
Discrete multifunctional sequence-defined oligomers with controlled chirality†
Jie
Li
a,
Maxime
Leclercq
b,
Mathieu
Fossepré
 b,
Mathieu
Surin
*b,
Karine
Glinel
a,
Alain M.
Jonas
*a and
Antony E.
Fernandes
*ac
b,
Mathieu
Surin
*b,
Karine
Glinel
a,
Alain M.
Jonas
*a and
Antony E.
Fernandes
*ac
aInstitute of Condensed Matter and Nanosciences, Bio- and Soft Matter, Université catholique de Louvain, 1348 Louvain-la-Neuve, Belgium
bLaboratory for Chemistry of Novel Materials, Centre of Innovation and Research in Materials and Polymers (CIRMAP), University of Mons - UMONS, 20 Place du Parc, 7000 Mons, Belgium. E-mail: mathieu.surin@umons.ac.be
cCertech, Rue Jules Bordet 7180 Seneffe, Belgium
First published on 29th May 2020
Abstract
Discrete sequence-defined oligomers are synthetic mimics of short peptides, with possible applications in catalysis, information storage, drug delivery or self-assembly. Here, we report on the efficient and stereocontrolled synthesis of sequence-defined poly(triazole-urethane) oligomers incorporating a very large range of functional side groups, including alkyl, phenyl, pyridyl, hydroxyl, amine, imidazole and carboxylic acid functions. The route involves the alternation of chloroformate-mediated carbamate bond formation and Cu-catalyzed azide–alkyne cycloaddition reactions, expeditiously leading to oligomers up to octamers by an iterative exponential growth strategy. Additionally, the chirality of the monomers is straightforwardly controlled and maintained during the elongation process, leading to optically-active multifunctional oligomers of controlled molecular configuration, as confirmed by 1H NMR, ESI-MS/MS and circular dichroism. Our strategy provides a versatile platform to efficiently synthesize discrete oligomers with programmable stereochemistry, sequentiality and multifunctionality.
Introduction
Sequence and stereoregularity are central to the functioning of biomacromolecules such as proteins or nucleic acids. Reaching a similar degree of control over primary structure and chirality in synthetic polymers has been a long endeavor,1 including the catalytic control of tacticity,2,3 the development of reversible deactivation routes for the control of molar mass,4–7 and the more recent emergence of synthetic strategies towards sequence-defined polymers.8–19 For that latter purpose, iterative stepwise methods, including iterative sequential growth (ISG) and iterative exponential growth (IEG), have generally been developed.20–24 In particular, IEG strategies provide rapid access to longer precision oligomers as the chain length is doubled after each iterative cycle, even though they are restricted to the production of symmetric products in which not every repeat unit is freely selectable. Hence, monomers bearing orthogonal protective groups, or latent functional groups,17,25,26 are activated divergently, releasing complementary-activated units, to be convergently reunified. It is however possible to go beyond the repetitive or palindromic primary structures typically obtained by IEG. For instance, the IEG+ strategy developed by Johnson and coll. combines both exponential chain growth and side chain functionalization.15,27,28Although such routes are effective in terms of sequence control, they rarely achieve a simultaneous control over stereochemistry, and most often have a limited tolerance towards the more reactive functional side groups typically found in natural polymers, responsible for catalytic activity for instance. Since one of the possible fields of application of discrete sequence-defined polymers is precisely multifunctional or cascade catalysis, as we demonstrated recently by showing that sequence order controls the catalytic activity of trifunctional catalytic oligomers,29,30 it becomes critical to develop more tolerant routes leading to chains of controlled sequence. Additionally, controlled stereochemistry, as typically present in enzymes, is also a very desirable feature for catalysis with the aim to further control the tridimensional arrangement of cooperative catalytic sites.
Here, we concentrate on the development of a synthetic route to reach this goal, and propose an innovative strategy leading to precision oligomers of controlled sequence and stereochemistry capable to incorporate a wide range of functional groups – such as those found in proteins – based on the copper-catalyzed azide–alkyne cycloaddition (CuAAC) “click” reaction.31 The CuAAC reaction, due to its high efficiency and chemoselectivity, was previously demonstrated to be a promising tool for the synthesis of sequence-defined polymers.12,23,25,27,32–35 This archetypal “click” reaction was for instance successfully used to obtain sequence-defined and stereocontrolled polymers, yet mainly limited to side groups of lesser reactivity.15,27,28,33 Building onto these previous works, we demonstrate a new strategy leading to discrete stereocontrolled and sequence-defined oligomers capable of accommodating a wide range of functional side groups alike natural peptides, albeit at a lower chain density.
Experimental
Experimental and simulation details are given in the ESI.†Results and discussion
The main building blocks were prepared starting from TBDMS-protected glycidyl propargyl ether 1 either in its racemic (rac-1) or enantiopure ((R)-1 or (S)-1) forms (TBDMS = tert-butyldimethylsilyl) (Scheme 1a).27 To prove the validity of our concept, the strategy was first developed starting from racemic rac-1. Side groups were inserted following opening of the oxirane ring of rac-1 with sodium azide and subsequent CuAAC reaction of 2 with alkynes 3a–d to provide functional monomers 4a–d with overall yields varying between 87% and quantitative. Noteworthy, alkynes 3a–d bearing respectively phenyl, pyridyl, methoxy and t-butyl groups were carefully selected to provide unique, unambiguous 1H NMR footprints in order to trace the composition of the oligomers.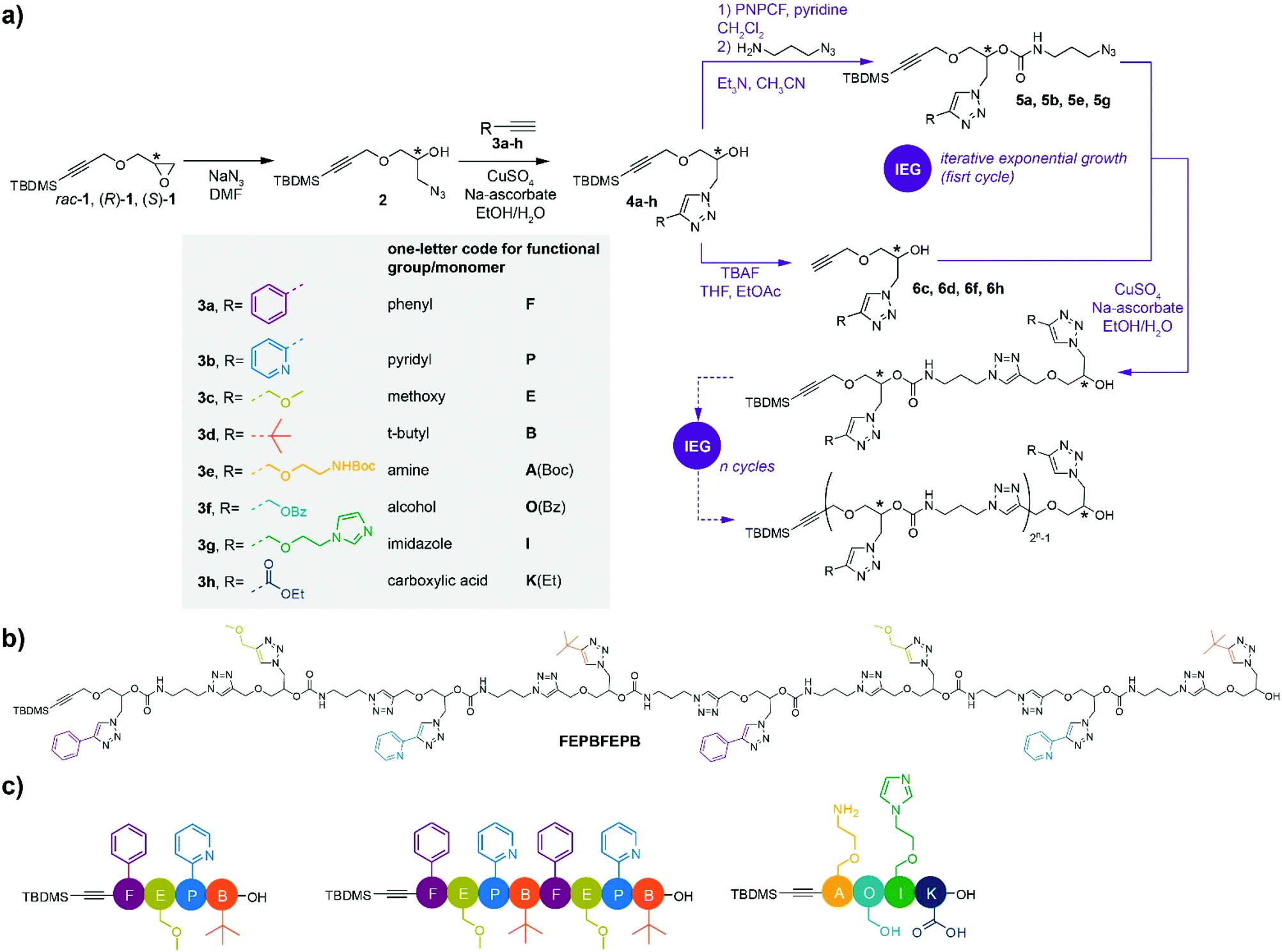 | ||
| Scheme 1 . Iterative exponential growth (IEG) strategy to generate discrete multifunctional stereocontrolled sequence-defined oligomers. (a) Synthetic scheme and one-letter code. (b) Structure of octamer FEPBFEPB obtained after 3 IEG cycles. (c) Simplified illustration of the obtained oligomers FEPB, FEPBFEPB and AOIK. | ||
From this, key to our strategy is the efficient re-introduction of the azide group and release of the free alkyne to enable chain elongation through CuAAC reaction. In this purpose, the secondary alcohol of building blocks 4a and 4b was first treated with p-nitrophenyl chloroformate (PNPCF) to yield the activated carbonate intermediate that was directly reacted with 3-azidopropylamine. This one pot procedure provides nearly quantitative yields of the monomers 5a and 5b bearing a stable carbamate bond within the main chain (92 and 87% yield, respectively).
Compared to previous work based on similar chemistry,15,27,28 our approach essentially differs by exchanging the functionalities used for installing the side chain groups and extending the chain backbone, which is a minor modification; and, much more crucially, by replacing ester groups by the considerably more stable carbamate groups, which is required for the installation of a much wider range of functional groups on the chains than could be done before. Indeed, we have recently shown that ester groups are not suitable in such a scheme, being not stable enough under hydrolytic and TBAF (tetrabutylammonium fluoride) conditions.29,36
Meanwhile, removal of the TBDMS group in 4c and 4d using TBAF provided free alkynes 6c and 6d (71 and 90% yield, respectively). Coupling of the obtained azides and alkynes yielded the dimeric species that allowed pursuing an IEG strategy towards higher oligomers. For clarity, a simple one-letter code was adopted for the naming of the different functional side groups/monomers that is inspired from the nomenclature used for natural amino acids (Scheme 1a).37 For instance, reaction of 5a with 6c under CuAAC conditions gives dimer FE (93% yield) bearing a phenyl (F) and a methoxy (E) side group, while reaction of 5b with 6d bearing a pyridyl (P) and tert-butyl (B) side group, respectively, afforded dimer PB (80% yield). A subsequent IEG cycle provided tetramer FEPB (72% yield); the later allowing access to octamer FEPBFEPB (Scheme 1b) in 57% yield through another IEG cycle. The overall yield of the octamer, starting from the protected monomers 4a–d, was 27%, providing access to milligrams of chains in our conditions. Of note, we did not face solubility issues for our oligomers despite their high content in triazole groups, possibly due to the functional similarity of such groups with amides.38
The stacked 1H NMR spectra of monomers 4a–d show that the signals corresponding to the pendant phenyl, pyridine, methoxy and tert-butyl protons do not overlap, as anticipated (Fig. 1a–d). Additionally, the triazole proton experiences different chemical environments in monomers 4a–d, which translates into different chemical shifts; this provides an additional way to identify the monomer units after their insertion in the oligomers. Precisely, the1H NMR spectra of tetramer FEPB and octamer FEPBFEPB evidence the presence of the expected characteristic peaks of each monomeric unit together with the additional resonances corresponding to the parent azidopropylamine moiety in the main chain (Fig. 1e and f).
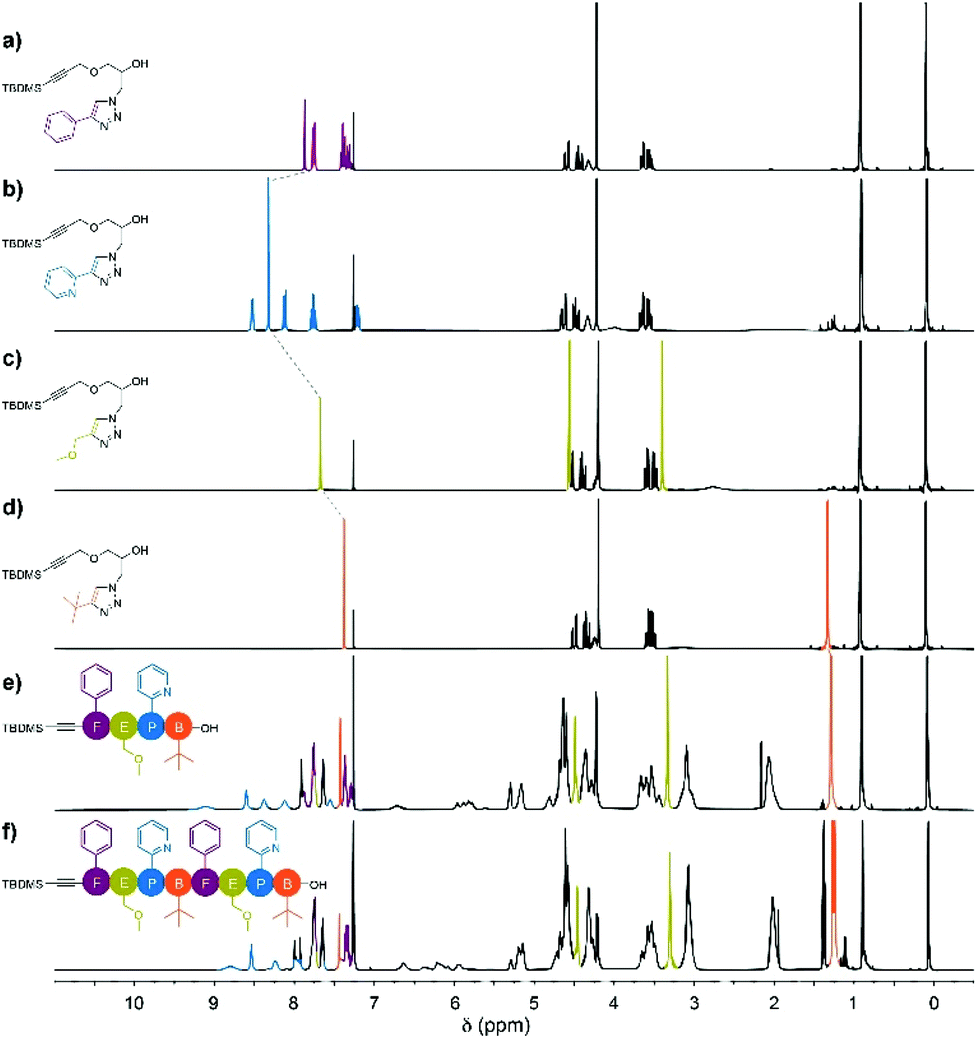 | ||
| Fig. 1 Stacked 1H NMR (300 MHz, CDCl3) of (a) 4a, (b) 4b, (c) 4c and (d) 4d; the dashed line connects resonances of the triazole proton within the different monomers. Stacked 1H NMR (500 MHz, CDCl3) of (e) tetramer FEPB and (f) octamer FEPBFEPB. | ||
In addition to 1H NMR analysis, electrospray ionization mass spectrometry (ESI-MS) confirmed the structure of the monomers and oligomers (ESI†). For instance, ESI-MS of the octamer FEPBFEPB displayed peaks at m/z = 2952.5932, 1476.8147, 984.8832, 738.9161 and 591.3353 corresponding to the theoretical values for [M + H]+, [M + 2H]2+, [M + 3H]3+, [M + 4H]4+ and [M + 5H]5+, respectively (Fig. S128†) The molecular sequence of the oligomers was deciphered using tandem mass spectrometry (ESI-MS/MS).
Cleavage of the ether bond in the main chain, yielding to two series of complementary fragments, was detected as the most prominent fragmentation mechanism in tetramer FEPB (Fig. S134†) and octamer FEPBFEPB (Fig. 2). According to this mechanism, the sequence of FEPBFEPB reading from left to right were recorded with m/z = 536.32, 887.50, 1271.70, 1634.91, 2018.06, 2369.11 and 2753.04 corresponding respectively to the sequence F, FE, FEP, FEPB, FEPBF, FEPBFE and FEPBFEP. Additionally, reading the sequence from the right side displayed a peak at m/z = 182.14 corresponding to monomer B. Thus, the sequence in the octamer FEPBFEPB was unraveled by ESI-MS/MS, which was confirmed from the right to left side as well. Overall, the fragmentation patterns unambiguously confirm that all the synthesized oligomers have the desired sequence.
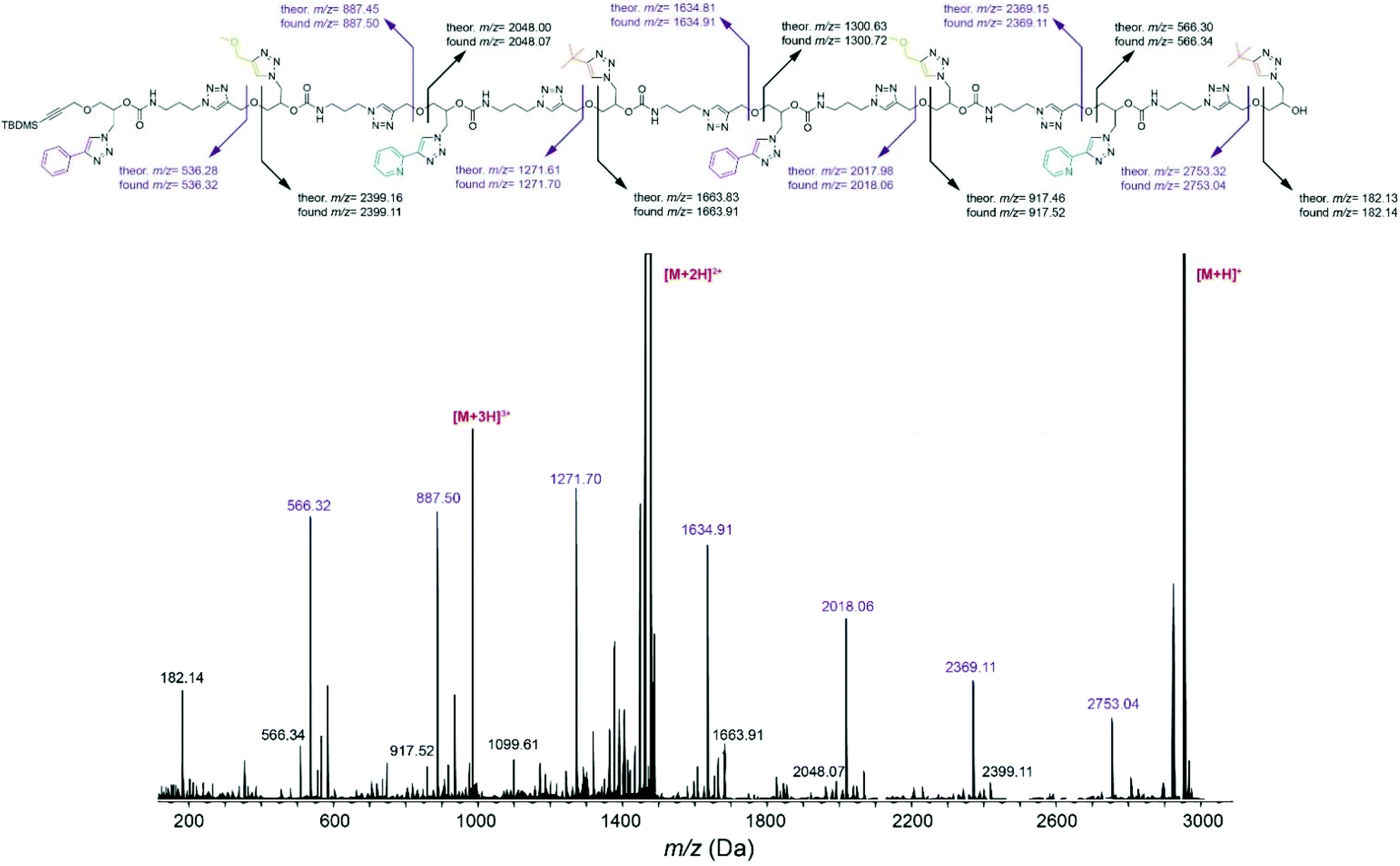 | ||
| Fig. 2 ESI-MS/MS sequencing of octamer FEPBFEPB obtained after collisional activation of the [M + H]+ precursor at m/z = 2952 Da. | ||
In order to further illustrate the versatility of our strategy, oligomers comprising a wider variety of functional side groups were synthesized. Thus, inspired by functional side groups found in natural amino acids, monomers comprising pendant amine (A), hydroxyl (O), imidazole (I) and carboxylic acid (K) side groups were synthesized (Scheme 1). Owing to the very chemistry involved in our approach, it was necessary to protect the nucleophilic side groups in order not to interfere with the elongation steps. Hence, A, O and K were protected with tert-butoxycarbonyl (Boc), benzoyl (Bz) and ethyl (Et) groups, respectively. Following the IEG procedure described above, tetramer A(Boc)O(Bz)IK(Et) was obtained in 35% overall yield after two IEG cycles. Final treatment of A(Boc)O(Bz)IK(Et) with trifluoroacetic acid and KOH provided the targeted AOIK tetramer in quantitative yield without hydrolysis of the main chain.
The structure of AOIK and its protected precursor A(Boc)O(Bz)IK(Et) was confirmed by 1H NMR (Fig. S99–S101†) and ESI-MS (Fig. S129–S130†). As previously, the sequence information was confirmed by tandem mass spectrometry (Fig. S139† for AOIK, and S138 for its protected version). Interestingly, the negative mode ESI-MS/MS was successfully developed to trace the sequence from the right to the left side of AOIK owing to the contribution of carboxylic acid group in the side chain (Fig. S140†). Especially, preservation of the integrity of the oligomers (composition and sequence) after complete cleavage of the protecting groups was confirmed, demonstrating the tolerance of our synthetic strategy to a wide variety of chemistries and functional groups.
Although synthetic methods are flourishing to access sequence-defined oligomers, a very limited number of synthetic strategies have been reported to control both sequence and chirality,15,27,28,39 a situation that is ever more challenging when multiple functional groups are involved within the chain. Therefore, a set of enantiopure monomers bearing F, E, P and B side groups was synthesized starting from chiral TBDMS-protected glycidyl propargyl ether (R)-1 or (S)-127 (Scheme 1a). The enantiomeric excess (ee) of the monomers was traced by chiral HPLC, in which 4a was carried out as the example (Fig. S141†). The ee of (R)-4a and (S)-4a were 90 and 88%, respectively, showing that the chirality was well controlled under the synthetic processes. Stereocontrolled sequence-defined oligomers (R,R,R,R)-FEPB, (S,S,S,S)-FEPB and (R,R,R,R,S,S,S,S)-FEPBFEPB were straightforwardly prepared employing the IEG strategy developed for rac-1 (Scheme 1a). As expected, their 1H NMR, ESI-MS and ESI-MS/MS spectra were identical to the ones of the racemic oligomers (ESI†). The maximum content of the (R,R,R,R,S,S,S,S)-FEPBFEPB octamer in oligomers of lower length was estimated from the ratio of 1H NMR peaks between 0.86 and 0.96 ppm (t-butyl silyl-) and 5.00–5.34 ppm (protons of the asymmetric carbons of all monomer units except the terminal one); it was found to be at most 5% by number, and very likely much lower. HPLC confirmed the very high purity of the stereocontrolled tetramers and octamer (Fig. S149–S151†).
Circular dichroism (CD) spectroscopy provided qualitative information on the stereochemistry of the chains. The CD spectra of monomers 4a–d and dimers FE and PB (Fig. S142†) with (R)- and (S)-configurations show opposite signals as expected for enantiomers. Likewise, the CD spectra of dilute solutions of the tetramers (R,R,R,R)-FEPB and (S,S,S,S)-FEPB show opposite signals peaking at 212 nm (Fig. 3a and Fig. S143†), confirming that the two tetramers have mirror configurations. In contrast, octamer (R,R,R,R,S,S,S,S)-FEPBFEPB exhibits a significantly weaker CD signal (Fig. 3a) because it contains identical amounts of tetrameric (S) and (R) sequences; the non-suppression of the CD signal results from the fact that the octamer nevertheless lacks centers and planes of symmetry. Overall, the CD results thus clearly indicate that the stereochemistry is utterly controlled, and racemization does not occur during the IEG process. Of note, since the enantiomeric excess of our starting monomer units is currently ca. 90% only, our oligomers are still mixtures of a series of stereochemical configurations, which does not prevent them from displaying a strong circular dichroism. An even narrower distribution of stereochemical configuration could be obtained by starting from a more enantiopure form of 2, as discussed elsewhere.14
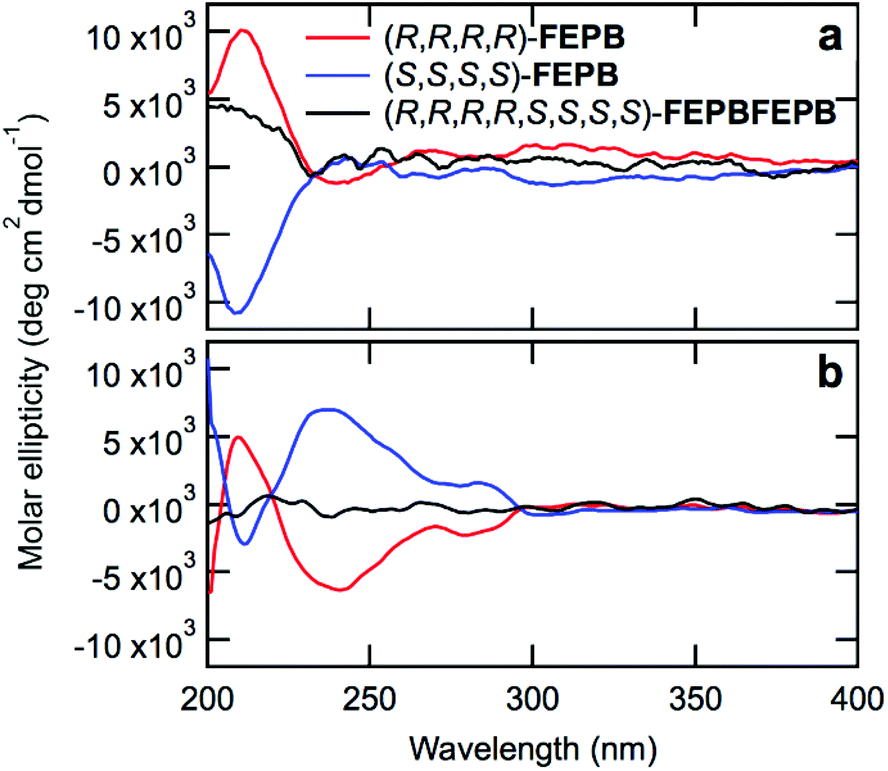 | ||
| Fig. 3 Circular dichroism spectra of (R,R,R,R)-FEPB (red), (S,S,S,S)-FEPB (blue) and (R,R,R,R,S,S,S,S)-FEPBFEPB (black) in CH3CN. Concentrations are (a) 30 µM and 15 µM for the tetramers and octamer, respectively; (b) 150 µM and 75 µM for the tetramers and octamer, respectively. The spectra were smoothed (Savitzky–Golay, 13-point, second order polynomial). | ||
Interestingly, at a five times higher concentration (Fig. 3b and Fig. S144†), the CD spectra of (R,R,R,R)- and (S,S,S,S)-FEPB display additional mirror peaks at 238 and 283 nm, which is likely due to aggregation of the chains and suggests that the aggregated states of the oligomers remain chiral. As before, the (R,R,R,R,S,S,S,S)-FEPBFEPB octamer only exhibits a weak signal. To further understand the aggregation behavior of the oligomers, all-atom Molecular Dynamics (MD) simulations were carried out from the single chain level to assemblies of a few chains (see ESI† for computational details). MD simulations of a single chain provided structural and dynamical aspects of the octamer (R,R,R,R,S,S,S,S)-FEPBFEPB.
The octamer rapidly folds into a compact globular structure (Fig. 4a), characterized by a low radius of gyration (Rg) of 7.4 Å in average, very low compared to the end-to-end distance of the fully elongated conformation (around 100 Å, see ESI S145†). The stability of Rg profile over time (ESI S146†) indicates that the globular folding is preserved on the microsecond timescale. Although a total of 93 hydrogen bonds were detected for all conformations of the MD, most of them are not persistent: 88 H-bonds are present for less than 10% of the MD time, and none was detected for more than 50% of the MD time (see ESI S147†). This large number of possible H-bonds arises from each repeat unit containing one carbamate group, plus two triazole groups which are good mimetics of secondary amide groups with respect to H-bonding donor/acceptor capabilities.38,40 The network of H-bonds is constantly evolving and important oscillations in the number of H-bonds are observed throughout MD time (Fig. 4b), varying from 0 to 7 H-bonds with an average of only 2–3 H-bonds per conformation. Therefore, H-bonds are only weakly participating to the compact globular folding of the octamer. In contrast, the numerous aromatic moieties located on the side chains and along the backbone, labeled on top of Fig. 4, provide many possibilities of π-stacking interactions. The distances between the centers-of-mass (COM) of all depicted aromatic pairs were estimated. As for H-bonds, the network of aromatic interactions is dynamic and a criterion (at least 50% of MD time at a distance below 5.0 Å) was considered to point out the most persistent aromatic interactions. Some of these most persistent stacking interactions are depicted on the final MD snapshot in Fig. 4c and d. With this criterion, six π-stacking interactions are persistent: one is involving two triazole moieties of the backbone (labelled as 13 and 15 in Fig. 4), two interactions concern backbone/side-chain aromatic moieties (5a–12 and 5b–14 pairs) and three interactions are occurring between side-chains (5a–8, 3a–6, and 3b–6). These persistent π-stacking interactions between aromatic groups far in the octamer sequence but close in space evidently contribute to stabilize the compact globular conformations of the oligomer.
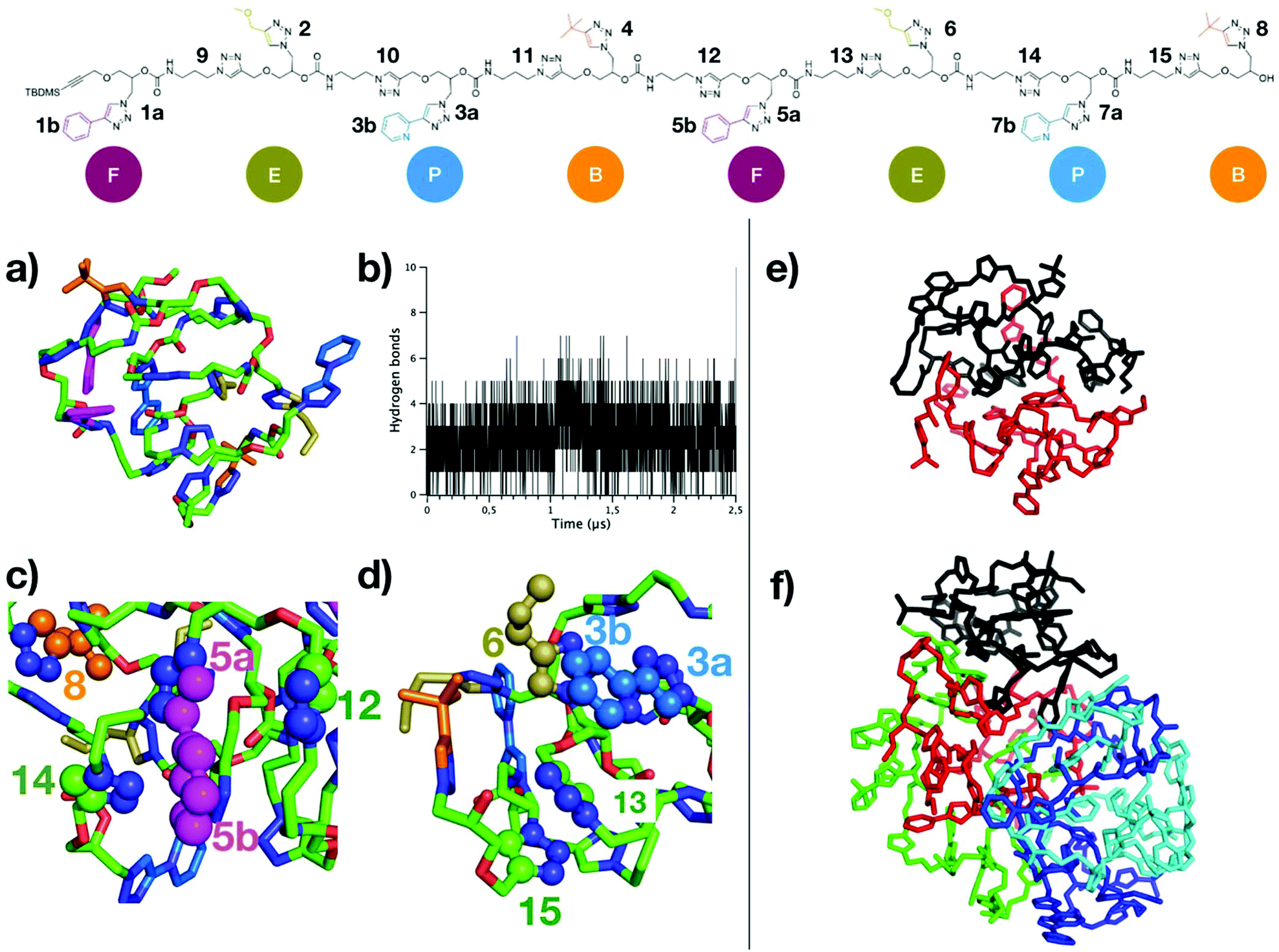 | ||
| Fig. 4 (a) Final MD snapshot representative of the folded conformation of (R,R,R,R,S,S,S,S)-FEPBFEPB octamer. (b) Number of hydrogen bonds along MD time. (c) Proximity of specific aromatic moieties (in spheres) that are involved in some of the most persistent π-stacking interactions (numbering refer to the labels shown on the top chemical structure). Aromatic groups labelled as 5a interact with 12, and 5b interacts with 14. (d) Both 3a and 3b interact with 6, and 13 interacts with 15. (e–f) Final MD snapshots of assemblies of 2 chains and 5 chains are depicted in (e) and (f), respectively. Each octamer is shown in a one color and only heavy atoms are represented for the sake of clarity. | ||
MD simulations involving either two chains or five chains were performed to investigate the driving force and dynamics for the aggregation of the octamer. For both MD simulations, octamer chains rapidly form a supramolecular complex that remains stable on the microsecond time scale, i.e. no dissociation between the chains were observed. Interestingly, self-assemblies of 2 chains or 5 chains do not show any intertwining between the chains (see MD snapshots in Fig. 4e and f). In comparison to the MD simulations of a single chain, only a minor increase of the average Rg is observed in the assembly of two chains (ESI S146†). The compactness of both chains in the assembly is similar as the average Rg of both chains converge to identical values. In the assembly of 5 chains, compact conformations are again observed, but with a slightly higher average Rg (around 10 Å). Concerning the dynamics of the octamer, the flexibility profile along the chain of the octamer (as estimated with Root-Mean-Square Fluctuations estimates, see ESI S148†) differs for the single chain and for the assemblies. Importantly, the flexibility of a single octamer chain can be discriminated inside the aggregate. For example, the octamer #1 corresponding to the chain colored in black in Fig. 4f is more rigid than the other octamers, because of its more compact folding (lower average Rg). Altogether, our results show that (R,R,R,R,S,S,S,S)-FEPBFEPB octamers fold into very compact structures, mainly stabilized by persistent π-stacking interactions, whereas hydrogen bonds are not persistent. Given the numerous possibilities of stacking interactions, stable assemblies of several chains are observed in which structural aspects and the dynamics of octamers can be influenced by the number of chains within the assembly. Altogether, this indicates that the oligomers studied in this work are prone to compact folding and assembly into quaternary structures because of the numerous π-stacking interactions.
Conclusions
In conclusion, we report on a straightforward and efficient approach for the synthesis of discrete multifunctional stereocontrolled sequence-defined oligomers. The method provides rapid access to optically-active poly(triazole-urethane) oligomers bearing triazole-linked side chains, with a broad range of possible functional side groups reminiscent of those found in proteins, albeit at a smaller linear density. Another difference is that the folding and aggregation of oligomers is most likely driven by π-stacking interactions, as revealed by MD simulations, whereas the numerous possible H-bonding interactions play a much more minor role. These oligomers offer attractive perspectives for multifunctional catalysis and self-assembly, applications for which octameric sequences are more than long enough as we demonstrated previously.29Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Fonds de la Recherche Scientifique - FNRS and the Fonds Wetenschappelijk Onderzoek under EOS project no. 30650939 for financial support. A.E.F. acknowledges the European Regional Development Fund (ERDF) and Wallonia Operational Program “Wallonia-2020.EU”. M.L. and M.S. acknowledge the Wallonia Region and FNRS under grant no. F.4532.16. The authors acknowledge Gabriella Barrozino-Consiglio for 500 MHz NMR analysis, Raoul Rozenberg for APCI-MS, Hervé Degand for ESI-MS/MS analysis and Laurent Collard for HPLC analysis. Computational resources have been provided by the Consortium des équipements de Calcul Intensif (CéCI), funded by the FNRS under grant 2.5020.11. KG and MS are Senior Research Associates of the F. R. S.–FNRS.References
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 1–14 CrossRef.
- G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed.
- L. E. Rosebrugh, V. M. Marx, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10032–10035 CrossRef CAS PubMed.
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef CAS PubMed.
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- N. Badi and J. F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- E. Maron, J. H. Swisher, J. Haven, T. Y. Meyer, T. Junkers and H. G. Börner, Angew. Chem., Int. Ed., 2019, 58, 10747–10751 CrossRef CAS PubMed.
- W. Konrad, C. Fengler, S. Putwa and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 7133–7137 CrossRef CAS PubMed.
- R. Dong, R. Liu, P. R. J. Gaffney, M. Schaepertoens, P. Marchetti, C. M. Williams, R. Chen and A. G. Livingston, Nat. Chem., 2019, 11, 136–145 CrossRef CAS PubMed.
- S. Celasun, D. Remmler, T. Schwaar, M. G. Weller, F. Du Prez and H. G. Borner, Angew. Chem., Int. Ed., 2019, 58, 1960–1964 CrossRef CAS PubMed.
- C. Yang, J. P. Flynn and J. Niu, Angew. Chem., Int. Ed., 2018, 57, 16194–16199 CrossRef CAS PubMed.
- S. Martens, A. Landuyt, P. Espeel, B. Devreese, P. Dawyndt and F. Du Prez, Nat. Commun., 2018, 9, 4451 CrossRef PubMed.
- Z. Huang, B. B. Noble, N. Corrigan, Y. Chu, K. Satoh, D. S. Thomas, C. J. Hawker, G. Moad, M. Kamigaito, M. L. Coote, C. Boyer and J. Xu, J. Am. Chem. Soc., 2018, 140, 13392–13406 CrossRef CAS PubMed.
- M. R. Golder, Y. Jiang, P. E. Teichen, H. V. Nguyen, W. Wang, N. Milos, S. A. Freedman, A. P. Willard and J. A. Johnson, J. Am. Chem. Soc., 2018, 140, 1596–1599 CrossRef CAS PubMed.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376–8383 CrossRef CAS PubMed.
- M. Porel, D. N. Thornlow, N. N. Phan and C. A. Alabi, Nat. Chem., 2016, 8, 590–596 CrossRef CAS PubMed.
- S. Wang, Y. Tao, J. Wang, Y. Tao and X. Wang, Chem. Sci., 2019, 10, 1531–1538 RSC.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- O. I. Paynter, D. J. Simmonds and M. C. Whiting, J. Chem. Soc., Chem. Commun., 1982, 20, 1165–1166 RSC.
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10617–10622 CrossRef CAS PubMed.
- F. Amir, Z. Jia and M. J. Monteiro, J. Am. Chem. Soc., 2016, 138, 16600–16603 CrossRef CAS PubMed.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef PubMed.
- J. Niu, R. Hili and D. R. Liu, Nat. Chem., 2013, 5, 282–292 CrossRef CAS PubMed.
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS PubMed.
- J. C. Barnes, D. J. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- Y. Jiang, M. R. Golder, H. V. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369–9372 CrossRef CAS PubMed.
- P. Chandra, A. M. Jonas and A. E. Fernandes, J. Am. Chem. Soc., 2018, 140, 5179–5184 CrossRef CAS PubMed.
- P. Chandra, A. M. Jonas and A. E. Fernandes, ACS Catal., 2018, 8, 6006–6011 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- M. Ciaccia, D. Nunez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2019, 141, 10862–10875 CrossRef CAS PubMed.
- N. F. Konig, A. Al Ouahabi, S. Poyer, L. Charles and J. F. Lutz, Angew. Chem., Int. Ed., 2017, 56, 7297–7301 CrossRef PubMed.
- D. Núñez-Villanueva, M. Ciaccia, G. Iadevaia, E. Sanna and C. A. Hunter, Chem. Sci., 2019, 10, 5258–5266 RSC.
- R. Szweda, C. Chendo, L. Charles, P. N. W. Baxter and J.-F. Lutz, Chem. Commun., 2017, 53, 8312–8315 RSC.
- P. Chandra, A. M. Jonas and A. E. Fernandes, RSC Adv., 2019, 9, 14194–14197 RSC.
- M. Saffran, Biochem. Educ., 1998, 26, 116–118 CrossRef CAS.
- Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674–1689 RSC.
- N. Franz, L. Menin and H.-A. Klok, Eur. J. Org. Chem., 2009, 2009, 5390–5405 CrossRef.
- M. Corredor, J. Solà and I. Alfonso, in Targets in Heterocyclic Systems, 2017, vol. 21, ch. 1, pp. 1–22 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00537a |
| This journal is © The Royal Society of Chemistry 2020 |