
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inherently degradable cross-linked polyesters and polycarbonates: resins to be cheerful
Theona
Șucu
 a and
Michael P.
Shaver
*ab
a and
Michael P.
Shaver
*ab
aSchool of Natural Sciences, Department of Materials, The University of Manchester, Manchester, M13 9PL, UK. E-mail: michael.shaver@manchester.ac.uk
bHenry Royce Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 1st October 2020
Abstract
Thermosets are an important class of materials that provide excellent temperature and solvent resistance; however, their high dimensional stability precludes degradation or reprocessing. While traditional thermoplastics can be mechanically and chemically recycled, these pathways are often elusive for resins due to their intrinsic structure. The renewed demand for sustainable polymers from public, industry and government stakeholders has increased research into (bio)degradable thermoplastics, but thermosets have often been overlooked. Aliphatic polyesters and polycarbonates are the cornerstone of biodegradable polymers, yet offer an arguably greater potential in thermosets as end-of-life options are more limited for these materials. This review summarises the most recent advances in the synthesis and characterisation of degradable thermosetting polyester and polycarbonate materials, including partially degradable systems derived from renewable resources such as itaconic acid or isosorbide. The review is organised by synthetic methodology including one-pot reactions and multi-step approaches making use of pre-polymers. Photo-cross-linking and high-energy irradiation are also discussed as emerging synthetic strategies.
 Theona Şucu | Theona was born and raised in Ploiești, Romania before moving to Scotland where she was awarded a first class Masters in Chemistry from the University of Edinburgh in 2019. Her degree included a year-long industrial placement at Solvay, USA and a research placement in the group of Dr Jennifer Garden. These experiences in industry and academia sparked her interest in polymer science and she started her PhD at the University of Manchester soon after graduation. As part of the Sustainable Materials Innovation Hub, Theona's research focuses on designing monomers for the cross-linking of biodegradable polymers through ring-opening copolymerisation techniques. |
 Michael P. Shaver | Mike is Professor of Polymer Science in the School of Natural Sciences at the University of Manchester where he leads initiatives in sustainable polymers, plastics and materials for the School and for the Henry Royce Institute, the UK's national advanced materials science centre since 2019. Following a PhD in his native Canada from the University of British Columbia and an NSERC Post-Doctoral Fellowship at Imperial College London, he began his independent research career at the University of Prince Edward Island before moving to the University of Edinburgh in 2012 where he was a Chancellor's Fellow, Reader and finally Professor of Polymer Chemistry. He is Director of the Sustainable Materials Innovation Hub at the University of Manchester, working on both fundamental projects in monomer design and functional polymers and industrial projects in plastic packaging and sustainable, functional materials. He was awarded the MacroGroup Young Polymer Scientist award (2015), two Canada Foundation for Innovation Leadership Awards (2010, 2012) as well as Fellowships in the Royal Society of Chemistry (2017) and Institute of Materials Minerals and Mining (2019). |
1. Introduction
The global demand for thermoplastics and thermosets continue to grow; production reached 359 million tonnes 2018.1 The low cost and versatility of these materials, arising from their light weight, intrinsic durability, ease of synthesis and resistance to degradation, drives market growth. These factors have created an inherent societal dependence on “plastics”, a term public and government stakeholders have used to classify both types of polymeric materials. The manufacture and use of most modern plastics rely on petroleum feedstocks, a non-renewable resource with politically-charged market dynamics, while their resilience and the lack of recycling infrastructure results in the generation of unprecedented amounts of waste, much of which is poorly managed.For thermoplastics, significant research on biodegradable polymers, such as poly(lactic acid), have promised superior environmental performance through both source, as they are derived from biological feedstocks, and end-of-life, through degradation or depolymerisation.2,3 Just as with mechanical recycling, however, this process requires careful control. Exposure to heat, light, ultrasound, microorganisms or certain chemicals such as acids or bases can all trigger degradation, alter chemical composition and progressively decrease the molecular weight of polymer chains.4,5 The inherently complex process and slow degradation in the environment of release means that for “green” credentials to be realised this is best done in a managed process.6 However, if degradation is enabled in the environment of release, ecotoxicological impacts of both plastics and their degradation products are understood, and application underpinned by life cycle assessment, these can be exceptionally sustainable materials. Comparison to traditional mechanical and emerging chemical recycling pathways for thermoplastics remain a challenge.
Thermosets, however, cannot be mechanically recycled. The high dimensional structure of thermosets precludes reprocessing. Degradation – biological or otherwise – thus represents potentially sustainable end-of-life pathway for these resins. As such, this review explores the recent developments in the design, development and understanding of degradable cross-linked polyester and polycarbonate resins as an essential part of a sustainable plastics future.
2. Polyesters and polycarbonates
Some of the most prevalent thermoplastics are polyesters and polycarbonates. Poly(ethylene terephthalate), PET, and bisphenol-A bridged polycarbonates, PC (Fig. 1a) are two such oxygen-containing polymers, with applications spanning packaging to rigid plastic to engineering materials. The rigid aromatic functionalities in the polymer backbone can impart superior physical and chemical properties such as high glass transition temperature (Tg) and gas impermeability. Environmental degradation of these commodity plastics is not facile, necessitating managed mechanical or chemical recycling infrastructure for which there is inconsistent global access.7–9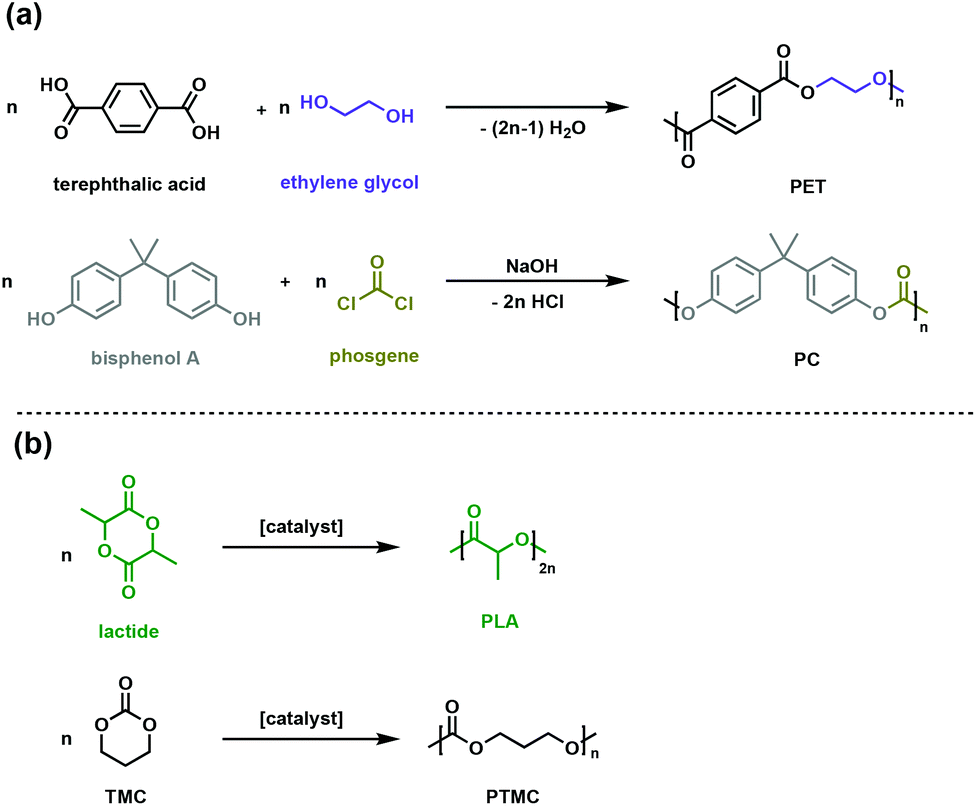 | ||
| Fig. 1 Synthesis routes for (a) conventional polyesters (PET) and polycarbonates (PC) (b) aliphatic biodegradable polyesters (PLA) and polycarbonates (PTMC). | ||
One of the goals of contemporary polymer research is to develop alternative polymers that are durable and fit-for-purpose, yet more easily degraded in the environment of release. In pursuit of such materials, the synthesis of biodegradable polyesters and polycarbonates, particularly when generated from renewable resources, has garnered significant attention. Monomeric precursors to polyesters and polycarbonates can be sourced from biomass or waste gas through a variety of biological or chemical transformations.10–12 A variety of polymerisation methods are now available to synthesise polymers with an array of backbone topologies and side-chain functionalities. Compared to petroleum-derived polyesters and polycarbonates, for which degradation kinetics are exceedingly slow, the ester and carbonate linkages in the novel polymer backbones can be enzymatically or chemically degraded, offering new options for the life cycle of these materials.10,13,14 Poly(lactic acid) (PLA) is the most commercially viable sustainable polyester and is produced through the ring opening polymerisation of renewable lactide (Fig. 1b).15 PLA has suitable mechanical and physical properties as a sustainable substitute for packaging and textiles, while its ability to be absorbed and degraded in vivo enables numerous biomedical applications.15–17
Polycarbonates typically have the high impact strength and excellent mechanical properties needed for challenging engineering applications like airplane windows and safety glass. The conventional synthesis of polycarbonates involves the use of toxic and energy-intensive phosgene (Fig. 1a).18,19 New synthetic strategies use dialkyl carbonates, which are easily synthesised through a dehydrative condensation of alcohols and CO2.19,20 While useful industrially, the energy cost and lack of control have given rise to the ROP of cyclic carbonates (such as trimethylene carbonate, TMC, Fig. 1b) as an attractive method to achieve well-defined polycarbonates with high molar masses.21
Due to their synthetic versatility and diverse properties, aliphatic polyesters and polycarbonates have been some of the most widely studied compounds of rather great potential as degradable thermoplastics. The decomposition of labile linkages could enable recovery of the starting materials or formation of different smaller molecules that could be readily metabolised or environmentally processed. Degradation of these compounds can be either hydrolytic or enzymatic. While both ester and carbonate groups are susceptible to hydrolysis, polyesters usually undergo bulk degradation whereas surface erosion predominates for polycarbonates.22 The short chains between ester or carbonate moieties enable faster degradation profiles for these polymers, although they are rarely completely assimilated into the environment. Enzymatic degradation is a viable alternative to hydrolysis, but it is usually less effective in highly crystalline systems. While promising, there remain concerns with synthetic degradable thermoplastics. As degradation profiles depend upon reaction conditions, biodegradable materials are often incorrectly disposed, rejected from composting facilities and landfilled or worse littered. This suggests that, for thermoplastics, a balance between environmental pollution, fossil-fuel depletion, plastic leaching and cost are needed to ensure sustainability.
The permanent molecular architecture of thermosets precludes reprocessing and mechanical recycling. The lack of traditional waste management, coupled with the higher value potential applications, suggest that degradable materials could more readily offer a sustainable pathway at end-of-life. There is thus an urgent need to extend these linear frameworks and synthetic strategies to degradable cross-linked polymers, enabling new applications (vide infra).
3. Cross-linked polymers
Conventional linear polymers become malleable and flow at their Tg and when the temperature is reduced, they retain their new form and are therefore known as thermoplastics (Fig. 2a). In contrast, cross-linking polymers set their form during synthesis, as the bonds that form between the individual chains cannot be re-moulded. These are referred to as thermosets (Fig. 2b); the term plastic now traditionally encompasses both thermoplastics and thermosets.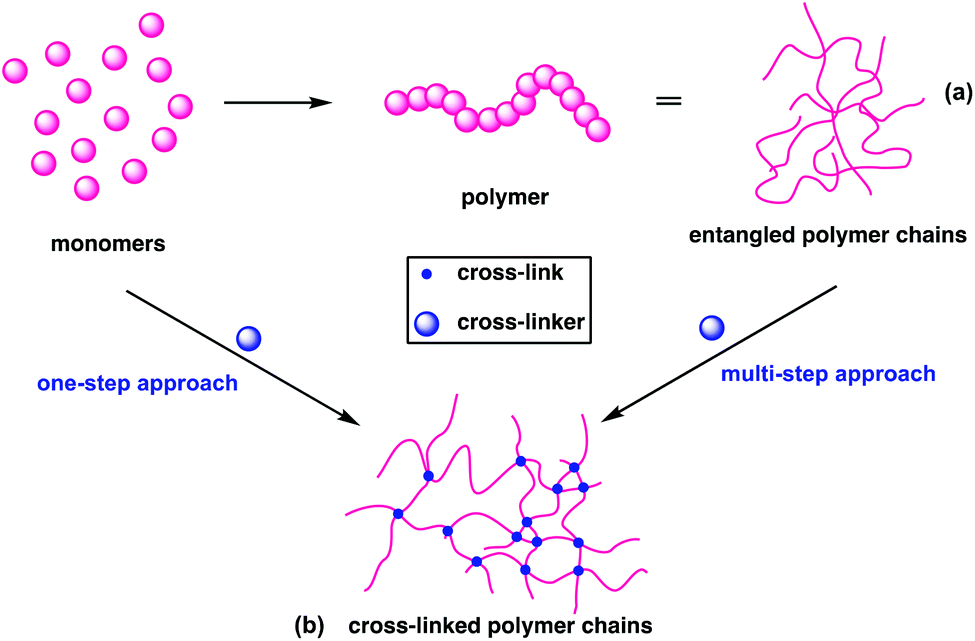 | ||
| Fig. 2 (a) Schematic representation of polymers (b) schematic of cross-linked polymer architecture. | ||
Thermosets can be created in one of two ways: in a one-pot reaction, through the copolymerisation of monomers with cross-linker or through the addition or presence of cross-links to a thermoplastic (Fig. 2), as is demonstrated in the curing of natural rubbers.23 The thermoset industry continued to grow, with cross-linked polymers now constituting 15–20% of all polymers produced.24 Their high dimensional stability, along with high thermal, mechanical and environmental resistance have found application as insulators, adhesives, coatings, foams and automotive parts.25 This stability is a challenge at end of life, as functions such as shape reconfiguration, self-healing and recycling are elusive for traditional cross-linked polymers.26
Why pursue research at the interface between these well-developed fields? Aliphatic polyesters and polycarbonates have excellent mechanical and thermal properties for linear polymers, and offer a unique advantage of hydrolytic or enzymatic degradation at end-of-life. Their poor solvent resistance, low thermal stability and significant stress softening mean that some applications are often impossible.27 Cross-linking these degradable systems could impart additional dimensional stability to the polymers, enhancing tensile and impact strength,28 producing materials with improved elasticity and shape retention with reduced creep.29 Research in this area lags behind other cross-linked systems like epoxy resins and polyurethanes, which are explicitly outside the scope of this review. This review will thus provide a detailed summary of the recent developments in the synthesis and characterisation of degradable cross-linked polyesters and polycarbonates from fundamental challenges to potential applications. Components are identified by monomer (M#), cross-linking agent (C#) and polymer (P#), recognising that a single monomer and cross-linker may produce several polymeric structures, sometimes in sequence.
3.1. One-step methodologies to degradable cross-linked polymers
Cross-linked polyesters have been extensively studied by Hillmyer and coworkers, who employed different types of bifunctional cross-linking agents in tandem copolymerisation and cross-linking processes.30 One-step methodologies eliminate processing steps and allow for the direct conversion of monomers to cross-linked polymers. A bis(cyclic carbonate) (C1, Fig. 3) was used to synthesise cross-linked elastomers prepared from poly(β-methyl-δ-valerolactone) (P1), using a triazabicyclodecene (TBD) catalyst. The monomer, β-methyl-δ-valerolactone, (M1) and cross-linker were predicted to have similar reactivity, which would improve the cross-linking efficiency. Because of C1's solubility in M1, the reaction was performed in bulk. As the reaction proceeded, the mixtures formed gels, whose thermal properties were investigated. Issues arose with monomer equilibrium concentration, so residual M1 had to be extracted before the samples were subjected to any tests. C1 incorporation did not affect the thermal properties of the new cross-linked materials, the Tg for all resins being between −47 and −49 °C, comparable with the value of −52 °C measured for linear P1. Remarkably, at low cross-linking densities, the materials exhibited higher tensile strength and elongation, along with substantial strain hardening compared to conventional elastomers such as vulcanised natural rubber. When increasing the cross-linking density, the materials showed similar extension properties to common elastomers, followed by a decrease in tensile strength. Moreover, higher plateau moduli along with better hysteresis recovery were observed at higher cross-linker loading. There was, however, no clear correlation between the cross-linking content and ultimate tensile strength. The synthesised elastomers were shown to be chemically recyclable, with 91% recovery of the pure M1 monomer. The materials were resilient to degradation under physiologically relevant conditions, whereas an acidic environment (HCl, 1 M) promoted network degradation at elevated temperatures (60 °C). Basic conditions (NaOH, 1 M), perhaps counterintuitively, did not prove to be optimal for degradation, suggesting that the carbonate moieties imparted by the cross-linker contributed additional chemical resistance in the cross-linked polymer. However, the recyclability and degradation under specific acidic conditions, along with the superior mechanical properties suggest promise for the sustainability and viability of these materials as rubber replacements.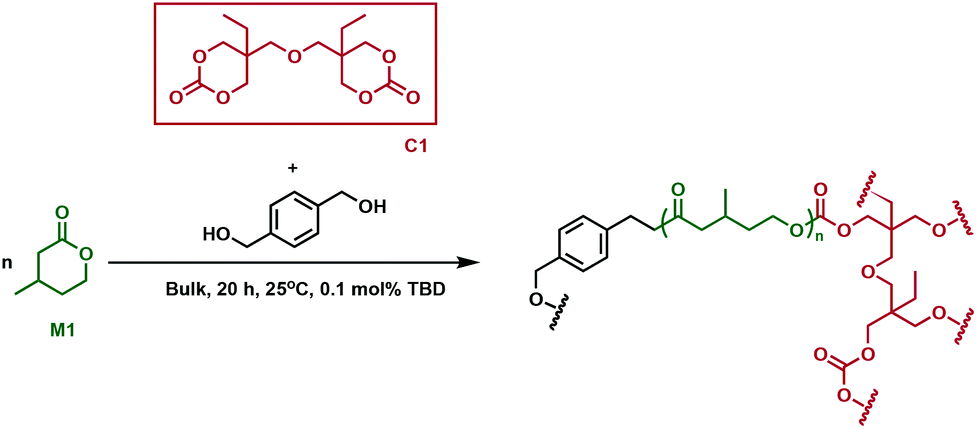 | ||
| Fig. 3 Synthesis of cross-linked P1 using cross-linker C1.30 | ||
In related work, Yang et al. investigated the in vivo enzymatic degradation of poly(ε-caprolactone) (P2) networks prepared by cross-linking with the aforementioned C1, as described in Fig. 3.31 The enzymatic degradation was performed using Thermomyces lanuginosus lipase. Cross-linking allowed for increased form-stability and decreased crystallinity compared to the homopolymer, while affording a decreased rate of degradation. Previous studies have shown that the degradation rate of cross-linked polyesters such as P2 and poly(D,L-lactide-co-ε-caprolactone) (P3) is reduced compared to the homopolymers, due to a decrease in the water molecule penetration into the networks.32 This can be exploited when targeting cross-linked P2 implants as controlled drug release agents, since the amount of acidic degradation products is reduced.31 The same group reported the synthesis of poly(trimethylene carbonate) (P4) networks through a one-step methodology involving the ring-opening copolymerization of C1 with trimethylene carbonate (M2) and/or ε-caprolactone (M3) using tin(II) 2-ethylhexanoate, Sn(Oct)2, as catalyst, as shown in Fig. 4. The networks were touted as biodegradable, but no study was reported in this paper to confirm this property.33
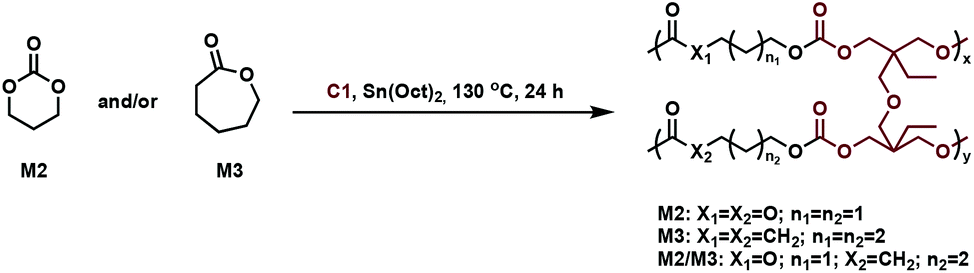 | ||
| Fig. 4 Synthesis of cross-linked P4 networks.33 | ||
An important class of cross-linkers are bis(ε-caprolactone)s, with two such comonomers studied in detail for the synthesis of degradable cross-linked polymers. Originally patented by Starcher and coworkers in 1963,34 Pitt, Palmgren and coworkers have further investigated bis(ε-caprolactone-4-yl) (C2) and a related molecule, 2,2-bis(ε-caprolactone-4-yl)propane (C3) as cross-linking agents for the synthesis of elastomers.35,36 These molecules allow for in situ cross-linking, thus eliminating prepolymer synthesis. Cross-linked aliphatic polyesters obtained through the Sn(Oct)2 catalysed ROP of 1,5-dioxepan-2-one (M4) in the presence of tetrafunctional C2 and C3 (Fig. 5) were investigated.36 Efficient cross-linking was highly dependent on the reactivity ratios of the comonomers, with C2 being more reactive than C3 towards M4. Monomer solubility was problematic, leading to high concentrations of cross-linkers (8–28 mol%) being required, thus restricting the ability to fully control the mechanical properties.36,37 The incorporation of C2 and C3 in the growing polymer chain resulted in the formation of multifunctional cross-links, allowing the synthesis of strong elastomeric polymers (Fig. 5). The Tg of the cross-linked poly(1,5-dioxepan-2-one) (P5) increased compared to its homopolymer. This is anticipated, since cross-linking reduces the available free volume and the chain requires more energy input to become mobile, shifting the Tg to higher values. C3 exhibits a more flexible structure compared to C2, hence imparting a longer physical cross-link and increasing the relative amount of free volume available. Therefore, the C3 cross-linked P5 demonstrated a lower Tg than the C2 cross-linked analogue. These properties demonstrated clear potential for applications where degradable elastomeric biomaterials are desirable, although environmental degradation remains unexplored.
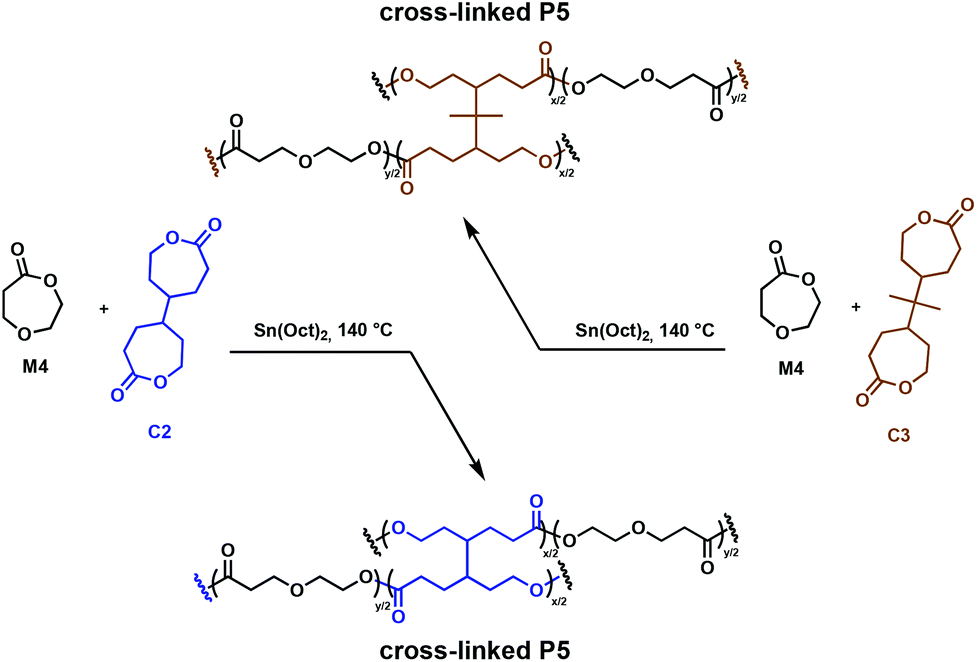 | ||
| Fig. 5 Synthesis of cross-linked P5 polymers using C2 cross-linkers.36 | ||
The “design to degrade” approach may tackle resin accumulation in the environment, but the functionality in the cross-links can also facilitate direct reprocessing or mechanical recycling. The incorporation of dynamic bonds in the polymer network can undergo associative exchange reactions such as vulcanisation, olefin metathesis, urethane exchange, siloxane exchange or boronic acid exchange.38–42 The most promising, and relevant, chemistry to promote recyclability is the transesterification of esters in the presence of free alcohols, typically catalysed by organocatalysts or Lewis acids.43 It has been postulated that Brønsted acids, known to catalyse the transesterification in small molecules, could also be utilised as catalysts in dynamic covalent bond exchange. These reprocessable networks are often called vitrimers, and can additionally promote reformability and self-healing. Bates and coworkers developed a one-pot synthesis of polyester vitrimers using C2, that enables in situ cross-linking (Fig. 6).44 This approach does not rely on post-polymerisation reactions and the networks are exclusively formed of ester linkages, preventing competitive exchange reactions in the cross-links, such as urethanes.45 While typical thermally-activated transesterification reactions catalysed by Lewis acids occur at temperatures higher than 100 °C, Brønsted acids promote facile exchange at temperatures as low as 25 °C. The comonomer, γ-methyl-ε-caprolactone (M5), gives rise to an amorphous polymer with a very low Tg of −55 °C. This allows dynamic mechanical thermal analysis (DMTA) study of the cross-linked polymer to be performed above the topology freezing transition temperature (Tv) and Tg at room temperature. A variety of Brønsted acid catalysts with pKa values ranging from −12 to 0.81 were embedded in the polyester vitrimers. A higher pKa allowed for faster stress relaxation times and lower activation energies. Overall, this quantitative study on the associated kinetics and thermodynamics of the system should prove foundational for novel acid-catalysed vitrimer systems.44
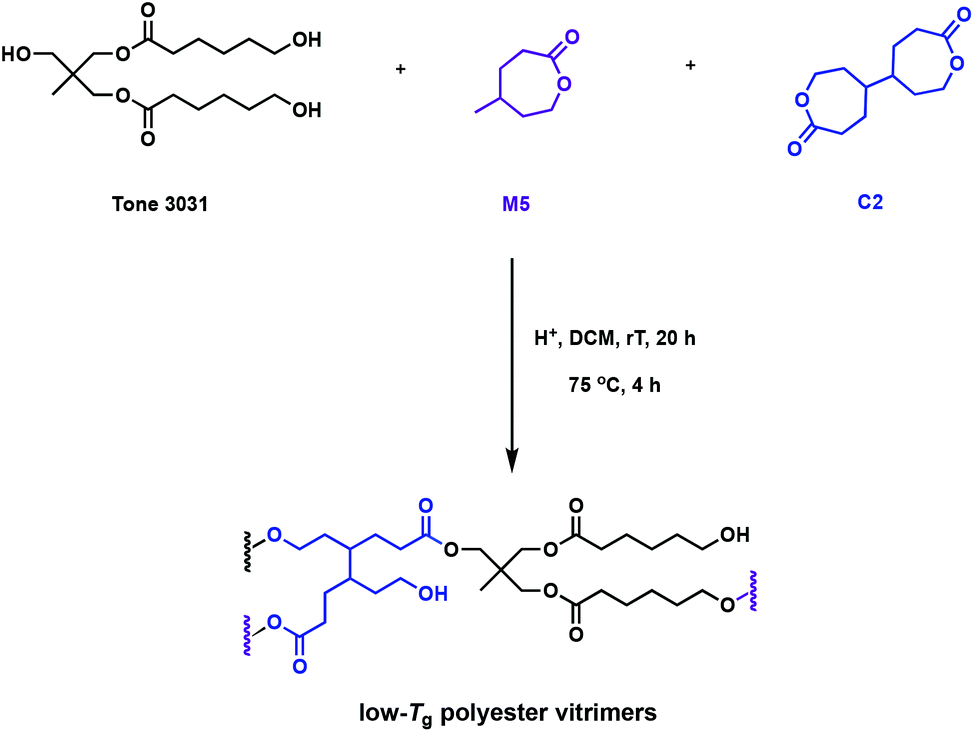 | ||
| Fig. 6 Synthesis of polyester vitrimers from M5 using cross-linking agent C2.44 | ||
Pennings and coworkers reported the cross-linking of lactones such as M2, M3 and L-lactide (M6) using a tetrafunctional spiro-bis-dimethylene-carbonate (C4) (Fig. 7).37C4 was prepared from diethylene carbonate and pentaerythritol.46 Copolymerisations were carried out in bulk at 110 °C, employing Sn(Oct)2 as a catalyst to obtain a novel class of cross-linked materials, ranging from rubbery to strong and tough semi-crystalline networks.37 Molecular architecture, degree of cross-linking and chain branching impact chain mobility and possible conformations. An increase in cross-link density reduces the available free volume and chain mobility is hindered. Consequently, crystallisation was impeded in P6, as the cross-linked materials showed a large melting point depression, from 192 to 177 °C. A significant reduction in heat of fusion was also observed with increasing cross-linking density. With increasing chain interconnectivity, tensile strength of P6 networks was measured at 65–70 MPa depending on the C4 concentration, versus 60 MPa for the homopolymer. The same trend was observed for impact strength values. The changes in the melting point and heat of fusion resulting from cross-linking with C4 were not as significant when M2 and M3 were used, implying a heavy dependence on the chosen comonomer. Furthermore, because C4 and M3 exhibit a significant difference in reactivity, cross-linking was found to be inefficient. Copolymerisation of C4 with M2 led to tougher and stronger cross-linked polycarbonate networks compared to the homopolymer counterparts, thus reinforcing the need for a reactivity match between the monomers. Their degradation products were found to be non-toxic and therefore showed promise for biomedical applications. The cross-linked materials, however, did not lose mass within 20 weeks, and 88% of the initial mass was still present after 47 weeks. The resultant degradation times are thus too long for biomedical applications, causing similar in vivo issues as P6,47 although this may open up other applications outside the biomedical realm where robustness is an asset.
 | ||
| Fig. 7 Synthesis of cross-linked P2, P4 and P6 employing C4 as a cross-linking agent.37 | ||
3.2. Multi-step approaches to cross-linked polymers
An alternative approach to synthesise cross-linked polymers is through a multi-step approach with a prepolymer that further reacts with a multifunctional cross-linker. This methodology allows fine-tuning of the prepolymer in order for specific properties to be imparted to the final material, but it is in general more time-consuming. Dove and coworkers explored an alternative way to make P6 networks by employing a flexible, bicyclic lactide derivative (C5) as a cross-linking agent.48C5 was synthesised through a thiol–ene reaction (i.e. radical addition of the thiol across the alkene moiety) between an allyl-functionalised M6 and 1,6-hexanedithiol. In the presence of 1,1,3,3-tetramethylguanidine (TMG), linear or branched P6 prepolymers were cross-linked with the bifunctional lactide derivative C5 (Fig. 8). Notably, the networks displayed low Tg values of only 30 °C, contradicting expectations that reduced mobility would lead to a higher Tg upon cross-linking.48 This observation was attributed to the long aliphatic chain in C5, which imparts greater mobility than classic shorter linkers such as disulfide, thus reducing Tg compared to the original P6 oligomers.48–50 Compared to P6, the thermosets showed reduced tensile strength (1.4 to 3.9 MPa vs. 59 MPa) and Young's moduli (13 to 34 MPa vs. 3500 MPa) due to the amorphous nature of the cross-linked network. Notably, the materials’ elongation at break values were found to be between 26 and 268%, higher than the 7% for P6. The networks were thus applicable where highly ductile P6 networks are desired, such as fibres, a remarkable accomplishment for a notoriously brittle homopolymer.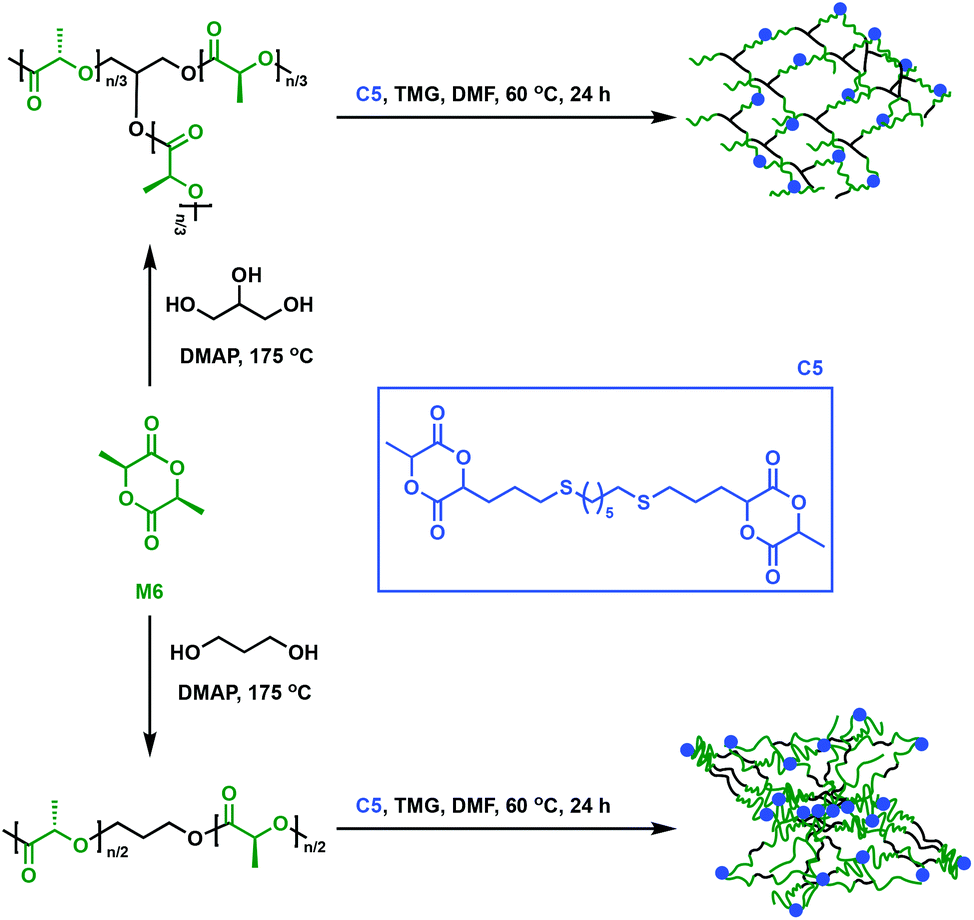 | ||
| Fig. 8 Synthesis of cross-linked P6 using C5 as a cross-linker.48 | ||
Amsden and collaborators employed C2 and C3 in a two-step strategy to synthesise polymer networks using 3-arm P3 (Fig. 9).51 The P3 star polymers were previously reported to have lower melt viscosities compared to their linear counterparts. Injection moulding at lower temperatures is thus more facile, requiring less energy and aiding processing.52 The desired applications for these systems were drug delivery and tissue engineering. The target elastomers consequently required a Tg < 37 °C in order to be rubbery, a property commonly associated with amorphous polymers. Therefore, a copolymer between M3 (Tg = −60 °C) and D,L-lactide, M7, (Tg = 68 °C) was prepared through ROP using glycerol as an initiator and Sn(Oct)2 as a catalyst.51,53 As the crystalline regions in the cross-linked polymer would likely increase both the network degradation time and the rate of bioabsorption,35 crystallinity was not a highly desired property for this study.45,46 Installing a C3 cross-link reduces the P3 crystallisable sequences, therefore allowing the cross-linked polymer to be enzymatically degraded. Release began within two weeks and approached 80% completion in 100 weeks, making biodegradation of the cross-linked polymer more efficient than for the linear crystalline homopolymer that undergoes long induction periods prior to mass loss.51,54,55
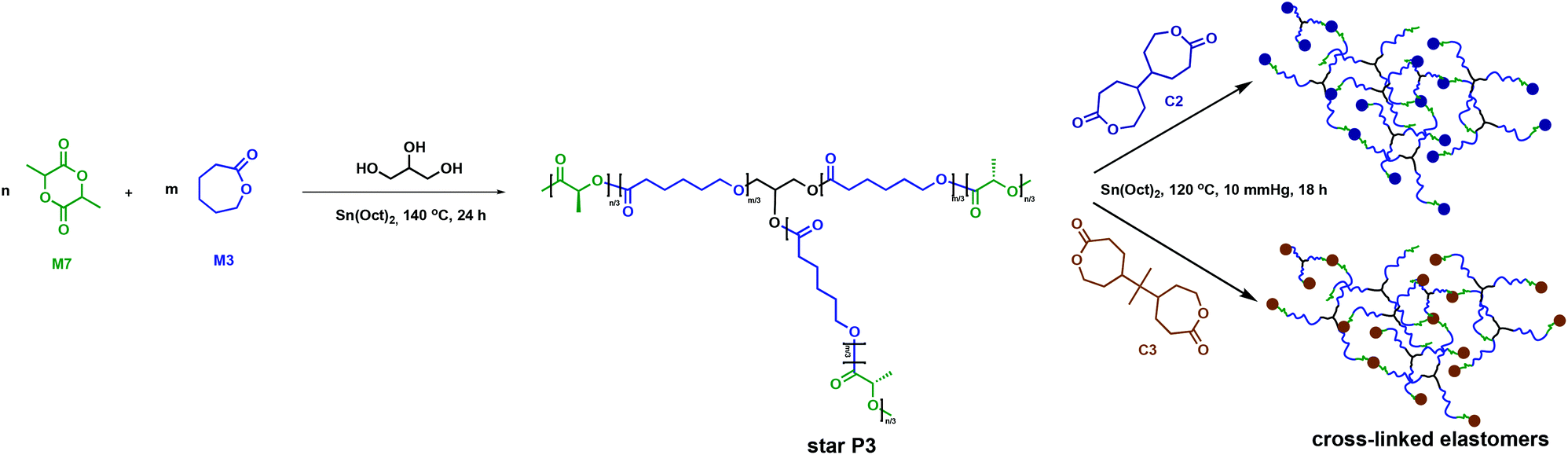 | ||
| Fig. 9 Synthesis of cross-linked elastomers employing C2 and C3 as cross-linkers.51 | ||
The C2 and C3 cross-linked polymers synthesised by both Amsden et al. and Pitt et al. displayed either a low degree of crystallinity or were amorphous.35,56 Different ratios of C3 and prepolymers were used to synthesise four elastomers with different mechanical properties. With physical properties such as Young's modulus (0.55–1.55 MPa), ultimate stress (0.21–0.60 MPa) and ultimate strain (65–154%) of the same order as elastin,57 this family of elastomers has been suggested as potential tissue engineering scaffolds for soft tissues. The cross-linking efficiency improved with increasing C3 content, forming a tough elastomer, whereas a low cross-linking density promoted faster degradation. In vitro degradation studies were performed in phosphate buffer solutions at pH = 7.4, and showed that no sample had degraded completely after 12 weeks. However, as the ultimate tensile stress showed a logarithmic decrease it was suggested that the degradation kinetics are first order.
Hillmyer and coworkers also used a bifunctional bis(β-lactone), C6, in their complementary studies.58C6 was synthesised through carbonylation of the diepoxide derived from 1,5-hexadiene. The lactone comonomer, M5, as well as the C6, can potentially be obtained from renewable feedstocks, hence addressing the circular economy goals.58,59 The polyester elastomers were prepared in two steps: first, a star-shaped poly(γ-methyl-ε-caprolactone) (P7) was prepared through ROP catalysed by Sn(Oct)2 in the presence of pentaerythritol initiator, followed by cross-linking with C6 using the same tin catalyst (Fig. 10). Even though Sn(Oct)2 is not reported as a particularly active catalyst for the ROP of butyrolactones,60,61 it generated β-hydroxyesters at the junctions between star-shaped P7, opening the β-lactone through the hydroxy end groups. Reminiscent of the above-mentioned elastomers, the properties of this cross-linked elastomer were competitive with a commercial rubber band. Degradation was tested under environmentally-relevant conditions, with full hydrolysis of the samples observed across a range of temperatures.
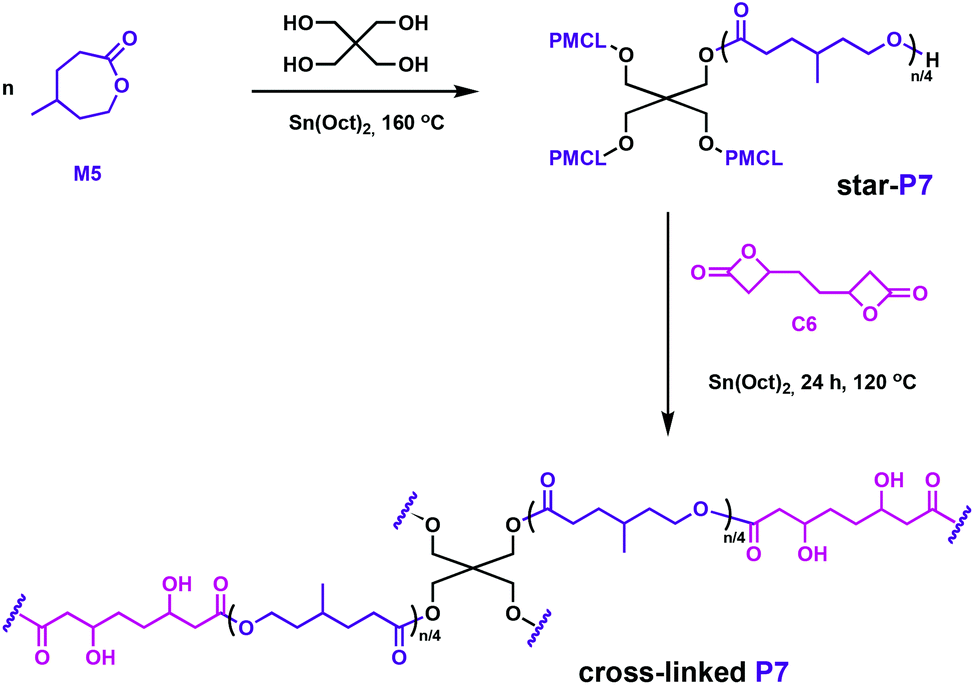 | ||
| Fig. 10 Synthesis of polyester networks using a bifunctional linker, C6.58 | ||
Snyder et al. designed and prepared polycarbonate vitrimers using an inexpensive Ti(IV) catalyst.62 In the same way that transesterification can be exploited with polyesters, carbonates undergo transcarbonation. Consequently, polycarbonate vitrimers could potentially develop into a sustainable class of degradable, reprocessable cross-linked polymers. In order for the networks to undergo transcarbonation, both carbonates and primary hydroxyl groups have to be incorporated in the polymer, achieved in one step using C1 and 1,4-butanediol in the presence of titanium(IV) isopropoxide (Ti(OiPr)4) (Fig. 11). The starting materials solution cast into molds; solvent evaporation afforded transparent colourless networks. The stress relaxation rates were similar to polyester vitrimers, whereas the activation energy of stress relaxation was found to vary as a function of cross-linking density. These networks were ground and reprocessed at various temperatures and pressures (140–160 °C, 5–10 MPa). Higher reprocessing temperatures led a significantly lower Tg in the final materials, whereas a lower temperature retained the Tg for the yielded networks. Recovery of the tensile strength and plateau storage modulus were used to assess their reprocessability. Values between 71–133% after reprocessing support these claims. Acid-catalysed hydrolysis of the networks recovers 80% pure di(trimethylolpropane) and induces Ti(OiPr)4 hydrolysis to form non-toxic TiO2.62 Although room-temperature Tg values and initiator volatility remain challenges, both polyester and polycarbonate vitrimers are materials that could feasibly tackle the end-of-life challenges of conventional thermosets.
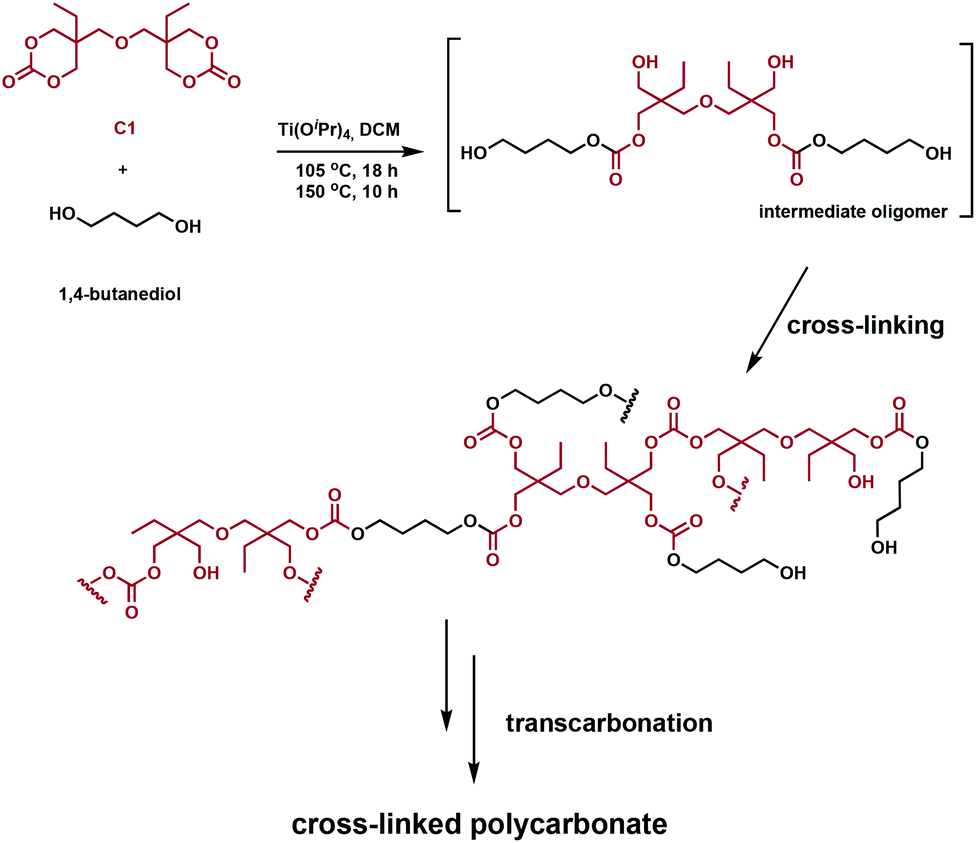 | ||
| Fig. 11 Synthesis of a cross-linked polycarbonate network from C1 and 1,4-butanediol.62 | ||
Zhao et al. translated these polycarbonate vitrimers into paper composites, that are synthesised via in situ cross-linking inside cellulose networks.63 Vitrimeric paper composites with various cross-linker to diol ratios exhibited enhanced fracture strengths of 20 to 42.7 MPa, compared to 8 MPa for untreated paper. Comparable strain values were observed over a wide temperature range (25–120 °C). Moreover, the vitrimeric component imparts reprocessability, shape-memory and self-healing properties to the paper. Both the polycarbonate precursors and cellulose can be easily recovered under mildly acidic conditions. This novel material has since been further explored as transparent coatings for silver nanowire conductive paper, offering improved thermal stabilities compared to the analogues produced through traditional methods.63
3.3. Photo-induced cross-linking
In soft tissue engineering, drug delivery and dentistry, photo-cross-linking has gained a lot of interest due to fast curing rates at ambient temperatures. In vivo studies of biodegradable resins have shown facile, controlled material solidification along with important, beneficial clinical outcomes such as minimal scarring, risk of infection or treatment costs.29,64,65 Different groups employed a multi-step approach to photo-cross-linked networks by using telechelic degradable prepolymers. These could be synthesised through the ROP of M6 in the presence of Sn(Oct)2, with various alcohols (glycerol, hexanediol, sorbitol) as initiators. Following functionalisation of the hydroxy end-groups with acrylates, cross-linking was performed through radical polymerisation, giving rise to novel P6-based tissue engineering scaffolds.66–68 A similar methodology was used by Seppälä and coworkers who reported the synthesis of cross-linked P3 elastomers (Fig. 12).69 Telechelic P3 star-shaped oligomers were synthesised, followed by chain-end functionalisation with methacrylic anhydride. Cross-linking afforded elastomers with excellent form stability, particularly when a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar feed of M3:M7 was used. In vitro hydrolytic degradation rates depend on the oligomer composition, with networks containing high amounts of M3 showing slower kinetics compared to the copolymers having higher M7 contents. There remains untapped potential for these random copolymers in biomedicine,69 although complementary toxicity studies are essential.
:1 molar feed of M3:M7 was used. In vitro hydrolytic degradation rates depend on the oligomer composition, with networks containing high amounts of M3 showing slower kinetics compared to the copolymers having higher M7 contents. There remains untapped potential for these random copolymers in biomedicine,69 although complementary toxicity studies are essential.
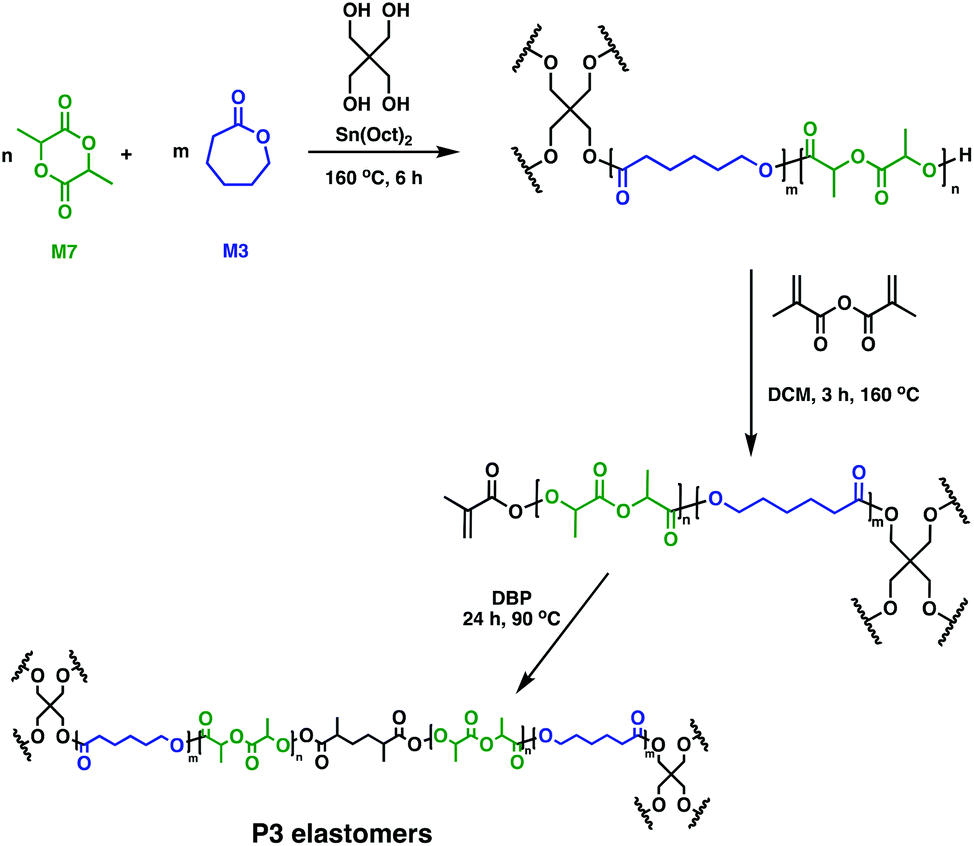 | ||
| Fig. 12 Synthesis of double-bond-cured P3 elastomers.69 | ||
Introduction of acrylate end groups does introduce non-degradable components and the potential for trace acrylate impurities, with the associated toxicity risk.66,70 However, Storey et al. reported that methacrylate-terminated M3 and M7-based prepolymers can be homopolymerised or copolymerised with methyl methacrylate, styrene and 2-methylene-1,3-dioxepane (M8) to yield non-toxic networks suitable for implantation.71 A high degree of cross-linking was reported, with <10% sol contents in all cases, reducing the hazard from residual olefinic monomers; degradability studies were not performed.
Polyesters are typically hydrolysed to afford acid products, which, depending on application, can be problematic due to potential side effects. In contrast, carbonates usually degrade into carbon dioxide and compounds containing hydroxyl moieties, enabling the design of materials that break down into bioactive molecules.72–75 To exploit this, high molecular weight P4 films of pentaerythritol triacrylate and a photoinitiator (Irgacure 369) were photo-cross-linked to give porous flexible scaffolds.76 Human mesenchymal stem cells were shown to readily adhere and proliferate on these scaffolds, highlighting the exciting potential of these materials for biomedical applications.76 Similarly, low molecular weight methacrylate-terminated P4 oligomers were synthesised and used as cross-linking aids along with Irgacure 2959.66 Upon UV irradiation, transparent films formed and it was found that the cross-link density was easily tuned by varying the exposure time (30–300 min), while maintaining high gel contents. The elasticity of these materials was found to be between 7.1 and 7.5 MPa, similar to 6.5 MPa for the non-cross-linked P4 scaffolds. These materials are cyto-compatible and while their erosion times are longer than for the linear counterparts, the degradation products were rendered non-toxic.65 In order to improve creep resistance, P4 oligomers were end-functionalised with fumaric acid monoethyl ester and further photopolymerised (Fig. 13).77 The resulting networks had low Tg values between −14.7 and −18.1 °C and were flexible, with Young's moduli varying between 1.25–2.12 MPa and 0% permanent deformation under cyclic loading experiments. While P4-type networks do degrade enzymatically,78,79 no detailed degradation studies were presented for this system. End-functionalisation was analogously employed for 3-armed hydroxyl-terminated P2, P4 and P6 oligomers. Photo-cross-linking with a 2,2-dimethoxy-2-phenylacetophenone (DMPA) initiator yielded networks with high gel contents up to 96%.80
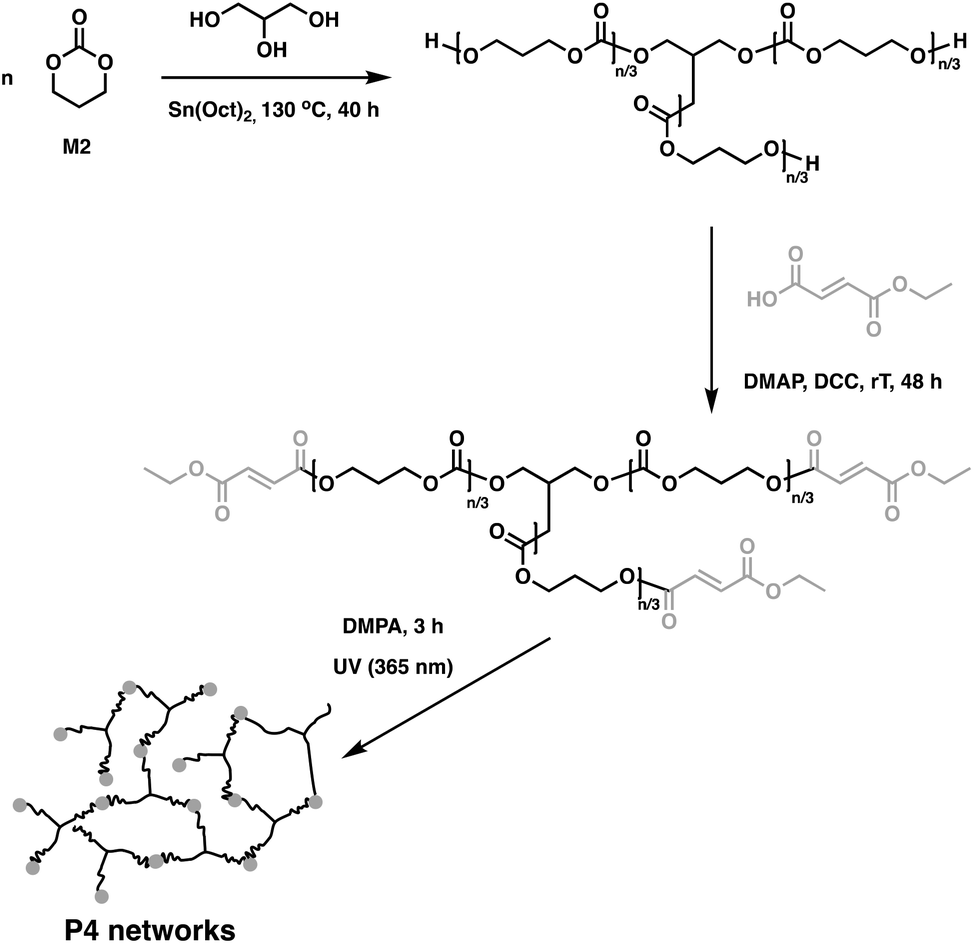 | ||
| Fig. 13 Synthesis of P4 networks through UV cross-linking of fumaric acid monoethyl ester-functionalised P4 oligomers.77 | ||
Further functionality is challenging to incorporate, as no sites for grafting traditional moieties, such as poly(ethylene glycol) (P8), heparin or collagen, exist on the linear polymer precursors. Timbart and Amsden circumvented this challenge with a novel star polymer, prepared via melt ROP of 2-oxepane-1,5-dione (M9) and M3 in the presence of Sn(Oct)2 and glycerol (Fig. 14).81 The acrylated star-copolymers were photo-cross-linked in the presence of DMPA. Post-polymerisation functionalisation through the remaining ketone M9 was realised through hydrazine modification of the networks was successful, producing elastomer films containing both hydroxyl and amine functional groups. This gives anchors for a variety of functional groups to load molecules, vary hydrolytic degradation rates or tune hydrophilicity.
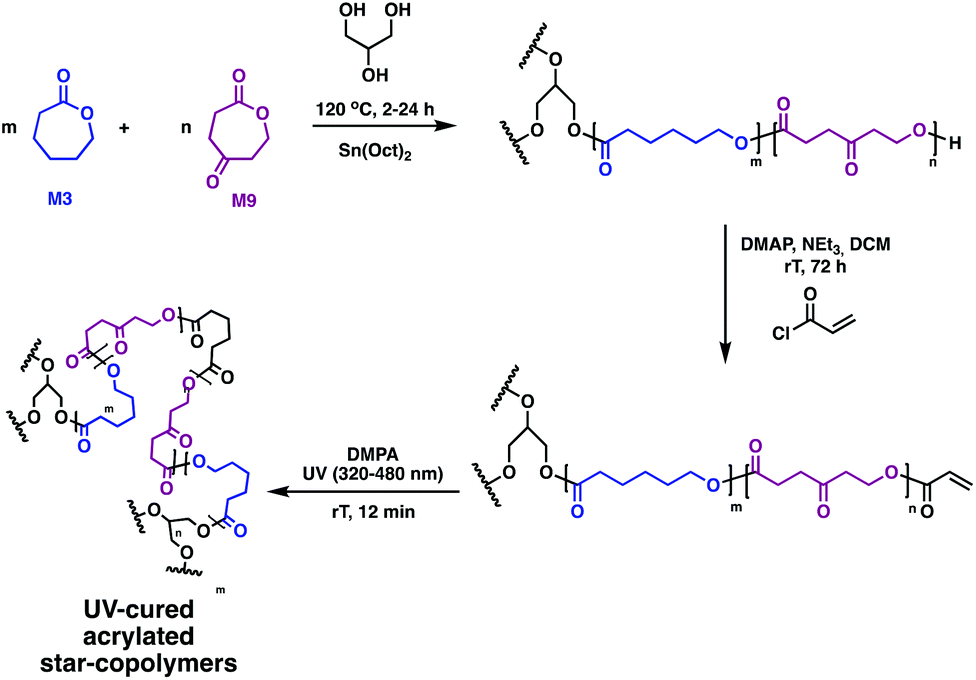 | ||
| Fig. 14 Synthesis of biodegradable photo-cross-linked elastomers based on M9 and M3.81 | ||
3.4. Cross-linking through gamma and electron beam irradiation
Gamma and electron beam irradiation have been widely applied to modify the properties of polymers and biomaterials.82–84 These high-energy irradiation methods modify the surface and bulk properties of the materials, with the bonus of simultaneously sterilizing them.82 While these methodologies proved ineffective for polymers P2, P6 or poly(propylene carbonate), biodegradable elastomeric P4 networks can efficiently form using this strategy.85–88 High molecular weight P4 was synthesised through the conventional ROP of M2 with Sn(Oct)2 as catalyst, followed by compression moulding under an inert atmosphere to afford films. 60Co gamma-irradiation at doses of 25, 50 and 100 kGy was used to cure the P4 films, yielding flexible bioelastomers.89 Grijpma demonstrated that the resulting networks had gel contents between 50–70%, an increase proportional to the irradiation dose. The degree of swelling decreased with increasing irradiation dose, allowing the study to conclude that the cross-linking density increased with an increasing irradiation. Materials with moduli between 4.6 and 5.8 MPa were obtained, lower than the 6.8 MPa observed for non-irradiated P4. Yield stress of 1.1–1.6 MPa and a high elongation at break of 580 to 820% were obtained, with the results being of the same order of magnitude to that of the P4 homopolymer. It was found that cross-linking improves the permanent deformation values, from 4.1% for the homopolymer, to values between 0.2 and 1% for the resins. Furthermore, increasing the irradiation dose causes an increase in gel contents and modulus, whereas the maximum tensile stress and elongation at break do not show significant changes. The elastic networks were used as a substrate for culturing various relevant cells, such as human umbilical vein endothelial cells, smooth muscle cells or mesenchymal stem cells. As expected, the biocompatibility of the gamma-irradiation-cured P4 elastomeric networks was bolstered. Furthermore, tubular, porous scaffolds were prepared using these materials with the flexibility and elongation at break allowing the materials to be used in blood vessel constructs, as shown in Fig. 15.89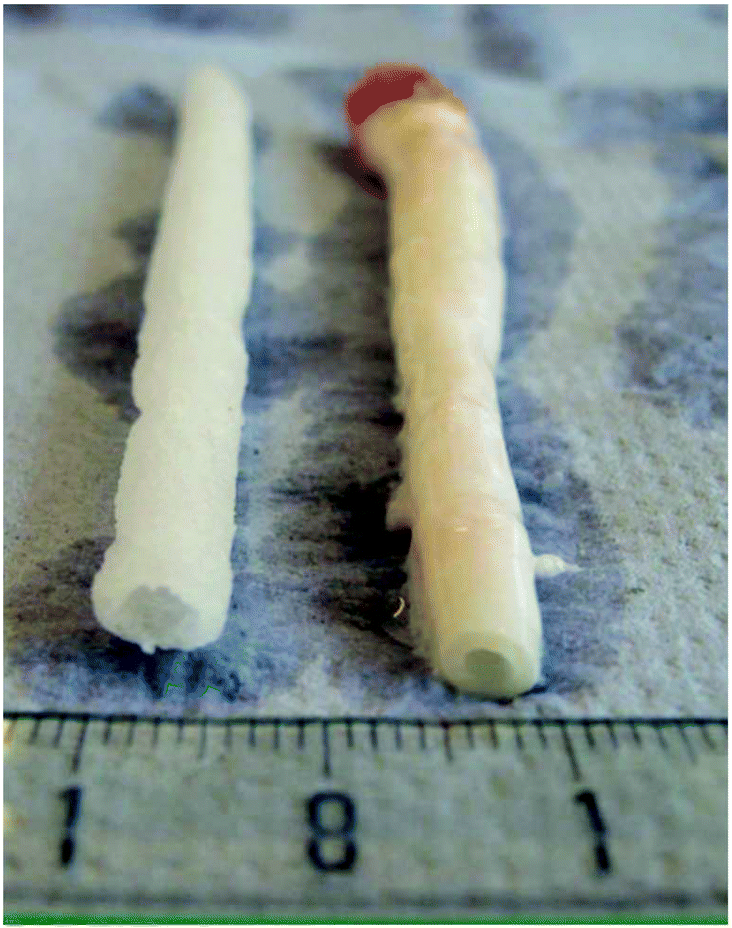 | ||
| Fig. 15 Porous scaffold (left) synthesised from cross-linked P4 and porcine carotid artery (right).89 Reprinted from ref. 89, Copyright 2020, with permission from Elsevier. | ||
In later works by Bat et al., pentaerythritol triacrylate was introduced during the 60Co gamma-irradiation curing of high molecular weight P4.90 Degradation studies were carried out in vitro with cholesterol esterase, an enzyme which catalyses cholesterol esters hydrolysis.91 The degradation rate of the cross-linked polymer was found to be much slower than for typical networks not containing pentaerythritol triacrylate, with a mass loss of 13% vs. 64% over three weeks. Cross-linking trials were extended to poly(3-hydroxyalkanoate)s (PHAs), employing electron beam irradiation to cure a microbial PHA produced using Pseudomonas oleovorans on a mixture of n-octane and 1-octene. The moduli and cross-link density of the networks were varied by altering the ratios of the carbon sources, along with using an extensive range of electron beam irradiation doses, between 30 and 500 kGy. The resins are shown to degrade via enzymatic hydrolysis and subsequent surface erosion, a degradation mode resembling that of crystalline PHA.92
4. Degradable resins from itaconic acid
An emerging route to novel renewable resins is the use of bio-based polysaccharides or lignocellulosic feedstocks as platform molecules.93,94 An economically viable building block that was extensively studied is itaconic acid (M10). On industrial scale, this aliphatic dicarboxylic acid building block is synthesised via carbohydrate fermentation aided by Aspergillul terreus.95,96 Owing to its two carboxylic acid moieties, α,β-unsaturated double bond and short chain, it has proven to be a precursor for a myriad of applications, such as flame retardants, coatings, paints and latexes.97,98With the global unsaturated polyester resin market being valued at 11.63 billion USD in 2019,99 there is plenty of room for itaconic acid to expand its scope as a sustainable alternative.93 This platform molecule was recently employed by Hillmyer and collaborators in a variety of high yielding, catalytic transformations to afford a saturated diol and diester and an unsaturated diester.93 All three monomers synthesised were easily purified and employed in step-wise polymerisations to yield high molecular weight polyesters in high yields. The fully M10-derived triblock polymer was cross-linked using thiol–ene “click” chemistry with the potentially renewable cross-linker, pentaerythritol tetrakis(3-mercaptopropionate) (C7) (Fig. 16). This afforded resins with mechanical properties dependant on the proportion of saturated and unsaturated diols used. The synthesised networks also displayed promising base-catalysed hydrolysis at ambient temperature, hinting at the potential of these degradable, high-value polymers.93 Dai et al. reported the use of M10 along with other bio-based platform chemicals, 2,5-furandicarboxylic acid (M11), succinic acid (M12) and 1,3-propanediol (M13), to afford a range of fully bio-based unsaturated polyesters via melt polymerisation.100 Notably, M11 is obtained through the oxidation hydroxymethylfurfural, which is derived from sugars or polysaccharides,101,102 and its structural rigidity imparts superior thermomechanical properties to the resultant polyesters.103 These polyesters were thermally cured to give Tg values between 73.5–141.7 °C and flexural strengths of 41.9–116.8 MPa, matching fossil-fuel derived polyester networks.100
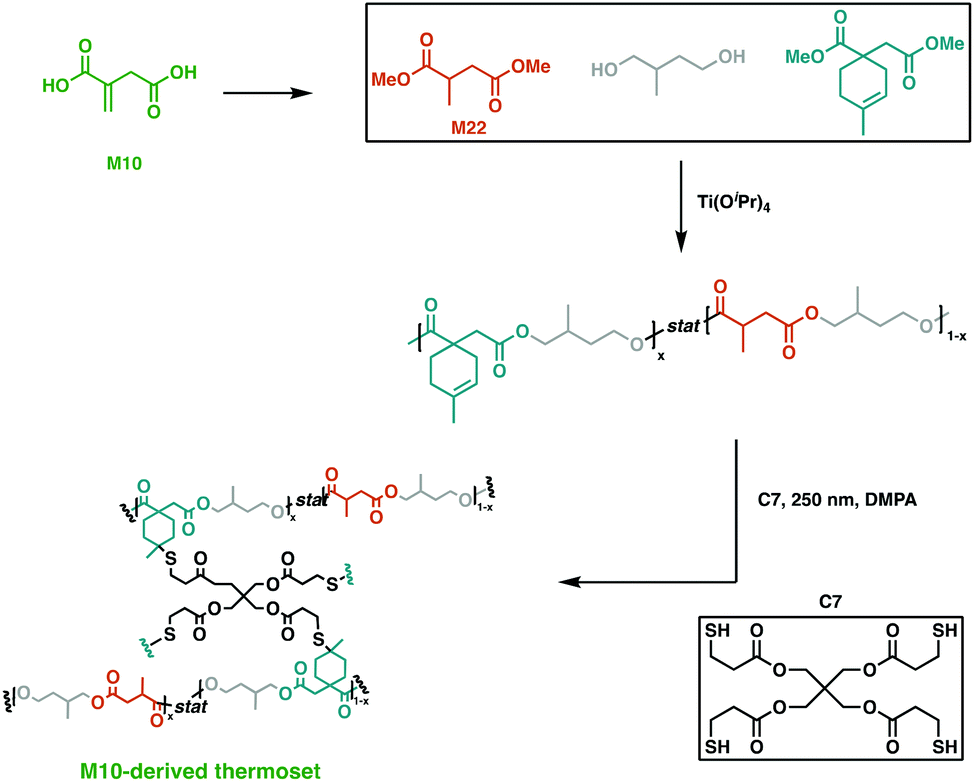 | ||
| Fig. 16 Cross-linking of M10-derived triblock polymers using C7.93 | ||
M10 has also been employed in a biobased vulcanised unsaturated aliphatic polyester elastomer in order to toughen the brittleness of P6.104 The bio-based unsaturated polyester elastomer was synthesised through a polycondensation esterification reaction between four aliphatic monomers: M10, M13, 1,4-butanediol (M14) and sebacic acid (M15). A thermoplastic vulcanizate was then prepared, facilitated by the double bond moieties of the M10 components. Dynamic vulcanisation of the unsaturated polyester, using dicumyl peroxide (DCP) as the initiator in a melted P6 matrix, gave a well dispersed system. Significantly, P6 chains were grafted onto the vulcanised unsaturated polyester elastomer, improving the compatibility between the vulcanised phase and the matrix. Dynamic vulcanisation led to a decrease in tensile toughness of the P6 blend, from 110 MJ m−3 to 84 MJ m−3. However, the impact toughness of this material was enhanced up to 586.6 J m−1, a 31-fold increase compared to the homopolymer. These materials were suggested to have the potential to replace some conventional petroleum-based plastics, arguing that the monomers were fully bio-based,104 but an investigation into the fate of these products at end-of-life would be complementary to these claims. Although the ester bonds in the polymer backbone are labile and they have been shown to hydrolyse under specific conditions, the cross-links may inhibit degradability.
The resemblance of M10 to (meth)acrylic acids, commonly used cross-linkers in UV-cured coatings opens up new applications.105 By exploiting multiple distinct bio-based monomers such as M10, M14 and D,L-lactic acid (M16), Harlin and coworkers employed condensation polymerisation protocols to synthesise barrier coatings for commercial cardboard (Fig. 17).106 The double-bond functionality of M10 allows the free-radical cross-linking of poly(D,L-lactic acid-co-1,4-butanediol-co-itaconic acid) (P9). The cross-linking efficiency was tested with a variety of acrylate and methacrylate cross-linkers, along with radical and UV initiators. Using a P8 diacrylate together with dilauroyl peroxide yielded a cross-linked P9 with 77% gel content in only 15 minutes. An aqueous dispersion of the synthesised resin was coated on commercial folding boxes. The water vapor transmission rate was measured, showing 50% smaller values than the commercially available extruded P6 coatings. These coatings are completely oil-resistant and their use has been implemented on pilot scales using line coaters, suggesting that this material could therefore offer exciting applications in the food packaging sector.106 A more straightforward methodology for synthesising M10-based polyester coatings was reported by Brännström et al.107 A series of bio-based unsaturated polyesters were synthesised through polycondensation in bulk using M10, M12 and M14. The polyester films were UV-cured and the variation of molecular weight and amounts of residual M10 and M12 were assessed to tune the properties of the cross-linked coatings.107 A challenge with M10 is that isomerisation to 2-methyl fumaric acid occurs at temperatures of 180 °C.108 This inhibits effective polymerisation, because the 2-methylfumaric acid isomer is significantly less reactive than M10; the polymerisation conditions must thus be well-controlled to reach high monomer conversion and avoid isomerisation.107,108 DMA studies indicated that cured M10-based polyesters show increased storage modulus and hardness with increasing molecular weights. Compared to M12, M10 has a lower reactivity due to the conjugated carboxylic acid moiety, resulting in a faster reaction rate for higher mole ratios of M12. However, M12 incorporation into the polymer led to materials with lower modulus and hardness compared to the UV-cured M10-based coatings.107
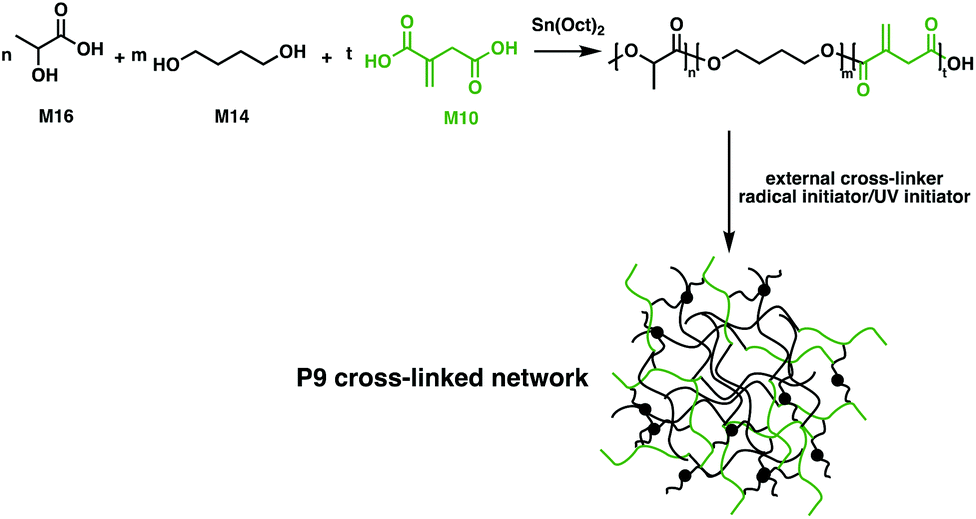 | ||
| Fig. 17 Three component cross-linked networks for coating applications.106 | ||
A similar approach was described by Dai et al. who reported the synthesis of unsaturated polyesters from M10 and three different diols, namely M14, ethylene glycol (M17) and 1,6-hexanediol (M18). These polyesters were UV-cured to afford bio-based waterborne coatings with excellent water resistance for this type of M17-based coating.109 This study, however, was lacking information regarding the degree of curing and comparisons with the unsaturated polyesters. The same group published a more comprehensive report on cross-linked networks based on the same aforementioned unsaturated polyesters.109,110 The thermosetting resins were prepared following a copolymerisation of the unsaturated polyesters with acrylated epoxidized soybean oil (M19), another bio-based platform molecule, allowing the coatings to have superior flexibility compared to what was previously reported. The two coatings were taken further in a paper that reports the aim to improve the adhesion of the previously reported coatings by incorporating glycerol (M20) in the unsaturated polyester chain.111 Moreover, through further incorporation of M19, bio-based, UV-curable and water dispersible coatings were afforded, with outstanding flexibility, high adhesion and hardness along with excellent solvent resistance.111
While there has been considerable work on itaconic acid-based resins, materials with competitive prices and consistent performance remain elusive. Trying to meet these industrial requirements, Panic et al. used esters of M10 as reactive diluents, whereas the prepolymers were synthesised using M10 and 1,2-propylene glycol (M21) through a polycondensation reaction. The cured networks showed Tg values in the range of 65–118 °C and displayed moderate stiffness (270–660 MPa), whereas the storage moduli were found to vary between 370 and 1400 MPa. Compared to the resins synthesised using styrene as a reactive diluent, the dimethyl itaconate (M22) resins had lower elastic moduli, with comparable other properties. There is thus potential for M10 and its derivatives to be used as bio-based resources for unsaturated polyester resins.112
5. Isosorbide-based resins
While aliphatic polyesters like P6 show great potential as biodegradable polymers, bio-based feedstocks such as sugars also hold promise for materials development due to their high functionality and large scale production capability.113 A key platform chemical derived from sugars is isosorbide (M23), a fused bi-heterocyclic diol that thanks to its high stability and functionality has reached industrial scale production114–116 through glucose hydrogenation to sorbitol, followed by a selective dehydration step to afford M23.114,116–119 The bicyclic monomer has been incorporated in a plethora of different polymers including the commercial plastic Durabio™, a M23-based non-biodegradable copolycarbonate for use in their automotive parts.120–126 While most work has focussed on linear polymers, M23-based cross-linked resins have also been explored.Copolyesters of M23 with M10 and M12 have been made via polycondensation in toluene at 140 °C using microwave irradiation.127 These fully bio-based cross-linked polyesters showed shape memory properties following cross-linking with M22 in the presence of a free radical initiator (2,2-azobis(2,4-dimethylvaleronitrile, V65).127 Copolyesters of M10, M14, M15 and M23 have also been synthesised and characterised (Fig. 18).128 The prepolymers had low Tg values, varying between −51 and −35 °C and Tm values between 34 and 60 °C. With the double bond functionality in M10, the polyesters were cross-linked in the presence of DCP. The bio-based networks gave improved shape memory properties, with recovery rates near 100%. Moreover, these materials were bio-compatible in cytotoxicity studies paving the way for medical implants.128
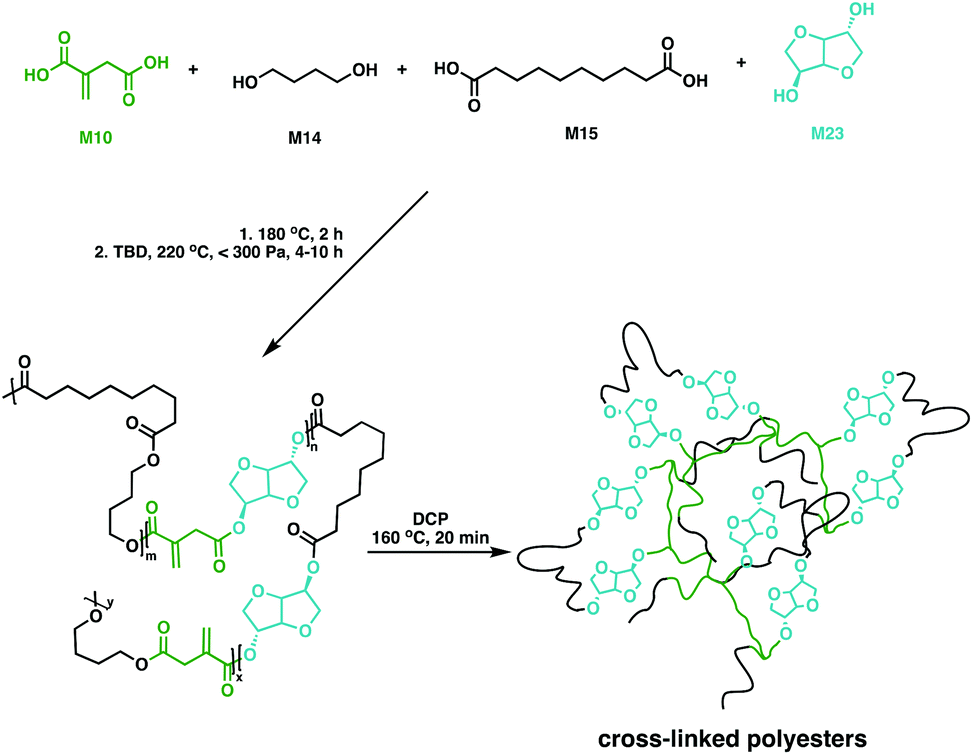 | ||
| Fig. 18 Shape-memory cross-linked copolyesters synthesised using M10, M14, M15 and M23.128 | ||
M23-containing polyester and polycarbonate resins have also been employed in the coatings industry. Copolyesters of M12, M23 (18–36 wt%) and various diols (M13, M14, M20 and 1,2-propanediol, M24) were synthesised, with Tg values of 3 to 30 °C. Melamine was used to cross-link the prepolymers and while the Tg values of the novel networks were low, the coatings showed high resistance to photodegradation and hydrolysis.129M23 was also incorporated at 10–70 mol% in polyester coatings containing M11, M15 and various diols, with the Tg increasing from −46 to 35 °C with increasing M23 content.130M23-based copolycarbonates have also been reported by Noordover et al., in a study exploring the melt polycondensation of M23 with M13 or M14, using triphosgene and diphenyl carbonate.131 Curing the synthesised copolycarbonates using conventional polyisocyanate cross-linkers yielded coatings with good mechanical properties. While the polymer architecture and composition were carefully tailored in these reports, (bio)degradation was largely unexplored, and needs to be critically evaluated to support potential sustainability and closed-loop life-cycle applications.
Thiol–ene chemistry has been used to cross-link M23-based resins through allyl ether-functionalisation of the monomer, followed by subsequent polymerisation with a multifunctional mercaptopropionate.132–135 This atom economic methodology afforded potentially biodegradable elastomers with variable Tg values, ranging from −10 to −2 °C.132,134,135 Backbone degradability can also be introduced in a polymer through carbonate linkages through the synthesis of poly(thioether-co-carbonate) (P10) networks using quinic acid. An alloc-functionalisation strategy of the quinic acid lactone (M25) was exploited, along with a photocatalysed thiol–ene methodology to synthesise polycarbonate networks.75 Wooley and coworkers have since used the potential of previous studies as impetus to report the synthesis of a M23-based cross-linked polycarbonate (Fig. 19).132,136 Isosorbide dialloc (M26) was synthesised from M23, and it was copolymerised with trimethylolpropane tris(3-mercaptopropionate) (C8) through thiol–ene cross-linking. The alloc group, an allyl carbonate, imparted degradability to M23 along with providing the double bond for cross-linking. Post-cure thermal treatment (at 100 °C) did not impact thermomechanical properties, suggesting post-cure heating was unnecessary. The optically transparent elastomers had Tg values below room temperature, moduli of 1.9–2.8 MPa and elongation at break of 220–344%. In terms of degradation, the materials demonstrated a 92% mass loss under basic conditions (1 M NaOH) at elevated temperatures (60 °C), but only a 4% mass loss under physiologically-relevant conditions over 60 days. These novel cross-linked systems have the properties to be good candidates for both biomedical applications and renewable plastics.136
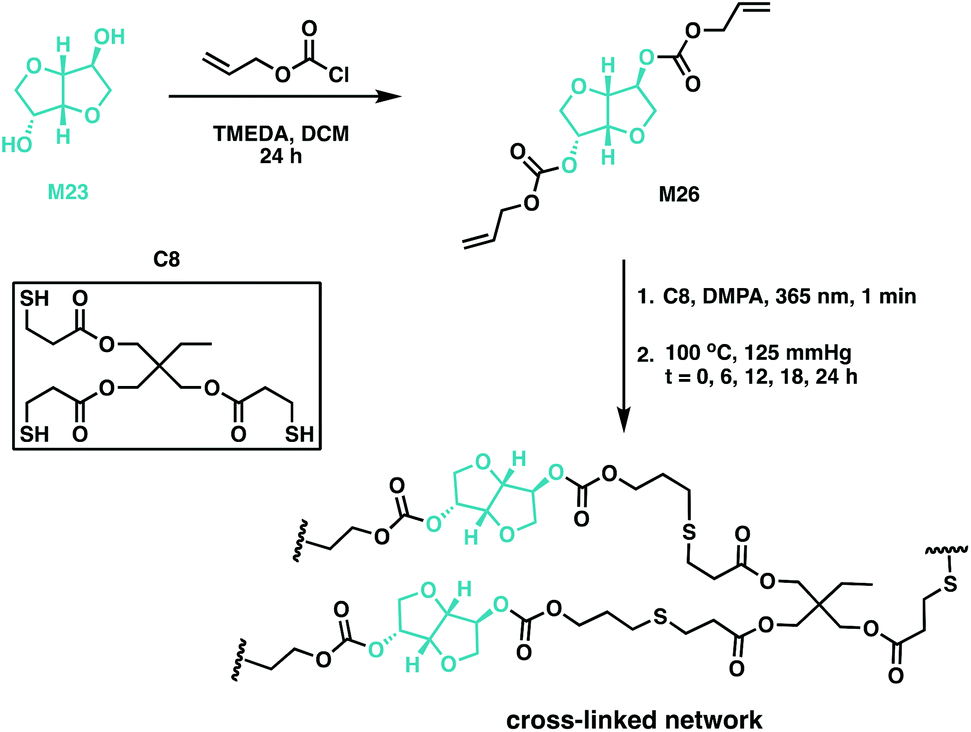 | ||
| Fig. 19 Synthesis of M26 from M23, along with the formation of cross-linked networks using C8 as a cross-linking agent.136 | ||
M23 can also be used in unsaturated polyesters is as a reactive diluent, after chemical modification.137 Most examples of renewable unsaturated polyester systems report styrene as reactive diluent for cross-linking, due to its low viscosity, high reactivity and good miscibility. Replacing this suspected carcinogen is a key target for bio-based unsaturated polyester thermosets.138–140 A M23 derivative, isosorbide methacrylate (M27), was used for a M19-based matrix due to the rigidity imparted to the cured resin. Besides imparting a high flexural modulus (28.4 MPa vs. 4.8 MPa for epoxidized oil) to the cured networks, M27 also allowed the formulation of a network with a high bio-based content, along with imparting superior processability to any other soybean oil-based resins due to the lower viscosity.141 Odelius and coworkers, inspired by the rigidity of M23, utilized it along with M14 maleic and succinic anhydride (M28 and M29) to synthesise a series of bio-based unsaturated polyester thermosets with tailorable chemical structures and Tg values varying from 57 to 84 °C. The storage moduli of 0.5–3.0 GPa suggest that these materials can match the thermomechanical properties of petroleum-derived alternatives.137 While no degradation studies are reported for these M23-based resins, two studies do examine degradability using cyclic anhydride-functionalised M23.115,142 Wilbon et al. employed microwave-assisted condensation of M23 with M29 to afford the corresponding diacid that was reacted with M20 or oligomeric poly(ethylene oxide) (P11) in the presence of Sn(Oct)2 to yield a novel family of thermosets. Cross-linking with M20 in the absence of solvent led to a high modulus (7.9 MPa at 100 °C) resin, having a Tg of 55 °C, while P11 incorporation afforded more flexible thermosets. The resins were easily degraded under acidic and basic conditions, with increasing P11 content enhancing the degradation rate. Reprocessing was also possible, although the resulting materials had diminished mechanical properties.115 A similar reaction between M23 and M28 afforded a trimer that was used to cross-link epoxidized sucrose soyate, yielding hard but flexible thermosets with excellent coating performance. In addition, the resins were fully degraded under basic conditions at 50 °C, while exhibiting high stability to acids.142
6. Conclusions and future perspectives
Thermosets make up one-third of overall polymer production, and while they offer high thermal and dimensional stability their end of life pathways are often limited to a landfill or incinerator. For a circular plastics future it is paramount to place sustainability at the core of polymer design from cradle to grave. But to design these materials is a real challenge: the thermomechanical properties of cross-linked materials responsible for their resistance and robustness are the same that preventing traditional recycling and degradation. While biodegradation of thermoplastics competes with mechanical and chemical recycling strategies, biodegradation is indeed one of the powerful potential tools that could control polymer fate in resins. Degradable thermosets built from polyester and polycarbonate functionalities are thus of growing importance. However, in order for these novel materials to be accepted and employed over existing resins, it is imperative that they match or exceed the properties of their petroleum-derived competitors.It is clear that no single monomer would match all performance requirement be suitable for all scenarios. Because of the currently accessible properties, applications have focussed on biomedicine and coatings. Most of the materials discussed in this review are inherently low modulus thermosets and therefore the demanding material requirements needed in the automotive or aerospace fields remain elusive. Important targets for these materials would be robust/reusable packaging and injection moulded parts or composites which require better temperature resistance, hardness and tensile strength.
These degradable cross-linked materials have been built from both ring-opening polymerisation and polycondensation reactions. Ring-opening methodologies afforded important precedent setters of degradable cross-linked polymers obtained from renewably derived aliphatics such as poly(lactic acid), poly(ε-caprolactone) or poly(trimethylene carbonate), but mechanical properties were often limited to low moduli and high elasticity, focussing applications to the biomedical field. Polycondensation methodologies reported were not as robust, but allowed incorporation of bio-based chemical feedstocks such as itaconic acid or isosorbide that could incorporate non-degradable cross-linking strategies, often yielding resins suitable for a wider array of applications. In composing this review, it was clear that a more consistent and systematic approach to evaluating sustainability and biodegradability was needed, as closed-loop life cycles are an essential part of our materials future.
The formation of sustainable resins through cross-linking is key to achieving networks with high dimensional stability, reduced creep and relaxation times. This requires an interplay between a variety of tunable components to achieve the desired architecture. Dynamic cross-linking is possible, but combining simple monomers and catalysts with robust and controlled exchange reactions in a truly sustainable way remains a challenge. Synthesising degradable cross-linked polymers demands controlled polymerisation methods, either through one-pot or multi-step approaches, under mild, scalable and cost-effective operating conditions. While industrial chemistry will underpin commercial efforts, this remains an area of research where creative synthetic chemistry is still foundational in developing novel classes of monomers and cross-linkers to target new applications. These are reasons – or resins if you excuse the pun – to be cheerful: degradable bio-based polyester and polycarbonate thermosets have a bright future ahead.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for funding from the EPSRC (EP/S025200/1) and Henry Royce Institute for Advanced Materials (EP/R00661X/1, EP/S019367/1, EP/P025021/1 and EP/P025498/1).References
- PlasticsEurope, Plasts. – Facts 2019, 2019, p. 14 Search PubMed.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- A. L. Andrady, J. Macromol. Sci., Part C, 1994, 34, 25–76 CrossRef.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- Y. Tokiwa, B. P. Calabia, C. U. Ugwu and S. Aiba, Int. J. Mol. Sci., 2009, 10, 3722–3742 CrossRef CAS.
- A. Salerno and C. D. Pascual, RSC Adv., 2013, 3, 17355–17363 RSC.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- H. Tsuji and K. Suzuyoshi, Polym. Degrad. Stab., 2002, 75, 347–355 CrossRef CAS.
- H. Tsuji and K. Suzuyoshi, Polym. Degrad. Stab., 2002, 75, 357–365 CrossRef CAS.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- E. Castro-Aguirre, F. Iñiguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- A. A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS.
- G. L. Gregory, M. Ulmann and A. Buchard, RSC Adv., 2015, 5, 39404–39408 RSC.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- S. M. Guillaume and J. F. Carpentier, Catal. Sci. Technol., 2012, 2, 898–906 RSC.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- J. A. Brydson, Plastics Materials, Butterworth Heinemann, Oxford, 1999 Search PubMed.
- Statistics on the Plastic Resins Industry, https://plastics.americanchemistry.com/Jobs/EconomicStatistics/Plastics-Statistics/, (accessed April 2020).
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- A. Watts, N. Kurokawa and M. A. Hillmyer, Biomacromolecules, 2017, 18, 1845–1854 CrossRef CAS.
- A. J. Nijenhuis, D. W. Grijpma and A. J. Pennings, Polymer, 1996, 37, 2783–2791 CrossRef CAS.
- H. Liu and J. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1051–1083 CrossRef CAS.
- Q. Liu, L. Jiang, R. Shi and L. Zhang, Prog. Polym. Sci., 2012, 37, 715–765 CrossRef CAS.
- J. P. Brutman, G. X. De Hoe, D. K. Schneiderman, T. N. Le and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11097–11106 CrossRef CAS.
- L. Yang, J. Li, Y. Jin, M. Li and Z. Gu, Polym. Degrad. Stab., 2015, 112, 10–19 CrossRef CAS.
- R. Chapanian, M. Y. Tse, S. C. Pang and B. G. Amsden, J. Biomed. Mater. Res., Part A, 2010, 92, 830–842 Search PubMed.
- L. Q. Yang, B. He, S. Meng, J. Z. Zhang, M. Li, J. Guo, Y. M. Guan, J. X. Li and Z. W. Gu, Polymer, 2013, 54, 2668–2675 CrossRef CAS.
- P. S. Starcher, S. W. Tinsley and B. Phillips, US Pat, 3072680, 1963 Search PubMed.
- C. G. Pitt, R. W. Hendren, A. Schindler and S. C. Woodward, J. Controlled Release, 1984, 1, 3–14 CrossRef CAS.
- R. Palmgren, S. Karlsson and A. C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1635–1649 CrossRef CAS.
- D. W. Grijpma, E. Kroeze, A. J. Nijenhuis and A. J. Pennings, Polymer, 1993, 34, 1496–1503 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read De Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS.
- H. Krimm and H. J. Buysch, European Patent, 0057360, 1984 Search PubMed.
- D. W. Grijpma and A. J. Pennings, Macromol. Chem. Phys., 1994, 195, 1649–1663 CrossRef CAS.
- P. K. Kuroishi, K. R. Delle Chiaie and A. P. Dove, Eur. Polym. J., 2019, 120, 109192 CrossRef CAS.
- L. E. Nielsen and R. F. Landel, Mechanical properties of polymers and composites, Marcel Dekker Inc, New York, 1994 Search PubMed.
- J. E. Báez, Á. Marcos-Fernández and P. Galindo-Iranzo, J. Polym. Res., 2011, 18, 1137–1146 CrossRef.
- H. M. Younes, E. Bravo-Grimaldo and B. G. Amsden, Biomaterials, 2004, 25, 5261–5269 CrossRef CAS.
- W. W. Graessley and J. Roovers, Macromolecules, 1979, 12, 959–965 CrossRef CAS.
- J. E. Mark, Polymer Data Handbook, Oxford University Press, Oxford, 2009 Search PubMed.
- C. G. Pitt, F. I. Chasalow, Y. M. Hibionada, D. M. Klimas and A. Schindler, J. Appl. Polym. Sci., 1981, 26, 3779–3787 CrossRef CAS.
- G. G. Pitt, M. M. Gratzl, G. L. Kimmel, J. Surles and A. Sohindler, Biomaterials, 1981, 2, 215–220 CrossRef.
- B. Amsden, S. Wang and U. Wyss, Biomacromolecules, 2004, 5, 1399–1403 CrossRef CAS.
- J. Gosline, M. Lillie, E. Carrington, P. Guerette, C. Ortlepp and K. Savage, Philos. Trans. R. Soc., B, 2002, 357, 121–132 CrossRef CAS.
- G. X. De Hoe, M. T. Zumstein, B. J. Tiegs, J. P. Brutman, K. McNeill, M. Sander, G. W. Coates and M. A. Hillmyer, J. Am. Chem. Soc., 2018, 140, 963–973 CrossRef CAS.
- F. O. Ayorinde, G. Osman, R. L. Shepard and F. T. Powers, J. Am. Oil Chem. Soc., 1988, 65, 1774–1777 CrossRef CAS.
- O. Coulembier, P. Degée, C. Barbaud, P. Guérin and P. Dubois, Polym. Bull., 2004, 52, 41–48 CrossRef CAS.
- X.-Q. Liu, M.-X. Wang, Z.-C. Li and F.-M. Li, Macromol. Chem. Phys., 1999, 200, 468–473 CrossRef CAS.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- W. Zhao, Z. Feng, Z. Liang, Y. Lv, F. Xiang, C. Xiong, C. Duan, L. Dai and Y. Ni, ACS Appl. Mater. Interfaces, 2019, 11, 36090–36099 CrossRef CAS.
- C. J. Bettinger, Pure Appl. Chem., 2009, 81, 2183–2201 CAS.
- E. Bat, T. G. Van Kooten, J. Feijen and D. W. Grijpma, Acta Biomater., 2011, 7, 1939–1948 CrossRef CAS.
- F. P. W. Melchels, J. Feijen and D. W. Grijpma, Biomaterials, 2009, 30, 3801–3809 CrossRef CAS.
- J. L. Escobar Ivirico, M. Salmerón-Sánchez, J. L. Gómez Ribelles and M. Monleón Pradas, Colloid Polym. Sci., 2009, 287, 671–681 CrossRef CAS.
- D. K. Han and J. A. Hubbell, Macromolecules, 1997, 30, 6077–6083 CrossRef CAS.
- A. O. Helminen, H. Korhonen and J. V. Seppälä, Macromol. Chem. Phys., 2002, 203, 2630–2639 CrossRef CAS.
- S. M. Oskui, G. Diamante, C. Liao, W. Shi, J. Gan, D. Schlenk and W. H. Grover, Environ. Sci. Technol. Lett., 2016, 3, 1–6 CrossRef CAS.
- R. F. Storey, S. C. Warren, C. J. Allison, J. S. Wiggins and A. D. Puckett, Polymer, 1993, 34, 4365–4372 CrossRef CAS.
- T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS.
- Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma and J. Feijen, Biomaterials, 2006, 27, 1741–1748 CrossRef CAS.
- A.-C. Albertsson and M. Eklund, J. Appl. Polym. Sci., 1995, 57, 87–103 CrossRef CAS.
- L. A. Link, A. T. Lonnecker, K. Hearon, C. A. Maher, J. E. Raymond and K. L. Wooley, ACS Appl. Mater. Interfaces, 2014, 6, 17370–17375 CrossRef CAS.
- E. Bat, B. H. M. Kothman, G. A. Higuera, C. A. van Blitterswijk, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 8696–8705 CrossRef CAS.
- Q. Hou, D. W. Grijpma and J. Feijen, Acta Biomater., 2009, 5, 1543–1551 CrossRef CAS.
- M. A. Foks, K. A. J. Dijkhuis, D. W. Grijpma, L. A. Brouwer, M. J. A. van Luyn and J. Feijen, J. Controlled Release, 2005, 101, 325–327 CAS.
- R. Chapanian, M. Y. Tse, S. C. Pang and B. G. Amsden, Biomaterials, 2009, 30, 295–306 CrossRef CAS.
- D. W. Grijpma, Q. Hou and J. Feijen, Biomaterials, 2005, 26, 2795–2802 CrossRef CAS.
- L. Timbart and B. G. Amsden, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 8191–8199 CrossRef CAS.
- Q. Liu, L. Jiang, R. Shi and L. Zhang, Prog. Polym. Sci., 2012, 37, 715–765 CrossRef CAS.
- A. P. Pêgo, D. W. Grijpma and J. Feijen, Polymer, 2003, 44, 6495–6504 CrossRef.
- P. H. S. Kumara, N. Nagasawa, T. Yagi and M. Tamada, J. Appl. Polym. Sci., 2008, 109, 3321–3328 CrossRef CAS.
- H. Mitomo, A. Kaneda, T. M. Quynh, N. Nagasawa and F. Yoshii, Polymer, 2005, 46, 4695–4703 CrossRef CAS.
- T. M. Quynh, H. Mitomo, N. Nagasawa, Y. Wada, F. Yoshii and M. Tamada, Eur. Polym. J., 2007, 43, 1779–1785 CrossRef CAS.
- F. Yoshii, D. Darwis, H. Mitomo and K. Makuuchi, Radiat. Phys. Chem., 2000, 57, 417–420 CrossRef CAS.
- Y. S. Qin, Q. W. Ma, X. H. Wang, J. Z. Sun, X. J. Zhao and F. S. Wang, Polym. Degrad. Stab., 2007, 92, 1942–1947 CrossRef CAS.
- Y. Song, M. M. J. Kamphuis, Z. Zhang, L. M. T. Sterk, I. Vermes, A. A. Poot, J. Feijen and D. W. Grijpma, Acta Biomater., 2010, 6, 1269–1277 CrossRef CAS.
- E. Bat, J. Feijen and D. W. Grijpma, Biomacromolecules, 2010, 11, 2692–2699 CrossRef CAS.
- D. Y. Hui and P. N. Howles, J. Lipid Res., 2002, 43, 2017–2030 CrossRef CAS.
- G. J. M. de Koning, H. M. M. van Bilsen, P. J. Lemstra, W. Hazenberg, B. Witholt, H. Preusting, J. G. van der Galiën, A. Schirmer and D. Jendrossek, Polymer, 1994, 35, 2090–2097 CrossRef CAS.
- J. T. Trotta, A. Watts, A. R. Wong, A. M. Lapointe, M. A. Hillmyer and B. P. Fors, ACS Sustainable Chem. Eng., 2019, 7, 2691–2701 CrossRef CAS.
- T. J. Farmer, J. W. Comerford, A. Pellis and T. Robert, Polym. Int., 2018, 67, 775–789 CrossRef CAS.
- T. Willke and K. D. Vorlop, Appl. Microbiol. Biotechnol., 2001, 56, 289–295 CrossRef CAS.
- M. Okabe, D. Lies, S. Kanamasa and E. Y. Park, Appl. Microbiol. Biotechnol., 2009, 84, 597–606 CrossRef CAS.
- A. A. El-Imam and C. Du, J. Biodiversity Bioprospect Dev., 2014, 1, 1000119 Search PubMed.
- S. Kumar, S. Krishnan, S. K. Samal, S. Mohanty and S. K. Nayak, Polym. Int., 2017, 66, 1349–1363 CrossRef CAS.
- Unsaturated Polyester Resin Market Size Industry Report 2027, https://www.grandviewresearch.com/industry-analysis/unsaturated-polyester-resin-upr-market, (accessed May 2020).
- J. Dai, S. Ma, N. Teng, X. Dai, X. Shen, S. Wang, X. Liu and J. Zhu, Ind. Eng. Chem. Res., 2017, 56, 2650–2657 CrossRef CAS.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- H. Ait Rass, N. Essayem and M. Besson, Green Chem., 2013, 15, 2240–2251 RSC.
- A. F. Sousa, M. Matos, C. S. R. Freire, A. J. D. Silvestre and J. F. J. Coelho, Polymer, 2013, 54, 513–519 CrossRef CAS.
- G. C. Liu, Y. S. He, J. B. Zeng, Q. T. Li and Y. Z. Wang, Biomacromolecules, 2014, 15, 4260–4271 CrossRef CAS.
- T. Robert and S. Friebel, Green Chem., 2016, 18, 2922–2934 RSC.
- T. Mehtiö, A. Anghelescu-Hakala, J. Hartman, V. Kunnari and A. Harlin, J. Appl. Polym. Sci., 2017, 134, 1–8 CrossRef.
- S. Brännström, E. Malmström and M. Johansson, J. Coat. Technol. Res., 2017, 14, 851–861 CrossRef.
- C. S. Marvel and T. H. Shepherd, J. Org. Chem., 1959, 24, 599–605 CrossRef CAS.
- J. Dai, S. Ma, X. Liu, L. Han, Y. Wu, X. Dai and J. Zhu, Prog. Org. Coat., 2015, 78, 49–54 CrossRef CAS.
- J. Dai, S. Ma, Y. Wu, L. Han, L. Zhang, J. Zhu and X. Liu, Green Chem., 2015, 17, 2383–2392 RSC.
- J. Dai, S. Ma, Y. Wu, J. Zhu and X. Liu, Prog. Org. Coat., 2015, 87, 197–203 CrossRef CAS.
- V. V. Panic, S. I. Seslija, I. G. Popovic, V. D. Spasojevic, A. R. Popovic, V. B. Nikolic and P. M. Spasojevic, Biomacromolecules, 2017, 18, 3881–3891 CrossRef CAS.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, Macromolecules, 2014, 47, 498–505 CrossRef CAS.
- D. J. Saxon, A. M. Luke, H. Sajjad, W. B. Tolman and T. M. Reineke, Prog. Polym. Sci., 2020, 101, 101196 CrossRef CAS.
- P. A. Wilbon, J. L. Swartz, N. R. Meltzer, J. P. Brutman, M. A. Hillmyer and J. E. Wissinger, ACS Sustainable Chem. Eng., 2017, 5, 9185–9190 CrossRef CAS.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- J. A. Galbis, M. D. G. García-Martín, M. V. De Paz and E. Galbis, Chem. Rev., 2016, 121, 1450–1463 Search PubMed.
- D. Juais, A. F. Naves, C. Li, R. A. Gross and L. H. Catalani, Macromolecules, 2010, 43, 10315–10319 CrossRef CAS.
- W. C. Shearouse, L. M. Lillie, T. M. Reineke and W. B. Tolman, ACS Macro Lett., 2015, 4, 284–288 CrossRef CAS.
- H. Kang, X. Li, J. Xue, L. Zhang, L. Liu, R. Xu and B. Guo, RSC Adv., 2014, 4, 19462–19471 RSC.
- Durabio™, https://www.m-chemical.co.jp/en/products/departments/mcc/sustainable/product/1201026_7964.html, (accessed June 2020).
- A. M. Nelson and T. E. Long, Polym. Int., 2012, 61, 1485–1491 CrossRef CAS.
- S. Chatti, G. Schwarz and H. R. Kricheldorf, Macromolecules, 2006, 39, 9064–9070 CrossRef CAS.
- M. Yokoe, A. O. I. Keigo and M. Okada, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2312–2321 CrossRef CAS.
- O. Goerz and H. Ritter, Polym. Int., 2013, 62, 709–712 CrossRef CAS.
- H. Kang, M. Li, Z. Tang, J. Xue, X. Hu, L. Zhang and B. Guo, J. Mater. Chem. B, 2014, 2, 7877–7886 RSC.
- A. Caretto, V. Passoni, N. Brenna, M. Sitta, L. Ogliosi, G. Catel, S. Turri and G. Griffini, ACS Sustainable Chem. Eng., 2018, 6, 14125–14134 CrossRef CAS.
- M. Lomelí-Rodríguez, J. R. Corpas-Martínez, S. Willis, R. Mulholland and J. A. Lopez-Sanchez, Polymers, 2018, 10, 1–19 CrossRef.
- B. A. J. Noordover, D. Haverman, R. Duchateau, R. A. T. M. van Benthem and C. E. Koning, J. Appl. Polym. Sci., 2011, 121, 1450–1463 CrossRef CAS.
- C. Lorenzini, A. Haider, I. K. Kang, M. Sangermano, S. Abbad-Andalloussi, P. E. Mazeran, J. Lalevée, E. Renard, V. Langlois and D. L. Versace, Biomacromolecules, 2015, 16, 683–694 CrossRef CAS.
- C. Lorenzini, D. L. Versace, C. Gaillet, C. Lorthioir, S. Boileau, E. Renard and V. Langlois, Polymer, 2014, 55, 4432–4440 CrossRef CAS.
- R. A. Ortiz, A. Y. R. Martínez and A. E. G. Valdez, J. Biobased Mater. Bioenergy, 2012, 6, 36–41 CrossRef CAS.
- T. Modjinou, D. L. Versace, S. Abbad-Andallousi, N. Bousserrhine, J. Babinot, V. Langlois and E. Renard, ACS Sustainable Chem. Eng., 2015, 3, 1094–1100 CrossRef CAS.
- T. S. Kristufek, S. L. Kristufek, L. A. Link, A. C. Weems, S. Khan, S.-M. Lim, A. T. Lonnecker, J. E. Raymond, D. J. Maitland and K. L. Wooley, Polym. Chem., 2016, 7, 2639–2644 RSC.
- Y. Xu, G. Hua, M. Hakkarainen and K. Odelius, Biomacromolecules, 2018, 19, 3077–3085 CrossRef CAS.
- C. J. Weschler, Atmos. Environ., 2009, 43, 153–169 CrossRef CAS.
- Q. Li, S. Ma, X. Xu and J. Zhu, in Unsaturated Polyester Resins, ed. S. Thomas, M. Hosur and C. J. Chirayil, Elsevier, Amsterdam, 2019, ch. 20, pp. 515–555 Search PubMed.
- J. E. C. Lerner, T. Kohajda, M. E. Aguilar, L. A. Massolo, E. Y. Sánchez, A. A. Porta, P. Opitz, G. Wichmann, O. Herbarth and A. Mueller, Environ. Sci. Pollut. Res., 2014, 21, 9676–9688 CrossRef CAS.
- W. Liu, T. Xie and R. Qiu, ACS Sustainable Chem. Eng., 2017, 5, 774–783 CrossRef CAS.
- S. Ma, D. C. Webster and F. Jabeen, Macromolecules, 2016, 49, 3780–3788 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |