
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum(II) non-covalent crosslinkers for supramolecular DNA hydrogels†
Kaka
Zhang
 and
Vivian Wing-Wah
Yam
*
and
Vivian Wing-Wah
Yam
*
Institute of Molecular Functional Materials, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, PR China. E-mail: wwyam@hku.hk
First published on 5th March 2020
Abstract
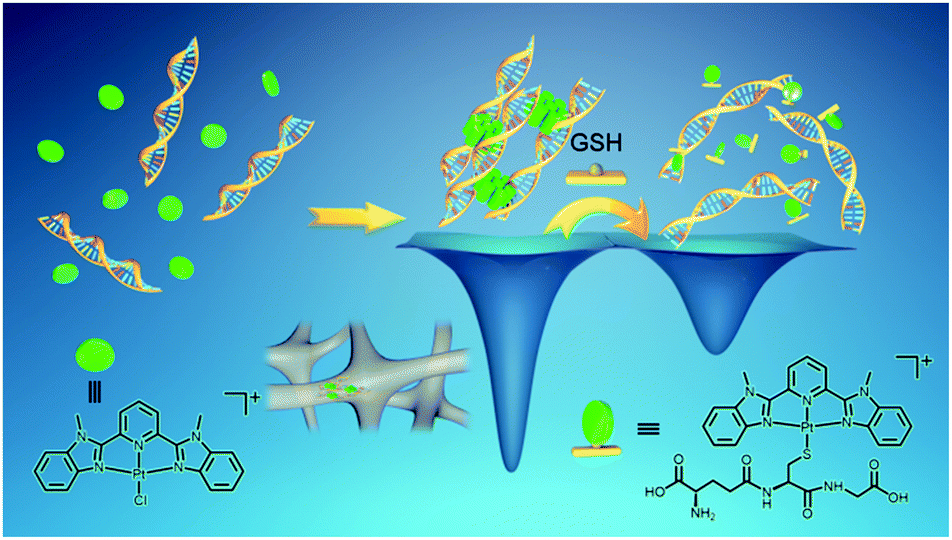
Manipulation of non-covalent metal–metal interactions allows the fabrication of functional metallosupramolecular structures with diverse supramolecular behaviors. The majority of reported studies are mostly designed and governed by thermodynamics, with very few examples of metallosupramolecular systems exhibiting intriguing kinetics. Here we report a serendipitous finding of platinum(II) complexes serving as non-covalent crosslinkers for the fabrication of supramolecular DNA hydrogels. Upon mixing the alkynylplatinum(II) terpyridine complex with double-stranded DNA in aqueous solution, the platinum(II) complex molecules are found to first stack into columnar phases by metal–metal and π–π interactions, and then the columnar phases that carry multiple positive charges crosslink the negatively charged DNA strands to form supramolecular hydrogels with luminescence properties and excellent processability. Subsequent platinum(II) intercalation into DNA competes with the metal–metal and π–π interactions at the crosslinking points, switching on the spontaneous gel-to-sol transition. In the case of a chloro (2,6-bis(benzimidazol-2′-yl)pyridine)platinum(II) complex, with [Pt(bzimpy)Cl]+ serving as a non-covalent crosslinker where the metal–metal and π–π interactions outcompete platinum(II) intercalation, the intercalation-driven gel-to-sol transition pathway is blocked since the gel state is energetically more favorable than the sol state. Interestingly, the ligand exchange reaction of the chloro ligand in [Pt(bzimpy)Cl]+ with glutathione (GSH) has endowed the complexes with enhanced hydrophilicity, decreasing the planarity of the complexes, and turning off the metal–metal and π–π interactions at the crosslinking points, leading to GSH-triggered hydrogel dissociation.
Introduction
Platinum(II) polypyridine complexes of d8 electronic configuration and square-planar geometry have attracted growing attention because of their intriguing spectroscopic and luminescence properties, as well as their diverse supramolecular behaviors associated with non-covalent metal–metal and π–π interactions.1 These platinum(II) complexes can form highly ordered extended linear chains or oligomeric structures and display rich polymorphism in the solid state.1,2 Metal–metal and π–π interactions serve as the main driving force to induce the aggregation of platinum(II) complexes in the solution state and direct the fabrication of platinum(II) supramolecular assemblies such as molecular architectures,3 liquid crystals4 and well-defined nanostructures.5 Polyelectrolytes are also found to interact with platinum(II) complexes electrostatically and induce the aggregation of platinum(II) complexes via metal–metal and π–π interactions, giving rise to drastic UV-vis absorption and luminescence changes.6 When coupled to chemical reactions, these polyelectrolyte-induced aggregation systems have been used for chemo/biosensing applications, but neither well-defined nanostructures nor metallosupramolecular gels have been observed in these systems.6 The covalent tethering of platinum(II)–polypyridine units to block copolymers can produce pH- or temperature-responsive metallopolymers, whereas formation of metallosupramolecular gels has not been observed so far in such systems.7 The fabrication of supramolecular gels of platinum(II) complexes via metal–metal and π–π stacking interactions in both organic and aqueous media has also been observed.8 To date, the stimuli-responsive behaviors of these platinum(II) supramolecular gels in reported studies remain limited to thermal responsiveness.8The manipulation of non-covalent metal–metal interactions has been demonstrated to be an efficient pathway for the fabrication of metallosupramolecular structures and the modulation of their supramolecular behaviors and functions.1–8 The majority of the reported metallosupramolecular systems are designed and governed by thermodynamics, with supramolecular systems involving pathway complexity and fuel-driven processes that exhibit intriguing kinetics much less reported (vide infra).
Investigation and control of kinetics in supramolecular systems have emerged as an important topic in supramolecular chemistry since it can provide a deep and comprehensive understanding of the behaviors of supramolecular systems which is inaccessible by the study of supramolecular systems based on thermodynamics alone.9,10 In the supramolecular systems involving pathway complexity, metastable supramolecular intermediates would appear first and would then be converted to stable products upon subsequent incubation, which gives rise to interesting spectroscopic and morphological changes as a function of time.9 It has been shown in reported studies that helical aggregates at the early stage of supramolecular assembly possess opposite chirality to those at the final stage, so the investigation of the kinetics in supramolecular systems is indispensable for the study of the helical aggregation mechanism and the chirality transformation kinetics.9a Fuel-driven and reaction-driven supramolecular systems10 have been reported to construct supramolecular hydrogels with material properties such as lifetimes and mechanical strengths controlled by the consumption rates of chemical fuels and reaction kinetics rather than by thermodynamic stability.10c,d In such systems, the investigation of temporal properties is of crucial importance since the material properties are a function of time and show drastic changes at different fuel levels and reaction rates.10 In living systems, the realization of biological functions requires the cooperation of a series of biochemical processes with programmed temporal properties, such as the blood coagulation cascade by which blood changes from a liquid to a gel.11 In artificial systems, it has been reported recently that the balance of temporal properties between monomer exchanges and interfiber association in peptide amphiphile systems can lead to the formation of hydrogels with bundled structures, enhanced mechanical properties and improved functions when used as a bioactive matrix.12 In short, based on the improved understanding of supramolecular systems, the manipulation of kinetics in supramolecular systems shows promising applications in devising novel functional supramolecular materials.9–12 Our group as well as others have observed interesting kinetic behaviors in several platinum(II) supramolecular assembly systems.5i,m,13 A recent study in our laboratory showed intriguing kinetic pathways and morphological transformation as a function of time in a co-assembly system of platinum(II) complexes and block copolymers.13a The morphologies and properties of the co-assembly system have been found to be controlled by the manner by which thermal energy was input into the system.13a
Here we report a serendipitous finding of platinum(II) complexes serving as non-covalent crosslinkers for the fabrication of supramolecular DNA hydrogels. Upon mixing the alkynylplatinum(II) terpyridine complex with double-stranded DNA in aqueous solution, the platinum(II) complex molecules are found to first stack into columnar phases by metal–metal and π–π interactions, and then the columnar phases that carry multiple positive charges crosslink the negatively charged DNA strands to form supramolecular hydrogels with luminescence properties and excellent processability. Subsequent platinum(II) intercalation into DNA competes with the metal–metal and π–π interactions at the crosslinking points, switching on the spontaneous gel-to-sol transition. In the case of a chloro (2,6-bis(benzimidazol-2′-yl)pyridine)platinum(II) complex, with [Pt(bzimpy)Cl]+ serving as a non-covalent crosslinker where the metal–metal and π–π interactions outcompete platinum(II) intercalation, the intercalation-driven gel-to-sol transition pathway is blocked since the gel state is energetically more favorable than the sol state. Interestingly, the ligand exchange reaction of the chloro ligand in [Pt(bzimpy)Cl]+ with glutathione (GSH) has endowed the complexes with enhanced hydrophilicity, decreasing the planarity of the complexes, and turning off the metal–metal and π–π interactions at the crosslinking points, leading to GSH-triggered hydrogel dissociation.
Results and discussion
Platinum(II) complexes as non-covalent crosslinkers for supramolecular DNA hydrogels
The original objective of the present study was to investigate the intercalation of alkynylplatinum(II) terpyridine complexes (Scheme 1) into double-stranded DNA. When complex 1 (0.15 mM) was mixed with salmon sperm DNA (smDNA, [base pair] = 1.35 mM) in aqueous solution, surprising formation of gel-like precipitates with a brown color was found. The gel-like precipitates disappeared after stirring the mixture for tens of minutes to give a clear yellow solution finally. Room-temperature incubation of the gel-like precipitates in the assembly mixture overnight without stirring also led to gel dissociation into a clear yellow solution. The UV-vis absorption spectrum of the gel-like precipitates isolated from the mixture before their disappearance is found to show an emergence of a low-energy metal–metal-to-ligand charge transfer (MMLCT) absorption band at 602 nm (Fig. 1a), as well as the switching on of a triplet MMLCT (3MMLCT) near-infrared emission at 790 nm (Fig. 1b) when compared to that of the monomeric form of complex 1 in acetonitrile solution. These observations suggest the aggregation of platinum(II) complexes and the formation of non-covalent metal–metal and π–π interactions within the gel-like precipitates.5j,6a The UV-vis spectrum of the final clear yellow solution shows a red shift of the absorption band with respect to complex 1 in acetonitrile (Fig. 1a), while its emission spectrum shows a typical triplet metal-to-ligand charge transfer (3MLCT) emission band similar to that of complex 1 in acetonitrile solution (Fig. 1b). According to literature reported studies,1i,14 these observations suggest the intercalation of the platinum(II) complex molecules into double-stranded DNA in the final yellow solution. The transient presence of the gel-like precipitates and the intriguing transformation of platinum(II) complexes from monomeric forms through aggregation into intercalated forms have motivated us to investigate the structure and the formation mechanism of these gel-like precipitates and the gel-to-sol transition behaviors of the platinum(II)–DNA supramolecular system.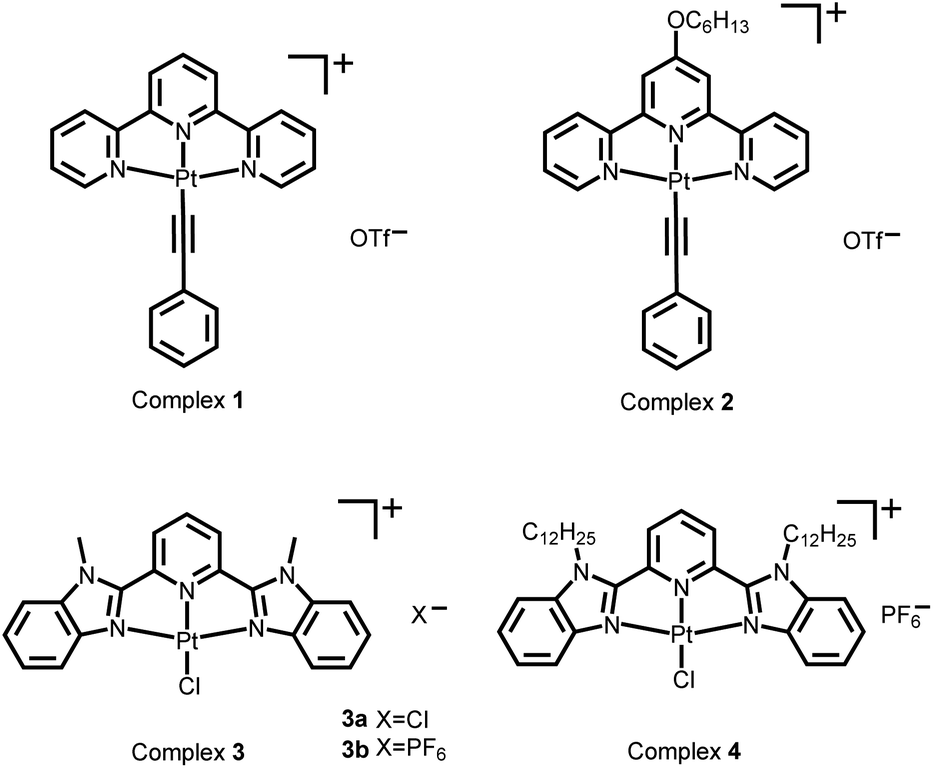 | ||
| Scheme 1 Chemical structures of the platinum(II) complexes. | ||
 | ||
| Fig. 1 (a) UV-vis absorption and (b) steady-state emission spectral changes of the supramolecular system of complex 1 (0.15 mM) and smDNA ([base pair] = 1.35 mM) during the transformation from platinum(II) monomers through the platinum(II)–DNA gel state to the platinum(II) intercalation sol state. | ||
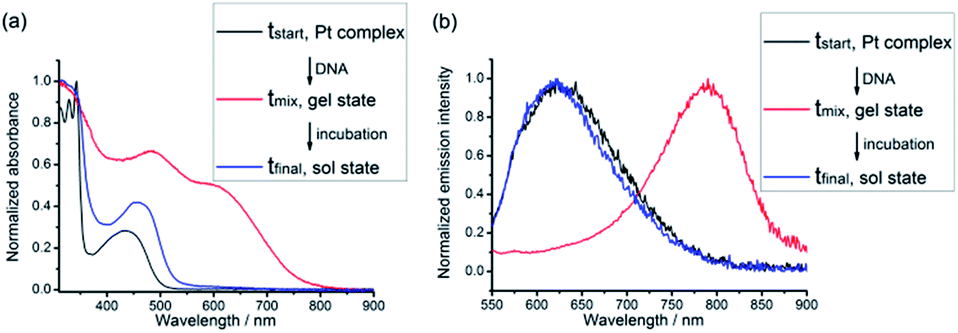
Hydrogels can be obtained with an isolation yield of approximately 70% by mixing complex 1 (1 mg mL−1) and smDNA (1 mg mL−1) in aqueous solution at a platinum(II)/base pair molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (see the Experimental section in the ESI†). After isolation from the assembly mixture, the hydrogels are found to be stable in water. The 1–DNA gel has a water content of approximately 90% and exhibits a fibrous network structure under TEM observation (vide infra). No hydrogel formation is observed when complex 1 with a concentration below 0.1 mM is mixed with smDNA at a platinum(II)/base pair molar ratio of 1:1. Another alkynylplatinum(II) terpyridine complex 2 and [Pt(bzimpy)Cl]+ complexes 3a, 3b and 4 also show hydrogel formation when mixed with aqueous solutions of smDNA. Fig. 2a shows that star-shaped hydrogels of complex 3a and smDNA can be obtained with tailorable sizes by using a silicone mold. The red hydrogels can be encapsulated by agarose hydrogels to form core–shell star-shaped hydrogels (Fig. 2b and c), which display red emission under 365 nm UV irradiation (Fig. 2d).
:1 (see the Experimental section in the ESI†). After isolation from the assembly mixture, the hydrogels are found to be stable in water. The 1–DNA gel has a water content of approximately 90% and exhibits a fibrous network structure under TEM observation (vide infra). No hydrogel formation is observed when complex 1 with a concentration below 0.1 mM is mixed with smDNA at a platinum(II)/base pair molar ratio of 1:1. Another alkynylplatinum(II) terpyridine complex 2 and [Pt(bzimpy)Cl]+ complexes 3a, 3b and 4 also show hydrogel formation when mixed with aqueous solutions of smDNA. Fig. 2a shows that star-shaped hydrogels of complex 3a and smDNA can be obtained with tailorable sizes by using a silicone mold. The red hydrogels can be encapsulated by agarose hydrogels to form core–shell star-shaped hydrogels (Fig. 2b and c), which display red emission under 365 nm UV irradiation (Fig. 2d).
 | ||
| Fig. 2 (a) A star-shaped 3a–DNA gel prepared by using silicone mold A. (b) A core–shell star-shaped hydrogels prepared by cooling a hot agarose solution in the presence of 3a–DNA gel in silicone mold D. (c and d) The core–shell star-shaped hydrogels in water under ambient light (c) and under 365 nm UV irradiation (d). Inset in (a) shows the star-shaped silicone molds A–D with different sizes. | ||
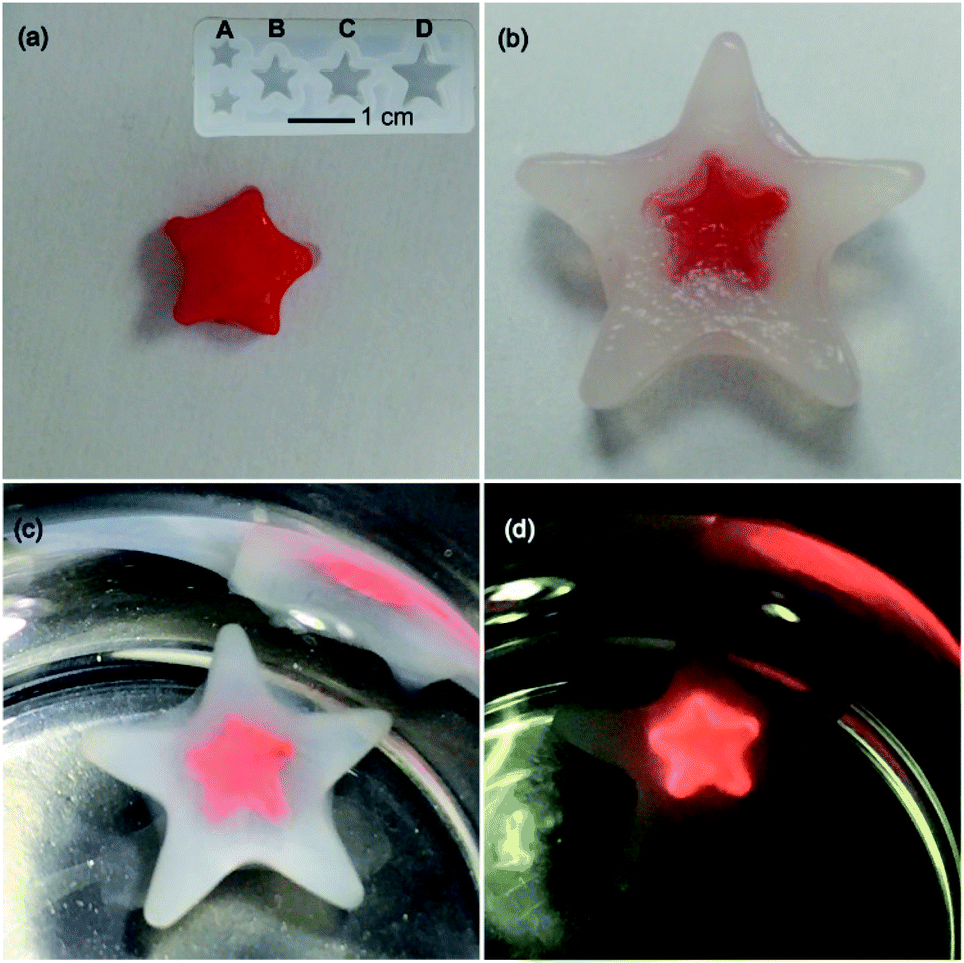
The UV-vis absorption spectra of the platinum(II)–DNA hydrogels, 1–DNA gel and 2–DNA gel, show the emergence of low-energy MMLCT absorption bands with peak maxima at 603 nm and 564 nm, respectively, as well as new low-energy 3MMLCT emission bands at 791 nm and 749 nm in the emission spectra, respectively (Fig. 3a and b).5j,6a For 3a–DNA gel and 4–DNA gel, low-energy MMLCT absorption bands appear in the region of 470 to 630 nm in the UV-vis absorption spectra, and their emission spectra exhibit low-energy 3MMLCT emission bands with peak maxima at 645 nm and 674 nm, respectively (Fig. 3c and d).15
 | ||
| Fig. 3 (a and c) UV-vis absorption and (b and d) steady-state emission spectra of the platinum(II)–DNA hydrogels in the dried state. | ||
TEM images show that 1–DNA gel and 2–DNA gel possess fibrous network structures (Fig. 4a and d). The weight percentages of platinum elements in the dried gel have been determined by inductively coupled plasma-mass spectrometry (ICP-MS) to be 5% and 7% for 1–DNA gel and 2–DNA gel, respectively. These results indicate that DNA strands are the major component and consist of a continuous phase in the platinum(II)–DNA hydrogels. Selected area electron diffraction (SAED) study of a fibrous network of 1–DNA gel shows a ring-like pattern that corresponds to a d-spacing of 0.34 nm (Fig. 4b). SAED study of a single nanofiber of 1–DNA gel exhibits a pair of diffraction arcs that correspond to a d-spacing of 0.34 nm along the nanofiber long axis (Fig. 4c). A similar single-fiber SAED pattern is observed in the study of 2–DNA gel (Fig. 4e). These observations of characteristic diffraction arcs indicate the formation of non-covalent metal–metal and π–π interactions between the platinum(II) complexes along the nanofiber long axis. The powder X-ray diffraction (PXRD) pattern of the dried 2–DNA gel shows a series of Bragg peaks in the region of 5° < 2θ < 18° (Fig. 4f). The distances obtained from the Bragg peaks followed the ratios of 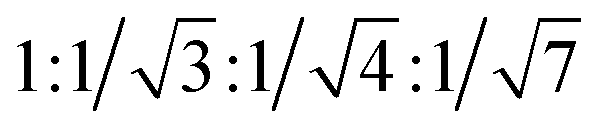 that can be indexed to (100), (110), (200) and (210) reflections, characteristic of a hexagonal packing of molecular columns of platinum(II) complexes.
that can be indexed to (100), (110), (200) and (210) reflections, characteristic of a hexagonal packing of molecular columns of platinum(II) complexes.
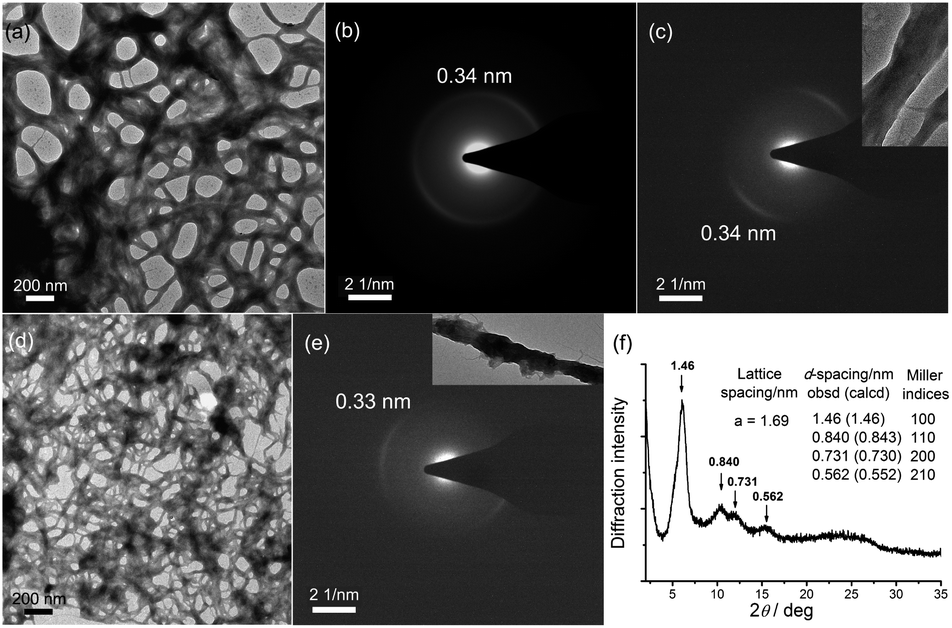 | ||
| Fig. 4 (a) TEM image, (b) SAED pattern of the fibrous network and (c) single-fiber SAED pattern of 1–DNA gel in the dried state. (d) TEM image, (e) single-fiber SAED pattern and (f) PXRD pattern of 2–DNA gel in the dried state. | ||
Based on the above results and analysis (see also Text S1 and Fig. S1 in the ESI†), it is proposed that, during hydrogel formation, the platinum(II) complexes would first stack into columnar phases by metal–metal and π–π interactions, and then the platinum(II) columnar phases that carry multiple positive charges would crosslink the DNA strands by electrostatic attractions to form supramolecular hydrogels with luminescence properties and excellent processability (Scheme 2). The platinum(II) columnar phases are preferentially oriented along the nanofiber long axis to maximize the electrostatic attractions with the DNA strands (Scheme 2). Therefore, the platinum(II)–DNA hydrogels are supramolecular hydrogels formed and stabilized by non-covalent metal–metal and π–π interactions and electrostatic attractions.
 | ||
| Scheme 2 Supramolecular system of alkynylplatinum(II) terpyridine complexes and double-stranded DNA. The wells represent the qualitative energy profile of the system. In the platinum(II)–DNA system at a complex 1/base pair molar ratio of 1:9, upon the mixing of complex 1 and DNA aqueous solution, the platinum(II) complexes are found to first stack into columnar phases and then non-covalently crosslink the DNA strands by electrostatic attractions. Subsequently, due to the presence of excessive DNA in the assembly mixture, platinum(II) intercalation into DNA base pairs can compete with non-covalent metal–metal and π–π interactions between the platinum(II) complexes at the crosslinking points of the hydrogels, switching on the spontaneous transition from the off-pathway gel state to the on-pathway sol state. The platinum(II) stacking and electrostatic crosslinking are kinetically favored, while the platinum(II) intercalation into DNA base pairs is thermodynamically stable. | ||
Platinum(II) intercalation-driven gel-to-sol transition
As described above, after purification from the assembly mixture, the supramolecular DNA hydrogels are found to be stable in water. Surprisingly, although the hydrogels are formed by non-covalent interactions, the purified hydrogels exhibit thermal stability even in boiling water. The purified hydrogels are also found to be stable in water at pH 4 and pH 10 or with 150 mM NaCl at room temperature. In contrast, the as-formed hydrogels of the alkynylplatinum(II) terpyridine complex and DNA show transient presence in the assembly mixture where DNA is in excess as shown in Fig. 1. To study the spontaneous sol-to-gel transformation in detail, freshly purified hydrogels of the alkynylplatinum(II) terpyridine complex and smDNA have been immersed in fresh smDNA solution at room temperature (see the Experimental section). The colorless smDNA solution gradually turns yellow due to the release of platinum(II) complexes from the hydrogels. In situ UV-vis spectra of the yellow solutions show the increase of absorption bands at 452 nm and 444 nm for 1–DNA gel and 2–DNA gel, respectively, with the increase of incubation time (Fig. 5). The 1–DNA gel and 2–DNA gel have a half-life of 8 min and 14 min in smDNA solution, respectively. The –OC6H13 moiety on the platinum(II) complexes can provide additional hydrophobic stabilizing interactions for the 2–DNA gel and may hinder the platinum(II) intercalation into DNA base pairs to some extent, leading to slower release kinetics of 2–DNA gel in DNA solution.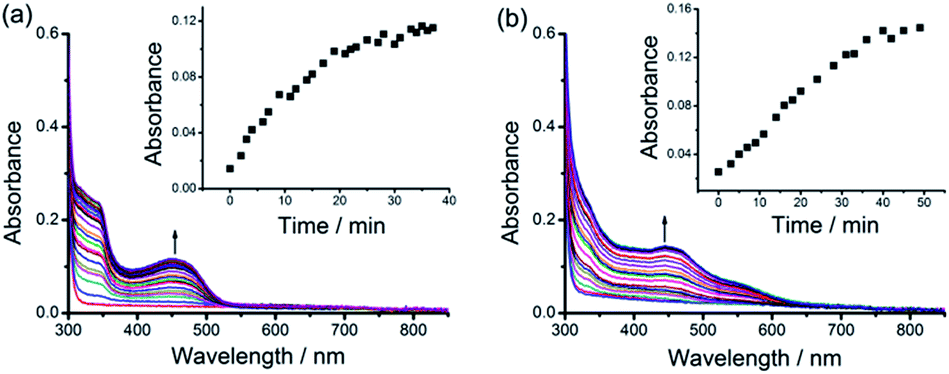 | ||
| Fig. 5 UV-vis absorption spectral changes of (a) 1–DNA gel and (b) 2–DNA gel upon incubation in 0.5 mg mL−1 smDNA at room temperature. Insets show the plots of UV-vis absorption at 452 nm and 444 nm versus incubation time for (a) 1–DNA gel and (b) 2–DNA gel, respectively. | ||
The interesting gel-to-sol transition behaviors have been further studied by using a combination of various spectroscopic techniques. UV-vis spectral changes upon DNA titration into the platinum(II)–DNA system are found to show a decrease of the MMLCT absorption band at 604 nm and an increase of the MLCT absorption band at 456 nm (Fig. 6a). Upon increasing the DNA/platinum ratios, the emission spectral titration studies have been found to exhibit a growth of the 3MLCT emission band at 620 nm with the diminution of the 3MMLCT emission in the region of 770 to 950 nm (Fig. 6b). The circular dichroism (CD) spectral changes upon addition of complex 1 into DNA aqueous solution show a remarkable change of the Cotton effect, with the diminution of the positive band at 278 nm and the growth of a new negative band at 274 nm (Fig. 6c). Similar changes have been observed in the CD spectral titration studies of complex 2 in DNA solution (Fig. 6d), indicative of the platinum(II) intercalation into double-stranded DNA chains with reference to literature reported studies.14 All the spectroscopic studies point to the fact that, upon the immersion of 1–DNA gel and 2–DNA gel into DNA solution, the microenvironment of the alkynylplatinum(II) terpyridine complexes changes from the aggregated form with metal–metal interactions to the platinum(II) intercalation form with DNA base pairs. This can explain the intriguing gel-to-sol transition as shown in Fig. 1. In the platinum(II)–DNA system at a complex 1/base pair molar ratio of 1/9, upon the mixing of complex 1 and DNA aqueous solution, the platinum(II) complexes are found to first stack into columnar phases and then non-covalently crosslink the DNA strands by electrostatic attractions (Scheme 2). Subsequently, due to the presence of excessive DNA in the assembly mixture, platinum(II) intercalation into DNA base pairs can compete with non-covalent metal–metal and π–π interactions between the platinum(II) complexes at the crosslinking points of the hydrogels, switching on the spontaneous transition from the off-pathway gel state to the on-pathway sol state (Scheme 2). The platinum(II) stacking and electrostatic crosslinking are kinetically favored, while the platinum(II) intercalation into DNA base pairs is thermodynamically stable. The hydrogel formation and subsequent gel-to-sol transition exhibit pathway complexity in the platinum(II)–DNA supramolecular system, which have not been observed before in metallosupramolecular systems.
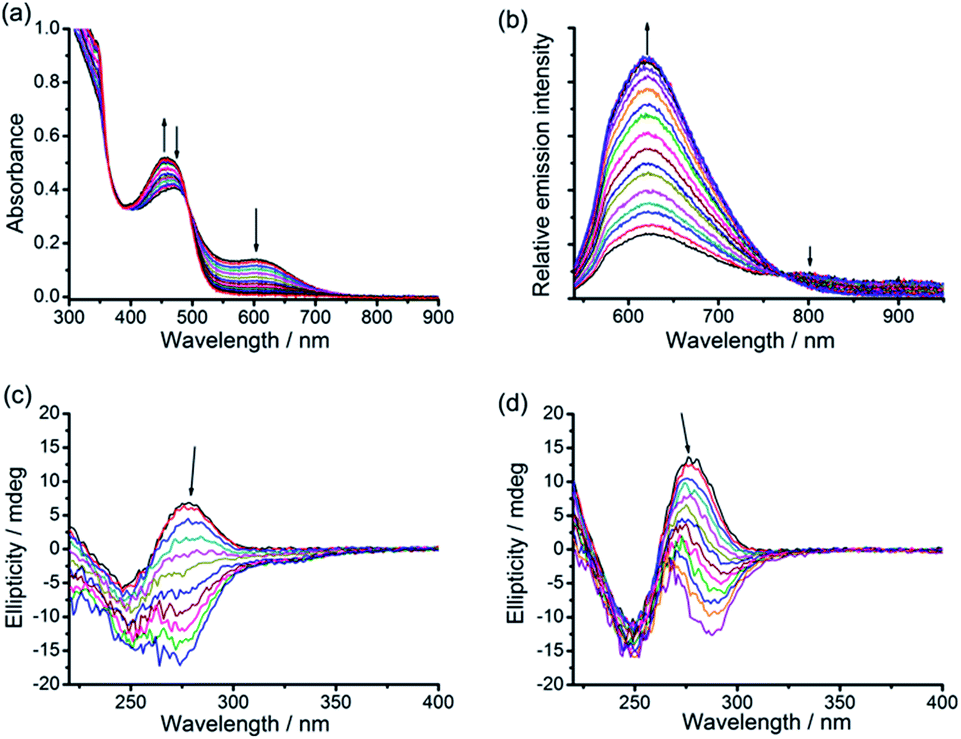 | ||
| Fig. 6 (a) UV-vis absorption and (b) steady-state emission (λex = 492 nm) spectral changes of the 1–DNA supramolecular system ([complex 1] = 0.15 mM) upon DNA titration at DNA base pair/platinum(II) molar ratios from 1.7 to 4.2. (c and d) CD spectral changes of DNA with increasing platinum(II)/base pair molar ratio from 0 to 0.3 in 1–DNA (c) and 2–DNA (d) supramolecular systems. | ||
The stability of platinum(II)–DNA hydrogels, 3a–DNA gel, 3b–DNA gel and 4–DNA gel, in DNA solution has also been tested, and it turns out that these [Pt(bzimpy)Cl]+–DNA hydrogels are kept intact by incubation of the hydrogels in smDNA solution at room temperature for one week. Spectroscopic studies indicate the absence of platinum(II) intercalation in the [Pt(bzimpy)Cl]+–DNA supramolecular systems (Text S2 and Fig. S2 in the ESI†).15 In the case of [Pt(bzimpy)Cl]+ complexes as non-covalent crosslinkers, although the DNA is in excess, the strong tendency for the formation of metal–metal and π–π interactions between [Pt(bzimpy)Cl]+ complexes due to their large π-surface, as well as the additional hydrophobic interactions of C12 alkyl chains on complex 4, can outcompete the platinum(II) intercalation into DNA base pairs. Therefore, the gel state in the [Pt(bzimpy)Cl]+–DNA systems is energetically more favorable than the sol state, which can explain the enhanced stability of the [Pt(bzimpy)Cl]+–DNA hydrogels in DNA solution. These observations suggest that the intercalation-driven gel-to-sol transition pathway can be switched on or off by rational design of the metal complexes in supramolecular systems.
GSH-triggered hydrogel dissociation
Platinum(II) is one of the soft metal centers with a high binding affinity towards thiol. The endogenous thiol-containing molecule, glutathione (GSH), exists at a concentration of up to approximately 10 mM in animal cells and is often overproduced in tumor microenvironments.16 GSH has been frequently employed as a redox stimuli trigger in reported studies,16b,17 while the utilization of GSH based on platinum(II)–thiol coordination in tuning material properties has been much less explored.The stability of the [Pt(bzimpy)Cl]+–DNA hydrogels in aqueous solution of GSH has been investigated (Fig. 7). ICP-MS measurements show a gradual release of platinum from 3b–DNA gel upon incubation of the hydrogels in 10 mM GSH aqueous solution. Conversely, in the control experiment, 3b–DNA gel does not show any platinum release after incubation in pure water for one week. Gradual platinum release has also been observed by ICP-MS upon incubation of 3a-DNA in 10 mM GSH solution. 3a-DNA is also stable in pure water. Interestingly, 4–DNA gel incorporated with C12 alkyl chains on [Pt(bzimpy)Cl]+ complexes has been found to show insignificant platinum release in both pure water and 10 mM GSH solution. Besides, 2–DNA gel of alkynylplatinum(II) terpyridine complexes is also found to be stable in 10 mM GSH solution. These observations indicate that the stability of platinum(II)–DNA hydrogels in GSH solution can be modulated by the structure of the platinum(II) complexes.
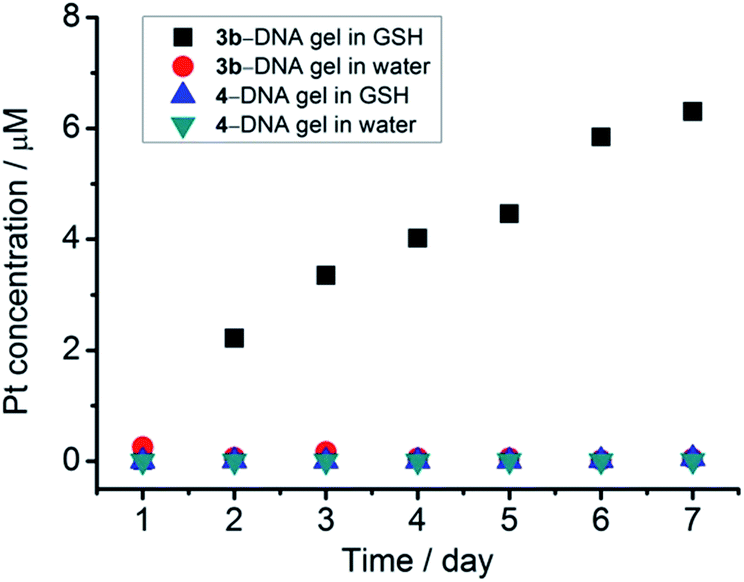 | ||
| Fig. 7 Platinum release from [Pt(bzimpy)Cl]+–DNA hydrogels by incubation of the hydrogels in 10 mM GSH aqueous solution at room temperature. | ||
Since GSH cannot react with smDNA in aqueous solution, NMR experiments of the 3a–GSH reaction system in D2O were performed (Fig. 8, Text S3, Fig. S3 and S4 in the ESI†) to investigate the mechanism of GSH-triggered dissociation of the [Pt(bzimpy)Cl]+–DNA hydrogels. The new peaks at δ 2.61 ppm and 2.82 ppm in the 1H NMR spectrum of the purified product from 3a–GSH reaction mixture that can be assigned to β-Cys protons (Fig. 8a), together with the large downfield shift of β-Cys carbon from δ 25.3 ppm to 33.6 ppm in the 13C{1H} NMR spectrum (Fig. 8b), indicated the involvement of the sulfur in the coordination of GSH to platinum(II). The 195Pt{1H} NMR spectrum with a peak at δ −3063 ppm (ref. 18) and the HR ESI mass spectra of the reaction mixture further confirmed the platinum(II)–thiol coordination (Fig. 8c and S5 in the ESI†), that is, the ligand exchange reaction of the chloro ligand in complex 3a with glutathione. The emergence of a set of signals in the region of aromatic protons in both 1H NMR and 13C{1H} NMR spectra (Fig. 8a and b), when compared to the NMR spectrum of complex 3a in D2O (Fig. 8a), indicated the deaggregation of the platinum(II) complexes due to the enhanced hydrophilicity provided by the GS moiety after platinum(II)–thiol coordination.
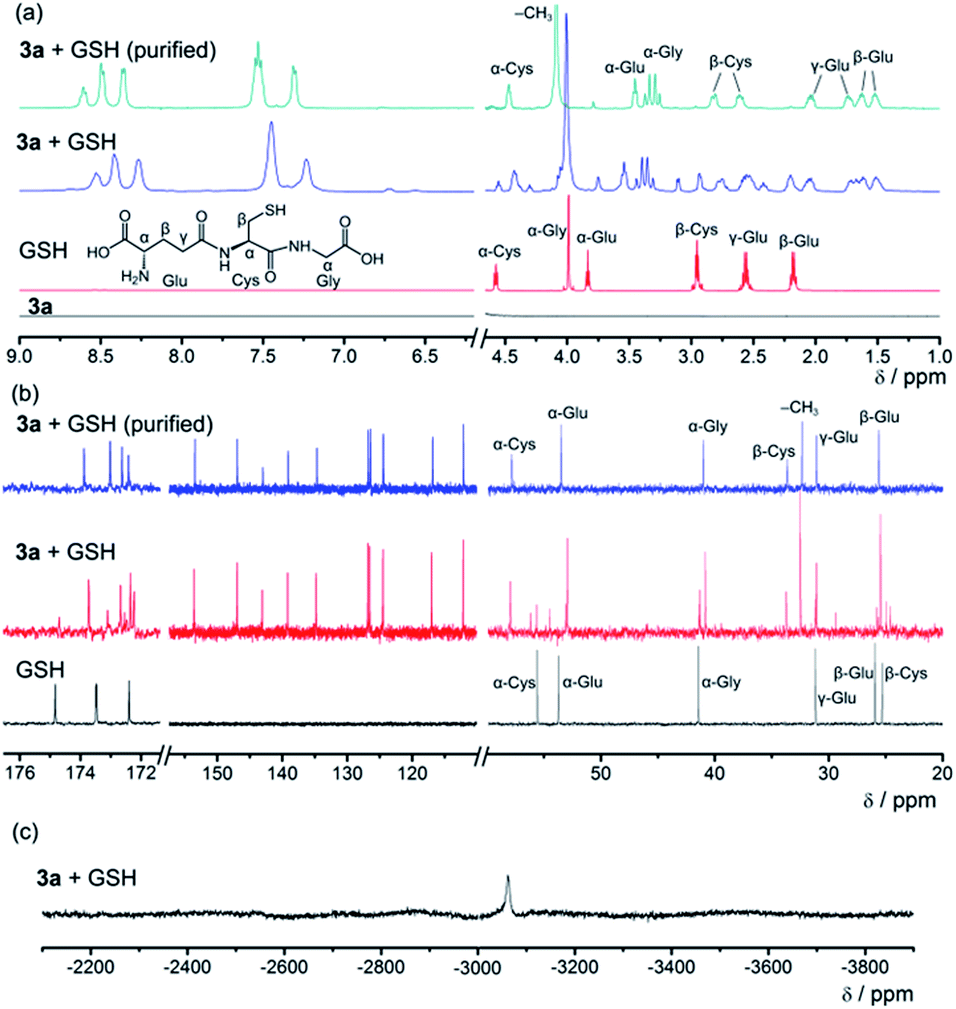 | ||
| Fig. 8 (a) 1H NMR, (b) 13C{1H} NMR and (c) 195Pt{1H} NMR spectra of the 3a–GSH reaction system in D2O. | ||
When the aqueous solution of [Pt(bzimpy)(GS)]+ (the purified product from the 3a–GSH reaction mixture) is mixed with smDNA, no hydrogel formation is observed. Therefore, in the case of 3a–DNA gel, it is the ligand exchange reaction of the chloro ligand in [Pt(bzimpy)Cl]+ with GSH that endows the complex molecules with enhanced hydrophilicity, decreases the planarity of the complex molecules, and turns off the metal–metal and π–π interactions at the crosslinking points, leading to GSH-triggered gradual dissociation of the hydrogels (Scheme 3). Complex 4 is found to have a higher dimerization constant in a mixed solvent of dimethylformamide–water (1:9, v/v) at room temperature (9.6 × 105 M−1) than complex 3b (3.8 × 105 M−1) as determined by UV-vis measurements. 4–DNA gel with C12 alkyl chains on [Pt(bzimpy)Cl]+, which possesses enhanced hydrogel stability and prevents GSH diffusion into the hydrogels due to the hydrophobic nature of the C12 alkyl chains, shows insignificant release in both pure water and GSH solution. These [Pt(bzimpy)Cl]+–DNA supramolecular hydrogels exhibit tunable gel-to-sol transition behaviors by the structures of the platinum(II) non-covalent crosslinkers and the reaction kinetics of the ligand exchange reactions. Unlike the reported studies where GSH has been used as a redox stimuli trigger,16b,17 the gel-to-sol transition behaviors in the present study are based on platinum(II)–thiol coordination between the platinum(II) complexes and GSH.
 | ||
| Scheme 3 Supramolecular system of [Pt(bzimpy)Cl]+ and double-stranded DNA. The platinum(II) complexes stack into columnar phases and then non-covalently crosslink the DNA strands by electrostatic attractions to form hydrogels. In GSH aqueous solution, the ligand exchange reaction of the chloro ligand in [Pt(bzimpy)Cl]+ with GSH endows the complexes with enhanced hydrophilicity, decreasing the planarity of the complexes, and turning off the metal–metal and π–π interactions at the crosslinking points, leading to energetically unfavorable hydrogel dissociation. The wells represent the qualitative energy profile of the system. | ||
Conclusions
In summary, the present study shows a serendipitous finding of platinum(II) non-covalent crosslinkers for the fabrication of supramolecular DNA hydrogels with controlled kinetic properties and tunable gel-to-sol transition behaviors (Scheme 4). Upon mixing the alkynylplatinum(II) terpyridine complex with double-stranded DNA in aqueous solution, the platinum(II) complexes have been found to first stack into columnar phases by metal–metal and π–π interactions, and then the platinum(II) columnar phases that carry multiple positive charges crosslink negatively charged DNA chains by electrostatic attractions to form supramolecular hydrogels with luminescence properties and excellent processability. Subsequently, platinum(II) intercalation into DNA competes with the metal–metal and π–π interactions between the platinum(II) complexes at the crosslinking points of the supramolecular DNA hydrogels, switching on the spontaneous transition from the off-pathway gel state to the on-pathway sol state (Scheme 4). The rate of the gel-to-sol transition has been found to decrease with the hydrophobicity of the alkynylplatinum(II) terpyridine complexes in the hydrogels. With [Pt(bzimpy)Cl]+ as a non-covalent crosslinker where the metal–metal and π–π interactions are stronger than platinum(II) intercalation, the gel state is energetically more favorable than the sol state, and thus the intercalation-driven gel-to-sol transition pathway can be blocked. Interestingly, the ligand exchange reaction of the chloro ligand in [Pt(bzimpy)Cl]+ with GSH has been found to endow the complexes with enhanced hydrophilicity, decreasing the planarity of the complexes, and turning off the metal–metal and π–π interactions at the crosslinking points, leading to energetically unfavorable hydrogel dissociation that is triggered by GSH (Scheme 4). The rate of GSH-triggered hydrogel dissociation has been found to decrease with the hydrophobicity of the [Pt(bzimpy)Cl]+ complexes. In view of the flexibility of the modular approach of the present study, the biocompatibility of the DNA matrix, as well as the intriguing luminescence properties and anti-cancer properties of metal complexes, the supramolecular DNA hydrogels may exhibit promising characteristics for biomedical applications.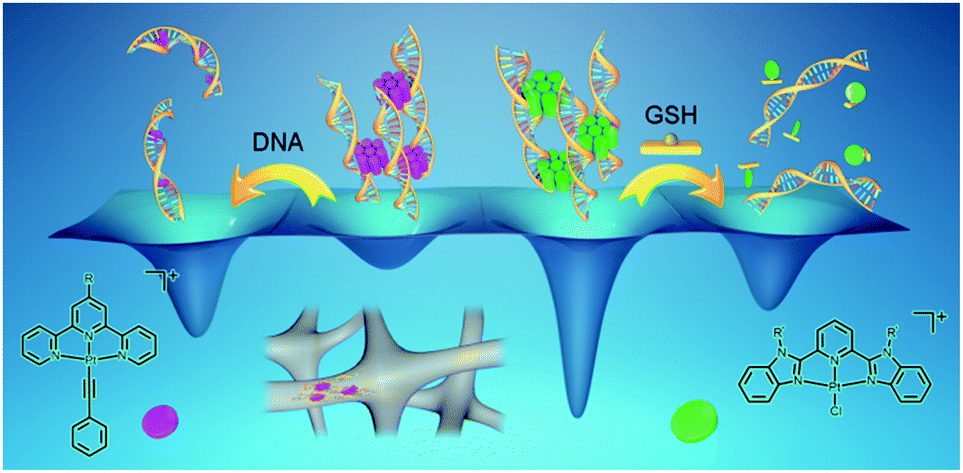 | ||
| Scheme 4 Schematic diagram showing platinum(II) non-covalent crosslinkers for supramolecular DNA hydrogels. By varying the structures of the platinum(II) non-covalent crosslinkers, as well as the incorporation of platinum(II) intercalation and ligand exchange reactions into the systems, supramolecular DNA hydrogels with controlled kinetic properties and tunable gel-to-sol transition behaviors have been produced. The wells represent the qualitative energy profile of the system. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. W.-W. Y. acknowledges support from The University of Hong Kong under the University Research Committee (URC) Strategically Oriented Research Theme (SORT) on Functional Materials for Molecular Electronics. This work has been supported by a General Research Fund (GRF) grant from the Research Grants Council of Hong Kong Special Administrative Region, P. R. China (HKU17306219). Dr M. C. L. Yeung, Dr S. Y. L. Leung and Mr V. C. H. Wong are gratefully acknowledged for providing the platinum(II) samples. We also thank the Electron Microscope Unit at The University of Hong Kong for technical support. The Beijing Synchrotron Research Facility (BSRF) is also thanked for providing beamline time (beamline 4B9A) in the synchrotron radiation X-ray diffraction facilities for powder X-ray diffraction studies.Notes and references
- (a) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529 CrossRef CAS; (b) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446 CrossRef CAS; (c) V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145 CrossRef CAS; (d) J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591 CrossRef CAS; (e) H.-K. Yip, L.-K. Cheng, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933 RSC; (f) R. H. Herber, M. Croft, M. J. Coyer, B. Bilash and A. Sahiner, Inorg. Chem., 1994, 33, 2422 CrossRef CAS; (g) W. B. Connick, R. E. Marsh, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1997, 36, 913 CrossRef CAS; (h) R. Buchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 711 RSC; (i) K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS; (b) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS PubMed; (c) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef CAS PubMed; (d) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and B. W.-K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169 CrossRef CAS PubMed; (b) S. Y.-L. Leung, A. Y.-Y. Tam, C.-H. Tao, H. S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2012, 134, 1047 CrossRef CAS PubMed; (c) A. Gross, T. Moriuchi and T. Hirao, Chem. Commun., 2013, 49, 1163 RSC; (d) Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Chem. Sci., 2012, 3, 1185 RSC; (e) Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 14117 CrossRef CAS PubMed; (f) A. K.-W. Chan, W. H. Lam, Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 690 CrossRef CAS PubMed; (g) F. K. W. Kong, A. K. W. Chan, M. Ng, K. H. Low and V. W. W. Yam, Angew. Chem., Int. Ed., 2017, 56, 15103 CrossRef CAS PubMed.
- (a) W. Lu, Y. Chen, V. A. L. Roy, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621 CrossRef CAS PubMed; (b) V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286 CrossRef CAS PubMed.
- (a) C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136 CrossRef CAS PubMed; (b) H.-L. Au-Yeung, S. Y.-L. Leung, A. Y.-Y. Tam and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17910 CrossRef CAS PubMed; (c) S. Y.-L. Leung, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845 CrossRef CAS PubMed; (d) H. L.-K. Fu, S. Y.-L. Leung and V. W.-W. Yam, Chem. Commun., 2017, 53, 11349 RSC; (e) V. C.-H. Wong, C. Po, S. Y.-L. Leung, A. K.-W. Chan, S. Yang, B. Zhu, X. Cui and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 657 CrossRef CAS PubMed; (f) M.-Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M.-H. So, S. S.-Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895 CrossRef CAS PubMed; (g) Y. Chen, K. Li, W. Lu, S. S.-Y. Chui, C.-W. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909 CrossRef CAS PubMed; (h) W. Lu, S. S.-Y. Chui, K.-M. Ng and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 4568 CrossRef CAS PubMed; (i) C. Po and V. W.-W. Yam, Chem. Sci., 2014, 5, 4868 RSC; (j) K. Zhang, M. C.-L. Yeung, S. Y.-L. Leung and V. W.-W. Yam, Chem, 2017, 2, 825 CrossRef CAS; (k) K. Zhang, M. C.-L. Yeung, S. Y.-L. Leung and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11844 CrossRef CAS PubMed; (l) M. H.-Y. Chan, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 7637 CrossRef CAS PubMed; (m) A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2015, 8, 10 CrossRef PubMed.
- (a) C. Yu, K. M.-C. Wong, K. H.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791 CrossRef CAS PubMed; (b) C. Yu, K. H.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19652 CrossRef CAS PubMed; (c) C. Y.-S. Chung and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 18775 CrossRef CAS PubMed; (d) M. C.-L. Yeung and V. W.-W. Yam, Chem. Sci., 2013, 4, 2928 RSC; (e) A. S.-Y. Law, M. C.-L. Yeung and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 41143 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, Y. Hu, K. H.-Y. Chan and C. Y.-S. Chung, Chem. Commun., 2009, 6216 RSC; (b) C. Y.-S. Chung and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 13182 CrossRef CAS PubMed; (c) H. K. Cheng, C. Y.-S. Chung, K. Zhang and V. W.-W. Yam, Chem.–Asian J., 2017, 12, 1509 CrossRef CAS PubMed.
- (a) A. Y.-Y. Tam, K. M.-C. Wong, G. Wang and V. W.-W. Yam, Chem. Commun., 2007, 2028 RSC; (b) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef CAS PubMed; (c) F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J.-P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659 CrossRef CAS PubMed; (d) X. Xiao, W. Lu and C.-M. Che, Chem. Sci., 2014, 5, 2482 RSC; (e) M. H.-Y. Chan, M. Ng, S. Y.-L. Leung, W. H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 8639 CrossRef CAS PubMed; (f) Y. Li, A. Y.-Y. Tam, K. M.-C. Wong, W. Li, L. Wu and V. W.-W. Yam, Chem.–Eur. J., 2011, 17, 8048 CrossRef CAS PubMed; (g) A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC; (h) L. Geng, X. Yu, Y. Wang, Y. Wu, J. Ren, F. Xue and T. Yi, Nanoscale, 2019, 11, 4044 RSC.
- (a) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492 CrossRef CAS PubMed; (b) P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540 CrossRef CAS PubMed; (c) F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469 CrossRef CAS PubMed; (d) T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2016, 9, 493 CrossRef PubMed; (e) E. E. Greciano, B. Matarranz and L. Sánchez, Angew. Chem., Int. Ed., 2018, 57, 4697 CrossRef CAS PubMed; (f) S. Ogi, C. Grzeszkiewicz and F. Würthner, Chem. Sci., 2018, 9, 2768 RSC.
- (a) J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825 CrossRef CAS PubMed; (b) J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502 CrossRef CAS PubMed; (c) J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. van der Mee, C. B. Minkenberg, E. Mendes, J. H. van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433 CrossRef CAS PubMed; (d) J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075 CrossRef CAS PubMed; (e) E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111 CrossRef CAS PubMed; (f) C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119 CrossRef CAS PubMed; (g) M. Endo, T. Fukui, S. H. Jung, S. Yagai, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2016, 138, 14347 CrossRef CAS PubMed; (h) C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491 RSC; (i) A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089 CrossRef CAS PubMed; (j) B. S. L. Collins, J. C. M. Kistemaker, E. Otten and B. L. Feringa, Nat. Chem., 2016, 8, 860 CrossRef CAS; (k) M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235 CrossRef CAS PubMed.
- K. G. Mann, K. Brummel-Ziedins, T. Orfeo and S. Butenas, Blood Cells, Mol., Dis., 2006, 36, 108 CrossRef CAS PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 362, 808 CrossRef CAS PubMed.
- (a) K. Zhang, M. C.-L. Yeung, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 9594 CrossRef CAS PubMed; (b) B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534 CrossRef CAS PubMed; (c) A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernández, J. Am. Chem. Soc., 2019, 141, 5192 CrossRef CAS PubMed; (d) L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344 CrossRef CAS PubMed.
- (a) P. J. Bond, R. Langridge, K. W. Jennette and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4825 CrossRef CAS PubMed; (b) K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777 CrossRef CAS PubMed; (c) I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834 CrossRef CAS PubMed; (d) H. Kurosaki, N. Yamakawa, M. Sumimoto, K. Kimura and M. Goto, Bioorg. Med. Chem. Lett., 2003, 13, 825 CrossRef CAS PubMed.
- (a) L. J. Grove, J. M. Rennekamp, H. Jude and W. B. Connick, J. Am. Chem. Soc., 2004, 126, 1594 CrossRef CAS PubMed; (b) A. Y.-Y. Tam, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, Chem.–Eur. J., 2008, 14, 4562 CrossRef CAS PubMed.
- (a) N. Traverso, R. Ricciarelli, M. Nitti, B. Marengo, A. L. Furfaro, M. A. Pronzato, U. M. Marinari and C. Domenicotti, Oxid. Med. Cell. Longevity, 2013, 972913 Search PubMed; (b) Y. Liang, L. Li, R. A. Scott and K. L. Kiick, Macromolecules, 2017, 50, 483 CrossRef CAS PubMed; (c) P. Papadia, N. Margiotta, A. Bergamo, G. Sava and G. Natile, J. Med. Chem., 2005, 48, 3364 CrossRef CAS PubMed.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- A. K. Fazlur-Rahman and J. G. Verkade, Inorg. Chem., 1992, 31, 2064 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supporting text and figures. See DOI: 10.1039/c9sc05910e |
| This journal is © The Royal Society of Chemistry 2020 |