
Random alloy and intermetallic nanocatalysts in fuel cell reactions
Junming
Zhang†
,
Linfan
Shen†
,
Yanxia
Jiang
 * and
Shigang
Sun
*
* and
Shigang
Sun
*
State Key Laboratory of Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, People's Republic of China. E-mail: yxjiang@xmu.edu.cn; sgsun@xmu.edu.cn
First published on 31st August 2020
Abstract
Fuel cells that use small organic molecules or hydrogen as the anode fuel can power clean electric vehicles. From an experimental perspective, the possible fuel cells’ electrocatalytic reaction mechanisms are obtained through in situ electrochemical spectroscopy techniques and density functional theory calculations, providing theoretical guidance for further development of novel nanocatalysts. As advanced nanocatalysts for fuel cells’ electrochemical reactions, alloy nanomaterials have greatly improved electrocatalytic activity and stability and have attracted widespread attention. Enhanced electrocatalytic performance of alloy nanocatalysts could be closely related to the synergistic effects, such as electronic and strain effects. Depending on the arrangement of atoms, alloys can be classified into random alloy and intermetallic compounds (ordered structure). Intermetallic compounds generally have lower heats of formation and stronger heteroatomic bonding strength relative to the random alloy, resulting in high chemical and structural stability in either full pH solutions or electrochemical tests. Here, we summarize the latest advances and the structure–function relationship of noble metal alloy nanocatalysts, among which Pt-based catalysts are the main ones, as well as comprehensively understand why they significantly affect the electrocatalytic performance of fuel cells. Novel alloy nanocatalysts with a robust three-phase interface to achieve efficient charge and mass transfer can obtain desirable activity and stability in the electrochemical workstation tests, and is expected to acquire a higher power density on fuel cell test systems with harsh test conditions.
1. Introduction
Research on the electrocatalytic energy conversion processes has been increasing due to the reduction in energy reserves and concerns over sustainable technological developments such as fuel cells (FCs).1–3 In the past few decades FCs have made great progress, however their commercialization has been impeded by factors such as high cost, insufficient life expectancy, and low power density. Heterogeneous electrocatalysts play a crucial role in FCs as a result of improving the rate, efficiency and selectivity of the chemical transformations involved.4,5 The electrocatalytic performance of almost all nanocatalysts is heavily dependent on the binding strength between the reactive species and the active sites on the catalyst surface.6 Therefore, the rational design of heterogeneous electrocatalysts by controlling their structure–function relationships has received extensive attention. Among all the single metals used in FC systems, platinum is considered to be the most effective electrocatalyst; however, its high cost and sensitivity to CO poisoning hamper its practical application.7–9 At present, to incorporate other metals into Pt atoms is an effective strategy for adjusting the surface electronic effects and spin density distribution, increasing CO tolerance, and enhancing the electrocatalytic performance.10,11 Improved synthesis approaches for various alloy nanomaterials have been explored to realize the practical application, which has been demonstrated in some excellent reviews.12–14 Generally, as-synthesized Pt-based or Pd-based alloys have a face-centered cubic (fcc) structure, and two atoms randomly occupy the crystal lattice, whereas intermetallic compounds possess unique properties, including well-defined atomic ratios and long-range ordered atomic arrangement of the entire nanoparticle, in which the key for controlling ordered structures is usually through appropriate high-temperature treatment.15–19 Pt-based or Pd-based intermetallic compounds have specific crystalline phases, such as face-centered tetragonal (fct),20,21 simple cubic (sc),22,23 body-centered cubic (bcc),24,25 hexagonal close packed (hcp),26,27 and so on, which can be visually confirmed through powder X-ray diffraction (XRD) and cs-corrected transmission electron microscopy (TEM). Here, we emphasize the structure–function relationship of the latest noble metal alloy nanocatalysts and their high-efficiency applications in various fuel cell electrochemical reactions.With the development of synthetic methods, the study of the structure–function relationship can be conducted in more detail. Electrocatalytic reactions are quite sensitive to the surface structure of nanocatalysts, thereby metal nanomaterials with different sizes, shapes, and compositions can directly regulate the electrocatalytic activity and durability.28–30 For the particle size of catalysts, reducing the particle size results in a maximum surface-to-volume ratio, and more reactive sites appear on a given alloy. Meier et al. studied the relationship between particle size and electrochemical proton reduction performance by scanning tunneling microscopy combined with electrochemical tests.31 As the particle size of Pd nanoparticles decreases from 200 to 6 nm, the catalytic activity is enhanced by more than two orders of magnitude. Thus, an appropriate particle size is important for electrocatalytic reactions, and 2–5 nm Pt-based alloy nanocatalysts have better oxygen reduction reaction (ORR) activity.32 Smaller nanoparticles are more likely to form alloys due to the faster diffusion between heteroatoms. When the particle size of alloys is increased to a certain size, the solubility of heteroatoms decreases, accompanied by the appearance of surface segregation. Except for some surfactants and nanocarbon materials, mesoporous silica or zeolitic imidazolate frameworks also were used as a coating to control nanoparticles with a smaller size at higher annealing temperature, such as PtZn nanoparticles.33,34
Beyond that, metallic nanomaterials with specific crystal facets have also received extensive interest due to their enhanced electrocatalytic properties.35 Nanocrystals with specific crystal facets can provide some active sites similar to edges, corners, kinks, and so on.36–38 And the shape of nanocrystals strongly depends on thermodynamic and kinetic parameters. For example for fcc Pt, the surface energy order is γ(111) < γ(100) < γ(110) < γ(hkl), where (hkl) represents high-index faces with at least one Miller index greater than 1.39 From a thermodynamic point of view, crystal faces with high surface energy are unstable; the catalysts usually obtained by the simple liquid-phase synthesis method are spherical nanoparticles, thus it is difficult to synthesize catalysts with special crystal planes. However, Sun et al. have developed an electrochemical square wave potential method to synthesize polyhedral Pt nanocrystals enclosed by {730}, {210} or adjacent high-index crystal faces, which exhibited higher electrocatalytic activity than common nanostructures.40–42 Excavated cubic PtIr alloys with high {710} crystal faces were also successfully synthesized in a deep eutectic solvent by the electrochemical method.43
In addition, controlling the relationship between alloy composition and electrocatalytic performance is more complicated, because factors such as atomic ratio, crystal phase, and the degree of ordering all affect the electrocatalytic performance. For example, Pt100−xFex alloys with various compositions were prepared by chemical reduction, in which the Pt50Fe50 nanocatalysts exhibited the highest electrocatalytic activity and long-term stability towards formic acid electrooxidation.44 Pt3Zr alloy with an hcp crystal structure was obtained by annealing fcc Pt3Zr nanoparticles at 1000 °C, the former exhibited better electrocatalytic performance toward the ethanol oxidation reaction (EOR) than the latter and commercial Pt/C.45 The characterization results of hard X-ray photo-emission spectroscopy showed that the hcp structure resulted in a stronger interaction between Pt and Zr atoms, and increased the surface energy which is beneficial for the electrocatalytic oxidation process. Moreover, Pd3Pb intermetallic compound with a nanowire network structure was synthesized, and its catalytic performance for the ORR and EOR was enhanced compared with Pd black.46 For the EOR, the mass activity of the intermetallic Pd3Pb was about twice that of Pd black, and it also exhibited excellent resistance to methanol and CO as the cathode catalyst. The higher electrocatalytic performance was partially attributed to the defect-rich porous architecture and ordered structure that provided abundant active sites, accelerated mass diffusion rates, and improved the electronic effect. In brief, the design principles (i.e., size, shape, composition, etc.) of catalyst materials are fully developed for the efficient FC reaction.
Among them, the synergistic effects are known to achieve the above advantages: strain effects and electronic effects.47–51 In Pt-based alloys, Pt atoms can be purposefully controlled to accumulate on the surface of the alloy and then form surface strain, which can markedly affect the corresponding electrochemical activity. As the thickness of the Pt-shell increases, this strain effect decreases. According to the matching degree of lattice constant, the strain can be classified into compressive strain and tensile strain.52 The compressive strain will weaken the interaction between the catalyst surface and adsorbates.53 Conversely, the tensile strain can cause the orbital overlap of the outer metal to decrease, the d-band center of Pt to move upwards, and the interaction between the adsorbate and catalytic surface to enhance. For instance, polycrystalline Pt5Gd electrodes were sputter-cleaned in an ultra-high vacuum chamber, and its ORR activity increased by 5-fold that of pure Pt due to the compressive strain effect.54 DFT calculations indicated that the compressive strain would reduce the binding strength of OHads species and provide an optimum OH binding energy of 0.1 eV, which is 2% weaker than Pt (111). Fcc PtCu alloy shells were deposited on PdCu intermetallic seed to prepare faceted core–shell (i.e. intermetallic-random alloy) nanocatalysts with high compressive strain, which greatly improved the ORR performance.55 Selecting intermetallic nanoparticles as substrates can not only provide a strain effect, but also inhibit the corrosion of Cu atoms during electrochemical tests. The PtPb/Pt core–shell nanostructure with large biaxial strain also exhibited high ORR mass activity reaching 4.3 A mgPt−1 at 0.9 V.56 DFT calculations showed that the tensile strain optimized the adsorption energy of O species. As suggested by the previous literature, the compressive strain helps to weaken the bonding strength of the OH species, while the tensile strain will optimize the strength of the Pt–O bond. Furthermore, the electrons of alloys are transferred from lower-electronegative atoms to higher-electronegative atoms, which cause the d-band center to deviate from the Fermi level and change the electron structure of surface atoms.57–59 For example, PdCu3 intermetallic compound is electrochemically dealloyed to form a Cu-free surface with a lower d-band center and oxygen binding energy than the PdCu3 and Pt (111) nanostructures, which supports the excellent electrocatalytic performance for the ORR.60 Alloys alter the surface coordination environment and electronic structure of the active sites, thereby optimizing the interaction between the adsorbates and the catalyst surface. Both the strain and electronic effects of alloys can cause the same change of atomic orbital overlap and d-band center shift, so the two effects are difficult to distinguish and are often interrelated. Adding an oxophilic metal into Pt atoms can make the adsorption potential of OH species lower than that of Pt, and then enhance the resistance to CO poisoning and electrocatalytic activity through the strain and electronic effects.
Regardless of any electrocatalytic reactions of FCs, the goals of achieving high activity and durability are the same, which can be realized by using Pt-based alloy nanocatalysts. Although random alloys exhibit enhanced electrocatalytic performance, the rapid dissolution of non-noble components can result in performance decay. As mentioned above, converting a random alloy to the intermetallic compound is an effective method to solve this problem. For example, random and ordered PtCu3 alloys with the same size, composition, and particle distribution were prepared to compare their electrocatalytic performance.61 Due to high Cu content in the alloys, the initial spherical nanoparticles become more faceted after 500 cycles, while the potentiostatic treatment will cause the spherical nanoparticles to become porous. After the cyclic voltammetry test of the 40th cycle, the electrochemical signals of Cu disappeared and a protective Pt-shell was formed rapidly for the intermetallic structure, while Cu below the surface was sequentially exposed on the surface of the random alloy. The ICP test found that the amount of Cu leaching from the random alloy was always higher than that of the intermetallic catalyst. Sun et al. have transformed the fcc-PtFe to the fct-PtFe by coating a MgO layer on the fcc-PtFe nanoparticles, which would prevent nanoparticles from sintering during the annealing process.62 Then they compared the stability of fcc-PtFe/C and fct-PtFe/C nanocatalysts by simple acid leaching in H2SO4 solution. After immersing for 1 h, the Fe content in fcc-PtFe/C decreased by 36.5% while fct-PtFe/C showed only a small Fe change (3.3% loss). Taking Abruña's work as another example, a core–shell structured PtCo nanocatalyst with an intermetallic core and a 2–3 atomic-layer-thick Pt shell (Pt3Co/Pt) was reported.63 The specific activity of the ORR on Pt3Co/Pt was increased by 200% and 300% relative to random Pt3Co alloy and Pt/C. After 5000 cycles on Pt3Co/Pt, the ORR activity loss was minimal, and the intermetallic structure remained intact. High activity and stability can be ascribed to the strain and electronic effects, leading to optimal surface–adsorbate interactions. Another method is the purposeful dealloying from Pt-based nanocatalysts, and two types of dealloying methods, namely chemical etching and electrochemical methods, can be found in the literature. After auxiliary metals are selectively leached from the near-surface region by designed pretreatment, the Pt-rich shell structure can be obtained, such as Pt25Cu75 and Pt25Ni75.64,65 As-prepared Pt-based nanoparticles have defective Pt layers after being dealloyed; further annealing treatment at 400 °C can smoothen Pt layers to form a Pt-skin surface.66–68 The application of dealloying methods and the improvement in the activity and stability will be described in detail in the ORR section below. Finally, surface segregation is another effective strategy to engineer the skin structure and control the alloys’ surface properties by reducing the surface free energy. Many factors can greatly alter the surface energy, including temperature, adsorbate, and even substrates.69 Sun's group synthesized well-dispersed PtBi intermetallic compound by a combination of conventional impregnation and hydrogen annealing, in which the mass activity for HCOOH electrooxidation was 10.2 and 28.1 times those of Pd black and Pt/C catalyst, respectively.70 In subsequent work, they regulated the surface atomic composition of intermetallic PtBi and obtained the Bi-rich surface structure.71 Both PtBi alloys are superior formic acid oxidation reaction (FAOR) electrocatalysts, and the electrochemical in situ Fourier transform infrared spectroscopy (FTIRS) results display that the FAOR on the Bi-rich surface is through the non-CO pathway. Moreover, the Pt3Ni/PC alloy with adjustable surface composition, namely a Pt-skin and Ni-rich structure, was prepared via a controlled thermal treatment method.72 Pt-skin and Ni-rich Pt3Ni/PC nanostructures enhanced the catalytic activity for the ORR and FAOR, respectively, both of which are three times as active as the Pt/C catalyst. The controlled heat treatment method can be used to design catalysts with various nanostructures, such as intermetallic compounds, Pt-skin catalysts, non-precious metal-rich catalysts, etc. These nanostructures can be used for various electrocatalytic reactions to adjust the reaction pathway and the molecular adsorption mode.
So far, with the improvement of nanotechnology and characterization methods, researchers can purposely synthesize alloy nanocatalysts with specific physical properties and excellent electrochemical performance. Engineering the surface/near-surface nanostructure is a promising strategy to improve the electrocatalytic performance of alloys, which can maximize the exposure and utilization of active sites. In each section of this article, based on the combination of in situ electrochemical spectroscopy techniques and classical theoretical calculations, we discuss molecular adsorption modes, chemical bond cleaving, reaction energy barriers, coordination environments, underlying reaction mechanisms, etc. Then, the corresponding challenges in the five electrocatalytic reactions are proposed, and how can they be addressed? The development and application of Pt-based alloy nanocatalysts in recent years are discussed in detail. In particular, the electronic effect, strain effect, and ordered structure of alloys are introduced to emphasize their role in improving the electrocatalytic performance, and the progress of tuning strategies and quantification methods is also described. Furthermore, some typically used strategies to improve the alloy nanostructure are also summarized, including annealing treatment, dealloying strategies, the galvanic replacement reaction, and so on, which provide useful clues for guiding the synthesis of nanocatalysts with excellent performance and durability. Designing advanced alloy nanomaterials with unique effects and structures is expected to realize the commercialization of FCs. We clarified the future prospects of alloy nanocatalysts, which are both challenges and opportunities for energy conversion.
2. Small organic molecule electrooxidation reaction
For low-temperature FCs, besides hydrogen, various small organic molecules (SOMs) can be used as anodic fuels, such as methanol, ethanol, formic acid, and so on.73,74 As mentioned above, the issue of low efficiency of platinum catalysts can be addressed by selecting an inexpensive or earth-rich element to form alloys, which are the most versatile and effective catalysts in the anode and cathode of FCs.75–77 For Pt-based alloys, besides strain effects and electronic effects, changes in the distance between Pt–Pt atoms and the number of adjacent Pt atoms also can adjust the bonding strength of reaction intermediates and optimize the reaction activation energy. In this section, the goal is to understand the electrocatalytic reaction mechanisms of the methanol oxidation reaction (MOR), the ethanol oxidation reaction (EOR), and the formic acid oxidation reaction (FAOR), the latest development trend of noble metal alloy catalysts, as well as the significance of the structure–activity relationship for designing advanced electrocatalysts.2.1 MOR
Methanol is a relatively stable, economical, and easy-to-transport liquid that can be fed directly into the cell as a fuel (DMFCs).78,79 The primary reason for the low efficiency of DMFC anode catalysts is that the COads intermediate firmly adheres to the active sites, requiring a higher removal potential. For Pt-based nanocatalysts, it is generally accepted, despite the controversy, that the MOR has two parallel pathways: indirect CO poisoning and direct formate pathways (Fig. 1A).80,81 A cyanide-modified Pt (111) electrode was used as a chemically modified electrode to study the role of atomic ensembles in the electrooxidation of methanol molecules.81 Electrochemical tests and in situ FTIRS indicated that two adjacent platinum atoms are the smallest atomic ensemble for the MOR to CO2 through the non-CO pathway. They also clarified that formate might be formed via the reaction between methanol-derived adsorbates and bulk water. The formation of CO via the indirect oxidation pathway requires at least three contiguous Pt atoms, and this result can help design a non-CO poisoning catalyst. For instance, the reaction free energy on different sites for each MOR electrochemical step was calculated by DFT, where PtZn (111), stepped PtZn (211) and Pt24Zn24 cluster represented terrace, edge, and corner sites, respectively.34 All three PtZn models underwent the direct oxidation pathway, and the MOR on corner sites had smaller barriers than that on edge and terrace sites. Unlike the PtZn system, the Pt (111) underwent the indirect oxidation pathway, in which CO* was formed after continuous cleavage of C–H and O–H bonds, followed by oxidation to form CO2. Compared with Pt atoms, the bonding strength of Zn atoms with OH* is higher, as a result the PtZn underwent a CO-free pathway (CH2O* → CH2O* + OH* → H2COOH* → COOH* → CO2). | ||
| Fig. 1 (A) MOR reaction pathways. Reproduced with permission from ref. 81. Copyright 2006, American Chemical Society. (B) In situ ATR-SEIRAS of preadsorbed M-13COL on the Ru/Pt (M-13COL refers to the linearly bonded CO generated by methanol), and the integrated IR intensity of the M-13COL-Pt and M-13COL-Ru as a function of electrode potential. Reproduced with permission from ref. 82. Copyright 2015, John Wiley & Sons Ltd. (C) COads and CH3OHads band integrals as a function of potential in both positive and negative scans on Pt/C and PtRu/C, respectively. Reproduced with permission from ref. 84. Copyright 2012, Royal Society of Chemistry. | ||
For PtRu bimetallic catalysts, the Watanabe–Motoo bifunctional mechanism is generally accepted. However, in 2015, Tong et al. used Ru decorated Pt thin films (Ru/Pt) as catalysts to propose a new revised bifunctional mechanism.82 They combined in situ FTIRS with control experiments, which offered flow-cell operation and diagnostic selectivity by isotope labeling, to show that COL–Pt was not part of the reactions. In situ attenuated total-reflection-surface-enhanced IR reflection absorption spectroscopy (ATR-SEIRAS) experiments showed the onset oxidation potential of methanol molecules coinciding with formate appearance, lower than that of COL–Pt oxidation and higher than that of COL–Ru (Fig. 1B). Therefore the bifunctional mechanism (Pt-CO + Ru-OH → Pt* + Ru* + CO2 + H+ + e−) has nothing to do with enhanced MOR at a lower overpotential (<0.5 V vs. RHE). It is worth noting that the revised reaction mechanism requires that the free Ru* and PtRu* reaction sites must be adjacent to each other and be available, and PtRu boundary sites catalyze the oxygen insertion reaction that leads to the formation of formate and undergoes the direct oxidation pathway. Furthermore, there was a dispute over the attribution of the MOR inverse-sweep peak, and recently in situ FTIRS techniques have provided experimental evidence. In 1992, Goodenough's group reported that the forward sweep peak (If) of the MOR was attributed to methanol oxidation, while the backward sweep peak (Ib) was attributed to the oxidation of residual intermediate carbon species.83 After this work, many researchers used the peak ratio (If/Ib) to estimate the CO-tolerance. The origin of Ib on commercial Pt/C and PtRu/C catalysts for the MOR was investigated in-depth through cyclic voltammetry (CV) tests and in situ FTIRS.84 The COads and methanolads band integrals from FTIRS as a function of potential for Pt/C and PtRu/C, along with the corresponding MOR CV, are shown in Fig. 1C. For Pt/C, the methanol molecules have been dissociated and adsorbed on the catalyst surface at a lower potential (<−0.25 V vs. Ag/AgCl), and generated the COads intermediate. As the potential increases, the amount of COads and methanolads increases slightly until about 0.3 V. Then, the MOR current increases sharply between 0.3 and 0.75 V, along with the decrease of the number of COads and methanolads. When the potential is higher 0.8 V, no COads species adsorb on the surface of the catalyst. PtRu/C has a similar change as described above with the Pt/C catalyst. However, the complete oxidation on PtRu/C occurs at a lower potential than Pt/C, and the amount of COads is much smaller than that produced in the positive, indicating that CO oxidation is easier on PtRu/C. There was a significant inverse correlation between the MOR current and the amount of CH3OHads on the Pt/C and PtRu/C catalysts during the MOR, but the amount of COads did not have this correlation. This indicates that both If and Ib are attributed to the direct oxidation of methanol, regardless of the oxidation of the remaining carbonaceous species. Moreover, electrochemical impedance spectroscopy tests showed that the reason for the CV hysteresis between the forward sweep peak and backward sweep peak is that the rate-determining step has changed, from the OH adsorption caused by water dissociation to dehydrogen of methanol.85 They also emphasized that If/Ib is irrelevant to the CO tolerance, but actually related to the oxophilic degree of Pt-based nanocatalysts, such as the results in Fig. 1C.
PtRu is still the best bimetallic catalyst for the MOR, the coexisting electronic effect and the bifunctional mechanism can stimulate the chemisorption and dissociation of methanol molecules at a lower potential.86 A highly active and durable Pt72Ru28 porous flower-like alloy, which was assembled from nanoparticles below 4.0 nm, was reported by Wang's group.87 The specific and mass activity can reach 4.19/3.54-fold and 4.27/5.0-fold enhancement compared to those on commercial Pt black and Pt50Ru50 black, respectively. At present, its electrocatalytic performance is the highest under the same test conditions reported in the literature. Moreover, PtRu nanocrystals with a controllable shape (nanowires, nanorods, and nanocubes) were synthesized, and these {100} and {111} terminated PtRu nanocrystals were used to investigate the facet-dependent MOR electrocatalytic performance.88 The {111}-terminated PtRu nanowires showed the highest mass activity, which was 2.28 and 4.32 times higher than those of PtRu nanocubes and commercial Pt/C, respectively, indicating that the {111} facets possess higher MOR activity relative to the {100} facets. Furthermore, the PtRu nanocatalyst (PtRu/PC-H) was annealed at a high temperature (700 °C, PtRu/PC-H) to enhance the alloying degree, thereby further improving the electrocatalytic performance of the MOR.89 The PtRu/PC-H alloy has a higher 2θ value than that of the Pt/PC nanocatalyst in XRD, which means the occurrence of lattice contraction, and the enhancement of alloying degree after annealing. It is interesting that at annealing temperatures up to 700 °C, the as-synthesized nanocatalysts still have a small particle size (∼2.82 nm) and excellent dispersibility, which may be attributed to the confined effect of porous carbon (PC) supports on nanoparticles. For the MOR, the onset oxidation potential of PtRu/PC-H shifts to a more negative value than that of the Pt/PC nanocatalyst, and the mass activity (1674.2 mA mgPt−1) of PtRu/PC-H is about 4.08 times that of Pt/PC (Fig. 2A). Moreover, membrane electrode assembly (MEA) tests using PtRu/PC-H as the anode catalyst with a low Pt loading (1 mg cm−2) exhibit a maximum power density of 83.7 mW cm−2, which is approximately 3.1 times that of the commercial Pt/C catalyst (Fig. 2B). As seen from the in situ FTIRS in Fig. 2C, PtRu/PC-H alloy generates CO2 (2345 cm−1) at a lower potential of −150 mV than those on PtRu/PC-L (0 mV) and Pt/PC (50 mV) nanocatalysts, dramatically alleviating the COads poisoning on active sites. In addition, the PtRu/PC-H nanocatalyst can easily adsorb and dissociate methanol molecules, and has a stronger bond cleaving ability of C–H and O–H.
 | ||
| Fig. 2 (A) MOR in 0.5 M CH3OH + 0.1 M HClO4 solution for PtRu/PC-H, PtRu/PC-L, and Pt/PC nanocatalysts at a scan rate of 50 mV s−1. (B) Steady-state polarization and power density curves for fuel cells employing PtRu/PC-H, PtRu/PC-L, commercial PtRu/C, and commercial Pt/C as anode catalysts. (C) In situ FTIRS for methanol electrooxidation on three nanocatalysts, the reference spectra were acquired at −0.27 V. Reproduced with permission from ref. 89. Copyright 2019, Elsevier BV. | ||
Pt3Ti possesses a large enthalpy of formation (ΔHf = −298 kJ mol−1), while the value of PtRu is only a few kJ mol−1. Bardi and co-workers reported that the CO adsorption energy on the pure Pt surface was 5 times higher than that on the Pt3Ti surface, implying that Pt3Ti may have an increased CO poisoning tolerance.90 DiSalvo et al. synthesized fcc-Pt3Ti samples with a smaller size (about 3 nm) in tetrahydrofuran; then they were transformed into the intermetallic compound with a large size (∼37 nm) by annealing above 400 °C.91 CO stripping experiment shows that the CO oxidation peaks of Pt3Ti nanoparticles are much weaker than those of pure Pt and PtRu samples, suggesting that the CO affinity of Pt3Ti catalysts is very low and the catalyst surface is difficult to poison with CO. The Pt3Ti intermetallic compound exhibits substantially the highest electrocatalytic performance for methanol and formic acid compared to pure Pt, PtRu, and fcc-Pt3Ti, which could be attributed to the lower CO affinity, ordered structure, and lower d-band center of Pt atoms. Moreover, they used KCl by-product as an encapsulant to control and synthesize a small-sized Pt3Ti intermetallic compound (∼6 nm) under high-temperature annealing.92 By increasing the annealing temperature, the ΔG of forming Pt3Ti alloy is changed to be negative, that is, it is favorable for the formation of alloy. The mass activity of the Pt3Ti/C intermetallic compound towards the MOR is greatly enhanced, which is about three times higher than that of Pt/C. The chronopotentiometric test shows that the intermetallic compound undergoes a small potential change for 1 h at 1 mA cm−2, which may be attributed to the strong anti-toxicity and stable surface structure. Shanmugam et al. also fabricated Pt8Ti intermetallic compound supported on N-doped carbon (Pt8Ti–TiO2/C), which was used as the anode catalyst for the DMFC single-cell test.93 Pt8Ti–TiO2/C displayed 10 times higher specific activity than that of the commercial Pt/C catalyst, due to the presence of the intermetallic compound and corrosion-resistant TiO2. More importantly, in the MEA test, the maximum power density of the Pt8Ti–TiO2/C anode catalyst is 48.7 mW cm−2 at 333 K, which is 1.6 times that of the Pt/C-based anode catalyst. The better MEA test result is in good agreement with the result observed for the half-cell MOR.
Screening studies of intermetallic compounds for the MOR have been mentioned in reported literature, and have revealed that various ordered structures exhibit better MOR performance relative to random alloys.94,95 Recently, Li et al. prepared the Pt3Ga intermetallic nanocatalyst by co-reducing Pt(acac)2 and GaCl3, and then they tuned the strain of Pt by chemical etching to form 2–3 atomic-layer Pt-skin on the Pt3Ga intermetallic compound (Pt3Ga/Pt).96Fig. 3A shows the typical XRD patterns of XC-72 supported Pt3Ga/Pt and Pt nanocrystals. Except for the carbon peak at around 25°, the other five diffraction peaks are attributed to the fcc structure in Pt nanocrystals. Pt3Ga/Pt has six additional ordered diffraction peaks of (100), (110), (210), (211), (300) and (310), which match well with the Pt3Ga intermetallic compound. Interestingly, the peak positions of Pt3Ga/Pt have a positive shift compared to that of Pt nanocrystals, indicating that the Pt atoms cause the unit cell contraction phenomenon. The fast Fourier transform (FFT) pattern of a single Pt3Ga/Pt shows the simultaneous existence of the (100) and (110) superlattice spots (Fig. 3B), which is consistent with the results obtained by XRD. The corresponding unit cell is Ga and Pt atoms located at the eight vertices and the centers of six facets of the cube, respectively. Through high-angle annular dark-field scanning TEM (HAADF-STEM), it can be seen that Ga and Pt atoms are arranged orderly in the nanoparticles, and the atoms of near-surface are all Pt atoms. Through theoretical calculation, the tensile strain of Pt-skin is 3.2% along the [001] direction, while the strain along the [100]/[010] direction is negligible. The mass activity of the Pt3Ga/Pt nanocatalyst is 3.7 and 1.9 times that of Pt nanocrystals and commercial Pt/C, respectively (Fig. 3C). After 1000 potential cycles, Pt3Ga/Pt nanocatalyst had a negligible loss in mass activity, demonstrating its excellent stability. DFT calculations are also used to study the reaction mechanism of the MOR on the surface of tensile strained and unstrained Pt nanocrystals (Fig. 3D). On both sample surfaces, the dehydrogenation of methanol is more likely to cleave the C–H bonds than the O–H bond, but the tensile strained surface is more energetically favorable than the unstrained surface. Thus, the increased activity is attributed to the stronger binding of OH* and the lower activation energy of water. Also, the strong p–d hybridization of PtGa alloy can redistribute the electrons of two atoms, weaken the bonding strength of COads, and suppress the diffusion of lattice atoms.97 PtGa alloy exhibits a lower d-band center and increased vacancy formation energy relative to pure Pt, implying that the accumulation of electrons around Pt atoms results in enhanced oxidation resistance and structural stability. Therefore, the overpotential and reaction rate of the MOR on platinum have been improved.
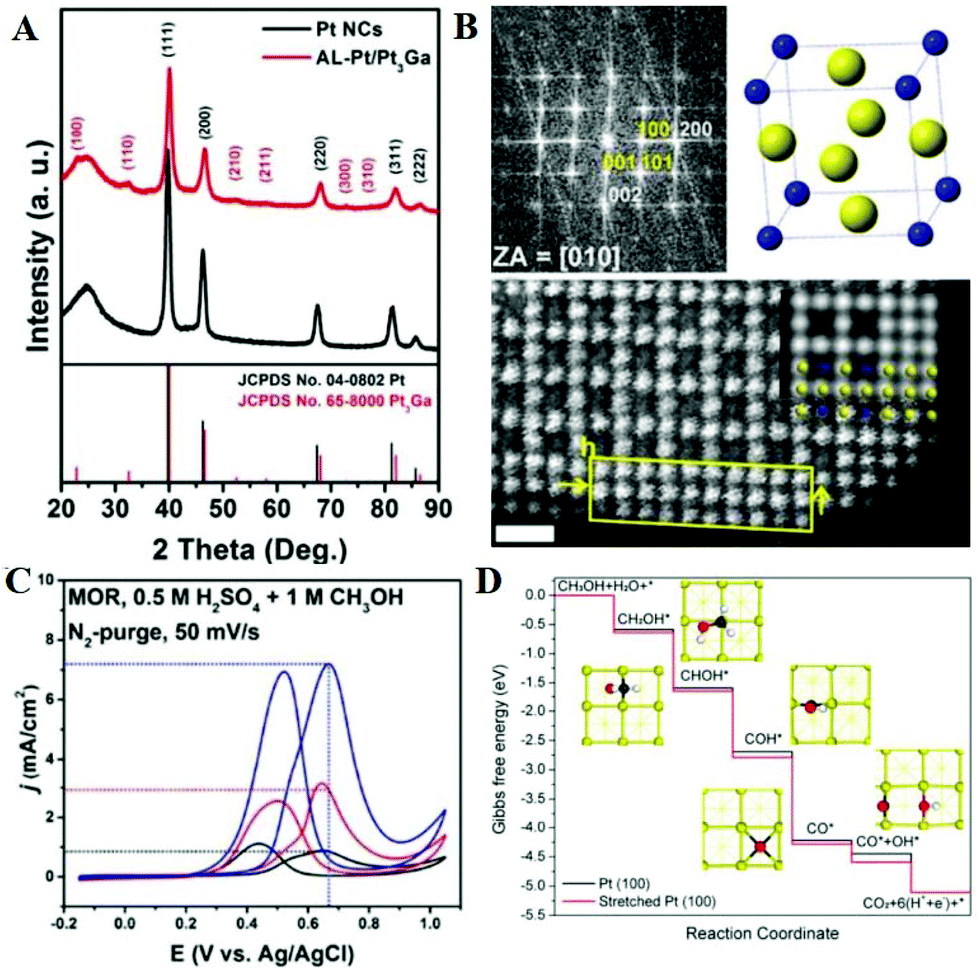 | ||
| Fig. 3 (A) XRD patterns of XC-72 supported Pt3Ga/Pt and Pt nanocrystal samples. (B) FFT pattern of the Pt3Ga/Pt; the unit cell of the intermetallic Pt3Ga phase, yellow and blue spheres represent Pt and Ga atoms, respectively; enlarged high-resolution HAADF-STEM images. (C) CVs of three catalysts in 1 M methanol + 0.5 M H2SO4 solution at a sweep rate of 50 mV s−1. (D) Calculated reaction pathways of the MOR on unstrained Pt (100) and stretched Pt (100) surfaces at pH = 0.25, U = 0.88 V with respect to the RHE. The insets show the optimized structures of intermediates (Pt: yellow, C: black, O: red, H: white). Reproduced with permission from ref. 96. Copyright 2018, American Chemical Society. | ||
Alloy nanomaterials allow for further reducing the Pt loading, enhancing activity and resistance of CO poisoning, as well as increasing durability. These excellent performances are related to the promoted C–H and O–H bond activation, which may be caused by the strain effect, electronic effect, crystal type, ordered structure etc. Intermetallic compounds can separate active sites, increase the rate of methanol oxidation, and improve catalyst poisoning because CO generation from the methanol molecule requires three adjacent Pt atoms. Compared with stable metals (Au, Pd, etc.), oxophilic metals (Ru, Ti, Ga, Cu, Co, etc.) are more likely to provide adsorbed OH* species after alloying with Pt and more effective to electrooxidize methanol molecules. There is still a big gap between the intrinsic activity of Pt-based alloy catalysts and the power density of MEA.
2.2 EOR
Ethanol is of confocal interest because it is non-toxic, has a higher energy density (8.01 kW h kg−1) relative to methanol (6.09 kW h kg−1), and is easy to transport and store, which may lead to a carbon-neutral economy.13,98,99 Ethanol oxidation can occur at 84 mV under thermodynamic conditions.100 However, due to the lack of effective catalysts to promote the complete oxidation of ethanol to CO2 without heat loss, ethanol fuels have not been widely used except for petroleum additives in traditional combustion systems. The C–C bond cleavage in ethanol molecules requires a very high dissociating energy (∼350 kJ mol−1).101 Thus, electrocatalytic oxidation of ethanol to carbon dioxide has a lower selectivity than acetic acid or acetaldehyde, and Pt nanocatalysts have a CO2 selectivity of only 0.5–7.5%.102 The efficiency of direct ethanol fuel cells (DEFCs) is significantly decreased because the rate of cleaving of C–C bonds is very low.103,104Fig. 4A shows some possible pathways for the oxidation of ethanol molecules to acetaldehyde, acetic acid and CO2.105 The EOR through the C1 and C2 pathways is widely accepted. For ethanol molecules, the adsorption of α-C–H on catalysts is considered as the initial step, and the dehydrogenation energy barrier of α-C–H is much lower than that of the O–H bond.106 The reaction between acetyl and OH species is an important step to form acetic acid (CH3CO + OH → CH3COOH), but the C–C bond cleavage is the determining step in the formation of CO2 (CH2CO → CH2 + CO). Therefore, the acetic acid formation process requires the assistance of OH species, that is the presence of oxygen-containing species could enhance the obstacles towards C–C bond cleavage (Fig. 4B).105 Relative to the clean Pt surface, the presence of OH and O species will reduce the rate of C–C bond cleavage by about two orders of magnitude and six orders of magnitude, respectively. Adzic et al. deposited a Pt monolayer (PtML) on different single-crystal surface substrates using the Cu underpotentially deposition method to study the strain and electronic effect on the EOR. During the synthesis, five single crystal surfaces (i.e. Au (111), Pd (111), Ir (111), Rh (111), and Ru (0001)) were used as substrates.107 By changing the substrate, the strain and electronic effects of the PtML catalysts are tailored. Fig. 4C shows the EOR curves of Pt (111) and PtML electrocatalysts, and the activity of PtML/Au (111) is four times that of Pt (111), which is caused by tensile strain, the upshift of the d-band center, and the easy formation of OHads. From the in situ infrared reflection absorption spectrum (IRRAS), it can be seen the signals of acetic acid and acetaldehyde without the CO signal (Fig. 4D), indicating that PtML/Au (111) mainly oxidizes ethanol through the C2 pathway. The following two paragraphs introduce excellent alloy nanocatalysts reported in recent years, which can oxidize ethanol molecules through C1 and C2 pathways, respectively. | ||
| Fig. 4 (A) Reaction pathways for the formation of acetaldehyde, acetic acid, and CO2. (B) Effect of surface oxidants (O, OH) on the key step of C–C bond cleavage. The bars are the barriers of the key step of CO formation without oxidant (green), in the presence of OH (red), and in the presence of O (brown). C in gray, O in red, and H in white. Reproduced with permission from ref. 105. Copyright 2012, John Wiley & Sons Ltd. (C) Positive voltammetric scans for Pt (111) and PtML supported on five different substrates in 0.1 M HClO4 containing 0.5 M ethanol with the scan rate of 10 mV s−1. (D) In situ IRRAS spectra recorded during the EOR on the PtML/Au (111) electrode. Reproduced with permission from ref. 107. Copyright 2012, American Chemical Society. | ||
Surface/near-surface engineering of alloys can effectively adjust the selectivity of ethanol electrooxidation and the optimal adsorption mode of intermediates, for example, the C–C bond cleavage activation energy of Pt (100) is 2.1 times lower than that of Pt (111).108 Sun's group explored the EOR mechanism of PtRh nanocubes in detail. Pt1Rh1 alloy nanocubes show an ultrahigh mass activity, 2.48 times higher than that of commercial Pt/C at 0.4 V vs. SCE.109 The α-C and β-C of ethanol molecules adsorbed on the neighboring Pt and Rh atoms to form CH2CO species. This nanostructure displays efficient C–C bond cleavage and better CO2 selectivity, due to the incorporation of Rh into the Pt lattice and the preferential (100) orientation. In situ FTIRS using isotopically-labeled ethanol further confirms that the CO2 selectivity of Pt1Rh1 is 16.2 times higher than that on commercial Pt/C. Abruña et al. reported the synthesis of PtSn with a cubic core–shell morphology (more than 90% of the total).110 Calculated by X-ray photoelectron spectroscopy (XPS), the atomic ratio of PtSn is 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :76 and tin oxide accounts for 79% of the total Sn, implying that a low density and amorphous thin layer on the exterior nanocube is mainly tin oxide. In Fig. 5A, scanning transmission electron microscopy-electron energy loss spectroscopy (STEM-EELS) composition maps with simultaneous annular dark-field (ADF) images of Pt and Sn indicate that cubic PtSn contains a Pt-rich core surrounded by an Sn-rich shell that is approximately 2–3 nm thick. They further built a Pt-rich surface by electrochemical etching, sweeping 5000 cycles in acidic medium. Compared with before cycling samples, nanoparticles maintained the cubic morphology and the Pt-skin layer formed after cycling, which is a Pt-richcore/Sn-richsubsurface/Ptskin structure. This structure exhibits the highest electrocatalytic performance under potential cycling (Fig. 5B). This exceptional activity could be attributed to the electronic effect, strain effect, as well as high density Pt (100) surface sites, favoring the C–C bond cleavage. PtIr nanocubes with precisely controlled Ir-skin atomic layers were obtained through colloidal synthesis.111 Pt38Ir and Pt17Ir nanocubes have one and two thin surface atomic layers of Ir, respectively. Pt38Ir/C exhibits the largest current density compared to that of Pt17Ir/C and Pt/C catalysts, suggesting that Ir-skin with only one atomic layer is more favorable for high EOR activity. Besides, Pt38Ir/C also shows 4.5 times higher CO2 selectivity at 0.85 V vs. RHE than that of Pt/C, which can be attributed to the combination of the crystal structure and electronic effect (Fig. 5C). The chronoamperometric test and in situ IRFTS are combined to study the generation of CO2 (Fig. 5D). The Pt38Ir/C sample started to convert ethanol to carbon dioxide at 0.45 V vs. RHE after 200 s, but CO2 could not be detected on the Pt/C catalyst even after 600 s. The DFT results also show that the PtIr (100) surface is more conducive to the C–C bond cleavage and the adsorption of *CxHyO/CxHy species. Moreover, Ta is more electropositive and oxophilic, and Pt3Ta intermetallic compound containing a larger enthalpy of formation (ΔHf = −59.5 kJ mol−1) compared with PtFe (ΔHf = −13.6 kJ mol−1) can act as an effective catalyst.112 Pt3Ta intermetallic compound exhibited superior electrocatalytic performance compared to Pt3Sn nanocatalysts for the EOR.113 The Pt3Ta sample efficiently cleaves C–C bonds and completely oxidizes ethanol to CO2. After 10000 cycles, Pt3Ta retains 85% of the initial electrochemically active surface areas (ECSA) (Fig. 5E). The promoted C–C bond cleavage leads to the adsorption of COads intermediates on the Pt3Ta surface, and the high coverage of oxygenous species on the surface of Ta atoms facilitated the oxidation of COads to CO2. MEA tests demonstrate that the power density of the Pt3Ta intermetallic compound was more than 2 times higher than that of Pt nanoparticles (Fig. 5F). Furthermore, a core–shell nanostructure with a Pt-shell on the intermetallic PtBi core was synthesized by the single-atom self-assembly method, and PtBi/Pt was supported on the graphene matrix.114 The mass activity of PtBi/Pt for ethanol oxidation is 8.26 times higher than that of Pt/C due to the tensile strain effect. The in situ FTIRS and DFT results prove that the PtBi/Pt nanocatalyst can achieve the complete oxidation of ethanol molecules and obtain C1 products.
:76 and tin oxide accounts for 79% of the total Sn, implying that a low density and amorphous thin layer on the exterior nanocube is mainly tin oxide. In Fig. 5A, scanning transmission electron microscopy-electron energy loss spectroscopy (STEM-EELS) composition maps with simultaneous annular dark-field (ADF) images of Pt and Sn indicate that cubic PtSn contains a Pt-rich core surrounded by an Sn-rich shell that is approximately 2–3 nm thick. They further built a Pt-rich surface by electrochemical etching, sweeping 5000 cycles in acidic medium. Compared with before cycling samples, nanoparticles maintained the cubic morphology and the Pt-skin layer formed after cycling, which is a Pt-richcore/Sn-richsubsurface/Ptskin structure. This structure exhibits the highest electrocatalytic performance under potential cycling (Fig. 5B). This exceptional activity could be attributed to the electronic effect, strain effect, as well as high density Pt (100) surface sites, favoring the C–C bond cleavage. PtIr nanocubes with precisely controlled Ir-skin atomic layers were obtained through colloidal synthesis.111 Pt38Ir and Pt17Ir nanocubes have one and two thin surface atomic layers of Ir, respectively. Pt38Ir/C exhibits the largest current density compared to that of Pt17Ir/C and Pt/C catalysts, suggesting that Ir-skin with only one atomic layer is more favorable for high EOR activity. Besides, Pt38Ir/C also shows 4.5 times higher CO2 selectivity at 0.85 V vs. RHE than that of Pt/C, which can be attributed to the combination of the crystal structure and electronic effect (Fig. 5C). The chronoamperometric test and in situ IRFTS are combined to study the generation of CO2 (Fig. 5D). The Pt38Ir/C sample started to convert ethanol to carbon dioxide at 0.45 V vs. RHE after 200 s, but CO2 could not be detected on the Pt/C catalyst even after 600 s. The DFT results also show that the PtIr (100) surface is more conducive to the C–C bond cleavage and the adsorption of *CxHyO/CxHy species. Moreover, Ta is more electropositive and oxophilic, and Pt3Ta intermetallic compound containing a larger enthalpy of formation (ΔHf = −59.5 kJ mol−1) compared with PtFe (ΔHf = −13.6 kJ mol−1) can act as an effective catalyst.112 Pt3Ta intermetallic compound exhibited superior electrocatalytic performance compared to Pt3Sn nanocatalysts for the EOR.113 The Pt3Ta sample efficiently cleaves C–C bonds and completely oxidizes ethanol to CO2. After 10000 cycles, Pt3Ta retains 85% of the initial electrochemically active surface areas (ECSA) (Fig. 5E). The promoted C–C bond cleavage leads to the adsorption of COads intermediates on the Pt3Ta surface, and the high coverage of oxygenous species on the surface of Ta atoms facilitated the oxidation of COads to CO2. MEA tests demonstrate that the power density of the Pt3Ta intermetallic compound was more than 2 times higher than that of Pt nanoparticles (Fig. 5F). Furthermore, a core–shell nanostructure with a Pt-shell on the intermetallic PtBi core was synthesized by the single-atom self-assembly method, and PtBi/Pt was supported on the graphene matrix.114 The mass activity of PtBi/Pt for ethanol oxidation is 8.26 times higher than that of Pt/C due to the tensile strain effect. The in situ FTIRS and DFT results prove that the PtBi/Pt nanocatalyst can achieve the complete oxidation of ethanol molecules and obtain C1 products.
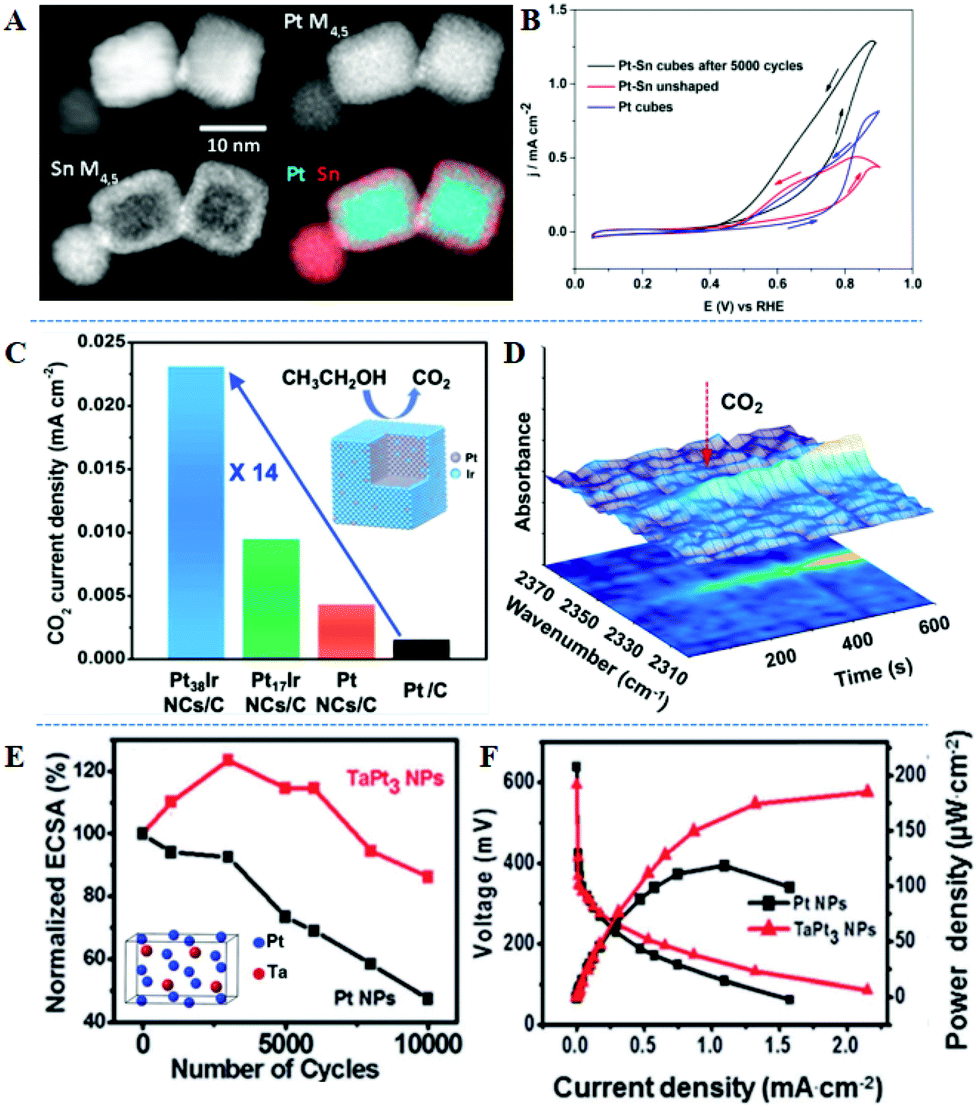 | ||
| Fig. 5 (A) ADF-STEM image of PtSn nanoparticles, the EELS maps of Pt and Sn, and the composite PtSn map with Sn in red and Pt in cyan. (B) Comparison of EOR curves at a scan rate of 20 mV s−1. Reproduced with permission from ref. 110. Copyright 2018, American Chemical Society. (C) Specific activity. (D) In situ IRRAS spectra recorded during the chronoamperometry test at 0.45 V vs. RHE for the EOR on Pt38Ir/C. Reproduced with permission from ref. 111. Copyright 2019, American Chemical Society. (E) Variation of the ECSA of the TaPt3 and Pt nanoparticles as a function of potential cycles. (F) I–V profiles and power density profiles of TaPt3 and Pt nanoparticles, obtained at room temperature in 1 M ethanol solution. Reproduced with permission from ref. 113. Copyright 2015, Royal Society of Chemistry. | ||
Although the C1 pathway can achieve higher electrical efficiency, the EOR is generally performed through the C2 pathway. Thus, OH species are provided to facilitate the oxidation of intermediates such as acetaldehyde. Considerable efforts have been made to optimize Pt-based nanocatalysts with the desired composition and structure. For instance, the ion irradiation technique was used to tune the defects and interfaces of intermetallic PtPb nanoplates.115 These defects like the dislocations, subgrain boundaries, and amorphization can be controlled by changing the energy and variety of incident ions as well as the location of the beam spot. Engineered PtPb nanoplates with different defects and interfaces show the volcano-like electrocatalytic activity for the EOR as a function of ion irradiation fluence. Optimized PtPb nanoplates exhibit high EOR activity, due to the fact that this structure could undoubtedly create active sites by breaking the electron–hole symmetry. PtNi octahedral nanocrystals with an average edge length of 10 nm were synthesized.116 The specific activity of the nanocrystal is 2.4 and 3.7 times that of conventional Pt2Ni/C and Pt/C toward the EOR, respectively, indicating that the (111) facet could improve the electrocatalytic performance. In situ FTIRS measurements show that the absorbance peak intensity of acetic acid/CO2 on octahedra is 7.6 and 1.4 times higher than those of commercial Pt/C and conventional Pt2Ni/C, respectively, implying that the octahedra show faster kinetics in the C2 pathways. Moreover, Pt3Co@Pt/PC with a 1–2 Pt-atomic-skin structure was successfully prepared by a simple controlled thermal treatment method.117 The d-spacing of the edge and center is 2.27 and 2.22 Å (Fig. 6A), which is consistent with Pt and Pt3Co (111), respectively, implying the nanocatalyst with a Pt-skin surface. Fig. 6B shows the line-profile analysis; the image confirms that the nanoparticle surface contains a large amount of Pt compared to Co and the thickness of Pt-skin is ∼0.5 nm. For the EOR, the mass activity of Pt3Co@Pt/PC is 2.5 times higher than that of Pt/C (Fig. 6C), due to the bimetallic synergetic effect and unique structural advantages. To understand the impact of the strain effect on ethanol, in situ FTIRS contributes to recognition of the intermediates and final products, and identification of the selectivity, as shown in Fig. 6D and F. The ratio of CO2/CH3COOH of Pt/C first increases but decreases subsequently when the applied potentials increase. Compared with Pt/C, the value of CO2/CH3COOH for Pt3Co@Pt/PC is low (Fig. 6E), which suggests that the Pt3Co@Pt/PC nanocatalyst prefers breaking the α-C–H bond producing CH3COH rather than cleaving the β-C–H bond yielding CH2COH and obtains C2 products (such as CH3COOH).
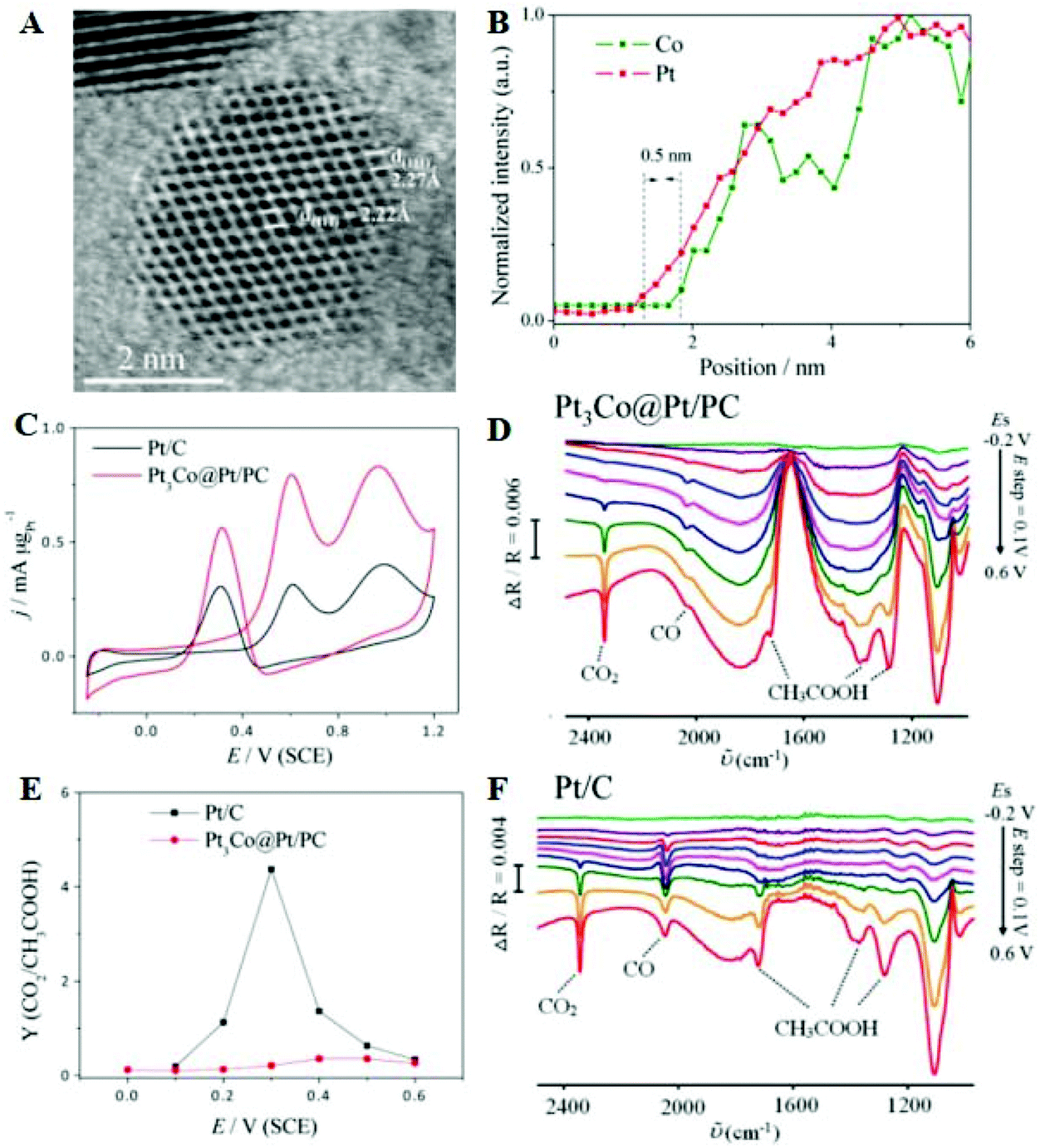 | ||
| Fig. 6 (A) Atomic-resolution ADF-STEM image and (B) line-profile analysis of Pt3Co@Pt/PC nanoparticles. (C) CVs in 0.1 M CH3CH2OH + 0.1 M HClO4 solution. (D) and (F) In situ FTIRS of Pt3Co@Pt/PC and Pt/C in 0.1 M CH3CH2OH + 0.1 M HClO4. (E) Ratio of integrated intensities of CO2 and intensities of CH3COOH as a function of working potential. Reproduced with permission from ref. 117. Copyright 2016, American Chemical Society. | ||
In brief, for Pt-based catalysts, C–C is more likely to be cleaved at low potentials, and the COads intermediate produced through the C1 pathway needs to be eliminated by oxygen-containing species (e.g. OH and O species). However, the production of oxygen-containing species on the surface of the catalyst will cause the C1 pathway to be blocked and the emergence of C2 species. Thus, it is important to regulate the bonding strength and the amount of oxygen-containing species on the catalyst surface to improve the C–C bond cleavage. Furthermore, the stable intermetallic structure is of great concern, and the ordering degree can be controlled by adjusting the heat treatment procedure. Since annealing is the most versatile and effective method for converting a random alloy to an intermetallic compound, it is critical to figure out the ideal heat treatment conditions (eg, annealing temperature, heating rate, lasting time and gas atmosphere) for the phase conversion.118 We have learned a lot about phase diagrams of bulk alloys from textbooks, but the practical electrocatalysts for fuel cells are often used on the nanoscale, which exhibit different behavior from the bulk form. The selectivity of ethanol oxidation products depends largely on the crystalline orientation of electrocatalysts. Therefore, it is more meaningful to study the ordered structure at the nanoscale level, and to obtain more advanced nanocatalysts that promote the C1 pathway, such as intermetallic compounds with the (100) crystal planes and strain effects.
2.3 FAOR
The theoretical energy density of formic acid oxidation is three times lower than that of methanol, but formic acid has many advantages as a fuel; for example, it is non-flammable, inexpensive, easily available, and stable over a wide temperature range.119–121 Moreover, direct formic acid fuel cells (DFAFCs) have minimal anodic fuel crossover through the membrane, which is a known issue in DMFCs that can cause cathode catalyst fouling.122 FAOR is a 2-electron transfer reaction, which can be proceeded by two different reaction mechanisms, i.e. the direct (dehydrogenation) pathway and the indirect (dehydration) pathway.123–126 As shown in Fig. 7A, this involves HCOOH cleaving bonds to form a HCOOH-derivative or adsorbed CO, which is finally oxidized to carbon dioxide. Murray and co-workers combined experimental and theoretical results to prove the reason why Pt3Pb/Pt nanocrystals could enhance the electrooxidative activity for formic acid relative to Pt.127 For Pt (111), the first dehydrogenation step to form COOH* or HCOO* has the same activation energy barrier (0.72 eV), but HCOO* requires a much higher activation energy barrier than COOH* (1.23 vs. 0.71 eV) during the second dehydrogenation step, so that the stable HCOOads intermediates will occupy most of the active sites and reduce the overall reaction rate. For Pt3Pb/Pt (111), all activation energy barriers are reduced compared to the Pt (111), while the adsorption strengths of HCOOH are enhanced, suggesting that the FAOR activity may be enhanced. Too high or low binding strength for intermediate species on the catalyst surface will hinder the kinetic process, so it is important to adjust an optimal value. Quan et al. also calculated the free energy of PtSn and PtBi catalysts for oxidizing formic acid through HCOO* and COOH* intermediates.128 The DFT calculations on the PtSn model surface indicate that the dehydration pathway is easier to perform than the dehydrogenation pathway because the CO* formation energy is as low as 0.04 eV, so PtSn has a lower activity for FAOR (Fig. 7B). Compared with PtSn, the CO* formation energy on the PtBi model surface is much higher, indicating that the addition of Bi atoms plays a key role in promoting the dehydrogenation pathway (Fig. 7C). The rate-determining step for formic acid dehydrogenation on PtBi and PtSn surfaces is the first and the second dehydrogenation, respectively. Moreover, Herrero et al. reported that the current density of the Bi adatom-modified Pt (111) electrodes was 30–40 times higher than that of the unmodified surface towards the FAOR, and the enhancement mechanism of the Bi atoms was proved.129 They pointed out that both Bi and Pt atoms are reactive sites. Formate chemisorbed on the Bi atoms by deprotonation of the formic acid molecules, and then the C–H bond of HCOO is cleaved on the adjacent Pt site to form CO2; Bi atoms play a decisive role in the adsorption of formic acid molecules and the further dehydrogenation of the formate intermediate. Although Pt-based catalysts generally oxidize formic acid through the CO pathway, some improved synthesis methods can change the reaction pathway. For instance, butylphenyl-functionalized Pt nanoparticles (Pt-BP) can greatly inhibit CO adsorption.130 Electrochemical in situ FTIRS shows that the Pt-BP catalyst effectively blocked the CO poisoning pathway and enhanced the electrocatalytic activity towards the FAOR through the third-body effect (Fig. 7D). Furthermore, regulating the morphology of metal nanocrystals can also change the reaction pathway, such as PtPb single-crystalline nanodendrites.131 This nanostructure shows excellent activity for the FAOR and the anti-CO-poisoning ability. In Fig. 7E, three HCOOH peaks and one CO2 peak can be seen, indicating that the as-prepared single crystal structure oxidizes formic acid through the direct route. Thus the main methods to improve the FAOR performance include weakening the dehydrogenation barrier, enhancing the adsorption strength of reactant molecules, and eliminating CO poisoning.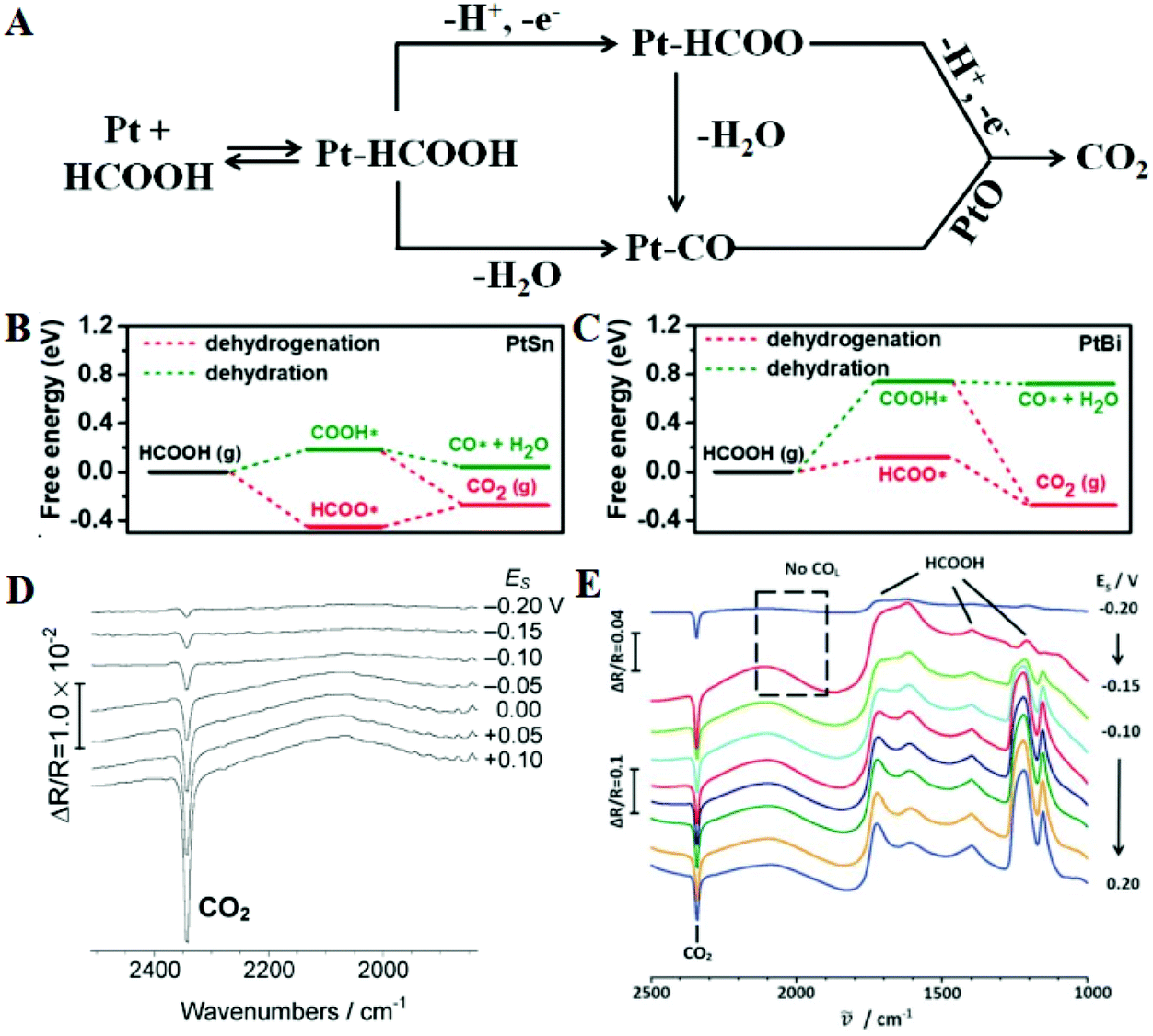 | ||
| Fig. 7 (A) Dual-path mechanism for formic acid oxidation on the surface of a Pt electrode. The free energy diagrams for dehydrogenation and dehydration steps on the (100) surface of PtSn (B), and PtBi (C), respectively. Reproduced with permission from ref. 128. Copyright 2019, Wiley-Blackwell. In situ FTIR spectra of formic acid oxidation on Pt-BP nanocatalysts (D) and PtPb nanodendrites (E) in HCOOH + 0.1 M H2SO4, ER = 0.25 V vs. SCE. Reproduced with permission from ref. 130. Copyright 2012, Royal Society of Chemistry. Reproduced with permission from ref. 131. Copyright 2016, Royal Society of Chemistry. | ||
Considering the FAOR of most Pt-based catalysts via the indirect pathway, adding oxophilic metals and engineering the surface/near-surface structure can promote CO oxidation or weaken CO adsorption by providing available OH* species at lower potentials, which help to improve the reaction rate and elelctrocatalytic performance. Li et al. provided a kinetically controlled method to tune the surface structure of nanocrystals, and cubic, concave cubic, and defect-rich cubic Pt3Sn nanocrystals were synthesized.132 Electrochemical tests confirmed the excellent FAOR performance of defect-rich intermetallic Pt3Sn, verifying the close relationship between the catalytic activity and surface defect. Moreover, to verify the relationship between composition and crystal structure, fcc-PtAg alloys with different compositions are synthesized.133 After Pt25Ag75 and Pt51.6Ag48.4 samples are annealed at 700 °C, their crystal structures are Ag-rich fcc alloy and intermetallic compound, respectively. Pt51.6Ag48.4 has a long-range compositional order stacking with 85% on the tetrahedral sites (hcp) and 15% on the octahedral sites (ccp). If the amount of Pt is above Pt51.6Ag48.4, the annealed bimetal separates into a Pt-rich fcc alloy and intermetallic compound. In contrast, if the Pt feeding ratio goes below 51.6/48.4 but above 25/75, an intermetallic compound and an Ag-enriched fcc-alloy are obtained. The Pt51.6Ag48.4 intermetallic compound has a 29 times higher specific activity at 0.4 V vs. RHE compared with Pt/C. PtAu/Pt intermetallic core/dendritic shell nanocrystals supported on the N-doped graphene support were synthesized by the wet-chemical route.134 This catalyst displays excellent FAOR activity via the direct pathway, because of the large accessible surface active sites, electronic effects, as well as the introduction of a stable N-doped graphene support. PtTe and N-PtTe intermetallic nanoparticles supported on reduced graphene oxide (rGO) were prepared by a two-step method, including the wet chemistry method and subsequent annealing under 5% H2/Ar or NH3, respectively.135 The as-prepared N-PtTe/rGO shows the highest FAOR activity compared to those of commercial Pt/C and PtTe/rGO catalysts, due to the synergetic effects between the electroactive Pt and Te atoms on the surface, in which Te helps to activate OH* and allows Pt to easily oxidize the COads species to CO2. Importantly, N-PtTe/rGO shows more excellent durability than the other two catalysts.
PtxCu100−x nanocubes were also successfully prepared using a colloidal approach, and the effects of the morphology and composition on the electrocatalytic properties for the FAOR were investigated. Pt80Cu20 nanocubes display the best electrocatalytic activity and remarkable long-term stability.136 It is well known that PtBi is the most promising candidate for Pt-based binary alloys. Sun et al. have showed the catalytic activity of tetrahexahedral (THH) Pt nanocrystals, which are bound by high-index facets and possess a superior electrocatalytic activity. The performance of THH Pt nanocrystals can be further improved significantly by decorating Bi adatoms yielding the third body effect and electronic effect.137 In comparison with bare THH Pt nanocrystals, the catalytic activity of the Bi decorated THH Pt nanocrystals has been drastically enhanced as high as 65 times at 0.4 V vs. RHE towards the FAOR. Recently, Mirkin et al.138 developed an effective alloying–dealloying shape-regulating process to prepare a series of monometallic and bimetallic alloy THH particles, the {210} planes of which were exposed and confirmed by simulation and experiment. These THH particles were synthesized on silicon wafers or carbon supports using trace elements (such as Sb, Bi, Pb, and Te) to stabilize high-index facets. As shown in Fig. 8A, four Pt-based THH with a particle size of about 500 nm are prepared; the evaporation or dealloying process of foreign metals is critical for generating the near-perfect THH particles. Simultaneously, this solid-phase synthesis route is useful for reestablishing the important THH structure from irregular nanoparticles. Fig. 8B shows that Bi-modified THH-shaped PtBi nanoparticles have exposed planes of {210} and the proportion of Bi is 1.2%. As is well known, the rank order of specific surface energies is (111) < (100) < (110) < (210). However, the DFT calculation results confirm that the specific surface energies of these facets changed drastically after foreign metal modification, and the specific surface energy of Pt (210)-foreign-metal is the lowest. Electrochemical tests of all catalysts are carried out in Ar-saturated 0.5 M H2SO4 solution (Fig. 8C); these curves show the absorption and desorption signals of H. Compared with commercial Pt/C and Pt-control catalysts, Pt-based THH displays better catalytic performance toward the FAOR (Fig. 8D). In FAOR curves, the peak I at ∼0.5 V corresponds to the oxidation of formic acid via the dehydrogenation pathway, and peak II at ∼0.9 V corresponds to the oxidation of COads formed via the dehydration pathway. Moreover, the specific activity of the THH PtBi particles made from a commercial Pt/C catalyst is about 20 times that of the Pt/C catalyst at 0.5 V vs. RHE. Furthermore, Ding et al. developed an ultra-thin layer (around 100 nm) structured anode consisting of an atomic layer Bi@Pt supported on nanoporous gold, which facilitates the mass transfer and improves the utilization of Pt.139 The maximum power density of MEA using this anode catalyst reaches ∼80 mW cm−2, which is twice that of the Pt/C catalyst.
 | ||
| Fig. 8 (A) Representative SEM images of THH Pt, and Pt particles synthesized through Bi, Pb, and Te modification. (B) STEM images, EDS elemental maps, TEM images, and the corresponding diffraction patterns of the PtBi particles. (C) CVs of THH-shaped Pt–M (M = Sb, Bi, Pb, or Te) catalysts, Pt control sample, and commercial Pt/C catalyst in Ar-saturated 0.5 M H2SO4 solution. (D) Polarization curves of formic acid oxidation for different catalysts. Reproduced with permission from ref. 138. Copyright 2019, American Association for the Advancement of Science. | ||
Alternatively, Pd-based nanocatalysts can directly oxidize HCOOH to carbon dioxide via the dehydrogenation pathway.140–142 Tang et al. have developed a novel carbon-supported cyanogel (C@cyanogel)-derived strategy to fabricate the Pd3Fe/C intermetallic compound; this method could effectively suppress the movement of Pd and Fe atoms in the crystal and benefit the formation of an ordered structure.143 The as-prepared intermetallic Pd3Fe/C exhibited high electrocatalytic activity (696.4 mA mg−1) for the FAOR relative to the fcc-Pd3Fe/C (493.9 mA mg−1) and commercial Pd/C (364.6 mA mg−1) nanocatalysts, which can be ascribed to the ordering structure and electronic effect. Hollow PdAg nanostructures with exposed [100] facets can be obtained by a galvanic replacement between Ag nanocubes and Pd precursor.144 The atomic ratio of the as-synthesized PdAg samples is controlled by changing the addition amount of the K2PdCl4 precursor, and the nanobox gradually becomes a hollow structure with the increase of Pd content. The FAOR activity of these samples showed an intuitive volcanic relationship with the Pd content, and PdAg alloys with a Pd content of 0.317 show the highest activity due to electronic effects and enhanced HCOO adsorption. PdCu nanosheets (thickness is about 2.8 nm) were prepared by the wet chemical method, and post-treatment using ethylenediamine (EN) can remove the surfactant on the catalyst surface.145 After ligand exchange experiments, EN exchanges oleic acid on the nanosheets. EN is an electron donor, so EN-treated samples are helpful for the adsorption of electron-deficient intermediates. The EN-treated sample has a Pd/Cu atomic ratio of 20:3, and the highest ECSA and mass activity, namely about 139.8 m2 gPd−1 and 1655.7 mA mgPd−1, respectively. The above examples demonstrate the advantages of Pd-based catalysts towards FAOR, but the stability of Pd-based catalysts is poor, and the electrocatalytic activity is not as high as Pt-based catalysts.
Adding other oxophilic metals into Pt atoms could improve the FAOR performance through the electronic effect, strain effect, and ordered structure and more importantly, optimize the binding strength between the active sites and the reaction intermediates. Moreover, alloys have a lower affinity for the CO poisoning intermediate, so formic acid is more likely to be directly oxidized to CO2via the dehydrogenation pathway.
3. Hydrogen-oxygen fuel cell electrocatalytic reactions
Clean and efficient alternative FC technologies have been extensively researched and developed in generation plants and automotive vehicles.146 The anodic hydrogen oxidation reaction (HOR) of FCs is one of the best-studied reactions, and the reaction is very rapid in the presence of a Pt-based catalyst.147 One of the greatest problems with HOR in acidic medium is the deactivation of Pt-based catalysts by small amounts of CO; CO present in the fuel as an impurity as the industrial hydrogen is produced by reforming hydrocarbons.148 Realization of the hydrogen economy depends on efficient hydrogen production, storage, and utilization. In alkaline environments, the HOR kinetics are more sluggish by about 2 orders of magnitude than in acid medium, most likely due to the co-adsorption of cations on active sites.55,149 At the cathode, a higher overpotential is required to meet the actual current density, accompanied by an open-circuit voltage significantly below 1.23 V. To overcome the problem of slow ORR kinetics, the cathode Pt loading needs to be increased to 8 times of the anode to achieve the desired FC performance.56,150 A high power density (>1.0 W cm−2) in MEA is the key to FC vehicle operation, but it is often limited due to low mass and charge transfer efficiency. Electrocatalysts used in practical FCs are limited by the harsh testing conditions, so it is necessary to develop efficient and more durable nanomaterials.151–153 The goal of this section is to provide an overview of noble metal alloys used in the ORR and HOR, with a summary highlighting the future directions for this field.3.1 ORR
In contrast to the anodic counterpart, the largest obstacle to the practical use of FCs is the sluggish reaction kinetics for the cathodic ORR.154–157 In 2020, the target mass activity and acceptable durability for the U.S. Department of Energy are >0.44 A mg−1 at 900 mViR-free and <40% loss in mass activity after 30000 cycles, respectively.158 Researchers around the globe are therefore investing much effort to develop both active and durable industrial electrocatalysts beyond the state-of-the-art Pt/C reported to date.159–164 Enhanced activity of alloy catalysts is due to the reduced adsorption energy of the blocked OHads by the electronic effect, strain effect, and ordered structure, resulting in a greater number of active sites available for the ORR. A long-standing problem in the study of the ORR has been to determine the reaction mechanism behind the electrocatalytic enhancement. The ORR is a complex process, in which the adsorption of OOH*, O* and OH* species is a key factor in kinetics. Three predominant ORR pathways are shown in Fig. 9A.165,166 The ORR on the Pd (111) surface is carried out through the dissociation and adsorption of O2 molecules in the presence of a hydrated proton (Fig. 9B).167 Protonation of Oads to form OHads on the surface of Pd (111) has been suggested as the main rate-determining step. The kinetics of the ORR is strongly related to the exposed crystal plane of the nanocatalyst, and the activity of Pt (hkl) increases in the sequence of (100) < (110) < (111).168In situ electrochemical surface-enhanced Raman scattering (EC-SERS) was used to obtain direct evidence at the Pt (111) surfaces, reported by Tian's group.169 The in situ EC-SERS spectrum on the Pt (111) electrode from 1.1 V to 0.5 V is shown in Fig. 9C. The Raman signals at around 732 and 933 cm−1 were ascribed to νs. and νs. (ClO4−), respectively, and
and νs. (ClO4−), respectively, and 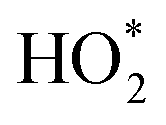 intermediates are stably adsorbed on the Pt (111) surface. As shown in Fig. 9D, the Raman band at 732 cm−1 appears at a potential below 0.8 V, and increases as the potential is made less positive. Therefore, the ORR reaction mechanism is as follows: adsorbed O2 on the Pt (111) surface forms the
intermediates are stably adsorbed on the Pt (111) surface. As shown in Fig. 9D, the Raman band at 732 cm−1 appears at a potential below 0.8 V, and increases as the potential is made less positive. Therefore, the ORR reaction mechanism is as follows: adsorbed O2 on the Pt (111) surface forms the  intermediate through proton and electron transfer, and then dissociates into OH* and O* intermediates, and finally the OH* species is converted to H2O (Fig. 9E). From the above description, we can find that the reaction mechanism and the rate-determining steps are largely dependent on the active sites. In principle, the optimal electronic and geometric structures with the appropriate intermediate binding energy will lead to better catalytic performance.
intermediate through proton and electron transfer, and then dissociates into OH* and O* intermediates, and finally the OH* species is converted to H2O (Fig. 9E). From the above description, we can find that the reaction mechanism and the rate-determining steps are largely dependent on the active sites. In principle, the optimal electronic and geometric structures with the appropriate intermediate binding energy will lead to better catalytic performance.
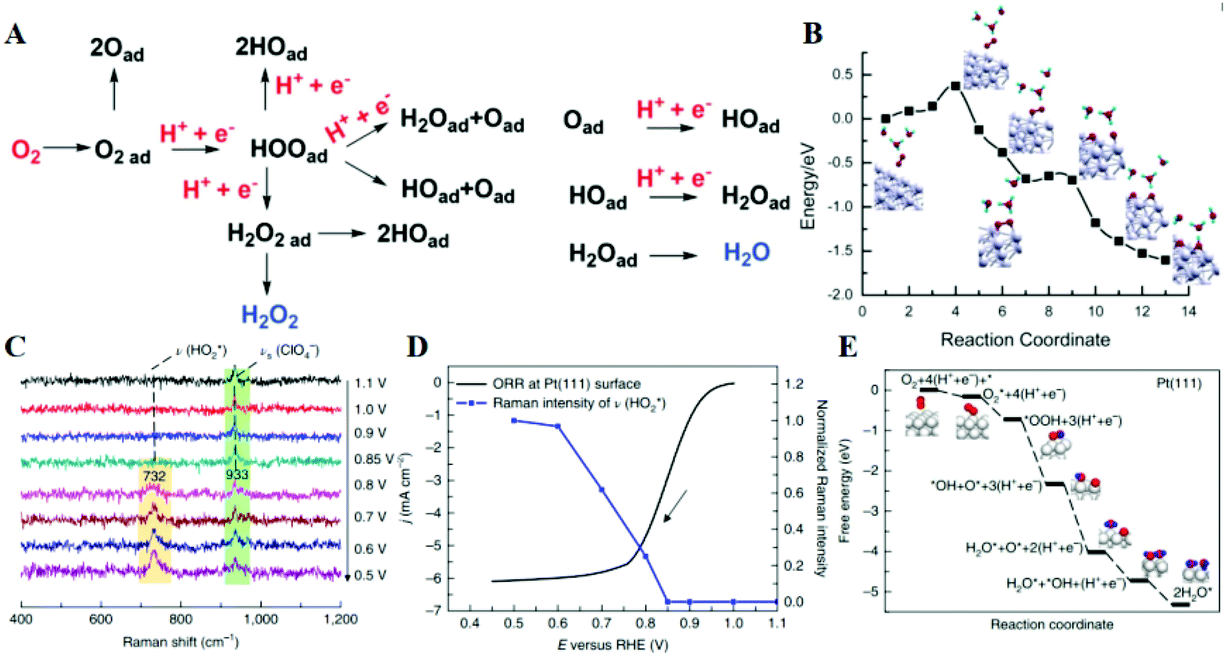 | ||
| Fig. 9 (A) Schematic illustration of the possible ORR pathways. Reproduced with permission from ref. 166. Copyright 2019, Wiley-Blackwell. (B) Minimum-energy paths on the Pd (111) surface in the presence of hydrated proton. Reproduced with permission from ref. 167. Copyright 2013, American Chemical Society. (C) In situ EC-SERS spectra of the ORR system at a Pt (111) electrode surface in a 0.1 M HClO4 solution saturated with O2. (D) Normalized EC-SERS intensities of the stretching mode of O–OH around 732 cm−1 at different potentials. (E) The proposed ORR mechanism at Pt (111) surfaces in 0.1 M HClO4 solution and relevant Gibbs free energy (eV). Reproduced with permission from ref. 169. Copyright 2018, Springer Nature. | ||
Many advances have been made in the design of nanocatalysts to improve the activity, durability, and cost of Pt-based nanomaterials. One-dimensional bunched PtNi alloy nanospheres (BNSs) were synthesized, and then Ni species were selectively etched in acidic solution to form bundled nanocages (BNCs) with a Pt-skin.170 Except that the precursor of Ni was not introduced, Pt nanowires (NWs) were obtained using a similar method. The TEM image (Fig. 10A) shows one-dimensional and highly dispersed PtNi-BNCs, whose length can reach hundreds of nanometers. Internal voids and dark walls with an average thickness of 2.2 nm are observed in the enlarged TEM image (Fig. 10B). And in the HRTEM images, the lattice fringe of 0.221 nm corresponds to the (111) plane of the PtNi alloy. In addition, Fig. 10C shows the appearance of high-index (211) and (311) facets with high-density steps and edges. STEM-EDS elemental mapping (Fig. 10D) further confirms the Pt-skin structure with 2–3 atomic layers of the wall in BNCs. Fig. 10E shows the ORR activity of four catalysts: PtNi-BNCs/C, PtNi-BNSs/C, Pt NWs/C, and Pt/C; the BNC nanostructure exhibits the highest mass and specific activity of 3.52 A mgPt−1 and 5.16 mA cmPt−2, which are about 17 and 14 times higher than commercial Pt/C, respectively. The PtNi-BNCs/C catalyst also exhibits excellent durability, after 50000 cycles, the activity decay is negligible (less than 1.5%) (Fig. 10F). This work also studied the synergistic effect of strain and the coordination number on the d-band center through in situ X-ray absorption fine structure (XAFS) technology and DFT calculations, indicating that PtNi alloy nanostructures have weaker Pt–O bonds and the optimal adsorption energies compared to Pt. In addition, compared with PtNi-BNSs/C, both the Pt–Ni coordination number and the Pt–Pt bond length for PtNi-BNCs/C catalysts are slightly reduced. Sun et al. used 8 nm PtFe intermetallic nanoparticles as a model catalyst to form core–shell FePt/Pt nanoparticles with about 2 atomic layers of Pt shell, in which Fe has better stability.66 This FePt/Pt intermetallic compound showed excellent mass activity and better durability for the ORR in 0.1 M HClO4 solution (at 60 °C) and in the MEAs at 80 °C. According to the half-cell ORR test, the mass activity is 0.7 A mg−1 at 0.9 V; and the mass activity shows no obvious loss after 30000 fuel cell testing cycles between 0.6 V and 0.95 V in the MEAs, reaching the DOE 2020 target. In 2019, they reported CoPt/Pt nanoparticles with a tetragonal intermetallic core and 2–3 atomic layers of strained Pt shell as a highly active and durable ORR catalyst.171 The structure was prepared by converting 9 nm fcc-CoPt into a CoPt intermetallic compound (ordered degree: 88%), followed by acid etching at 60 °C and annealing at 400 °C. In Fig. 10G, the STEM image shows that the structure is formed with the core containing alternative layers of Pt and Co and the shell being 2–3 atoms thick of Pt. ICP analysis shows that the Co composition in CoPt/Pt dissolved 10.2% in the 24-hour etching, while fcc-CoPt lost 69.4% during the 7 hours of etching time, which further demonstrates that the CoPt/Pt nanoparticles could effectively protect Co from acid corrosion. The specific and mass activity of CoPt/Pt is ∼38 and ∼19 times, respectively, that of commercial Pt/C. In the MEA test, the mass activity of CoPt/Pt reaches 0.56 A mgPt−1 with 19% loss at 80 °C after 30000 cycles, exceeding the target of DOE 2020. For associative mechanisms, the formation of HOO* (A2) is uphill in free energy along with the protonation steps of O* (A4) and HO* (A5), as shown in Fig. 10H. For the dissociative mechanism, the two protonation steps of O* (D2) and HO* (D3) are uphill in free energy diagrams (Fig. 10I). With the decrease of CoPt/Pt overlayer thickness, the enhancement of the electronic effect further reduces the overpotential in the dissociation mechanism. For PtFe and PtCo systems, A2 and D3 are the potential limiting steps for the association mechanism and dissociation mechanism, respectively. DFT calculations indicate that the electronic effect and biaxial strain weaken the binding of all oxygenated intermediates on the Pt surface, resulting in an enhanced ORR performance. Wu et al. developed a novel method that used ZIF-derived carbon as a Co source and C support to prepare the Pt3Co intermetallic nanocatalyst via the thermal treatment of Pt nanoparticles supported on ZIF-derived carbon.172 Co atoms in ZIF-derived carbon can diffuse into Pt nanoparticles through high-temperature annealing to form the Pt3Co intermetallic compound, and the Co content in ZIF and annealing temperature are critical to the formation of intermetallic nanostructures. Moreover, Huang et al. prepared hexagonal PtBi intermetallic nanoplates using the colloidal chemistry method, which exhibited high tolerance over chemical fuels (such as CH3OH, HCOOH, and CO).173 In general, the ORR activity can be improved by adjusting the adsorption energy of the atomic O, and the upshift or downshift of the Pt d-band center has a great influence on this adsorption energy.
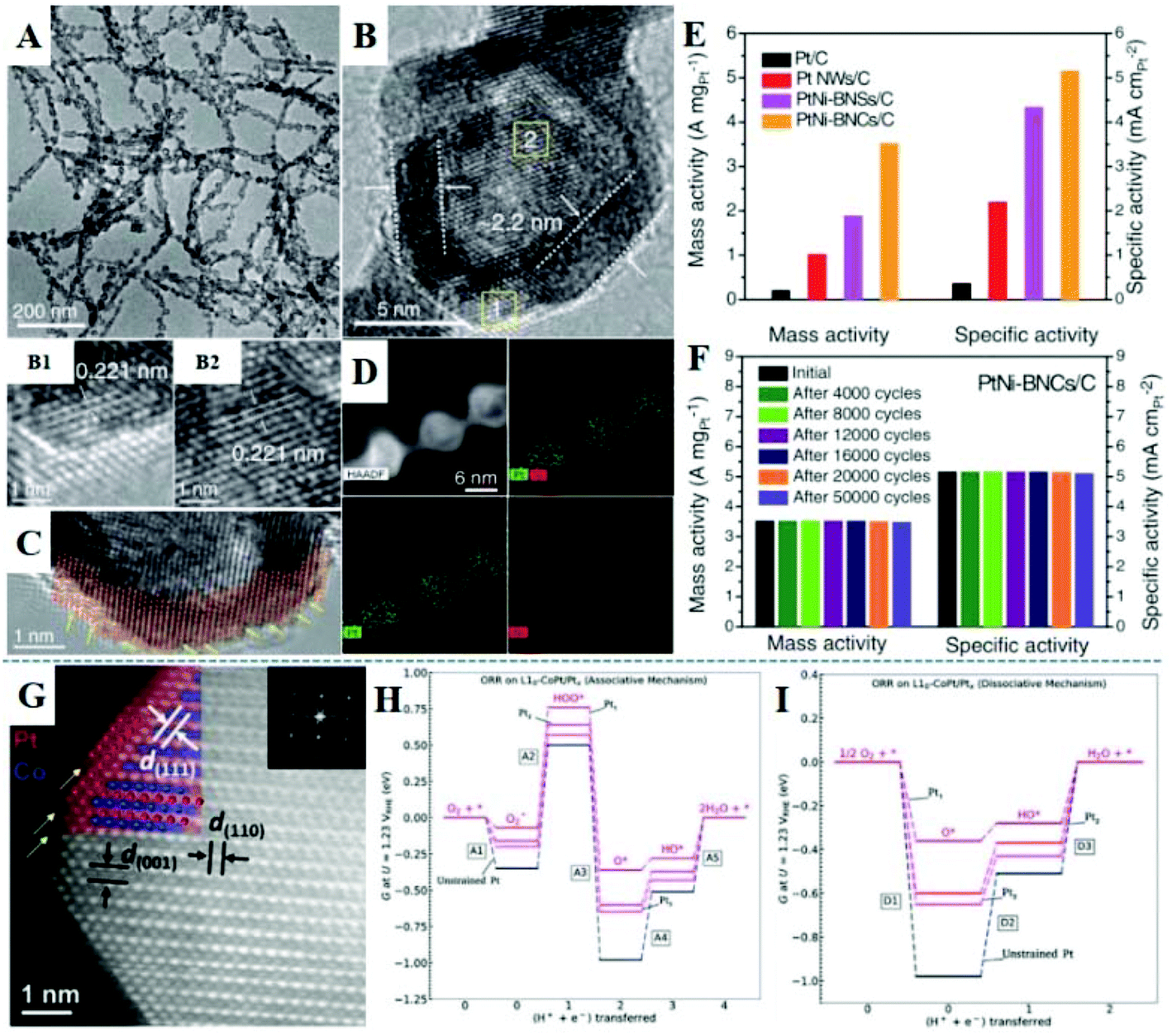 | ||
| Fig. 10 (A) TEM images of PtNi-BNCs. (B) Enlarged TEM image and the corresponding HRTEM images of the areas marked by yellow squares. (C) Atomic-resolution HRTEM image. (D) HAADF-STEM images. (E) Mass and specific activities of Pt/C, Pt NWs/C, PtNi-BNSs/C, and PtNi-BNCs/C at 0.9 V vs. RHE. (F) Mass and specific activity evolutions of PtNi-BNCs/C. Reproduced with permission from ref. 170. Copyright 2019, American Association for the Advancement of Science. (G) Enlarged STEM image of L10–CoPt/Pt nanoparticles, Pt is colored in red and Co is colored in blue. The free energy diagram calculated via the DFT method on the associative pathway (H) and on the dissociative pathway (I) for the L10–CoPt/Ptx (111) surface (x = 1–3 Pt overlayers) and the unstrained Pt (111) surface. Reproduced with permission from ref. 171. Copyright 2019, Cell Press. | ||
In order to reduce the cost of nanocatalysts, Pd-based alloys are used instead of Pt-based catalysts, and great efforts have been made to enhance the catalytic activity of Pd-based alloys to reach the level of Pt-based catalysts.174,175 Pd3Pb intermetallic compound was synthesized using a modified impregnation–reduction and heat treatment approach, and the insoluble KCl by-products could prevent the agglomeration of nanoparticles during the annealing process.176 The experimental results show that increasing the annealing temperature and time is beneficial to form the intermetallic structure. Samples annealed at 600 °C for 24 hours are fully ordered because the ratios of the (110) to (111) peak are very close to the reference bulk X-ray pattern. The mass activity of the Pd3Pb/C intermetallic compound is 168.9 mA mgPd−1 at 0.9 V, which is ∼2–4 times higher relative to random Pd3Pb/C, Pd/C, and Pt/C samples. Fcc-FePd and fct-FePd alloys were obtained by reduction annealing of core–shell Pd-Fe3O4 nanoparticles, and then intermetallic PdFe was subjected to Fe etching to form an fct-FePd/Pd structure.177 When using acetic acid to etch nanoparticles at a controlled temperature (from 25 to 70 °C), the thicknesses of the Pd shell can be controlled to 0.27, 0.65 or 0.81 nm, thereby tuning the compressive strain and the binding energy of oxygen. Among the three different types of core–shell FePd/Pd samples studied, the fct-FePd/Pd-0.65 exhibits the best activity and stability for the ORR, which originates from the desired Pd lattice compression in the 0.65 nm Pd shell induced by the fct-FePd core. Atomic ordering nanoparticles exhibit significantly enhanced catalytic performance for the ORR due to the electronic effect and ordered structure. Moreover, the addition of gold atoms can cause PdCo nanoparticles to be an ordered structure during the annealing process at 800 °C, and the PdCo has a rhombohedral structure decorated with Au clusters.178 PdMo alloy with ultrathin nanosheets (0.88 nm) was synthesized by a wet chemical method.179 The nanostructure can achieve a larger ECSA and higher atomic utilization, resulting in the mass activity towards the ORR 327 times higher than that of commercial Pd/C. DFT calculations show that alloying, strain, and size effects all help to increase the ORR activity of PdMo nanoplatelets, as they can adjust the electronic structure to optimize oxygen binding. Research on Pd-based catalysts has also made some progress, but their electrocatalytic activity and stability still cannot reach the level of Pt-based catalysts.
Post-treatment of Pt-based or Pd-based nanoparticles to form a Pt-rich structure will result in weaker binding of adsorbates, especially OHads, enhancing the catalytic activity and durability, as proven by the above systems. Peter Strasser et al. reviewed the progress in their preparation, structural characterization, and electrocatalytic performance of the dealloyed Pt-based core–shell structure.180,181 Illustration of basic synthesis approaches is shown in Fig. 11A. The approaches of electrochemical dealloying, acid leaching, thermal annealing, and surface segregation have been mentioned above. In particular, alloy nanostructures with a Pt shell or Au-doped structure can be achieved by means of galvanic displacement. This brings the benefits of controlling the shell thickness and surface strain, thereby increasing the atomic efficiency. Abruña and Wang et al. have done a lot of work in the field, with K2PtCl4 or KAuCl4 as the precursor of the galvanic replacement reaction. For example, PdCo@Pd/C core–shell nanoparticles were successfully synthesized using an adsorbate-induced surface segregation method.182 The electrocatalytic activity of the PdCo@Pd/C catalyst towards the ORR is lower than that of Pt/C. However, the activity and stability were enhanced, after a small amount of Pt was deposited on the surface of PdCo@Pd/C nanoparticles to form a Pt-decorated PdCo@Pd/C nanocatalyst. The simple method significantly reduces the loading of Pt. Moreover, the ORR performance of PdFe nanoparticles is reasonably improved by the structural transition from the fcc-PdFe/C to fct-PdFe/C.183 To further enhance the activity of fct-PdFe/C, a Pt layer was decorated on the nanoparticle surface forming an fct-PdFe@Pt/C structure (Fig. 11B), because the equilibrium electrode potential of the PtCl42−/Pt couple is more positive than those of the Fe2+/Fe and PdCl42−/Pd couples. The mass and specific activity of fct-PdFe@Pt/C is 23.5 and 140 times higher compared to Pt/C, respectively, which can be ascribed to the strain effect and weakened oxygen affinity. Then, a series of PdFe@Pd/C nanoparticles with a tunable core composition were synthesized and decorated with ultralow amounts of Pt.184 The bimetallic nanoparticles with optimal atomic ratio achieved 14 times higher mass activity than that of Pt/C. Compressive strain and electronic effects appear to be competitive for ORR activity and suggest a volcanic curve between activity and composition. This work thoroughly investigates the minimal Pt/Pd required for the monolayer coverage of Pt, and presented a formula to calculate the Pt layer thickness based on Pt content and nanoparticle size (Fig. 11C). Based on this calculation formula, the Pt/Pd atomic ratio of 10 nm PdFe@Pt particles must be larger than 1:2.7 to obtain a Pt monolayer with 100% coverage, which is also true for AuCu@Pt185 and Pd6CoCu@Pt/C.186 The PdZn/C intermetallic nanocatalyst with several atomic-layer Pd shells was transformed by random PdZn/C annealing at 500 °C, and the ORR activity was increased by three times relative to Pd/C.187 Also, Au was incorporated into the PdZn/C intermetallic compound (Au–PdZn/C) by a galvanic replacement (Au:Pd = 1:40), since AuCl43−/Au has a more positive redox potential than those of the PdCl42−/Pd and Zn2+/Zn couple. Au–PdZn/C exhibits a homogeneous elemental distribution of Au, Pd and Zn based on the corresponding EDX elemental line profiles (Fig. 11D), suggesting that Au is uniformly distributed through the entire particle. Unlike the Pt galvanic replacement method, Au not only galvanically replaced Pd and Zn on the surface but also entered the PdZn lattice to form a ternary alloy. After 30000 potential cycles, the mass activity of Au–PdZn/C decreased by less than 10%, due to the ordered structure and stabilization effect of Au atoms.
 | ||
| Fig. 11 (A) Illustration of the basic synthesis approaches for the preparation of Pt-skin core–shell nanoparticles (green balls: Pt, yellow balls: a less noble metal). (B) Schematic illustration of intermetallic PdFe@Pt/C, and the elemental maps of Pd, Fe, and Pt. Reproduced with permission from ref. 183. Copyright 2018, Elsevier BV. (C) Theoretical calculations of the Pd/Pt atomic ratio as a function of the particle size and the shell thickness (x); solid lines are Pd@Pt core–shell particles, while dashed lines are PdFe@Pt. Reproduced with permission from ref. 184. Copyright 2018, American Chemical Society. (D) STEM image and the corresponding EDX elemental maps of Pd, Zn, and Au. Reproduced with permission from ref. 187. Copyright 2019, American Chemical Society. | ||
In short, the platinum atom is still the most active site for the ORR, so the design and research on metal catalysts have attempted to mimic the geometric, electronic, and ordering structures of Pt. The above structures can be used to change the adsorption energy of reaction intermediates, thereby enhancing the electrocatalytic performance for the ORR, and the Pt-skin structure is an excellent configuration. Although the MEA test results of some Pt-based cathode catalysts have reached the DOE 2020 target, further efforts are needed in terms of the cost, activity and durability of nanocatalysts.
3.2 HOR
The HOR is the counter-reaction of the ORR in a hydrogen-oxygen fuel cell, which can directly convert chemical energy to electricity, and has gained more and more attention. For PEMFCs, only very small amounts of Pt are required due to its extremely high activity for the HOR. However, the rate of the HOR is so high in acid that the conventional rotating disk electrode (RDE) measurements cannot provide sufficiently high hydrogen mass-transport rates, due to the Nernstian diffusion overpotential.188 Until then, 100-fold lower exchange current density values for Pt in acid were reported erroneously, generally based on RDE measurements, a microelectrode would be required to quantify kinetic rates of the HOR in acid unambiguously.189 Indeed, in the face to the development and application of fuel cells, anode catalysts with a high CO tolerance are essential to reduce the cost of and simplify the stationary fuel cell system. In practice, Pt-based catalysts are extensively used in many electrocatalytic processes, such as we mentioned before, but they presented a weak selectivity. Currently, in industry, most of H2 was produced from hydrocarbon reforming from fossil fuels cost-efficiently, while small amounts of CO were introduced in reformed H2. Impurities can strongly adsorb on the Pt surface, thereby affecting the PEMFC performance and durability. Even low concentrations (∼10 ppm) of CO drastically reduce the performance of PEMFCs.190 Anti-poisoning technology is critical for applications that use reformate hydrogen as a fuel. Hence, it's urgent to design Pt-based catalysts that can tolerate these poisonous species. Typically, two different ways were proposed to enhance the CO tolerance of nanocatalysts: (1) combining Pt with oxophilic metals or metallic oxides to promote CO oxidation, since the second metal can reduce the CO adsorption or OHads can accelerate the electrooxidation of CO. The classical CO electro-oxidation reaction model is the Langmuir–Hinselwood (L–H) mechanism, which can be summarized as follows:191| COads + OHads → COOHads | (a) |
| COOHads + OHads → CO2 + H2O | (b) |
Another CO oxidation in this onset region is relying on an Eley–Rideal mechanism where the adsorbed CO reacts with bulk water (or OH−) and not as evidence that OH− has adsorbed onto the catalyst surface;192 (2) the diameters of CO molecules ∼2–5 Å, allowing them to adsorb on isolated metal atoms created by molecular architectures.
The improvement of PtRu nanoparticles is one of the most promising roads to obtaining highly tolerant anode catalysts. Takeguchi et al. optimized the preparation of PtRu catalysts using a rapid quenching method and realized thoroughly mixed PtRu nanoparticles (Fig. 12A).193 An MEA using this sample as an anode catalyst exhibits higher performance than that with conventional PtRu catalysts at a high concentration of CO. The addition of Ru contributes to CO removal, resulting in the enhancement of the efficiency of residential fuel cell systems. Zhang et al. synthesized the surface composition of Pt3Co/C catalysts with Co-increased, the intermetallic compound, and Pt-increased by the surface segregation approach.191 Due to the differences in the surface atomic distribution and alloying extent, the nanocatalysts show different CO poisoning tolerance in the order of Co-increased > intermetallic compound > Pt-increased. They found that the electronic effect plays a major role in weakening CO adsorption on Pt3Co/C nanocatalysts and thus promoting CO oxidation to form the COOHads intermediate consistent with the Langmuir–Hinselwood mechanism, and the oxophilic effect promotes the oxidation of the COOHads intermediate.
 | ||
| Fig. 12 (A) Schematic illustration of CO tolerance on the PtRu alloy. Reproduced with permission from ref. 193. Copyright 2012, American Chemical Society. (B) Arrhenius plots for the apparent rate constant kapp for the HOR (CO-free) at the Nafion-coated supported catalysts. Reproduced with permission from ref. 196. Copyright 2017, American Chemical Society. (C) The proposed model for the reduced form DAcPy adsorbed on Pt (111) from the side view. (D) CO tolerance test of commercial PtRu in 0.5 M H2SO4 solution with (red line) and without (green line) 10 mM DAcPy. Reproduced with permission from ref. 198. Copyright 2018, Royal Society of Chemistry. | ||
However, the decrease in anode performance by the dissolution of a 3d transition metal from anode catalysts has been shown to contribute to degradation in long-term operation. There is a desire for improved durability of the anode catalyst against a high concentration of CO and/or a transient increase in the CO concentration. In particular, an anode catalyst that could tolerate a high concentration of CO could eliminate the need for a selective CO cleaner, and hence help to reduce the cost of and simplify the stationary PEMFC system. Hsieh et al. have designed well-defined Ru@Pt nanoparticles with complete, uniform Pt shells providing an opportunity to achieve the CO tolerance and corrosion resistance needed for commercializing reformate-fed PEMFCs, which was further confirmed by MEA. Their results suggest that CO tolerance can be improved by reducing the CO adsorption energy by tuning the d-band of the Pt shell.194 Similarly, PtCo alloy with a Pt-skin structure shows highly CO-tolerant HOR activity and stability, and this enhancement is ascribed to the weak Hads and COads adsorption introduced by Co.195 They further examined the effect of the non-precious-metal species M (M = Fe, Co, Ni) in Pt2AL–Pt–M/C on the CO tolerance at 70 and 90 °C. The HOR activities increased in the order Pt < Pt2AL–PtNi < Pt2AL–PtCo < Pt2AL–PtFe, which were ascribed to the decreasing H adsorption strength on the (111) terraces (Fig. 12B).196 Igarashi et al. found that the surfaces of all non-precious metal alloys are composed of a thin Pt layer with an electronic structure different from that of pure Pt, indicating an increased 5d vacancy of Pt in the layers of the CO-tolerant alloys. In situ FTIR spectra further demonstrate the weak bond strength between the Pt skin layer and CO, suggesting that the oxidation sites are not blocked by CO due to its enhanced H2 mobility.197
Recently, a CO-tolerant anode catalyst using an organic complex as a co-catalyst is also attractive. DAcPy-modified Pt exhibits higher CO tolerance than most frequently used PtRu alloy catalysts. This molecular architecture acts as a canopy, under which Pt atoms can be accessible for small-sized H2, but not for relatively large CO and H2S (Fig. 12C and D).198 This novel concept for a CO-tolerant anode catalyst using an organic complex as a co-catalyst is an attractive way for the improvement of CO tolerance. Poor tolerance to CO impurity is a drawback of PEMFCs which makes the fuel cell system complicated and expensive. The tolerance to CO impurity has attracted attention not only for the stationary applications but also for use in transportation from the viewpoint of hydrogen purity. The development of innovative and practical technology would be greatly appreciated.
Additionally, the HOR in alkaline media is more attractive and promising with the development of an alkaline membrane (OH−-exchange membranes) for the replacement of the noble-metal intensive PEM technology. In an alkaline electrolyte, non-noble metal catalysts are very active and stable for the ORR and the OER.188,189 However, for yet unclear reasons, the kinetics of the HOR in the base is 1–2 orders lower in acidic pH, and large amounts of Pt are needed to catalyze the HOR in alkaline medium.199 Correspondingly, it is under extensive debate on how the transition metals promote the alkaline HOR kinetics of Pt.200–203 Understanding such a scientific foundation and underlying principles of alkaline electrochemistry is a critical challenge to make alkaline membrane fuel cells (AMFCs) more practical. There are currently two different views of the catalytic roles of the second metal in promoting the HOR of Pt surfaces. Despite the extensive debates within the thoughts, it is generally believed that the HOR proceeds via the Tafel–Volmer or Heyrovsky–Volmer mechanism in base with the Volmer step as the rate-determining step:17,204,205
| Tafel step: H2 + 2* ↔ 2Hads |
| Heyrovsky step: H2 + OH− + * ↔ Hads + H2O + e− |
| Volmer step: Hads + OH− ↔ * + H2O + e− |
The classical model regards the hydrogen binging energy (HBE) as the only descriptor of the HOR, as represented by the Sabatier principle, that too strong or too weak of Pt–Hads interaction is the disadvantage of HOR activity.206 This view is accessible in an acid electrolyte because of the very fast reaction kinetics, however, it has been doubtful in alkaline media because of the 1–2 orders lower kinetics. The first point supports the classical model. Normally, the adsorption/desorption peak potential (Epeak) can reflect the Pt–Hads interaction (−FEpeak = ΔH) (Fig. 13A). Sheng et al. found that the HBE shows a monotonic decrease with the hydrogen oxidation activity, demonstrating that Hads is the only reaction descriptor for the HOR on monometallic platinum.207 Abruna et al. found that HOR activity on Pt/C and Ir/C is better than that on Ru/C and Pd/C. This is in accordance with the fact that the H adsorption/desorption process on Pt/C and Ir/C is more reversible than that on Ru/C and Pd/C (Fig. 13B).208 As shown in Fig. 13C, the peak power density is improved to 1.0 W cm−2 for PtRu/C as the HOR catalyst for the AMFCs, in comparison to 0.6 W cm−2 when using Pt/C as the anode catalyst. Wang et al. ascribed such a remarkable improvement to electronic effects, because OHads can generate on specific sites of the PtRu/C surface at more positive potentials than on the Pt/C surface. Rather, alloying with Ru weakens the Pt–Hads interaction resulting from the electronic effect demonstrated by the CVs in combination with DFT calculation, which thus supports the HBE theory.209 Lu et al. studied the alkaline HOR mechanism by using pristine Pt/C, PtNi/C and PtNi/C after acid treatment (acid-PtNi/C) as the model catalysts.210 XPS and CVs demonstrate that PtNi/C and acid-PtNi/C have similar HBE values, but weaker than Pt/C. And the OH adsorption on PtNi/C is much stronger than that on acid-PtNi/C and Pt/C. The alkaline HOR activity follows the order of acid-PtNi/C ≈ PtNi/C > Pt/C. Hence, the main reason for the enhanced HOR activities of PtNi alloys may be that the electronic effect weakens the Pt-Had interaction. Ru@Pt catalysts prepared from submonolayer to multilayer Pt coverage were used as a model system to distinguish between the bifunctional mechanism and HBE theory. It shows that the HOR activity of fully Pt-covered Ru is more active than those of partially covered Ru@Pt nanoparticles in 0.1 M NaOH. Therefore, as far as basic HOR is concerned, the role of Ru as an auxiliary metal to modify the electronic structure of Pt atoms is more important than its influence on the cleave the water. Moreover, the most active Ru@Pt core–shell nanoparticles show 4–5 times enhancement of HOR activity as compared to Pt/C, further confirming the role of lowering the Hads energy barrier in the improvement of HOR activity in base.205
 | ||
| Fig. 13 (A) Steady-state CVs of Pt collected in different pH solutions. Reproduced with permission from ref. 207. Copyright 2015, Nature Publishing Group. (B) HOR polarization curves for Pt/C, Pd/C, Ir/C, and Ru/C. Reproduced with permission from ref. 208. Copyright 2017, American Chemical Society. (C) Cell performance of the AMFCs using the Pt/C or PtRu/C as the anode catalyst. Reproduced with permission from ref. 209. Copyright 2015, Royal Society of Chemistry. (D) HOR/HER polarization curves for Pt-poly (black curves) and PtRu alloys with 50% Ru (dashed grey) and 90% Ru (dashed red). The inset shows the comparison between activities at 50 mV on monometallic and bimetallic surfaces for the HOR. Reproduced with permission from ref. 211. Copyright 2013, Nature Publishing Group. (E) HOR polarization curves of Pt/C with 0, 3, 6, and 9 μL doped 5 mM RuCl3 and Pt1Ru1/C. Reproduced with permission from ref. 212. Copyright 2017, John Wiley & Sons Ltd. (F) HER/HOR polarization curves of the Pt disk with 0%, 5%, 15%, and 35% coverage of Ni. Reproduced with permission from ref. 213. Copyright 2019, Elsevier BV. | ||
It is still uncertain the real influence of the second metal on the alkaline HOR activity of Pt surfaces. Another view is proposed by Markovic's group that they ascribed the improvement of alkaline HOR activity to the oxophilic effect. In their study, among different catalysts, both oxophilic metal (Ir) and Pt alloy (Pt0.1Ru0.9) that have an optimal balance between the active sites that contributed to the H2 adsorption/dissociation and to the adsorption of OHad show the maximum HOR activity, in which the oxophilic sites can facilitate the adsorption of OHads species, and then react with the hydrogen intermediates (Hads) (Fig. 13D).211 Similarly, tracing Ru on commercial Pt/C can dramatically promote the HOR activity in the alkaline electrolyte. In situ EXAFS demonstrates the presence of OHads on the surface Ru sites in the HOR potential region and accordingly verifies the bifunctional mechanism for the HOR in alkaline medium (Fig. 13E).212 Ruthenium-based RuxMy/C (M = Pt or Pd) alloy catalysts also were synthesized to investigate the HOR in alkaline electrolytes. The exchange current density on a Pt-rich Ru0.20Pt0.80/C catalyst (1.42 mA cm−2) is nearly 3 times that of Pt (0.490 mA cm−2). Furthermore, the Tafel slope of the RuxPty alloy in 0.1 M KOH is ∼30 mV dec−1, in contrast to ∼125 mV dec−1 of Pt/C, demonstrating that H2 dissociative adsorption is the rate-limiting step. However, RuxPdy/C alloys do not change kinetics obviously. These results were attributed to the interplay of ligand and bifunctional effects. Recently, by using a bulk Pt surface decorated with different coverage Ni(OH)2 and Pt–Ni model catalysts as model catalysts, Sun's group found that the rate of the HER is controlled by both the Hads and OHads while the rate of the HOR is mainly determined by near-optimal Hads (Fig. 13F).213 Particularly, the electronic structure of the Ni(OH)2/Pt disk will be changed with the coverage of Ni(OH)2 increasing.
Overall, bimetallic systems offer a unique opportunity for designing a new generation of nanomaterials for the HOR in alkaline environments. These nanomaterials can be designed to be as active as the best catalysts in acidic media; an understanding of the fundamental mechanism will benefit the discovery of active as well as cost-effective anode catalysts for AMFCs.
4. Conclusion and outlook
In this review, we have presented the recent advancements in understanding the reaction mechanisms and designing nanocatalysts in FC devices. An in-depth understanding of the electrocatalytic reaction mechanisms can guide the synthesis of optimal electrocatalysts, thereby optimizing the bonding strength between intermediate species and active sites, and enhancing the power density of fuel cells. Here, this paper outlines the latest Pt-based alloy nanomaterials, which are important electrocatalysts for electrochemical energy conversion. These alloy nanomaterials with controlled size, shape, and composition have been widely studied as highly active and stable electrocatalysts in an attempt to reduce the required precious metal loading. Improved activity and stability of alloy catalysts can be clearly seen in the electrochemical tests and MEA systems, where the electronic effect, strain effect and ordering structure can change the activation energy barrier and reaction kinetics. Considering the obvious progress in alloy nanomaterials in terms of resistance to poisoning, stability, and activity, we are confident that more exciting work will emerge in this field.Although alloys have made great progress in the field of electrocatalysis, there are still some challenges that seriously hinder their development. First, the electrocatalytic performance of shape-controlled nanocatalysts is often superior to that of spherical nanoparticles due to the surface sensitivity of electrochemical reactions. However, intermetallic nanocatalysts often are obtained by the annealing method, which has to minimize surface energy; the control of ordered Pt-based or Pd-based shape has been met with limited success. Therefore, rationally designing intermetallic nanocatalysts with special crystal planes may enhance the activity and selectivity of fuel cell electrocatalytic reactions. Second, the outermost surface of heterogeneous catalysts maintains the actual structure in the fuel cell electrocatalytic reactions, because the nanocatalyst surface will contact the gas and electrolyte solution (such as H2, O2, HClO4, KOH etc.) during the reaction to cause the surface reconstruction. Third, although more desirable catalysts have been designed in the research of the rotary disk electrode (RDE), some problems occur in the actual fuel cell test systems, for example, the catalysts can hardly reach the performance of RDE, the anodic fuels pass through the membrane to the cathode, and some non-precious metal ions damage the proton exchange membrane (such as Fe-catalysed Fenton reactions), etc. Therefore, the practical application of alloy nanomaterials in fuel cells still has a long way to go.
With the development of materials engineering technologies and theoretical calculations, a large number of highly active alloy catalysts have been successfully designed. The goal of these synthetic strategies is either to improve the binding strength of reaction intermediates or to reduce the reaction energy barrier. The emergence of various high-performance alloy nanocatalysts has laid the foundation for the practical application of fuel cells, and spotlights some insights into the future directions. (1) Some electrocatalytic reaction mechanisms still lack experimental evidence and the reasons for the slow kinetic processes are still ambiguous. The solution to these problems is crucial for the design of highly active and durable nanocatalysts. Many in situ characterization technologies (such as FTIRS, TEM, XRD, XPS, NMR, Raman, XAFS etc.) can monitor the information such as reaction species, structural changes, surface states and adsorption modes, which reveal the underlying reaction mechanism and control of active sites. (2) From the perspective of the electrochemical reaction process, the transport of reactive species through the electric double layer/water layer is very important, and few studies have proposed specific strategies in these aspects. The electronic structure of the electric double layer is closely related to the catalytic surface, so the dynamic changes of the catalyst surface in the electrolyte solution are also the main research directions in the future. (3) For the design of alloy materials, the surface structure of the catalyst must be adjusted due to surface oxidation and dissolution of non-precious metals during long-term operation. In addition to the intermetallic compounds mentioned in this article, the controlled synthesis of thin-layer carbon film-coated alloy nanoparticles, non-metal alloy nanomaterials, and high entropy alloy materials can also enhance the stability. (4) Additionally, the long-term operation of fuel cells also needs to figure out the degradation mechanism of nanomaterials, which has rarely been discussed. In general, the understanding of relevant phenomena in fundamental research, and the development of advanced alloy materials are essential for the future widespread utilization of fuel cells in daily life.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by grants from the National Key Research and Development Program of China (2017YFA0206500), and the National Natural Science Foundation of China (21773198 and U1705253).References
- J. T. L. Gamler, H. M. Ashberry, S. E. Skrabalak and K. M. Koczkur, Adv. Mater., 2018, 30, 1801563–18015681 Search PubMed.
- B.-W. Zhang, H.-L. Yang, Y.-X. Wang, S.-X. Dou and H.-K. Liu, Adv. Energy Mater., 2018, 8, 1703597–1703613 Search PubMed.
- H.-H. Li and S.-H. Yu, Adv. Mater., 2019, 31, 1803503 Search PubMed.
- J. K. Norskov, T. Bligaard, B. Hvolbaek, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 Search PubMed.
- D. R. Rolison, Science, 2003, 299, 1698–1701 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, 4998–5009 Search PubMed.
- E. Antolini, Energy Environ. Sci., 2009, 2, 915–931 Search PubMed.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 Search PubMed.
- J. Jiang, W. Ding, W. Li and Z. Wei, Chem, 2019, 6, 1–17 Search PubMed.
- M. V. Lebedeva, V. Pierron-Bohnes, C. Goyhenex, V. Papaefthimiou, S. Zafeiratos, R. R. Nazmutdinov, V. Da Costa, M. Acosta, L. Zosiak, R. Kozubski, D. Muller and E. R. Savinova, Electrochim. Acta, 2013, 108, 605–616 Search PubMed.
- X.-Y. Lang, G.-F. Han, B.-B. Xiao, L. Gu, Z.-Z. Yang, Z. Wen, Y.-F. Zhu, M. Zhao, J.-C. Li and Q. Jiang, Adv. Funct. Mater., 2015, 25, 230–237 Search PubMed.
- W. Wang, F. Lv, B. Lei, S. Wan, M. Luo and S. Guo, Adv. Mater., 2016, 28, 10117–10141 Search PubMed.
- L. Wu, A. Mendoza-Garcia, Q. Li and S. Sun, Chem. Rev., 2016, 116, 10473–10512 Search PubMed.
- C. Zhu, D. Du, A. Eychmuller and Y. Lin, Chem. Rev., 2015, 115, 8896–8943 Search PubMed.
- S. Furukawa and T. Komatsu, ACS Catal., 2016, 7, 735–765 Search PubMed.
- M. Luo, Y. Sun, L. Wang and S. Guo, Adv. Energy Mater., 2017, 7, 1602073–1602085 Search PubMed.
- X. Yu, J. Zhao, L.-R. Zheng, Y. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, ACS Energy Lett., 2018, 3, 237–244 Search PubMed.
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997–1606025 Search PubMed.
- B. Zhang, G. Fu, Y. Li, L. Liang, N. S. Grundish, Y. Tang, J. B. Goodenough and Z. Cui, Angew. Chem., 2020, 59, 7857–7863 Search PubMed.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y. H. Chung, H. Kim, B. S. Mun, K. S. Lee, N. S. Lee, S. J. Yoo, D. H. Lim, K. Kang, Y. E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 Search PubMed.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 Search PubMed.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850–11859 Search PubMed.
- Z. Cui, L. Li, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 7278–7281 Search PubMed.
- K. Jiang, P. Wang, S. Guo, X. Zhang, X. Shen, G. Lu, D. Su and X. Huang, Angew. Chem., Int. Ed., 2016, 55, 9030–9035 Search PubMed.
- C. Wang, D. P. Chen, X. Sang, R. R. Unocic and S. E. Skrabalak, ACS Nano, 2016, 10, 6345–6353 Search PubMed.
- S. Maksimuk, S. Yang, Z. Peng and H. Yang, J. Am. Chem. Soc., 2007, 129, 8684–8685 Search PubMed.
- J. Shim, J. Lee, Y. Ye, J. Hwang, S.-K. Kim, T.-H. Lim, U. Wiesner and J. Lee, ACS Nano, 2012, 6, 6870–6881 Search PubMed.
- L. Gan, C. Cui, M. Heggen, F. Dionigi, S. Rudi and P. Strasser, Science, 2014, 346, 1502–1506 Search PubMed.
- J. Liang, F. Ma, S. Hwang, X. Wang, J. Sokolowski, Q. Li, G. Wu and D. Su, Joule, 2019, 3, 956–991 Search PubMed.
- P. Yang, X. Yuan, H. Hu, Y. Liu, H. Zheng, D. Yang, L. Chen, M. Cao, Y. Xu, Y. Min, Y. Li and Q. Zhang, Adv. Funct. Mater., 2018, 28, 1704774–1704781 Search PubMed.
- J. Meier, J. Schiøtz, P. Liu, J. K. Nørskov and U. Stimming, Chem. Phys. Lett., 2004, 390, 440–444 Search PubMed.
- K. Elbert, J. Hu, Z. Ma, Y. Zhang, G. Chen, W. An, P. Liu, H. S. Isaacs, R. R. Adzic and J. X. Wang, ACS Catal., 2015, 5, 6764–6772 Search PubMed.
- Z. Qi, Y. Pei, T. W. Goh, Z. Wang, X. Li, M. Lowe, R. V. Maligal-Ganesh and W. Huang, Nano Res., 2018, 11, 3469–3479 Search PubMed.
- Z. Qi, C. Xiao, C. Liu, T. W. Goh, L. Zhou, R. Maligal-Ganesh, Y. Pei, X. Li, L. A. Curtiss and W. Huang, J. Am. Chem. Soc., 2017, 139, 4762–4768 Search PubMed.
- Y. Kang and C. B. Murray, J. Am. Chem. Soc., 2010, 132, 7568–7569 Search PubMed.
- Q. Chen, Y. Yang, Z. Cao, Q. Kuang, G. Du, Y. Jiang, Z. Xie and L. Zheng, Angew. Chem., Int. Ed., 2016, 55, 9021–9025 Search PubMed.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 Search PubMed.
- N. S. Porter, H. Wu, Z. Quan and J. Fang, Acc. Chem. Res., 2013, 46, 1867–1877 Search PubMed.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186–202 Search PubMed.
- S. Liu, N. Tian, A. Y. Xie, J. H. Du, J. Xiao, L. Liu, H. Y. Sun, Z. Y. Cheng, Z. Y. Zhou and S. G. Sun, J. Am. Chem. Soc., 2016, 138, 5753–5756 Search PubMed.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 Search PubMed.
- Z. Y. Zhou, Z. Z. Huang, D. J. Chen, Q. Wang, N. Tian and S. G. Sun, Angew. Chem., Int. Ed., 2010, 49, 411–414 Search PubMed.
- Y. J. Mao, L. Wei, X. S. Zhao, Y. S. Wei, J. W. Li, T. Sheng, F. C. Zhu, N. Tian, Z. Y. Zhou and S. G. Sun, Chem. Commun., 2019, 55, 9335–9338 Search PubMed.
- W. Chen, J. Kim, S. Sun and S. Chen, Langmuir, 2007, 23, 11303–11310 Search PubMed.
- G. V. Ramesh, R. Kodiyath, T. Tanabe, M. Manikandan, T. Fujita, N. Umezawa, S. Ueda, S. Ishihara, K. Ariga and H. Abe, ACS Appl. Mater. Interfaces, 2014, 6, 16124–16130 Search PubMed.
- Q. Shi, C. Zhu, C. Bi, H. Xia, M. H. Engelhard, D. Du and Y. Lin, J. Mater. Chem. A, 2017, 5, 23952–23959 Search PubMed.
- T. Bligaard and J. K. Nørskov, Electrochim. Acta, 2007, 52, 5512–5516 Search PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 Search PubMed.
- P. Liu and J. K. Nørskov, Phys. Chem. Chem. Phys., 2001, 3, 3814–3818 Search PubMed.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 Search PubMed.
- L. Rößner and M. Armbrüster, ACS Catal., 2019, 9, 2018–2062 Search PubMed.
- B. T. Sneed, A. P. Young and C. K. Tsung, Nanoscale, 2015, 7, 12248–12265 Search PubMed.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 Search PubMed.
- M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, U. Gronbjerg, B. P. Knudsen, A. K. Jepsen, J. Rossmeisl, I. E. Stephens and I. Chorkendorff, J. Am. Chem. Soc., 2012, 134, 16476–16479 Search PubMed.
- C. Wang, X. Sang, J. T. L. Gamler, D. P. Chen, R. R. Unocic and S. E. Skrabalak, Nano Lett., 2017, 17, 5526–5532 Search PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 Search PubMed.
- K. A. Kuttiyiel, S. Kattel, S. Cheng, J. H. Lee, L. Wu, Y. Zhu, G.-G. Park, P. Liu, K. Sasaki, J. G. Chen and R. R. Adzic, ACS Appl. Energy Mater., 2018, 1, 3771–3777 Search PubMed.
- J.-M. Zhang, J.-J. He, X.-Q. Wang, Y.-J. Fan, X.-J. Zhang, J.-P. Zhong, W. Chen and S.-G. Sun, Int. J. Hydrogen Energy, 2019, 44, 28709–28719 Search PubMed.
- J.-M. Zhang, R.-X. Wang, R.-J. Nong, Y. Li, X.-J. Zhang, P.-Y. Zhang and Y.-J. Fan, Int. J. Hydrogen Energy, 2017, 42, 7226–7234 Search PubMed.
- T. Gunji, S. H. Noh, F. Ando, T. Tanabe, B. Han, T. Ohsaka and F. Matsumoto, J. Mater. Chem. A, 2018, 6, 14828–14837 Search PubMed.
- N. Hodnik, C. Jeyabharathi, J. C. Meier, A. Kostka, K. L. Phani, A. Recnik, M. Bele, S. Hocevar, M. Gaberscek and K. J. Mayrhofer, Phys. Chem. Chem. Phys., 2014, 16, 13610–13615 Search PubMed.
- J. Kim, Y. Lee and S. Sun, J. Am. Chem. Soc., 2010, 132, 4996–4997 Search PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 Search PubMed.
- J. Snyder, I. McCue, K. Livi and J. Erlebacher, J. Am. Chem. Soc., 2012, 134, 8633–8645 Search PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 Search PubMed.
- J. Li, Z. Xi, Y. T. Pan, J. S. Spendelow, P. N. Duchesne, D. Su, Q. Li, C. Yu, Z. Yin, B. Shen, Y. S. Kim, P. Zhang and S. Sun, J. Am. Chem. Soc., 2018, 140, 2926–2932 Search PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 Search PubMed.
- C. Wang, M. Chi, D. Li, D. Strmcnik, D. van der Vliet, G. Wang, V. Komanicky, K. C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Am. Chem. Soc., 2011, 133, 14396–14403 Search PubMed.
- H. Liao, A. Fisher and Z. J. Xu, Small, 2015, 11, 3221–3246 Search PubMed.
- B.-W. Zhang, C.-L. He, Y.-X. Jiang, M.-H. Chen, Y.-Y. Li, L. Rao and S.-G. Sun, Electrochem. Commun., 2012, 25, 105–108 Search PubMed.
- X. Zhang, S. Yu, L. Qiao, W. Zheng and P. Liu, J. Chem. Phys., 2015, 142, 194710 Search PubMed.
- B.-W. Zhang, Z.-C. Zhang, H.-G. Liao, Y. Gong, L. Gu, X.-M. Qu, L.-X. You, S. Liu, L. Huang, X.-C. Tian, R. Huang, F.-C. Zhu, T. Liu, Y.-X. Jiang, Z.-Y. Zhou and S.-G. Sun, Nano Energy, 2016, 19, 198–209 Search PubMed.
- E. Antolini, RSC Adv., 2016, 6, 3307–3325 Search PubMed.
- T. Gunji, T. Tanabe, A. J. Jeevagan, S. Usui, T. Tsuda, S. Kaneko, G. Saravanan, H. Abe and F. Matsumoto, J. Power Sources, 2015, 273, 990–998 Search PubMed.
- W. Huang, X. Kang, C. Xu, J. Zhou, J. Deng, Y. Li and S. Cheng, Adv. Mater., 2018, 30, 1706962–1706967 Search PubMed.
- T. Liu, K. Wang, Q. Yuan, Z. Shen, Y. Wang, Q. Zhang and X. Wang, Nanoscale, 2017, 9, 2963–2968 Search PubMed.
- K. J. J. Mayrhofer and M. Arenz, Nat. Chem., 2009, 1, 518–519 Search PubMed.
- H. Liao, J. Zhu and Y. Hou, Nanoscale, 2014, 6, 1049–1055 Search PubMed.
- J. Zheng, D. A. Cullen, R. V. Forest, J. A. Wittkopf, Z. Zhuang, W. Sheng, J. G. Chen and Y. Yan, ACS Catal., 2015, 5, 1468–1474 Search PubMed.
- D. Cao, G. Q. Lu, A. Wieckowski, S. A. Wasileski and M. Neurock, J. Phys. Chem. B, 2005, 109, 11622–11633 Search PubMed.
- A. Cuesta, J. Am. Chem. Soc., 2006, 128, 13332–13333 Search PubMed.
- D. J. Chen and Y. J. Tong, Angew. Chem., Int. Ed., 2015, 54, 9394–9398 Search PubMed.
- R. Mancharan and J. B. Goodenough, J. Mater. Chem., 1992, 2, 875–887 Search PubMed.
- A. M. Hofstead-Duffy, D.-J. Chen, S.-G. Sun and Y. J. Tong, J. Mater. Chem., 2012, 22, 5205–5208 Search PubMed.
- D. Y. Chung, K.-J. Lee and Y.-E. Sung, J. Phys. Chem. C, 2016, 120, 9028–9035 Search PubMed.
- T. Takeguchi, T. Yamanaka, K. Asakura, E. N. Muhamad, K. Uosaki and W. Ueda, J. Am. Chem. Soc., 2012, 134, 14508–14512 Search PubMed.
- W.-Y. Zhao, B. Ni, Q. Yuan, P.-L. He, Y. Gong, L. Gu and X. Wang, Adv. Energy Mater., 2017, 7, 1601593–1601600 Search PubMed.
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 Search PubMed.
- J. Zhang, X. Qu, Y. Han, L. Shen, S. Yin, G. Li, Y. Jiang and S. Sun, Appl. Catal., B, 2019, 263, 118345–118353 Search PubMed.
- U. Bardi, D. Dahlgren and P. N. Ross, J. Catal., 1986, 100, 196–209 Search PubMed.
- H. Abe, F. Matsumoto, L. R. Alden, S. C. Warren, H. D. Abruña and F. J. DiSalvo, J. Am. Chem. Soc., 2008, 130, 5452–5458 Search PubMed.
- Z. Cui, H. Chen, M. Zhao, D. Marshall, Y. Yu, H. Abruna and F. J. DiSalvo, J. Am. Chem. Soc., 2014, 136, 10206–10209 Search PubMed.
- J. Sanetuntikul, K. Ketpang and S. Shanmugam, ACS Catal., 2015, 5, 7321–7327 Search PubMed.
- E. Casado-Rivera, D. J. Volpe, L. Alden, C. Lind, C. Downie, T. Vázquez-Alvarez, A. C. D. Angelo, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2004, 126, 4043–4049 Search PubMed.
- Y. Kang, J. B. Pyo, X. Ye, T. R. Gordon and C. B. Murray, ACS Nano, 2012, 6, 5642–5647 Search PubMed.
- Q. Feng, S. Zhao, D. He, S. Tian, L. Gu, X. Wen, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 2773–2776 Search PubMed.
- L. Gao, X. Li, Z. Yao, H. Bai, Y. Lu, C. Ma, S. Lu, Z. Peng, J. Yang, A. Pan and H. Huang, J. Am. Chem. Soc., 2019, 141, 18083–18090 Search PubMed.
- S. Song and P. Tsiakaras, Appl. Catal., B, 2006, 63, 187–193 Search PubMed.
- P. J. Rheinländer, J. Herranz, J. Durst and H. A. Gasteiger, J. Electrochem. Soc., 2014, 161, F1448–F1457 Search PubMed.
- T. Herranz, M. Ibáñez, J. L. Gómez de la Fuente, F. J. Pérez-Alonso, M. A. Peña, A. Cabot and S. Rojas, ChemElectroChem, 2014, 1, 885–895 Search PubMed.
- S. Sarkar, R. Jana, S. Prasad, U. V. Waghmare, B. Thapa, S. Sampath and S. C. Peter, Chem. Mater., 2015, 27, 7459–7467 Search PubMed.
- H. Wang, Z. Jusys and R. J. Behm, J. Phys. Chem. B, 2004, 108, 19413–19424 Search PubMed.
- Z. Liang, L. Song, S. Deng, Y. Zhu, E. Stavitski, R. R. Adzic, J. Chen and J. X. Wang, J. Am. Chem. Soc., 2019, 141, 9629–9636 Search PubMed.
- Q. Wang, G. Q. Sun, L. H. Jiang, Q. Xin, S. G. Sun, Y. X. Jiang, S. P. Chen, Z. Jusys and R. J. Behm, Phys. Chem. Chem. Phys., 2007, 9, 2686–2696 Search PubMed.
- R. Kavanagh, X. M. Cao, W. F. Lin, C. Hardacre and P. Hu, Angew. Chem., Int. Ed., 2012, 51, 1572–1575 Search PubMed.
- Z. F. Xu and Y. Wang, J. Phys. Chem. C, 2011, 115, 20565–20571 Search PubMed.
- M. Li, P. Liu and R. R. Adzic, J. Phys. Chem. Lett., 2012, 3, 3480–3485 Search PubMed.
- H.-F. Wang and Z.-P. Liu, J. Am. Chem. Soc., 2008, 130, 10996–11004 Search PubMed.
- F. Zhu, K. Tu, L. Huang, X. Qu, J. Zhang, H. Liao, Z. Zhou, Y. Jiang and S. Sun, Electrochim. Acta, 2018, 292, 208–216 Search PubMed.
- R. Rizo, R. M. Aran-Ais, E. Padgett, D. A. Muller, M. J. Lazaro, J. Solla-Gullon, J. M. Feliu, E. Pastor and H. D. Abruña, J. Am. Chem. Soc., 2018, 140, 3791–3797 Search PubMed.
- Q. Chang, S. Kattel, X. Li, Z. Liang, B. M. Tackett, S. R. Denny, P. Zhang, D. Su, J. G. Chen and Z. Chen, ACS Catal., 2019, 9, 7618–7625 Search PubMed.
- A. R. Miedema, R. Boom and F. R. De Boer, J. Less-Common Met., 1975, 41, 283–298 Search PubMed.
- R. Kodiyath, G. V. Ramesh, E. Koudelkova, T. Tanabe, M. Ito, M. Manikandan, S. Ueda, T. Fujita, N. Umezawa, H. Noguchi, K. Ariga and H. Abe, Energy Environ. Sci., 2015, 8, 1685–1689 Search PubMed.
- B.-W. Zhang, W.-H. Lai, T. Sheng, X.-M. Qu, Y.-X. Wang, L. Ren, L. Zhang, Y. Du, Y.-X. Jiang, S.-G. Sun and S.-X. Dou, J. Mater. Chem. A, 2019, 7, 5214–5220 Search PubMed.
- Y. Sun, Y. Liang, M. Luo, F. Lv, Y. Qin, L. Wang, C. Xu, E. Fu and S. Guo, Small, 2018, 14, 1702259–1702266 Search PubMed.
- J. E. Sulaiman, S. Zhu, Z. Xing, Q. Chang and M. Shao, ACS Catal., 2017, 7, 5134–5141 Search PubMed.
- B.-W. Zhang, T. Sheng, Y.-X. Wang, X.-M. Qu, J.-M. Zhang, Z.-C. Zhang, H.-G. Liao, F.-C. Zhu, S.-X. Dou, Y.-X. Jiang and S.-G. Sun, ACS Catal., 2016, 7, 892–895 Search PubMed.
- M. R. Zamanzad Ghavidel and E. B. Easton, Appl. Catal., B, 2015, 176–177, 150–159 Search PubMed.
- D. J. Chen, Z. Y. Zhou, Q. Wang, D. M. Xiang, N. Tian and S. G. Sun, Chem. Commun., 2010, 46, 4252–4254 Search PubMed.
- T. Ghosh, B. M. Leonard, Q. Zhou and F. J. DiSalvo, Chem. Mater., 2010, 22, 2190–2202 Search PubMed.
- D. Sun, L. Si, G. Fu, C. Liu, D. Sun, Y. Chen, Y. Tang and T. Lu, J. Power Sources, 2015, 280, 141–146 Search PubMed.
- K. Jiang, H. X. Zhang, S. Zou and W. B. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 Search PubMed.
- E. Casado-Rivera, Z. Gál, A. C. D. Angelo, C. Lind, F. J. DiSalvo and H. D. Abruña, ChemPhysChem, 2003, 4, 193–199 Search PubMed.
- L. Chen, J. Zhu, C. Xuan, W. Xiao, K. Xia, W. Xia, C. Lai, H. L. Xin and D. Wang, J. Mater. Chem. A, 2018, 6, 5848–5855 Search PubMed.
- X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286–293 Search PubMed.
- M. Osawa, K. Komatsu, G. Samjeske, T. Uchida, T. Ikeshoji, A. Cuesta and C. Gutierrez, Angew. Chem., Int. Ed., 2011, 50, 1159–1163 Search PubMed.
- Y. Kang, L. Qi, M. Li, R. E. Diaz, D. Su, R. R. Adzic, E. Stach, J. Li and C. B. Murray, ACS Nano, 2012, 6, 2818–2825 Search PubMed.
- S. Luo, W. Chen, Y. Cheng, X. Song, Q. Wu, L. Li, X. Wu, T. Wu, M. Li, Q. Yang, K. Deng and Z. Quan, Adv. Mater., 2019, 31, 1903683–1903689 Search PubMed.
- J. V. Perales-Rondon, A. Ferre-Vilaplana, J. M. Feliu and E. Herrero, J. Am. Chem. Soc., 2014, 136, 13110–13113 Search PubMed.
- Z. Y. Zhou, J. Ren, X. Kang, Y. Song, S. G. Sun and S. Chen, Phys. Chem. Chem. Phys., 2012, 14, 1412–1417 Search PubMed.
- X. Qu, Z. Cao, B. Zhang, X. Tian, F. Zhu, Z. Zhang, Y. Jiang and S. Sun, Chem. Commun., 2016, 52, 4493–4496 Search PubMed.
- H. Rong, J. Mao, P. Xin, D. He, Y. Chen, D. Wang, Z. Niu, Y. Wu and Y. Li, Adv. Mater., 2016, 28, 2540–2546 Search PubMed.
- Y. T. Pan, Y. Yan, Y. T. Shao, J. M. Zuo and H. Yang, Nano Lett., 2016, 16, 6599–6603 Search PubMed.
- H. Xu, B. Yan, S. Li, J. Wang, C. Wang, J. Guo and Y. Du, Chem. Eng. J., 2018, 334, 2638–2646 Search PubMed.
- L. An, H. Yan, B. Li, J. Ma, H. Wei and D. Xia, Nano Energy, 2015, 15, 24–32 Search PubMed.
- D. Xu, S. Bliznakov, Z. Liu, J. Fang and N. Dimitrov, Angew. Chem., Int. Ed., 2010, 49, 1282–1285 Search PubMed.
- Q. S. Chen, Z. Y. Zhou, F. J. Vidal-Iglesias, J. Solla-Gullon, J. M. Feliu and S. G. Sun, J. Am. Chem. Soc., 2011, 133, 12930–12933 Search PubMed.
- L. Huang, M. Liu, H. Lin, Y. Xu, J. Wu, V. P. Dravid, C. Wolverton and C. A. Mirkin, Science, 2019, 365, 1159–1163 Search PubMed.
- R. Wang, J. Liu, P. Liu, X. Bi, X. Yan, W. Wang, Y. Meng, X. Ge, M. Chen and Y. Ding, Nano Res., 2014, 7, 1569–1580 Search PubMed.
- S. Hu, F. Munoz, J. Noborikawa, J. Haan, L. Scudiero and S. Ha, Appl. Catal., B, 2016, 180, 758–765 Search PubMed.
- R. Jana, U. Subbarao and S. C. Peter, J. Power Sources, 2016, 301, 160–169 Search PubMed.
- Z. Xi, J. Li, D. Su, M. Muzzio, C. Yu, Q. Li and S. Sun, J. Am. Chem. Soc., 2017, 139, 15191–15196 Search PubMed.
- Z. Liu, G. Fu, J. Li, Z. Liu, L. Xu, D. Sun and Y. Tang, Nano Res., 2018, 11, 4686–4696 Search PubMed.
- D. Liu, M. Xie, C. Wang, L. Liao, L. Qiu, J. Ma, H. Huang, R. Long, J. Jiang and Y. Xiong, Nano Res., 2016, 9, 1590–1599 Search PubMed.
- N. Yang, Z. Zhang, B. Chen, Y. Huang, J. Chen, Z. Lai, Y. Chen, M. Sindoro, A. L. Wang, H. Cheng, Z. Fan, X. Liu, B. Li, Y. Zong, L. Gu and H. Zhang, Adv. Mater., 2017, 29, 1700769–1700774 Search PubMed.
- Q. Liu, L. Du, G. Fu, Z. Cui, Y. Li, D. Dang, X. Gao, Q. Zheng and J. B. Goodenough, Adv. Energy Mater., 2019, 9, 1803040–1803046 Search PubMed.
- R. F. Service, Science, 2007, 315, 172–172 Search PubMed.
- A. U. Nilekar, S. Alayoglu, B. Eichhorn and M. Mavrikakis, J. Am. Chem. Soc., 2010, 132, 7418–7428 Search PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 Search PubMed.
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 Search PubMed.
- X. Li and K. Lu, Science, 2019, 364, 733–734 Search PubMed.
- B.-A. Lu, T. Sheng, N. Tian, Z.-C. Zhang, C. Xiao, Z.-M. Cao, H.-B. Ma, Z.-Y. Zhou and S.-G. Sun, Nano Energy, 2017, 33, 65–71 Search PubMed.
- Z. Wang, X. Yao, Y. Kang, L. Miao, D. Xia and L. Gan, Adv. Funct. Mater., 2019, 29, 1902987–1902995 Search PubMed.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 Search PubMed.
- V. Čolić and A. S. Bandarenka, ACS Catal., 2016, 6, 5378–5385 Search PubMed.
- X. X. Du, Y. He, X. X. Wang and J. N. Wang, Energy Environ. Sci., 2016, 9, 2623–2632 Search PubMed.
- L. Zou, J. Li, T. Yuan, Y. Zhou, X. Li and H. Yang, Nanoscale, 2014, 6, 10686–10692 Search PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 Search PubMed.
- T. Cheng, X.-Y. Lang, G.-F. Han, R.-Q. Yao, Z. Wen and Q. Jiang, J. Mater. Chem. A, 2016, 4, 18878–18884 Search PubMed.
- J. Greeley, I. E. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552–556 Search PubMed.
- D. Zhang, F. Wu, M. Peng, X. Wang, D. Xia and G. Guo, J. Am. Chem. Soc., 2015, 137, 6263–6269 Search PubMed.
- E. Antolini, Appl. Catal., B, 2017, 217, 201–213 Search PubMed.
- S. Prabhudev, M. Bugnet, C. Bock and G. A. Botton, ACS Nano, 2013, 7, 6103–6110 Search PubMed.
- Y. Qin, M. Luo, Y. Sun, C. Li, B. Huang, Y. Yang, Y. Li, L. Wang and S. Guo, ACS Catal., 2018, 8, 5581–5590 Search PubMed.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 Search PubMed.
- Z. Zhao, C. Chen, Z. Liu, J. Huang, M. Wu, H. Liu, Y. Li and Y. Huang, Adv. Mater., 2019, 31, 1808115–1808130 Search PubMed.
- L. Ou and S. Chen, J. Phys. Chem. C, 2013, 117, 1342–1349 Search PubMed.
- G. Wang, Z. Yang, Y. Du and Y. Yang, Angew. Chem., Int. Ed., 2019, 58, 15848–15854 Search PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2018, 4, 60–67 Search PubMed.
- X. Tian, X. Zhao, Y.-Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850–856 Search PubMed.
- J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu and S. Sun, Joule, 2019, 3, 124–135 Search PubMed.
- X. X. Wang, S. Hwang, Y. T. Pan, K. Chen, Y. He, S. Karakalos, H. Zhang, J. S. Spendelow, D. Su and G. Wu, Nano Lett., 2018, 18, 4163–4171 Search PubMed.
- Y. Feng, Q. Shao, F. Lv, L. Bu, J. Guo, S. Guo and X. Huang, Adv. Sci., 2020, 7, 1800178–1800185 Search PubMed.
- D. Sun, Y. Wang, K. J. T. Livi, C. Wang, R. Luo, Z. Zhang, H. Alghamdi, C. Li, F. An, B. Gaskey, T. Mueller and A. S. Hall, ACS Nano, 2019, 13, 10818–10825 Search PubMed.
- D. Wang, H. L. Xin, H. Wang, Y. Yu, E. Rus, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Chem. Mater., 2012, 24, 2274–2281 Search PubMed.
- Z. Cui, H. Chen, M. Zhao and F. J. DiSalvo, Nano Lett., 2016, 16, 2560–2566 Search PubMed.
- G. Jiang, H. Zhu, X. Zhang, B. Shen, L. Wu, S. Zhang, G. Lu, Z. Wu and S. Sun, ACS Nano, 2015, 9, 11014–11022 Search PubMed.
- K. A. Kuttiyiel, K. Sasaki, D. Su, L. Wu, Y. Zhu and R. R. Adzic, Nat. Commun., 2014, 5, 5185–5192 Search PubMed.
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 Search PubMed.
- R. Chattot, O. Le Bacq, V. Beermann, S. Kuhl, J. Herranz, S. Henning, L. Kuhn, T. Asset, L. Guetaz, G. Renou, J. Drnec, P. Bordet, A. Pasturel, A. Eychmuller, T. J. Schmidt, P. Strasser, L. Dubau and F. Maillard, Nat. Mater., 2018, 17, 827–833 Search PubMed.
- P. Strasser and S. Kühl, Nano Energy, 2016, 29, 166–177 Search PubMed.
- D. Wang, H. L. Xin, Y. Yu, H. Wang, E. Rus, D. A. Muller and H. D. Abruña, J. Am. Chem. Soc., 2010, 132, 17664–17666 Search PubMed.
- W. Xiao, M. A. L. Cordeiro, G. Gao, A. Zheng, J. Wang, W. Lei, M. Gong, R. Lin, E. Stavitski, H. L. Xin and D. Wang, Nano Energy, 2018, 50, 70–78 Search PubMed.
- Y. Xiong, Y. Yang, F. J. DiSalvo and H. D. Abruna, J. Am. Chem. Soc., 2018, 140, 7248–7255 Search PubMed.
- G. Wang, B. Huang, L. Xiao, Z. Ren, H. Chen, D. Wang, H. D. Abruna, J. Lu and L. Zhuang, J. Am. Chem. Soc., 2014, 136, 9643–9649 Search PubMed.
- S. Liu, W. Xiao, J. Wang, J. Zhu, Z. Wu, H. Xin and D. Wang, Nano Energy, 2016, 27, 475–481 Search PubMed.
- Y. Yang, W. Xiao, X. Feng, Y. Xiong, M. Gong, T. Shen, Y. Lu, H. D. Abruna and D. Wang, ACS Nano, 2019, 13, 5968–5974 Search PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 Search PubMed.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1529–B1536 Search PubMed.
- S. J. Lee, S. Mukerjee, E. A. Ticianelli and J. McBreen, Electrochim. Acta, 1999, 44, 3283–3293 Search PubMed.
- Z.-C. Zhang, X.-C. Tian, B.-W. Zhang, L. Huang, F.-C. Zhu, X.-M. Qu, L. Liu, S. Liu, Y.-X. Jiang and S.-G. Sun, Nano Energy, 2017, 34, 224–232 Search PubMed.
- S. St John, R. W. Atkinson, R. R. Unocic, T. A. Zawodzinski and A. B. Papandrew, J. Phys. Chem. C, 2015, 119, 13481–13487 Search PubMed.
- T. Takeguchi, T. Yamanaka, K. Asakura, E. N. Muhamad, K. Uosaki and W. Ueda, J. Am. Chem. Soc., 2012, 134, 14508–14512 Search PubMed.
- Y. C. Hsieh, Y. Zhang, D. Su, V. Volkov, R. Si, L. Wu, Y. Zhu, W. An, P. Liu, P. He, S. Ye, R. R. Adzic and J. X. Wang, Nat. Commun., 2013, 4, 2466 Search PubMed.
- G. Shi, H. Yano, D. A. Tryk, M. Matsumoto, H. Tanida, M. Arao, H. Imai, J. Inukai, A. Iiyama and H. Uchida, Catal. Sci. Technol., 2017, 7, 6124–6131 Search PubMed.
- G. Shi, H. Yano, D. A. Tryk, A. Iiyama and H. Uchida, ACS Catal., 2017, 7, 267–274 Search PubMed.
- H. Igarashi, T. Fujino, Y. Zhu, H. Uchida and M. Watanabe, Phys. Chem. Chem. Phys., 2001, 3, 306–314 Search PubMed.
- T. Wang, Z.-X. Chen, S. Yu, T. Sheng, H.-B. Ma, L.-N. Chen, M. Rauf, H.-P. Xia, Z.-Y. Zhou and S.-G. Sun, Energy Environ. Sci., 2018, 11, 166–171 Search PubMed.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509 Search PubMed.
- X. Wang, Y. Zhu, A. Vasileff, Y. Jiao, S. Chen, L. Song, B. Zheng, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2018, 3, 1198–1204 Search PubMed.
- J. Chen, J. Wang, J. Chen and L. Wang, J. Mater. Sci., 2017, 52, 13064–13077 Search PubMed.
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 Search PubMed.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 Search PubMed.
- Y. Cong, B. Yi and Y. Song, Nano Energy, 2018, 44, 288–303 Search PubMed.
- J. N. Schwämmlein, B. M. Stühmeier, K. Wagenbauer, H. Dietz, V. Tileli, H. A. Gasteiger and H. A. El-Sayed, J. Electrochem. Soc., 2018, 165, H229–H239 Search PubMed.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 Search PubMed.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 5848 Search PubMed.
- H. Wang and H. D. Abruna, J. Am. Chem. Soc., 2017, 139, 6807–6810 Search PubMed.
- Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2015, 8, 177–181 Search PubMed.
- S. Lu and Z. Zhuang, J. Am. Chem. Soc., 2017, 139, 5156–5163 Search PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 Search PubMed.
- J. Li, S. Ghoshal, M. K. Bates, T. E. Miller, V. Davies, E. Stavitski, K. Attenkofer, S. Mukerjee, Z.-F. Ma and Q. Jia, Angew. Chem., Int. Ed., 2017, 56, 15594–15598 Search PubMed.
- L.-F. Shen, B.-A. Lu, X.-M. Qu, J.-Y. Ye, J.-M. Zhang, S.-H. Yin, Q.-H. Wu, R.-X. Wang, S.-Y. Shen, T. Sheng, Y.-X. Jiang and S.-G. Sun, Nano Energy, 2019, 62, 601–609 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |