
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-catalytic depolymerization of polyethylene terephthalate (PET)†
Kayla R.
Delle Chiaie
 ,
Fergus R.
McMahon
,
Esme J.
Williams
,
Matthew J.
Price
and
Andrew P.
Dove
*
,
Fergus R.
McMahon
,
Esme J.
Williams
,
Matthew J.
Price
and
Andrew P.
Dove
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: A.Dove@bham.ac.uk
First published on 17th February 2020
Abstract
Limiting our plastic waste and finding greener, more sustainable solutions for disposal is currently an environmental priority. Polyethylene terephthalate (PET), one of the more prominent single-use plastics, has recently been under investigation for chemical recycling as a means to ameliorate the environmental impact. This work reports a dual-catalytic approach to the chemical recycling of PET, aiming to combine inexpensive, readily available Lewis acid–base pairs to exhibit cooperative catalytic activity.
Introduction
The world's production of synthetic plastics is continuously and exponentially increasing as these materials have become ubiquitous in everyday life.1,2 This is a consequence of these materials being low cost and functional. Unfortunately, many plastics are single-use and thus waste from these materials has been building up in the environment to create a global problem. One such polymer, polyethylene terephthalate (PET) is a major part of the world's synthetic plastic production (33 million tons in 2015).3–5 Unfortunately, the combination of the slow natural degradation of PET in the environment and poor disposal management has contributed to the global issue of plastic pollution. As PET production continues to increase, recycling strategies must also adapt.6–11 While mechanical recycling of PET is well exploited, ultimately it leads to a deterioration in mechanical properties as a consequence of thermal degradation, contamination, and discoloration. In turn, the recycled material reduces in quality and does not perform as well as virgin PET.Recently, chemical recycling of PET has been investigated as an innovative solution to the treatment of post-consumer PET.12–15 Chemical recycling involves depolymerisation of the end-use plastic down to its constituent monomers that are then suitable for repolymerization (with mechanical properties comparable to the virgin material), resulting in a closed-loop cycle for the material.16–20 Specifically, glycolysis of PET leading to bis(2-hydroxyethylene)terephthalate (BHET) has been shown to be suitable for depolymerization and repolymerization (Fig. 1).13,21–25
 | ||
| Fig. 1 Glycolysis of PET to form BHET. | ||
Chemical depolymerization of PET has been studied with a variety of both metal-based and organic catalysts.4,13,14,20,25 Previously, we have shown that the depolymerization process benefits from catalyst systems composed of ionic acid–base pairs that outperformed base catalysis alone.25,26 As such, this has led us to focus subsequent studies on the effect of combining organic bases with Lewis acidic metal complexes. The benefit of using a dual-catalytic system with a metal-based Lewis acid component is that many different variables can be tuned including Lewis acidity of the metal, size of the metal, and metal ligands/counter anions.20,27
Results and discussion
Previous dual-catalytic depolymerization of PET with organic salts required formation of an ionic salt precursor prior to introduction into depolymerization conditions.25 Early investigation of PET glycolysis using 4-dimethylaminopyridine (DMAP), a popular catalyst for transesterification reactions in ester-based polymers, with readily available MgCl2 and a pre-formed DMAP–MgCl2 complex showed comparable performance (see ESI Fig. S1†). Most notably, even in these initial studies, the depolymerizations were rapid and efficient.28 In turn, all subsequent depolymerisations were run using in situ addition of both catalyst components. A simple screening of metal halides while retaining DMAP as base showed that metal identity has a dramatic effect on depolymerization activity (Table 1) with many metal halides affording little or no conversion (Table 1, entries 7–13). This could be due to a myriad of factors not limited to solubility, metal size, and coordination. These results are in line with similar trends seen for the dual-catalytic ROP of lactones.29 The complexation with DMAP is likely to have a dramatic effect on the activation of the polyester backbone and subsequent nucleophilic attack needed for depolymerization. If the Lewis acid–base interaction is too strong there may not be enough “free” acid and base to enable depolymerization. Similarly, if the interaction is too weak then the catalyst components will not be stabilized and decompose under depolymerization conditions.| Entry | Lewis acid | Conversion after 3 hours |
|---|---|---|
| a Each depolymerization was run with 0.15 equivalents of catalyst and 20 equivalents of ethylene glycol at 180 °C. | ||
| 1 | None | 0.55 |
| 2 | MgCl2 | 0.62 |
| 3 | MgBr2 | 0.60 |
| 4 | MgI2 | 0.71 |
| 5 | ZnBr2 | 0.69 |
| 6 | ZnI2 | 0.54 |
| 7 | BiCl3 | 0.13 |
| 8 | YCl3 | 0.08 |
| 9 | AlCl3 | — |
| 10 | FeCl3 | — |
| 11 | SnCl4 | — |
| 12 | TiCl4 | — |
| 13 | Bi(OTf)3 | — |
| 14 | Zn(OTf)2 | 0.41 |
| 15 | Zn(OAc)2 | 0.89 |
| 16 | Mg(OTf)2 | 0.57 |
| 17 | Mg(OAc)2 | 0.68 |
Conversely, comparing different halogen anions had a much smaller effect. Increasing the size of the halide from MgCl2 to MgI2 showed less than a 10% enhancement. Similar results were obtained with ZnI2 and ZnBr2. A more significant difference was observed when the anions are changed to oxygen-based moieties such as triflate (OTf) or acetate (OAc). Since magnesium is a hard metal it is expected to bind more strongly with oxygen-based ligands leading to a slight decrease in activity (Table 1, entries 16 and 17). Alternatively, zinc as a soft metal appears to have a slight increase in activity when moving from halide ligands to oxygen-based ligands. Notably, for both metals, triflate complexes show lower activity than the comparable acetate complexes. More in depth kinetic studies revealed that the triflates and halides appear to have an induction period early in the reaction (see ESI Fig. S2†). This led us to hypothesize that in some cases the metal ligands may be replaced by carboxylates that form the chain end or transesterification location in the polymer. It would stand to reason that this exchange or formation of the active species would be more facile with metal acetate complexes. Since Zn(OAc)2 showed the most promising reactivity and synergistic effect (see Fig. 2) with DMAP, it was screened with various base co-catalysts.
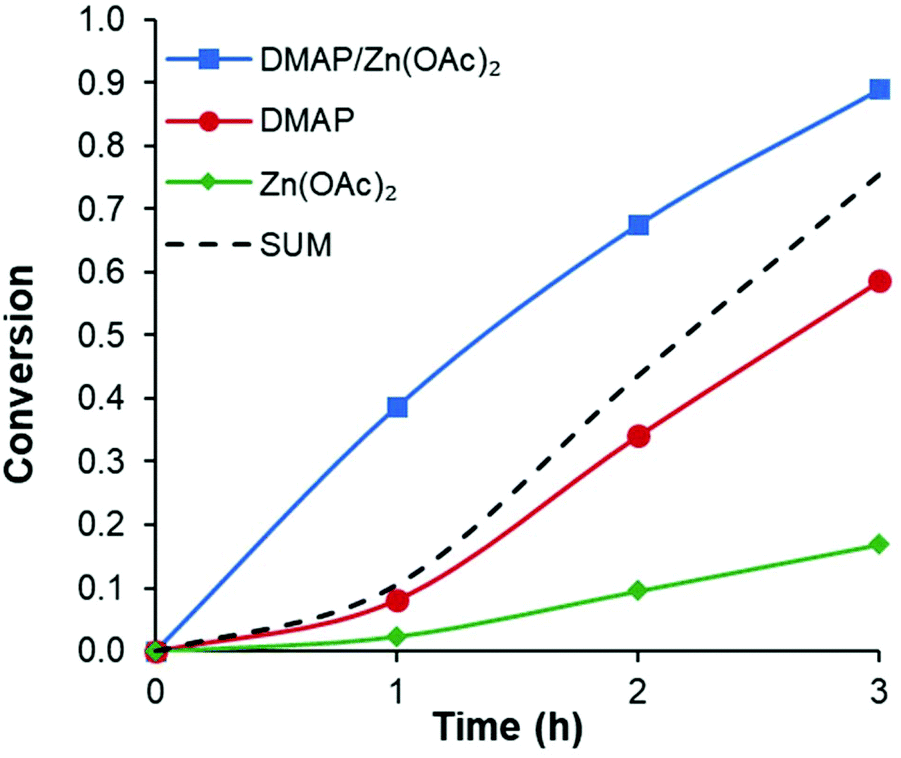 | ||
| Fig. 2 Example of synergistic effect with the combination of DMAP and Zn(OAc)2. Sum of both catalysts alone shown for reference. | ||
Combinations of Zn(OAc)2 with varying bases showed a wide range of reactivity from 55% to 96% conversion at 3 h (Fig. 3). As a consequence of the complex relationship between the basicity and complexation of these bases there is not a clear trend. It appears that bases with pKa's ranging from 9–12 are the most active. As the bases get stronger a decrease in reactivity is observed (for example TBD and BEMP). Alternatively, when the base is too weak a marked decrease in activity is observed (for example PPh3, Fig. 3). Additionally, complexation in an equimolar relationship to the Lewis acid appears to be important. This can be seen when DABCO (two basic sites) is compared to ABCO (one basic site). A similar phenomenon has been shown to affect the temperature stability of monobasic and dibasic ionic salts. There is a clear “Goldilocks Effect” for acid–base coordination and activity.26 Additionally, a synergistic effect is seen with all of the bases weaker than TBD and BEMP (see ESI Fig. S4–S15†). It is hypothesized that these two strong and stable bases do not benefit from the addition of the Lewis acid due to their already remarkable reactivity. Additionally, no synergistic effect is seen with the more nucleophilic bases (i.e. DABCO and ABCO); it is possible that these bases are operating via an alternative mechanism.
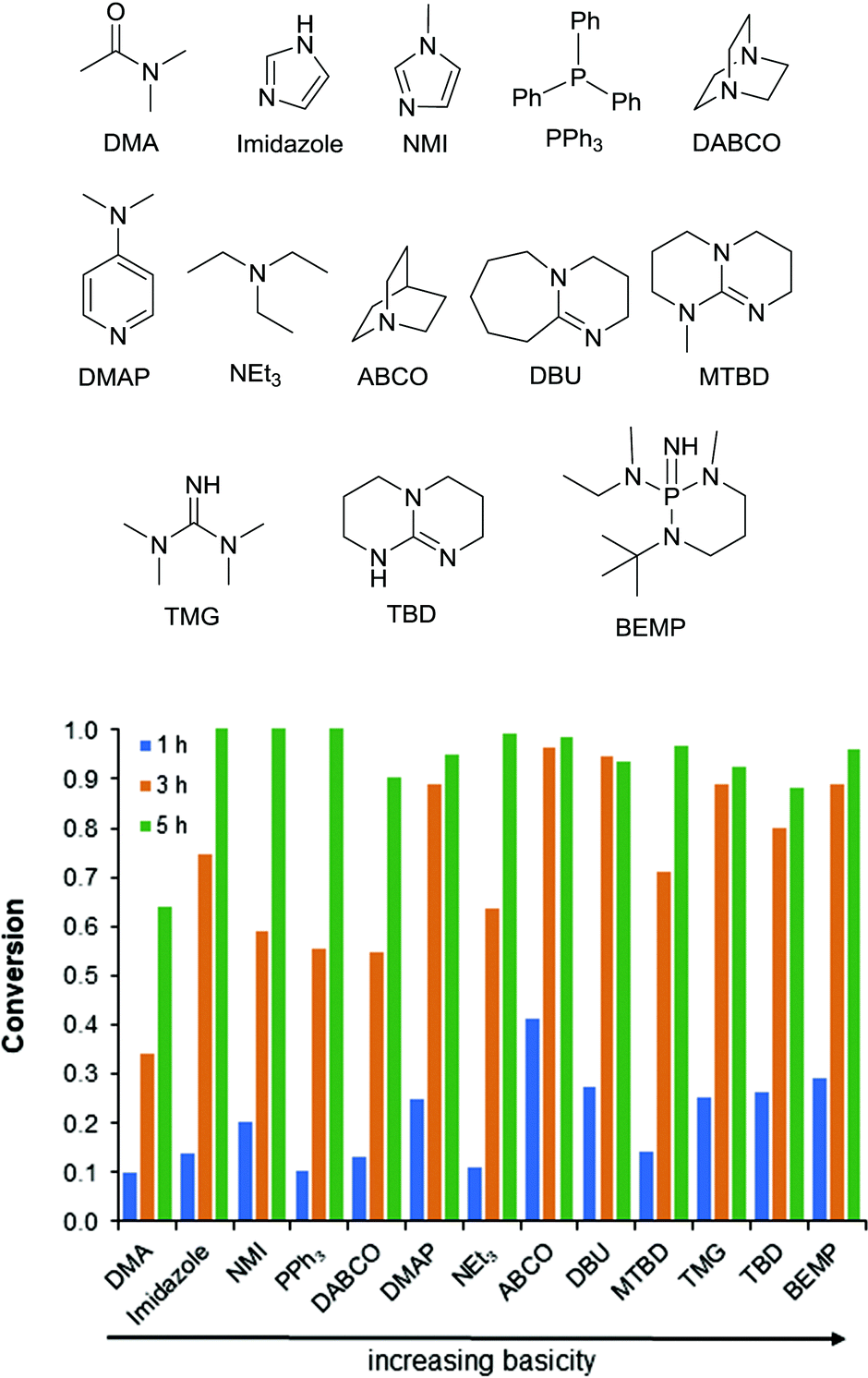 | ||
| Fig. 3 Activity of Zn(OAc)2 with various bases for the depolymerization of PET. Bases are ordered left-to-right by basicity. | ||
Currently, attempts to isolate and reuse the catalyst for subsequent depolymerizations have shown some promise (see ESI Fig. S3†). The main limitation appears to be decomposition of the catalyst components over these reaction times at high temperatures.
Conclusions
Overall, there is an apparent enhancement of depolymerization reactivity when a combination of Lewis acid and base are employed as the catalyst, similar to the organic salt systems. It is clear that tuning the strength of the acid–base interaction is the key to influencing the subsequent catalytic activity and stability. If the catalyst components bind too tightly this hinders the reactivity. However, if binding is too weak, there is no enhancement of activity or catalyst stability. Future studies will focus on fine tuning the acid–base interactions and finding a dual-catalytic system that can be recycled for many depolymerizations and ultimately lead to a closed-loop PET cycle.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The University of Birmingham is thanked for the award of a Global PhD Scholarship to M. J. P.References
- S. Matar, in Chemistry of Petrochemical Processes, Woburn, 2nd edn, 2001, pp. xi–xii Search PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3750 CrossRef CAS.
- W. T. S. Huck, Nature, 2011, 472, 425–426 CrossRef CAS.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, J. Waste Manage., 2009, 29, 2625–2643 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, Energy Convers. Manage., 2016, 115, 308–326 CrossRef CAS.
- C. Jehanno, M. M. Perez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- G. L. Colomines, J. J. Robin and G. Tersac, Polymer, 2005, 46, 3230–3247 CrossRef CAS.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610–1616 RSC.
- P. Hodge, Chem. Rev., 2014, 114, 2278–2312 CrossRef CAS PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559–586 CrossRef CAS.
- J. Y. Chen, C. F. Ou, Y. C. Hu and C. C. Lin, J. Appl. Polym. Sci., 1991, 42, 1501–1507 CrossRef CAS.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Muller, F. Ruiperez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1606–1611 CrossRef CAS.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- A. M. Al-Sabagh, F. Z. Yehia, A. M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A. R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- F. Ahmadnian, F. Velasquez and K. H. Reichert, Macromol. React. Eng., 2008, 2, 513–521 CrossRef CAS.
- C.-H. Chen, C.-Y. Chen, Y.-W. Lo, C.-F. Mao and W.-T. Liao, J. Appl. Polym. Sci., 2001, 80, 943–948 CrossRef CAS.
- J. W. Chen and L. W. Chen, J. Appl. Polym. Sci., 1999, 73, 35–40 CrossRef CAS.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3292–3300 CrossRef CAS.
- A. Basterretxea, C. Jehanno, D. Mecerreyes and H. Sardon, ACS Macro Lett., 2019, 8, 1055–1062 CrossRef CAS.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC.
- Excess ethylene glycol was used to enable the collection of experimental data in a reasonable time frame on the small reaction scale used in this study.
- S. Naumann, P. B. V. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01920k |
| This journal is © The Royal Society of Chemistry 2020 |