
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-photon absorption and two-photon-induced isomerization of azobenzene compounds†
Marta Dudek
 *,
Nina Tarnowicz-Staniak
,
Marco Deiana
,
Ziemowit Pokładek
,
Marek Samoć
and
Katarzyna Matczyszyn
*
*,
Nina Tarnowicz-Staniak
,
Marco Deiana
,
Ziemowit Pokładek
,
Marek Samoć
and
Katarzyna Matczyszyn
*
Advanced Materials Engineering and Modelling Group, Faculty of Chemistry, Wroclaw Unviersity of Science and Technology, Wyb. Wyspianskiego 27, 50-370 Wroclaw, Poland. E-mail: marta.ziemianek-dudek@pwr.edu.pl; katarzyna.matczyszyn@pwr.edu.pl
First published on 6th November 2020
Abstract
The process of two-photon-induced isomerization occurring in various organic molecules, among which azobenzene derivatives hold a prominent position, offers a wide range of functionalities, which can be used in both material and life sciences. This review provides a comprehensive description of nonlinear optical (NLO) properties of azobenzene (AB) derivatives whose geometries can be switched through two-photon absorption (TPA). Employing the nonlinear excitation process allows for deeper penetration of light into the tissues and provides opportunities to regulate biological systems in a non-invasive manner. At the same time, the tight focus of the beam needed to induce nonlinear absorption helps to improve the spatial resolution of the photoinduced structures. Since near-infrared (NIR) wavelengths are employed, the lower photon energies compared to usual one-photon excitation (typically, the azobenzene geometry change from trans to cis form requires the use of UV photons) cause less damage to the biological samples. Herein, we present an overview of the strategies for optimizing azobenzene-based photoswitches for efficient two-photon excitation (TPE) and the potential applications of two-photon-induced isomerization of azobenzenes in biological systems: control of ion flow in ion channels or control of drug release, as well as in materials science, to fabricate data storage media, optical filters, diffraction elements etc., based on phenomena like photoinduced anisotropy, mass transport and phase transition. The extant challenges in the field of two-photon switchable azomolecules are discussed.
 Marta Dudek | Marta Dudek is a research assistant at Wroclaw University of Science and Technology (Poland), where she obtained her PhD degree in 2019 in the field of Materials Engineering under the guidance of Prof. Katarzyna Matczyszyn. She gained additional experience during several internships at University of Strasbourg (France), École Normale Supérieure in Cachan (France), Humboldt University (Germany) and at Institute de Sciences Chimiques de Rennes (France). Her scientific interests involve the design, synthesis and characterization of new azobenzene photo-switches as well as their application in biological settings. |
 Nina Tarnowicz-Staniak | Nina Tarnowicz-Staniak received her MSc degree in Materials Science from Wrocław University of Science and Technology in 2018. Currently, she is a PhD candidate in Materials Science under the supervision of Prof. Katarzyna Matczyszyn. Her research experience involved also several internships in Université de Rennes 1 (France), University of St Andrews (Scotland), and Donostia International Physics Center (Spain). Her research interest is focused on plasmonic gold and bimetallic nanoparticles, their modification, and application in photo-driven processes such as photocatalysis. |
 Marco Deiana | Marco Deiana received his BSc in Chemistry from Università di Cagliari (Italy). He obtained both his MSc degree in Medicinal Chemistry and PhD in the field of Materials Engineering at Wrocław University of Science and Technology (Poland) under the supervision of Prof. Katarzyna Matczyszyn. Currently, he is a postdoctoral researcher at Umeå University (Sweden). His research interests are at the interface of chemistry, biochemistry and molecular biology and mainly combine spectroscopy and microscopy tools to characterize new photonic materials for DNA-related diagnostic and therapeutic applications. |
 Ziemowit Pokladek | Ziemowit Pokladek received his MSc and PhD in Chemistry from Wroclaw University of Science and Technology (Poland), under the guidance of Prof. Katarzyna Matczyszyn and Prof. Piotr Mlynarz. Currently his is a Synthesis Team Leader at R&D department of PCC Exol. His scientific interest involves the synthesis and molecular modeling of new photoswichable azobenzene and correlation between structure and properties of organic molecules. |
 Marek Samoć | Marek Samoć is a physical chemist specializing in nonlinear optical (NLO) materials and effects, nano-photonics, biophotonics as well as the electrical properties of molecular materials. He has extensive experience in investigations of NLO effects using short pulse lasers. After graduating from the Technical University of Wroclaw, Poland, he worked at NRC Canada in 1979/80, at Dartmouth College in 1987/88, at SUNY Buffalo in 1988/89 and at the Laser Physics Centre, ANU, Canberra in 1991–2008. In 2008, he moved back to Poland and is now Professor of Materials Science at WUST. He is a corresponding member of the Polish Academy of Sciences, Polish Academy of Knowledge and a member of European Academy of Sciences. |
 Katarzyna Matczyszyn | Katarzyna Matczyszyn is a professor at Wroclaw University of Science and Technology (Poland). After obtaining her PhD in physical chemistry in Wroclaw, she gained her research experience in Politecnica de Madrid (Spain), CEA Saclay (France), Universite Pierre et Marie Curie in Paris (France), Ecole Normale Superieure de Cachan (France), Australian National University in Canberra (Australia). Since 2008 she works in the group of Prof. Marek Samoc. Her research interests focus on linear and non-linear spectroscopic and microscopic studies of photoactive materials and biomaterials including photochromic molecules, liquid crystals (mostly lyotropic), plasmonic nanostructures, DNA and photodynamic therapy. |
1. Introduction
Light is an ideal external stimulus for manipulation of matter on the molecular level.1–4 It is eco-friendly, the response can be ultrafast, and, crucially, it can provide remote control of phenomena and processes with high temporal and spatial precision.1,5 Therefore, light-responsive molecules play a key role in the ongoing quest for selective and specific molecular agents with the ability to provide instantaneous reaction upon irradiation.6,7 A particularly important group of such compounds are azobenzenes (ABs), which have several advantages as molecular photoswitches. Firstly, their synthesis is relatively simple, secondly, their chemical scaffold is easily amenable to structural modifications, thirdly, their photochemical stability is usually high, and finally, there is a possibility of use of various wavelengths for the induction of the isomerization process.4,8–10 The mechanism of photoswitching in AB relies on the trans-to-cis isomerization, which induces the change of their various physicochemical properties such as absorption spectrum (photochromism), geometry, polarizability, dipole moment, etc.8,10,11 Most often the wavelengths used to trigger the trans-to-cis isomerization of various AB derivatives are in the near-UV spectral region.6,10,12In general, the benefit sought from the scenario of employing the photochromic processes would be that light could penetrate a material or tissue in an essentially non-invasive way, with the limitations to the spatial resolution coming only from Abbe's law of diffraction, in practice the resolution being approximately half of the used wavelength. But, very importantly, there are limitations to the light penetration depth that depend on absorption and scattering of the light beam. In biological systems, short wavelength (UV) irradiation is prone to much scattering and may be toxic, due to various photochemical processes, thus it is not really non-invasive.7,13 In general, in any applications, the scattering on small structures may lead to the light beams being poorly penetrating.14,15 Accordingly, there is a great interest in the development of photochromic switches sensitive to either visible or infrared light irradiation.7,16–18 While there has been remarkable progress in this field, the synthesis of such molecules still remains challenging.19 In this situation, nonlinear optics comes in handy as the two-photon excitation of the photochromic molecules can be less invasive than the usual one-photon excitation, since it may employ photons of half of the energy needed for one-photon excitation, typically in the near infrared region. Thus, one way of addressing the deficiencies of traditional photoswitching is through the development of two-photon absorbing photochromic molecules.20,21
The approach of using two-photon absorption can reduce material or cell damage and ensures better penetrability through the tissue due to lowering both the absorption and scattering.21,22 Moreover, due to the use of tightly focused beams, the two-photon absorption can provide excitation of a very small volume of the sample (down to femtoliters) which assures high spatial resolution and very selective addressing of the molecules as well as enables optical sectioning for three-dimensional imaging.23–25 While the laser beam spot size attainable at twice longer wavelength is larger than that in the case of one-photon excitation, this is partly compensated by the fact that the absorption process itself depends on the square of the light intensity, thus making the excitation region smaller. Two-photon absorption is less perturbing to e.g. living cells due to the reduced phototoxicity acquired throughout the sample.26,27 The ability to localize the excitation at a certain depth in the sample minimizes its degradation by scattered excitation and emission photons.28 In response to the need for photoswitches addressable by the two-photon excitation, we summarize in the present review the current progress on such switches based on the AB moiety.
2. Azo compounds
The aromatic azo compounds were discovered in the middle of the nineteenth century, but their remarkable photochemistry was not described until 1937.29,30 While azo dyes exist in two configurations: trans and cis, most often the more thermodynamically favored one is the trans form. Due to the fact that these isomers have different geometries, their properties differ significantly, e.g. the unsubstituted trans isomer azobenzene molecule is almost flat and has no dipole moment while the cis form presents an angular geometry and a dipole moment of 3.0 D.10,31 Intriguing features of azobenzene and its derivatives result from the structure of the AB molecule, which consists of two aromatic rings linked by an azo group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–). The isomerization process from trans to cis form of an azobenzene motif usually occurs when it is exposed to UV light, while the reverse reaction can be achieved either thermally or by irradiation with visible light (Fig. 1).6,10 In view of their photochemical properties and the possibility to control them by an external stimulus such as light, the photochromic molecules based on AB motif are desirable for various applications.4,32,33 Unfortunately, the fact that the overwhelming majority of such molecules require photons in the UV range to switch from the trans into the cis form,34,35 is usually a constraint for biomedical or solar-energy-related applications.6,11 However, it turns out that substitution of AB with proper functional groups may influence the position of the absorption bands, resulting in the possibility of using longer wavelength photons to switch the azo motif.6
N–). The isomerization process from trans to cis form of an azobenzene motif usually occurs when it is exposed to UV light, while the reverse reaction can be achieved either thermally or by irradiation with visible light (Fig. 1).6,10 In view of their photochemical properties and the possibility to control them by an external stimulus such as light, the photochromic molecules based on AB motif are desirable for various applications.4,32,33 Unfortunately, the fact that the overwhelming majority of such molecules require photons in the UV range to switch from the trans into the cis form,34,35 is usually a constraint for biomedical or solar-energy-related applications.6,11 However, it turns out that substitution of AB with proper functional groups may influence the position of the absorption bands, resulting in the possibility of using longer wavelength photons to switch the azo motif.6
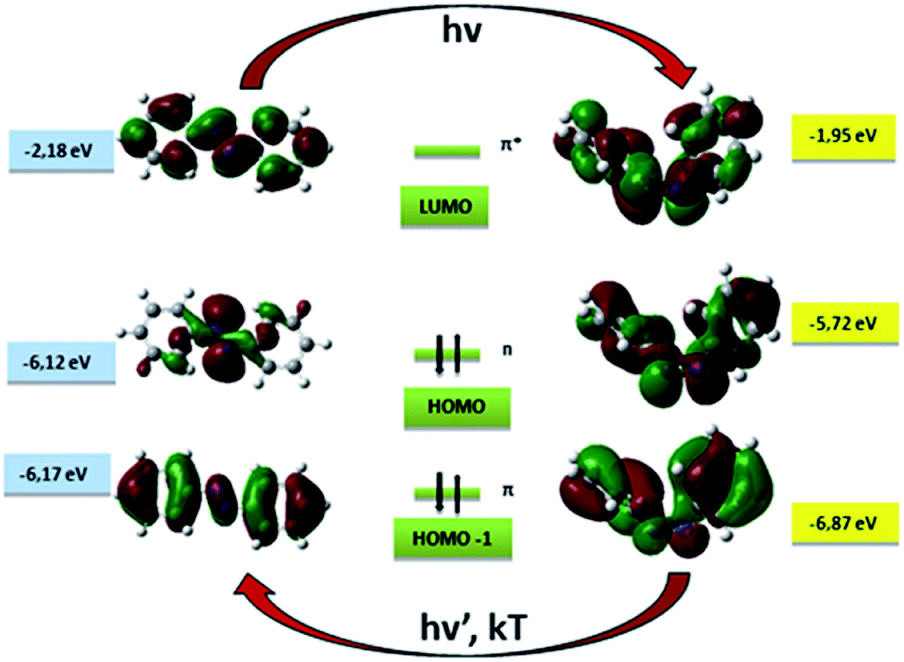 | ||
| Fig. 1 Molecular orbitals for trans and cis isomers of AB with corresponding energies in eV. The arrows indicate that the isomerization process can be driven by light for trans–cis transition and light or heat in the cis–trans case. | ||
2.1. One-photon induced isomerization of azobenzene – how substitution can modify photoswitching wavelengths
To expand the potential practical use of photochromic azobenzene switches there have been attempts to design AB derivatives switchable with Vis/NIR photons by taking advantage of the fact that substituents on the phenyl rings may strongly influence the position and shapes of azobenzene absorption bands.7,11,31 Recently, several modifications of the AB structure have led to achieving complete photoregulation with visible light. One approach concerns the decrease of the HOMO–LUMO gap and hence red-shift the AB absorption band. It can be achieved by extending π conjugation or introducing electron-donating groups (EDGs) and/or electron-withdrawing groups (EWGs) at ortho or para position in relation to the azo group.8,31,36 A drawback of this strategy is a considerable decrease of thermal half-life of the cis isomer, hence cis-to-trans isomerization is mostly driven by thermal relaxation.31,37In ABs, the energy of the n → π* transition is comparable for both isomers, so the trans and cis bands associated with this transition are overlapping. Therefore, another strategy involves the separation of the n → π* transition for trans and cis isomers. In 2009, Siewertsen et al. synthesized a cyclic azobenzene derivative in which, for the first time, large splitting of the n → π* bands originating from geometrical constraints, was observed.38 Thanks to this large separation of the bands, the cyclic azobenzene could be switched quantitatively in both directions using only visible light, namely irradiation in the green and blue (490 nm and 404 nm, respectively).38 Two years later, Woolley's group followed the strategy of introducing the methoxy group at the ortho position. Separation of the bands observed at lower energies resulted, in that case, from the difference in the interaction of lone electron pairs of oxygen and nitrogen depending on the geometric structure of AB (Fig. 2A). When the AB is in the trans form, the electrostatic repulsion of lone electron pairs is observed, while in the twisted geometry of the cis form these interactions are much weaker.39 It is important to note that in this particular example of the molecule (Fig. 2A), the pH has strong influence on the UV-Vis absorption spectra as pKa for the protonated species was found to be ∼3.8. At pH above 5.0 the spectra were essentially unaffected. Moreover, there is a smaller separation of the trans and cis n–π* bands in aqueous solution, which affects the lower trans/cis ratio at PSSs.39 Shortly thereafter, Bleger et al. exchanged methoxy groups with fluorine (Fig. 2B) and also detected separation of the AB bands present in the visible region of the spectrum.40 Such a splitting of the n → π* band is due to the strong negative induction effect of fluorine, and substitution of AB in the ortho position with this substituent causes a reduction of the electron density of the –NN– bond and thus reduces the orbital energy of lone electron pairs on nitrogen atoms.40 Importantly, this strategy leads to almost complete trans-to-cis as well as cis-to-trans photoisomerization (∼90% in both directions).7,40 Replacing F with Cl or Br also leads to similar electronic transitions as for fluorinated ABs.41–43 It is important to note that ortho-fluorinated azobenzene derivatives present a bistable character and, in comparison to azobenzene modified with methoxy group, they maintain almost planar geometry of the trans form and are sterically less demanding.40,44 This makes them likely to fit for photobiological applications in which intermolecular π-stacking interactions with the host molecule are required.45
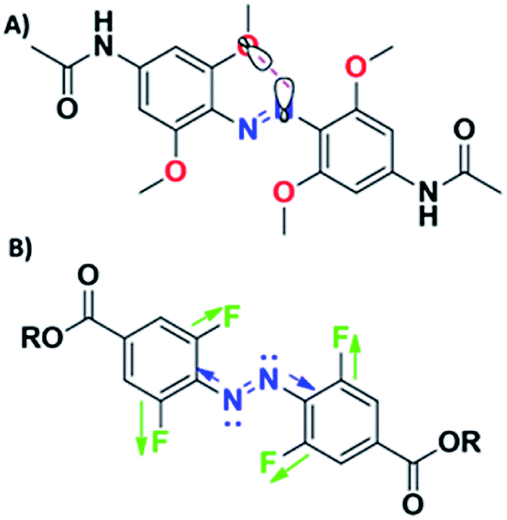 | ||
| Fig. 2 Modification of azobenzene molecule at ortho position can influence the energy of the n → π* transition of isomers. (A) Trans–cis isomerization can be driven with green light (530–560 nm), cis–trans isomerization could be then induced by using blue light (450–460 nm).39 (B) In the case of ortho-fluorinated azobenzene n → π* transitions of isomers are separated by up to 50 nm, so trans–cis isomerization can be driven with λ > 500 nm light and the reverse process can be performed with 410 nm.40 | ||
3. Nonlinear optics
3.1. Introduction to nonlinear optics
The advent of the laser was a milestone in the development of modern optics and photonics. The research on interactions of laser light with matter has led to the discovery of novel phenomena. In particular, at high light intensities available from short pulse lasers, there is a need to introduce nonlinear terms into the description of the response of matter to the electric field of the electromagnetic wave. This is the basis of nonlinear optical processes. In the context of this review, the relevant NLO process is nonlinear absorption, especially the two-photon absorption.At low electric field strengths, such as present in low intensity electromagnetic wave it suffices to consider the response of a medium in terms of the electric polarization vector P being proportional to the strength E of the electric field:
| P = ε0χE | (1) |
In the high intensity regime, the conventional description of NLO processes is that the polarization of the medium is considered to be a power series with respect to the incident electric field:
| P = ε0(χ(1)E + χ(2)E2 + χ(3)E3 + …) | (2) |
| P(ω) = P(1)(ω) + P(3)(ω) + P(5)(ω) + … = ε0[χ(1)(ω)E(ω) + χ(3)(−ω:ω, − ω, ω)E(ω)E*(ω)E(ω) + χ(5)(−ω:ω, − ω, ω, − ω, ω)E(ω)E*(ω)E(ω)E*(ω)E(ω) + …] | (3) |
3.2. Two-photon absorption
The concept of two-photon absorption was introduced in 1931 by Nobel Prize laureate Maria Göppert-Mayer.46 However, because of the unavailability of high light intensity sources, her theoretical prediction of molecular transition between two eigenstates via simultaneous absorption of two photons has not been experimentally proven until the invention of the laser 30 years later.47 The first research regarding TPA was published in 1961 by Kaiser and Garrett48 and concerned the frequency upconversion fluorescence (at 425 nm) induced by TPA (occurring at 694.3 nm) in CaF2:Eu2+ crystal. Since then also higher-order multi-photon excitation (MPE) phenomena (due to three-photon, four-photon, etc. processes) have been observed.It is important to note that MPE concerns not only the absorption of a number of photons but also the related physical and chemical effects, e.g. induced polymerization, induced isomerization, dissociation of molecules or enhanced changes of the physical parameters of the material. Indeed, MPE found applications in different areas of photonics, including multi-photon pumped lasing, multi-photon absorption based optical limiting, high-precision 3D micro-fabrication, high-density 3D optical data storage and more.
From the quantum mechanics point of view the process of a transition between two states, induced by the absorption of two photons is often depicted as involving a virtual intermediate state. Such a state is essentially extremely short lived and thus unobservable. On the other hand, there exist processes known as sequential absorption whose behaviour may to a certain extent mimic that of the true multiphoton absorption processes, but that do involve real intermediate states having measurable occupancy and lifetimes. An important difference between the instantaneous multiphoton absorption and the sequential processes is also apparent in the involved selection rules. While the selection rules for sequential processes reflect the probabilities of consecutive transitions, the probability of a true TPA process is a reflection of the properties of the initial and final state alone coupled by the electric field of the electromagnetic wave. It follows that, e.g. in centrosymmetric systems, the selection rules are vastly different: there is a so-called mutual exclusion principle stating that the excited states reachable by one-photon absorption are not available to two-photon absorption and the other way round. For the three-photon absorption, on the other hand, the selection rules are the same as for one-photon absorption. One needs to mention that there are also nondegenerate MPA processes possible, i.e. ones involving photons of different energies rather than identical ones, however, they are beyond the scope of this review.
3.3. Parameters describing two-photon absorption
The attenuation of the light beam traveling through a medium can be expressed as:
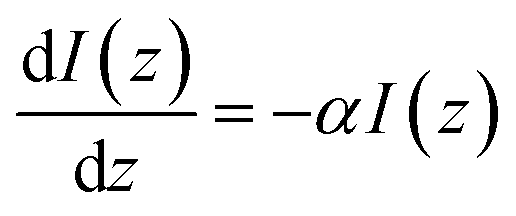 | (4) |
Under the conditions of nonlinear absorption, one usually modifies this equation as:
 | (5) |
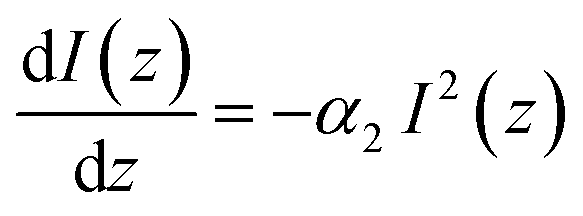 | (6) |
 | (7) |
The TPA coefficient α2 is a macroscopic parameter, dependent not only on the wavelength of the incident light but also on the concentration of the species (e.g. molecules, nanoparticles etc.) exhibiting two-photon absorption. α2(λ) can be described as:
| α2(λ) = σ2(λ)Nhν = σ2(λ)NAchν | (8) |
The two-photon absorption cross-section σ2 is a microscopic parameter characterizing an individual absorber, and is usually expressed in units called GM (the abbreviation of Göppert-Mayer):
| 1 GM = 10−50 cm4 s per photon | (9) |
In a similar way to the macroscopic two-photon absorption coefficient being related to the imaginary part of χ(3)(−ω:ω, −ω, ω), the TPA cross section is proportional to the imaginary part of the microscopic quantity called the cubic hyperpolarizability γ(−ω:ω, −ω, ω).
The hyperpolarizability as well as the cross-section values σ2 can in principle be calculated based on the complete data on the eigenstates and transition parameters (e.g. transition dipoles corresponding to the transitions) of the NLO species, however, quite often obtaining these information, especially for large systems, requires tedious computations. Fortunately, both the macroscopic parameter α2 and microscopic parameter σ2 can be derived from the experiments.
3.4. Two-photon absorbing materials
The research in the field of nonlinear optics has been focused not only on the fundamental studies but even more on the development of new materials exhibiting optimized nonlinear optical properties.50 At the same time, another crucial goal has been to find applications for those materials that merge the optimal combination of their inherent properties and the nonlinear optical properties. Many materials that exhibit linear absorption in certain spectral ranges, and thus also should absorb nonlinearly at the wavelengths multiplied by the number of absorbed simultaneously photons (provided the selection rules do not prevent it), are simply characterized by disappointingly low values of the multi-photon absorption cross-sections which diminishes their further utility in this area.In the early days of TPA studies, it has been realized that there are two classes of promising materials presenting strong TPA, namely bulk semiconductors and organic dyes, the latter usually studied in solutions. This scope has been largely expanded with the advent of nanotechnology and, indeed, it has been found that many nanosystems exhibit attractive nonlinear absorption properties. The development of MPA-related disciplines, such as bioimaging,51–53 microfabrication54,55 and 3D data storage56–58 also had an impact on the overall advancement in the field.
There are four basic requirements for the two-photon absorbing materials:
(i) insignificant linear absorption in the required spectral range;
(ii) large nonlinear absorption coefficients;
(iii) sufficient physicochemical stability;
(iv) durability under high-intensity laser irradiation.
The ease of modification of structures of organic and organometallic materials has been long perceived as the pathway to high nonlinear absorption coefficients, although such materials may be less robust than e.g. semiconductors. Organic chromophores (dyes, oligomers, polymers) have been widely studied as an interesting class of materials exhibiting MPA properties. Most of the π-conjugated organic dyes are transparent in the NIR region of the spectrum, which, combined with their linear absorption in the UV-Vis range, makes them good candidates as multi-photon active materials. It has been proven that the multi-photon activity of organic chromophores is directly linked to their specific molecular design.50,59,60 In particular, there is a correlation between intramolecular charge-transfer and the TPA capability of a molecule. Therefore, the most important motifs in the structure of widely studied nonlinear chromophores have been strong π-electron donor (D) and acceptor (A) groups linked with a π-conjugated bridge. A and D groups can be arranged either symmetrically (i.e. A–π–A or D–π–D) or asymmetrically (A–π–D) and by varying the length of the bridge one can tremendously increase the TPA cross-section of a molecule, introducing states with extended charge separation (Fig. 3). The effectiveness of the charge transfer (and thus also the magnitude of the TPA cross-section) can be also further increased by the co-planarity of the bridge.
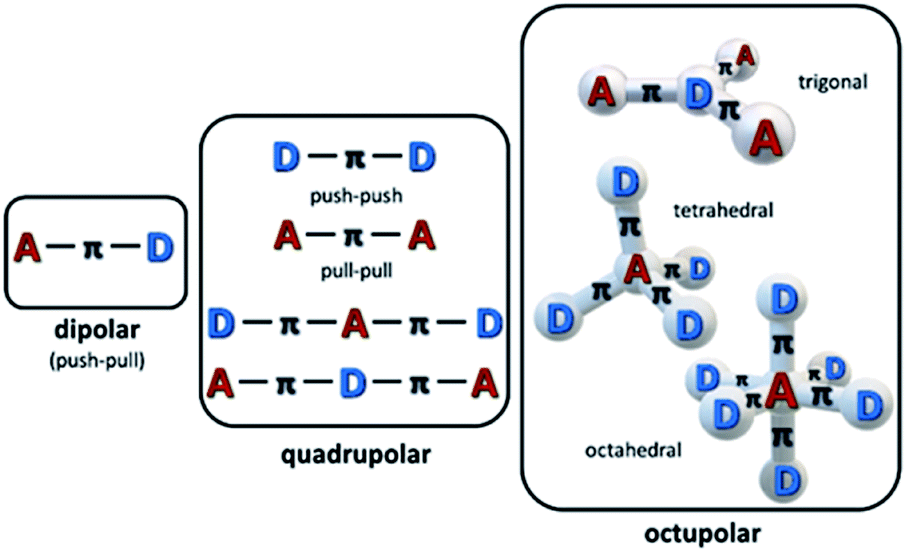 | ||
| Fig. 3 Schematic depiction of basic structural motifs of multi-photon absorbing chromophores. | ||
One may mention that there are several additional strategies to even further enlarge the TPA cross-sections of the chromophores,61 including the multibranched design of the molecules,62–64 which leads to the larger number of the active, multi-photon absorbing centres with the possibility of their mutual interactions, metallation of the structures as well as aggregation induced enhancements.
3.5. Z-scan – the principal technique for two-photon absorption measurements in azobenzenes
Among the measurements leading to the determination of TPA (and higher-order absorption) spectra of molecules one can distinguish direct and indirect methods. The very popular two-photon excited fluorescence (TPEF) technique is an indirect method based on the secondary effect of the TPA process which is the resulting fluorescence.65,66 Typically, in this technique one uses a reference dye with known TPA spectrum and by comparing its fluorescence with that of an investigated sample the determination of two-photon absorption cross-section spectra is possible. As most of the azobenzene derivatives are not fluorescent, in this review we will focus on the direct methods used to determine the third-order nonlinear optical properties of molecules. Currently, the Z-scan technique67 is by far the most popular method of characterization of nonlinear absorption and nonlinear refraction of materials owing to the relatively simple construction of the experimental setup and straightforward interpretation of the obtained data. Moreover, it is important to note that a multitude of data is obtained from just a single measurement run. The name of this single-beam technique is derived from the fact that its principle is that the investigated sample is moved along the z-axis of a focused laser beam. In this method, the transmittance of a non-linear medium as a function of its position (z) relative to the position of the focal plane of the lens is measured in two alternative ways. Either the power of the whole beam is being measured and this is called the open-aperture (OA) Z-scan trace or an aperture is placed in the far field and the transmittance through this aperture is recorded, which is called the closed-aperture (CA) trace.The principle of Z-scan has been described in numerous papers.68 Briefly, when the sample is located far from the focal point (z = 0), the transmittance in both OA and CA case is relatively constant. However, with approaching the focal point, the beam is being modified by NLO processes, which gain more importance as the intensity increases due to the beam spot size decreasing. The nonlinear absorption causes decrease of the transmittance and this is seen in both OA and CA signal, but there is also beam modification due to nonlinear refraction, which is equivalent to the sample acting as an induced positive or negative lens. In effect, the CA signal, in addition to the nonlinear absorption caused effects, shows a pattern of transmittance changes that is indicative of the sign and magnitude of nonlinear refraction. As seen in Fig. 4, through the use of a beam splitter (BS) one can perform the CA and OA scans simultaneously. The nonlinear refractive index and nonlinear absorption coefficient of the sample can be calculated based on the shape of the CA and OA signals, however, the light intensity needs to be known, which involves knowledge of the laser pulse energy, its temporal behavior (pulse duration) and geometrical shape. As it may pose a difficulty, to simplify the determination of the third-order nonlinear properties, in the laboratory of the authors of this review the measurements are typically carried out in a relative manner employing a reference, which is a fused silica plate for which the nonlinear parameters are well known. Moreover, when carrying studies of nonlinear materials in the form of solutions, an additional Z-scan measurement for the solvent used to dissolve the investigated compounds needs to be performed to provide the background for the determination of nonlinear refractive parameters. The experimental data obtained for the reference, solvent, and sample can be further analyzed using the equations derived by Sheik-Bahae et al.67 to retrieve the values of the parameters describing the nonlinear refraction and nonlinear absorption of a sample.
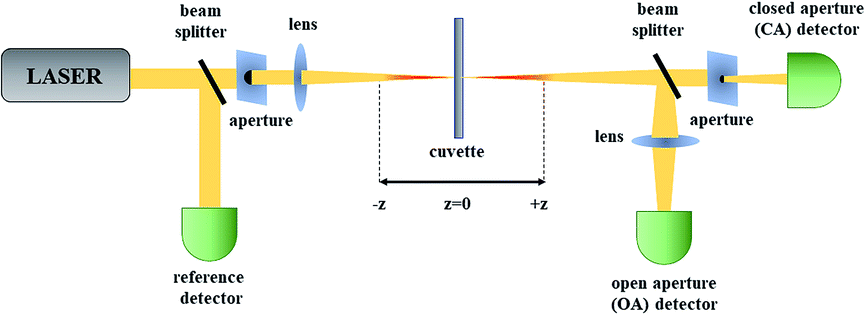 | ||
| Fig. 4 The scheme of a Z-scan setup. OA – open aperture for measurement of nonlinear absorption; CA – closed aperture for measurement of nonlinear refractive index. | ||
4. Two-photo absorption in azo dyes
The third-order nonlinear optical phenomena in azobenzene derivatives such as Disperse Red69–72 and polymers functionalized with AB derivatives were already investigated in the 1990s.73 However, the first systematic studies of TPA processes for the unsubstituted AB and AB derivatives were those of Mendonça and co-workers.71,72 First, they examined eight azo compounds with electron-donating (EDG) or/and electron-withdrawing (EWG) groups substituted at the para position to establish the correlation between molecular structure and the TPA process in the investigated molecules.72 They have shown that the unsubstituted AB is a relatively weak two-photon absorber (Fig. 5). Substitution of AB with EDG/EWG groups enhanced NLO properties and this effect is more pronounced when both donor and acceptor substituents are present (push–pull system).70,72,74 To fully characterize the influence of substituent on NLO properties of AB derivatives Kamounah et al. synthesized azobenzenes modified with groups of different strength of the donor and/or acceptor.69 It is known that the TPA cross-section for organic molecules depends on the intramolecular charge-transfer (ICT) efficiency in push/pull type structures (D/A–π–D/A) as well as the length of the π-conjugation.60 Indeed, the NLO properties were improved as the substituent polarity increased. The decrease of third-order nonlinear properties was more pronounced when the acceptor was exchanged with a weaker one comparing to the design in which the donor was exchanged with a weaker one.69 It is noteworthy that the calculations were in a good agreement with experimental data.75 The decrease in two-photon absorption cross-section (σ2) of the lowest π–π* two-photon absorbing states was found to result mostly from the decrease in value of the dipole moment of the excited states.76,77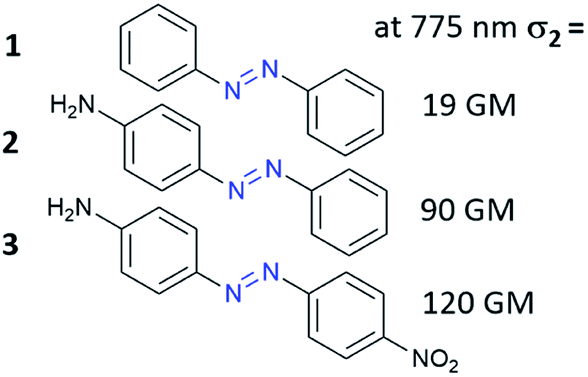 | ||
| Fig. 5 Two-photon absorption cross-section of AB can be enhanced by donor/acceptor substituents. The values of the two-photon absorption cross section were determined by Mendonça and co-workers using the Z-scan technique with femtosecond laser pulses at 775 nm.72 | ||
Further development of the work presented by Mendonça and co-workers has led to yet another molecular strategy to increase the TPA cross-section which relied on extending the π-conjugation.78 Unfortunately, the synthesized diazobenzene compounds did not stand out in terms of more significant two-photon absorption cross-section. Despite this, it has been confirmed that the main factor influencing σ2 is the presence of strong EDG and EWG groups.78 The impact on NLO properties of the extended π-conjugated system can be easily evaluated by the coupling of AB with different moieties like e.g. triphenylamine, pyrene, fluorene, carbazole. Mendonça and co-workers synthesized triphenylamine modified with EDG and EWG substituents as well as triphenylamine modified with both EDG group and the azo motif (as EWG group).79 One-photon absorption spectra of the investigated compounds exhibit a band at 340 nm and a band at 500 nm that stems from transition to a charge-transfer excited state. For all studied molecules, in the case of TPA spectra, a monotonous increase of the cross-section (towards blue) and a band around 650 nm were observed. Moreover, triarylamine modified with azo unit exhibited an additional band centred around 900 nm which was assigned to the presence of the AB motif which influences the molecular symmetry allowing the first-excited state (π–π*) to be reached by one- and two-photon absorption.79 It is worth mentioning that such a modification of the triarylamine structure by azo motif derivatives enables TPA in the so-called first biological window (i.e. the region of 650–1000 nm where the absorption of light by tissues is relatively low) with two-photon absorption cross-section of a molecule being around 200 GM. Another way to gain enhancement of NLO properties from the π-conjugation includes the incorporation of a π-bridge between two ABs terminated with donor groups.80 This approach allowed to obtain AB derivatives with improved TPA properties, with σ2 greater than 1200 GM (Fig. 6A).80 Recently, based on this strategy of extension of π-conjugated bridge, the enhanced TPA cross-sections were observed when bithienyl, pyrenyl, or anthryl groups were introduced to an azo moiety through conjugation linkage (CN) (Fig. 6B).81 The values of σ2 estimated for those molecules by the Z-scan technique ranged from 580 GM to 830 GM and were the highest for molecules modified with bithienyl and pyrenyl.81 Likewise, fluorene derivatives with good π-conjugation were used to enhance NLO properties of AB. Such compounds exhibit significant two-photon absorption, comparable to that of Disperse Orange III (DO3) (although there is no information about the exact value of the TPA cross-section).82
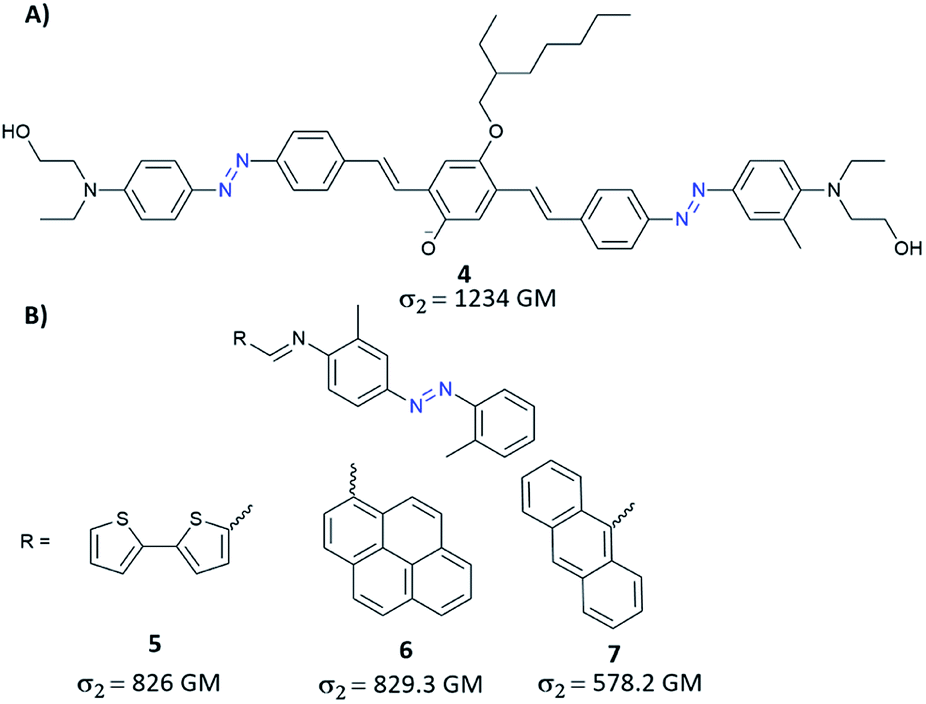 | ||
| Fig. 6 Representative examples of azobenzene derivatives with increased length of π-conjugation bridge. The values of the two-photon absorption cross section were determined using the Z-scan technique with femtosecond laser pulses at 780 nm (A)80 or 600 nm (B).81 | ||
Unfortunately, in most of the extant data, enhanced NLO properties go along with simultaneous significant reduction of the thermal stability of the cis isomer. Hence certain ideas emerged to obtain bistable azobenzenes with enhanced third-order NLO properties.62 Our group decided to synthesize isocyanurate derivatives with AB arms terminated by various groups (NO2, H, NMe2). As expected, the highest value of σ2 was observed for a compound with the donor group and the lowest σ2 was measured for a compound terminated with H. The three molecules exhibited improved NLO properties in comparison to the simple AB62,72 and, importantly, the cis isomer of two out of three investigated compounds (with NO2 or H) showed satisfactory thermal stability at room temperature.62
Another strategy to enhance NLO properties in azo compounds is to incorporate metal into the molecular structure (Fig. 7).83–85 There are two ways of introducing the metal atoms into the azo molecules: (i) metal is coordinated by the nitrogen coming from the azo moiety,83–86 (ii) metal is coordinated by another part of the azo derivatives excluding –NN– bond.87–89 Enhancement of the third-order nonlinearity in the first case (i) can be related to intramolecular electron delocalization coming from charge transfer from metal to ligand (MLCT transition) or from ligand to metal (LMCT transition).84 Up to date, a few complexes of azo derivatives with metals like Cu,86 Co,84 Ni86 have been successfully synthesized and their NLO properties have been measured. Unfortunately, there is no information about their isomerization properties. As mentioned above, the second way to incorporate metal into the structure of the azo derivative relies on the coordination of metal atom by AB derivatives excluding azo bond. Mendonça et al. synthesized a series of salen complexes possessing two azo dye units and examined them in the context of their NLO properties.87–89 They found no differences in TPA cross-section between the ligand and its complex with metal. Moreover, NLO responses of the investigated compounds indicated an additive effect of chromophores showing nondetrimental dipole–dipole interaction between the chromophores.88
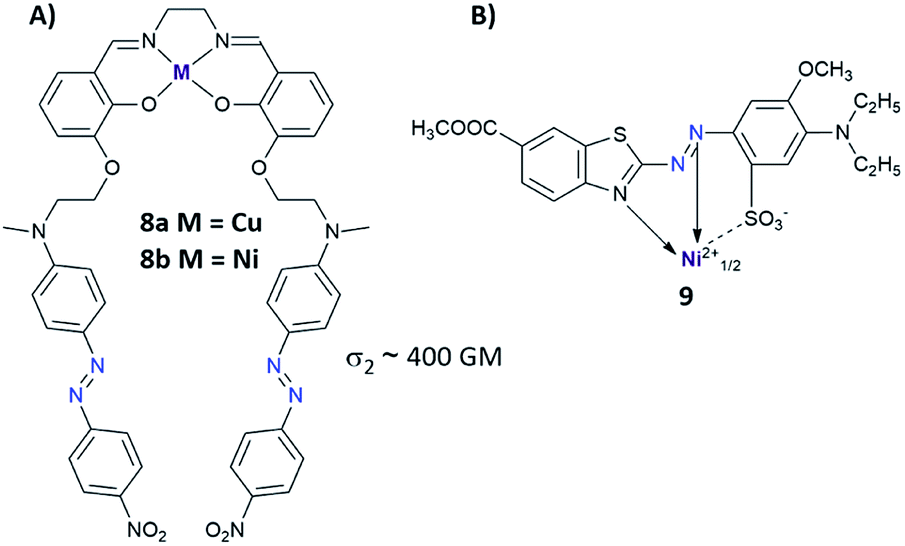 | ||
| Fig. 7 (A) Structure of the salen complexes tethered with two azo dye units. The values of the two-photon absorption cross section were determined from 700 nm to 1550 nm using the Z-scan technique with femtosecond laser pulses. The TPA maxima were found at approximately 1000 nm for the investigated molecules 8a and 8b.87 (B) Structural example of azo metal chelate dye.83 | ||
The values of the two-photon absorption cross section as well as the structures of the AB derivatives studied in that respect are presented in Table S1.†
5. Two-photon induced isomerization
While photoisomerization of molecular switches is usually carried out through one-photon excitation,31 the two-photon activation of this process can offer additional possibilities.21,90 Two-photon excitation may provide a 3D control of the process60,91,92 as only the molecules close to the focus point of the laser beam are excited due to the fact that TPA probability depends on the square of the excitation intensity.60,67,93 Since two-photon excitation takes place at longer wavelengths, this improves the penetration depth and reduces photochemical damage of material which is important for biological applications. Two-photon induced isomerization is a consequence of two-photon absorption process which may be performed in both direct (Fig. 8A) and indirect (Fig. 8B) way.7,94 | ||
| Fig. 8 Two-photon induced isomerization triggers interconversion between two switching forms (A) and (B). (A) Direct photoexcitation, (B) energy transfer from antenna (donor) to the switch. S0: ground state, S1: first singlet excited state, RET: resonance energy transfer. | ||
5.1. Direct two-photon excitation of photoswitches
Various aspects of two-photon induced photochemistry have been studied, including diverse fields such as dimerization reaction,95 singlet oxygen generation,96 or microfabrication.97 The photoinduced effects in azo-containing materials triggered by multi-photon absorption, such as reversible anisotropic third-harmonic generation or photoinduced birefringence also have been reported.98,99 Advanced studies of the two-photon induced isomerization process of azo chromophores were carried out by Magennis et al.94 They identified photoproducts and described kinetic parameters of the photoreactions induced by either one-photon or two-photon excitation.94 The data indicated that the same isomerization pathway of an azobenzene derivative can be evoked by both one- and two-photon excitation and the efficiencies of photoisomerization are comparable for both routes.94 Almost 10 years later, Moreno et al. published an article about an isomerization of ortho-fluorinated AB derivatives (Fig. 9, compound 10) induced by either one- or two-photon process.90 The two-photon absorption cross-section was calculated from bleach signal and found to be equal 7.1 GM. Two-photon excitation of trans ortho-fluoroazobenzene derivative populates simultaneously both S2 and higher Sn states because of sequential reabsorption S2 → Sn of the pump. However, the bleach signal and the isomerization products remain to be of two-photon origin as indicated by bleach anisotropy data and from pump power dependence of the isomerization kinetics. The photostationary state (PSS) (trans-to-cis isomerization) reached by two-photon excitation had a lower percentage of the cis isomer (60%) compared to 90% of the cis form after one-photon excitation.90 Guo and co-workers also observed higher population of the cis isomer at PSS (trans-to-cis isomerization) under one-photon excitation.100,101 Also, amino-substituted ABs were found to undergo trans-to-cis isomerization under nonlinear excitation and this process produced, as previously observed, a lower cis form content in comparison to the linear excitation.102 As this type of azo-compounds are known for very fast thermal relaxation to more thermodynamically stable trans isomer, no light is needed to switch the cis form back to its initial state. In turn, Passlick et al. synthesized compound 11 (Fig. 9) and demonstrated that such a bistable type of switch can undergo two-photon induced isomerization in both directions (trans-to-cis and cis-to-trans) and thus it reaches a high cis/trans ratio at the PSS state.103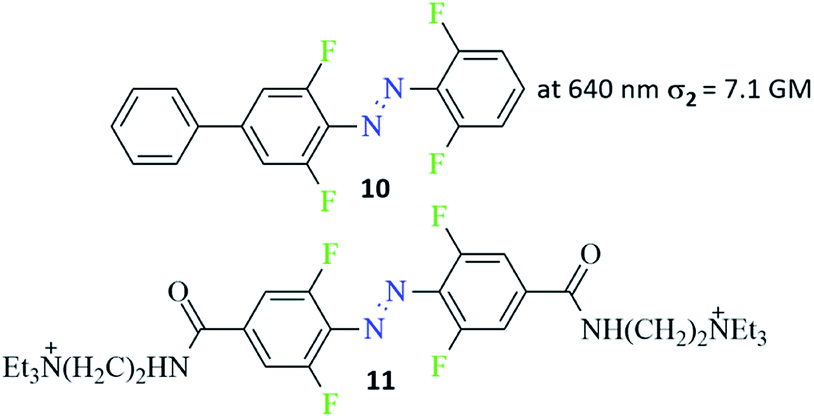 | ||
| Fig. 9 Structure of bistable azobenzene derivatives switchable in the nonlinear regime.90,103 The value of the two-photon absorption cross section was determined from bleach signal.90 | ||
5.2. Indirect two-photon excitation of photoswitches
As there is enormous interest in the development of molecular photoswitches that can be addressed entirely by Vis/NIR light, indirect ways of photoexcitation of azo chromophores have been designed.7,104 Undoubtedly, photoexcitation of azobenzene via energy transfer from a light-harvesting molecule (i.e. an antenna) with a high two-photon absorption cross-section is the most common method.7,105The Förster resonant energy transfer process (FRET) relies on the excitation of a donor molecule and subsequent energy transfer from the excited electronic state to a nearby molecule called an acceptor, which is simultaneously brought into its electronically excited state in a resonant process.106 If the acceptor molecule is emissive, that is it has non-zero fluorescence quantum yield, then it will fluoresce, otherwise the excitation energy will be converted to heat or consumed in photochemical processes, thus the isomerization process can occur (Fig. 8B).7,106 Several criteria must be fulfilled to observe this process. The absorption spectrum of the acceptor has to overlap with the emission spectrum of the donor. The donor and acceptor molecules also need to be relatively close and oriented in such a way that the interaction is optimized. The Förster critical distance (R0), which corresponds to the separation between D and A at which the FRET is 50% efficient, depends on the integral of the overlapped spectra, according to the eqn (10):
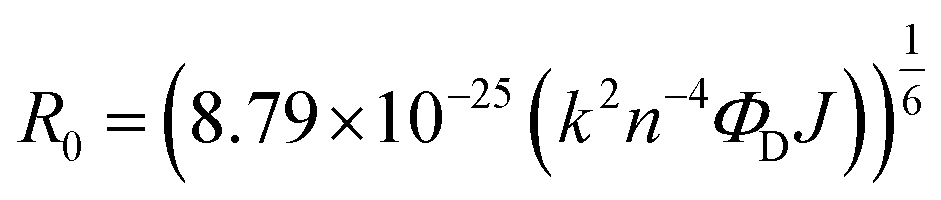 | (10) |
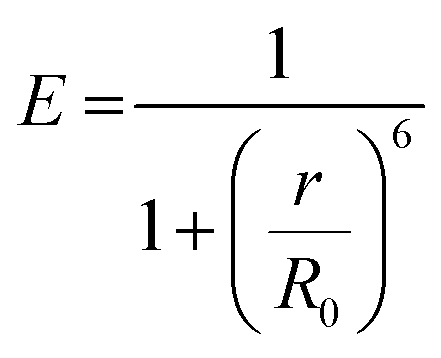 | (11) |
Typically, r should be in the range 1–10 nm to observe FRET, while at shorter (less than 1 nm) distances other energy transfer mechanisms may occur.
Two-photon induced isomerization can occur either by intramolecular107 or intermolecular105 FRET process. Typically, the molecular design of the system may include a well-known two-photon absorber antenna like naphthalene,107 pyrene,22 triphenylamine,108 or anthracene105 derivatives, a spacer (for intramolecular FRET) and a molecular switch (Fig. 10).
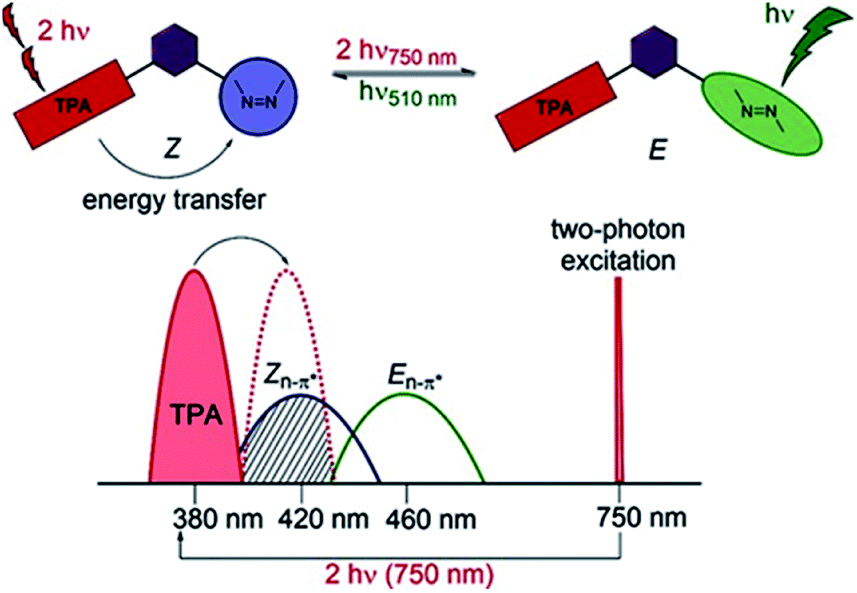 | ||
| Fig. 10 Illustration of the operating principle of indirect switching based on FRET process. Reproduced from ref. 108 with permission from John Wiley and Sons. | ||
An example of intermolecular resonant energy transfer was described by Croissant et al. who synthesized mesoporous silica nanoparticles combining a two-photon fluorophore and an azo chromophore.105 Nonlinear excitation of the two-photon absorber followed by intermolecular energy transfer to trans azobenzene derivative caused photoisomerization of azobenzene from trans to cis isomer.105 The energy-transfer quantum yield was found to be very high (95–99%), however, the authors did not give the exact ratio of cis and trans isomer at PSS.
A more widespread route of energy transfer is an intramolecular process. Izquierdo-Serra et al. obtained AB derivatives modified with naphthalene and they induced two-photon isomerization of azo chromophore directly (740 nm) and indirectly (880 nm).107 They showed that the response of the system is similar in both cases. However, these specific AB derivatives suffered from low thermal stability of the cis isomer. This fact inspired the design of a new AB derivative with a long thermal lifetime of the cis state.22 The pyrene unit was chosen as a crucial unit, since it manifests significant two-photon absorption under NIR irradiation109 and emits in the same spectral region where the AB core absorbs.22 However, the selective excitation of the pyrene motif can pose some problems since the absorption spectrum of pyrene overlaps with that of the AB core and the emission spectra of the fluorophore overlap with the absorption spectra of both isomers. Unfortunately, although a pyrene-modified AB chromophore was designed to be active for nonlinear excitation, there is no experimental data that would confirm that.22 Significant progress towards indirect two-photon induced isomerization of bistable azo chromophores was achieved by Hecht and co-workers (Fig. 10).108 They synthesized ortho-fluorinated AB derivatives modified with triphenylamine – a moiety known to be a good two-photon absorber – linked to AB by thiophene and phenylene group in order to red-shift the absorption band of the antenna to the region where the AB exhibits negligible absorption. Such a molecular design enables trans-to-cis isomerization with green light and led to high photoconversion, with 92% of cis isomer in PSS. Back reaction could be induced either by one or two-photon excitation. Irradiation at 380 nm resulted in 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24 trans/cis ratio at PSS while the two-photon excited process (at 750 nm) resulted in 65:35 trans/cis ratio at PSS. It needs to be stressed that irradiation condition (time, power, wavelength) have pivotal influence on the photochromic reaction. Under the experimental conditions used by the authors, the PSSE was achieved after 35 min of irradiation. Moreover, the thermal stability of the cis form of the molecule was found to be as long as 522 days at room temperature.108 It is worth to notice that bistable photoswitches provide an excellent starting point for designing two-photon active photoswitches as it is less difficult, by prolonged irradiation, to produce large steady state fraction of the cis isomer.42,110,111
:24 trans/cis ratio at PSS while the two-photon excited process (at 750 nm) resulted in 65:35 trans/cis ratio at PSS. It needs to be stressed that irradiation condition (time, power, wavelength) have pivotal influence on the photochromic reaction. Under the experimental conditions used by the authors, the PSSE was achieved after 35 min of irradiation. Moreover, the thermal stability of the cis form of the molecule was found to be as long as 522 days at room temperature.108 It is worth to notice that bistable photoswitches provide an excellent starting point for designing two-photon active photoswitches as it is less difficult, by prolonged irradiation, to produce large steady state fraction of the cis isomer.42,110,111
5.3. Plasmon enhanced two-photon induced isomerization
A relatively novel approach to increase the cross-section of multi-photon absorbers is based on using materials with the ability to localize light, such as photonic crystals,112,113 microspheres,114 or noble metal nanoparticles. The latter materials exhibit a phenomenon called Localized Surface Plasmon Resonance (LSPR), which refers to the excitation of collectively oscillating conduction band electrons of the metallic nanostructure by incident light at the resonant frequency.115,116 As a result, a strong localization of the electromagnetic fields can occur in the vicinity of the nanostructure. This phenomenon can be employed in various photochemical processes, including photocatalysis and photochromism. This approach has been proposed to overcome the need for using high irradiances in order to induce multiphoton absorption, and hence to facilitate the application of multiphoton absorbers in biophotonics and biomedical applications.117 Tsuboi and co-workers demonstrated a successful two-photon ring-opening reaction of diarylethene (DE) derivative assisted by 20 nm gold nanospheres and induced by near-infrared continuous-wave laser light in an ultrathin polymer film.118 Wu and co-workers complemented the study by investigating DE derivative ring-opening reaction in a solution system, using micrometer-sized reaction chambers.159 The reaction was assisted by the ordered arrays of gold nanoblock dimers. Even stronger enhancements can occur near the fractal clusters (clusters exhibiting random fractal geometry) of metal (e.g. silver) nanoparticles.120The plasmon-enhanced two-photon induced isomerization of AB molecules has also been demonstrated. Prasad and co-workers created an inorganic–organic interface by the adsorption of non D/A modified AB moiety on the surface of silver nanoparticles.104 The azobenzene was linked to a peptide-based metal binding ligand, constituting a hybrid moiety that was further used for both synthesis and stabilization of spherical silver nanoparticles (4.3 nm in diameter, LSPR band at 430 nm overlapping the n → π* band of the AB). The inorganic–organic interface of these hybrid nanoparticles could be reconfigured using two-photon induced isomerization, and the LSPR enhancement selectively promoted cis → trans isomerization at excitation wavelengths from 700 to 1000 nm (Fig. 11). Further experiments confirmed that the origin of the phenomenon was not thermal, but electronic.121,122
 | ||
| Fig. 11 Overlapping between the LSPR band of plasmonic nanoparticles (blue) and absorption bands of azobenzene (black, here: n → π* band of azo-based ligand depicted above the spectra) may contribute to the selective enhancement of photoswitching (here only cis → trans two-photon induced isomerization is promoted). Reproduced from ref. 104 with permission from Royal Society of Chemistry. | ||
However, as presented by Sivapalan and co-workers, plasmon-induced two-photon absorption cross-section enhancement is distance-dependent.121,122 The authors were able to modulate the distance between gold nanorods and dye molecules (AF-348-3A) by surface modification of nanoparticles with polyelectrolyte coating, applying up to eight layers of polyelectrolyte on top of the CTAB bilayer (which translates to 15 nm distance between the chromophore and the bare gold surface).122 Moreover, it is crucial to note that the distance dependence of the enhancement factors is different for the on- and off-resonant conditions, i.e. whether the LSPR band of the nanoparticle is at the wavelength of the excitation laser or away from it.
Photochromic molecules have been proposed as promising materials for optical memory devices and optical switches. Hence, their two-photon sensitivity enhanced by the presence of plasmonic materials can potentially enrich the nanophotonic devices. The enhanced two-photon induced isomerization of chromophores can be used for the writing process, while one-photon isomerization can be used for either reading or erasing procedure.123 Moreover, plasmons can also enhance other two-photon driven phenomena, such as two-photon induced polymerization, as presented by Ueno and co-workers.119 On the other hand, two-photon absorbing azopolymers can be used for the near-field observations of hot spots generated e.g. in plasmonic oligomers.
6. Application of direct/indirect two-photon excitation of azo chromophore
6.1. Biological applications
The most intense studies concerning two-photon induced isomerization of azobenzenes are related to the fields of photopharmacology and synthetic optogenetics.21,124 Optogenetics is a novel approach developed for the remote studies of neurons that relies on the use of synthetic light-responsive chromophores to employ photon absorption to modulate structure and function of the target molecule.124,125 The first demonstration of the use of AB derivatives toward photo-manipulation was conducted on the nicotinic cholinergic receptor (nACh) in the late 1960s.126 To date, significant modifications of AB derivatives have been employed to improve their properties as well as to develop new strategies for AB-mediated photo-control of the receptor and channel function.127 Especially important approach concerns illumination with Vis/NIR light to trigger isomerization process. In the majority of systems UV and Vis light are involved to provoke geometry change of AB, which, besides cytotoxicity, is not suitable for deep tissue penetration due to strong absorption and scattering from endogenous components. The use of NIR light to stimulate isomerization process is challenging, however the evolution of nonlinear optics enables adoption of infrared light to switch molecular switches between the cis and trans states. In 2014 the two-photon excitation of azobenzene derivatives was used to control ion channel (Fig. 12).107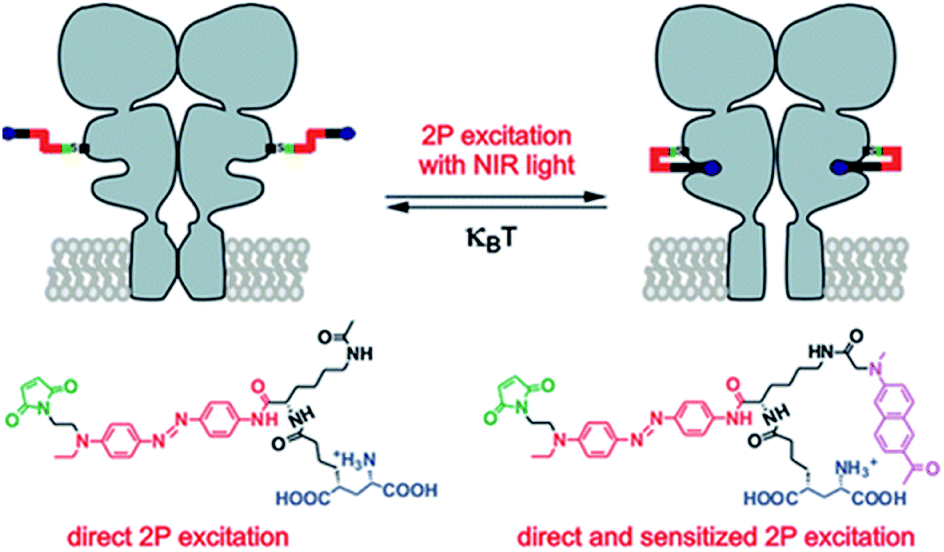 | ||
| Fig. 12 TPE with NIR light induces glutamate recognition and the channel is opening by trans–cis isomerization, which leads to ion flow across the membrane. Irradiation with Vis light or thermal relaxation of AB revert the process. Reproduced from ref. 107 with permission from American Chemical Society. | ||
Azobenzenes called MAG, MAG2P and MAGA2P, their structures involving maleimide–azobenzene–glutamate units, were tested regarding their one- and two-photon excitation toward control of the activity of glutamate receptor (Fig. 13).124 Previous works showed that an ionotropic glutamate receptor can be controlled by switching MAG (12) derivatives between two states with violet and green light.128 The real challenge here was to improve the two-photon absorption cross-section of MAG and for that reason MAG2P (13) and MAGA2P (14) were designed and synthesized. The direct/indirect two-photon excitation of azo chromophore leads to trans-to-cis isomerization of MAG2P and MAGA2P, which opens the channel. However, as the thermal stability of 13 and 14 is very low (the involved lifetimes are in the ms range) channel closure takes place just after the illumination is switched off (in contrast to 12 which requires green light irradiation to switch back to the trans state). The advantage here comes from the fact that just one source of light is required to switch MAG2P and MAGA2P, however under two-photon excitation, MAG achieves higher current amplitudes than newly designed compounds.107
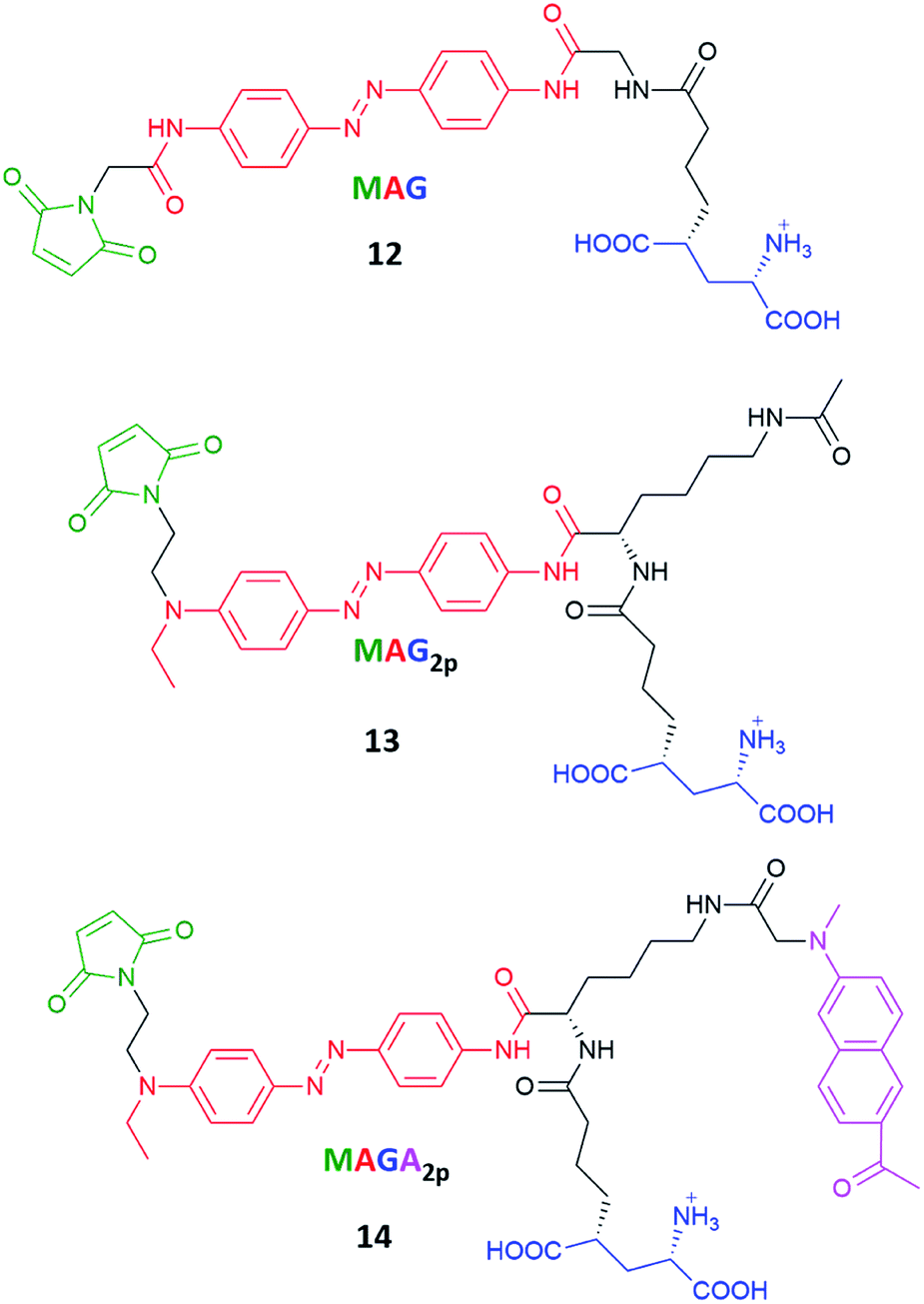 | ||
| Fig. 13 Structures of the photoswitchable tethered ligands applied to the two-photon control of LiGluR (light-gated glutamate receptors).107,128 | ||
A few months later Isacoff's group demonstrated results concerning the two-photon absorption cross-section in a wide range of wavelengths (600 nm–900 nm) for molecules from the MAG family (Fig. 14). They also checked the efficacy of two-photon excitation followed by activation of the GluK2 kainate receptor, LiGluR.102 L-MAG0 (15, amino-azobenzene type10) was tested under one- and two-photon excitation conditions towards photocontrol of opening and closing of the channel. The OPE process is more efficient, because the photocurrents achieved under two-photon excitation of azo derivatives never exceeded more than 15% of photocurrents induced under one-photon activation from the same cell.102 However, for L-MAG0460 (16, push–pull type azobenzene10) the response under TPE was found to be equal to 40% in comparison to OPE of the same neuron.102 Finally, ABs (17) from MAG family with both high two-photon absorption cross-section and slow thermal back-isomerization were obtained. This was achieved by synthesis of the electronically asymmetric AB derivative (17) with weak mesomeric EDG (azo-NHCO–) and EWG (azo-CONH) as well as strong inductive EWG (F). Particularly important here is the fact that biological activities under one- and two-photon excitations are comparable.21 Gorostiza and co-workers showed, employing TPE of alloswitch (AB derivative), reversible silencing of the activity of metabotropic glutamate 5 (mGlu5) receptor in intact brain tissue.23 Moreover, they calculated the axial plane selectivity of TPE of a photoswitchable ligand in brain slices and they noticed that such a value corresponds to physiologically relevant dimensions, such as dendritic spines.23
 | ||
| Fig. 14 Structures of the photoswitchable tethered ligands applied to the two-photon control of LiGluR.100 | ||
Besides the two-photon control of light-gated glutamate receptors (ionotropic and metabotropic), nonlinear excitation of an azo moiety was used to photocontrol activity of hippocampal CA1 neuron. A tetra-fluorinated azobenzene derivative with quaternary ammonium salt (Fig. 9, compound 11) was synthesized. This molecule was studied as a potential ion channel antagonist. The isomerization process was stimulated by both OPE and TPE, showing that the linear geometry of trans isomer enables better blocking of the ion channel in comparison to the efficiency of the bent cis isomer. OPE of AB makes the blocking process more efficient.103
Isomerization process of molecular switches can be also applied as an attractive strategy to develop the light-responsive nanocarriers for the on-demand release of the drugs with spatial control. In the majority of cases, upon UV irradiation of azobenzene containing nanocarriers, the polarity of the nanoparticles is changed, which is accompanied by the photoisomerization (trans-to-cis) and disassembly of nanoparticles to enhance the release rate and efficiency of drugs payload. However, as mentioned above, it is crucial to implement NIR radiation instead of UV. As a proof of concept Zink et al. designed two-photon fluorophores with a significant two-photon absorption cross-section, suitable for photoisomerization of azobenzene derivatives (trans-to-cis) via the FRET mechanism.105 The nanoimpeller groups present in the porous framework allow the drug (anticancer drug, camptothecin (CPT)) to be trapped. The two-photon induced isomerization triggered by irradiation with NIR light leads to the CPT being kicked out from the pores and ultimately results in cancer cells killing under in vitro conditions.105 These promising results led to subsequent attempts at creating drug nanocarriers controlled by light. The next report concerned also systems based on mesoporous silica nanoparticles (MSN) and AB (grafted on the surface of the nanoparticles) with a high specific surface area and pore volume for TPE-triggered drug delivery in cancer cells.129 As previously mentioned, the Förster resonance energy transfer process was used to induce isomerization. The fluorophore suitable for FRET was a paracyclophane derivative (CF).129 The cargo (CPT) was loaded in the pores of the nanoparticles consisting of MNS, CF, and azobenzene derivatives and the supramolecular complexation of β-cyclodextrin was performed to cap the porous surface and to create a specific kind of the nanovalve. The two-photon induced isomerization (trans-to-cis) of azobenzene led to the opening of the nanovalves and release of the drug (Fig. 15). This in vitro study demonstrated excellent biocompatibility, stability, and photocontrolled delivery in cancer cells under two-photon excitation.105,129
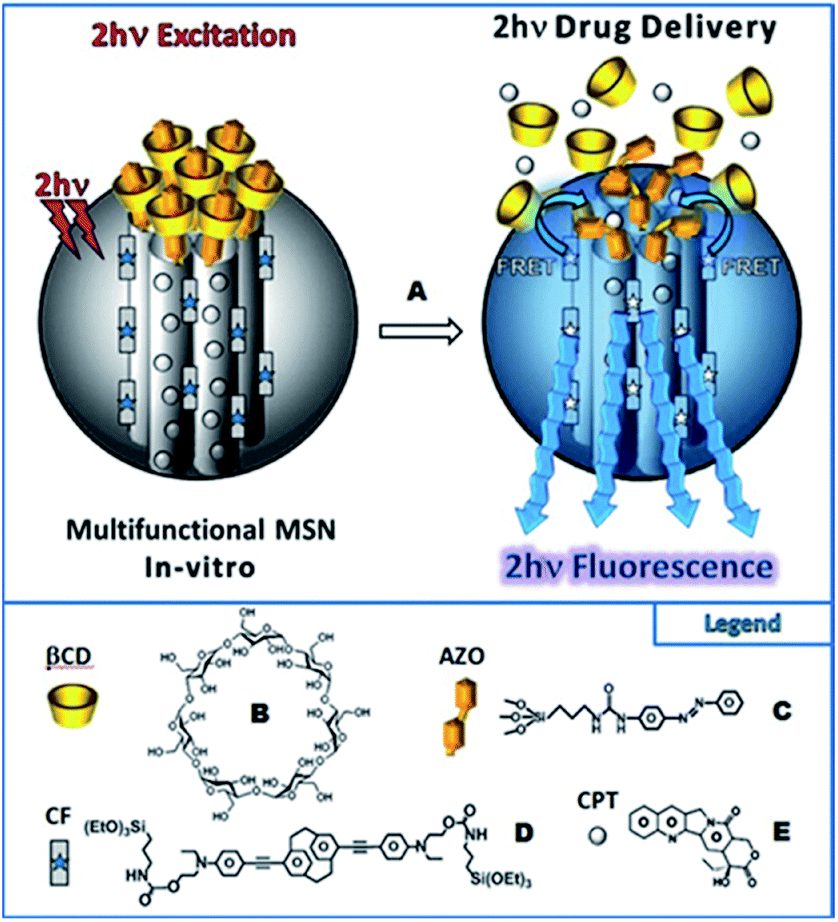 | ||
| Fig. 15 Schematic representation of the two-photon-triggered multifunctional nanovalve (A). The two-photon irradiation triggers the release of the camptothecin anticancer drugs (E), via the photo-isomerization of azobenzene moieties (C) grafted on the surface of so-called CF fluorophore (D) modified MSN, and bonded with β-cyclodextrin (B). Reproduced from ref. 129 with permission from John Wiley and Sons. | ||
Recently, Zhu et al. reported a NIR sensitive gated assembly of supramolecular conjugated unimicelles utilized as a drug carrier.130 The constructed system was based on host–guest recognition between β-cyclodextrin-grafted hyperbranched conjugated polymer (HCP) and azobenzene-functionalized poly(ethylene glycol) (AZO-PEG), and, ultimately the anticancer drug doxorubicin (DOX) was encapsulated inside. The working principle of such a system is based on photoisomerization of AB motif from trans to cis form triggered by NIR light via FRET strategy, which leads to disassembly of micelles as a result of leaving of AZO-PEG from the cavity of β-cyclodextrin and finally the DOX is released in the cells (Fig. 16). It has been shown that the intracellular release of the drug could be controlled by near-infrared light irradiation, moreover, the released drug could be visualized when displaced from cytoplasm to nucleus by confocal laser scanning microscopy.130
 | ||
| Fig. 16 Schematic depiction of azo unit photoisomerization induced by (A) ultraviolet (UV) light, and (B) NIR light via two-photon (TP)-excited FRET of a conjugated polymer. (C) The mechanism of the NIR-triggered photoisomerization of azo by TP-FRET. (D) The preparation of supramolecular conjugated HCP-PEG unimicelles and their NIR-triggered drug release in cancer cells. Reproduced from ref. 130 with permission from Royal Society of Chemistry. | ||
6.2. Application of TPE of azobenzene derivatives in materials science
In the last few years, there has been a growing interest in using azo compounds in materials science owing to their potential device applications (data storage, optical filters, diffraction elements, ordering layers) based on properties such as photoinduced anisotropy, mass transport and phase transitions.131–133 The light-induced modifications of macroscopic properties of AB based materials result mostly from the isomerization process of its molecule. However, the majority of the studies based on the photoinduced geometry change of photochromic motif have been carried out with one-photon excitation, although it has been realized that localized material modulation can be driven in the system with a high degree of spatial resolution through the two-photon induced isomerization.24 Thus, while the photoinduced phase transition studies in azo derivative/doped liquid crystals have been carried out mostly with OPE,134,135 the advantages of TPE for better control of the phase transitions, especially with respect to three-dimensional spatial resolution have been noted.131 It has been shown that 4-n-heptyl-4-n-propylazobenzene (DAAB-307) can undergo two-photon induced trans-to-cis isomerization and cause nematic-to-isotropic phase transition of an AB LC.131 A disadvantage of using TPE is, however, that, due to the need for high light intensities which require focused beams, only a very limited volume of a material can be addressed at one time.A much studied phenomenon has been light-induced birefringence, sometimes also called orientational hole burning, which is directly associated with photo-orientation of AB motif.136 This phenomenon relies on selective absorption of light by azo molecules and their further isomerization and rotation perpendicularly to the direction of the polarization of light.136 Absorption of the excitation light depends on the alignment of the direction of its polarization with the transition moment of the absorbing molecule. Hence, illumination of the azo-based material with linearly polarized light induces trans-to-cis-to-trans isomerization leading to decrease of the population of molecules with their transition dipole along the light polarization direction and the increase of the population of molecules with their transition moments perpendicular to it (both in plane and out of plane). Consequently, optical anisotropy is induced and the material displays birefringence.136,137 Induced birefringence achieved by photoisomerization and subsequent molecular reorientation allows azopolymers to be potentially used as surface-relief gratings138 or in optical storage media.139
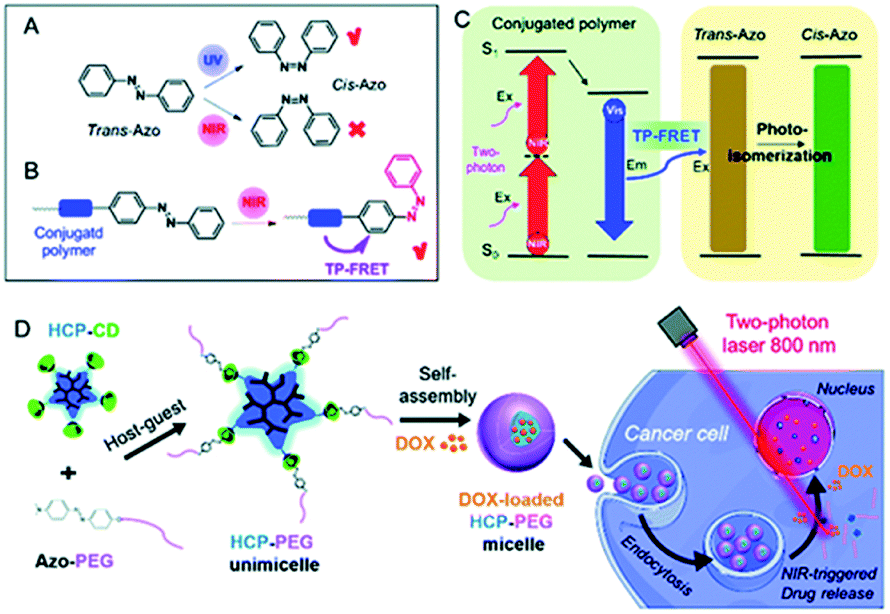
One of the first attempts to the data recording by polarized nonlinear excitation of the AB motif (Disperse Red 1, 18) was carried out by Kawata and co-workers.140 They showed that, by using pure poly(methyl methacrylate) (PMMA), no written bit can be observed, however the presence of AB derivatives enables recording bits. Before irradiation, the sample is isotropic, and only nonlinear excitation of AB induces isomerization and thus anisotropy appears. However, the molecules can bleach over time, as it was described in the case of compound 18.140 These results became a motivation for Mendonça and coworkers who decided to examine two-photon induced anisotropy in PMMA films doped with 19 (DR13).141 Interestingly, two-photon excitation of 19 induces reversible birefringence of the material and no photobleaching of the chromophore was observed (Fig. 17).141
 | ||
| Fig. 17 Volumetric birefringence pattern recorded via two-photon absorption. (a) Schematic representation of optical storage experimental setup. (b) When the sample orientation is set at 45° with respect to the polarizer axis the pattern can be visualized. (c) When the sample is placed parallel to the polarizer axis the picture cannot be seen. Based on the ref. 141. | ||
In order to further explore the optical storage properties of azobenzene-based materials, they prepared PMMA films containing salen dyes (SD) and SD derivatives with metals such as Cu and Ni (Fig. 7).This enabled them to study the influence of the metal on the optical storage features.142 The differences were observed under OPE, the longer writing times were required for the organometallic compounds in comparison with molecules without metal.142 Moreover, it has been demonstrated that light-induced orientation by one- or two-photon-induced isomerization of ABs (20) (Fig. 18) in PMMA films is characterized by similar dynamics of isomerization and the degree of molecular orientation is comparable for both ways of excitation.143
 | ||
| Fig. 18 Structures of photoswitches used as guest molecules in polymer films. DR = Disperse Red, DO = Disperse Orange.70,143 | ||
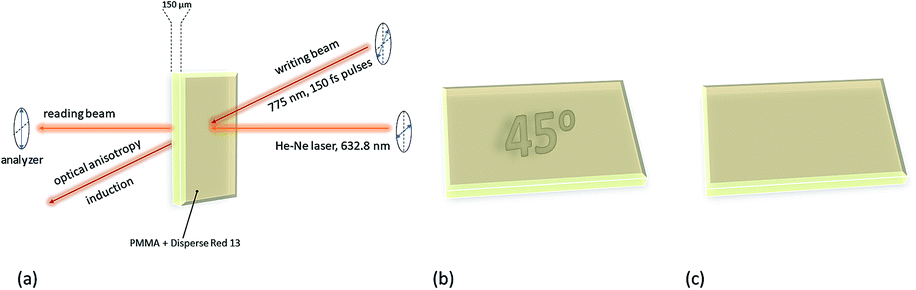
While much work has been done on guest–host polymer films containing azo dyes, the two-photon-induced birefringence can also be observed for azochromophores attached covalently to the polymeric chain.133 An example of such a polymer is polythiophene bearing an azo motif attached to the polymer backbone.133 It has been shown that the two-photon induced birefringence is two orders of magnitude higher than that reported for guest–host films, probably due to smaller mobility of the azo unit after the proper orientation and restrained dye aggregation.144 However, the optically induced birefringence was an order of magnitude higher when one-photon excitation was used.133 Another way to enhance birefringence and thermal stability of the material relies on using bis-azobenzene chromophores. It is expected that such a system should have better storage effect, including higher signal contrast, better readout properties, and larger storage stability.145 Zhang et al. synthesized a copolymer containing bis-azobenzene chromophores used for two-photon-induced polarization storage.145 They proved that the data readout effect depends on the direction of reading polarization and that two letters can be encoded in the same region of a given layer by modification of the two polarization directions of recording beams, which makes this material a promising candidate for high-density optical storage.145 Furthermore, the possibility of rewriting new data after erasing the previous information has been checked using the same material (Fig. 19).146 It was found out that, indeed, the data can be recorded by linearly polarized light and then erased by either circularly or linearly polarized light, however in the case of the erasing, the linear polarization of the beam needs to be perpendicular to that of the recording beam. Finally, new data could be subsequently rewritten in the same material area after erasing.146 Those findings have been also confirmed by other studies.147 Moreover, the growth and decay of the birefringence were examined as functions of the pulse intensities for azo-dye bearing polyimide.148 It has been shown that the time constant associated with a fast process of birefringence growth decreased with the power, contrary to time constant associated with the slow process which appeared to be not dependent on the writing power.148
 | ||
| Fig. 19 Optical microscopy images of the copolymer containing bis-azobenzene thin film after recording (a)–(c), after erasing (d), and after rewriting (e)–(f) in the same region. Reproduced from ref. 146 with permission from Elsevier. | ||
Two-photon excitation of azobenzene derivatives can be used to induce nanomovement in azopolymers and can potentially lead to high-resolution topographic pattern formation.11,14 It is known that trans-to-cis-to-trans isomerization cycles induced by one-photon absorption of light can lead to mass migration on the free surface of the azopolymer films, which further leads to the creation of surface relief gratings (SRG).138,149–156 This phenomenon occurs on azopolymers when they are illuminated by a light pattern associated with light intensity gradients perpendicular to the light polarization.132 The first demonstration of the possibility of a light-driven mass migration by means of a two-photon absorption process was provided by Kawata et al.14 They used poly(Disperse Red 1 methacrylate) (DR1-PMA) as a light-responsive material and studied the effect of the incident light polarization and the position of the laser focus on surface deformation of the polymer induced by mass movement resulting from the two-photon isomerization.14 It has been shown that the polymer moves in the direction of the irradiation light polarization and that the induced surface pattern strongly depends on the Z-position of the laser focus.14 Based on the mass transport phenomenon in azopolymers associated with photoisomerization under the TPA excitation, Ambrosio et al. also fabricated gratings on a film of azopolymer (Fig. 20).24 By means of nonlinear excitation of the azo motif (at 800 nm) they created structures with sizes down to 250 nm, which is below the half-wavelength diffraction limit of the laser beam. They were also able to create diffraction gratings with periods of 500 nm, 1 μm, and 2 μm and each pattern covered an area of 1.2 × 0.5 mm2.132 Two years later the same group used an azopolymer to show the formation of gratings (mass migration activated mainly by TPA of azo moiety) with subwavelength periodicity and featuring height up to 100 nm, depending on the incident pulse energy. The structures could be formed when the incident pulse energy threshold was around 50 μJ per pulse. Moreover, they demonstrated that the features could be partially erased by exposure to incoherent UV light.24
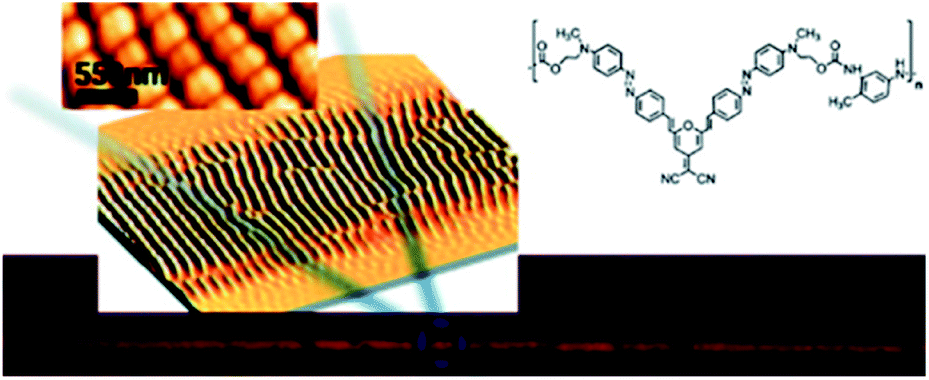 | ||
| Fig. 20 Optically induced spontaneous formation of periodic two-dimensional patterns on newly synthesized polymers based on Y-shape azobenzene molecules. Reproduced from ref. 24 with permission from American Chemical Society. | ||
Huang et al. noticed that surface deformation (namely, any surface features formed when the light intensity exceeds an effective critical value) should be taken into account during the multi-dimensional bits recording process.157 They recorded bits by gradually increasing the exposure dose and observed that the bits could be recorded due to birefringence, however, when the recording laser reached the effective critical intensity, large-scale mass migration could be produced and the surface deformation would modulate the recorded bits. Although those are characterized by higher stability, nevertheless one has to sacrifice the erasing performance (unless heating the material above Tg).157 Those features are advantageous since, by manipulating the recording energies, different kind of data storage can be produced. Recording mechanisms based on two-photon induced birefringence can be applied to multi-dimensional reversible storage, when the information needs to be manipulated and updated frequently and the mechanism involving surface deformation is more suitable for long-term data memory in the optical disc.158
7. Conclusions
As discussed in the present review, the applications of photochromic molecules, especially in biological context, are often limited by the need to employ relatively short wavelength irradiation (UV/Vis) in order to perform the switching between the forms of such molecules. While progress has been made in designing and synthesizing photochromic molecules based on azobenzene, for which the switching can be performed entirely in the visible range, unfortunately, azobenzenes characterized by red-shifted electronic transitions possess very low thermal stability of the cis form. An alternative is to perform the photoisomerization under nonlinear NIR excitation. Two-photon excitation has, indeed, been employed for isomerization of ABs and it holds much promise for the use in material science and biology, with huge benefits in localized drug release, controlling of ion channel flow as well as 3D data storage, optical filters etc. In most cases, these applications have been demonstrated by using ABs with low and modest TPA cross-sections, however the efficiency of two-photon induced isomerization can be boosted when TPA cross-sections are increased by well-known design principles of enhancing the π-electron conjugation and donor–acceptor character of the molecules.While using one-photon absorption for the photochromic process is obviously simple and convenient for processing large areas of the relevant materials, nonlinear absorption has many advantages, among them the possibility of limiting the excitation with very high precision to small volumes and minimizing the light-induced damage thanks to the use of low energy NIR photons.
In this review we have summarized the current state of art concerning nonlinear optical properties of azobenzene derivatives (for both molecules and polymers), quantified as two-photon absorption cross section, which enable the two-photon induced isomerization and we showed the possibilities where nonlinear switching can be used. We highlighted the efforts towards designing of AB derivatives able to undergo two-photon induced isomerization and we also outlined the alternate way of exploiting nonlinear optics for switching: through the separation of the roles of a two-photon absorber (the antenna or the donor) and the photoswitch (the acceptor). We believe that this review will contribute to indicate opportunities for designing better NLO photoswitches and expand application of AB derivatives in the wider range. As can be seen, many labs around the world are working intensely to address these issues. We anticipate that next years will bring more rapid development in this area as it becomes widely appreciated that two photons can be better than one.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of WUST are acknowledged as well as NCN Preludium grant UMO-2018/29/N/ST5/00944, and the support from the BioTechNan (NCBiR) program of interdisciplinary PhD studies at Wrocław University of Science and Technology. M. S. acknowledges the NCN Harmonia grant UMO-2016/22/M/ST4/00275.References
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 CrossRef CAS.
- R. J. Mart and R. K. Allemann, Chem. Commun., 2016, 52, 12262–12277 RSC.
- Z. L. Pianowski, Chem.–Eur. J., 2019, 25, 5128–5144 CrossRef CAS.
- Y. Kamiya and H. Asanuma, Acc. Chem. Res., 2014, 47, 1663–1672 CrossRef CAS.
- A. S. Lubbe, T. van Leeuwen, S. J. Wezenberg and B. L. Feringa, Tetrahedron, 2017, 73, 4837–4848 CrossRef CAS.
- A. Beharry and A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef.
- M. Dudek, Z. Pokladek, M. Deiana and K. Matczyszyn, Dyes Pigm., 2020, 108501 CrossRef CAS.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 CrossRef CAS.
- P. Weis and S. Wu, Macromol. Rapid Commun., 2018, 39, 1700220 CrossRef.
- D. Pijper and B. L. Feringa, Soft Matter, 2008, 4, 1349–1372 RSC.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS.
- H. Ishitobi, S. Shoji, T. Hiramatsu, H.-B. Sun, Z. Sekkat and S. Kawata, Opt. Express, 2008, 16, 14106–14114 CrossRef CAS.
- Z. Sekkat, H. Ishitobi, M. Tanabe, T. Hiramatsu and S. Kawata, Single-, two-, and multi-photon driven molecular motion and nanopatterning in azo-polymer films, SPIE, 2011 Search PubMed.
- A. Mourot, M. A. Kienzler, M. R. Banghart, T. Fehrentz, F. M. E. Huber, M. Stein, R. H. Kramer and D. Trauner, ACS Chem. Neurosci., 2011, 2, 536–543 CrossRef CAS.
- D. Bléger, Z. Yu and S. Hecht, Chem. Commun., 2011, 47, 12260–12266 RSC.
- M. Dudek, M. Deiana, Z. Pokladek, K. Pawlik and K. Matczyszyn, Chem.–Eur. J., 2018, 24, 18963–18970 CrossRef CAS.
- M. S. Maier, K. Hüll, M. Reynders, B. S. Matsuura, P. Leippe, T. Ko, L. Schäffer and D. Trauner, J. Am. Chem. Soc., 2019, 141, 17295–17304 CrossRef CAS.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef.
- G. Cabré, A. Garrido-Charles, M. Moreno, M. Bosch, M. Porta-de-la-Riva, M. Krieg, M. Gascón-Moya, N. Camarero, R. Gelabert, J. M. Lluch, F. Busqué, J. Hernando, P. Gorostiza and R. Alibés, Nat. Commun., 2019, 10, 907 CrossRef.
- M. Gascón-Moya, A. Pejoan, M. Izquierdo-Serra, S. Pittolo, G. Cabré, J. Hernando, R. Alibés, P. Gorostiza and F. Busqué, J. Org. Chem., 2015, 80, 9915–9925 CrossRef.
- S. Pittolo, H. Lee, A. Lladó, S. Tosi, M. Bosch, L. Bardia, X. Gómez-Santacana, A. Llebaria, E. Soriano, J. Colombelli, K. E. Poskanzer, G. Perea and P. Gorostiza, Proc. Natl. Acad. Sci., 2019, 116, 13680–13689 CrossRef CAS.
- A. Ambrosio, P. Maddalena, A. Carella, F. Borbone, A. Roviello, M. Polo, A. A. R. Neves, A. Camposeo and D. Pisignano, J. Phys. Chem. C, 2011, 115, 13566–13570 CrossRef CAS.
- R. M. Williams, D. W. Piston and W. W. Webb, FASEB J., 1994, 8, 804–813 CrossRef CAS.
- T. J. Thomas, H. A. Tajmir-Riahi and T. A. A. Thomas, Amino Acids, 2016, 48, 2423–2431 CrossRef CAS.
- A. Ustione and D. W. Piston, J. Microsc., 2011, 243, 221–226 CrossRef CAS.
- R. K. P. Benninger and D. W. Piston, Curr. Protoc. Cell Biol., 2013, 59, 4.11.1–4.11.24 CrossRef.
- G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS.
- J. Griffiths, Chem. Soc. Rev., 1972, 1, 481–493 RSC.
- H. M. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- M. Dudek, M. Deiana, Z. Pokladek, P. Mlynarz, M. Samoc and K. Matczyszyn, Nanoscale, 2018, 10, 11302–11306 RSC.
- X. Wang, J. Huang, Y. Zhou, S. Yan, X. Weng, X. Wu, M. Deng and X. Zhou, Angew. Chem., Int. Ed., 2010, 49, 5305–5309 CrossRef CAS.
- M. Zhou, X. Liang, T. Mochizuki and H. Asanuma, Angew. Chem., Int. Ed., 2010, 49, 2167–2170 CrossRef CAS.
- H. Nishioka, X. Liang, H. Kashida and H. Asanuma, Chem. Commun., 2007, 4354–4356 RSC.
- R. Yin, W. Xu, M. Kondo, C.-C. Yen, J.-i. Mamiya, T. Ikeda and Y. Yu, J. Mater. Chem., 2009, 19, 3141–3143 RSC.
- Z. Mahimwalla, K. G. Yager, J. Mamiya, A. Shishido, A. Priimagi and C. J. Barrett, Polym. Bull., 2012, 69, 967–1006 CrossRef CAS.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS.
- D. Bleger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef CAS.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS.
- A. Rullo, A. Reiner, A. Reiter, D. Trauner, E. Y. Isacoff and G. A. Woolley, Chem. Commun., 2014, 50, 14613–14615 RSC.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem.–Eur. J., 2014, 20, 16492–16501 CrossRef CAS.
- L. Agnetta, M. Bermudez, F. Riefolo, C. Matera, E. Claro, R. Messerer, T. Littmann, G. Wolber, U. Holzgrabe and M. Decker, J. Med. Chem., 2019, 62, 3009–3020 CrossRef CAS.
- M. Göppert-Mayer, Ann. Phys., 1931, 401, 273–294 CrossRef.
- T. H. Maiman, Nature, 1960, 187, 493–494 CrossRef.
- W. Kaiser and C. G. B. Garrett, Phys. Rev. Lett., 1961, 7, 229–231 CrossRef CAS.
- M. S. Malcuit, R. W. Boyd, L. W. Hillman, J. Krasinski and C. R. Stroud, J. Opt. Soc. Am. B, 1984, 1, 73–75 CrossRef CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS.
- M. Grzybowski, V. Hugues, M. Blanchard-Desce and D. T. Gryko, Chem.–Eur. J., 2014, 20, 12493–12501 CrossRef CAS.
- H.-Y. Ahn, S. Yao, X. Wang and K. D. Belfield, ACS Appl. Mater. Interfaces, 2012, 4, 2847–2854 CrossRef CAS.
- E. Maçôas, G. Marcelo, S. Pinto, T. Cañeque, A. M. Cuadro, J. J. Vaquero and J. M. G. Martinho, Chem. Commun., 2011, 47, 7374–7376 RSC.
- K.-S. Lee, R. H. Kim, D.-Y. Yang and S. H. Park, Prog. Polym. Sci., 2008, 33, 631–681 CrossRef CAS.
- J. Gu, W. Yulan, W.-Q. Chen, X.-Z. Dong, X.-M. Duan and S. Kawata, New J. Chem., 2007, 31, 63–68 RSC.
- A. S. Dvornikov, E. P. Walker and P. M. Rentzepis, J. Phys. Chem. A, 2009, 113, 13633–13644 CrossRef CAS.
- C. C. Corredor, Z.-L. Huang and K. D. Belfield, Adv. Mater., 2006, 18, 2910–2914 CrossRef CAS.
- K. Ogawa and Y. Kobuke, Org. Biomol. Chem., 2009, 7, 2241–2246 RSC.
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS.
- B. Gao, L. M. Mazur, M. Morshedi, A. Barlow, H. Wang, C. Quintana, C. Zhang, M. Samoc, M. P. Cifuentes and M. G. Humphrey, Chem. Commun., 2016, 52, 8301–8304 RSC.
- Z. Pokladek, M. Dudek, O. Mongin, R. Métivier, P. Mlynarz, M. Samoc, K. Matczyszyn and F. Paul, ChemPlusChem, 2017, 82, 1372–1383 CrossRef CAS.
- Z. Pokladek, N. Ripoche, M. Betou, Y. Trolez, O. Mongin, J. Olesiak-Banska, K. Matczyszyn, M. Samoc, M. G. Humphrey, M. Blanchard-Desce and F. Paul, Chem.–Eur. J., 2016, 22, 10155–10167 CrossRef CAS.
- Y. Gautier, G. Argouarch, F. Malvolti, B. Blondeau, N. Richy, A. Amar, A. Boucekkine, K. Nawara, K. Chlebowicz, G. Orzanowska, M. Dudek, K. Matczyszyn, M. Samoc, M. Blanchard-Desce, O. Mongin, J. Waluk and F. Paul, ChemPlusChem, 2020, 85, 411–425 CrossRef CAS.
- W. Denk, J. Strickler and W. Webb, Science, 1990, 248, 73–76 CrossRef CAS.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS.
- M. Sheik-Bahae, A. A. Said, T. Wei, D. J. Hagan and E. W. V. Stryland, IEEE J. Quantum Electron., 1990, 26, 760–769 CrossRef CAS.
- R. Medishetty, J. K. Zaręba, D. Mayer, M. Samoć and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 RSC.
- L. Antonov, K. Kamada, K. Ohta and F. S. Kamounah, Phys. Chem. Chem. Phys., 2003, 5, 1193–1197 RSC.
- L. De Boni, L. Misoguti, S. C. Zílio and C. R. Mendonça, ChemPhysChem, 2005, 6, 1121–1125 CrossRef CAS.
- C. R. Mendonça, D. S. dos Santos, L. De Boni, O. N. Oliveira and S. C. Zilio, Synth. Met., 2001, 121, 1489–1490 CrossRef.
- L. De Boni, J. J. Rodrigues, D. S. dos Santos, C. H. T. P. Silva, D. T. Balogh, O. N. Oliveira, S. C. Zilio, L. Misoguti and C. R. Mendonça, Chem. Phys. Lett., 2002, 374, 684 CrossRef.
- F. Cherioux, P. Audebert, H. Maillotte, L. Grossard, F. E. Hernandez and A. Lacourt, Chem. Mater., 1997, 9, 2921–2927 CrossRef CAS.
- B. Derkowska-Zielinska, K. Matczyszyn, M. Dudek, M. Samoc, R. Czaplicki, A. Kaczmarek-Kedziera, V. Smokal, A. Biitseva and O. Krupka, J. Phys. Chem. C, 2019, 123, 725–734 CrossRef CAS.
- P. Krawczyk, A. Kaczmarek, R. Zaleśny, K. Matczyszyn, W. Bartkowiak, M. Ziółkowski and P. Cysewski, J. Mol. Model., 2009, 15, 581–590 CrossRef CAS.
- K. Ohta, L. Antonov, S. Yamada and K. Kamada, J. Chem. Phys., 2007, 127, 084504 CrossRef.
- P. Krawczyk, J. Mol. Model., 2010, 16, 659–668 CrossRef CAS.
- A. A. Andrade, S. B. Yamaki, L. Misoguti, S. C. Zilio, T. D. Z. Atvars, O. N. Oliveira and C. R. Mendonça, Opt. Mater., 2004, 27, 441–444 CrossRef CAS.
- E. Piovesan, L. De Boni, E. Ishow and C. R. Mendonça, Chem. Phys. Lett., 2010, 498, 277–280 CrossRef CAS.
- R. D. Breukers, S. Janssens, S. G. Raymond, M. D. H. Bhuiyan and A. J. Kay, Dyes Pigm., 2015, 112, 17–23 CrossRef CAS.
- W. Xu, W. Wang, J. Li, Q. Wu, Y. Zhao, H. Hou and Y. Song, Dyes Pigm., 2019, 160, 1–8 CrossRef CAS.
- C.-C. Chu, Y.-C. Chang, B.-K. Tsai, T.-C. Lin, J.-H. Lin and V. K. S. Hsiao, Chem.–Asian J., 2014, 9, 3390–3396 CrossRef CAS.
- Y. Z. Gu, F. X. Gan, S. Q. Wang and H. J. Xu, Opt. Commun., 2001, 197, 501–505 CrossRef CAS.
- S. Wu, W. Qian, Z. Xia, Y. Zou, S. Wang, S. Shen and H. Xu, Chem. Phys. Lett., 2000, 330, 535–540 CrossRef CAS.
- S. Wang, S. Shen and H. Xu, Dyes Pigm., 2000, 44, 195–198 CrossRef CAS.
- Y. Zhang, Q. Gan, S. Wang and G. Yang, J. Inorg. Organomet. Polym. Mater., 2012, 22, 48–53 CrossRef.
- U. M. Neves, L. De Boni, Z. Ye, X. R. Bu and C. R. Mendonça, Chem. Phys. Lett., 2007, 441, 221–225 CrossRef CAS.
- Z. Ye, L. D. Boni, U. M. Neves, C. R. Mendonça and X. R. Bu, Tetrahedron Lett., 2009, 50, 1371–1373 CrossRef CAS.
- U. M. Neves, L. De Boni, C. R. Mendonça, Z. Ye and X. R. Bu, Advances in Optical Sciences Congress, Honolulu, Hawaii, 2009 Search PubMed.
- J. Moreno, M. Gerecke, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, D. Bléger, S. Hecht and S. A. Kovalenko, J. Phys. Chem. B, 2015, 119, 12281–12288 CrossRef CAS.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CrossRef CAS.
- V. Grande, C.-A. Shen, M. Deiana, M. Dudek, J. Olesiak-Banska, K. Matczyszyn and F. Würthner, Chem. Sci., 2018, 9, 8375–8381 RSC.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS.
- S. W. Magennis, F. S. Mackay, A. C. Jones, K. M. Tait and P. J. Sadler, Chem. Mater., 2005, 17, 2059–2062 CrossRef CAS.
- A. S. Dvornikov, H. Bouas-Laurent, J.-P. Desvergne and P. M. Rentzepis, J. Mater. Chem., 1999, 9, 1081–1084 RSC.
- W. G. Fisher, W. P. Partridge Jr, C. Dees and E. A. Wachter, Photochem. Photobiol., 1997, 66, 141–155 CrossRef CAS.
- S. Kawata, H.-B. Sun, T. Tanaka and K. Takada, Nature, 2001, 412, 697–698 CrossRef CAS.
- V. M. Churikov, J. T'Suen Lin, H. Hsien Wu, J. Hung Lin, T.-H. Huang and C.-C. Hsu, Opt. Commun., 2002, 209, 451–460 CrossRef CAS.
- J. Si, J. Qiu, J. Guo, G. Qian, M. Wang and K. Hirao, Appl. Opt., 2003, 42, 7170–7173 CrossRef CAS.
- Z. Wei, L. He, Z. Chi, X. Ran and L. Guo, Spectrochim. Acta, Part A, 2019, 206, 120–125 CrossRef CAS.
- X. Ran, H. Wang, L. Shi, J. Lou, B. Liu, M. Li and L. Guo, J. Mater. Chem. C, 2014, 2, 9866–9873 RSC.
- E. C. Carroll, S. Berlin, J. Levitz, M. A. Kienzler, Z. Yuan, D. Madsen, D. S. Larsen and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E776–E785 CrossRef CAS.
- S. Passlick, M. T. Richers and G. C. R. Ellis-Davies, Angew. Chem., Int. Ed., 2018, 57, 12554–12557 CrossRef CAS.
- C.-K. Lim, X. Li, Y. Li, K. L. M. Drew, J. P. Palafox-Hernandez, Z. Tang, A. Baev, A. N. Kuzmin, M. R. Knecht, T. R. Walsh, M. T. Swihart, H. Ågren and P. N. Prasad, Nanoscale, 2016, 8, 4194–4202 RSC.
- J. Croissant, M. Maynadier, A. Gallud, H. Peindy N'Dongo, J. L. Nyalosaso, G. Derrien, C. Charnay, J.-O. Durand, L. Raehm, F. Serein-Spirau, N. Cheminet, T. Jarrosson, O. Mongin, M. Blanchard-Desce, M. Gary-Bobo, M. Garcia, J. Lu, F. Tamanoi, D. Tarn, T. M. Guardado-Alvarez and J. I. Zink, Angew. Chem., Int. Ed., 2013, 52, 13813–13817 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, 3 edn, 2006 Search PubMed.
- M. Izquierdo-Serra, M. Gascon-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, E. Ferrer, R. Alibes, F. Busque, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136, 8693–8701 CrossRef CAS.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef CAS.
- C.-Z. Wang, R. Zhang, K. Sakaguchi, X. Feng, X. Yu, M. R. J. Elsegood, S. J. Teat, C. Redshaw and T. Yamato, ChemPhotoChem, 2018, 2, 749–756 CrossRef CAS.
- F. Riefolo, C. Matera, A. Garrido-Charles, A. M. J. Gomila, R. Sortino, L. Agnetta, E. Claro, R. Masgrau, U. Holzgrabe, M. Batlle, M. Decker, E. Guasch and P. Gorostiza, J. Am. Chem. Soc., 2019, 141, 7628–7636 CrossRef CAS.
- S. Samanta, H. I. Qureshi and G. A. Woolley, Beilstein J. Org. Chem., 2012, 8, 2184–2190 CrossRef CAS.
- J. Y. Ye, M. Ishikawa, Y. Yamane, N. Tsurumachi and H. Nakatsuka, Appl. Phys. Lett., 1999, 75, 3605–3607 CrossRef CAS.
- J. Y. Ye and M. Ishikawa, Opt. Lett., 2008, 33, 1729–1731 CrossRef.
- S. Lecler, S. Haacke, N. Lecong, O. Crégut, J.-L. Rehspringer and C. Hirlimann, Opt. Express, 2007, 15, 4935–4942 CrossRef CAS.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- I. Cohanoschi and F. E. Hernández, J. Phys. Chem. B, 2005, 109, 14506–14512 CrossRef CAS.
- Y. Tsuboi, R. Shimizu, T. Shoji and N. Kitamura, J. Am. Chem. Soc., 2009, 131, 12623–12627 CrossRef CAS.
- K. Ueno, S. Juodkazis, T. Shibuya, Y. Yokota, V. Mizeikis, K. Sasaki and H. Misawa, J. Am. Chem. Soc., 2008, 130, 6928–6929 CrossRef CAS.
- W. Wenseleers, F. Stellacci, T. Meyer-Friedrichsen, T. Mangel, C. A. Bauer, S. J. K. Pond, S. R. Marder and J. W. Perry, J. Phys. Chem. B, 2002, 106, 6853–6863 CrossRef CAS.
- S. T. Sivapalan, J. H. Vella, T. K. Yang, M. J. Dalton, R. N. Swiger, J. E. Haley, T. M. Cooper, A. M. Urbas, L.-S. Tan and C. J. Murphy, Langmuir, 2012, 28, 9147–9154 CrossRef CAS.
- S. T. Sivapalan, J. H. Vella, T. K. Yang, M. J. Dalton, J. E. Haley, T. M. Cooper, A. M. Urbas, L.-S. Tan and C. J. Murphy, J. Phys. Chem. Lett., 2013, 4, 749–752 CrossRef CAS.
- X. Liu, X. Jia, M. Fischer, Z. Huang and D. R. Smith, Nano Lett., 2018, 18, 6181–6187 CrossRef CAS.
- S. Kellner and S. Berlin, Appl. Sci., 2020, 10, 805 CrossRef CAS.
- S. Berlin and E. Y. Isacoff, EMBO Rep., 2017, 18, 677–692 CrossRef CAS.
- J. Bieth, S. M. Vratsanos, N. Wassermann and B. F. Erlanger, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 1103–1106 CrossRef CAS.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef.
- M. A. Kienzler, A. Reiner, E. Trautman, S. Yoo, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2013, 135, 17683–17686 CrossRef CAS.
- J. Croissant, A. Chaix, O. Mongin, M. Wang, S. Clément, L. Raehm, J.-O. Durand, V. Hugues, M. Blanchard-Desce, M. Maynadier, A. Gallud, M. Gary-Bobo, M. Garcia, J. Lu, F. Tamanoi, D. P. Ferris, D. Tarn and J. I. Zink, Small, 2014, 10, 1752–1755 CrossRef CAS.
- Y. Huang, L. Shen, D. Guo, W. Yasen, Y. Wu, Y. Su, D. Chen, F. Qiu, D. Yan and X. Zhu, Chem. Commun., 2019, 55, 6735–6738 RSC.
- T. V. Truong, C.-Y. Chen, N. V. Tabiryan and Y. R. Shen, J. Opt. Soc. Am. B, 2007, 24, 2623–2626 CrossRef CAS.
- A. Ambrosio, E. Orabona, P. Maddalena, A. Camposeo, M. Polo, A. A. R. Neves, D. Pisignano, A. Carella, F. Borbone and A. Roviello, Appl. Phys. Lett., 2009, 94, 011115 CrossRef.
- D. S. Correa, M. R. Cardoso, V. C. Gonçalves, D. T. Balogh, L. De Boni and C. R. Mendonça, Polymer, 2008, 49, 1562–1566 CrossRef CAS.
- K. Matczyszyn and J. Sworakowski, J. Phys. Chem. B, 2003, 107, 6039–6045 CrossRef CAS.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2016, 116, 15089–15166 CrossRef CAS.
- J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817–1846 CrossRef CAS.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS.
- H. Nakano, T. Takahashi, T. Kadota and Y. Shirota, Adv. Mater., 2002, 14, 1157–1160 CrossRef CAS.
- A. Natansohn, P. Rochon, J. Gosselin and S. Xie, Macromolecules, 1992, 25, 2268–2273 CrossRef CAS.
- M. Maeda, H. Ishitobi, Z. Sekkat and S. Kawata, Appl. Phys. Lett., 2004, 85, 351–353 CrossRef CAS.
- C. R. Mendonça, U. M. Neves, L. De Boni, A. A. Andrade, D. S. dos Santos, F. J. Pavinatto, S. C. Zilio, L. Misoguti and O. N. Oliveira, Opt. Commun., 2007, 273, 435–440 CrossRef.
- M. R. Cardoso, U. M. Neves, L. Misoguti, Z. Ye, X. R. Bu and C. R. Mendonça, Opt. Mater., 2006, 28, 1118–1122 CrossRef CAS.
- H. Ishitobi, Z. Sekkat and S. Kawata, J. Chem. Phys., 2006, 125, 164718 CrossRef.
- D. Brown, A. Natansohn and P. Rochon, Macromolecules, 1995, 28, 6116–6123 CrossRef CAS.
- Z. Zhang, Y. Hu, Y. Luo, Q. Zhang, W. Huang and G. Zou, Opt. Commun., 2009, 282, 3282–3285 CrossRef CAS.
- D. Hu, Z. Zhang, Y. Hu, Y. Luo, Q. Zhang and W. Huang, Opt. Commun., 2011, 284, 802–806 CrossRef CAS.
- Y. Hu, Z. Zhang, Y. Chen, W. Huang and Q. Zhang, J. Laser Micro/Nanoeng., 2010, 5, 64–67 CrossRef CAS.
- A. Kozanecka-Szmigiel, K. Switkowski, E. Schab-Balcerzak and E. Grabiec, Appl. Phys. B: Lasers Opt., 2011, 105, 851–855 CrossRef CAS.
- C. Fiorini, N. Prudhomme, G. de Veyrac, I. Maurin, P. Raimond and J. M. Nunzi, Synth. Met., 2000, 115, 121–125 CrossRef CAS.
- P. Lefin, C. Fiorini and J.-M. Nunzi, Pure Appl. Opt., 1998, 7, 71–82 CrossRef CAS.
- R. Kirby, R. G. Sabat, J.-M. Nunzi and O. Lebel, J. Mater. Chem. C, 2014, 2, 841–847 RSC.
- P. Lefin, C. Fiorini and J.-M. Nunzi, Opt. Mater., 1998, 9, 323–328 CrossRef CAS.
- H. Nakano, T. Tanino and Y. Shirota, Appl. Phys. Lett., 2005, 87, 061910 CrossRef.
- J. Kumar, L. Li, X. L. Jiang, D.-Y. Kim, T. S. Lee and S. Tripathy, Appl. Phys. Lett., 1998, 72, 2096–2098 CrossRef CAS.
- T. Fukuda, H. Matsuda, T. Shiraga, T. Kimura, M. Kato, N. K. Viswanathan, J. Kumar and S. K. Tripathy, Macromolecules, 2000, 33, 4220–4225 CrossRef CAS.
- R. Barillé, P. Tajalli, S. Zielinska, E. Ortyl, S. Kucharski and J. M. Nunzi, Appl. Phys. Lett., 2009, 95, 053102 CrossRef.
- Y. Hu, Y. Chen, J. Li, D. Hu, J. Chu, Q. Zhang and W. Huang, Appl. Phys. Lett., 2012, 101, 251116 CrossRef.
- Y. Hu, D. Wu, J. Li, W. Huang and J. Chu, Opt. Express, 2016, 24, 23557–23565 CrossRef.
- B. Wu, K. Ueno, Y. Yokota, K. Sun, H. Zheng and H. Misawa, J. Phys. Chem. Lett., 2012, 3(11), 1443–1447 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07693g |
| This journal is © The Royal Society of Chemistry 2020 |