
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of a chiral artificial enzyme used for enantioselective catalysis in live cells†
Ya
Zhou
ab,
Weili
Wei
a,
Fengchao
Cui
ab,
Zhengqing
Yan
a,
Yuhuan
Sun
ab,
Jinsong
Ren
 ab and
Xiaogang
Qu
*ab
ab and
Xiaogang
Qu
*ab
aLaboratory of Chemical Biology and State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: xqu@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, Anhui 230026, China
First published on 23rd September 2020
Abstract
Nanozymes as a newcomer in the artificial enzyme family have shown several advantages over natural enzymes such as their high stability in harsh environments, facile production on large scale, long storage time, low costs, and higher resistance to biodegradation. However, compared with natural enzymes, it is still a great challenge to design a nanozyme with high selectivity, especially high enantioselectivity. It is highly desirable and demanding to develop chiral nanozymes with high and on-demand enantioselectivity for practical applications. Herein, we present an unprecedented approach to construct chiral artificial peroxidase with ultrahigh enantioselectivity. Inspired by the structure of the natural enzyme horseradish peroxidase (HRP), we have constructed a series of stereoselective nanozymes (Fe3O4@Poly(AA)) by using the ferromagnetic nanoparticle (Fe3O4 NP) yolk as the catalytic core and amino acid-appended chiral polymer shell as the chiral selector. Among them, Fe3O4@Poly(D-Trp) exhibits the highest enantioselectivity. More intriguingly, their enantioselectivity will be readily reversed by replacing D-Trp with L-Trp. The selectivity factor is up to 5.38, even higher than that of HRP. Kinetic parameters, dialysis experiments, and molecular simulations together with activation energy reveal that the selectivity originates from the D-/L-Trp appended polymer shell, which can result in better affinity and catalytic activity to D-/L-tyrosinol. The artificial peroxidases have been used for asymmetric catalysis to prepare enantiopure D- or L-enantiomers. Besides, by using fluorescent labelled FITC-tyrosinolL and RhB-tyrosinolD, the artificial peroxidases can catalyze green or red fluorescent chiral tyrosinol to selectively label live yeast cells among yeast, S. aureus, E. coli and B. subtilis bacterial cells. This work opens a new avenue for better design of stereoselective artificial enzymes.
Introduction
The design and synthesis of artificial enzymes or enzyme mimetics with high activity and selectivity have been the focus in the pursuit of alternatives for natural enzymes since the middle of the last century.1 In the past 10 years, as a new generation of artificial enzymes, nanomaterials with enzyme–mimetic activity, named nanozymes, have attracted great interest because of their many advantages over natural enzymes, including high stability in high temperature and harsh environments, facile production on a large scale, long storage time, low costs, and higher resistance to biodegradation.2 Among them, the Fe3O4 nanozyme is a classical nanomaterial with intrinsic peroxidase-like activity and was first reported by Yan and coworkers in 2007.3 Different types of nanozymes have been developed and successfully used for biosensing,3a,4 cancer therapy,5 combating bacteria and biofilms,6 detoxification,7 and cytoprotection.8 Although promising, compared with natural enzymes, rational design and synthesis of nanozymes are still challenging,9,10 especially for the design of a chiral nanozyme with high selectivity.11 Therefore, it is important and necessary to develop chiral nanozymes with high and on-demand enantioselectivity for biosynthesis and practical applications.12In natural enzymes, such as horseradish peroxidase (HRP, Scheme 1a, left), stereoselective reactions can be realized in the catalytic site (heme) with the assistance of proximally arrayed L-amino acid residues.13 The utilization of chiral amino acids as chiral selectors has also been proved to be effective for the construction of chiral nanozymes.11d,e However, the previously reported chiral nanozymes relied on the direct modification of amino acids on the surface of nanoparticles. The density of the chiral selector is relatively low, resulting in low selectivity.11d,e Besides, part of the active sites are covered by direct modification, which can hinder their activity.11d,e Thus, a new modification strategy is desirable for the design of chiral nanozymes. The yolk–shell structure has been confirmed to be powerful for the construction of catalysts and this structure can avoid direct covalent bonding of amino acids blocking the central catalytic site of nanozymes.14 In addition, plenty of chiral amino acids can be introduced into the polymer shell to realize high efficiency chiral recognition.15 Inspired by the natural enzyme and advantages of the yolk–shell structure, a yolk–shell chiral nanozyme is designed by using Fe3O4 nanoparticle (NP) yolk as the catalytically active peroxidase-mimic and amino acid-appended polymer shell as the selector for chiral recognition (Fe3O4@Poly(AA), where Poly(AA) refers to polyacrylate(amino acid), Scheme 1a, right). The Poly(AA) shell allows selective access of chiral tyrosinol to the surface of peroxidase-like cores, thus yielding enantioselective artificial peroxidase. Scheme 1b illustrates well the synthesis process of Fe3O4@Poly(AA) via four steps, including preparing Fe3O4 NPs, coating Fe3O4 with a silica shell (Fe3O4@SiO2), modifying Fe3O4@SiO2 with a chiral polymer shell (Fe3O4@SiO2@Poly(AA)), and removing the silica shell to form yolk–shell Fe3O4@Poly(AA).
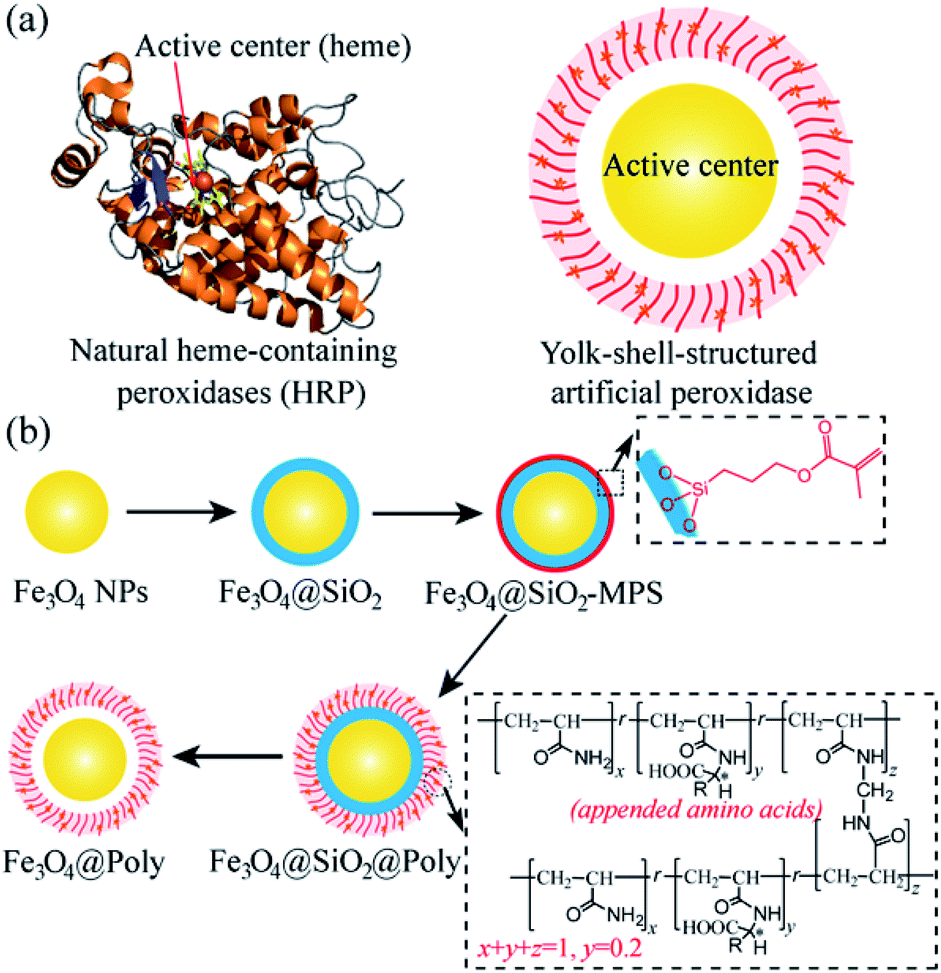 | ||
| Scheme 1 (a) Representative structures of natural heme-containing peroxidase (left) and yolk–shell-structured artificial peroxidase (right). (b) Preparation procedures of the yolk–shell-structured artificial peroxidase. | ||
Results and discussion
Fe3O4 NPs were first prepared via a hydrothermal process by using citrate as a stabilizer. Then, Fe3O4 NPs were coated with a SiO2 shell and modified with 3-(trimethoxysilyl)propylmethacrylate (MPS) to introduce carbon–carbon double bonds. As shown in Fig. 1a and b, Fe3O4 NPs with an average diameter of ca. 200 nm were homogeneously coated with a SiO2 shell of about 25 nm thickness. Next, amino acid D-/L-tryptophan (Trp), D-/L-phenylalanine (Phe), D-/L-aspartic acid (Asp), and D-/L-histidine (His) appended polymer monomers were synthesized, respectively, and their corresponding 1H nuclear magnetic resonance (NMR) spectra were shown in the ESI.† Then Fe3O4@SiO2 was coated with different amino acid appended polymers. By taking the L-Trp appended polymer as an example, the formation of the polymer shell on the Fe3O4@SiO2 was first characterized by transmission electron microscopy (TEM). Obviously, after coating the polymer shell, the surface became rough (Fig. 1c). The thickness of the polymer shell was about 8.5 nm as determined by dynamic light scattering (DLS) at room temperature (Fig. S1†). The mid-layer silica of Fe3O4@SiO2@Poly(L-Trp) was further selectively removed by etching in a highly concentrated NaOH solution to form yolk–shell structured Fe3O4@Poly(L-Trp) nanoparticles. As shown in Fig. 1d, a thin polymer coating around the surface of Fe3O4 NPs was observed by TEM, which was consistent with the results of DLS determination (Fig. S1†). The TEM elemental mapping images shown in Fig. 1e were further used to confirm the preparation of yolk–shell structured Fe3O4@Poly(L-Trp). Silica and polymer shells in Fe3O4@SiO2@Poly(L-Trp) were confirmed by the representative elements Si and N. As shown in Fig. 1f, NaOH selective etching led to the disappearance of Si and retention of N, further confirming the formation of yolk–shell Fe3O4@Poly(L-Trp). The existence of the chiral polymer could also be supported by the obviously enhanced UV-vis absorption and circular dichroism (CD) spectra (Fig. S2 and S3†).The X-ray photoelectron spectroscopy (XPS) spectrum in Fig. S4† showed minimal interaction between Fe3O4 and the amino acid polymer, further confirming the successful synthesis of the yolk–shell Fe3O4@Poly(L-/D-Trp). 3,3,5,5-tetramethylbenzidine (TMB) was used to measure the catalytic activity of Fe3O4@Poly(AA) in the presence of H2O2. All the catalytic activities of Fe3O4@Poly(AA) were found to be similar to that of bare Fe3O4 NPs, as reported previously.3 Thus, yolk–shell Fe3O4@Poly(AA) nanoparticles still exhibit peroxidase activity (Fig. S5†). Notably, the core-shell-structured Fe3O4@SiO2@Poly(AA) showed no catalytic activity due to the blocking of the catalytically active site (data not shown), suggesting the necessity of a yolk–shell structure. Taking Fe3O4@Poly(L-Trp) as an example, it had peroxidase activity in a wide pH range (Fig. S5†).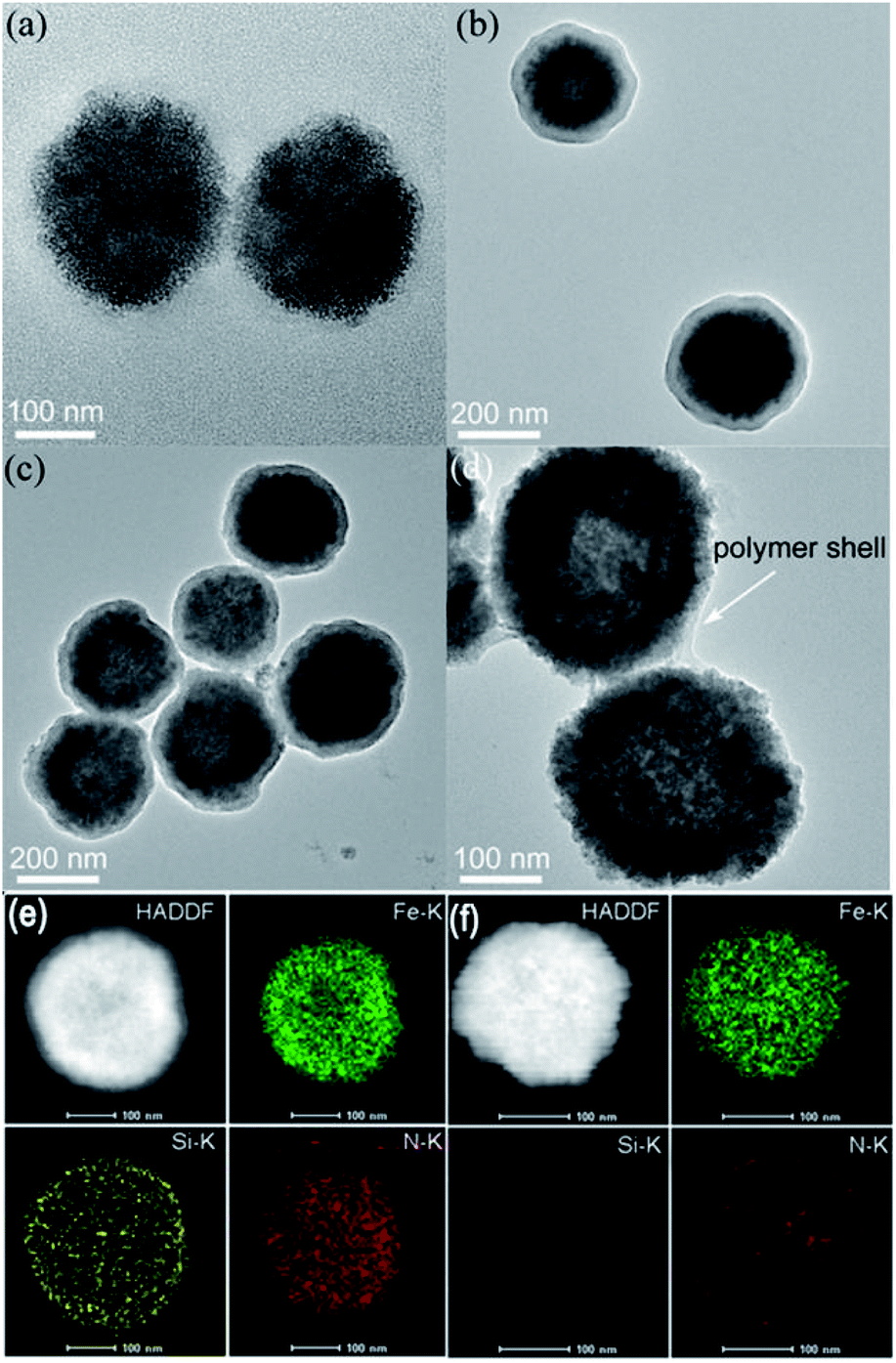 | ||
| Fig. 1 TEM images of (a) Fe3O4 NPs, (b) Fe3O4@SiO2, (c) Fe3O4@SiO2@Poly(L-Trp), and (d) yolk–shell Fe3O4@Poly(L-Trp). (e) and (f) show the TEM elemental mapping images of Fe3O4@SiO2@Poly(L-Trp) and yolk–shell Fe3O4@Poly(L-Trp), respectively. | ||
In the following, the catalytic activities of Fe3O4@Poly(AA) toward a specific chiral substrate (L-/D-tyrosinol) were further studied by monitoring the time-dependent absorbance at 320 nm (Fig. 2a). The initial rates of the oxidation of L-/D-tyrosinol enantiomers could reveal the enantioselectivity of Fe3O4@Poly(AA). Fig. 2b showed the initial rates for the oxidation of L-and D-tyrosinol with Fe3O4@Poly(L-Trp), Fe3O4@Poly(D-Trp) and bare Fe3O4, respectively. Fe3O4@Poly(D-Trp) showed a better catalytic activity for D-tyrosinol than L-tyrosinol. The ratio calculated using the initial rates of oxidation of the D enantiomer and L enantiomer for Fe3O4@Poly(D-Trp) was 2.38. Interestingly, the enantioselectivity was reversed completely by using Fe3O4@Poly(L-Trp) with a ratio of 0.37, which was consistent with the first principles of stereochemistry.16 This indicated that the enantioselectivity of our designed nanozyme could be reversed readily by changing the chirality of the polymer shell. Similarly, the oxidation of D-/L-tyrosinol enantiomers by a series of Fe3O4@Poly(AA) was also investigated and the corresponding results were shown in Fig. 2c. As shown in Fig. 2c, Fe3O4@Poly(Phe) and Fe3O4@Poly(Asp) showed moderate enantioselectivity, while Fe3O4@Poly(His) showed poor enantioselectivity. In contrast, bare Fe3O4 did not show any stereoselectivity for oxidizing L-or D-tyrosinol.
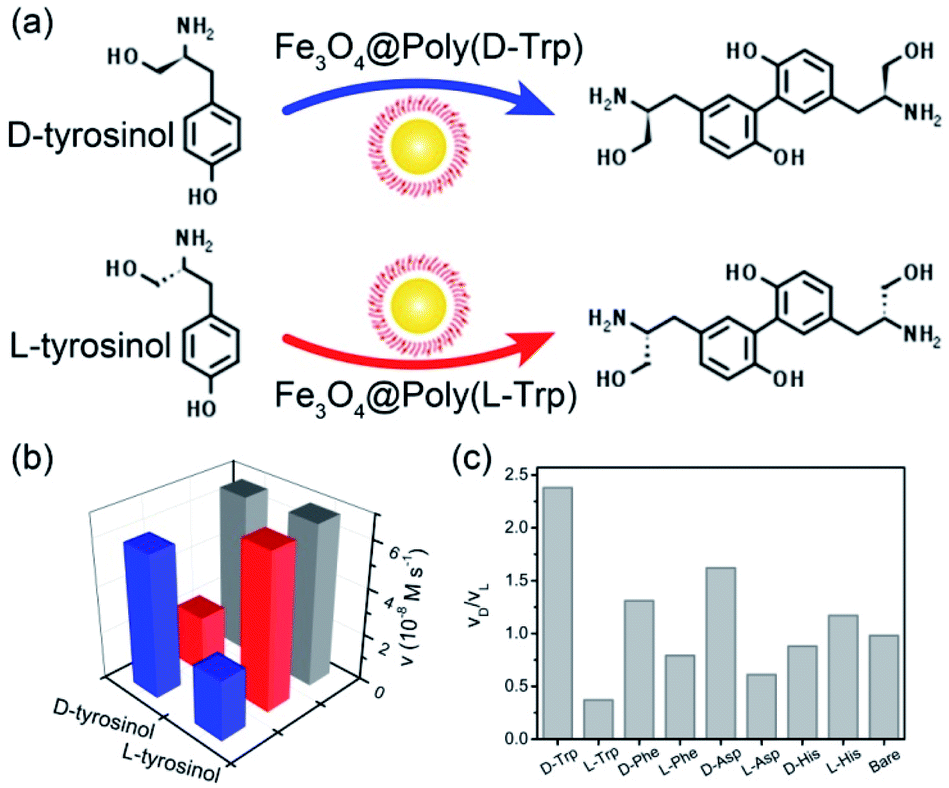 | ||
| Fig. 2 (a) The enantioselective oxidation of chiral tyrosinol catalyzed by Fe3O4@Poly(L-/D-Trp). (b) The initial velocity for the oxidation of tyrosinol enantiomers with Fe3O4@Poly(D-Trp) (blue), Fe3O4@Poly(L-Trp) (red) and bare Fe3O4 (gray). (c) The ratio of initial rates for the oxidation of the D enantiomer to L enantiomer. | ||
To better understand the stereoselectivity of Fe3O4@Poly(Trp) towards tyrosinol enantiomers, we analyzed and compared the kinetic parameters based on the Michaelis–Menten model according to the saturation curves in Fig. S6.† The results were summarized in Table S1.† The smaller the KM, the higher the binding affinity between enzymes and substrates. The catalytic number (kcat) indicates the ability of enzymes to catalyze one certain substrate. kcat/KM is usually applied for describing the catalytic efficiency of the enzyme. For Fe3O4@Poly(D-Trp), kcat values for D- and L-tyrosinol were (16.44 ± 0.40)×103 s−1 and (10.78 ± 1.27)×103 s−1, respectively, indicating the higher catalytic ability of Fe3O4@Poly(D-Trp) for oxidizing D-tyrosinol rather than L-tyrosinol. Moreover, a lower KM value was observed for D-tyrosinol than for L-tyrosinol, implying stronger binding affinity of Fe3O4@Poly(D-Trp) for D-tyrosinol. The higher catalytic activity and binding affinity to D-tyrosinol resulted in higher catalytic efficiency toward D-tyrosinol. As decided by kcat/KM values, Fe3O4@Poly(D-Trp) was 5.38 times more active for D-tyrosinol than for L-tyrosinol. As for Fe3O4@Poly(L-Trp), the preference was reversed to L-tyrosinol due to the better catalytic ability and binding affinity to L-tyrosinol. Thus, the selectivity of the yolk–shell nanozyme originated from the higher activity and affinity to D-/L-tyrosinol and the preference was related to the chirality of the polymer. The selectivity factor of the yolk–shell nanozyme was higher than those of many chiral nanozymes reported before, such as graphene oxide-based peroxidase,11a gold nanoparticle-based glucose oxidase,11b gold nanoparticle-based phosphorylase,11c ceria nanoparticle-based oxidase11d and gold nanoparticle-based peroxidase11e (Table S2†). In addition, in the oxidation of tyrosinol enantiomers, the Fe3O4@Poly(D-Trp) had a higher selectivity factor than HRP, which was reported to be 4.77 according to a previous report (Table S2†).13c The improvement of enantioselectivity could be attributed to the strategies of the surface modifications.9b,17 Compared with the previously reported chiral nanozymes, in which the catalytic sites were partly covered by the direct modification of the chiral selector even though the density of the chiral selector was relatively low, our designed yolk–shell structure allowed highly dense modification of amino acids as the chiral selector but not direct covalent bonding to the central catalytic site. This enhanced the selectivity without blocking the catalytic site of the nanozyme.15 This design endowed the artificial peroxidase with high and on-demand enantioselectivity.
The selective binding affinity and catalytic activity of Fe3O4@Poly(AA) toward L-/D-tyrosinol were further confirmed. We first conducted dialysis experiments to reveal the binding preference.18 Fe3O4@Poly(AA) in the dialysis tube was dialyzed against racemic mixtures of tyrosinol, and circular dichroism was used to monitor the dialysate for enrichment of the enantiomer with weaker interaction with Fe3O4@Poly(AA). As shown in Fig. S7,† the dialysate for bare Fe3O4 was racemic, indicating no binding preference to tyrosinol enantiomers. The dialysate for Fe3O4@Poly(L-Trp) and Fe3O4@Poly(D-Trp) was rich in D-tyrosinol and L-tyrosinol, respectively, which obviously indicated the higher affinity of Fe3O4@Poly(L-Trp) to L-tyrosinol and Fe3O4@Poly(D-Trp) to D-tyrosinol. Here we proposed a possible catalytic process: D-/L-tyrosinol first diffused through the Poly(D-/L-Trp) shell, then reacted with the hydroxyl radicals, which were formed from the Fe3O4 NP-catalyzed decomposition of hydrogen peroxide. The obtained phenoxy radical could then couple with another D-/L-tyrosinol to give the final products. We supposed that the selectivity should be more relevant to the diffusion process of D-/L-tyrosinol since the bare Fe3O4 NPs displayed no selectivity to the tyrosinol enantiomers. The diffusion of L-/D-tyrosinol into the inner catalytic core through the Poly(D-/L-Trp) shell could be divided into three processes: adsorption, transport and desorption. We then examined the role of L-/D-tyrosinol adsorption/desorption using a patch of Poly(D-/L-Trp) shell models by molecular simulation methods (Fig. S8†). The amino acid-appended polymer shell could selectively transport L-/D-tyrosinol from the outer solution into the inner catalytic yolk. Therefore, the adsorption of L-/D-tyrosinol on the outer surface of the Poly(D-/L-Trp) shell and the desorption from the inner surface of the Poly(D-/L-Trp) shell could give information about the binding preference. In order to clearly demonstrate this fact, we investigated the top 3 binding structures of L-/D-tyrosinol with the outer and inner surface of Poly(D-/L-Trp) (shown in Fig. S9†), respectively. The detailed illustration in Fig. 3 depicted the best binding mode of L-/D-tyrosinol on the outer surface of the Poly(D-/L-Trp) shell. We also examined their corresponding average adsorption free energy (AFE) on the outer surface and desorption free energy (DFE) from the inner surface of Poly(D-/L-Trp) (listed in Table S3†) based on the top 3 binding models. It was found that the AFE of D-tyrosinol on the outer surface of Poly(D-Trp) was much lower than that of L-tyrosinol by 2.9 kcal mol−1, indicating that Poly(D-Trp) was more prone to adsorb D-tyrosinol, not L-tyrosinol. Similarly, Poly(L-Trp) was more favourable for the adsorption of L-tyrosinol with a free energy difference of 1.8 kcal mol−1 compared to D-tyrosinol. More interestingly, we also noted that the desorption of D-tyrosinol from Poly(D-Trp) and L-tyrosinol from Poly(L-Trp) was easier than the opposite configuration because of the lower binding free energy. Nevertheless, it was also found that the differences of adsorption free energy were distinctly larger than those of desorption free energy, indicating that the adsorption process played an important role in the enantioselectivity of Poly(D-/L-Trp). Furthermore, we also decomposed the binding free energy into individual energy components to shed light on the dominant interaction for driving D-tyrosinol and L-tyrosinol binding to Poly(D-/L-Trp), as presented in Table S4.† We found that van der Waals interactions provided a major contribution to the difference of adsorption free energy of D-tyrosinol and L-tyrosinol on the outer surface of Poly(D-Trp) and Poly(L-Trp). This indicated that van der Waals interactions were closely correlated with the enantioselectivity of Fe3O4@Poly(D-/L-Trp). Electrostatic interactions, determined by the hydrogen bond interactions, were favourable for the adsorption of D-tyrosinol on the outer surface of Poly(D-Trp), while it showed unfavourable contributions to the adsorption of L-tyrosinol on the outer surface of Poly(L-Trp). The results of molecular simulation were consistent with dialysis experiments, and demonstrated that Poly(D-Trp) and Poly(L-Trp) contributed to the binding preference of Fe3O4@Poly(D-/L-Trp) to D-/L-tyrosinol. It is worth mentioning that the transport dynamics of L-/D-tyrosinol within the Poly(D-/L-Trp) shell should also play an important role in the catalytic enantioselectivity, which will be studied in the future. Then, the activation energy (Ea) was calculated according to the Arrhenius equation19 (Fig. S10†). For Fe3O4@Poly(D-Trp), the Ea values for D-tyrosinol and L-tyrosinol were estimated to be 65.1 ± 2.3 kJ mol−1 and 86.6 ± 2.4 kJ mol−1, respectively. It was obvious that in the presence of Fe3O4@Poly(D-Trp), the energy barrier for the oxidation of D-tyrosinol was lower. In contrast, Fe3O4@Poly(L-Trp) exhibited a lower energy barrier for the oxidation of L-tyrosinol. The results confirmed undoubtedly the higher catalytic activity of Fe3O4@Poly(D-Trp) to D-tryosinol and Fe3O4@Poly(L-Trp) to L-tyrosinol, which was consistent with the kinetic parameters.
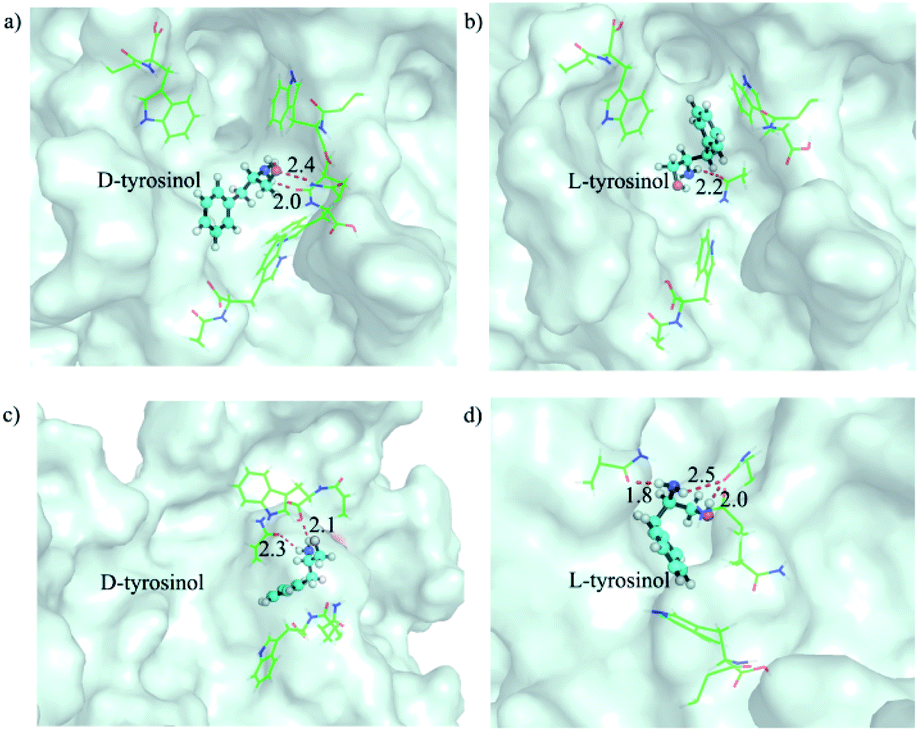 | ||
| Fig. 3 The representative binding models of D and L-tyrosinol on the outer surface of the (a, b) Poly(D-Trp) and (c, d) Poly(L-Trp) shell. The outer surfaces of Poly(D-/L-Trp) are shown as pale cyan surfaces. D-/L-tyrosinol is shown as a ball_and_stick model. The residues stabilizing D and L-tyrosinol by hydrophobic or hydrogen bond interactions are shown as lines. Hydrogen bonds are shown as red dashes and their distances are noted. | ||
The performance of Fe3O4@Poly(L-/D-Trp) in enantioselective catalysis was further studied by high-performance liquid chromatography (HPLC). As shown in Fig. S11,† in the presence of Fe3O4@Poly(L-Trp), L-tyrosinol was almost consumed to generate the product with an 89% yield. The main product was found to be L-dityrosinol according to the 1H NMR spectra in Fig. S12a.† In contrast, the yield for the reaction of D-tyrosinol was just 32%. For Fe3O4@Poly(D-Trp), D-tyrosinol was almost transformed to D-dityrosinol with a 92% yield as shown in Fig. S11d and S12b,† while the yield for the reaction of L-tyrosinol was only 29%. The results displayed the catalytic selectivity of our nanozymes toward L-/D-tyrosinol. The chiral nanozyme was then applied for the oxidation of racemic tyrosinol. As shown in Fig. S13,† with Fe3O4@Poly(L-Trp), L-tyrosinol was almost consumed while D-tyrosinol was in excess, indicating the higher conversion of L-tyrosinol. As for Fe3O4@Poly(D-Trp), D-tyrosinol was almost consumed while L-tyrosinol was in excess, indicating the higher conversion of D-tyrosinol. The oxidation of the racemic substrate further proved that the oxidation of tyrosinol by Fe3O4@Poly(L-/D-Trp) was enantioselective. Besides, the enantiomeric excess was up to 99% toward D-tyrosinol with the catalysis of Fe3O4@Poly(L-Trp). This meant that enantiopure D-tyrosinol was prepared. Similarly, enantiopure L-tyrosinol was prepared from racemate tyrosinol with the catalysis of Fe3O4@Poly(D-Trp). Significantly, the results suggested the potential ability of our artificial peroxidase for preparation of optically pure compounds.
Then, we verified whether the catalytic activity and selectivity toward L-/D-tyrosinol of our designed nanozymes can be realized in living cells. Live/dead cell staining was first performed. As shown in Fig. S14,† the nanozyme and H2O2 showed minimal influence on the cell viability. Klibanov et al. demonstrated that reactive oxygen species can catalyze tyrosinol to form free phenol radicals, subsequently covalently linked to the tyrosine residues on the surface of yeast cells.20 For visualizing these processes, we modified the L-/D-tyrosinol with FITC and Rhodamine B (RhB), marked as FITC-tyrosinolL and RhB-tyrosinolD. Yeast cells were treated with FITC-tyrosinolL and RhB-tyrosinolD in the presence of bare Fe3O4, Fe3O4@Poly(L-Trp) or Fe3O4@Poly(D-Trp), respectively (Scheme S1†). As shown in Fig. 4, for the control group, negligible fluorescence was observed in the yeast cells. For the bare Fe3O4 group, both green and red fluorescence were observed, which suggested that bare Fe3O4 NPs had no enantioselectivity towards tyrosinol. For the Fe3O4@Poly(L-Trp) group, the green fluorescence was much stronger than the red fluorescence, indicating the high enantioselectivity of Fe3O4@Poly(L-Trp) toward L-tyrosinol. For the Fe3O4@Poly(D-Trp) group, strong red fluorescence was observed on the surface of yeast cells, indicating the high enantioselectivity of Fe3O4@Poly(D-Trp) towards D-tyrosinol. Flow cytometry (Fig. 4b) also showed similar results, further supporting the confocal fluorescence data.
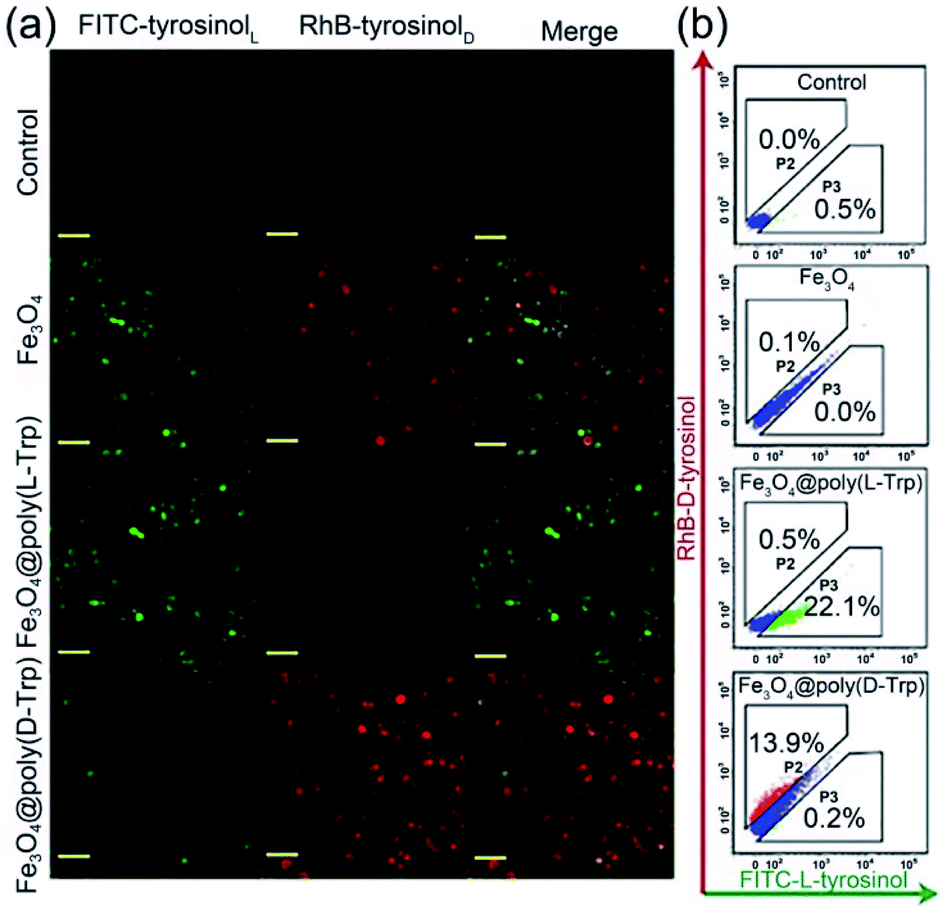 | ||
| Fig. 4 (a) Confocal fluorescence microscopy and (b) flow cytometry analysis results of yeast cells treated with H2O2 (100 μM), FITC-tyrosinolL and RhB-tyrosinolD with no nanozyme (control), bare Fe3O4, Fe3O4@Poly(L-Trp) or Fe3O4@Poly(D-Trp), respectively. (Scale bars: 20 μm.) | ||
We also attempted to label bacterial cells, such as S.aureus, E.coli and B.subtilis with FITC-tyrosinolL/RhB-tyrosinolD by using the same assay as we did for yeast cells. As shown in Fig. S15–20,† neither green emission nor red emission was observed in all groups including the control, bare Fe3O4, Fe3O4@Poly(L-Trp) and Fe3O4@Poly(D-Trp). These results indicated that the surface of S.aureus, E.coli and B.subtilis cells were difficult to label with a tyrosinol-based fluorescent agent. Next four different microbial cells (yeast, S.aureus, E.coli and B.subtilis) were treated with Fe3O4@poly(L-Trp), H2O2, FITC-tyrosinolL and RhB-tyrosinolD to demonstrate the specificity of the labelling process towards yeast. As shown in Fig. 5, only yeast cells showed green fluorescence among these cells because of the abundant tyrosine residues on the surface of yeast cells.20 The different labelling results on S.aureus, E.coli and B.subtilis might be due to the different structures and chemical compositions of their cell walls. The specific feature of the Gram-positive bacterial cell wall (S. aureus) is that it is composed of teichoic/lipoteichoic acids. The characteristic components of the Gram-negative bacterial cell wall (E.coli and B.subtilis) are lipopolysaccharides.21 Fewer tyrosine residues exist on the bacterial cell wall compared with the yeast cells. Thus fewer fluorescent agents could be labelled on their surface.
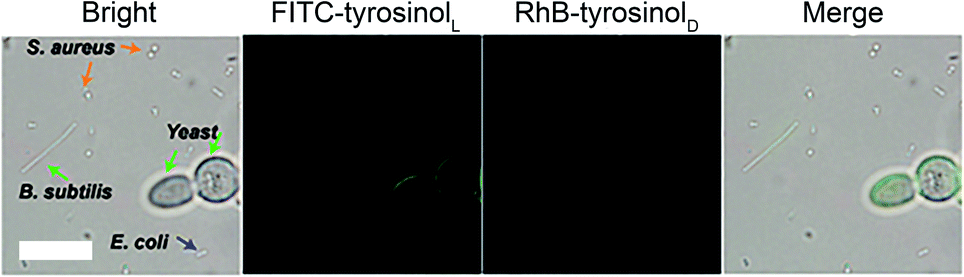 | ||
| Fig. 5 Fe3O4@Poly(L-Trp) NPs catalyze FITC-L-tyrosinol to label yeast cell with specificity. Fluorescence images of different microbial cells (yeast, S.aureus, E.coli and B.subtilis) after treatment with Fe3O4@Poly (L-Trp), H2O2 (100 μM), FITC-tyrosinolL and RhB-tyrosinolD. (Scale bars: 10 μm.) | ||
Conclusions
In summary, we present an unprecedented approach to integrate the active yolk artificial peroxidase Fe3O4 NP and D- or L-amino acid-appended chiral selective polymer shell for construction of a series of yolk–shell structured artificial peroxidases. Different from the chiral nanozymes with direct surface modification, the chiral artificial peroxidases exhibit high enantioselectivity. Among them, Fe3O4@Poly(D-Trp) shows the highest selectivity factor of 5.38, which is even higher than that of HRP. The enantioselectivity is readily reversed by replacing D-Trp with L-Trp. Kinetic parameters, dialysis experiments, and molecular simulations together with activation energy reveal that the D-/L-Trp appended polymer shell results in better affinity and catalytic activity to D-/L-tyrosinol, thus leading to the selectivity of Fe3O4@Poly(D-Trp) to D-tryosinol and Fe3O4@Poly(L-Trp) to L-tryosinol, respectively. These artificial peroxidases could be used for preparing enantiopure D- or L-enantiomers. Besides, confocal fluorescence microscopy and flow cytometry analysis have demonstrated that fluorescent dye-modified chiral tyrosinol can selectively label yeast cells among S.aureus, yeast, E.coli and B.subtilis in the presence of our designed nanozyme. This work would promote rational design and synthesis of stereoselective artificial enzymes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Key R&D Program of China (2019YFA0709202), Natural Science Foundation of China (21533008, 91856205, 21820102009, 21871249), and Key Program of Frontier of Sciences (CAS QYZDJ-SSW-SLH052).Notes and references
- (a) X. Huang, X. Liu, Q. Luo, J. Liu and J. Shen, Chem. Soc. Rev., 2011, 40, 1171 RSC; (b) M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138 CrossRef CAS; (c) Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890 RSC; (d) E. Golub, H. B. Albada, W. C. Liao, Y. Biniuri and I. Willner, J. Am. Chem. Soc., 2016, 138, 164 CrossRef CAS; (e) D. S. M. Xavier Garrabou, B. I. M. Wicky and D. Hilvert, Angew. Chem., Int. Ed., 2018, 57, 5288 CrossRef.
- (a) F. Manea, F. B. Houillon, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2004, 43, 6165 CrossRef CAS; (b) H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060 RSC; (c) Y. Lin, J. Ren and X. Qu, Acc. Chem. Res., 2014, 47, 1097 CrossRef CAS; (d) M. Raynal, P. Ballester, A. Vidal-Ferran and P. Leeuwen, Chem. Soc. Rev., 2014, 43, 1734 RSC; (e) F. Mancin, L. J. Prins, P. Pengo, L. Pasquato, P. Tecilla and P. Scrimin, Molecules, 2016, 21, 1014 CrossRef.
- (a) L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577 CrossRef CAS; (b) L. Gao, K. Fan and X. Yan, Theranostics, 2017, 7, 3207 CrossRef CAS.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206 CrossRef CAS.
- (a) M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef; (b) P. Prasad, C. R. Gordijo, A. Z. Abbasi, A. Maeda, A. Ip, A. M. Rauth, R. S. DaCosta and X. Y. Wu, ACS Nano, 2014, 8, 3202 CrossRef CAS; (c) M. Song, T. Liu, C. Shi, X. Zhang and X. Chen, ACS Nano, 2016, 10, 633 CrossRef CAS; (d) J. Kim, H. R. Cho, H. Jeon, D. Kim, C. Song, N. Lee, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2017, 139, 10992 CrossRef CAS; (e) C. Yao, W. Wang, P. Wang, M. Zhao, X. Li and F. Zhang, Adv. Mater., 2018, 30, e1704833 CrossRef.
- (a) F. Natalio, R. Andre, A. F. Hartog, B. Stoll, K. P. Jochum, R. Wever and W. Tremel, Nat. Nanotechnol., 2012, 7, 530 CrossRef CAS; (b) Z. Chen, Z. Wang, J. Ren and X. Qu, Acc. Chem. Res., 2018, 51, 789 CrossRef CAS.
- (a) Y. Li, X. He, J. J. Yin, Y. Ma, P. Zhang, J. Li, Y. Ding, J. Zhang, Y. Zhao, Z. Chai and Z. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1832 CrossRef CAS; (b) A. Vernekar, T. Das and G. Mugesh, Angew. Chem., Int. Ed., 2016, 55, 1412 CrossRef CAS; (c) Y. Huang, Z. Liu, C. Liu, E. Ju, Y. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2016, 55, 6646 CrossRef CAS; (d) M. Soh, D. W. Kang, H. G. Jeong, D. Kim, D. Y. Kim, W. Yang, C. Song, S. Baik, I. Y. Choi, S. K. Ki, H. J. Kwon, T. Kim, C. K. Kim, S. H. Lee and T. Hyeon, Angew. Chem., Int. Ed., 2017, 56, 11399 CrossRef CAS.
- (a) X. Liu, Q. Wang, H. Zhao, L. Zhang, Y. Su and Y. Lv, Analyst, 2012, 137, 4552 RSC; (b) W. Zhang, S. Hu, J. J. Yin, W. He, W. Lu, M. Ma, N. Gu and Y. Zhang, J. Am. Chem. Soc., 2016, 138, 5860 CrossRef CAS; (c) N. Singh, M. A. Savanur, S. Srivastava, P. D'Silva and G. Mugesh, Angew. Chem., Int. Ed., 2017, 56, 14267 CrossRef CAS; (d) W. Li, Z. Liu, C. Liu, Y. Guan, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2017, 44, 13661 CrossRef.
- (a) K. Fan, H. Wang, J. Xi, Q. Liu, X. Meng, D. Duan, L. Gao and X. Yan, Chem. Commun., 2016, 53, 424 RSC; (b) B. Liu and J. Liu, Nano Res., 2017, 10, 1125 CrossRef CAS; (c) Z. Zhang, X. Zhang, B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 5412 CrossRef CAS.
- (a) S. Ghosh, P. Roy, N. Karmodak, E. D. Jemmis and G. Mugesh, Angew. Chem., Int. Ed., 2018, 57, 4510 CrossRef CAS; (b) G. Fang, W. Li, X. Shen, J. M. Perez-Aguilar, Y. Chong, X. Gao, Z. Chai, C. Chen, C. Ge and R. Zhou, Nat. Commun., 2018, 9, 129 CrossRef.
- (a) C. Xu, C. Zhao, M. Li, L. Wu, J. Ren and X. Qu, Small, 2014, 10, 1841 CrossRef CAS; (b) P. Zhan, Z.-G. Wang, N. Li and B. Ding, ACS Catal., 2015, 5, 1489 CrossRef CAS; (c) J. L. Chen, C. Pezzato, P. Scrimin and L. J. Prins, Chem.–Eur. J., 2016, 22, 7028 CrossRef CAS; (d) Y. Sun, N. Gao, J. Ren and X. Qu, Chem.–Eur. J., 2017, 23, 18146 CrossRef CAS; (e) Y. Zhou, H. Sun, H. Xu, S. Matysiak, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2018, 57, 16791–16795 CrossRef CAS.
- (a) T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4890 CrossRef; (b) M. Bartok, Chem. Rev., 2010, 110, 1663 CrossRef CAS; (c) Y. Zhang, J. Guo, L. Shi, Y. F. Zhu, K. Hou, Y. L. Zheng and Z. Y. Tang, Sci. Adv., 2017, 3, e1701162 CrossRef.
- (a) M. Deurzen, F. Rantwijk and R. A. Sheldon, Tetrahedron, 1997, 53, 13183 CrossRef; (b) W. A. Ute Hoch, R. Fell, C. R. Saha-Miiller and P. Schreier, J. Mol. Catal. A: Chem., 1997, 117, 321 CrossRef; (c) M. A. Gilabert, L. G. Fenoll, F. Garcia-Molina, P. A. Garcia-Ruiz, J. Tudela, F. Garcia-Canovas and J. N. Rodriguez-Lopez, Biol. Chem., 2004, 385, 1177 CAS.
- (a) E. Gross, F. D. Toste and G. A. Somorjai, Catal. Lett., 2015, 145, 126 CrossRef CAS; (b) S. Wu, J. Dzubiella, J. Kaiser, M. Drechsler, X. Guo, M. Ballauff and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 2229 CrossRef CAS.
- K. Sakaki, in Encyclopedia of Membranes, ed. E. Drioli and L. Giorno, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 1–2, DOI:10.1007/978-3-642-40872-4_1428-3.
- (a) S. H. Shabbir, C. J. Regan and E. V. Anslyn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10487 CrossRef CAS; (b) W. Wei, L. Wu, C. Xu, J. Ren and X. Qu, Chem. Sci., 2013, 4, 1156 RSC.
- (a) N. A. Rossi, I. Constantinescu, D. E. Brooks, M. D. Scott and J. N. Kizhakkedathu, J. Am. Chem. Soc., 2010, 132, 3423 CrossRef CAS; (b) J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O'Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Nat. Chem., 2017, 9, 537 CrossRef CAS.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655 CrossRef CAS.
- (a) H. Sun, A. Zhao, N. Gao, K. Li, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2015, 54, 7176 CrossRef CAS; (b) M. Menzinger and R. Wolfgang, Angew. Chem., Int. Ed., 1969, 8, 438 CrossRef CAS.
- (a) D. Lipovsek, E. Antipov, K. A. Armstrong, M. J. Olsen, A. M. Klibanov, B. Tidor and K. D. Wittrup, Chem. Biol., 2007, 14, 1176–1185 CrossRef CAS; (b) E. Antipov, A. E. Cho, K. D. Wittrup and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17694–17699 CrossRef CAS.
- (a) G. Bugla-Ploskonska, Postępy Mikrobiologii, 2008, 47, 191–197 CAS; (b) M. Rohde, Microbiol. Spectrum, 2019, 7, 21 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods in detail; kinetic parameters; comparison of kinetic parameters; the adsorption/desorption free energy; characterizations of materials; pH dependent activity of nanozymes; saturation curves; CD spectra for dialysis experiments; the shell and binding structure models; HPLC chromatograms; live/dead cell staining; fluorescence microscopy and flow cytometry analysis of the labelling of S. aureus, E. coli and B. subtilis. See DOI: 10.1039/d0sc03082a |
| This journal is © The Royal Society of Chemistry 2020 |