punctate antibody fluorescence (left micrographs in Fig. 6b, c and Fig. S9b, S13, ESI†). Finally, proof of concept transport of antibodies in animal tissues was studied in murine cornea culture (Fig. 6d–g). The 3D confocal micrographs of the transfected corneas showed an excellent transport profile and nuclei labelling for the corneal epithelial cells by MP1 (Fig. 6e). Again, control experiments in the absence of MP1 showed weaker endosomal fluorescence signal (Fig. 6d). Fluorescein diacetate/propidium iodide live/dead staining was employed to check viability and integrity of the plasma membrane and the confocal micrographs confirmed an excellent viability of the cornea cells in the presence of MP1, which was nearly identical to the staining of untreated corneas (Fig. 6f and Fig. S14a, ESI†). Triton X-100 showed the red stained micrographs, which confirmed the toxic profile of amphiphilic detergents (Fig. 6g and Fig. S14b, ESI†).
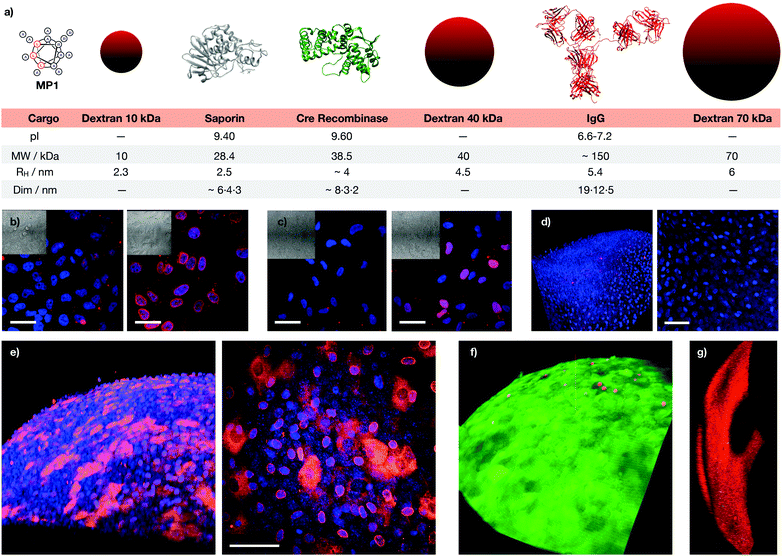 |
| | Fig. 6 Macromolecular cargos size and antibody transport in cells and tissues. (a) Table of the different cargos transported in this work (see ESI†). (b) HeLa cells were incubated with Mab414 and 0 (left) or 40 μM (right) MP1. (c) ARPE-19 cells incubated with Mab414 and 0 (left) or 50 μM (right) MP1. In (b and c), cells were incubated with the antibody for 1 h, followed by 2 h of incubation in serum supplemented media and fixation for immunofluorescence with an Alexa594 labelled secondary antibody (red); nuclei were stained with DAPI (blue). (d) Cornea incubated with Mab414. (e) Cornea incubated with Mab414 and 150 μM MP1. In (d and e), left panel is a 3D reconstruction of the tissue; right panel is the maximal projection of the stack. (f) Cytotoxicity in corneas treated with 150 μM MP1 by staining with propidium iodide (dead cells, red) and fluorescein diacetate (live cells, green). (g) Toxicity assay in cornea treated with Triton X-100. [Mab414] = 0.3 mg mL−1 in (b, d and e) and 0.15 mg mL−1 in (c). Scale bars are all 50 μm. | |
Conclusions
The objective of this work was to establish the potential of a rational strategy for the minimization of the number of cationic and hydrophobic residues in the development of membrane-targeted peptides capable of efficient but non-lytic transitory membrane disruption and macromolecular transport. The three key breakthroughs reported here are: (1) a rational strategy that allowed the discovery of oligoalanine peptide vehicles for protein transport with competitive efficiency compared to state of the art controls; (2) the non-toxic cytosolic delivery of exogenous monoclonal antibodies into animal tissues and (3) the assembly of supramolecular nanosized channels from a peptide sequence that is shorter than a lipid membrane. The discovery of penetrating peptide sequences showing different secondary structures such as random coils (e.g. CLIP6), alpha-helices (e.g. TP10, Pep-1) or beta-sheets (e.g. penetratin),5,62–64 might have led to the conclusion that a particular secondary structure would not be required for membrane translocation. In addition, despite the evidence of conformational changes from natural (e.g. melittin),65 synthetic (e.g. TP10, Pep-1),6,63,66 and recent rational strategies,67,68 the supramolecular control over the folding and assembly of small peptides with low cationic/amphiphilic character has not been sufficiently explored. The work described here introduces the selective non-lytic membrane perturbation from a peptide molecule shorter than a membrane leaflet. The peptide described here represents one of the simplest lineal peptide sequences with the minimum number of cationic and hydrophobic residues and yet capable of large protein (150 kDa) membrane transport, which allowed, to the best of our knowledge, the first example of antibody transport in animal tissues (cornea). It should be noted that since MP1 is not covalently bound to its cargo, the supramolecular dynamic interactions between peptide, membrane and cargo could influence the optimal peptide concentration for a particular cargo and in a particular membrane. It should be noted that the behaviour of these peptides could be different in artificial and in cell membranes. For example, the enhancement of endocytosis, the different membrane composition and the dynamic character of the cargos employed (i.e. protein shape) can influence the size-selective transport observed for MP1 in vesicles and cells. It should also be mentioned that in cell experiments, the transient membrane ruffling during endocytic or macropinocytic uptake of carrier and cargo might also lead to direct transport of the proteins.69,70 Control experiments with reported peptide vehicles10,12,38 confirmed MP1 as a competitive candidate for protein delivery with an excellent biocompatibility and efficiency as well as a structural simplicity. Beyond these particular properties, the importance of this work relies on the application of a rational design strategy to minimize the number of cationic and hydrophobic residues for the “en route” modulation of peptide amphiphilicity for non-destructive transient membrane permeation and cargo transport. Even though there is still no FDA approved penetrating peptide vehicle,71 this field is continuously improving in terms of carrier rational design and functional properties. The study presented here transversally covers the challenge of macromolecular transport from the molecular detail to the supramolecular level and up to the final functional applications in animal tissues. The simplicity of this new conceptual approach, together with the remarkable efficiency and broad scope of this methodology prompts new opportunities for the transport of macromolecules of biological interest and inspire the design and optimization of new synthetic membrane-targeted supramolecular transporters.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by the Spanish Agencia Estatal de Investigación (AEI) [SAF2017-89890-R], the Xunta de Galicia (ED431C 2017/25, 2016-AD031 and Centro Singular de Investigación de Galicia accreditation 2019–2022, ED431G 2019/03), the European Union (European Regional Development Fund – ERDF) and the ISCIII (RD16/0008/003). M. P. thanks the Xunta de Galicia (ED481A-2017/142), and M. J. and G. S. thank MINECO for their F. P. I. fellowships (BES-2015-071779; PRE2018-085973). R. G.-F. thanks a RyC (RYC-2016-20335), MINECO (RTI2018-098795-A-I00) and Xunta de Galicia (ED431F 2020/05). J. M. received a RyC (RYC-2013-13784), an ERC-Stg (DYNAP-677786), a HFSP (RGY0066/2017) and ISCIII (COV20/00297). All calculations were carried out at Centro de Supercomputación de Galicia (CESGA).
References
- G. Gasparini, E.-K. Bang, J. Montenegro and S. Matile, Chem. Commun., 2015, 51, 10389–10402 RSC.
- G. Wiedman, S. Y. Kim, E. Zapata-Mercado, W. C. Wimley and K. Hristova, J. Am. Chem. Soc., 2017, 139, 937–945 CrossRef CAS.
- I. Lostalé-Seijo and J. Montenegro, Nat. Rev. Chem., 2018, 2, 258–277 CrossRef.
- S. Y. Kim, A. E. Pittman, E. Zapata-Mercado, G. M. King, W. C. Wimley and K. Hristova, J. Am. Chem. Soc., 2019, 141, 6706–6718 CrossRef CAS.
- P. M. Fischer, Med. Res. Rev., 2007, 27, 755–795 CrossRef CAS.
- D. Kalafatovic and E. Giralt, Molecules, 2017, 22, 1929 CrossRef.
- M. Chiper, K. Niederreither and G. Zuber, Adv. Healthcare Mater., 2017, 347, 1701040 Search PubMed.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- A. Méndez-Ardoy, I. Lostalé-Seijo and J. Montenegro, ChemBioChem, 2018, 20, 488–498 CrossRef.
- M. C. Morris, J. Depollier, J. Mery, F. Heitz and G. Divita, Nat. Biotechnol., 2001, 19, 1173–1176 CrossRef CAS.
- S. Deshayes, M. Morris, F. Heitz and G. Divita, Adv. Drug Delivery Rev., 2008, 60, 537–547 CrossRef CAS.
- A. Erazo-Oliveras, K. Najjar, L. Dayani, T.-Y. Wang, G. A. Johnson and J.-P. Pellois, Nat. Methods, 2014, 11, 861–867 CrossRef CAS.
- J. Yang, A. Bahreman, G. Daudey, J. Bussmann, R. C. L. Olsthoorn and A. Kros, ACS Cent. Sci., 2016, 2, 621–630 CrossRef CAS.
- F. Versluis, I. Tomatsu, S. Kehr, C. Fregonese, A. W. J. W. Tepper, M. C. A. Stuart, B. J. Ravoo, R. I. Koning and A. Kros, J. Am. Chem. Soc., 2009, 131, 13186–13187 CrossRef CAS.
- J. Allen, K. Najjar, A. Erazo-Oliveras, H. M. Kondow-McConaghy, D. J. Brock, K. Graham, E. C. Hager, A. L. J. Marschall, S. Dübel, R. L. Juliano and J.-P. Pellois, ACS Chem. Biol., 2019, 14, 2641–2651 CrossRef CAS.
- A. Steinauer, J. R. LaRochelle, S. L. Knox, R. F. Wissner, S. Berry and A. Schepartz, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 512–521 CrossRef CAS.
- J. R. LaRochelle, G. B. Cobb, A. Steinauer, E. Rhoades and A. Schepartz, J. Am. Chem. Soc., 2015, 137, 2536–2541 CrossRef CAS.
- S. E. Miller, Y. Yamada, N. Patel, E. Suárez, C. Andrews, S. Tau, B. T. Luke, R. E. Cachau and J. P. Schneider, ACS Cent. Sci., 2019, 5, 1750–1759 CrossRef CAS.
- I. Lostalé-Seijo, I. Louzao, M. Juanes and J. Montenegro, Chem. Sci., 2017, 8, 7923–7931 RSC.
- K. Petkau-Milroy, M. H. Sonntag, A. H. A. M. van Onzen and L. Brunsveld, J. Am. Chem. Soc., 2012, 134, 8086–8089 CrossRef CAS.
- S. A. Bode, I. C. Kruis, H. P. J. H. M. Adams, W. C. Boelens, G. J. M. Pruijn, J. C. M. van Hest and D. W. P. M. Löwik, ChemBioChem, 2016, 18, 185–188 CrossRef.
- S. I. Lim, C. I. Lukianov and J. A. Champion, J. Controlled Release, 2017, 249, 1–10 CrossRef CAS.
- B. M. de Ronde, N. D. Posey, R. Otter, L. M. Caffrey, L. M. Minter and G. N. Tew, Biomacromolecules, 2016, 17, 1969–1977 CrossRef CAS.
- N. L. Benner, X. Zang, D. C. Buehler, V. A. Kickhoefer, M. E. Rome, L. H. Rome and P. A. Wender, ACS Nano, 2017, 11, 872–881 CrossRef CAS.
- E. Derivery, E. Bartolami, S. Matile and M. Gonzalez-Gaitan, J. Am. Chem. Soc., 2017, 139, 10172–10175 CrossRef CAS.
- S. Ulrich, Acc. Chem. Res., 2019, 52, 510–519 CrossRef CAS.
- H. Fernández-Caro, I. Lostalé-Seijo, M. Martínez-Calvo, J. Mosquera, J. L. Mascareñas and J. Montenegro, Chem. Sci., 2019, 10, 7923–7931 RSC.
- K. A. Mix, J. E. Lomax and R. T. Raines, J. Am. Chem. Soc., 2017, 139, 14396–14398 CrossRef CAS.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS.
- H. H. Wang and A. Tsourkas, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 22132 CrossRef CAS.
- I. Canton, M. Massignani, N. Patikarnmonthon, L. Chierico, J. Robertson, S. A. Renshaw, N. J. Warren, J. P. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, FASEB J., 2013, 27, 98–108 CrossRef CAS.
- C. Liu, T. Wan, H. Wang, S. Zhang, Y. Ping and Y. Cheng, Sci. Adv., 2019, 5, eaaw8922 CrossRef CAS.
- E. A. Prasetyanto, A. Bertucci, D. Septiadi, R. Corradini, P. Castro-Hartmann and L. De Cola, Angew. Chem., Int. Ed., 2016, 55, 3323–3327 CrossRef CAS.
- R. Mout, M. Ray, Y.-W. Lee, F. Scaletti and V. M. Rotello, Bioconjugate Chem., 2017, 28, 880–884 CrossRef CAS.
- P. Yuan, H. Zhang, L. Qian, X. Mao, S. Du, C. Yu, B. Peng and S. Q. Yao, Angew. Chem., Int. Ed., 2017, 56, 12481–12485 CrossRef CAS.
- M. Marsh and A. Helenius, Cell, 2006, 124, 729–740 CrossRef CAS.
- P. M. Matos, M. Marin, B. Ahn, W. Lam, N. C. Santos and G. B. Melikyan, J. Biol. Chem., 2013, 288, 12416–12425 CrossRef CAS.
- M. Akishiba, T. Takeuchi, Y. Kawaguchi, K. Sakamoto, H.-H. Yu, I. Nakase, T. Takatani-Nakase, F. Madani, A. Gräslund and S. Futaki, Nat. Chem., 2017, 9, 751–761 CrossRef CAS.
- N. J. Caron, S. P. Quenneville and J. P. Tremblay, Biochem. Biophys. Res. Commun., 2004, 319, 12–20 CrossRef CAS.
- A. Klimpel, T. Lützenburg and I. Neundorf, Curr. Opin. Pharmacol., 2019, 47, 8–13 CrossRef CAS.
- H. O. McCarthy, J. McCaffrey, C. M. McCrudden, A. Zholobenko, A. A. Ali, J. W. McBride, A. S. Massey, S. Pentlavalli, K.-H. Chen, G. Cole, S. P. Loughran, N. J. Dunne, R. F. Donnelly, V. L. Kett and T. Robson, J. Controlled Release, 2014, 189, 141–149 CrossRef CAS.
- A. Lamiable, P. Thévenet, J. Rey, M. Vavrusa, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2016, 44, W449–W454 CrossRef CAS.
- S. Marqusee, V. H. Robbins and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 5286–5290 CrossRef CAS.
- A. Chakrabartty, T. Kortemme and R. L. Baldwin, Protein Sci., 1994, 3, 843–852 CrossRef CAS.
- A. Ho, S. R. Schwarze, S. J. Mermelstein, G. Waksman and S. F. Dowdy, Cancer Res., 2001, 61, 474–477 CAS.
- M. Pazo, M. Juanes, I. Lostalé-Seijo and J. Montenegro, Chem. Commun., 2018, 54, 6919–6922 RSC.
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2003, 125, 14348–14356 CrossRef CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS.
- W. Li, F. Nicol and F. C. Szoka, Adv. Drug Delivery Rev., 2004, 56, 967–985 CrossRef CAS.
- F. Hullin-Matsuda, T. Taguchi, P. Greimel and T. Kobayashi, Semin. Cell Dev. Biol., 2014, 31, 48–56 CrossRef CAS.
- L. M. Reid, C. S. Verma and J. W. Essex, Drug Discovery Today, 2019, 24, 1821–1835 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS.
- L. A. Patel and J. T. Kindt, Soft Matter, 2016, 12, 1765–1777 RSC.
-
S. Matile and N. Sakai, The Characterization of Synthetic Ion Channels and Pores, Wiley-Blackwell, Weinheim, Germany, 2006 Search PubMed.
- H. D. Herce, A. E. Garcia, J. Litt, R. S. Kane, P. Martin, N. Enrique, A. Rebolledo and V. Milesi, Biophys. J., 2009, 97, 1917–1925 CrossRef CAS.
- T. M. Fyles, Acc. Chem. Res., 2013, 46, 2847–2855 CrossRef CAS.
- L. Li, T. Wan, M. Wan, B. Liu, R. Cheng and R. Zhang, Cell Biol. Int., 2015, 39, 531–539 CrossRef CAS.
- P. Aimar, M. Meireles and V. Sanchez, J. Membr. Sci., 1990, 54, 321–338 CrossRef CAS.
- G. Meinke, A. Bohm, J. Hauber, M. T. Pisabarro and F. Buchholz, Chem. Rev., 2016, 116, 12785–12820 CrossRef CAS.
- A. N. Zelikin, C. Ehrhardt and A. M. Healy, Nat. Chem., 2016, 8, 997–1007 CrossRef CAS.
- T. A. Slastnikova, A. V. Ulasov, A. A. Rosenkranz and A. S. Sobolev, Front. Pharmacol., 2018, 9, 1208 CrossRef CAS.
- S. H. Medina, S. E. Miller, A. I. Keim, A. P. Gorka, M. J. Schnermann and J. P. Schneider, Angew. Chem., Int. Ed., 2016, 55, 3369–3372 CrossRef CAS.
- M. Magzoub, L. E. G. Eriksson and A. Gräslund, Biochim. Biophys. Acta, 2002, 1563, 53–63 CrossRef CAS.
-
U. Langel, Cell-penetrating peptides: methods and protocols, Humana Press, Totowa, NJ, USA, 2011 Search PubMed.
- C. E. Dempsey and G. S. Butler, Biochemistry, 1992, 31, 11973–11977 CrossRef CAS.
- R. Rennert, I. Neundorf and A. G. Beck-Sickinger, Adv. Drug Delivery Rev., 2008, 60, 485–498 CrossRef CAS.
- S. Hyun, Y. Choi, H. N. Lee, C. Lee, D. Oh, D.-K. Lee, C. Lee, Y. Lee and J. Yu, Chem. Sci., 2018, 9, 3820–3827 RSC.
- W. He, X. Xing, X. Wang, D. Wu, W. Wu, J. Guo and S. Mitragotri, Adv. Funct. Mater., 2020, 1910566 CrossRef CAS.
- M. Akishiba and S. Futaki, Mol. Pharmaceutics, 2019, 16, 2540–2548 CrossRef CAS.
- H. Hirose, T. Takeuchi, H. Osakada, S. Pujals, S. Katayama, I. Nakase, S. Kobayashi, T. Haraguchi and S. Futaki, Mol. Ther., 2009, 20, 984–993 CrossRef.
- J. Xie, Y. Bi, H. Zhang, S. Dong, L. Teng, R. J. Lee and Z. Yang, Front. Pharmacol., 2020, 11, 697 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00103a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Giulia
Salluce
a,
Giulia
Salluce