
Electrolyte additives to enable nonaqueous polyelectrolyte solutions for lithium ion batteries
Kyle M.
Diederichsen
 ab and
Bryan D.
McCloskey
*ab
ab and
Bryan D.
McCloskey
*ab
aDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720, USA. E-mail: bmcclosk@berkeley.edu
bEnergy Storage and Distributed Resources Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 1st July 2019
Abstract
Nonaqueous polyelectrolyte solutions, in which a negatively charged macromolecule neutralized by lithium is dissolved in nonaqueous solvents, have shown promise as potential high transference number electrolytes. However, in battery-relevant carbonate solvents (ethylene carbonate/dimethyl carbonate blends), it has been shown that lithium ions do not readily dissociate from easily synthesized sulfonated polymers, despite the solvent's high dielectric constant (∼50). In this work, a range of additives are screened to improve conductivity, and we demonstrate that the addition of less than 5 vol% of 15-crown-5 achieves an order of magnitude conductivity increase by selectively improving lithium dissociation. Utilizing the optimized electrolyte, we show that polyelectrolyte solutions may be directly substituted for a standard electrolyte with commercial electrodes in a graphite/LiFePO4 cell, providing further motivation for future study of these new electrolytes.
 Bryan D. McCloskey | Bryan D. McCloskey is an Assistant Professor in the Department of Chemical and Biomolecular Engineering at the University of California, Berkeley, and holds a joint appointment as Faculty Chemical Engineer in the Energy Storage and Distributed Resources Division at Lawrence Berkeley National Laboratory. His laboratory focuses on characterization of fundamental electrochemical processes to provide guidance for the development of energy storage, electrocatalytic, and corrosion-resistant materials. He has co-authored more than 75 articles, holds 6 patents, and has won numerous awards for his research, including an NSF CAREER Award and the VW/BASF Science Award Electrochemistry. More information on Prof. McCloskey's group and research can be found at his group's website: http://www.mccloskeylab.com. |
Design, System, ApplicationLithium ion batteries have achieved significant market penetration, but further innovation is necessary to enable the next generation of fast charging, high energy density cells. A key component of any high energy density battery is the porous electrode, where the pores are wetted by the ion-transporting electrolyte to provide a high surface area for electrochemical reactions to occur. At high current rates, transport within these electrodes becomes a major limiting factor as it is possible to deplete the concentration of lithium in the electrolyte contained within the electrode. This problem could be overcome by designing an electrolyte in which the lithium is more mobile than the anion, as the high anion mobility in current electrolytes is the main cause of large concentration gradients. Polyelectrolyte solutions, where the anion is a large macromolecule, have been proposed as systems in which this discrepancy could be directly tuned due to the control of charge density and size of the anion. In a battery, the range of stable solvents is limited, and thus solubility and dissociation of ion groups on the polyelectrolyte are important problems. Here, additives are studied to improve the dissociation of lithium-sulfonate groups, a common anion for polyelectrolytes, in a battery-relevant solvent to overcome the low conductivity measured in prior studies. The success shown here could enable future understanding of transport within battery electrolytes, and thereby achieve higher energy density, faster charging cells. |
Introduction
Lithium-ion battery electrolytes have been engineered extensively to have high conductivity and form a stable, Li+-conductive passivation layer on the graphite electrode surface. A key remaining challenge in these electrolytes, however, is engineering control over the relative motion of anions to cations. Lithium is the electroactive species within the battery, but in all commercial electrolytes to date the anion is in fact more mobile than the lithium. This discrepancy is captured by the low cation transference number (t+) of the electrolyte, defined as the fraction of the total conductivity that arises due to lithium motion in the absence of any concentration gradient.1 It has been shown that battery rate capability and energy density could be improved with a high t+ electrolyte (HTNE), even if the total conductivity is reduced relative to the standard electrolyte.2,3Recently, nonaqueous polyelectrolyte solutions, in which a lithium-neutralized negatively charged polymer is fully dissolved in a solvent, have been proposed as potential HTNEs due to the bulky nature of the polyanion.3–6 These solutions have been investigated extensively in water due to their relevance to biological systems,7,8 but significantly less effort has been expended for nonaqueous polyelectrolyte solutions.9 For a battery application, the preferred solvent is a blend of ethylene carbonate (EC) and a linear carbonate like dimethyl carbonate (DMC). EC has a high dielectric constant and degrades into a stable graphite electrode passivation layer, while DMC is added to reduce the electrolyte melting temperature and lower the viscosity.10 We have recently investigated a model sulfonated polysulfone/poly(ethylene glycol) copolymer in this solvent and found that sulfonate groups on the polymer do not readily dissociate in the battery-relevant solvent, leading to very low conductivity when compared with a similar dielectric constant solvent that has been studied more frequently with polyelectrolytes, dimethylsulfoxide (DMSO).6
In this work, we investigate the possibility of using additives to enable dissociation of the lithium from the sulfonate group within the desired EC/DMC solvent. Additives are key to the performance of all current battery electrolytes, and have been studied extensively both as transport enhancers and for stability.11,12 Doyle in 2001 studied the possibility of using additives to enable higher conductivity in charged polymer membranes for battery applications, but we know of no study which has ever investigated additives for nonaqueous polyelectrolyte solutions, particularly with the goal of improving lithium ion dissociation and transport.13 Here we show that preferential lithium binding solvents, particularly crown ethers, are a promising means to enable lithium dissociation from sulfonate groups in EC/DMC, allowing fabrication of a full battery from commercially available electrodes.
Results and discussion
We select and screen several potential additives to promote lithium dissociation in Fig. 1. For this screening study, the control solution is 0.1 M of the sulfonated polysulfone-co-poly(ethylene glycol) shown in Scheme 1 and described fully in our previous work dissolved in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EC:DMC (vol/vol) solvent.6 The reported polymer molarity is the appended sulfonate ion molarity in each solution, or twice the molarity of the sulfonated sulfone repeat unit (given that each sulfone repeat unit has 2 sulfonate groups). 0.1 M polymer represents the maximum in conductivity for this polymer in EC/DMC, and the 2:1 ratio is selected due to issues with crystallization of EC at higher EC contents. Candidate additives are introduced at an equivalent molar ratio of lithium to additive for comparison here. Also shown is the addition of lithium bis(trifluoromethylsulfonimide) (LiTFSI), which was included in our previous study. As shown there, this solution has a conductivity roughly the same as the salt without polymer. Representative additives were selected that had a high donor number (DMSO),14 could coordinate to metal ions (imidazole),15 or had ether functionality (tetraglyme). Each of these additives indeed improves solution conductivity with minimal viscosity change, but not significantly. Stabilizing additives, vinylene carbonate16 and fluoroethylene carbonate,17 have a negligible effect on the conductivity or viscosity. Crown ethers are shown here to have the most impact on the conductivity, again with relatively minimal change in viscosity. The specific crown ethers chosen here are the same previously studied by Doyle,13 with the addition of dibenzo 24-crown-8 which is a commercially available crown of larger ring size. The magnitude of this impact is particularly apparent in comparison to dioxane and tetraglyme, which have similar functionality, and even water.
:1 EC:DMC (vol/vol) solvent.6 The reported polymer molarity is the appended sulfonate ion molarity in each solution, or twice the molarity of the sulfonated sulfone repeat unit (given that each sulfone repeat unit has 2 sulfonate groups). 0.1 M polymer represents the maximum in conductivity for this polymer in EC/DMC, and the 2:1 ratio is selected due to issues with crystallization of EC at higher EC contents. Candidate additives are introduced at an equivalent molar ratio of lithium to additive for comparison here. Also shown is the addition of lithium bis(trifluoromethylsulfonimide) (LiTFSI), which was included in our previous study. As shown there, this solution has a conductivity roughly the same as the salt without polymer. Representative additives were selected that had a high donor number (DMSO),14 could coordinate to metal ions (imidazole),15 or had ether functionality (tetraglyme). Each of these additives indeed improves solution conductivity with minimal viscosity change, but not significantly. Stabilizing additives, vinylene carbonate16 and fluoroethylene carbonate,17 have a negligible effect on the conductivity or viscosity. Crown ethers are shown here to have the most impact on the conductivity, again with relatively minimal change in viscosity. The specific crown ethers chosen here are the same previously studied by Doyle,13 with the addition of dibenzo 24-crown-8 which is a commercially available crown of larger ring size. The magnitude of this impact is particularly apparent in comparison to dioxane and tetraglyme, which have similar functionality, and even water.
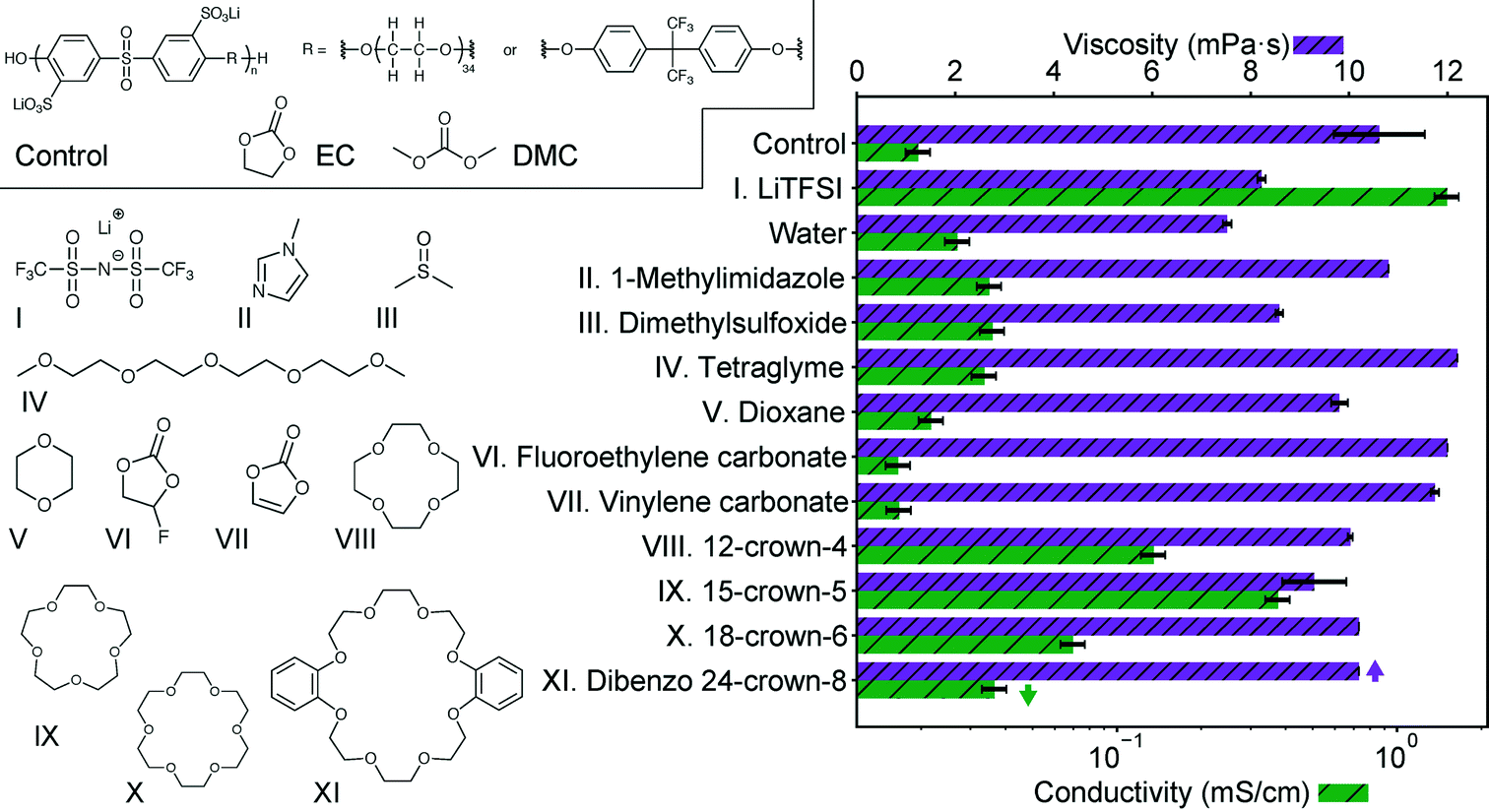 | ||
| Fig. 1 Conductivity and viscosity of a range of potential additives to improve conductivity of the control 0.1 M polymer in 2:1 (vol.) EC:DMC solution. Each additive is introduced at 0.1 M to match the lithium content of the solution. The structure of the polymer, EC and DMC in the control solution are also included. | ||
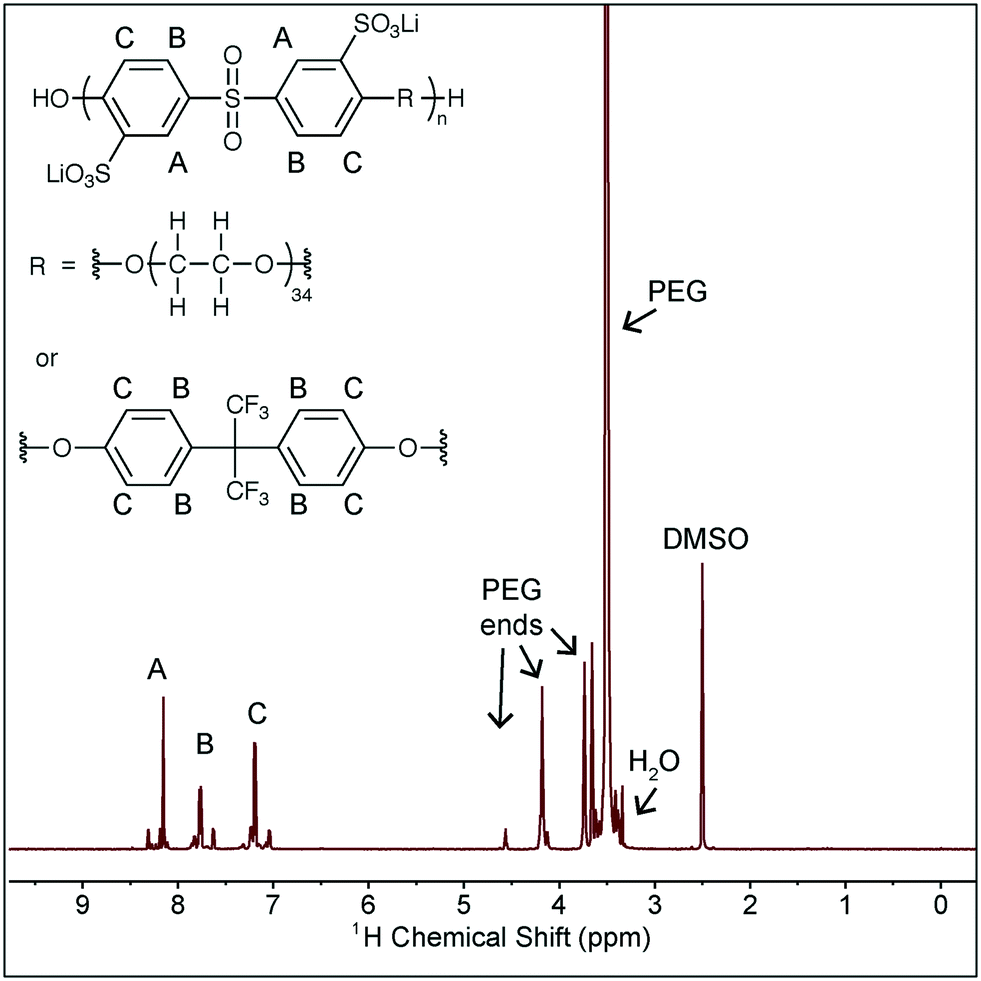 | ||
| Scheme 1 Structure of the polymer used in this study, and 1H NMR analysis of the polymer structure. The R group is ∼10 mol% of the flourinated monomer, as a tag for backbone diffusion measurement via19F NMR. | ||
The trend in conductivity with crown ether size (15-crown-5 > 12-crown-4 > 18-crown-6 > 24-crown-8) mirrors the trend observed by Doyle for EC/DMC swollen Nafion membranes with added crown ether.13 The close size match of the cavity formed by each crown and the metal ion is likely the cause of this trend as it has long been known that different metal ions bind preferentially to different crown ethers.18 This is further supported by past calorimetric measurements of crown ether complexation with lithium in several different solvents, which quantify the strength of binding. These measurements correspond directly to the trend in conductivity observed here, with 15-crown-5 having the strongest interaction.19 As conductivity must arise from charged species, this suggests addition of crown ether allows lithium to dissociate from the sulfonate group in solution, and the more strongly coordinated species provides a greater degree of dissociation. This ability to improve dissociation and solubility was recognized for battery electrolytes before,20 but in these cases addition of crown tends to ultimately be detrimental to transport because the lithium/crown complex is bulkier than simply lithium/solvent, lowering the transference number.10 This is significantly different than the poorly dissociated polyelectrolyte solution case, where the alternative to a crown/lithium complex is coordination with a very bulky slow-moving polymer. Importantly, crown ether additives have not been noted to change electrolyte stability significantly, having been used in the past to enhance the solid interface in PC electrolytes, and appearing to have similar stability as poly(ethylene oxide).10,21
In Fig. 2 we examine the effect of 15-crown-5 (the additive that provided the highest conductivity in our initial additive screening) concentration on the solution conductivity and viscosity. Furthermore, we quantify the influence of water on these measurements given its strong solvation properties (high dielectric constant and Lewis acidity), and its ubiquitous presence as an impurity in nonaqueous solvents. These solutions were prepared by diluting a higher concentration solution to 0.1 M Li+ either by adding EC/DMC or additive, keeping the Li+ concentration constant. In Fig. 2A, a clear maximum in conductivity appears near ∼4 vol% crown ether, corresponding to a crown to lithium ratio of 2. This conductivity is significantly higher than that observed for a similar ratio of water to lithium (Fig. 2B). A similar conductivity with water as the additive is not achieved until a much higher water volume. As the amount of crown ether in the solvent increases, conductivity begins to decrease. This could be a result of the slight increase in viscosity at higher concentration (the viscosity of 15-crown-5 alone is 22 mPa s) or result from the formation of larger solvation structures in solution, such as a single ion with two crown ethers.
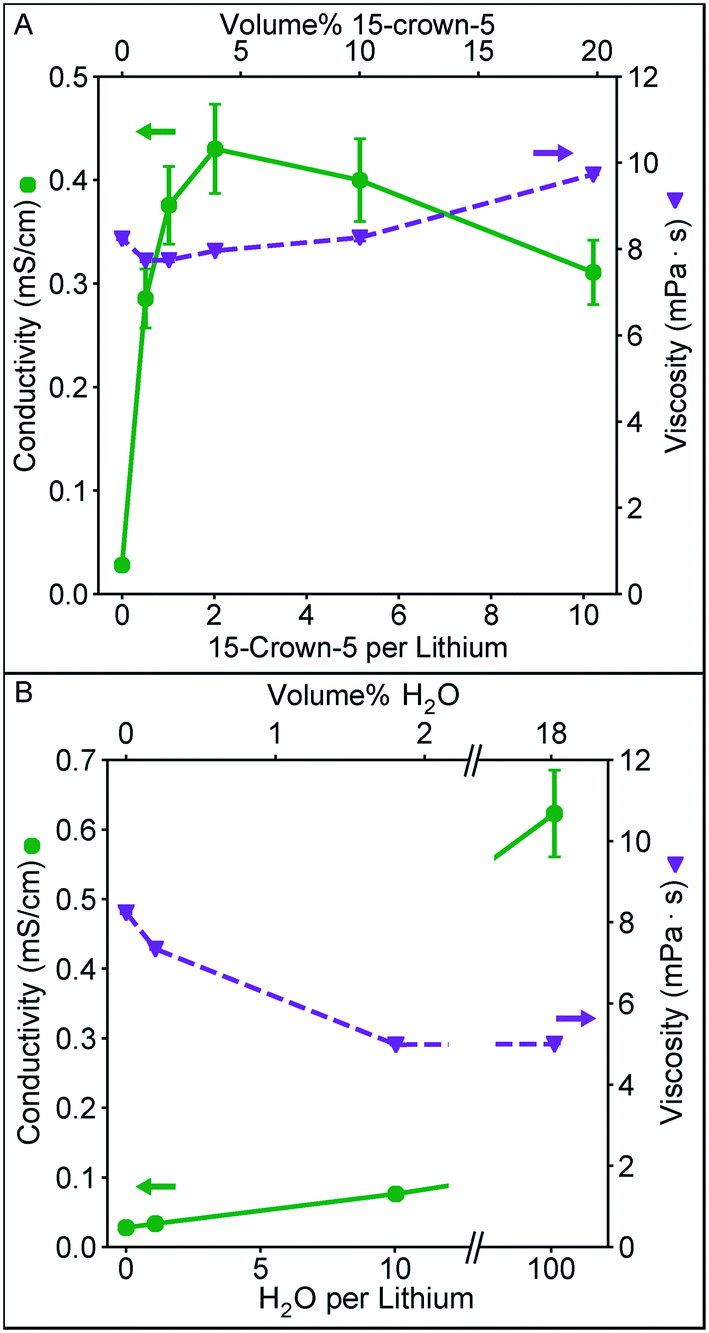 | ||
| Fig. 2 Conductivity and viscosity as a function of A) 15-crown-5 content and B) water content in a 0.1 M polymer in 2:1 (vol.) EC:DMC solution. The total lithium concentration is maintained constant in each solution by diluting from a higher concentration with either 15-crown-5, water, or EC/DMC. Viscosity error bars taken from repeat measurements of the same sample are smaller than the data points in this plot. | ||
The local solution structure and diffusion coefficients of the solution constituents are probed directly through NMR measurements summarized in Fig. 3. In Fig. 3A, diffusion coefficients measured through pulse field gradient NMR of EC, lithium, and the polymer backbone clearly demonstrate that the increase in conductivity observed in Fig. 2A are due to changes in lithium motion alone. Diffusion coefficients for DMC and the crown ether could not be measured independently as their 1H NMR peaks overlap. The polymer backbone diffusion coefficient is measured via19F NMR. It should be noted that the lithium diffusion coefficient measured by this technique averages all lithium species in solution. The trend in lithium diffusion coefficient directly aligns with the trend in conductivity in Fig. 2A, with a maximum in diffusion coefficient observed at 2 crowns per lithium. At higher crown content the decrease in lithium diffusion can be explained by the slight increase in bulk solution viscosity, as the relative decrease in EC diffusion is equivalent to the decrease in lithium diffusion.
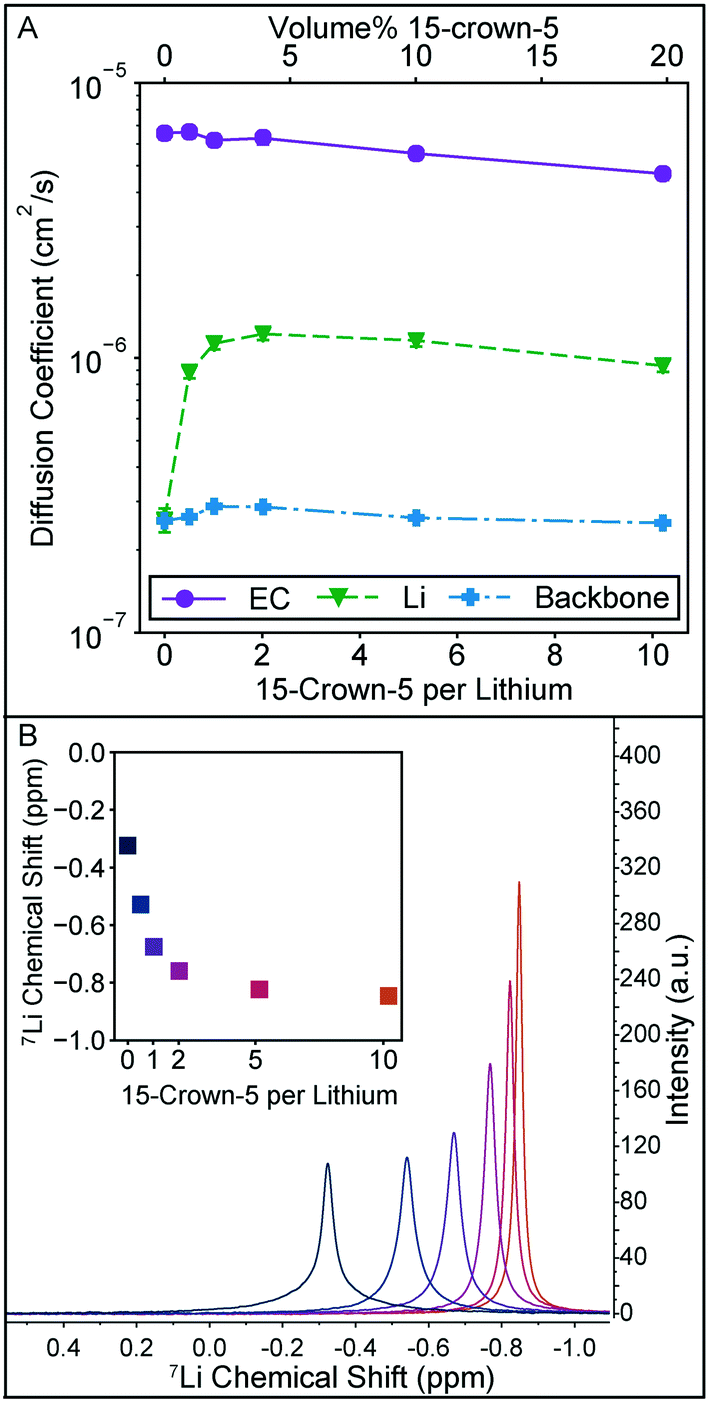 | ||
| Fig. 3 A) Diffusion coefficient for EC from the 1H spectra, 7Li diffusion coefficient, and polymer backbone diffusion coefficient measured from the 19F spectra for the set of solutions in Fig. 2A. Error bars, estimated from the diffusion calibration, are smaller than the data points. B) 7Li spectra for each 15-crown-5 containing solution in Fig. 2A and inset plot of the 7Li peak location. With increasing crown ether content, the peak narrows and shifts upfield. | ||
The increase in lithium diffusion is accompanied by a narrowing of the NMR peak width, as shown in Fig. 3B. Here the 7Li spectra of each solution is shown, with the chemical shift referenced to 9.7 m LiCl in D2O.22 Each of these peaks has a nearly equivalent area. NMR peak width can be a result of numerous interactions in solution, but polymers tend to have broad peaks due to slow molecular reorientation rates in solution. For this system where lithium can either be strongly associated with a polymer or more freely moving in solution, it is reasonable to associate narrowing with an average decrease in association of the lithium with the polymer. 7Li shift has also been used in previous studies on 15-crown-5 solvation with lithium, and the monotonic shift upfield is reminiscent of the trend seen for LiClO4 in propylene carbonate, and other solvents.19 Importantly, a sharp leveling off of the shift is not observed at a 1:1 ratio, and no change in the shift direction is observed. These observations would be consistent with extremely stable 1:1 complexes, or higher order (2:1) crown complexes. Together these observations on complexation and lithium diffusion indicate that the decrease in conductivity past 2:1 crown per lithium is not due to the formation of larger crown complexes, but instead is likely due to the increased viscosity at higher crown ether content.
As a proof of concept for the optimized electrolyte containing a 2:1 molar ratio of 15-crown-5 to lithium here, we fabricate a battery composed of commercially available electrode components. A graphite anode and LiFePO4 cathode were purchased from MTI Inc. and a coin cell battery was fabricated using a quartz fiber separator. We note that the majority of other proposed novel electrolytes would require optimization of electrode design, as dry polymer electrolytes, swollen charged membranes, and ceramic lithium conductors must be incorporated directly within porous electrodes to achieve high energy density. An advantage of a polyelectrolyte solution is the direct applicability to already available electrode designs. In Fig. 4, the second charge and discharge curve at C/20 is compared for an electrolyte with no additives, an electrolyte containing stabilizing additives, and the optimized 15-crown-5 containing electrolyte. Combining 15-crown-5 with the stabilizing additives achieves nearly 90% of the full theoretical capacity on the second discharge, while only 20 and 30% are achieved with no additives, or just stabilizing additives, respectively. The difference in capacity can likely be ascribed to the large increase in conductivity when 15-crown-5 is added to the electrolyte, thereby improving Li+ transport through the porous electrode and increasing active material utilization. While cyclability and rate capability are still under study, this initial result clearly demonstrates the ability of additives to enable polyelectrolyte solutions for battery application. A wide variety of sulfonated and other weakly dissociating charged polymers exist, but none have ever been tested for battery applications in solution.
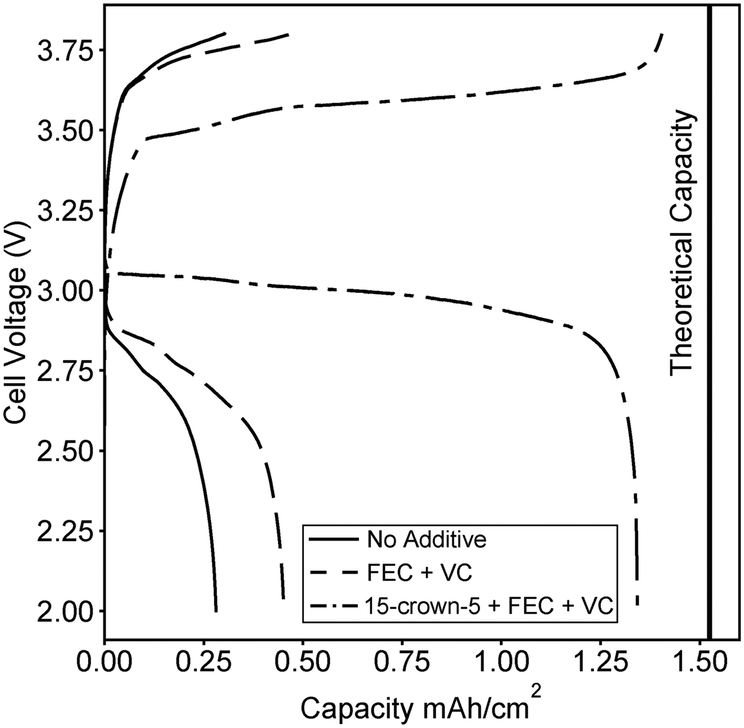 | ||
| Fig. 4 Second charge and discharge curves for batteries fabricated with the 0.1 M polymer in 2:1 (vol.) EC:DMC solution containing no additives, containing stabilizing additives FEC and VC, and containing 15-crown-5 and the stabilizing additives. The structure of each additive was given in Fig. 1. The calculated theoretical maximum capacity of the cell is also shown. | ||
These results also provide further motivation to fully characterize the transport properties of nonaqueous polyelectrolyte solutions utilizing fully rigorous transport theories and experiments, a significant undertaking. To date, no study has accurately measured the transference number of a lithium containing polyelectrolyte solution experimentally. Computational work in collaboration with our lab has identified a rich assortment of fundamental questions governing transport within these solutions.23 In particular, the true transference number may vary significantly from any simple estimate due to the complex relationship between diffusion and mobility in these systems. This complex relationship suggests that a unique opportunity exists with polyelectrolyte solutions to directly tune transference number by varying the fraction of charged groups on the polymer, changing the type of anion, or varying the solvent properties through additive engineering.
Conclusions
We have demonstrated that additives, particularly 15-crown-5, enable charge and discharge of a battery fabricated of commercially available electrodes and a polyelectrolyte solution that would otherwise have prohibitively low conductivity. We show that 15-crown-5 selectively solvates lithium ions in solution, allowing much higher diffusivity and thereby increasing solution conductivity. This application of crown ethers allows more direct quantification of crown to lithium ratios as compared with previous studies in membranes and is significantly different from historical applications of crown ethers in batteries which tend to increase solubility at the expense of lithium mobility. While significant work remains in quantifying the effects of crown ethers and polyelectrolytes on battery performance, we believe these initial results should spur further interest in the field. A polyelectrolyte solution could be exchanged directly with existing electrolytes in current cell designs, but also enable direct control over transference number. This could enable faster charge rates and higher energy densities, and provide better fundamental understanding of the effect of transference number on battery performance.Materials and methods
Materials
Battery grade ethylene carbonate, dimethyl carbonate, lithium bis(trifluoromethylsulfonimide), and tetraglyme were purchased from BASF and used as received. Fluorinated ethylene carbonate was kindly provided by Daikin, and all other additives were purchased from Sigma Aldrich and used as received. The polymer used in this study is a second batch of the polymer used in our prior work and the synthesis and characterization is described there.6 The structure of the polymer is shown in Scheme 1, along with NMR characterization. The peaks noted as A, B, C contain several peaks due to the different possible neighbor monomers and the slight shift that results from these different neighbors. The same is true for the PEG peak, where the peaks noted as “PEG ends” correspond to the PEG monomers at the end of the chain, within the copolymer. The presence of these peaks verifies the incorporation of PEG into the main polymer backbone. This NMR was taken after drying the polymer, and the very small H2O residual peak (equivalent to the residual H2O in DMSO-d6) confirms the polymer is in fact dry. This batch of the polymer has Mn = 6.7 kDa and a dispersity of 1.6 as measured against poly(ethylene oxide) standards (Sigma Aldrich) in an Agilent 1150 Series GPC with Waters Styragel HR3 and HR4 columns and NMP with 0.05 M LiBr as the mobile phase.Solution characterization
Conductivity was measured using a Mettler Toledo InLab 751-4 mm conductivity probe in vials kept at 25 °C by a dry heating/cooling block inside of an argon glovebox kept below 1 ppm water and 10 ppm oxygen (Vac Atmospheres). The conductivity probe was calibrated to known standards (Mettler Toledo) at 84 μS cm−1, 1413 μS cm−1 and 12.88 mS cm−1. Viscosity was measured with an electromagnetically spinning viscometer (EMS-1000, Kyoto Instruments) using a 2 mm aluminum ball at a rotation rate of 1000 rpm and 25 °C after the tube was sealed in the glovebox. Error on conductivity is estimated from several repeat measurements on separate samples to be around 10%. Viscosity error bars are taken from 10 repeat measurements of each individual sample.Nuclear magnetic resonance spectroscopy (NMR) measurements
Diffusion coefficients of each species were measured by pulse field gradient NMR on a Bruker Avance III 500 MHz instrument fitted with a 5 mm Z-gradient broadband probe and variable temperature unit maintained at 25 °C throughout the measurement. The gradient was calibrated to known values of H2O in D2O,24 H-DMSO in d6-DMSO,25 0.25 M LiCl in H2O,24 and 4 M LiCl in H2O.24 A recycle delay of at least 4xT1 was utilized for all measurements. Bruker's double stimulated bipolar gradient pulse sequence (dstebpgp3s) was used for all measurements to account for any convection. The signal decay as a function of gradient strength was fit to eqn (1), which includes adjustments for a slight change introduced to the pulse program in TopSpin 3.0, as compared with the original paper.26,27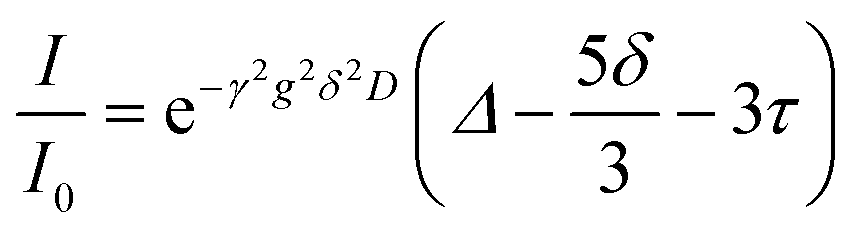 | (1) |
Coin cell preparation
Graphite (2.64 mAh cm−2) and LiFePO4 (1.52 mAh cm−2) electrode sheets were purchased from MTI Corp. and punched to 1.5 cm diameter electrodes inside of an argon glovebox (Vac Atmospheres). These were then assembled into a CR2032 coin cell with a 1.6 cm diameter quartz fiber (Whatman) separator and 200 μL of the electrolyte of interest. The electrolyte was dropped onto each electrode and the separator was fully soaked before being stacked and crimped closed. The cells were allowed to rest for 12 hours, before cycling at C/20 (0.076 mA cm−2). 3.8 and 2.0 V cutoffs were chosen for charge and discharge, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Vehicle Technologies Office, of the U.S. Department of Energy under Contract DE-AC02- 05CH11231, under the Advanced Battery Materials Research (BMR) Program. Work at the Molecular Foundry (NMR and ICP-OES chemical analysis) was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231. We are grateful for access to NMR facilities at the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. We thank David Babb of Dow Chemical Company for a helpful initial discussion that spurred this work.References
- J. Newman and K. E. Thomas-Alyea, Electrochemical Systems, 2004 Search PubMed.
- M. Doyle, T. F. Fuller and J. Newman, Electrochim. Acta, 1994, 39, 2073–2081 CrossRef CAS.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- K.-D. Kreuer, A. Wohlfarth, C. C. de Araujo, A. Fuchs and J. Maier, ChemPhysChem, 2011, 12, 2558–2560 CrossRef CAS PubMed.
- H. G. Buss, S. Y. Chan, N. A. Lynd and B. D. McCloskey, ACS Energy Lett., 2017, 2, 481–487 CrossRef CAS.
- K. M. Diederichsen, K. D. Fong, R. C. Terrell, K. A. Persson and B. D. McCloskey, Macromolecules, 2018, 51, 8761–8771 CrossRef CAS.
- M. Muthukumar, Macromolecules, 2017, 50, 9528–9560 CrossRef CAS PubMed.
- A. V. Dobrynin, Solutions of Charged Polymers, Elsevier B.V., 2012, vol. 1 Search PubMed.
- M. Hara, in Physical Chemistry of Polyelectrolytes, ed. T. Radeva, 2001, pp. 245–279 Search PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- M. Doyle, M. E. Lewittes, M. G. Roelofs and S. A. Perusich, J. Phys. Chem. B, 2001, 105, 9387–9394 CrossRef CAS.
- F. Cataldo, Eur. Chem. Bull., 2015, 4, 92–97 CAS.
- N. S. Schauser, G. E. Sanoja, J. M. Bartels, S. K. Jain, J. G. Hu, S. Han, L. M. Walker, M. E. Helgeson, R. Seshadri and R. A. Segalman, Chem. Mater., 2018, 30, 5759–5769 CrossRef CAS.
- B. Simon and J.-P. Boeuve, US Pat. US005626981A, SAFT, Romainville, France, 1997 Search PubMed.
- R. McMillan, H. Slegr, Z. Shu and W. Wang, J. Power Sources, 1999, 81–82, 20–26 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- A. J. Smetana and A. I. Popov, J. Solution Chem., 1980, 9, 183–196 CrossRef CAS.
- M. Morita, H. Hayashida and Y. Matsuda, J. Electrochem. Soc., 1987, 134, 2107 CrossRef CAS.
- G. Nagasubramanian, A. I. Attia and G. Halpert, J. Electrochem. Soc., 1992, 139, 9–12 CrossRef.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CAS.
- K. D. Fong, J. Self, K. M. Diederichsen, B. D. McCloskey and K. A. Persson, ACS Cent. Sci. DOI:10.1021/acscentsci.9b00406.
- M. Holz and H. Weingartner, J. Magn. Reson., 1991, 92, 115–125 CAS.
- M. Holz, X. Mao, D. Seiferling and A. Sacco, J. Chem. Phys., 1996, 104, 669–679 CrossRef CAS.
- A. Jerschow and N. Müller, J. Magn. Reson., 1997, 125, 372–375 CrossRef CAS.
- D. Sinnaeve, Concepts Magn. Reson., Part A, 2012, 40, 39–65 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |