
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing the effect of structure curations on the simulated CO2 separation performances of MOFs†
Sadiye
Velioglu
 ab and
Seda
Keskin
*a
ab and
Seda
Keskin
*a
aDepartment of Chemical and Biological Engineering, Koc University, Rumelifeneri Yolu, Sariyer, 34450, Istanbul, Turkey. E-mail: skeskin@ku.edu.tr; Tel: +90 212 338 1362
bInstitute of Nanotechnology, Gebze Technical University, Gebze, 41400, Kocaeli, Turkey. E-mail: sadiyevelioglu@gtu.edu.tr; Tel: +90 262 605 1757
First published on 24th April 2020
Abstract
Experimentally reported metal organic frameworks (MOFs) may have structural issues such as the presence of solvent molecules in their pores, missing hydrogen atoms in the frameworks and/or absence of charge balancing ions, which all require curation of structures before using them in molecular simulations. The development of computation-ready MOF databases significantly accelerated the assessment of CO2 adsorption by providing directly usable, curated crystal structures for molecular simulations. Each database followed different methods to curate MOFs which caused the same material to be reported with different structural features in databases. In order to understand the role of curated computation-ready MOF databases in the predicted CO2 separation performances of MOFs, we studied various MOFs commonly existing in databases but curated differently in terms of (i) removal of bound solvents, (ii) treatment of missing hydrogens, and (iii) retention of charge balancing ions (CBIs). We used molecular simulations to compute CO2/CH4, CO2/H2, and CO2/N2 mixture adsorption and predicted various separation performance metrics such as selectivity, regenerability (R%), and the adsorbent performance score (APS) for the curated computation-ready MOFs. Our results showed that the CO2 separation performances of MOFs and the identity of the best performing MOFs significantly change depending on the structure curation. For example, removal of coordinated solvents from MOFs resulted in higher simulated CO2 uptakes, selectivities, and APSs compared to the structures having solvents. On the other hand, the absence of CBIs in the frameworks resulted in overestimated CO2 uptakes, APSs, and R%, and underestimated CO2 selectivities compared to MOFs having CBIs. Based on these results, we suggested a path showing how to use the curated, computation-ready MOF structures in high-throughput molecular simulations.
1. Introduction
Metal organic frameworks (MOFs) are highly promising materials for various applications due to their various pore sizes and shapes, high porosities, large surface areas and structural tunabilities.1–3 A very large number of MOFs have been synthesized to date (93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 306 structures according to the Cambridge Structural Database (CSD), version 5.40) and this extensive pool of MOFs with diverse structural features offers tremendous opportunities for adsorption-based gas separation applications. However, characterization and gas adsorption measurements of every single MOF material are time- and resource-consuming when the fast-growing number of synthesized MOFs is considered. Therefore, identification of the best MOF materials for target gas separation is challenging. To this extent, computational methods offer a great opportunity for screening large numbers of MOFs in a time-efficient manner and short-listing the best candidates for further experimental testing. Molecular simulations in the early times only focused on a small number of MOFs but the number of materials examined in molecular simulations significantly increased with the advancement in computational methods and with the availability of comprehensive MOF databases.
306 structures according to the Cambridge Structural Database (CSD), version 5.40) and this extensive pool of MOFs with diverse structural features offers tremendous opportunities for adsorption-based gas separation applications. However, characterization and gas adsorption measurements of every single MOF material are time- and resource-consuming when the fast-growing number of synthesized MOFs is considered. Therefore, identification of the best MOF materials for target gas separation is challenging. To this extent, computational methods offer a great opportunity for screening large numbers of MOFs in a time-efficient manner and short-listing the best candidates for further experimental testing. Molecular simulations in the early times only focused on a small number of MOFs but the number of materials examined in molecular simulations significantly increased with the advancement in computational methods and with the availability of comprehensive MOF databases.
In high-throughput computational screening of MOFs, grand canonical Monte Carlo (GCMC) simulations have been widely used to describe the gas adsorption and separation performances of a very large number of MOFs and to define the top performing candidates. Results of computational studies are useful to direct experimental efforts, time and resources to the best performing materials. There are excellent examples in which promising MOFs identified by computational studies4–7 were synthesized and experimentally tested.4,5,8 For example, Ahmed et al.4 computationally screened nearly half a million MOFs for H2 storage, and proposed three MOFs that exceed the storage capacity of prototype MOF-5. They demonstrated a perfect agreement between experiments and simulations for both gravimetric and volumetric H2 adsorption isotherms of these MOFs up to 100 bar at 77 K. Moghadam et al.5 performed a computational study to screen almost 3000 MOFs to predict their deliverable O2 capacities, and experimentally synthesized and tested one of the top materials. They reported a very good agreement between the measured and calculated O2 adsorption isotherms up to 140 bar at 298 K. Computational studies proposed an MMIF (microporous metal–imidazolate framework) as a good candidate for adsorption-based6 and membrane7-based CO2/CH4 and CO2/N2 separations. Ahmadi et al.8 recently synthesized and characterized this MOF, incorporated it into Matrimid to measure the gas separation performance of the composite membrane and revealed the excellent potential of the MMIF for CO2 separation.
The main input of these molecular simulations is the crystallographic information file (CIF) of MOF materials. The synthesized MOFs are deposited into the CSD using a refcode, a six-letter reference code,9 but these experimentally reported CIFs generally contain information about the solvent molecules that result from the solvothermal synthesis of MOFs. Before performing molecular simulations, solvent molecules are removed from the pores to mimic the experimental activation procedure of MOFs. There are also other structural issues in the CSD-deposited CIFs such as the absence of hydrogen atoms, the presence of disordered atoms and/or the absence of charge balancing ions in the frameworks. These matters should be handled before using CIFs in molecular simulations to get accurate results about the gas adsorption properties of MOFs. On the other hand, identifying the CIFs having these problems and manually correcting them is not straightforward. Therefore, there has been a strong need for the development of a computation-ready MOF database that can provide solvent-free, curated CIFs which can facilitate the large-scale molecular simulations of MOFs.
The first database was the computation-ready, experimental (CoRE) MOF database,10 which was established by curating the MOF structures by removing solvents, retaining charge balancing ions (CBIs), and manually editing MOFs having missing hydrogen atoms and overlapping atoms. A set of 4764 computation-ready curated MOFs together with 345 unmodified MOFs were provided in the CoRE MOF database. This MOF database was recently updated to include 14142 3-D structures collected from the CoRE MOF database users, and the updates of the CSD in addition to the Web of Science search for the terms “porous organic polymer” and “metal–organic frameworks”.11 Solvent removal, optimization of some structures after the bound solvent removal, semi-automated restoration of disordered structures, and removal of duplicate structures were performed to curate MOFs.11 The second database was referred to as the CSD non-disordered MOF subset,12 which was provided with a Python script to remove solvents from the structures. A set of 54808 MOFs (CSD version 5.37) containing 1-D, 2-D, and 3-D MOFs was released. These two databases, the CoRE MOF and CSD non-disordered MOF subset, have been very useful since they provide the main input of molecular simulations, computation-ready CIFs of MOFs, and significantly contributed to exploring the gas adsorption properties of MOFs using molecular simulations. Many high-throughput computational screening studies used either of the databases to investigate the gas storage and/or gas separation potentials of MOF materials.12–26 Simon et al.27 recently provided an excellent review on molecular simulations of MOFs and discussed some shortcomings of computation-ready MOF databases. For example, the method used for solvent removal from MOFs could be too aggressive so that (i) the structural integrity of some MOFs without solvents may become questionable or (ii) the MOF could relax into a different form after the solvent removal or (iii) performing automatic solvent-removal on large numbers of MOFs can lead to chemically inaccurate structures by accidentally removing an essential part such as the ligand or CBIs of the framework. Our group recently showed that there are 3490 common MOFs in the intersection of two computation-ready databases, the CoRE MOF and CSD MOF subset, but simulated CH4 and H2 adsorption data of 387 MOFs significantly differ depending on from which database MOFs are taken.28 A detailed analysis of these structures showed that the two computation-ready databases curated structures differently in terms of solvent removal, treatment of missing hydrogen atoms and retention of CBIs. One important outcome was that the predicted CH4/H2 separation performances of the MOFs that are reported with different curated structures in the two databases significantly differ and this caused large variations in rankings and identification of the top performing materials for CH4/H2 separations. Considering the fact that CO2 separation is still the most widely studied application field of MOFs, understanding how the predicted CO2 separation performances of MOFs would be affected by different curations performed by databases is crucial. The simulated CO2 adsorption properties of MOFs strongly depend on the electrostatic interactions between the MOF atoms and CO2 molecules and the different treatments of the CBIs could have a remarkable effect on these interactions which may cause significant differences in the predicted CO2 adsorption and separation performances of MOFs in high-throughput screening studies.
Motivated by this, we aimed to reveal the role of using curated computation-ready MOF databases in accurately assessing the CO2 separation performance of MOFs from CO2/CH4, CO2/H2 and CO2/N2 mixtures. We focused on several representative MOFs that are common in both computation-ready MOF databases but reported with different structural features due to different structural modifications in terms of the (i) removal of bound solvents, (ii) treatment of missing hydrogen atoms, and (iii) retention of CBIs in the frameworks. Computation-ready, curated MOFs were compared with their corresponding experimental structures to elucidate different curations made by each database. We then performed molecular simulations for the differently curated MOFs and computed adsorption of the CO2/CH4, CO2/H2 and CO2/N2 mixtures in the MOFs at different pressures. Based on the molecular simulations results, the CO2 selectivity, working capacity, regenerability and adsorbent performance score of the MOFs were calculated and compared to examine the sensitivity of the predicted CO2 separation performances of the MOFs to the different structural modifications done in the establishment of computation-ready databases.
2. Computational details
The structural properties of MOFs, such as pore limiting diameters (PLDs), the largest cavity diameters (LCDs), surface areas, and pore volumes, were computed using Zeo++ (version 0.3) software.29 The pore volumes and surface areas were calculated using probe molecules with radii of 0 Å and 1.8 Å (representing N2), respectively, with a trial number of 50000. We specifically focused on MOFs having PLDs >3.8 Å so that all gas molecules that we studied in this work (CO2, CH4, H2, and N2) can be adsorbed within the MOFs’ pores. We focused on three mostly encountered structural curations in this work: (i) removal of bound solvents, (ii) treatment of missing hydrogen atoms, and (iii) retention of CBIs in the frameworks. Detailed information about 68 representative MOFs that we studied in this work, their refcodes, the types of solvents and CBIs in their frameworks, computed structural properties and all the simulation data for single-component and mixture gas uptakes is provided in the ESI.†
2.1 Removal of bound solvents
MOF structures deposited into the CSD may have bound and/or unbound solvent molecules in the pores due to the artifacts of X-ray powder diffraction (XRD) analysis. MOFs with solvent molecules dominate about 90% of the CSD.12 In order to assess the gas adsorption capacities of MOFs, residual solvent molecules should be evacuated from the structures via an activation process in the experiments. To mimic this process in computational studies, solvent molecules are mostly removed from the structures before performing molecular simulations. We specifically focused on MOFs having the most widely used solvents, water and dimethylformamide (DMF), in their structures and selected 27 MOFs from our previous study28 which were identified to have different structural modifications due to the removal of bound solvents. Briefly, the CoRE MOF database removed bound solvents from 16 MOFs, whereas the CSD MOF subset kept them in the structures. On the other hand, the CSD MOF subset removed bound solvents in the remaining 11 MOFs, while the CoRE MOF kept them. Additionally, to study the effect of structure optimization on simulation results, we optimized the computation-ready structures whose coordinated solvents were removed using Materials Studio 2017 R2 (MS).30 The Forcite module was used for energy minimization with a tolerance value of 10−4 kcal mol−1 and geometry optimization using the Smart algorithm with the maximum number of iterations of 1000. The convergence tolerances in geometry optimization were set as 10−6 Å, 10−5 kcal mol−1, and 10−4 kcal mol−1 Å for the displacement, energy and force, respectively. During this step, the cell parameters were also optimized. The Universal Force Field (UFF)31 was used for optimization of structures without coordinated solvents in MS.2.2 Treatment of missing hydrogen atoms
The difficulty in refinement of the weakly scattering X-rays of light elements such as hydrogen caused the deposition of MOF structures with missing hydrogen atoms. However, these missing hydrogen atoms should be present in the frameworks, with their probable configurations, to obtain an accurate crystallographic representation of the structures. In order to investigate the curation approach for handling the missing hydrogen atoms, 14 MOFs were selected from our previous study.28 The CoRE MOF database curated these MOFs by adding the missing hydrogen atoms of the framework, whereas the CSD MOF subset did not make any treatment for missing hydrogen atoms. Therefore, a comparison of the simulated CO2 uptakes of the MOFs taken from the two different computation-ready databases shows how the absence of hydrogens would affect the predicted CO2 separation performances of MOFs in high-throughput computational screening of MOFs.2.3 Retention of CBIs
Several MOFs have CBIs to make the overall framework charge neutral. Since CBIs are unbound in the framework, in some cases they may be incorrectly identified as solvents and removed during the automated solvent removal process.32 27 MOFs were chosen from our previous study28 to assess how accidental removal of CBIs during the automated solvent removal affects the predicted CO2 separation performances of the MOFs. There was only one MOF for which the CSD MOF subset removed CBIs. For the remaining MOFs, CBIs were removed during the curations performed by the CoRE MOF database. In high-throughput computational screening studies, MOFs, even the ones having CBIs, are assumed to be rigid to save significant computational time. However, the flexibility of the CBIs can be important as the change in the configuration and position of the ions within the framework would affect the CO2 adsorption properties of the MOFs. To investigate the effect of the flexibility of CBIs, we selected two MOFs and obtained three distinct snapshots from their GCMC simulations to examine the change in CO2 uptake. We first equilibrated CBIs in the selected computation-ready structures to find their most probable configurations by fixing the main framework and letting the CBIs move. For this step, the Dynamics task in the Forcite module of MS was used and a 500 ps molecular dynamics (MD) simulation was run in the NVE ensemble with a time step of 1 fs. The UFF31 and charge equilibration method (QEq)33 were used for the equilibration of the selected structures with CBIs in MS. In order to imitate the mobility of ions in the framework, we selected three representative snapshots with the lowest energy configurations from the trajectory of MD simulations and used them in GCMC simulations to compute the gas uptakes as explained in detail below.2.4 Molecular simulations of gas adsorption
We calculated the adsorption of single-component CO2, CO2/CH4: 50/50, CO2/H2: 15/85 and CO2/N2: 15/85 mixtures in MOFs by performing GCMC simulations at 0.1, 1 and 10 bar at 298 K. The GCMC simulations were carried out using RASPA simulation code.34 The single-site spherical Lennard-Jones (LJ) 12-6 potential was used to model H235 and CH436 molecules, whereas the three-site rigid LJ 12-6 potential was assigned for CO237 and N238 molecules with a dummy atom at the center of the mass in N2 to balance the net charges. The UFF was used for the potential parameters of MOF atoms.31 Each MOF was converted into P1 symmetry using MS and every length of the unit cell was adjusted to be >26 Å and the cut-off distance was set as the half of the unit cell length. The Lorentz–Berthelot mixing rules were employed. In order to calculate the electrostatic interactions between the adsorbate and framework atoms, the QEq method33 as implemented in RASPA was used to define the partial point charges of MOF atoms. The Ewald summation was used to compute the electrostatic interactions.39 For each MOF, single-component (mixture) gas adsorption simulations were carried out with 10000 initialization cycles and 100000 (200000) production cycles. The validity of the potentials, force fields and charge assignment method used in this work to compute the gas adsorption properties of MOFs was shown in our previous simulation studies40–42 where we represented the good agreement between our simulated and experimentally reported gas uptakes.
The results of the GCMC simulations were used to compute four different adsorbent evaluation metrics: adsorption selectivity (Si/j = (Ni/Nj)/(yi/yj)), working capacity (ΔNi = Nads,i − Ndes,i), adsorbent performance score (APS = Si/j × ΔNi) and percent regenerability (R% = ΔNi/Nads,i × 100). Here, N represents the gas uptake in mol gas per kg MOF, y is the bulk gas composition, subscripts i and j correspond to the gas species, and subscripts ads and des refer to the adsorption and desorption, respectively. The bulk gas compositions of the CO2/CH4: 50/50, CO2/H2: 15/85 and CO2/N2: 15/85 mixtures were set to represent industrial natural gas purification, hydrogen recovery and flue gas separation processes, respectively. All the adsorbent performance evaluation metrics were computed under both pressure swing adsorption (PSA) and vacuum swing adsorption (VSA) conditions to understand the impact of curation of MOF structures on the predicted gas uptake and separation properties of MOFs under different operating conditions. In order to mimic PSA, the adsorption and desorption pressures were set to 10 bar and 1 bar, respectively, for each gas mixture, whereas to study VSA, the adsorption and desorption pressures were set to 1 bar and 0.1 bar, respectively.
3. Results and discussion
3.1 Removal of the bound solvents
We compared the single-component CO2 uptakes of MOFs with and without solvent molecules at three different pressures as shown in Fig. 1(a). Each MOF was represented with the same color but with different symbols at each pressure. Fig. 1(a) shows that removal of the coordinated solvents leads to higher CO2 uptakes for all MOFs at all pressures. The deviations between the single-component CO2 uptakes of MOFs with and without solvents get smaller as the pressure increases. For comparison, we calculated the coefficient of determination, 0 ≤ R2 ≤ 1, using the formula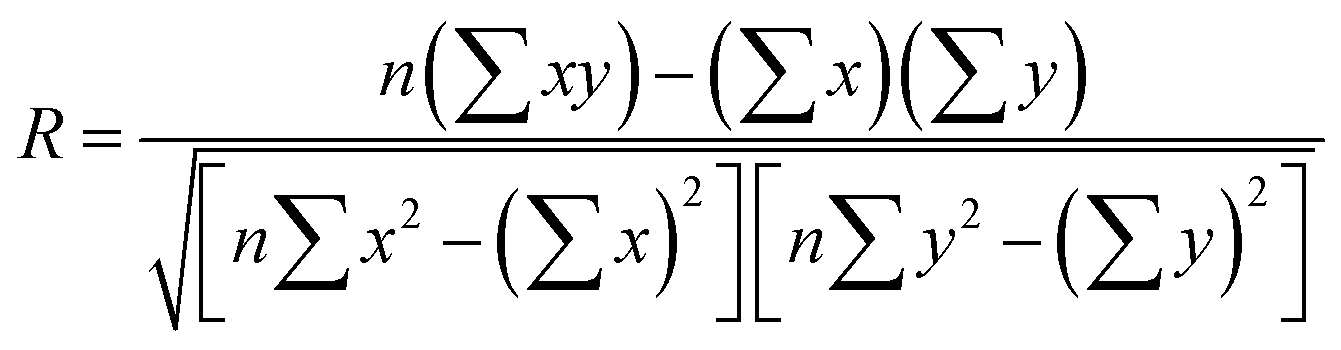 , where x(y) represents single-component CO2 uptakes calculated for a MOF with solvents (without solvents) and n is the total number of MOFs. At low pressures, the R2 values computed for the single-component CO2 uptakes of MOFs with and without solvents were low, 0.2 and 0.3 at 0.1 and 1 bar (see Fig. S1, ESI†), respectively. However, at 10 bar, the R2 value was calculated as 0.84. This is due to the adsorbate–framework interactions which become less pronounced as the pressure increases and the adsorbate–adsorbate interactions which start to dominate with the saturation of the most favorable adsorption sites.
, where x(y) represents single-component CO2 uptakes calculated for a MOF with solvents (without solvents) and n is the total number of MOFs. At low pressures, the R2 values computed for the single-component CO2 uptakes of MOFs with and without solvents were low, 0.2 and 0.3 at 0.1 and 1 bar (see Fig. S1, ESI†), respectively. However, at 10 bar, the R2 value was calculated as 0.84. This is due to the adsorbate–framework interactions which become less pronounced as the pressure increases and the adsorbate–adsorbate interactions which start to dominate with the saturation of the most favorable adsorption sites.
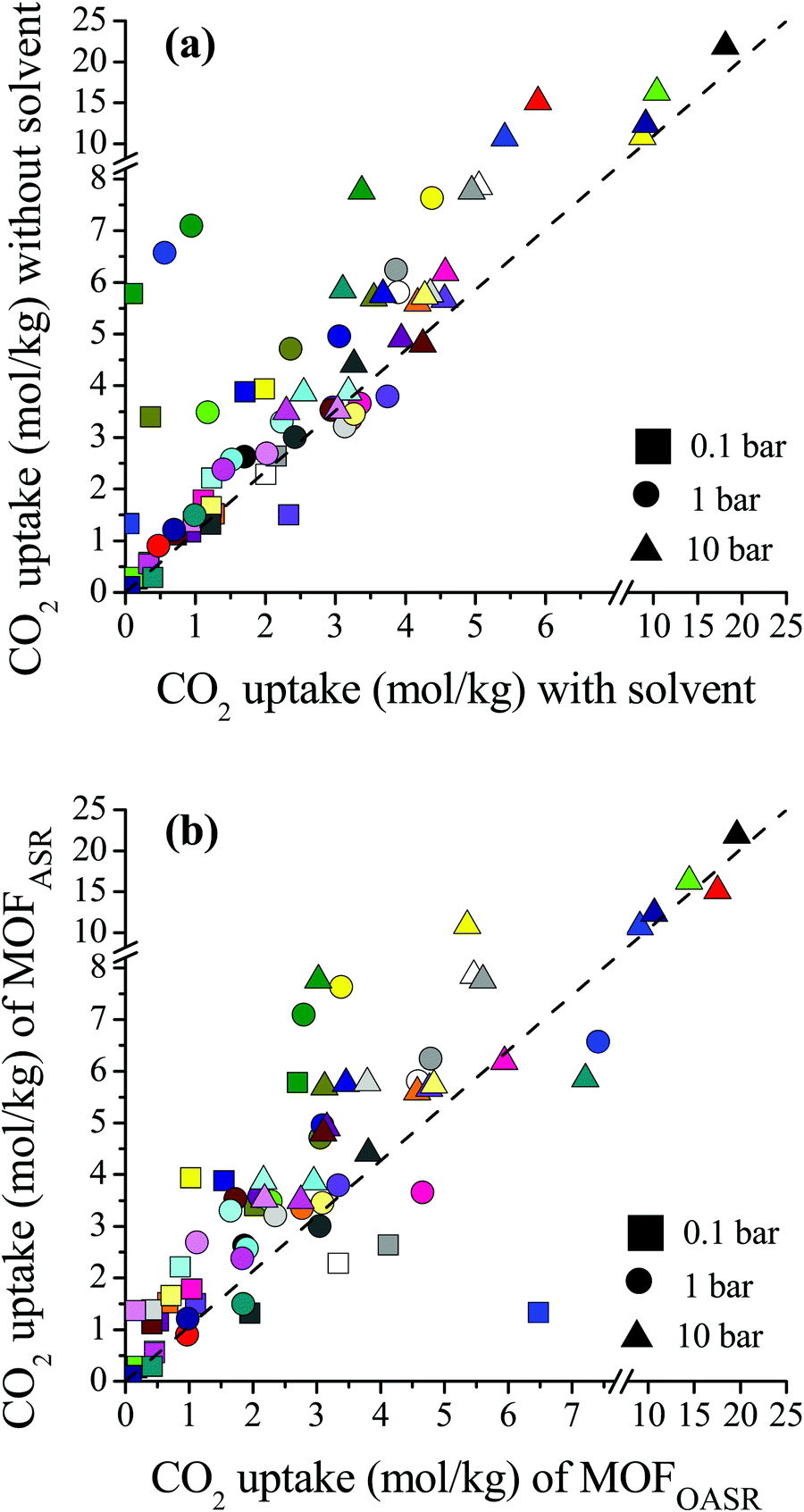 | ||
| Fig. 1 (a) Comparisons of the single-component CO2 uptakes of MOFs with and without coordinated solvent molecules. (b) Comparisons of the single-component CO2 uptakes of MOFs after solvent removal (MOFASR) and MOFs optimized after solvent removal (MOFOASR) at 0.1, 1 and 10 bar. Each MOF is represented with a different color. Detailed information about the refcodes of the MOFs, their solvent types and simulated CO2 uptakes at different pressures is given in Table S1 (ESI†). | ||
In order to quantitatively identify the increase in the CO2 uptakes after the solvent removal, we calculated the ratios of the simulated CO2 uptakes of MOFs without solvents to the simulated CO2 uptakes of MOFs with solvents as follows, 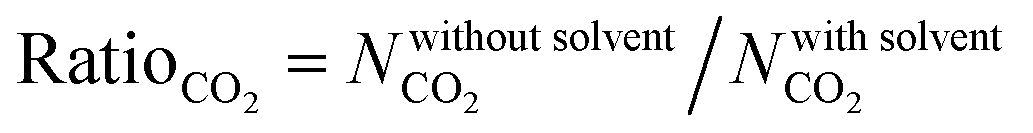 , and listed them in Table S1 (ESI†). If the simulated CO2 uptake of a MOF having coordinated solvent molecules is very similar to the one lacking the solvent, then the ratio gets a value around 1. While high values (RatioCO2 > 2.0) were observed in three MOFs at 1 bar, only two MOFs, PACZUQ and ECOLEP, were computed to have RatioCO2 > 2.0, 2.3 and 2.6, respectively, at 10 bar. Analysis of PACZUQ showed that the pore sizes of this MOF do not significantly change after the solvent removal but its accessible surface area increased from 249.4 to 371.3 m2 g−1. Removal of two water molecules which were located near the ligand made the nitrilotripropionic ligands of PACZUQ43 more accessible to CO2 molecules and increased the CO2 adsorption capacity. For ECOLEP, both the PLD and LCD increased (from 9.9 to 10.9 Å and from 10.2 to 11.6 Å, respectively) after the removal of three coordinated water molecules around each metal center. Both the increased pore sizes and the accessibility of tetrazolate-based organic ligand and metal clusters of ECOLEP44 enhanced its CO2 uptake. To clarify this, CO2 adsorption surface plots of PACZUQ and ECOLEP were obtained using the isosteric heat of adsorption of CO2 calculated from simulations performed at infinite dilution by iRASPA software.45 A comparison of the adsorption surfaces of PACZUQ with and without solvents is given in Fig. S2(a) (ESI†). CO2 molecules were adsorbed close to the nitrilotripropionic ligands after the solvent removal. However, the adsorption surface was oriented through the middle of the pores moving away from the ligand while the coordinated solvents were present. This indicates that removal of water molecules located near the ligands made the ligands more accessible to CO2 molecules and caused an increase in CO2 adsorption capacity. Similarly, for ECOLEP, Fig. S2(b) (ESI†) shows that CO2 molecules dominated only around the ligands of ECOLEP after solvent removal and caused an enhanced CO2 uptake. Our results also showed that the accessibility of adsorption sites in the framework has a more pronounced effect on the CO2 uptake than the pore size. For example, the PLD and LCD of ACUFEK dramatically increased from 7.6 to 9.0 Å, and from 13.2 and 15.6 Å, respectively, after the solvent removal but RatioCO2 was calculated to be 1.2, indicating that the CO2 uptake capacity of the MOF did not significantly change upon solvent removal. Nitrogen atoms on the triazine–triyltribenzoate ligand of ACUFEK46 were accessible for CO2 adsorption when the solvent was present and removal of the coordinated water molecules did not change the accessibility of these favorable adsorption sites.
, and listed them in Table S1 (ESI†). If the simulated CO2 uptake of a MOF having coordinated solvent molecules is very similar to the one lacking the solvent, then the ratio gets a value around 1. While high values (RatioCO2 > 2.0) were observed in three MOFs at 1 bar, only two MOFs, PACZUQ and ECOLEP, were computed to have RatioCO2 > 2.0, 2.3 and 2.6, respectively, at 10 bar. Analysis of PACZUQ showed that the pore sizes of this MOF do not significantly change after the solvent removal but its accessible surface area increased from 249.4 to 371.3 m2 g−1. Removal of two water molecules which were located near the ligand made the nitrilotripropionic ligands of PACZUQ43 more accessible to CO2 molecules and increased the CO2 adsorption capacity. For ECOLEP, both the PLD and LCD increased (from 9.9 to 10.9 Å and from 10.2 to 11.6 Å, respectively) after the removal of three coordinated water molecules around each metal center. Both the increased pore sizes and the accessibility of tetrazolate-based organic ligand and metal clusters of ECOLEP44 enhanced its CO2 uptake. To clarify this, CO2 adsorption surface plots of PACZUQ and ECOLEP were obtained using the isosteric heat of adsorption of CO2 calculated from simulations performed at infinite dilution by iRASPA software.45 A comparison of the adsorption surfaces of PACZUQ with and without solvents is given in Fig. S2(a) (ESI†). CO2 molecules were adsorbed close to the nitrilotripropionic ligands after the solvent removal. However, the adsorption surface was oriented through the middle of the pores moving away from the ligand while the coordinated solvents were present. This indicates that removal of water molecules located near the ligands made the ligands more accessible to CO2 molecules and caused an increase in CO2 adsorption capacity. Similarly, for ECOLEP, Fig. S2(b) (ESI†) shows that CO2 molecules dominated only around the ligands of ECOLEP after solvent removal and caused an enhanced CO2 uptake. Our results also showed that the accessibility of adsorption sites in the framework has a more pronounced effect on the CO2 uptake than the pore size. For example, the PLD and LCD of ACUFEK dramatically increased from 7.6 to 9.0 Å, and from 13.2 and 15.6 Å, respectively, after the solvent removal but RatioCO2 was calculated to be 1.2, indicating that the CO2 uptake capacity of the MOF did not significantly change upon solvent removal. Nitrogen atoms on the triazine–triyltribenzoate ligand of ACUFEK46 were accessible for CO2 adsorption when the solvent was present and removal of the coordinated water molecules did not change the accessibility of these favorable adsorption sites.
During the activation process, some MOF structures can change via relaxation when solvent molecules were evacuated as reported by experiments.47 This transformation may not lead to the collapse of the structure but it may cause a change in the structural properties and hence in the gas adsorption properties of MOFs. In order to investigate this issue, we optimized MOFs after solvent removal which eventually alters the unit cell parameters as well as the pore sizes of the materials. A comparison of the single-component CO2 uptakes of the MOF structures after solvent removal (MOFASR) and the CO2 uptakes of the MOFs which were optimized after solvent removal (MOFOASR) at 0.1, 1 and 10 bar, 298 K, is shown in Fig. 1(b). Optimization of the crystal structures generally resulted in decreased CO2 uptakes at each pressure, which can be attributed to the shrinkage in the pore size that hinders the accessibility of adsorption sites and limits the available pore space. On the other hand, CO2 adsorption in a few structures increased after the optimization. For example, the PLD and LCD values of LEJRIC slightly changed from 10.9 to 9.6 Å and from 11.6 to 10.5 Å, respectively, after optimization. Hindrance in the pore sizes of LEJRIC slightly affected the CO2 uptakes at moderate and high pressures, whereas at low pressure (0.1 bar) the optimized structure showed an increase in gas uptake which can be explained by the change in the charge environment of the framework. After the optimization process, the atomic charge assignment was repeated for all MOF structures. The charges of the Ni metal center were 1.1 and 2.4 e− for LEJRICASR and LEJRICOASR, respectively. The increase in the CO2 uptake of LEJRICOASR at low pressure can be attributed to the increase in the metal charge after the optimization which enhanced the electrostatic interactions between CO2 molecules and metal centers. Our GCMC simulations showed that these electrostatic interactions are mainly responsible for the CO2 uptake of LEJRIC since they contribute to 69% of the total energy. Overall, the optimization of the MOFs after the solvent removal may (a) change the pore sizes of the materials which affect the CO2 uptake for entropic reasons especially at high pressures and/or (b) change the charge environment of the framework which affects the CO2 uptake for energetic reasons especially at low pressures.
We then examined how the removal of solvent molecules influences the predicted adsorbent performance evaluation metrics of MOFs such as selectivity, the APS, and R% for CO2/CH4: 50/50, CO2/H2: 15/85 and CO2/N2: 15/85 separations. Comparisons of the CO2 uptakes for all three mixtures in MOFs with and without coordinated solvent molecules can be found in Fig. S3 (ESI†). Removal of solvent molecules increased the CO2 uptakes in the mixtures similar to the single-component gas adsorption results. For each mixture, comparisons of the performance metrics computed for the MOFs with and without solvent molecules are shown in Fig. 2. The CO2/CH4 selectivities of the MOFs were found to increase after the solvent removal as shown in Fig. 2(a). The reason is that while the CO2 uptakes increased, the CH4 uptakes did not change significantly after the solvent removal. There was an exception (WEM-family) for which the CO2/CH4 selectivities decreased after the solvent removal. The CH4 adsorption in the MOFs belonging to the WEM-family having solvents was almost negligible. This can be attributed to two reasons: (i) the PLDs of these MOFs are very close to the kinetic diameter of CH4, causing steric hindrance; and (ii) the CO2 adsorption of these MOFs is high, causing a competitive adsorption. Removal of coordinated water molecules from these MOFs created an extra free volume, enabling CH4 molecules to be adsorbed within the pores. We note that the CH4 adsorption even after the solvent removal was still very low compared to the other MOFs but this increase caused a decrease in CO2/CH4 selectivities.
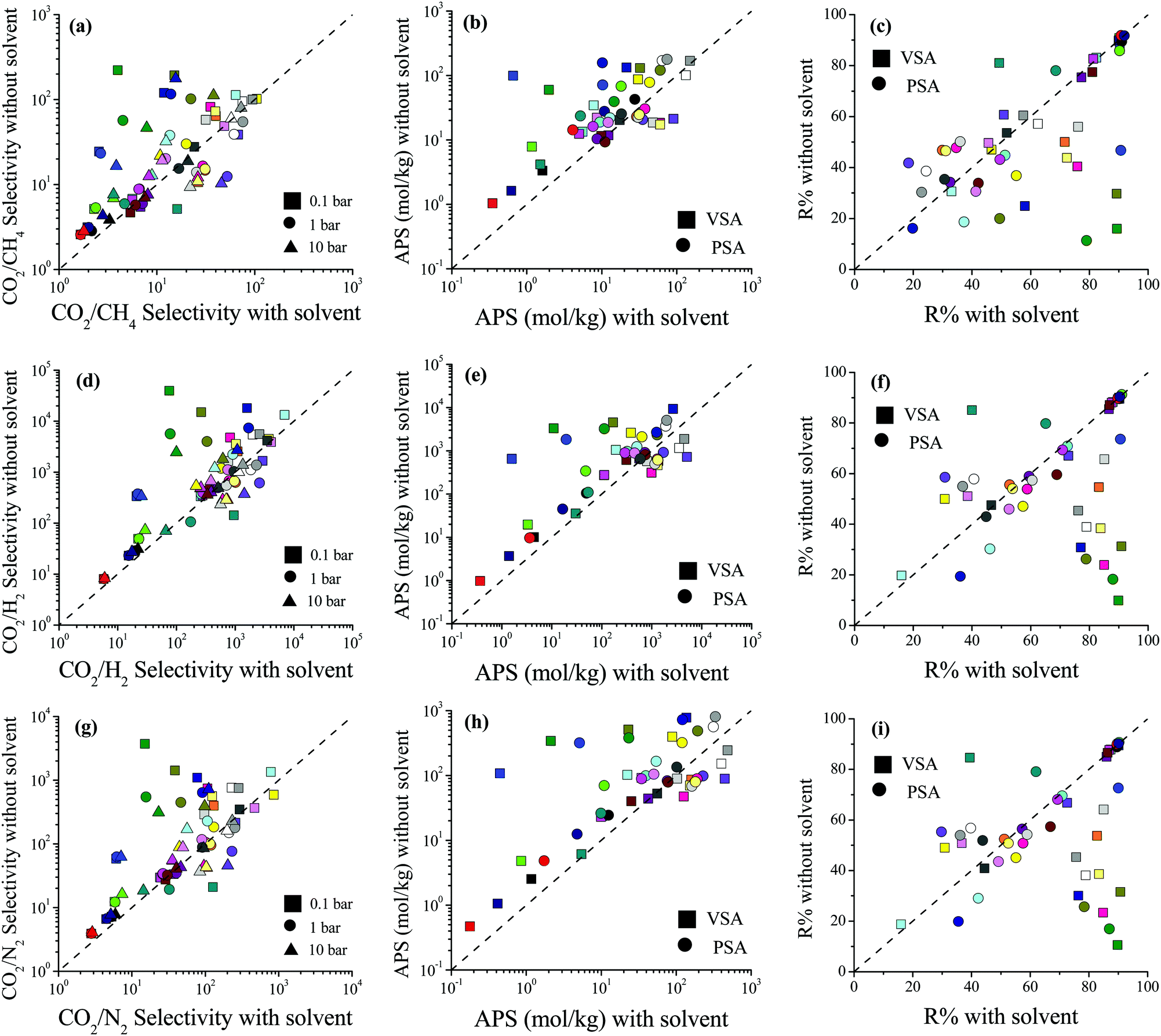 | ||
| Fig. 2 Comparisons of the selectivity, APS, and R% values of MOFs with and without coordinated solvent molecules for separation of (a–c) CO2/CH4: 50/50, (d–f) CO2/H2: 15/85 and (g–i) CO2/N2: 15/85 mixtures. | ||
The difference between the selectivities of MOFs with and without solvent molecules decreased as the pressure increased. In order to quantitatively identify the change in mixture selectivities after the solvent removal, we defined the ratio of the selectivities computed for MOFs without solvents to the selectivities of MOFs with solvents, 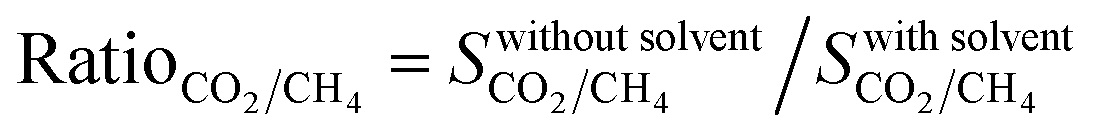 . PACZUQ, LEJRIC, OCIYUW, and RUWXAJ were calculated to have RatioCO2/CH4 > 3.0 at all pressures similar to their high RatioCO2 values. Due to the slight changes in CH4 uptake and significant increase in CO2 uptake, the CO2/CH4 selectivities of PACZUQ and LEJRIC increased. The reason for the high RatioCO2/CH4 values of OCIYUW and RUWXAJ is that while the CO2 uptakes increased, the CH4 uptakes decreased after the solvent removal due to the competitive adsorption between CO2 and CH4 for the preferential adsorption sites, metal centers. Fig. 2(b) compares the APSs of the MOFs with and without solvent molecules under VSA and PSA conditions for the CO2/CH4 mixture. Under both conditions, the MOFs without solvents exhibit higher APSs than the MOFs with coordinated solvents due to the increase in adsorption selectivity and capacity. After solvent removal, the calculated R% values of many MOFs sharply decreased as can be seen in Fig. 2(c). The highest reduction in R% belongs to PACZUQ, from 89.4 to 16.0% (from 79.0 to 11.4%) under VSA (PSA) conditions because of the significantly increased CO2 uptake after the solvent removal. Overall, Fig. 2(a–c) show that higher CO2/CH4 selectivities and higher APSs but lower R% values were predicted for the solvent-free MOFs compared to their counterparts with solvents. Similar conclusions are valid for the CO2/H2: 15/85 and CO2/N2: 15/85 separations as shown in Fig. 2(d–f) and (g–i).
. PACZUQ, LEJRIC, OCIYUW, and RUWXAJ were calculated to have RatioCO2/CH4 > 3.0 at all pressures similar to their high RatioCO2 values. Due to the slight changes in CH4 uptake and significant increase in CO2 uptake, the CO2/CH4 selectivities of PACZUQ and LEJRIC increased. The reason for the high RatioCO2/CH4 values of OCIYUW and RUWXAJ is that while the CO2 uptakes increased, the CH4 uptakes decreased after the solvent removal due to the competitive adsorption between CO2 and CH4 for the preferential adsorption sites, metal centers. Fig. 2(b) compares the APSs of the MOFs with and without solvent molecules under VSA and PSA conditions for the CO2/CH4 mixture. Under both conditions, the MOFs without solvents exhibit higher APSs than the MOFs with coordinated solvents due to the increase in adsorption selectivity and capacity. After solvent removal, the calculated R% values of many MOFs sharply decreased as can be seen in Fig. 2(c). The highest reduction in R% belongs to PACZUQ, from 89.4 to 16.0% (from 79.0 to 11.4%) under VSA (PSA) conditions because of the significantly increased CO2 uptake after the solvent removal. Overall, Fig. 2(a–c) show that higher CO2/CH4 selectivities and higher APSs but lower R% values were predicted for the solvent-free MOFs compared to their counterparts with solvents. Similar conclusions are valid for the CO2/H2: 15/85 and CO2/N2: 15/85 separations as shown in Fig. 2(d–f) and (g–i).
Here it is important to discuss the role of simulated R% values in high-throughput computational screening of MOFs. In large-scale screening of MOFs, different adsorbent performance evaluation metrics48 such as capacity,5,49 selectivity,21 and the APS22 have been used for the identification of the top adsorbents. We recently showed that focusing on MOFs having R% > 85% and then ranking them based on APSs is a good strategy to identify the most promising MOF adsorbents since several MOFs with high selectivities suffer from low R%.16 The results in Fig. 2 show the importance of solvent removal in the calculated R% values of the MOFs, and hence in the identification of the best materials for CO2 separations. Unless coordinated solvents were removed, very high R% values can be predicted for some MOFs which may cause inaccurate identification of these materials as top MOFs (false-positives).
We have so far discussed the impact of removal of solvent molecules from the MOFs’ pores on the simulated CO2 adsorption and separation potentials of the materials. It is also important to discuss the integrity and stability issues of structures which may occur after the solvent removal. The structural integrity and stability of the MOFs after the solvent removal can only be guaranteed by the experiments; therefore, we examined experimental synthesis papers of the MOFs considered in this work. We found information about the integrity of MOFs after the solvent removal only for two structures: OCIYUW50 was reported to show structural changes invoked by the loss of the solvents embedded in the structure at high temperature, 177 °C, and LEJRIC51 was reported to remain intact up to 190 °C. We also examined the thermal stability information of MOFs and our detailed comments are given in Table S1 (ESI†). We found that 13 MOFs were reported to be stable after solvent evaporation according to TGA (thermogravimetric analysis) patterns and/or statements about their thermal history provided in their synthesis papers. On the other hand, 12 MOFs suffered from the thermal stability problems once their solvents were removed. In other words, solvents of these MOFs should be kept during the curation process. The CoRE MOF database deleted solvents of 8 MOFs, and solvent removal script provided with the CSD MOF subset removed solvents from 4 MOFs. We finally note that newly developed activation methods like freeze-drying can be used instead of conventional heating and vacuum methods to remove solvents without damaging the stability of MOFs.52
3.2 Treatment of missing hydrogen atoms
We examined the impact of missing hydrogen atoms in MOF structures on the adsorption of single-component CO2 and adsorption of CO2/CH4: 50/50, CO2/H2: 15/85 and CO2/N2: 15/85 mixtures in 14 selected MOFs at 0.1, 1 and 10 bar, 298 K. Fig. 3 shows that the missing hydrogen atoms of the framework do not cause significant changes in the single-component CO2 uptakes and CO2 selectivities. RatioCO2 was defined as the ratio of the simulated CO2 uptake of MOFs having missing hydrogen atoms in the framework to the simulated CO2 uptake of MOFs without missing hydrogens in the framework,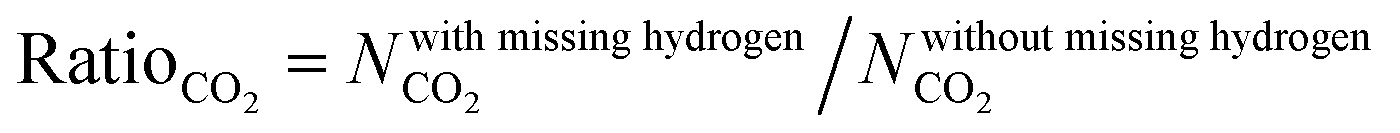 , and it was calculated to change between 0.99 and 1.3. As the pressure increases, RatioCO2 reaches 1, as can be seen from Table S3 (ESI†). Fig. 3(a) shows that the greatest difference between the MOFs with and without hydrogens is observed at low pressure (0.1 bar) as expected. At low pressures, the interactions between the adsorbates and MOF atoms are dominant as discussed above, whereas the structural features of MOFs such as surface area and porosity become important in terms of determining the gas uptake of a material at higher pressures.53–55 The greatest RatioCO2 value at 0.1 bar belongs to SABVUN (MIL-53).56 Once the missing hydrogen atoms were added, the electronic environment of the framework changed. While hydrogen atoms were missing in SABVUN, the coulombic interaction energy between the host and adsorbate molecules (SABVUN–CO2) was calculated as −30.1 kcal mol−1, which led to 1.6 mol kg−1 CO2 uptake at 0.1 bar. However, when there is no missing hydrogen atom, the coulombic interaction energy was −22.8 kcal mol−1, which led to 1.2 mol kg−1 CO2 at 0.1 bar. On the other hand, the SABVUN–CO2 van der Waals interaction energy, which contributed to 82% of the total interaction energy, changed slightly from −138.9 and −137.1 kcal mol−1 for SABVUN once the hydrogen atoms were completed, suggesting weak van der Waals interactions between the hydrogen atoms of SABVUN and CO2. The PLDs and LCDs of the MOFs with and without missing hydrogen atoms were almost the same, and therefore, RatioCO2 values of unity were obtained at high pressures as shown in Table S3 (ESI†). Since the gas uptakes were almost the same, the predicted APS and R% values of the MOFs computed for CO2/CH4: 50/50, CO2/H2: 15/85 and CO2/N2: 15/85 separations did not dramatically change in the absence of hydrogen atoms in the frameworks. Overall, the data shown in Table S4 (ESI†) reveal that using MOF structures with missing hydrogen atoms in the molecular simulations does not make a dramatic change in the identification of the top performing MOFs in high-throughput computational screening studies due to the weak van der Waals interactions between the hydrogen atoms of the framework and CO2 molecules.
, and it was calculated to change between 0.99 and 1.3. As the pressure increases, RatioCO2 reaches 1, as can be seen from Table S3 (ESI†). Fig. 3(a) shows that the greatest difference between the MOFs with and without hydrogens is observed at low pressure (0.1 bar) as expected. At low pressures, the interactions between the adsorbates and MOF atoms are dominant as discussed above, whereas the structural features of MOFs such as surface area and porosity become important in terms of determining the gas uptake of a material at higher pressures.53–55 The greatest RatioCO2 value at 0.1 bar belongs to SABVUN (MIL-53).56 Once the missing hydrogen atoms were added, the electronic environment of the framework changed. While hydrogen atoms were missing in SABVUN, the coulombic interaction energy between the host and adsorbate molecules (SABVUN–CO2) was calculated as −30.1 kcal mol−1, which led to 1.6 mol kg−1 CO2 uptake at 0.1 bar. However, when there is no missing hydrogen atom, the coulombic interaction energy was −22.8 kcal mol−1, which led to 1.2 mol kg−1 CO2 at 0.1 bar. On the other hand, the SABVUN–CO2 van der Waals interaction energy, which contributed to 82% of the total interaction energy, changed slightly from −138.9 and −137.1 kcal mol−1 for SABVUN once the hydrogen atoms were completed, suggesting weak van der Waals interactions between the hydrogen atoms of SABVUN and CO2. The PLDs and LCDs of the MOFs with and without missing hydrogen atoms were almost the same, and therefore, RatioCO2 values of unity were obtained at high pressures as shown in Table S3 (ESI†). Since the gas uptakes were almost the same, the predicted APS and R% values of the MOFs computed for CO2/CH4: 50/50, CO2/H2: 15/85 and CO2/N2: 15/85 separations did not dramatically change in the absence of hydrogen atoms in the frameworks. Overall, the data shown in Table S4 (ESI†) reveal that using MOF structures with missing hydrogen atoms in the molecular simulations does not make a dramatic change in the identification of the top performing MOFs in high-throughput computational screening studies due to the weak van der Waals interactions between the hydrogen atoms of the framework and CO2 molecules.
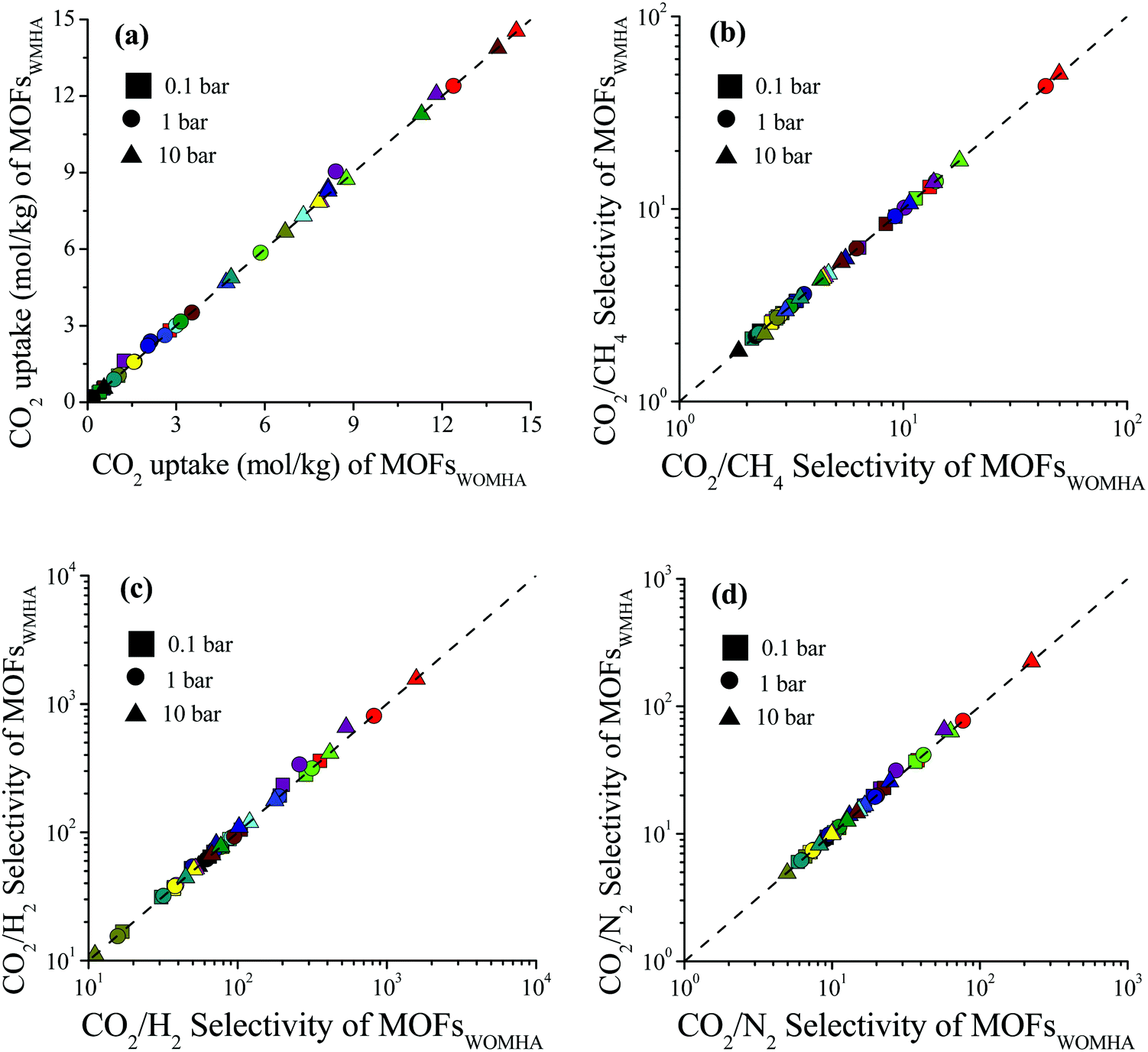 | ||
| Fig. 3 Comparisons of the (a) single-component CO2 uptakes and selectivities for the (b) CO2/CH4: 50/50, (c) CO2/H2: 15/85, and (d) CO2/N2: 15/85 separation performances of MOFs with missing hydrogen atoms (MOFWMHA) and without missing hydrogen atoms (MOFWOMHA). | ||
3.3 Retention of CBIs
Curation of CBIs during database construction is challenging because most of the CBIs are disordered and/or identifying the CBIs from the chemical formulae of MOFs deposited into the CSD is not simple. In some cases, CBIs can be accidentally deleted during the automated solvent removal and hence these MOFs are deposited without CBIs into the curated MOF databases. However, the presence of these ions is important to make the overall framework charge neutral and to provide framework stability. We examined several MOFs having small CBIs such as CHOO−, CH3HOO−, SO42−, CF3SO3−, and NO3− and there were a few MOFs with bulky CBIs, such as SiMo12O404+, [Cu(C3H7NO)6]2+, [Zn(C4H12NO)6]2+, [Fe(1,10-phen)3]2+, and [2-MepyH]+. The PLDs and LCDs of the MOFs having small CBIs slightly increase if ions are accidently removed. In cases where there are several small CBIs in the framework, both the PLD and LCD can increase. For instance, the PLD and LCD of MOXNUJ having CH3HOO− were calculated as 3.5 and 4.1 Å, whereas accidental removal of CBIs led to a PLD and an LCD of 7.1 and 7.3 Å, respectively. Removal of bulky CBIs also causes large differences in pore sizes but if the CBIs are located between the ligands, pore sizes are not much affected. For instance, structures of TEDGUG with and without large ion, [Zn(C4H12NO)6]2+, were found to have almost the same pore sizes due to the locations of the ions.A comparison of the single-component CO2 uptakes of 27 MOFs with and without CBIs at 0.1, 1 and 10 bar, 298 K, is given in Fig. 4(a). In order to quantitatively identify the change in CO2 uptakes depending on the presence of CBIs in the framework, we calculated the ratio of the simulated CO2 uptake of the MOFs without CBIs to the simulated CO2 uptake of the MOFs with CBIs, 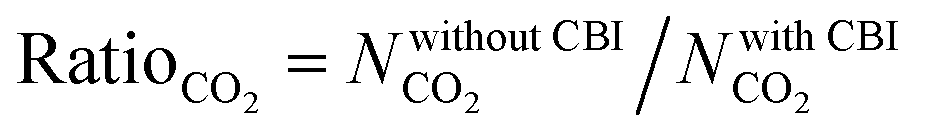 , and listed these values in Table S5 (ESI†). Accidental removal of CBIs generally caused overestimation of CO2 uptakes at all pressures due to the creation of available free space for gas adsorption. High RatioCO2 (>3.0) values were calculated for several MOFs. One of the large differences between the single-component CO2 uptakes of the MOFs with and without CBIs belongs to LIQCUK, which originates from the removal of several small CBIs, such as NO3−. The removal of CBIs created a new charge network within the framework which changes host–adsorbate coulombic interactions due to the change in the partial charge of the metal center. The MOF–CO2 electrostatic interaction energy was computed as −1.2 kcal mol−1 at 0.1 bar for LIQCUK having CBIs, whereas it was increased to −41.1 kcal mol−1 after the removal of CBIs. The effect of this new charge environment on CO2 adsorption decreases with increasing pressure, resulting in RatioCO2 of 5, 2 and 1.9 at 0.1, 1 bar and 10 bar, respectively. There were a few exceptions where removal of CBIs leads to decreased CO2 uptakes at low pressure. For example, the lowest RatioCO2 (0.27) was computed at 0.1 bar for VEHXEN having [Fe(1,10-phen)3]2+ and [2-MepyH]+ ions. Once the CBIs were removed, the total VEHXEN–CO2 interaction energy decreased from −38.5 to −5.5 kcal mol−1 since the CBIs acted as preferential adsorption sites for CO2 molecules.
, and listed these values in Table S5 (ESI†). Accidental removal of CBIs generally caused overestimation of CO2 uptakes at all pressures due to the creation of available free space for gas adsorption. High RatioCO2 (>3.0) values were calculated for several MOFs. One of the large differences between the single-component CO2 uptakes of the MOFs with and without CBIs belongs to LIQCUK, which originates from the removal of several small CBIs, such as NO3−. The removal of CBIs created a new charge network within the framework which changes host–adsorbate coulombic interactions due to the change in the partial charge of the metal center. The MOF–CO2 electrostatic interaction energy was computed as −1.2 kcal mol−1 at 0.1 bar for LIQCUK having CBIs, whereas it was increased to −41.1 kcal mol−1 after the removal of CBIs. The effect of this new charge environment on CO2 adsorption decreases with increasing pressure, resulting in RatioCO2 of 5, 2 and 1.9 at 0.1, 1 bar and 10 bar, respectively. There were a few exceptions where removal of CBIs leads to decreased CO2 uptakes at low pressure. For example, the lowest RatioCO2 (0.27) was computed at 0.1 bar for VEHXEN having [Fe(1,10-phen)3]2+ and [2-MepyH]+ ions. Once the CBIs were removed, the total VEHXEN–CO2 interaction energy decreased from −38.5 to −5.5 kcal mol−1 since the CBIs acted as preferential adsorption sites for CO2 molecules.
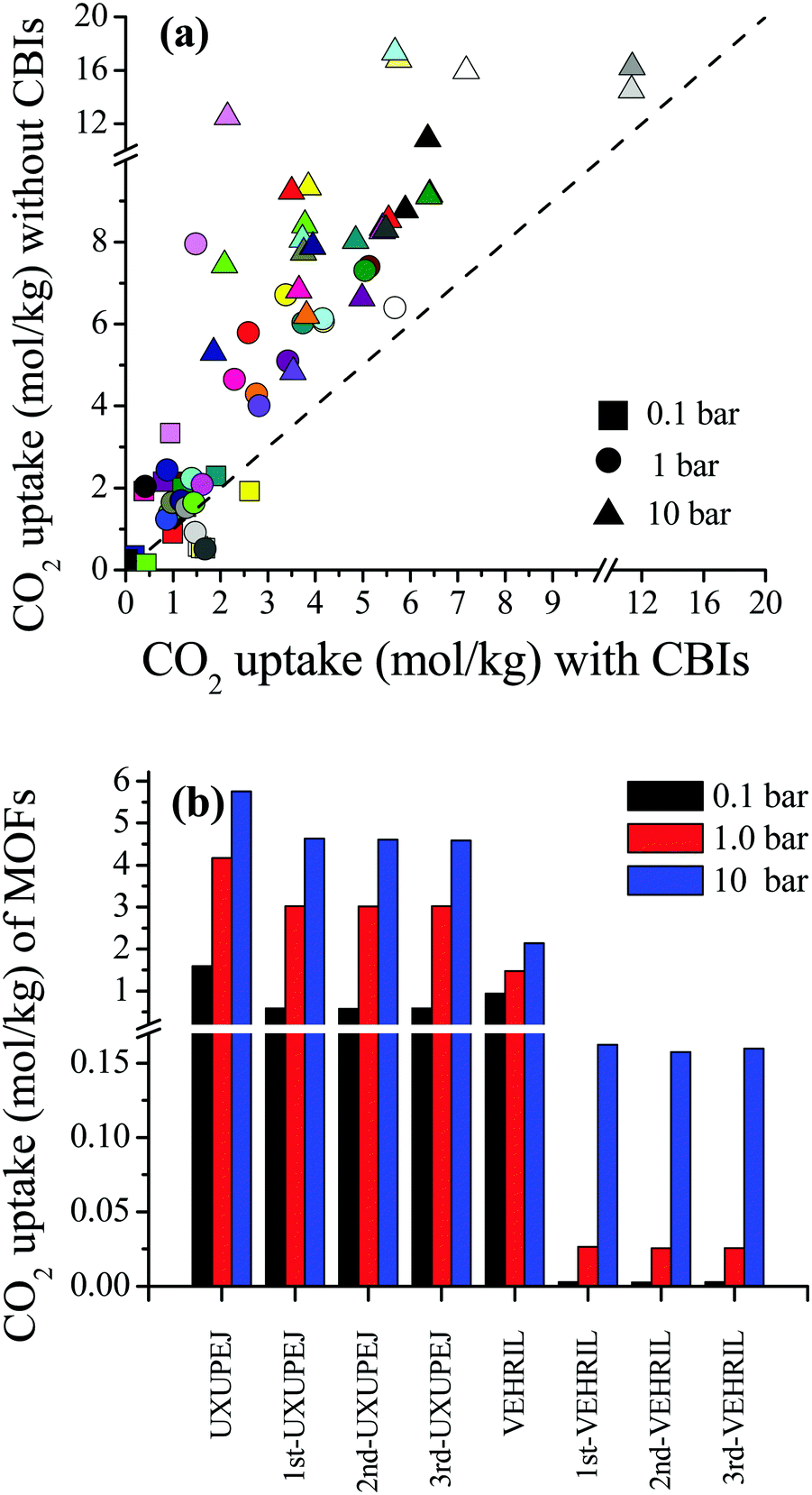 | ||
| Fig. 4 (a) Comparisons of the single-component CO2 uptakes of MOFs with and without CBIs at 0.1, 1 and 10 bar. Each MOF is represented with a different color. (b) Comparison of the single-component CO2 uptakes of UXUPEJ and VEHRIL having fixed and flexible CBIs. | ||
In high-throughput computational screening studies, MOFs have been assumed as rigid structures by neglecting flexibility due to the computational cost. Studies showed that identification of the location or distribution of CBIs inside frameworks is necessary to better understand the gas adsorption properties of MOFs.57,58 However, changing the configuration and position of the CBIs within the framework is computationally demanding due to the requirement of fully flexible modeling of ions. Therefore, we selected two representative MOFs, VEHRIL and UXUPEJ, to examine the impact of flexible modeling of CBIs on the simulated CO2 adsorption results of the MOFs. The reason for choosing these MOFs is that after the removal of SiMo12O404+ ions from VEHRIL, the simulated CO2 uptake at 10 bar significantly increases, whereas removal of CF3SO3− ions from UXUPEJ caused a significant decrease in the simulated CO2 uptake at 0.1 bar. Our results for the single-component CO2 uptakes of the MOFs with fixed and flexible CBIs are given in Fig. 4(b). Three distinct snapshots were taken from the MD simulations to observe the change in CO2 uptake. The single-component CO2 uptakes of UXUPEJ having fixed (flexible) CBIs were computed as 1.59 (0.58–0.60), 4.17 (3.02–3.07), and 5.76 (4.58–4.64) mol kg−1 at 0.1, 1, and 10 bar, respectively. The CO2 uptakes of VEHRIL with fixed CBIs were 0.94, 1.48, and 2.14 mol kg−1 at 0.1, 1, and 10 bar, respectively, whereas significantly lower CO2 uptakes, 0.0026–0.0028, 0.026–0.027, and 0.158–0.162 mol kg−1, were obtained from the simulations of VEHRIL having flexible CBIs. The more pronounced decrease in the CO2 uptake of VEHRIL compared to UXUPEJ can be attributed to the presence of a bulkier CBI, SiMo12O404+, which almost completely blocks the favorable adsorption sites, metal centers, after the equilibration of CBIs. These results showed that accommodation of ions close to the metal centers may result in decreased CO2 adsorption depending on the size of CBIs.
Selectivity, APS and R% comparisons of 27 MOFs with and without CBIs are given in Fig. 5 for CO2/CH4: 50/50, CO2/H2: 15/85 and CO2/N2: 15/85 mixtures at 0.1, 1 and 10 bar, 298 K, and the data are also given in Table S6 (ESI†). Fig. 5(a) shows that accidental removal of CBIs leads to a decrease in calculated CO2/CH4 selectivity. Although both the CO2 and CH4 uptakes increased after the removal of CBIs similar to the single-component CO2 adsorption, due to the higher increase in CH4 uptakes compared to CO2, the selectivities generally decreased. While the average RatioCO2 of 26 MOFs (except for XINWOU) computed for the CO2/CH4 mixture was 1.6, 1.7, and 2.0 at 0.1, 1, and 10 bar, respectively, the average RatioCH4 was comparably greater, 2.5, 3.1, and 3.5, respectively. The greatest decrease in selectivity was observed for XINWUO, which was computed to have RatioCO2/CH4 of 0.006, 0.010, and 0.033 at 0.1, 1, and 10 bar, respectively. The main reason for the dramatic increase in the CH4 uptakes of this MOF was the increase in its PLD and LCD. The removal of CBIs increased the PLD of XINWUO from 3.1 to 4.3 Å and the LCD from 3.6 to 6.3 Å and enabled CH4 adsorption. At low pressure, 0.1 bar, the XINWUO–adsorbate coulombic interaction energy was calculated as −10.0 kcal mol−1, whereas this energy was computed to be much smaller as −0.4 kcal mol−1 in the absence of CBIs, indicating the favorable effect of CBIs on the CO2 adsorption capacity of the MOF. Combination of decreased CO2 uptake and increased CH4 uptake caused a significant decrease in the CO2/CH4 selectivity of XINWUO after the accidental removal of CBIs. One exception was LIQCUK, with the highest RatioCO2/CH4 of 7.03, 3.55, and 2.19 at 0.1, 1 and 10 bar, respectively. Although the PLDs and LCDs of LIQCUK with and without CBIs were almost the same, its accessible surface area changed from 649.2 to 871.8 m2 g−1 when CBIs were removed which increased the CO2 uptakes without altering the CH4 adsorption at all pressures. While the van der Waals interaction energy computed between the adsorbates and LIQCUK having CBIs was five-times greater than that for LIQCUK without CBIs, there were two orders of magnitude differences in host–adsorbate coulombic interaction energy between LIQCUK–CO2 with and without CBIs, revealing the increase in CO2 uptake due to the dramatic increase in the strength of electrostatic interactions.
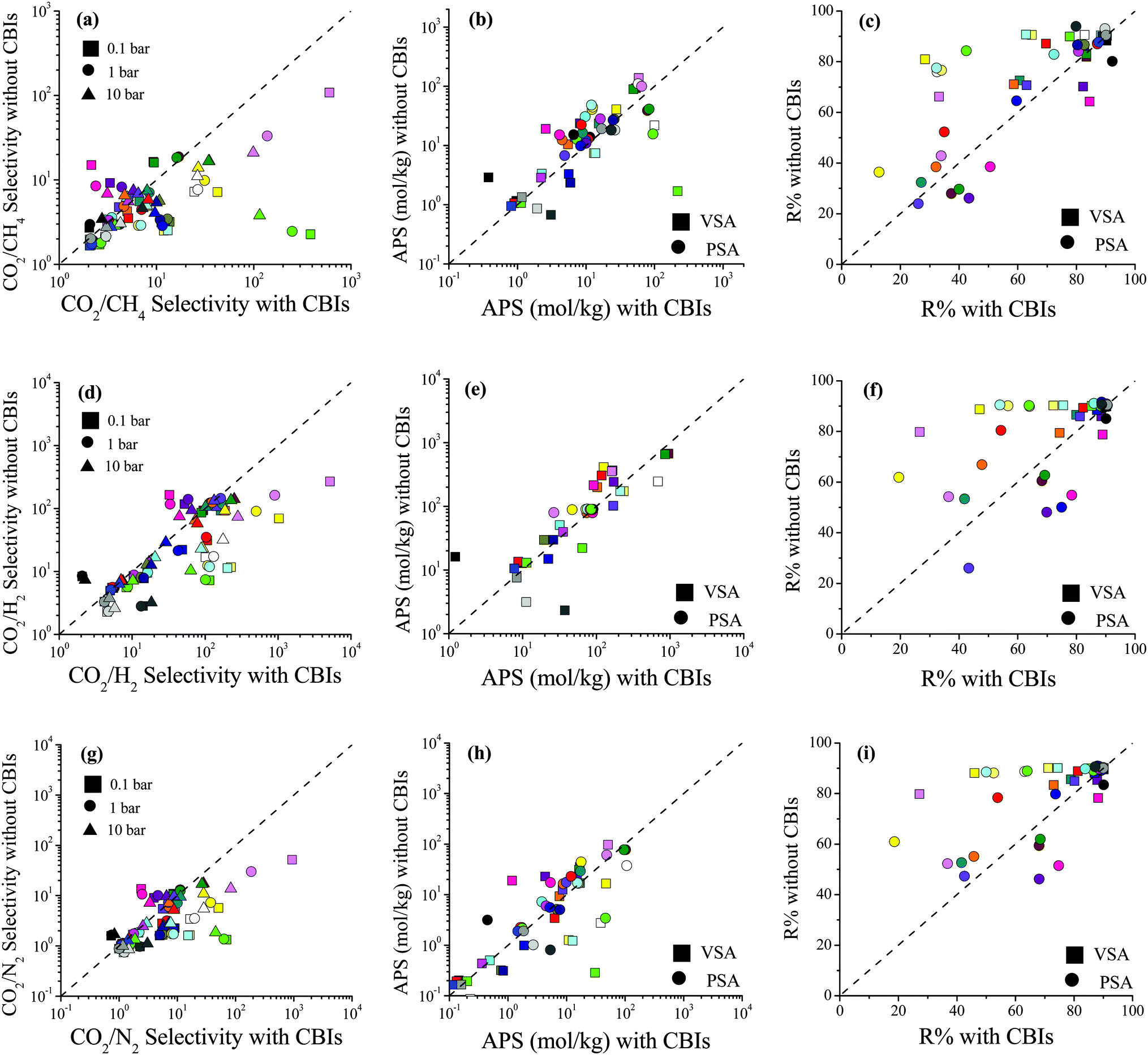 | ||
| Fig. 5 Comparisons of the selectivity, APS, and R% values of MOFs with and without CBIs for separation of (a–c) CO2/CH4: 50/50, (d–f) CO2/H2: 15/85 and (g–i) CO2/N2: 15/85 mixtures. | ||
Fig. 5(b) shows that accidental removal of CBIs generally caused overestimation of APSs for CO2/CH4 separation. Although a decrease in selectivity was observed with the removal of CBIs, due to the increase in CO2 working capacity, the APSs of MOFs without CBIs were generally predicted to be larger than those of MOFs with CBIs. The R% values of the MOFs with and without CBIs were compared for CO2/CH4 separation in Fig. 5(c). If CBIs were accidentally removed during the curation of databases, the R% values were overestimated. These comparisons showed that using the curated computational-ready MOF databases in molecular simulations may cause underestimation of CO2/CH4 selectivity and overestimation of the APS and R% if the CBIs of the MOFs are missing. Comparisons of the selectivity, APS and R% values of the MOFs with and without CBIs calculated for CO2/H2: 15/85 and CO2/N2: 15/85 separations are given in Fig. 5(d–f) and (g–i), respectively, and data show that the absence of CBIs caused lower selectivities and higher APSs and R%. Considering the selection strategy for the top performing MOFs as we discussed above, overestimation of the APS and R% would cause misleading predictions for the top materials (false-positives). All these results show that accurate treatment of CBIs in the construction of curated MOF databases is significant for high-throughput computational screening studies.
3.4 Using curated MOFs in molecular simulations
We finally provided a simple pathway in Fig. 6 to represent how to proceed with the curated computation-ready structures before using them in GCMC simulations to compute the CO2 adsorption properties of MOFs. Removal of coordinated solvent molecules is important to create available adsorption space for guest molecules and this leads to considerable increases in several adsorbent performance metrics such as CO2 selectivities and APSs. Here, it is very important to ensure that the integrity and stability of structures would not be adversely affected by the removal of solvents. If the stability of MOFs was ensured with the experiments, coordinated solvent molecules can be removed. We reported a decrease in CO2 uptakes after the optimization of structures due to decreases in pore sizes. Therefore, we suggest optimizing structures after the removal of coordinated solvents in order to make a more realistic prediction for the adsorbent performance metrics. Several MOFs suffer from missing hydrogen atoms in the databases and our simulation results showed that this drawback does not significantly affect the predicted adsorbent performance metrics of MOFs due to the weak interactions between hydrogens and adsorbate molecules. We still suggest completing the missing hydrogen atoms in the frameworks and optimizing their configurations to ensure the completeness of the structures. Perhaps the curation that requires the highest care is ensuring the presence of CBIs in the framework not only to ensure the integrity and stability of structures but also to make an accurate assessment of the gas uptakes and adsorbent performance metrics of MOFs. In high-throughput computational screening of MOFs where very large numbers of materials have been studied, considering the flexibility of the CBIs can be computationally very expensive, but when smaller numbers of MOFs are studied, optimizing the configuration of CBIs is important as we showed above. Here, a good strategy can be focusing on the best MOF materials identified from screening of rigid structures, selecting the ones having CBIs and then performing molecular simulations by letting the ions move.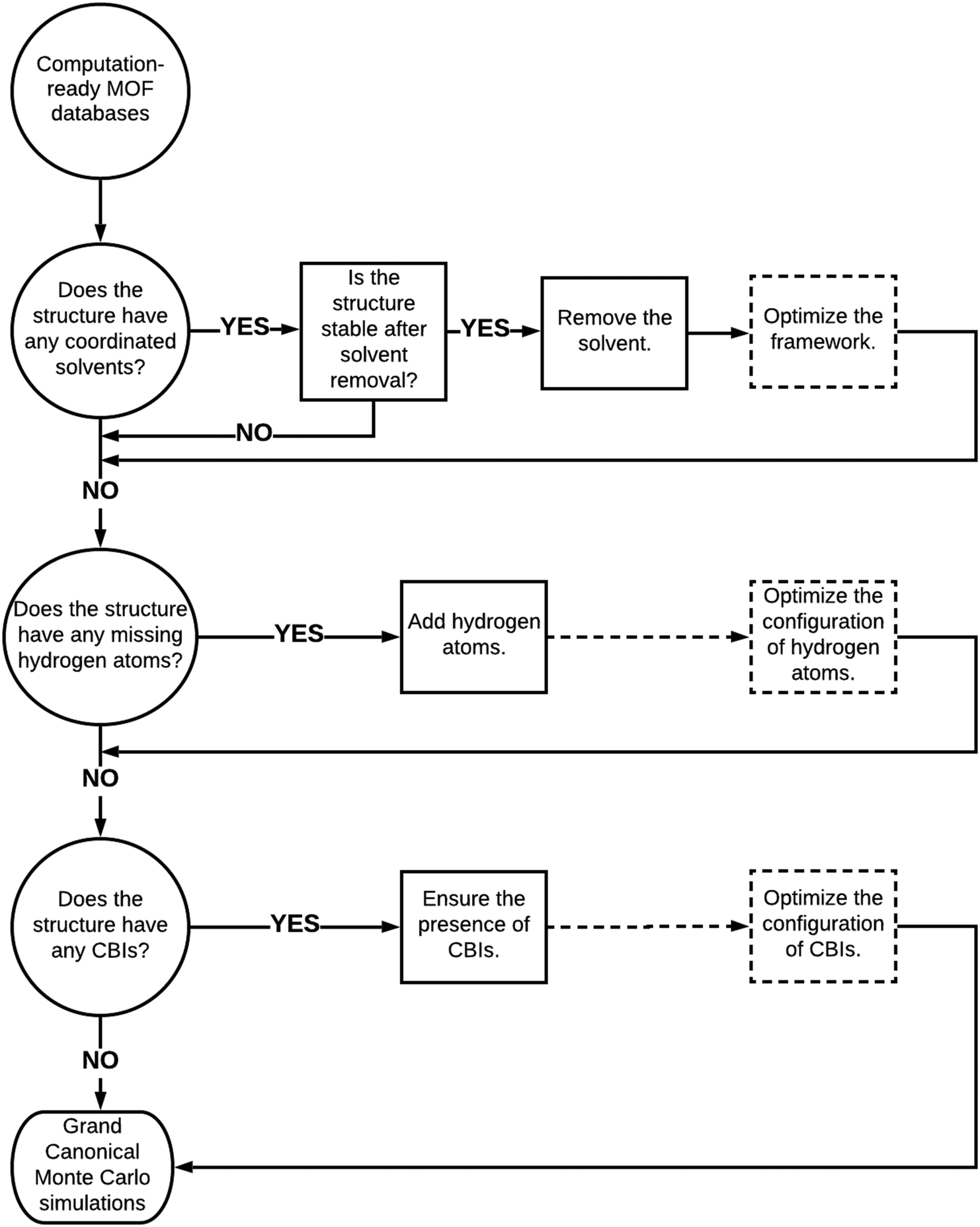 | ||
| Fig. 6 Proposed road to proceed with curated computation-ready structures before performing molecular simulations. Dashed boxes and dashed lines represent the optional paths. | ||
4. Conclusions
To reveal how structure curations affect the predicted CO2 adsorption and separation performances of MOFs, we studied 68 MOFs that were curated differently by computation-ready MOF databases in terms of removal of bound solvents, treatment of missing hydrogen atoms and retention of CBIs. Molecular simulations were performed to compute both the single-component and mixture adsorption in MOFs. The CO2/CH4, CO2/H2 and CO2/N2 separation performances of the MOFs were assessed by computing selectivity, APS and R%. Results showed that the simulated CO2 uptakes and selectivities of solvent-free MOFs were higher than those of MOFs containing solvents, which significantly affect the identification of the top performing MOFs. As the users of the curated computation-ready MOF databases, we should also be careful about the stability and/or integrity issues of the MOFs after the solvent removal, especially in search of MOFs exhibiting the maximum adsorption or separation capacity for target molecules. Our results showed that the problem of missing hydrogen atoms in the frameworks does not cause significant changes in the predicted gas uptakes and adsorbent performance metrics of the MOFs. In contrast to this, the absence of the CBIs which neutralize the charge distribution of the frameworks was found to have a dramatic effect on both the single-component and mixture gas uptake results. Missing CBIs during the curation of computation-ready MOFs led to overestimation of CO2 uptakes, APSs, and R%. Overall, our results highlight that curated, computation-ready MOF structures should be carefully examined before using them in molecular simulations considering the important consequences of the presence of coordinated solvent molecules and CBIs on the simulated gas uptakes. These results will guide the selection and use of curated computation-ready MOF structures from the existing databases for high-throughput molecular simulations of MOFs.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. K. acknowledges an ERC-2017-Starting Grant. This study received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (ERC-2017-Starting Grant, grant agreement no. 756489-COSMOS).References
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- M. Safaei, M. M. Foroughi, N. Ebrahimpoor, S. Jahani, A. Omidi and M. Khatami, TrAC, Trends Anal. Chem., 2019, 118, 401–425 CrossRef CAS.
- A. Ahmed, S. Seth, J. Purewal, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Nat. Commun., 2019, 10, 1568 CrossRef PubMed.
- P. Z. Moghadam, T. Islamoglu, S. Goswami, J. Exley, M. Fantham, C. F. Kaminski, R. Q. Snurr, O. K. Farha and D. Fairen-Jimenez, Nat. Commun., 2018, 9, 1378 CrossRef PubMed.
- S. Keskin, Ind. Eng. Chem. Res., 2011, 50, 8230–8236 CrossRef CAS.
- G. Yilmaz and S. Keskin, J. Membr. Sci., 2014, 454, 407–417 CrossRef CAS.
- M. Ahmadi, E. Taş, A. Kılıç, V. Kumbaracı, N. Talınlı, M. G. Ahunbay and S. B. Tantekin-Ersolmaz, ACS Appl. Mater. Interfaces, 2017, 9, 35936–35946 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- Y. G. Chung, J. Camp, M. Haranczyk, B. J. Sikora, W. Bury, V. Krungleviciute, T. Yildirim, O. K. Farha, D. S. Sholl and R. Q. Snurr, Chem. Mater., 2014, 26, 6185–6192 CrossRef CAS.
- Y. G. Chung, E. Haldoupis, B. J. Bucior, M. Haranczyk, S. Lee, H. Zhang, K. D. Vogiatzis, M. Milisavljevic, S. Ling and J. S. Camp, J. Chem. Eng. Data, 2019, 64, 5985–5998 CrossRef CAS.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- C. Altintas, I. Erucar and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10, 3668–3679 CrossRef CAS PubMed.
- Z. Qiao, K. Zhang and J. Jiang, J. Mater. Chem. A, 2016, 4, 2105–2114 RSC.
- B. J. Bucior, N. S. Bobbitt, T. Islamoglu, S. Goswami, A. Gopalan, T. Yildirim, O. K. Farha, N. Bagheri and R. Q. Snurr, Mol. Syst. Des. Eng., 2019, 4, 162–174 RSC.
- C. Altintas, G. Avci, H. Daglar, A. N. V. Azar, S. Velioglu, I. Erucar and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10(20), 17257–17268 CrossRef CAS PubMed.
- W. Yang, H. Liang, F. Peng, Z. Liu, J. Liu and Z. Qiao, Nanomaterials, 2019, 9, 467 CrossRef CAS PubMed.
- C. M. Simon, J. Kim, D. A. Gomez-Gualdron, J. S. Camp, Y. G. Chung, R. L. Martin, R. Mercado, M. W. Deem, D. Gunter and M. Haranczyk, Energy Environ. Sci., 2015, 8, 1190–1199 RSC.
- H. Zhang, P. Deria, O. K. Farha, J. T. Hupp and R. Q. Snurr, Energy Environ. Sci., 2015, 8, 1501–1510 RSC.
- Y. Basdogan, K. B. Sezginel and S. Keskin, Ind. Eng. Chem. Res., 2015, 54, 8479–8491 CrossRef CAS.
- S. Li, Y. G. Chung and R. Q. Snurr, Langmuir, 2016, 32, 10368–10376 CrossRef CAS PubMed.
- Y. G. Chung, D. A. Gómez-Gualdrón, P. Li, K. T. Leperi, P. Deria, H. Zhang, N. A. Vermeulen, J. F. Stoddart, F. You and J. T. Hupp, Sci. Adv., 2016, 2, e1600909 CrossRef PubMed.
- G. Avci, S. Velioglu and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10, 33693–33706 CrossRef CAS PubMed.
- A. N. V. Azar, S. Velioglu and S. Keskin, ACS Sustainable Chem. Eng., 2019, 7, 9525 CrossRef CAS PubMed.
- H. Daglar and S. Keskin, Adv. Theory Simul., 2019, 1900109 CrossRef CAS.
- H. Daglar and S. Keskin, J. Phys. Chem. C, 2018, 122, 17347–17357 CrossRef CAS PubMed.
- A. Sturluson, M. T. Huynh, A. R. Kaija, C. Laird, S. Yoon, F. Hou, Z. Feng, C. E. Wilmer, Y. J. Colón and Y. G. Chung, Mol. Simul., 2019, 45, 1082–1121 CrossRef CAS PubMed.
- C. Altintas, G. Avci, H. Daglar, A. N. V. Azar, İ. Erucar, S. Velioglu and S. Keskin, J. Mater. Chem. A, 2019, 7, 9593–9608 RSC.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- B. W. Dassault Systèmes BIOVIA, Release 2017; BIOVIA Pipeline Pilot, Release 2017, San Diego: Dassault Systèmes.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef CAS.
- C. Altintas, S. Velioglu and S. Keskin, ACS Appl. Mater. Interfaces, 2019, 11, 16266–16271 CrossRef CAS PubMed.
- C. E. Wilmer and R. Q. Snurr, Chem. Eng. J., 2011, 171, 775–781 CrossRef CAS.
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, Mol. Simul., 2016, 42, 81–101 CrossRef CAS.
- V. Buch, J. Chem. Phys., 1994, 100, 7610–7629 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569–2577 CrossRef CAS.
- J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS.
- K. Makrodimitris, G. K. Papadopoulos and D. N. Theodorou, J. Phys. Chem. B, 2001, 105, 777–788 CrossRef CAS.
- P. P. Ewald, Ann. Phys., 1921, 369, 253–287 CrossRef.
- Z. Sumer and S. Keskin, Ind. Eng. Chem. Res., 2016, 55, 10404–10419 CrossRef CAS.
- K. B. Sezginel, A. Uzun and S. Keskin, Chem. Eng. Sci., 2015, 124, 125–134 CrossRef CAS.
- Y. Basdogan and S. Keskin, CrystEngComm, 2015, 17, 261–275 RSC.
- Y. Xie, F. Y. Bai, Y. H. Xing, Z. Wang, H. Y. Zhao and Z. Shi, Z. Anorg. Allg. Chem., 2010, 636, 1585–1590 CrossRef CAS.
- M.-N. Li, D.-Y. Du, G.-S. Yang, S.-L. Li, Y.-Q. Lan, K.-Z. Shao, J.-S. Qin and Z.-M. Su, Cryst. Growth Des., 2011, 11, 2510–2514 CrossRef CAS.
- D. Dubbeldam, S. Calero and T. J. Vlugt, Mol. Simul., 2018, 44, 653–676 CrossRef CAS.
- D. Sun, S. Ma, Y. Ke, D. J. Collins and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 3896–3897 CrossRef CAS PubMed.
- C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC.
- K. Leperi, Y. G. Chung, F. You and R. Snurr, ACS Sustainable Chem. Eng., 2019, 7(13), 11529–11539 CrossRef CAS.
- Z. Qiao, Q. Xu and J. Jiang, J. Mater. Chem. A, 2018, 6, 18898–18905 RSC.
- A. B. Caballero, J. K. Maclaren, A. Rodríguez-Diéguez, I. Vidal, J. A. Dobado, J. M. Salas and C. Janiak, Dalton Trans., 2011, 40, 11845–11855 RSC.
- P. D. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvåg, Chem. Commun., 2006, 959–961 RSC.
- D. Kustaryono, N. Kerbellec, G. Calvez, S. Freslon, C. Daiguebonne and O. Guillou, Cryst. Growth Des., 2009, 10, 775–781 CrossRef.
- H. Frost and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 18794–18803 CrossRef CAS.
- H. Frost, T. Düren and R. Q. Snurr, J. Phys. Chem. B, 2006, 110, 9565–9570 CrossRef CAS PubMed.
- G. Avci, S. Velioglu and S. Keskin, J. Phys. Chem. C, 2019, 123(46), 28255–28265 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- M. De Toni, F.-X. Coudert, S. Paranthaman, P. Pullumbi, A. Boutin and A. H. Fuchs, J. Phys. Chem. C, 2012, 116, 2952–2959 CrossRef CAS.
- R. Babarao, J. Jiang and S. I. Sandler, Langmuir, 2008, 25, 5239–5247 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparisons of the single-component CO2 uptakes, selectivities, APSs, R% computed for separation of CO2/CH4: 50/50, CO2/H2: 15/85, and CO2/N2: 15/85 mixtures for MOFs (a) with and without coordinated solvent molecules, (b) with and without missing hydrogen atoms in the framework, and (c) with and without CBIs. See DOI: 10.1039/d0ma00039f |
| This journal is © The Royal Society of Chemistry 2020 |