
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu(I) diimine complexes as immobilised antibacterial photosensitisers operating in water under visible light†
Martin V.
Appleby
 a,
Peter G.
Walker
b,
Dylan
Pritchard
a,
Sandra
van Meurs
a,
Carly M.
Booth
a,
Craig
Robertson
a,
Michael D.
Ward
c,
David J.
Kelly
*b and
Julia A.
Weinstein
*a
a,
Peter G.
Walker
b,
Dylan
Pritchard
a,
Sandra
van Meurs
a,
Carly M.
Booth
a,
Craig
Robertson
a,
Michael D.
Ward
c,
David J.
Kelly
*b and
Julia A.
Weinstein
*a
aDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: Julia.Weinstein@sheffield.ac.uk
bDepartment of Molecular Biology and Biotechnology, University of Sheffield, UK. E-mail: D.Kelly@sheffield.ac.uk
cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
First published on 12th November 2020
Abstract
A complex of an Earth-abundant metal, copper, immobilised on silica offers a remarkably efficient way to kill bacteria in water under visible light, the first example of lighter transition metal complexes to do so. Photosensitisers which produce reactive oxygen species under light are emerging as an efficient way to kill microorganisms in water, yet the majority of photosensitising metal complexes are based on rare transition metals. Moreover, the efficiency of most solution-based photosensitisers is greatly compromised upon immobilisation on solid support, which is essential for safe treatment. Photosensitisers based on inexpensive metal complexes, such as those of Cu, Ni or Fe, usually have too short excited state lifetime to react efficiently with oxygen and are ineffective in production of reactive oxygen species. Here, we demonstrate that complexes of Cu(I) can be used as efficient photosensitisers for killing bacteria in water under visible light, when immobilised on surfaces, using as an example of [Cu(I)(xantphos)(dmp)]tfpb (1) [xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethyl-xanthine, dmp = 2,9-dimethyl-1,10-phenanthroline, and tfpb = tetrakis(3,5-bis-(trifluoromethyl)-phenyl)borate] immobilised on silica, 1-silica. In contrast to many short-lived Cu(I) complexes, a sterically-hindered coordination centre in 1 leads to a relatively long excited state lifetime of >200 ns, which enables 1 to efficiently photosensitise singlet oxygen (29%). Upon irradiation, 1-silica (55 μM) shows high antibacterial activity against both a Gram-negative bacterium E. coli, and a Gram-positive bacterium Staphylococcus aureus (S. aureus, the Methicillin Resistant strain MRSA 315), both of which commonly occur in water. 99.99% killing of S. aureus was observed after only 15 min of irradiation with 17.5 mW cm−2 light, with 99.9999% (‘complete’) killing achieved after 2 h. For E. coli, 99.99% killing was achieved after 2 h, and 99.9999% after 3 h of irradiation. Thus 1-silica exceeds the ≥99.99% threshold set by WHO for the “highly protective” antibacterial agents. This first example of an immobilised Cu(I) complex used for light-driven bacterial killing demonstrates the potential of Earth-abundant transition metal complexes as low-cost efficient photo-antibacterial agents.
Introduction
Clean water, sanitation and water scarcity are major issues worldwide, as highlighted in the UN Sustainable Development Goal 6.1 According to the UN, 1.8 billion people (25% of the global population) do not have access to a clean water source free from contamination.2 The situation could be much improved with proper water sanitation, however many regions where these problems occur lack access to proper water management systems and infrastructure. Developing point-of-use solutions/household water treatments to purify water on a local scale is one potential way of dealing with this problem.3,4 However, many current solutions are either very inefficient, or require large amounts of power.3–5There is a clear and urgent need to develop efficient, scalable, and ideally inexpensive antibacterial agents for water treatment. Utilizing sunlight for water purification could provide a cheap and clean way of providing water disinfection.
So far, this has been achieved via solar disinfection,6–8 which uses high energy UV light, that is absorbed by pathogens themselves including bacteria. Inspired by the ground-breaking developments in photodynamic therapy (PDT) of cancer,9 researchers are turning to development of photosensitisers, compounds that absorb light that bacteria do not absorb themselves, for use as antibacterial agents.10 Photosensitisers are ideally non-toxic to living organisms without light, but become toxic upon irradiation with light of an appropriate wavelength which promotes the photosensitiser into its excited state, PS*. One of the most common mechanisms of toxicity involves PS* reacting with dissolved oxygen, leading to production of reactive oxygen species (ROS). The two most damaging ROS are considered singlet oxygen (1O2) and the hydroxyl radical anion, as bacterial resistance to other ROS often occurs.11
The key requirements for a photosensitiser are efficient absorption of visible light, photostability, and an excited state with a sufficiently long lifetime to interact with oxygen dissolved in water by means of a bimolecular reaction; hundreds of nanoseconds is the lower limit of the excited state lifetimes needed to be practically relevant (see ESI† for estimation of lifetimes based on Stern–Volmer equation). Transition metal complexes with diimine ligands have been proven as excellent photosensitisers in applications such as anti-cancer PDT, as they possess a long-lived metal-to-ligand charge transfer (MLCT) excited state with triplet multiplicity. This triplet MLCT state is populated, usually with high efficiency, from the initially formed singlet excited state due to strong spin–orbit coupling of the central atom that promotes intersystem crossing.
Currently-used photosensitisers include organic compounds9,10 (which usually possess nanosecond excited-state lifetimes), porphyrins, and complexes of the 2nd and 3rd row transition metals (including Ir, Ru, Pt, Pd and Os)12–23 which are either rare, expensive, or both. More accessible alternatives are therefore beginning to be developed as photosensitisers.24–30 Metal ions such as Ag+,31,32 metal oxides such as ZnO,33 and complexes based on various metal ions including Cu(I),34 have shown to be toxic to bacteria without light;31,32,34 whilst inherently phototoxic metal nanoparticles have been used together with photosensitisers to promote the photoactivated production of ROS to initiate bacterial killing.33,35 Other well-known photosensitisers include porphyrins,36,37 such as derivatives of Zn(II) porphyrazine, or 5,10,15,20-tetrakis(1-methylpyridinium-4-yl)porphyrin tetra-iodide (tetra-Py+-Me), which have also been used for water treatment with good results: for example, tetra-Py+-Me at 10 μM achieved a reduction of 99.999% of E. coli after 120 min of irradiation with artificial white light (380–700 nm).
It is important to immobilise the photosensitiser on a support as this would eliminate the need to remove the sensitiser from water post-irradiation, but the immobilisation should not compromise the photosensitisation of ROS.38,39 Several organic photosensitisers, including BODIPY-derivatives immobilised on Nylon or polyacrylonitrile nanofibers,40 porphyrin derivatives immobilised on polyethylene elastomer or nanofibrillated cellulose (NFC) have shown efficient killing of bacteria, viruses, and other pathogens, often with 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10 efficiency, primarily on surfaces.41,42 A summary of the currently available immobilised photosensitisers is given in Table S9 in the ESI,† with the most relevant ones being used for comparison in the discussion section below.
log10 efficiency, primarily on surfaces.41,42 A summary of the currently available immobilised photosensitisers is given in Table S9 in the ESI,† with the most relevant ones being used for comparison in the discussion section below.
Most of the transition metal photosensitisers reported to date for water purification are, however, only active in solution. To develop them towards practical systems requires a 1st row transition metal complex that generates ROS efficiently under visible light, is photostable, not soluble in water, and can be immobilised on a solid support.
The class of compounds which is particularly promising in this regard are tetracoordinate Cu(I) diimine complexes, which absorb well in the UV/visible region due to an MLCT transition, and are potentially easy to modify at the periphery of the ligands, either to tune the energy of the absorption band or to attach functional groups which allow immobilisation onto a surface. However, Cu(I) diimine complexes often have excited state lifetimes that are too short to be of practical value for photosensitisation of ROS, often due to geometric distortion of a pseudo-tetrahedral ground-state geometry to a pseudo square-planar one in the MLCT excited state:43–48 this distortion creates a vacant coordination site43,44 for interaction with a solvent molecule (exciplex formation) which quenches the excited state,44 the process which has recently been observed directly by ultrafast spectroscopic methods.47,48 To “lock” the molecule in its tetrahedral ground-state geometry and prevent its geometry changing in the excited state, and thereby achieve long-lived excited states in Cu(I) complexes, sterically hindering ligands are required whose interlocking stops the angle between the two ligand planes from changing in the excited state.49
This approach was first demonstrated in 1980: the sterically-hindering ligand 2,9-dimethyl-1,10-phenanthroline (dmp) yields the complex [Cu(dmp)2]+ with an excited-state lifetime of 85 ns in deoxygenated DCM.44 There are now many examples of longer-lived excited states in Cu(I) complexes of various composition.30,45,49–55 Recent studies have shown that Cu(I) complexes containing one diimine ligand, and one diphosphine ligand such as 4,5-bis(diphenylphosphino)-9,9-dimethyl-xanthine (xantphos), which has a large bite angle and high steric hindrance, can have even longer excited state lifetimes (ns–μs).56–60 Cu(I) complexes are being increasingly used as photosensitisers in photocatalysis,24,25,30,51,61–73 and dyes-sensitised solar cells.74 They are also used as antitumor75–78 and antibacterial agents79–85 due to their innate dark toxicity. However, utilisation of their photosensitisation properties for PDT or antimicrobial PDT has been overlooked so far.
Here, we overturn the perception that only complexes of rare transition metals can be good photosensitisers, and that they do not work well when immobilised on a surface. We present the first example of a Cu(I) complex used as photosensitiser for antibacterial action in water, immobilised on silica support. We demonstrate the high light-induced antibacterial activity of the well-known complex [Cu(I)(xantphos)(dmp)]tfpb (1) [tfpb = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate]51,59,66,70,72 which has been previously examined as an oxygen sensor.59 The antibacterial action of 1 was tested on a Gram-negative bacterium, Escherichia coli (E. coli, strain MG1655),5,86,87 and a Gram-positive bacterium Staphylococcus aureus (S. aureus, the Methicillin Resistant strain MRSA 315), both of which commonly occur in water. We determine the yield of photosensitised 1O2 generation by 1, and demonstrate >99.9999% (>6log10) killing of both S. aureus and E. coli in presence of the complex 1 immobilised on silica particles, upon irradiation with blue light. To be classified as protective against bacteria according to WHO, an agent is required to achieve a ≥2log10 killing, and to be highly protective ≥4log10 is required, and this system comfortably exceeds both thresholds.87
Results and discussion
Complex 1 as its BF4− salt was synthesised following the known procedure, although the reaction was scaled up from 100 mg of product59 to 2.81 g. [Cu(CH3CN)4]BF4 was reacted with xantphos in DCM to form a solution of [Cu(xantphos)(CH3CN)2]BF4.This was followed by an addition of a solution of dmp in DCM, to yield [Cu(xantphos)(dmp)]BF4 as a yellow solid. The salt [Cu(xantphos)(dmp)]BF4 was re-dissolved in methanol, and a solution of Na(tfpb) in methanol solution added to yield complex 1 as a yellow solid, Fig. 1. The tfpb− counterion was chosen as it has been previously shown that the complex 1, with tfpb− as counterion, has a longer excited state lifetime in the solid state than does [Cu(xantphos)(dmp)]BF4.59 Neither salt is water-soluble, which is important to prevent the leaching of the compound from the solid support into water. Complex 1 was characterised by UV/Vis absorption spectroscopy, steady-state and time-resolved emission spectroscopy, high-resolution mass-spectrometry, and multinuclear NMR spectroscopy. The analytical data agree with those published previously.51,59,66,70,72
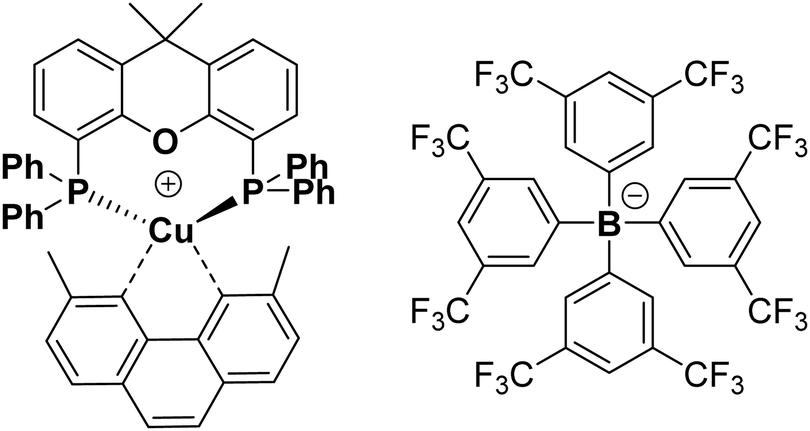 | ||
| Fig. 1 Chemical structure of complex 1 with counterion tfpb−. Structure of complex 1 obtained by single crystal X-ray crystallography, which is fully consistent with that published previously, is given in the ESI,† Fig. S8. CCDC 2012235. | ||
The properties of complex 1 are summarised in Table S1 in the ESI;† the absorption and emission spectra in acetonitrile solution, in the solid state, and when immobilised on silica, are shown in Fig. 2. The absorption maximum of 1 in acetonitrile is 378 nm, with a tail into the visible region; the emission maximum from the 3MLCT excited state occurs at ca. 550 nm. The excited state lifetime increases from 64 ns in aerated acetonitrile solution to 220 ns in deoxygenated solution. Assuming the concentration of dissolved of oxygen in acetonitrile to be 2.42 mM,88 the quenching constant of the excited state by oxygen, kq, was estimated as 4.6(5) × 109 M−1 s−1.
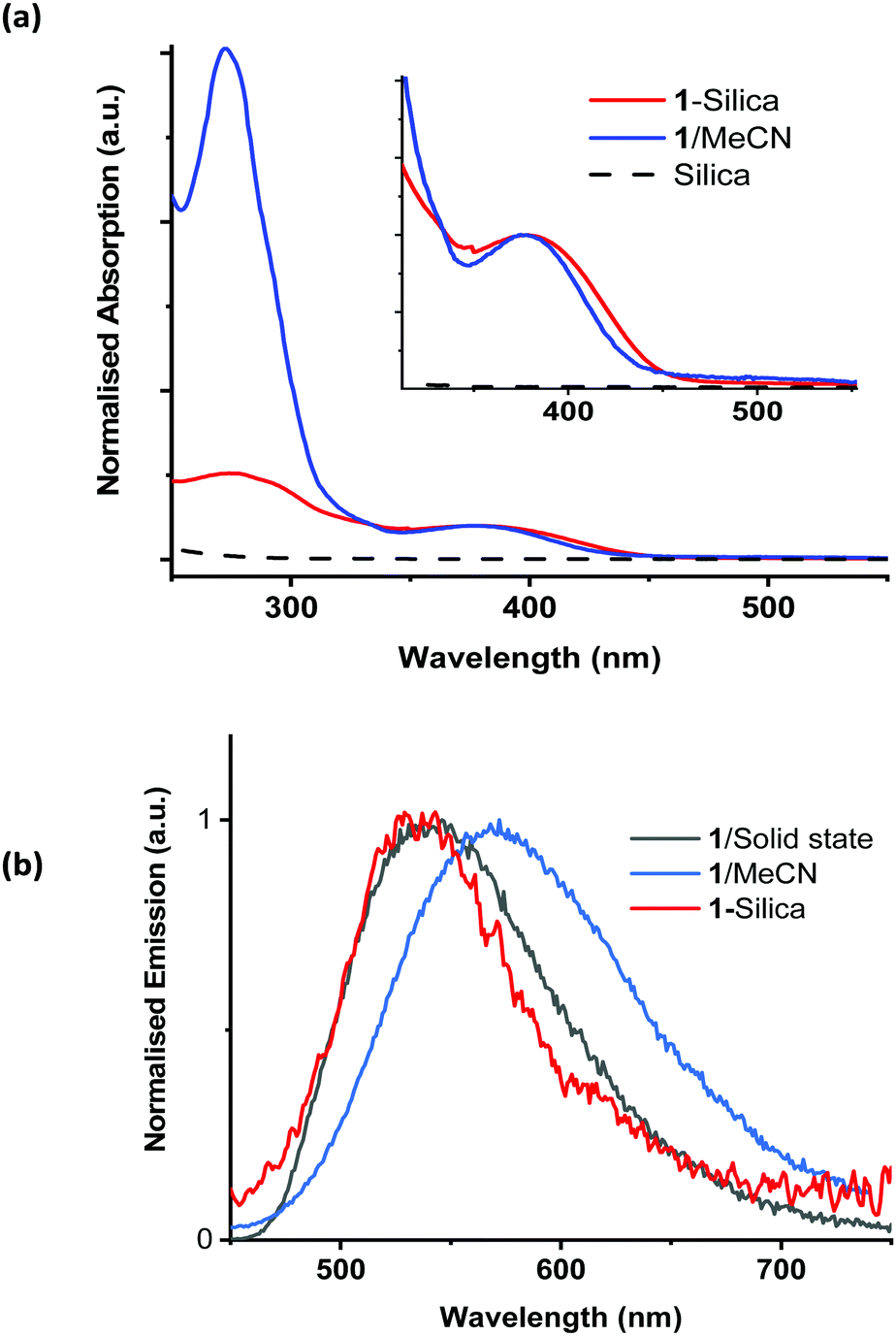 | ||
| Fig. 2 (a) Normalised (to OD at 380 nm) absorption spectra of 1 in MeCN and diffuse reflectance spectra of pure silica and 1-silica (55 μM); and (b) emission spectra of 1 in MeCN solution, in the solid state, and of 1-silica, excitation wavelength 400 nm. The MLCT absorption band of 1-silica is somewhat broader than that of 1 in solution, as is typical for solid-state spectra. A small shift to higher energies in the emission spectra of 1 in solid state and on silica vs. that in solution is observed, indicating emission from a relaxed configuration in solution. | ||
The quantum yield of singlet oxygen production,  by complex 1 in acetonitrile solution was measured directly, under excitation with a 355 nm (8 ns pulses) laser, by detecting the emission from photo-generated singlet oxygen at 1270 nm. The
by complex 1 in acetonitrile solution was measured directly, under excitation with a 355 nm (8 ns pulses) laser, by detecting the emission from photo-generated singlet oxygen at 1270 nm. The  value determined relative to a standard, perinaphthenone
value determined relative to a standard, perinaphthenone 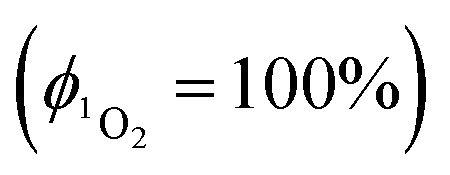 89 was 0.29 (±0.07), see Fig. S11 and S12 (ESI†). The relatively high quantum yield for singlet oxygen production explains the previously reported use of this compound as an oxygen sensor.59
89 was 0.29 (±0.07), see Fig. S11 and S12 (ESI†). The relatively high quantum yield for singlet oxygen production explains the previously reported use of this compound as an oxygen sensor.59
To avoid contaminating the water with the photosensitiser, it is beneficial to immobilise it on a solid support. The chosen support for these experiments was chromatography grade silica (40–63 μm, VWR chemicals), due to its high stability, ready availability, and low cost. The silica used had an average pore size of 60 Å and a surface area of 400 m2 g−1. Complex 1 was immobilised on silica (1-silica) by drying a concentrated solution of 1 in DCM onto the silica by slow rotary evaporation. For 1, 1 mg ml−1 corresponds to 583 μM. For 1-silica, prepared as described above, 1 mg ml−1 is equivalent to 11 μM of complex 1 (see Experimental part for detail). The size of the [Cu(xantphos)(dmp)]+ ion estimated from X-ray crystallographic data, Fig. S8 (ESI†), is smaller than the pore size of the silica, therefore some of the complex may have loaded inside the pores. The diffuse reflectance spectra, emission spectra, and emission excitation spectra all confirmed that the complex has been immobilised on the silica support (Fig. 2).
The presence of the photosensitiser on the silica substrate was also confirmed by solid state NMR spectroscopy, using the nuclei 13C and 31P (for the complex ion); 11B, 13C and 19F (for the anion); and 29Si (for the solid support). The spectra are shown in ESI,† Fig. S3–S7.
Comparative magic angle spinning (MAS) solid state NMR spectra were performed on complex 1, on silica gel without complex 1, and on 1-silica. In each of the high-abundance, high-sensitivity nuclei (11B, 19F and 31P) the signals were clearly retained from complex 1 to the 1-silica, even if slight shifts in position were observed on adsorption on the solid support and fine structure was lost. This was especially the case for the 31P spectra where the signal for complex 1 shows coupling to two NMR active Cu nuclei (63Cu and 65Cu),90 but when adsorbed on the silica support the signal is broadened to a wide envelope at the same chemical shift, giving confidence that the complex has been adsorbed. The reason for this broadening will partly be loss of crystallinity (tendency towards amorphous structure and associated shorter relaxation time) and partly due to signal-to-noise considerations, as the loading of complex 1 on the solid support is only 10 mg per 500 mg silica.
The same behaviour was observed for the low-abundance, low sensitivity nucleus 13C: a highly resolved 13C NMR spectrum for 1 in the solid state was changed to a severely broadened spectrum with comparable chemical shifts for 1-silica.
Direct determination of 1O2 production from 1-silica by measuring 1O2 emission at 1270 nm was not possible due to laser scattering from the silica particles. Instead, an indirect method that relies on the sensor Singlet Oxygen Sensor Green (SOSG, Invitrogen/Molecular Probes) was used: this sensor molecule is not emissive in solution, however, upon reaction with 1O2, a brightly emissive product is formed.91 The emission intensity of the sensor increased as the irradiation time of the suspension increased (Fig. S9 and S10, ESI†). The suspension was irradiated with a 405 nm diode with irradiance 10 mW cm−2 with the total radiant exposure equal to 6 J cm−2 after 600 s (Fig. 3). Whilst it is not possible to determine the 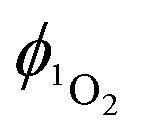 from 1-silica as the exact amount of the PS interacting with light cannot be determined accurately, the steady increase in probe emission intensity with irradiation time in the presence of a photosensitiser is significantly larger than upon irradiating the probe alone (Fig. 3). This observation confirms that adsorbed complex 1-silica produces 1O2 on light irradiation. A control experiment where SOSG was activated by a known 1O2 photosensitiser, [Ru(bpy)3]Cl2 (
from 1-silica as the exact amount of the PS interacting with light cannot be determined accurately, the steady increase in probe emission intensity with irradiation time in the presence of a photosensitiser is significantly larger than upon irradiating the probe alone (Fig. 3). This observation confirms that adsorbed complex 1-silica produces 1O2 on light irradiation. A control experiment where SOSG was activated by a known 1O2 photosensitiser, [Ru(bpy)3]Cl2 ( in aerated water 0.41)92 also confirms this conclusion.
in aerated water 0.41)92 also confirms this conclusion.
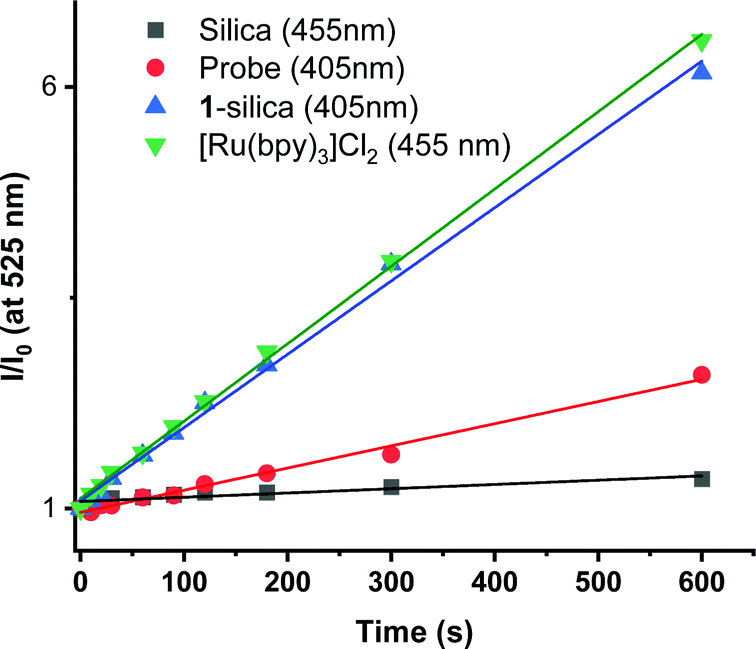 | ||
| Fig. 3 Relative emission intensity of SOSG probe (λmax = 525 nm) in water as a function of irradiation time. Probe (1 nM, -square-), [Ru(bpy)3]Cl2 in solution (1.56 μM, -upside down triangle-); silica suspension (5 mg ml−1, -circle-); 1-silica suspension (5 mg ml−1, 55 μM, -triangle-). | ||
S. aureus cultures were grown to mid-log growth phase before exposure to 405 nm light in 12-well plates with continuous shaking. For each experiment, bacterial viability reduction was compared against S. aureus cultures exposed without added photosensitiser as well as cultures treated with the well-characterised photosensitiser methylene blue (MB). No toxicity of the light alone for the S. aureus was observed, Fig. 4. A reduction from 1010 to 105 CFU ml−1 in presence of 50 μM of methylene blue was observed. Addition of 1 mg ml−1 (583 μM) of 1 (suspension) causes a 10-fold reduction of CFU in the dark or after irradiation with 405 nm light for 2 hours; thus small dark toxicity and no light toxicity at this concentration of 1 are observed. Increasing the concentration of 1 from 583 μM to 2917 μM causes significant dark toxicity, but no additional light toxicity, Fig. S13 (ESI†). It is clear that 1 alone displays some dark toxicity only at very large concentrations, but is not acting as a photosensitiser against S. aureus under any conditions studied.
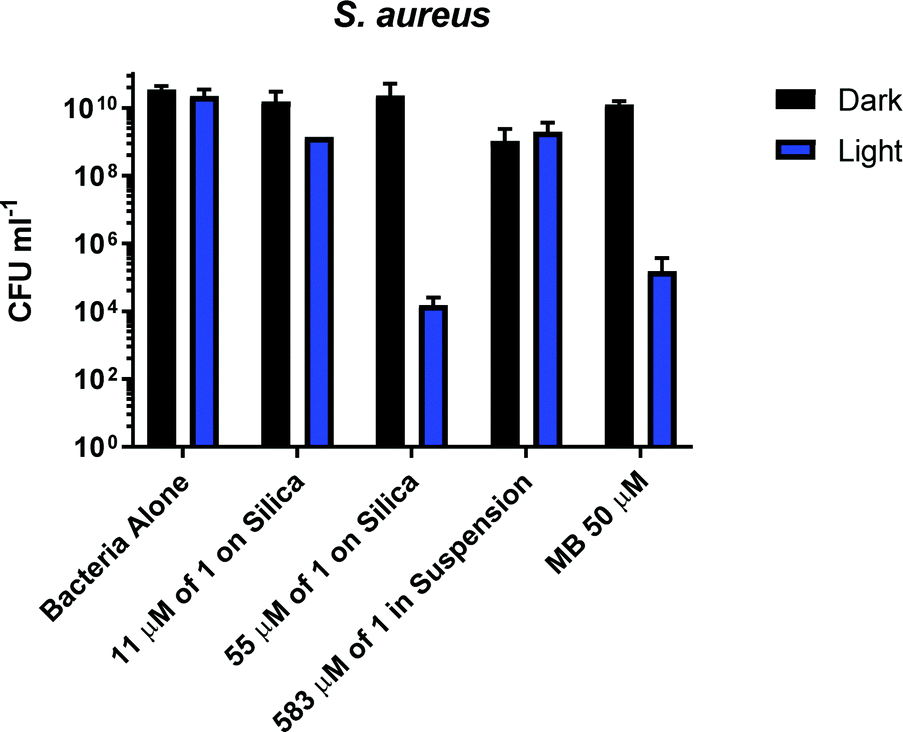 | ||
| Fig. 4 Bacterial viability assay measuring the colony forming units of S. aureus on its own and in the presence of 1-silica: 1 mg ml−1 (equivalent to 11 μM of complex 1) and 5 mg ml−1 (equivalent to 55 μM of complex 1), 5 mg ml−1 of complex 1 in suspension (583 μM) and methylene blue (MB) (50 μM). Assay without irradiation for 2 hours (black bars) and after irradiation for 2 hours with 405 nm light at 17.5 mW cm−2, total dose 126 J cm−2 (blue bars). | ||
In contrast to the free compound 1, immobilised 1-silica at 1 mg ml−1 or 5 mg ml−1 shows no dark toxicity towards S. aureus, Fig. 4. The lack of dark toxicity when the complex is immobilised on silica may be attributed to the much lower overall amount of the Cu-complex present. This also suggests that any leaching from the complex would have minimal effect on bacteria due to the highest loading (55 μM 1-silica) being only 1% of the concentration of 1 in suspension (583 μM) required to achieve ∼1log10 of killing, Fig. 4. Exposure of S. aureus to 405 nm light for 2 h in presence of 1-silica at 1 mg ml−1 leads to a 100-fold reduction in CFU, and to 6log10 reduction in presence of 5 mg ml−1 of 1-silica.
In order to identify the amount of 1-silica required for optimal bacterial killing, experiments were conducted at different concentrations, from zero to 40 mg ml−1. All samples had a starting CFU value of 106–107 and were irradiated with a 405 nm diode at 17.5 mW cm−2 for 30 min, Fig. 5. Fig. 5b shows that maximum killing of S. aureus is achieved using 5 mg ml−1 of 1-silica, and that any further increase in the amount of photosensitiser does not have an effect [note that 1 mg ml−1 was not causing any killing even at longer exposure times, Fig. 4]. In contrast, only a small reduction in CFU, up to a factor of 10, was observed for the Gram-negative E. coli at high concentrations of 1-silica after irradiation for 30 min, Fig. 5a. E. coli killing was be achieved at longer exposure times using 5 mg ml−1 of 1-silica (Fig. 6a). It has been reported previously that Gram-negative bacteria with an outer lipid/protein membrane are less susceptible to killing using exogenous photosensitisers such as 1-silica, as the outer membrane presents an additional barrier to protect the cell against extracellular generated ROS.93
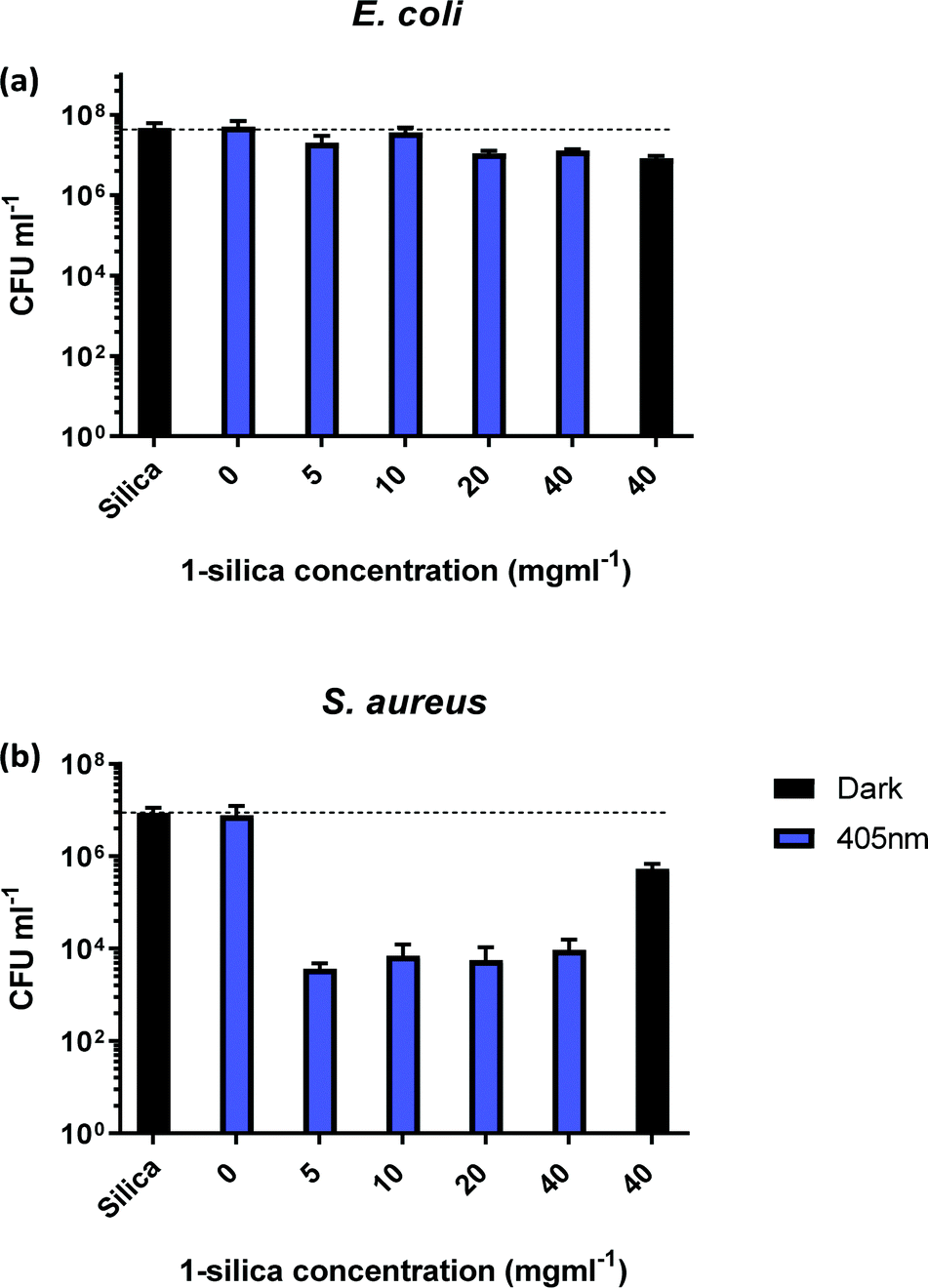 | ||
| Fig. 5 Colony forming units (CFU ml−1) for (a) E. coli and (b) S. aureus, as a function of exposure to different amounts of compound 1 adsorbed on silica (1-silica). Blue bars: irradiation with 405 nm light, 17.5 mW cm−2, 30 min. Black bars: no irradiation. All experiments performed in triplicate. 1 mg ml−1 of complex 1-silica is equivalent to 11 μM of complex 1. | ||
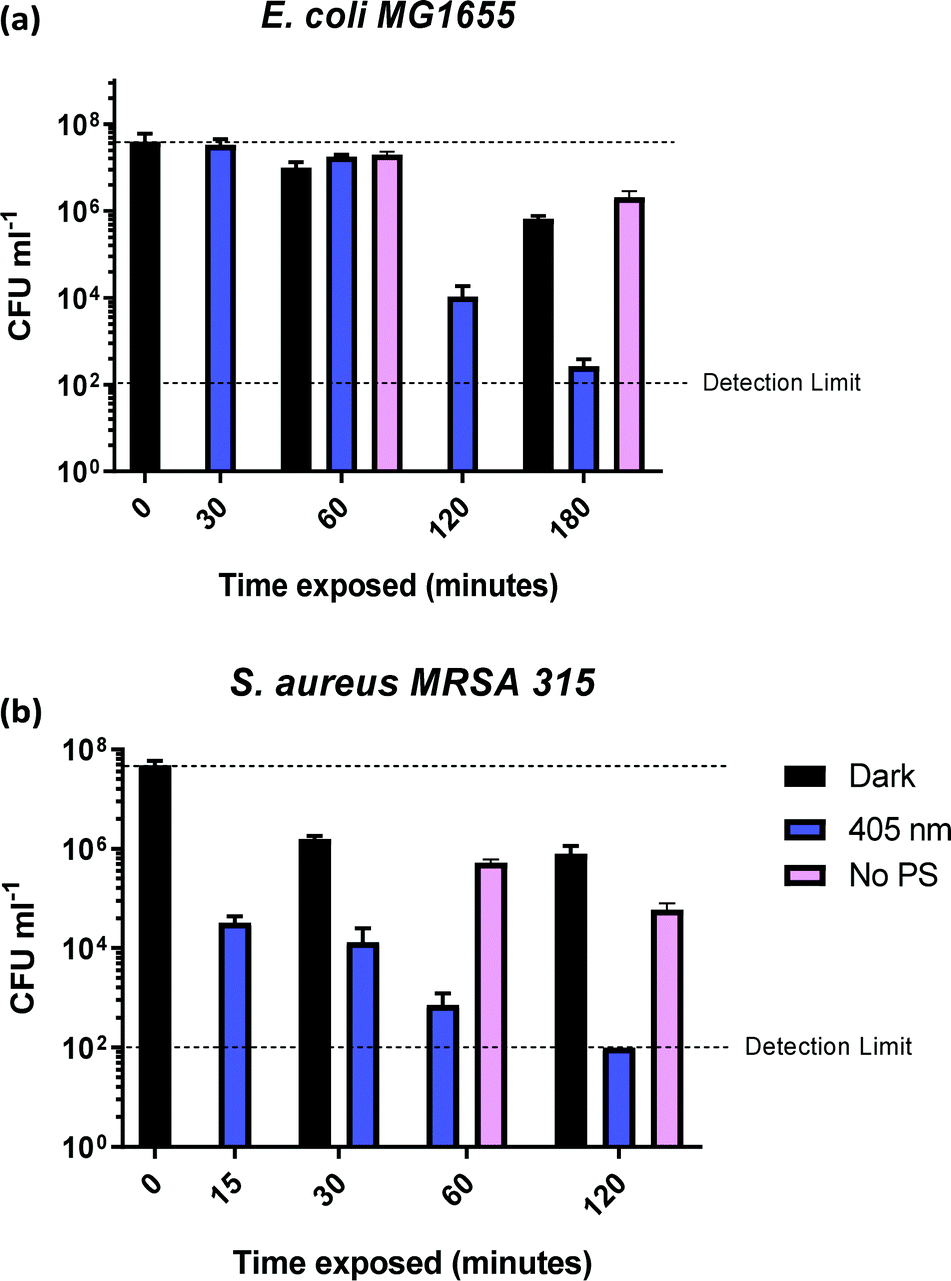 | ||
| Fig. 6 Colony forming units (CFU ml−1) for (a) E. coli MG1655 and (b) S. aureus MRSA 315, in presence of 5 mg ml−1 of 1-silica as a function of irradiation time with 405 nm light, 17.5 mW cm−2 (blue bars) and in the absence of 5 mg ml−1 of 1-silica (pink bars). | ||
In order to determine the time required to achieve complete killing of both bacterial strains at the 6log10 level, time-dependent measurements were performed (Fig. 6). The coloured bars in Fig. 6a show that E. coli killing (4log10 reduction) was observed after 2 h of irradiation with 405 nm light, and 5log10 reduction was achieved after 3 h of 405 nm irradiation.
No dark toxicity of 1-silica towards E. coli was observed after 1 h, with a 100-fold reduction in CFU after 3 hours. This result suggests that the killing of E. coli is due to a light-activated process. Killing of S. aureus was observed after only 15 min of exposure to 405 nm light, with a 4log10 reduction in CFU, Fig. 6b. Irradiation for 120 min led to reduction of S. aureus CFU to 102, the detection limit of the experiment, corresponding to a 6log10 reduction of S. aureus. The dark toxicity of 1-silica towards S. aureus was a 100-fold reduction in CFU after 30 min, a value which remained unchanged after 120 min. Thus, killing of S. aureus is also due to a light-activated process.
Finally, to establish the toxicity of light alone, killing of the bacteria when irradiated with 405 nm light in the presence and in the absence of the photosensitiser are compared in Fig. 6 (pink bars). Killing of E. coli to 6log10 is observed when the photosensitiser is present, with only 10-fold reduction in CFU using light irradiation alone. For S. aureus, light alone causes some killing at exposure times of 60 min and longer, but the presence of photosensitiser increases the light-induced killing 1000-fold, Fig. 6b. As both S. aureus and E. coli contain low levels of photo-excitable endogenous porphyrin molecules synthesised by the haem biosynthesis pathway, low levels of light-dependent killing by irradiation alone can be expected. However, neither S. aureus nor E. coli have sufficient concentrations of endogenous porphyrin molecules to allow for bacterial killing (4log10 reduction) using light alone, suggesting that the method of killing is via light activated 1-silica photosensitisation (Fig. 6a). It has been reported that Gram-negative bacteria with an outer lipid/protein membrane biolayer are less susceptible to killing using exogenous photosensitisers such as 1-silica as the outer membrane presents an additional permeability barrier to protect the cell against extracellular generated ROS.94
One possible light-dependent mechanism of bacterial killing with complex 1 is based on an oxidative burst that occurs upon illumination and will initially target extracellular structures in close proximity to the silica-immobilised photosensitiser which cannot penetrate the cell.95 The positively charged 1-silica will be brought to close proximity with the negatively charged bacterial surface to induce damage to extracellular cell envelope structures and biomolecules may be the primary site of damage, eventually causing death by cell lysis.
The lipophilic nature of 1 could also lead to potentially disruption of the bacterial lipid cell membranes (cytoplasmic membrane, and/or outer membrane in Gram-negative bacteria) in a light-independent manner. It is well-established that some lipophilic cations, for example of the triphenyl-phosphonium class96 can enter bacterial cells in a process driven by the cytoplasmic membrane potential (negative inside) and have even been conjugated to photosensitisers to increase their antibacterial effect in the cytoplasm.97 Multiple active transporters may also exist for many large cationic lipophilic molecules.98 The very small degrees of dark toxicity of 1 towards S. aureus and E. coli (Fig. 4–6) could be due to the direct membrane insertion, depolarisation and downstream cellular disruption caused by these processes should the compound be free in solution free rather than silica bound. In the latter case, it is clear that dark toxicity is much smaller than the degree of light-induced killing. We also note that whilst some dark toxicity has been observed at the 108 CFU ml−1 initial concentration of bacteria, no dark toxicity has been detected for 1010 CFU ml−1 initial concentration, even at 583 μM of 1 which would correspond to the ∼3.50 × 1017 molecules ml−1.
The stability of 1-silica in water was confirmed by time-dependent spectrophotometric investigation: 1-silica subjected to vigorous stirring and irradiation with light for up to 21 h did not show any leaching of the compound from silica support into water (within experimental uncertainty), see Part S6 in the ESI.† Therefore, it is unlikely that dark toxicity stems from an unbound photosensitiser.
While it is difficult to compare the efficiency of different photosensitisers already reported in the literature, due to the different conditions used, some observations can be made. It has previously been shown that 10 mg l−1 of methylene blue, MB, reduced the population of E. coli by 99.5% after 30 min of irradiation in solution, with sunlight with a total dose of 743 W m−2.94 However, immobilisation on multiple supports, including silica, led to greatly reduced bacteria killing potency of MB compared to that in solution. MB immobilised on polystyrene achieved a 97.5% reduction of CFU in 60 min of irradiation with white light,99 whilst for MB immobilised on silica (20 g l−1 of silica, 0.5 mg g−1 of MB on silica, equivalent to 31 μM of MB) only 65% reduction of CFU after 60 min (unknown irradiance) was observed, which will not be sufficient for practical use.99 In comparison, the 1-silica used at comparable concentrations (5 mg ml−1, equivalent to 55 μM of 1) achieves a 4 orders of magnitude more efficient killing of E. coli, >99.9999%.
Several immobilised photosensitisers have shown high killing under comparable conditions as 1-silica (Table S7, ESI†). For example, an organic photosensitiser BODIPY immobilised on Nylon leads to 99.9999% killing of MRSA under 400–700 nm light (30 min, 72 J cm−2), however it is photobleaching in the process.38 Photobleaching of BODIPY occurs at 118 J cm−2, half the total dose needed for 99.95% reduction of the Gram negative bacteria A. baumannii. Porphyrin derivatives, especially 5-(4-aminophenyl)-10,15,20-tris-(4-N-methylpyridinium)porphyrin (A3B3+) and its Zn-metallated derivative, (Zn-A3B3+) immobilised on polyethylene elastomer or nanofibrillated cellulose (NFC) have shown efficient killing of various pathogens, including MRSA and E. coli, with 99.9999% efficiency after 60 min of irradiation with a total dose of 234 and 288 J cm−2 respectively;41 (cf.1-silica which requires 120 min for MRSA and 180 min for E. coli to achieve 99.9999% killing with a total dose of 126 J cm−2 and 189 J cm−2 respectively).
Another organic photosensitiser, 9,10-anthraquinone-2-carboxylic acid bound to silica, ANT-SiO2,100 at a concentration of 700 μM caused reduction of 106 CFU ml−1 of E. coli after 110 min irradiation with 365 nm light, 3.85 mW cm−2 (total radiation exposure of 25.41 J cm−2), following an initial induction period of 60 min. In comparison, 90 min irradiation of TiO2 at 365 nm, 3.85 mW cm−2, led to a total inactivation of bacteria with no induction period. The difference between TiO2 (no induction time) and ANT-SiO2 photosensitiser on silica (induction time) in treatment of E. coli was attributed to the different ROS generated directly by TiO2 in comparison to the ROS produced via photosensitisation in ANT-SiO2.100
Recent work (2019/2020) on transition metal complexes for antibacterial action has included Re, Ir and Ru complexes in solution. Re-derivatives (5.8–11.6 μM, 365 nm light, 1 h, 3 J cm−2) were shown to inhibit bacterial growth for both E. coli and S. aureus.101 Ir(III) tris-diimine complexes of the structure [Ir(phen)2(R-phen)]3+, where R-phen = 3,8-dipyrenylphe-nanthroline and 3-pyrenylphenanthroline, caused 50% killing of S. aureus at 0.17 and 0.16 μM respectively when irradiated with visible light to a dose of 35 J cm−2.23 [Ru(bpy-TMEDA)3]8+, 15 μM, achieved a 6.87log10 reduction of S. aureus in PBS buffer after 20 min of irradiation at 470 nm, 22 mW cm−2 for a total dose of 27 J cm−2.102 Whilst some of these transition metal photosensitisers are very efficient in killing of bacteria, all of the above systems have been studied in solution and therefore cannot be compared directly to 1-silica.
Perhaps the closest comparison to 1-silica as a transition metal complex is [(4,7-diphenyl-1,10-phenanthroline)3Ru]2+, RDP2+, bound to porous silicone (pSil).18 The immobilised RDP2+ photo-sensitiser was used with a loading of 1–30 mg g−1 inside a micro-reactor with a Xe lamp and a cut-off filter letting through wave-lengths >373 nm. The total irradiation of 2 kW m−2 delivered for 9 h to water flowing with the rate of 15 ml h−1 resulted in an inactivation rate of E. coli of 1.1 × 105 CFU h−1 l−1.18 In our work, 1-silica achieved the rate of killing of ∼5 × 107 CFU h−1 l−1, under 405 nm irradiation with power density of 17.5 W m−2, although direct comparisons are difficult due to different setups used in our work and in that reported previously. Detailed summary of immobilised photosensitisers is given in Table S9 in the ESI.†
Overall, the data presented in Fig. 4–6 show that the bacterial killing is caused by a combination of the surface-bound photosensitiser 1-silica and light. Given the relatively high quantum yield of 1O2 production by compound 1 in solution (29%), singlet oxygen is likely the primary ROS involved in the killing of both bacteria, although contributions from other types of ROS cannot be ruled out.11
Conclusions
The first example of efficient killing of bacteria in water by an immobilised copper photosensitiser has been demonstrated. This simple, easy to make and easy to scale up copper(I) complex, which has a moderately long excited-state lifetime of 220 ns, has been shown to produce singlet oxygen upon irradiation with visible light in aqueous media. Significant killing of the Gram-positive bacterium, S. aureus (MRSA 315), with 99.99% killing observed after only 15 min, and 99.9999% (‘complete’) killing observed after 2 h. For a Gram-negative bacterium E. coli (MG1655), significant killing of 99.99% was achieved after 2 h of irradiation, and 99.9999% (‘complete’) killing after 3 h. Thus the complex immobilised on silica achieved significant light-induced killing of both Gram-positive and Gram-negative bacteria of a level considered “highly protective” by WHO standards (>4log10 reduction).87
This first example of application of Cu(I) complexes for photoactivated bacterial killing in water under visible light demonstrates the potential of this class of compounds as low-cost, immobilised antibacterial agents. These results could initiate future developments of Earth-abundant complexes for diverse antibacterial treatments (aPDT), such as water purification or surface disinfection, providing a long-sought replacement of rare transition metals.
Experimental
Reagents
Reagents were obtained from commercial sources and used without further purification unless stated otherwise. The starting materials of [Cu(CH3CN)4](BF4), dmp, xantphos and Na(tfpb) were purchased from Sigma-Aldrich. The solvents (DCM, MeOH, diethyl ether) used were purchased from commercial sources and were not purified further before use. Chromatography grade silica was purchased from VWR chemicals (particle size 40–63 μm, pore size 60 Å, surface area 400 m2 g−1). Singlet Oxygen Sensor Green (Molecular Probes) was purchased from ThermoFisher Scientific (https://www.thermofisher.com/order/catalog/product/S36002#/S36002).Synthesis of complex 1
[Cu(xantphos)(dmp)](tfpb) 1 was synthesised following the literature procedure.59 [Cu(CH3CN)4](BF4) (50 mg, 0.16 mmol) was added to a solution of xantphos (92 mg, 0.16 mmol) in DCM (20 ml). The solution was then stirred for 2 hours. Adding dmp (33 mg, 0.16 mmol) to the solution caused it to turn yellow. The solution was then stirred for a further hour. The product was precipitated by addition with diethyl ether and separated via vacuum filtration to give a bright yellow powder. This powder was then dissolved in MeOH (20 ml) and sodium tetrakis[3,5-bis(trifluoromethyl)-phenyl]borate (tfpb) (0.164 g, 0.185 mmol) was added. The solution was then stirred for 1 hour at room temperature. Upon addition of water, bright yellow crystals of complex 1 was yielded (0.114 g, 0.146 mmol). The method was later scaled up to produce 2.81 g, 1.64 mmol of 1.[Cu(xantphos)(dmp)]tfpb: 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.2 Hz, 2H), (7.74 s, 8H), 7.70 (s, 2H), 7.65 (dd, J = 7.8, 1.1 Hz, 2H), 7.51 (s, 4H), 7.41 (d, J = 8.3 Hz, 2H), 7.26–7.13 (m, 6H), 7.09–6.95 (m, 16H), 6.95–6.84 (m, 2H), 2.25 (s, 6H), 1.74 (d, J = 7.1 Hz, 6H). m/z (ES+) 849.6 (100%, M+).
Preparation of complex 1-silica
A solution of 10 mg of complex 1 in 10 ml of DCM was added onto 500 mg of silica, stirred, and rotary-evaporated. The resulting yellow powder was left to dry overnight, during which time no weight loss has been observed. The resulting concentration is 11 mM of complex 1 per gram of silica.Determination of singlet oxygen quantum yield using a singlet oxygen sensor green probe (SOSG, 2′,7′-dichlorofluorescin)
100 μg of SOSG probe was dissolved in 100 μL of MeOH to make a stock solution of SOSG of concentration ∼1.65 mM. For each experiment, 20 μl of the stock solution was added to 3 ml of water giving SOSG concentration of ∼11 nM. The 1.56 μM solution of [Ru(bpy)3]Cl2 in water was used. For the suspensions of complex 1 and 1-silica, 5 mg ml−1 was used.The experiment was conducted using standard 1 cm pathlength cuvettes, with 3 ml solution. The samples were stirred using a magnetic stirrer plate and irradiated with a 405 or 455 nm diode with an irradiance of 10 mW cm−2. The data were recorded using a Jobin-Yvon FluoroMax-4 Spectrofluorometer at the same time points for each sample (0, 10, 20, 30, 60, 90, 120, 180, 300 and 600 s). The emission intensity at the emission maximum of the probe (525 nm) was plotted against irradiation time as a ratio against the emission intensity at t = 0, before the irradiation.
Quantum yield of singlet oxygen production
Singlet oxygen was detected through measurement of the singlet oxygen emission band at ∼1275 nm. Complex 1 in acetonitrile solution (MeCN) was excited by the third harmonic of a Q-Switch Nd:YAG laser (λ = 355 nm, ∼8 ns pulse length, laser model LS-1231M from LOTISII). The time-resolved signal of 1O2 luminescence at 1275 nm was detected by a liquid nitrogen cooled InGaAs photodiode of ∅ 3 mm active area (J22D-M204-R03M-60-1.7, Judson Technologies). The output from the photodiode was coupled into a low-noise current amplifier (DLPCA-200, FEMTO Messtechnik GmbH). The amplifier output signal was recorded with a digital oscilloscope (TDS 3032B Tektronix) and transferred to a computer. To selectively detect the 1O2 emission, the high-contrast bandpass optical filter (1277 nm centre wavelength, 28 nm FWHM, custom-made by Izovac, Belarus) was fitted in front of the InGaAs photodiode. To increase the light collection efficiency, the spherical broadband mirror was set behind the sample to reflect the NIR emission through the sample towards the detector.The quantum yield of singlet oxygen production 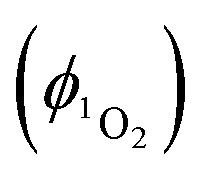 is determined by comparing the initial amplitude of the emission signal of 1O2 generated when irradiating the air-equilibrated solution of complex 1 and that of the standard (perinaphthenone,
is determined by comparing the initial amplitude of the emission signal of 1O2 generated when irradiating the air-equilibrated solution of complex 1 and that of the standard (perinaphthenone, 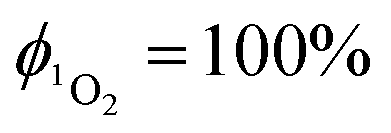 (acetonitrile)).89 The emission lifetime for 1O2 sensitised by the complex and the standard must be similar (within the range 70–90 μs in acetonitrile) to confirm that 1O2 does not react with the photosensitiser in its ground state. The optical densities of the complex and a standard are matched at 355 nm, and the same solvent was used for both compounds. The experiments are performed at a series of excitation energies ranging from 20 μJ to 500 μJ per pulse. The
(acetonitrile)).89 The emission lifetime for 1O2 sensitised by the complex and the standard must be similar (within the range 70–90 μs in acetonitrile) to confirm that 1O2 does not react with the photosensitiser in its ground state. The optical densities of the complex and a standard are matched at 355 nm, and the same solvent was used for both compounds. The experiments are performed at a series of excitation energies ranging from 20 μJ to 500 μJ per pulse. The  values are obtained in the low-energy limit whilst the intensity of the emission increases linearly with the laser power.
values are obtained in the low-energy limit whilst the intensity of the emission increases linearly with the laser power.
A correction is applied to the calculated initial intensities to account for small discrepancies in the optical density of the compound and standard solutions at 355 nm:
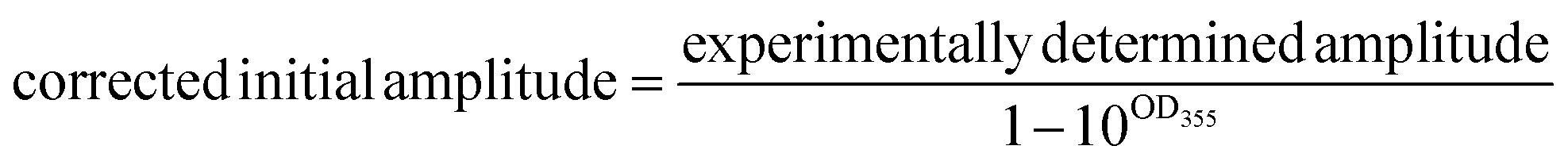 | (1) |
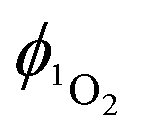 are calculated at each power by using eqn (2) and the average of the values stated is taken as the singlet oxygen yield.
are calculated at each power by using eqn (2) and the average of the values stated is taken as the singlet oxygen yield.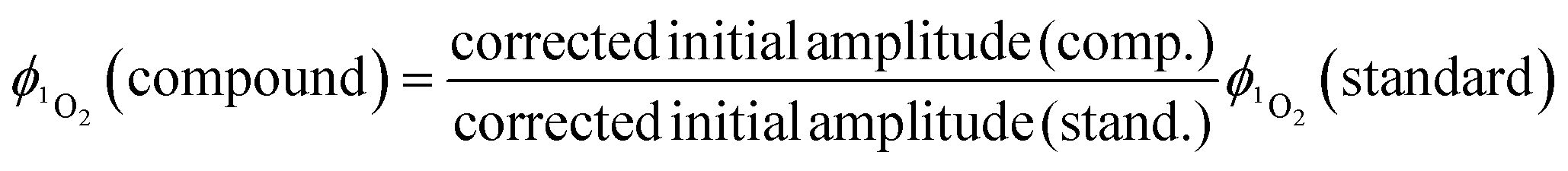 | (2) |
1H NMR spectroscopy in solution
1H spectra were measured using a 400 MHz Bruker Avance 400 spectrometer. The complexes were dissolved in spectroscopic grade deuterated chloroform (CDCl3) or acetonitrile (CD3CN) and calibrated against the residual solvent peak.Solid-state NMR spectroscopy
Solid-state NMR spectroscopy was carried out using a Bruker AVANCE III HD NMR spectrometer operating at 500.13 MHz 1H frequency, using 4 mm zirconia rotors and a magic angle spinning (MAS) rate of 10 kHz on a dual resonance (HX-type) MAS-probe. For those experiments using cross-polarisation (CP), a contact time of 2 ms was used and the relaxation delay for each sample was individually determined from a proton T1 measurement (setting the relaxation delay to 5 × T1 for the measurement). Spectra with high-power decoupling (31P) were acquired with a longer relaxation delay of 30 s (T1 not determined) and single-pulse experiments (19F) were acquired with 10 s relaxation delay (T1 not determined). Transients were collected until sufficient signal-to-noise was obtained. Values of the chemical shifts are referenced to adamantane in 13C (magnetic field set to place the higher shift resonance of adamantane at 38.48 ppm).Diffuse reflectance and UV-visible absorption spectroscopy
Diffuse reflectance spectra were recorded using a Varian Cary 5000 spectrophotometer. A Praying Mantis™ Diffuse Reflectance Accessory was used to hold the solid samples in the spectrophotometer. The UV-Vis spectra were recorded using a Cary 50 Bio Spectrometer and sample solutions in a quartz cuvette with a path length of 1 cm.Emission spectroscopy and emission lifetime experiments
Emission and excitation spectra were collected using a Horiba Jobin Yvon Fluromax-4 spectrofluorometer. Solutions were placed in 1 cm path length quartz cuvette. Emission lifetime experiments were conducted with an Edinburgh Instruments mini-tau Edinburgh Instrument set-up using 405 nm, 75 ps diode laser as an excitation source.Bacterial strains and growth conditions
S. aureus MRSA 315 and E. coli MG1655 were cultured aerobically at 37 °C in Luria-Bertani (LB) broth (Oxoid) sterilised by autoclaving. Liquid cultures were shaken at 200 rpm. Cultures were grown to mid-log growth phase (2–3 hours) before samples were collected.Light exposure viability assays
High intensity 405 nm light was produced by an LED with a nominal wavelength of 405 nm and a bandwidth of ∼20 nm at full half width maximum. Power density (J cm−2) was measured using a Thorlabs 5310C thermal power sensor at the position the samples were exposed, 5 cm away from the light source. Light sensitivity experiments were carried out on mid-log bacterial broth culture resuspended in sodium phosphate buffer (20 mM pH 7.2) to an OD600 of 0.1. For each condition tested, 1 ml of mid-log cell culture was added into 12 well plates containing the appropriate photosensitiser tested. Each condition was tested in triplicate with three technical replicates per repeat. Viability was calculated by counting the number of colony-forming units (C.F.U.) after appropriate dilution on LB agar plates, counting their number per ml.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (capital equipment award to Lord Porter Laser Laboratory), the BBSRC (DTP funding to P. J. W.), the Grantham Centre for Sustainable Futures (studentship to M. V. A.), and the University of Sheffield for support.Notes and references
- United Nations, 2015, 16301, pp. 1–35.
- World Health Organization and UNICEF, 2017, pp. 1–110.
- S. Loeb, R. Hofmann and J. H. Kim, Environ. Sci. Technol. Lett., 2016, 3, 73–80 CrossRef CAS.
- P. R. Hunter, Environ. Sci. Technol., 2009, 43, 8991–8997 CrossRef CAS.
- World Health Organization, 2011, pp. 1–543.
- V. Alipour, L. Rezaei, M. R. Etesamirad, S. Rahdar, M. R. Narooie, A. Salimi, J. Hasani, R. Khaksefidi, S. A. Sadat and H. Biglari, J. Global Pharma Technol., 2017, 9, 40–46 CAS.
- W. Heaselgrave and S. Kilvington, Acta Trop., 2011, 119, 138–143 CrossRef.
- A. Carratalà, A. D. Calado, M. J. Mattle, R. Meierhofer, S. Luzi and T. Kohn, Appl. Environ. Microbiol., 2016, 82, 279–288 CrossRef.
- D. Phillips, Pure Appl. Chem., 2011, 83, 733–748 Search PubMed; N. Manav, P. E. Kesavan, M. Ishida, S. Mori, Y. Yasutake, S. Fukatsu, H. Furuta and I. Gupta, Dalton Trans., 2019, 48, 2467–2478 RSC; J. M. Fernandez, M. D. Bilgin and L. I. Grossweiner, J. Photochem. Photobiol., B, 1997, 37, 131–140 CrossRef CAS ; see also , Coord. Chem. Rev., 2019, 379, 1–160 CrossRef (special issue, Ed. J. F. Lovell); see also J. Photochem. Photobiol., B, 2015, 150, 1–68 Search PubMed.
- M. Wainwright, T. Maisch, S. Nonell, K. Plaetzer, A. Almeida, G. P. Tegos and M. R. Hamblin, Lancet Infect. Dis., 2017, 17, e49–e55 CrossRef CAS; H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef; S. K. Sharma, T. H. Dai and M. R. Hamblin, in Antimicrobial Drug Discovery: Emerging Strategies, ed. A. Tegos and E. Mylonakis, 2012, CABI, Wallingford, pp. 310–322 Search PubMed.
- F. Vatansever, W. C. M. A. de Melo, P. Avci, D. Vecchio, M. Sadasivam, A. Gupta, R. Chandran, M. Karimi, N. A. Parizotto, R. Yin, G. P. Tegos and M. R. Hamblin, FEMS Microbiol. Rev., 2013, 37, 955–989 CrossRef CAS.
- F. Manjón, D. García-Fresnadillo and G. Orellana, Photochem. Photobiol. Sci., 2009, 8, 926–932 RSC.
- D. García-Fresnadillo, Y. Georgiadou, G. Orellana, A. M. Braun and E. Oliveros, Helv. Chim. Acta, 1996, 79, 1222–1238 CrossRef.
- M. Thandu, C. Comuzzi and D. Goi, Int. J. Photoenergy, 2015, 2015, 1–22 CrossRef.
- F. Manjón, M. Santana-Magaña, D. García-Fresnadillo and G. Orellana, Photochem. Photobiol. Sci., 2014, 13, 397–406 RSC.
- R. Bonnett, M. A. Krysteva, I. G. Lalov and S. V. Artarsky, Water Res., 2006, 40, 1269–1275 CrossRef CAS.
- A. I. Silverman, B. M. Peterson, A. B. Boehm, K. McNeill and K. L. Nelson, Environ. Sci. Technol., 2013, 47, 1870–1878 CrossRef CAS.
- M. E. Jiménez-Hernández, F. Manjón, D. García-Fresnadillo and G. Orellana, Sol. Energy, 2006, 80, 1382–1387 CrossRef.
- L. Villén, F. Manjón, D. García-Fresnadillo and G. Orellana, Appl. Catal., B, 2006, 69, 1–9 CrossRef.
- S. L. Rosado-Lausell, H. Wang, L. Gutiérrez, O. C. Romero-Maraccini, X. Z. Niu, K. Y. H. Gin, J. P. Croué and T. H. Nguyen, Water Res., 2013, 47, 4869–4879 CrossRef CAS.
- F. Manjón, M. Santana-Magaña, D. García-Fresnadillo and G. Orellana, Photochem. Photobiol. Sci., 2010, 9, 838–845 RSC.
- D. García-Fresnadillo, ChemPhotoChem, 2018, 2, 512–534 CrossRef.
- L. Wang, S. Monro, P. Cui, H. Yin, B. Liu, C. G. Cameron, W. Xu, M. Hetu, A. Fuller, S. Kilina, S. A. McFarland and W. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 3629–3644 CrossRef CAS.
- R. Giereth, W. Frey, H. Junge, S. Tschierlei and M. Karnahl, Chem. – Eur. J., 2017, 23, 17432–17437 CrossRef CAS.
- A. J. J. Lennox, S. Fischer, M. Jurrat, S. P. Luo, N. Rockstroh, H. Junge, R. Ludwig and M. Beller, Chem. – Eur. J., 2016, 22, 1233–1238 CrossRef CAS.
- S. Garakyaraghi, E. O. Danilov, C. E. McCusker and F. N. Castellano, J. Phys. Chem. A, 2015, 119, 3181–3193 CrossRef CAS.
- S. Garakyaraghi, P. D. Crapps, C. E. McCusker and F. N. Castellano, Inorg. Chem., 2016, 55, 10628–10636 CrossRef CAS.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2013, 52, 8114–8120 CrossRef CAS.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2015, 54, 6035–6042 CrossRef CAS.
- S. Garakyaraghi, C. E. McCusker, S. Khan, P. Koutnik, A. T. Bui and F. N. Castellano, Inorg. Chem., 2018, 57, 2296–2307 CrossRef CAS.
- J. R. Morones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5(190), 1–11 Search PubMed.
- A. V. Domínguez, R. A. Algaba, A. M. Canturri, Á. R. Villodres and Y. Smani, Antibiotics, 2020, 9(36), 1–10 Search PubMed.
- Y. Xiang, C. Mao, X. Liu, Z. Cui, D. Jing, X. Yang, Y. Liang, Z. Li, S. Zhu, Y. Zheng, K. W. K. Yeung, D. Zheng, X. Wang and S. Wu, Small, 2019, 15, 1900322 CrossRef.
- A. Kyzioł, A. Cierniak, J. Gubernator, A. Markowski, M. Jezowska-Bojczuk and U. K. Komarnicka, Dalton Trans., 2018, 47, 1981–1992 RSC.
- X. An, N. Naowarojna, P. Liu and B. M. Reinhard, ACS Appl. Mater. Interfaces, 2020, 12, 106–116 CrossRef CAS.
- M. Bartolomeu, S. Reis, M. Fontes, M. G. P. M. S. Neves, M. A. F. Faustino and A. Almeida, Water, 2017, 9, 630 CrossRef.
- L. Sobotta, J. Dlugaszewska, D. Ziental, W. Szczolko, T. Koczorowski, T. Goslinski and J. Mielcarek, J. Photochem. Photobiol., A, 2019, 368, 104–109 CrossRef CAS.
- F. Cieplik, D. Deng, W. Crielaard, W. Buchalla, E. Hellwig, A. Al-Ahmad and T. Maisch, Crit. Rev. Microbiol., 2018, 44, 571–589 CrossRef CAS.
- N. Maldonado-Carmona, T. S. Ouk, M. J. F. Calvete, M. M. Pereira, N. Villandier and S. Leroy-Lhez, Photochem. Photobiol. Sci., 2020, 19, 445–461 RSC.
- K. R. Stoll, F. Scholle, J. Zhu, X. Zhang and R. A. Ghiladi, Photochem. Photobiol. Sci., 2019, 18, 1923–1932 RSC.
- D. R. Alvarado, D. S. Argyropoulos, F. Scholle, B. S. T. Peddinti and R. A. Ghiladi, Green Chem., 2019, 21, 3424–3435 RSC.
- B. S. T. Peddinti, F. Scholle, R. A. Ghiladi and R. J. Spontak, ACS Appl. Mater. Interfaces, 2018, 10, 25955–25959 CrossRef CAS.
- M. W. Blaskie and D. R. Mcmillin, Inorg. Chem., 1980, 19, 3519–3522 CrossRef CAS.
- D. R. McMillin, J. R. Kirchhoff and K. V. Goodwin, Coord. Chem. Rev., 1985, 64, 83–92 CrossRef CAS.
- M. T. Miller, P. K. Gantzel and T. B. Karpishin, Inorg. Chem., 1999, 38, 3414–3422 CrossRef CAS.
- M. T. Miller, P. K. Gantzel and T. B. Karpishin, J. Am. Chem. Soc., 1999, 121, 4292–4293 CrossRef CAS.
- L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022–7034 CrossRef CAS.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS.
- M. Schmittel and A. Ganz, Chem. Commun., 1997, 999–1000 RSC.
- M. Sandroni, Y. Pellegrin and F. Odobel, Chimie, 2016, 19, 79–93 CrossRef CAS.
- M. Heberle, S. Tschierlei, N. Rockstroh, M. Ringenberg, W. Frey, H. Junge, M. Beller, S. Lochbrunner and M. Karnahl, Chem. – Eur. J., 2017, 23, 312–319 CrossRef CAS.
- K. Soulis, C. Gourlaouen, C. Daniel, A. Quatela, F. Odobel, E. Blart and Y. Pellegrin, Polyhedron, 2018, 140, 42–50 CrossRef CAS.
- M. T. Miller, P. K. Gantzel and T. B. Karpishin, Angew. Chem., Int. Ed., 1998, 37, 1556–1558 CrossRef CAS.
- M. T. Miller, P. K. Gantzel and T. B. Karpishin, Inorg. Chem., 1998, 37, 2285–2290 CrossRef CAS.
- M. T. Miller and T. B. Karpishin, Inorg. Chem., 1999, 38, 5246–5249 CrossRef CAS.
- S. Keller, F. Brunner, J. M. Junquera-Hernández, A. Pertegás, M. G. La-Placa, A. Prescimone, E. C. Constable, H. J. Bolink, E. Ortí and C. E. Housecroft, ChemPlusChem, 2018, 83, 143 CrossRef CAS.
- M. Alkan-Zambada, S. Keller, L. Martínez-Sarti, A. Prescimone, J. M. Junquera-Hernández, E. C. Constable, H. J. Bolink, M. Sessolo, E. Ortí and C. E. Housecroft, J. Mater. Chem. C, 2018, 6, 8460–8471 RSC.
- S. Keller, A. Prescimone, H. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí and C. E. Housecroft, Dalton Trans., 2018, 47, 14263–14276 RSC.
- C. S. Smith, C. W. Branham, B. J. Marquardt and K. R. Mann, J. Am. Chem. Soc., 2010, 132, 14079–14085 CrossRef CAS.
- S. M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS.
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC.
- Y. Yamazaki, T. Onoda, J. Ishikawa, S. Furukawa, C. Tanaka, T. Utsugi and T. Tsubomura, Front. Chem., 2019, 7, 288 CrossRef CAS.
- J. Kim, D. R. Whang and S. Y. Park, ChemSusChem, 2017, 10, 1883–1886 CrossRef CAS.
- C. Minozzi, A. Caron, J. C. Grenier-Petel, J. Santandrea and S. K. Collins, Angew. Chem., Int. Ed., 2018, 57, 5477–5481 CrossRef CAS.
- M. M. Cetin, R. T. Hodson, C. R. Hart, D. B. Cordes, M. Findlater, D. J. Casadonte, A. F. Cozzolino and M. F. Mayer, Dalton Trans., 2017, 46, 6553–6569 RSC.
- R. Giereth, I. Reim, W. Frey, H. Junge, S. Tschierlei and M. Karnahl, Sustainable Energy Fuels, 2019, 3, 692–700 RSC.
- B. Wang, D. P. Shelar, X. Z. Han, T. T. Li, X. Guan, W. Lu, K. Liu, Y. Chen, W. F. Fu and C. M. Che, Chem. – Eur. J., 2015, 21, 1184–1190 CrossRef CAS.
- B. J. McCullough, B. J. Neyhouse, B. R. Schrage, D. T. Reed, A. J. Osinski, C. J. Ziegler and T. A. White, Inorg. Chem., 2018, 57, 2865–2875 CrossRef CAS.
- Y. R. Zhang, X. Yu, S. Lin, Q. H. Jin, Y. P. Yang, M. Liu, Z. F. Li, C. L. Zhang and X. L. Xin, Polyhedron, 2017, 138, 46–56 CrossRef CAS.
- Y. Zhang, M. Heberle, M. Wächtler, M. Karnahl and B. Dietzek, RSC Adv., 2016, 6, 105801–105805 RSC.
- Y. Zhang, L. Zedler, M. Karnahl and B. Dietzek, Phys. Chem. Chem. Phys., 2019, 21, 10716–10725 RSC.
- Y. Zhang, P. Traber, L. Zedler, S. Kupfer, S. Gräfe, M. Schulz, W. Frey, M. Karnahl and B. Dietzek, Phys. Chem. Chem. Phys., 2018, 20, 24843–24857 RSC.
- O. Reiser, Acc. Chem. Res., 2016, 49, 1990–1996 CrossRef CAS.
- C. Dragonetti, M. Magni, A. Colombo, F. Fagnani, D. Roberto, F. Melchiorre, P. Biagini and S. Fantacci, Dalton Trans., 2019, 48, 9703–9711 RSC.
- R. Starosta, A. Bykowska, A. Kyziol, M. Plotek, M. Florek, J. Król and M. Jezowska-Bojczuk, Chem. Biol. Drug Des., 2013, 579–586 CrossRef CAS.
- U. K. Komarnicka, S. Kozieł, P. Zabierowski, R. Kruszyński, M. K. Lesiów, F. Tisato, M. Porchia and A. Kyzioł, J. Inorg. Biochem., 2020, 203, 110926 CrossRef CAS.
- V. Gandin, M. Porchia, F. Tisato, A. Zanella, E. Severin, A. Dolmella and C. Marzano, J. Med. Chem., 2013, 56, 7416–7430 CrossRef CAS.
- D. Mahendiran, N. Pravin, N. S. P. Bhuvanesh, R. S. Kumar, V. Viswanathan, D. Velmurugan and A. K. Rahiman, ChemistrySelect, 2018, 3, 7100–7111 CrossRef CAS.
- U. A. Khan, A. Badshah, M. N. Tahir and E. Khan, Polyhedron, 2020, 181, 114485 CrossRef CAS.
- O. Evangelinou, A. G. Hatzidimitriou, E. Velali, A. A. Pantazaki, N. Voulgarakis and P. Aslanidis, Polyhedron, 2014, 72, 122–129 CrossRef CAS.
- T. S. Lobana, J. K. Aulakh, H. Sood, D. S. Arora, I. Garcia-Santos, M. Kaur, C. E. Duff and J. P. Jasinski, New J. Chem., 2018, 42, 9886–9900 RSC.
- P. R. Chetana, B. S. Srinatha, M. N. Somashekar and R. S. Policegoudra, J. Mol. Struct., 2016, 1106, 352–365 CrossRef CAS.
- L. Tabrizi and H. Chiniforoshan, New J. Chem., 2017, 41, 10972–10984 RSC.
- M. K. Rauf, Imtiaz-ud-Din, A. Badshah, M. Gielen, M. Ebihara, D. de Vos and S. Ahmed, J. Inorg. Biochem., 2009, 103, 1135–1144 CrossRef CAS.
- P. R. Chetana, B. S. Srinatha, M. N. Somashekar, R. S. Policegoudra, R. Rao, B. Maity and S. M. Aradhya, Int. J. Pharm. Sci. Rev. Res., 2013, 21, 355–363 CAS.
- N. J. Ashbolt, Toxicology, 2004, 198, 229–238 CrossRef CAS.
- World Health Organization, 2011, pp. 1–68.
- C. Franco and J. Olmsted, Talanta, 1990, 37, 905–909 CrossRef CAS.
- R. Schmidt, C. Tanielian, R. Dunsbach and C. Wolff, J. Photochem. Photobiol., A, 1994, 79, 11–17 CrossRef CAS.
- F. Asaro, A. Camus, R. Gobetto, A. C. Olivieri and G. Pellizer, Solid State Nucl. Magn. Reson., 1997, 8, 81–88 CrossRef CAS.
- S. Kim, M. Fujitsuka and T. Majima, J. Phys. Chem. B, 2013, 117, 13985–13992 CrossRef CAS.
- C. Tanielian, C. Wolff and M. Esch, J. Phys. Chem., 1996, 100(16), 6555–6560 CrossRef CAS.
- F. Sperandio, Y.-Y. Huang and M. Hamblin, Recent Pat. Anti-Infect. Drug Discovery, 2013, 8, 108–120 CrossRef CAS.
- A. T. Cooper and D. Y. Goswami, J. Sol. Energy Eng., 2002, 124, 305–310 CrossRef CAS.
- T. Maisch, S. Hackbarth, J. Regensburger, A. Felgenträger, W. Bäumler, M. Landthaler and B. Röder, J. Ger. Soc. Dermatology, 2011, 9, 360–366 Search PubMed.
- D. G. Nicholls and S. J. Ferguson, Bioenergetics, 4th Edn, 2013 Search PubMed.
- R. Bresolí-Obach, I. Gispert, D. G. Peña, S. Boga, Ó. Gulias, M. Agut, M. E. Vázquez and S. Nonell, J. Biophotonics, 2018, 11, e201800054 CrossRef.
- S. Jindal, L. Yang, P. J. Day and D. B. Kell, BMC Microbiol., 2019, 19, 1–16 CrossRef CAS.
- A. Savino and G. Angeli, Water Res., 1985, 19, 1465–1469 CrossRef CAS.
- A. K. Benabbou, C. Guillard, S. Pigeot-Rémy, C. Cantau, T. Pigot, P. Lejeune, Z. Derriche and S. Lacombe, J. Photochem. Photobiol., A, 2011, 219, 101–108 CrossRef CAS.
- A. Frei, M. Amado, M. A. Cooper and M. A. T. Blaskovich, Chem. – Eur. J., 2020, 26, 2852–2858 CrossRef CAS.
- Y. Feng, W. Z. Sun, X. S. Wang and Q. X. Zhou, Chem. – Eur. J., 2019, 25, 13879–13884 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00642d |
| This journal is © The Royal Society of Chemistry 2020 |