
Tuning selectivity of CO2 hydrogenation by modulating the strong metal–support interaction over Ir/TiO2 catalysts†
Yaru
Zhang
ab,
Zhen
Zhang
c,
Xiaofeng
Yang
a,
Ruifeng
Wang
a,
Hongmin
Duan
a,
Zheng
Shen
a,
Lin
Li
a,
Yang
Su
a,
Runze
Yang
c,
Yongping
Zhang
c,
Xiong
Su
*a,
Yanqiang
Huang
 *a and
Tao
Zhang
d
*a and
Tao
Zhang
d
aCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: suxiong@dicp.ac.cn; yqhuang@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cChina Astronaut Research and Training Center, Beijing 100094, China
dState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 15th September 2020
Abstract
Exploration of highly selective catalysts for CO2 hydrogenation remains a great challenge since the reduction of CO2 over the supported metal catalysts may give rise to various products in response to the modulation of the chemical state of active sites. Herein, by varying the pretreatment temperature of iridium/titanium oxide (Ir/TiO2) catalysts, the selectivity of CO2 hydrogenation from CH4 to sole production of CO can be finely tuned. The change of product selectivity is achieved in such a way that the selectivity greatly depend on the formation of a reduced TiOx overlayer around Ir nanoparticles (NPs) as originated from the strong metal–support interaction (SMSI). With only a weak reduction treatment, the exposed Ir NPs without a TiOx coating promote CH4 production exclusively. After the catalyst undergoes a high temperature reduction, the evolution of the TiOx coating over Ir NPs shows a preference for CO production with an inhibition of further methanation. This study not only provides insights into the regulation of CO2 hydrogenation by SMSI, but also serves as an effective approach to tuning other catalytic processes.
Introduction
The ever-increasing threats of global warming and dramatic climate changes have drawn increasing attention in recent years. Carbon dioxide (CO2), as a major greenhouse gas, is being blamed for the ecological troubles. On the other hand, CO2 also serves as an ideal C1 source, and catalytic CO2 hydrogenation offers an effective strategy for the transformation of CO2 into high value-added chemicals1–3 and fuels4–6 by which the global carbon cycle can be sustained simultaneously.Catalytic CO2 hydrogenation at atmospheric pressure usually consists of two competitive processes over the supported Group VIII metal catalysts. One is direct CO2 methanation, known as the Sabatier reaction,7–9 and the other is CO2 hydrogenation to carbon monoxide (CO), known as the reverse water–gas shift (RWGS) reaction.10–12 Exploration of highly selective catalysts with the aim of acquiring desired products for each process still remains a great challenge. So far, enormous efforts have been devoted to tuning the selectivity of CO2 hydrogenation, including tuning the metal particle size,13–15 modifying the chemical state, changing the support type16,17 or crystal phase,18 and adding promoters.17,19 Despite these developments, intrinsic factors that govern the product selectivity remain less understood. As previously reported by Christopher et al., the product selectivity of CO2 hydrogenation greatly depended on the active sites, that is, atomically dispersed isolated rhodium (Rh) sites selected for CO production and Rh nanoparticles (NPs) selected for CO2 methanation.20 Our group has reported recently that, when decreasing the coordination number of metal sites to one, CO production was significantly enhanced by the inhibition of carbonyl dissociation and simultaneous facilitation of CO desorption.21 On the other hand, researchers have found that it was the chemical state of Ir species, rather than the coordination of metals, nanoparticles or single-atoms, that played a more important role in controlling product selectivity.22
Reducible oxides like titanium oxide (TiO2) and cerium oxide (CeO2) are widely used as supports in CO2 hydrogenation, as they give adequate assistance for CO2 activation.23–25 More importantly, strong metal–support interactions (SMSI) on reducible oxide-supported metal catalysts occur,26–28 where H2 and CO chemisorption can be suppressed and will further influence the catalytic CO2 hydrogenation processes. The intrinsic role of SMSI in tuning the selectivity of CO2 hydrogenation is worth studying in depth.
In this study, we succeeded in fabricating a highly dispersed Ir/TiO2 catalyst with resistance to agglomeration even when treated at high temperatures (up to 700 °C). By varying the reduction temperature, SMSI on Ir/TiO2 are modulated, which further plays a crucial role in tuning the product selectivity. With a weak reduction treatment at low temperatures (lower than 300 °C), metallic Ir NPs expose on the catalyst surface and promote CH4 production exclusively. After the catalyst undergoes a high temperature reduction, Ir NPs are entirely encapsulated by reduced TiOx species, which show a preference for CO production with an inhibition of further methanation. With this knowledge, we show that highly selective CO2 hydrogenation processes toward either of the two products can be achieved by tailoring the SMSI over the supported metal catalysts.
Results and discussion
Catalytic performance of Ir/TiO2-x catalysts
A series of Ir/TiO2 catalysts, with a 2.45 wt% Ir loading as detected by inductively coupled plasma optical emission spectroscopy (ICP-OES), were pretreated at different temperatures and were denoted as Ir/TiO2-x, where x refers to the reduction temperature (x = 200–700 °C). The catalytic CO2 hydrogenation tests of these catalysts were performed at 280 °C under atmospheric pressure. As shown in Fig. 1, the series of Ir/TiO2-x catalysts show a similar catalytic activity as reflected by the reaction rate, whereas a divergence in product selectivity from CH4 to CO is observed with an increase in the reduction temperature from 200 to 700 °C. Samples pretreated at temperatures lower than 300 °C exhibited complete methanation of CO2 conversion. An improved selectivity toward CO was observed as the reduction temperature increased, and a further increase of reduction temperature beyond 600 °C led to complete CO formation.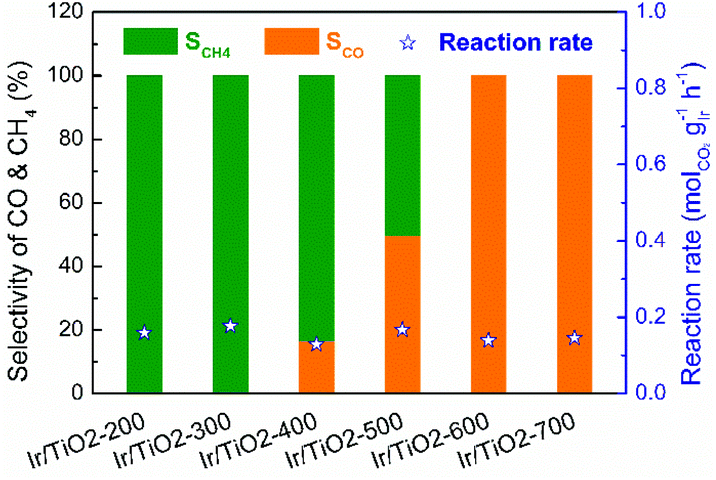 | ||
| Fig. 1 Catalytic performance of the series of Ir/TiO2-x catalysts for CO2 hydrogenation reactions. Reaction conditions: 280 °C, 0.1 MPa, space velocity = 9000 mL h−1 gcat−1, and H2/CO2/N2 = 70/20/10. | ||
The catalytic performance of CO2 hydrogenation over the Ir/TiO2-x catalysts was then compared with that of state-of-the-art catalysts. As shown in Table S1,† for the CO2 methanation reaction, the activity of the Ir/TiO2-200 catalyst is inferior to that of Ru- and the benchmark Ni-based catalysts since they always exhibit excellent hydrogenation properties. As for the reverse water–gas shift reaction, Ir/TiO2-700 exhibits an activity comparable to or higher than that of the reported catalysts. Nevertheless, the product selectivity on the Ir/TiO2 catalysts can be tuned from CH4 to exclusively CO just by varying the reduction temperature (Fig. 1 and Fig. S1†), which may help uncover the intrinsic factors that govern product selectivity and may offer greener pathways for the conversion of CO2.
In Fig. 2a, similar slopes of Arrhenius plots for CO2 conversion are observed, i.e., similar apparent activation energies (Ea) were calculated for CO2 activation over these Ir/TiO2-x catalysts. However, the slopes of Arrhenius plots for CH4 production greatly increased along with the increase of pretreatment temperature from 200 to 700 °C (Fig. 2b). A reverse trend is observed for the variation of Ea for CO production (Fig. S2†). From the calculated data, the Ea values for CH4 production manifest a significant increase from 39.6 to 233.9 kJ mol−1, while those for CO production appear to exhibit a modest decrease from 50.7 to 41.1 kJ mol−1 (Table S2†). Note that the Ea values for CO2 conversion all lie in a lower value range (40.5–47.1 kJ mol−1), indicative of the facile activation of CO2 over all Ir/TiO2-x catalysts. The increased barrier for CH4 production together with the decreased barrier for CO production offers a great opportunity for modulating the selectivity in the CO2 hydrogenation process. The tremendous shift of product selectivity together with the corresponding soaring increase of Ea for CH4 production greatly motivates us to investigate the intrinsic variation of Ir/TiO2-x catalysts as the pretreatment temperature varies.
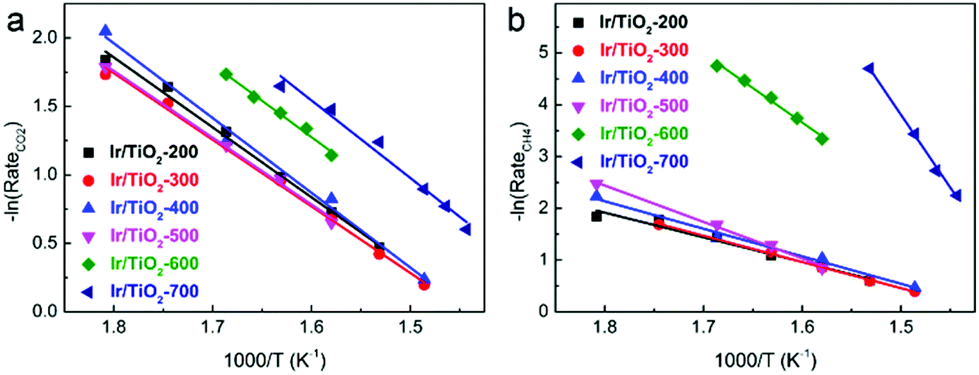 | ||
| Fig. 2 Arrhenius plots for (a) CO2 conversion and (b) CH4 production over Ir/TiO2-x catalysts. | ||
Structure identification of Ir/TiO2-x catalysts
The structure variations with the increasing pretreatment temperature of Ir/TiO2 catalysts were first studied by a set of characterization techniques. As shown in Fig. S3 and Table S3,† these Ir/TiO2-x catalysts possess similar BET surface areas and pore volumes. From XRD patterns, there is no characteristic peak for metallic Ir or IrO2 phases for all Ir/TiO2-x catalysts (Fig. S4†), indicative of a high dispersion of Ir species on the rutile TiO2 support. As observed from the HAADF-STEM images in Fig. 3, Ir nanoparticles in all these Ir/TiO2-x catalysts are highly dispersed and manifest a uniform size distribution with a diameter of 1.1–1.2 nm. The good stability of Ir NPs was primarily due to the employment of rutile TiO2, which possesses similar lattice parameters with those of rutile IrO2 (Table S4†). Upon calcination in air, IrO2 tends to spread onto TiO2 and facilely forms an IrxTi1−xO2 interphase between surface IrO2 and the TiO2 substrate (IrO2/IrxTi1−xO2/TiO2), which in turn resists the aggregation of Ir NPs even after a pretreatment at a temperature of 700 °C.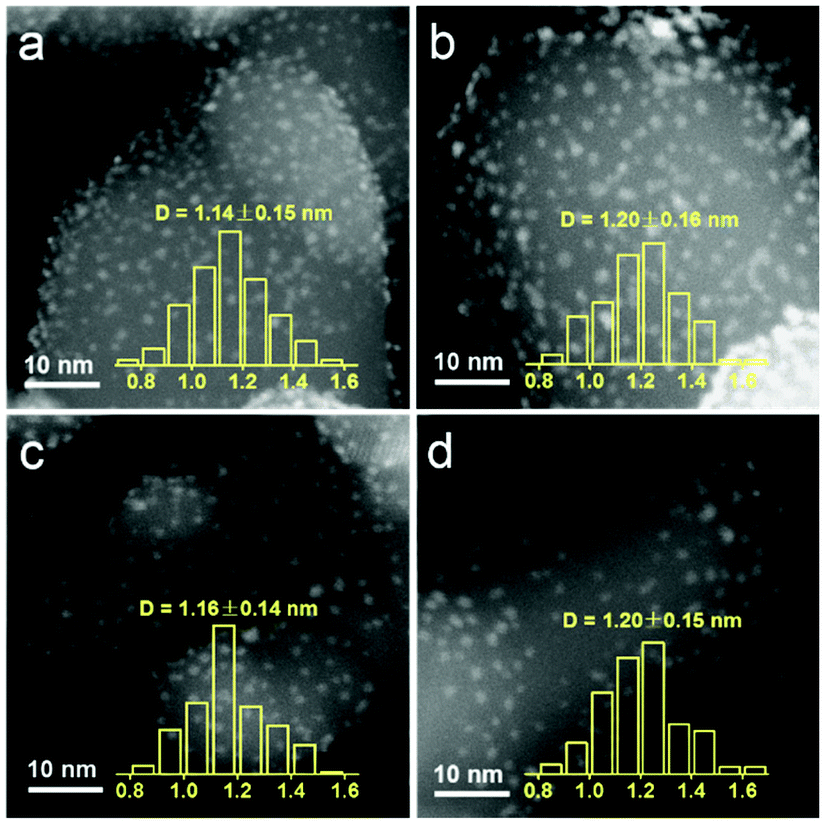 | ||
| Fig. 3 HAADF-STEM images of the series Ir/TiO2-x catalysts with the metal size distribution. (a) Ir/TiO2-200; (b) Ir/TiO2-400; (c) Ir/TiO2-600; and (d) Ir/TiO2-700. | ||
The reduction behavior of the as-prepared Ir/TiO2 catalyst was then acquired by a H2-TPR experiment. As shown in Fig. 4, two main reduction peaks (at 83 and 184 °C) are observed before 300 °C, which are attributed to the reduction of surface IrO2 and Ir species in the IrxTi1−xO2 interphase, respectively. The H2 consumption for the two stages (between 50–285 °C) was measured as 253.7 μmol g−1, which is approximately equivalent to the theoretical estimation of 254.9 μmol g−1 for IrO2 reduction (Table S5†). The total H2 consumption is 1.58 times that of theoretical estimation for full reduction of the IrO2 phase, indicative of the co-reduction of TiO2 in the Ir/TiO2 catalyst, which can be confirmed by the broad peaks at 320 and 530 °C. Benefiting from the formation of the IrxTi1−xO2 interphase, the reduction of TiO2 in Ir/TiO2 is greatly facilitated29 in comparison with that of pure TiO2 (at 671 °C) due to the facile hydrogen spillover. Therefore, for the Ir/TiO2 catalysts pretreated at temperatures less than 300 °C, i.e., the Ir/TiO2-200 and Ir/TiO2-300 samples, there are predominantly exposed metallic Ir NPs on the surface of TiO2 support. For the Ir/TiO2 catalysts pretreated at temperatures higher than 300 °C, apart from the reduction of IrO2, there is also co-reduction of TiO2. Accordingly, the degree of reduction of Ir/TiO2 was supposed to be improved with the increase of pretreatment temperature.
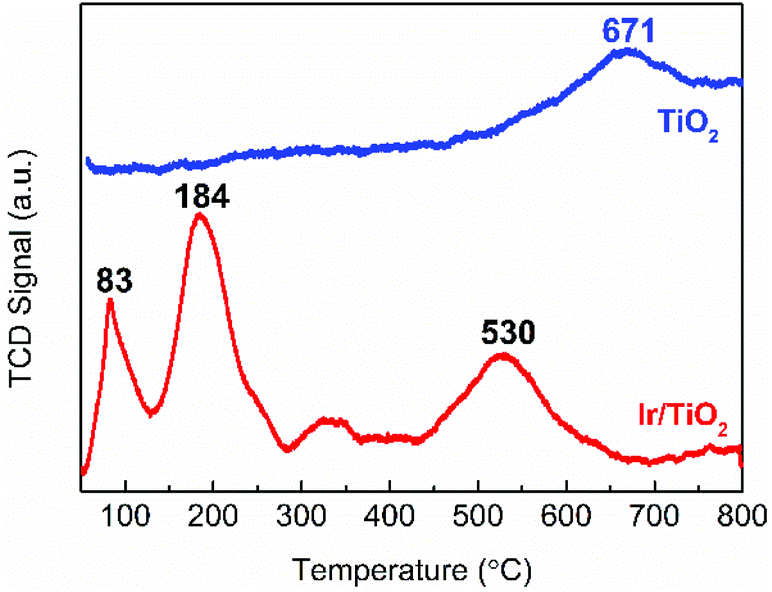 | ||
| Fig. 4 H2-TPR profiles of the Ir/TiO2 catalyst and the TiO2 support. | ||
As reported previously, the chemical state of Ir species plays a vital role in tuning the selectivity of CO2 hydrogenation, that is, metallic Ir NPs preferred CH4 production while partially oxidized Ir species favored CO production.22 In our case, the chemical states of Ir/TiO2-x catalysts were then investigated with X-ray photoelectron spectroscopy (XPS). As shown in Fig. 5a, the peaks approximately at 60.5 eV (Ir 4f7/2) and 63.3 eV (Ir 4f5/2) are attributed to metallic Ir species.30,31 In the Ir/TiO2-200 sample, both metallic Ir0 and partially oxidized Irδ+ species contribute to the observed binding energies of 61.4 eV (Ir 4f7/2) and 64.0 eV (Ir 4f5/2), which is due to the incomplete reduction of Ir NPs as confirmed by H2-TPR results. The shift in binding energy from 61.4 to 60.2 eV with the increase of reduction temperature suggests a gradual improvement of the degree of reduction of Ir NPs. Combined with the reaction data above, the improved degree of reduction failed to promote CH4 production, indicating that the chemical state of Ir species may not be the decisive factor on product selectivity. It thus motivates us to focus on other discrepancies originating from the variation of micro-structures in Ir/TiO2 influenced by different reduction treatments.
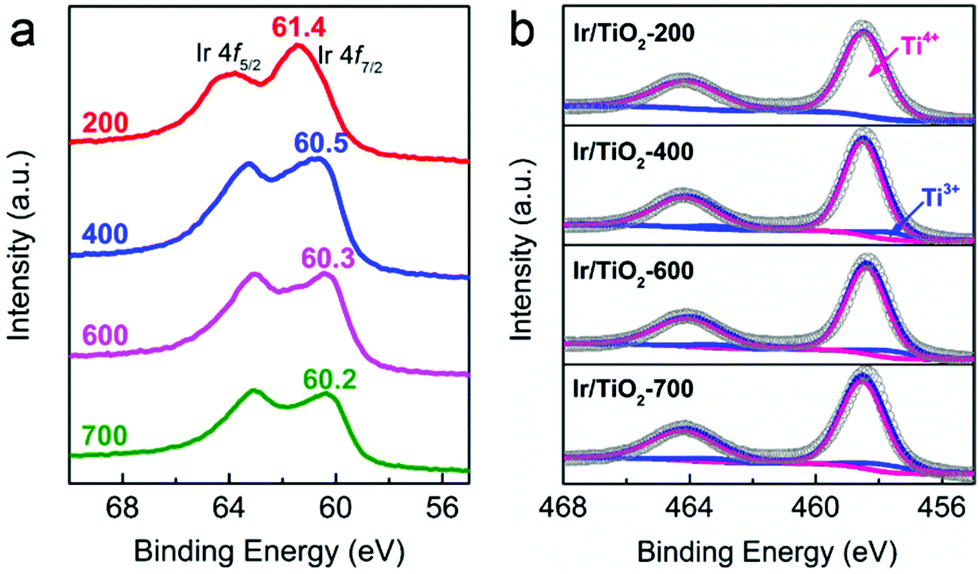 | ||
| Fig. 5 (a) Ir 4f and (b) Ti 2p XP spectra of Ir/TiO2-x catalysts. | ||
As previously reported by Tauster et al.,26–28 there are strong metal–support interactions (SMSI) over TiO2 supported Ir catalysts, in which reduced TiOx species migrates onto the metal surfaces and further forms a coating on the metal particles. Consequently, the chemisorption of CO and H2 can be severely suppressed, which usually alters the catalytic performance of CO2 hydrogenation. As indicated by the Ti 2p XP spectra (Fig. 5b and Table S6†), the increased Ti3+ concentration (from 1.1% to 9.7%) might be a reflection of the enhanced TiOx coating over Ir NPs when the pretreatment temperature was varied from 200 to 700 °C.
HRTEM observations provide intuitive information about the discrepancies in micro-structures with varying reduction temperature. As shown in Fig. 6, the Ir/TiO2-200 sample shows angular particles on the TiO2 support with a clear boundary due to a dominant reduction of IrO2 to metallic Ir, while an increased coating over Ir NPs is distinguishable with the pre-reduction temperature varying from 400 to 700 °C. This phenomenon is ascribed to a gradual migration of TiOx overlayer to the Ir NPs. Furthermore, CO was used as a probe in detecting the in situ diffuse reflectance infrared Fourier transform (DRIFT) spectra, with the aim of getting a qualitative comparison of the degree of exposure of Ir NPs after coating by TiOx overlayer. The main band appeared at 2080–2040 cm−1 in the carbonyl region (Fig. 7), which was attributed to the linear CO adsorption on Ir NPs.32 An obvious decay of the DRIFT intensity was observed as the pretreatment temperature varied from 300 to 700 °C, indicative of a reduced Ir exposure, which in turn demonstrated the growth of TiOx coating on Ir NPs. The intensity of CO adsorption on the Ir/TiO2-200 sample was found to be a little less than that of Ir/TiO2-300 probably due to the lower degree of reduction of Ir species in Ir/TiO2-200.
 | ||
| Fig. 6 HRTEM images of the series of Ir/TiO2-x catalysts. (a) Ir/TiO2-200; (b) Ir/TiO2-400; (c) Ir/TiO2-600; and (d) Ir/TiO2-700. | ||
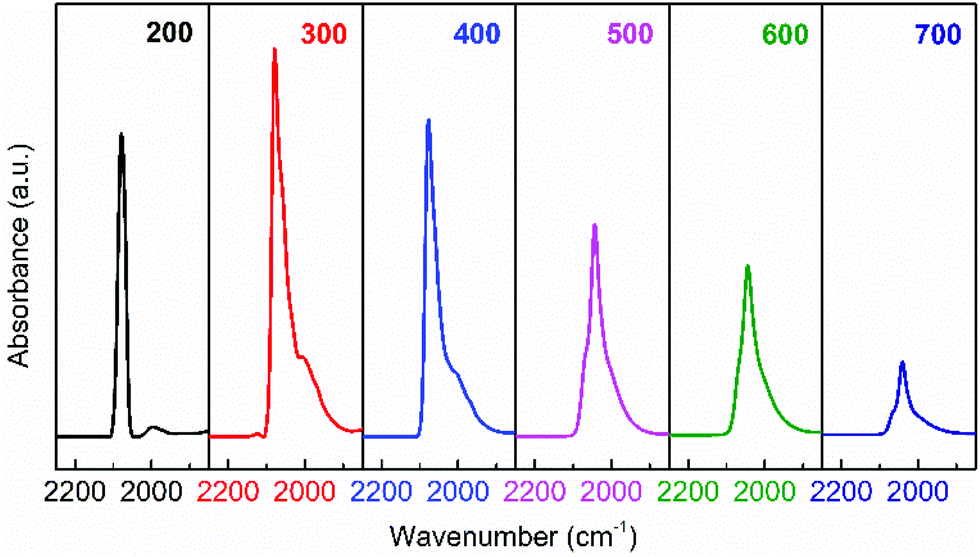 | ||
| Fig. 7 In situ DRIFT spectra obtained after CO adsorption and evacuation with helium at room temperature (25 °C), over the series of Ir/TiO2-x catalysts. | ||
H2 and CO chemisorption experiments were further conducted to estimate the dispersion of Ir species on Ir/TiO2-x catalysts. As shown in Table 1, the values determined by both H2 and CO probe molecules indicate the same tendency for different Ir/TiO2-x samples, that is, the dispersion of Ir decreases with the increase of reduction temperature from 300 to 700 °C. Ir/TiO2 catalysts pretreated at temperatures higher than 600 °C even lost their H2 or CO adsorption ability, which suggests an almost complete encapsulation of Ir NPs by the TiOx overlayer. Meanwhile, both H2 and CO uptakes on the Ir/TiO2-200 catalyst were found to be less than those on Ir/TiO2-300, primarily due to the insufficient reduction of Ir/TiO2-200, as indicated by H2-TPR and XPS results. The variation in Ir dispersion conforms with the results of HRTEM observations and DRIFT spectra and might be the most likely reason for the selectivity change in CO2 hydrogenation as all these Ir/TiO2-x catalysts manifest an analogous size distribution.
| Sample | H2 uptake (μmol gIr−1) | DIr by H2 | CO uptake (μmol gIr−1) | DIr by CO |
|---|---|---|---|---|
| Ir/TiO2-200 | 33.6 | 52.8% | 63.8 | 50.1% |
| Ir/TiO2-300 | 36.4 | 57.2% | 69.4 | 54.5% |
| Ir/TiO2-400 | 24.0 | 37.7% | 50.2 | 39.4% |
| Ir/TiO2-500 | 3.59 | 5.63% | 6.94 | 5.45% |
| Ir/TiO2-600 | 2.18 | 3.42% | 4.39 | 3.44% |
| Ir/TiO2-700 | 0.12 | 0.20% | 0.29 | 0.22% |
Structure evolution and the proposed catalytic mechanism of Ir/TiO2-x catalysts
On the basis of the above observations, a structure evolution of Ir/TiO2 catalysts at different reduction stages is then proposed (Fig. 8). Benefiting from the similar lattice parameters between IrO2 and rutile TiO2, highly dispersed Ir/TiO2 catalysts with a desired sintering resistance and similar particle size can be facilely fabricated. By varying the reduction temperature, the strong metal–support interaction in Ir/TiO2-x can be modulated accordingly, where a TiOx overlayer gradually appears and migrates on to Ir NPs, resulting in a shrinkage of the metallic Ir surface. The formation of TiOx coating by SMSI plays an important role in the selectivity change. For the Ir/TiO2-x catalysts treated at lower temperatures (x < 300 °C), there are dominant metallic Ir NPs, on which CO2 methanation can be facilitated as reported in our previous study.21 As for the samples treated with a moderate reduction (300 < x < 600 °C), the TiOx overlayer begins to migrate and coat the Ir surface and results in a shrinkage of the metallic Ir surface. In combination with the partially exposed Ir surfaces and a TiOx thin layer, both CO2 methanation and reverse water–gas shift (RWGS) processes will happen on the Ir/TiO2-x catalysts. With the increase of TiOx coating, the RWGS process producing CO is promoted accordingly. On the contrary, for the samples treated with an excessive reduction (x > 600 °C), Ir NPs are entirely encapsulated with the TiOx overlayer. The RWGS process on TiOx coating is facile to realize, while further hydrogenation to produce CH4 is completely suppressed due to the unreachable Ir surfaces.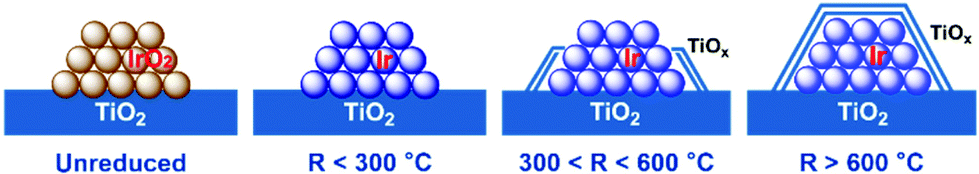 | ||
| Fig. 8 A schematic illustration of the structural evolution of Ir/TiO2-x catalysts. The gradually brighter color on Ir species suggests an improvement of the degree of reduction for Ir NPs. | ||
Considering the structural evolution in relation to the corresponding catalytic performance, CO probably serves as the intermediate during CO2 methanation over Ir/TiO2-x catalysts. As all these Ir/TiO2-x catalysts manifest similar reaction rates and Ea for CO2 conversion with a low value of ∼45 kJ mol−1, it is facile to realize CO2 activation. An increased barrier for CH4 production is observed as the reduction temperature increases, during which the encapsulation of Ir NPs results in an enhanced RWGS process to produce CO. When the reduction temperature is increased to modulate the metal-support interaction, the TiOx thin layer gradually migrates on to the surface of Ir NPs, which plays a vital role in further transformation of product selectivity from CH4 to CO.
Experimental
Catalyst preparation
Ir/TiO2 catalysts were prepared via a wetness impregnation method. In a typical synthesis, 0.42 g of the H2IrCl6 solution (38 wt%, 0.1418 g Ir per gram of solution, AR) was diluted to 50 mL with deionized water. Following this, 2.0 g of rutile TiO2 was added to the solution and the resulting suspension was dried in a 50 °C water bath with vigorous stirring through evaporation. The resulting solid was dried at 120 °C for 12 h, followed by calcination in air at 400 °C for 4 h. In order to remove the residual chlorides, the sample was then washed repeatedly with a dilute ammonia solution (1 mol L−1), followed by filtration and drying at 80 °C overnight. The sample thus obtained was denoted as the fresh Ir/TiO2 catalyst. The loading of Ir on the Ir/TiO2 catalyst was 2.45 wt% as detected by ICP. Prior to CO2 hydrogenation tests, the as-prepared Ir/TiO2 sample was reduced in situ in a H2 flow (20 mL min−1) at specific temperatures, denoting them as Ir/TiO2-x, where x indicates the reduction temperature (200, 300, 400, 500, 600 or 700 °C).Catalyst characterization
Powder X-ray diffraction (XRD) data were acquired using a PANalytical X'Pert-Pro X-ray diffractometer operated at 40 kV and 40 mA. Nitrogen physisorption was performed with a Micromeritics ASAP 2460 instrument at −196 °C. TEM observations, including high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and high-resolution transmission electron microscopy (HRTEM) images, were acquired using a JEOL JEM-2100F microscope operated at 200 kV. The Ir concentration in the Ir/TiO2 catalyst was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) with an ICP-OES 7300DV instrument.H2 temperature programmed reduction (H2-TPR) was performed with a Micromeritics AutoChem II 2920 apparatus. Prior to TPR measurement, the pristine Ir/TiO2 sample was pretreated with Ar at 200 °C for 60 min. After the temperature was reduced to 50 °C, a 10% H2/Ar flow was introduced into the reactor by heating the sample to 800 °C at a heating rate of 10 °C min−1. The signal was recorded online with a thermal conductivity detector (TCD).
X-ray photoelectron spectroscopy (XPS) data were obtained using a Thermo Fisher ESCALAB 250Xi instrument, employing monochromated Al Kα radiation (hυ = 1486.6 eV) as the X-ray source. The sample was pretreated at the desired temperature in a H2 flow and then held under an inert atmosphere, followed by rapid transfer to the sample chamber to minimize exposure to air. The results were calibrated by setting the C 1s adventitious carbon peak position to 284.6 eV.
In situ diffuse reflectance infrared Fourier transform (DRIFT) spectra were acquired using a Bruker Equinox 55 spectrometer recorded with a resolution of 4 cm−1. The sample was treated in situ under a H2 flow (20 mL min−1) at the desired temperature for 60 min. After cooling to room temperature (25 °C), the gas flow was purged with He for 30 min, following which the background spectrum was collected. Then, the He flow was switched to a 5 vol% CO in He flow (20 mL min−1) which was maintained until saturated adsorption was achieved. The system was purged with He to remove non-adsorbed CO and DRIFT spectra were collected, such that CO adsorption data at room temperature were obtained.
The exposure of Ir NPs was determined by CO and H2 chemisorption on a Micromeritics AutoChem II 2920 instrument. For the CO (or H2) chemisorption experiment, the sample was pretreated in a H2 flow at the desired temperature for 60 min, followed by purging with He (or Ar) for 30 min. After cooling down to 50 °C, a 5% CO in He (or 10% H2 in Ar) was injected into the reactor repeatedly until saturated adsorption is achieved. The dispersion of Ir was determined by assuming the CO/Ru (or H/Ru) adsorption stoichiometry to be 1:1.
Catalyst testing
CO2 hydrogenation reactions were performed in a fixed-bed quartz reactor at atmospheric pressure. Prior to each reaction, the Ir/TiO2 catalyst (0.15 g) was in situ reduced in a H2 flow (20 mL min−1) at the desired temperature for 2 h. After the reactor was cooled down, a feed gas with a H2/CO2 ratio of 3.5/1 (H2/CO2/N2 = 70/20/10 (v/v/v), N2 was used as an internal standard) was introduced into the reactor for CO2 hydrogenation tests. The reactions were carried out at 280 °C and 9000 mL gcat−1 h−1. After passing through an ice bath, the gaseous products were analyzed online using an A90 Echrom gas chromatograph equipped with a TDX-01 column connected to a thermal conductivity detector (TCD).The CO2 conversion, XCO2, was calculated using the following equation:
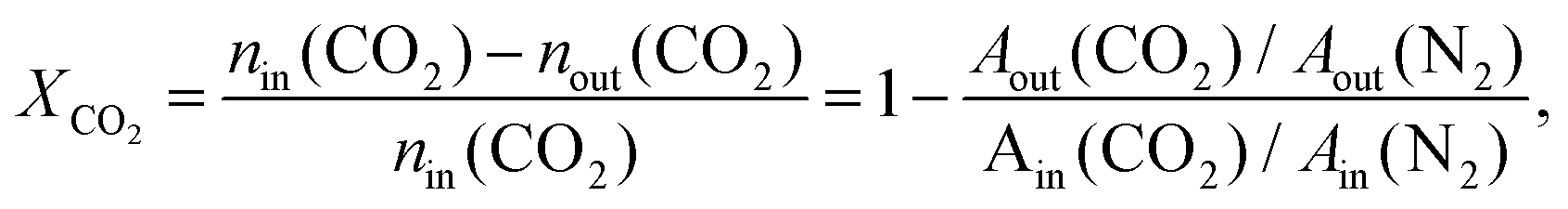
The reaction rate was calculated as:
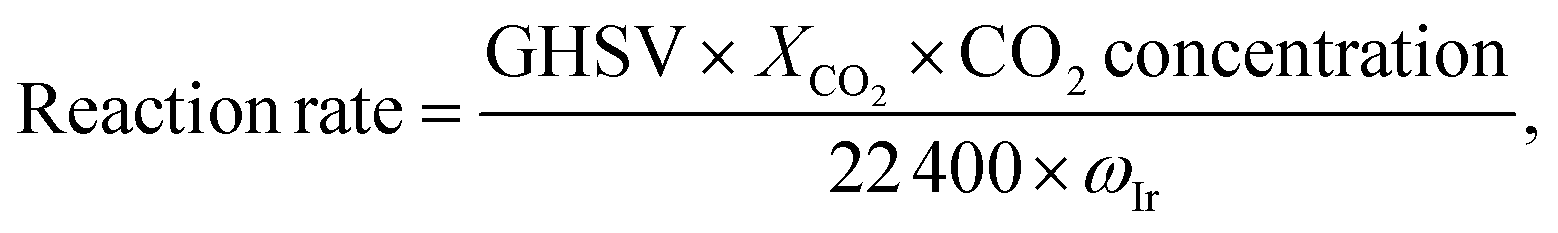
For the off-gas consisting only of CO (or CH4), the selectivity toward CO (or CH4) was assumed to be 100%. For the products in the off-gas consisting of both CO and CH4, the selectivity values for CO and CH4 (SCO and SCH4) were acquired by a normalization method:
| SCO + SCH4 = 1, |
where fCO/CH4 is the relative correction factor of CO to CH4, which was determined by the calibrating gas; and ACO and ACH4 refer to the chromatographic peak areas of CO and CH4, respectively.
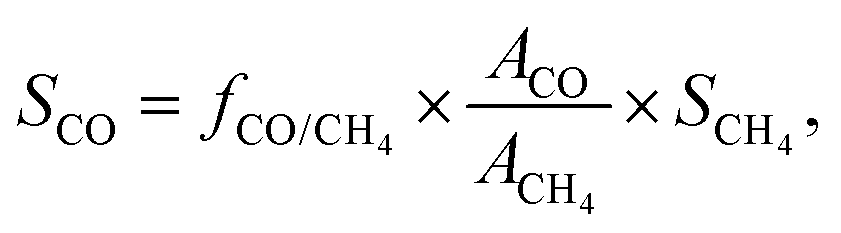
Conclusions
In conclusion, we have successfully fabricated a series of highly dispersed, similar sized Ir/TiO2 catalysts with sintering resistance to high temperature treatment. By varying the reduction temperature to modulate the metal–support interaction, the product selectivity of catalytic CO2 hydrogenation can be tuned from CH4 to CO. The reduced TiOx species that originated from SMSI plays a crucial role in tuning the product selectivity. Ir NPs encapsulated with a TiOx coating show a preference for CO production with an inhibition of further hydrogenation, while exposed Ir NPs without TiOx promote CH4 production exclusively. This study provides an understanding of the regulation of CO2 hydrogenation by SMSI and can serve as an effective approach to tailoring other catalytic processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFA0202804), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36030200), the National Natural Science Foundation of China (21978286, 21925803, 21776269), and the Youth Innovation Promotion Association CAS.Notes and references
- X. Wang, H. Shi and J. Szanyi, Nat. Commun., 2017, 8, 513–518 CrossRef.
- X. Jia, K. Sun, J. Wang, C. Shen and C.-J. Liu, J. Energy Chem., 2020, 50, 409–415 CrossRef.
- L. Wang, E. Guan, Y. Wang, L. Wang, Z. Gong, Y. Cui, X. Meng, B. C. Gates and F. S. Xiao, Nat. Commun., 2020, 11, 1033–1041 CrossRef CAS.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- J. Zhu, G. Zhang, W. Li, X. Zhang, F. Ding, C. Song and X. Guo, ACS Catal., 2020, 10, 7424–7433 CrossRef CAS.
- F. Wang, C. Li, X. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177–186 CrossRef CAS.
- K. Wang, W. Li, J. Huang, J. Huang, G. Zhan and Q. Li, J. Energy Chem., 2021, 53, 9–19 CrossRef.
- J. Gödde, M. Merko, W. Xia and M. Muhler, J. Energy Chem., 2021, 54, 323–331 CrossRef.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Chem. Commun., 2015, 51, 6988–6991 RSC.
- Y. Ma, Z. Guo, Q. Jiang, K.-H. Wu, H. Gong and Y. Liu, J. Energy Chem., 2020, 50, 37–43 CrossRef.
- R. Carrasquillo-Flores, I. Ro, M. D. Kumbhalkar, S. Burt, C. A. Carrero, A. C. Alba-Rubio, J. T. Miller, I. Hermans, G. W. Huber and J. A. Dumesic, J. Am. Chem. Soc., 2015, 137, 10317–10325 CrossRef CAS.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2449–2455 CrossRef CAS.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 13736–13745 CrossRef CAS.
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2016, 55, 7968–7973 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2094–2100 CrossRef CAS.
- W. Li, G. Zhang, X. Jiang, Y. Liu, J. Zhu, F. Ding, Z. Liu, X. Guo and C. Song, ACS Catal., 2019, 9, 2739–2751 CrossRef CAS.
- M. D. Porosoff and J. G. Chen, J. Catal., 2013, 301, 30–37 CrossRef CAS.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS.
- X. Chen, X. Su, H.-Y. Su, X. Liu, S. Miao, Y. Zhao, K. Sun, Y. Huang and T. Zhang, ACS Catal., 2017, 7, 4613–4620 CrossRef CAS.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS.
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227–237 CrossRef CAS.
- S. Li, G. Liu, S. Zhang, K. An, Z. Ma, L. Wang and Y. Liu, J. Energy Chem., 2020, 43, 155–164 CrossRef.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121–1125 CrossRef CAS.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- A. Gómez-Cortés, G. Díaz, R. Zanella, H. Ramírez, P. Santiago and J. M. Saniger, J. Phys. Chem. C, 2009, 113, 9710–9720 CrossRef.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- R. Jin, M. Peng, A. Li, Y. Deng, Z. Jia, F. Huang, Y. Ling, F. Yang, H. Fu, J. Xie, X. Han, D. Xiao, Z. Jiang, H. Liu and D. Ma, J. Am. Chem. Soc., 2019, 141, 18921–18925 CrossRef CAS.
- F. Solymosi, E. Novak and A. Molnar, J. Phys. Chem., 1990, 94, 7250–7255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterizations of Ir/TiO2 catalysts. See DOI: 10.1039/d0gc02302g |
| This journal is © The Royal Society of Chemistry 2020 |