
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of intimacy for metal-mesoporous solid acids catalysts for n-alkanes hydro-conversion†
Justine
Harmel
 ,
Lars I.
van der Wal
,
Jovana
Zečević
,
Petra E.
de Jongh
and
Krijn P.
de Jong
*
,
Lars I.
van der Wal
,
Jovana
Zečević
,
Petra E.
de Jongh
and
Krijn P.
de Jong
*
Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: K.P.deJong@uu.nl
First published on 6th March 2020
Abstract
Pt on both ordered mesoporous Al-SBA-15 and commercial amorphous mesoporous silica–alumina bi-functional catalysts were prepared and studied for n-heptane hydro-isomerization and n-hexadecane hydro-cracking. We investigated the influence of the location of the Pt nanoparticles and their proximity to the acid sites; by depositing Pt in a controlled manner either in the mesopores of the solid acid or on a γ-Al2O3 binder, surrounding the solid acid particles. We demonstrate that in the case of both ordered and amorphous mesoporous acidic support, a distance between the metal and acid sites of a hundreds of nanometers gives similar results compared to the closest intimacy (below 10 nm) between these functions. Due to the medium strength Brønsted acid sites of the Al-SBA-15, Pt–Al-SBA-15 exhibits higher activity than the corresponding Pt on commercial amorphous silica–alumina. These results suggest that, in the case of mesoporous solid acids, a fine control at the nanoscale of the proximity between the active sites is less important than the number and nature of the acid sites.
1. Introduction
Alkanes conversion via hydro-isomerization and hydro-cracking requires bifunctional catalysts and is a key reaction in oil refining used to improve the fuel quality of alkanes.1 In alkane hydro-isomerization, the metal sites of the bifunctional catalyst allow the (de)hydrogenation of alkanes/alkenes, while the acid sites promote the isomerization and cracking of alkenes intermediates. The activity, selectivity and stability of these bifunctional catalysts can be influenced by several parameters including the acid strength,2 the metal–acid balance3–7 and the degree of intimacy between metal and acid sites.5,8 The “intimacy criterion”, first introduced by Weisz, involves the distance between the metal and acid sites and its optimum range to keep the catalytic activity maximal and suggests that this distance should be less than 100 μm.9,10 High resolution characterization techniques now allow us to determine precisely the location of the active sites in the catalyst. Recently, it has been found by Zečević et al. that in the case of Pt/zeolite Y, a nanoscale intimacy i.e. Pt nanoparticles (NPs) being located outside of the zeolite micropores, on a γ-Al2O3 binder, results in an improved selectivity in the cracking of high molecular weight hydrocarbon feedstock.8 They demonstrated that localizing the Pt NPs on the γ-Al2O3 binder surrounding zeolite Y, resulting in a distance of up to several tens of nanometers between metal and the acid sites, enhanced the isomerization reaction and reduced secondary cracking. Locating the Pt NPs inside the zeolite micropores in closer contact with the acid sites produced more undesired cracking products (gas and naphtha). This finding deviates from the generally established concept that the closest proximity between the metal and acid sites would result in the best performance. These differences were tentatively explained by pore-mouth catalysis in the outer shell of the zeolite crystal dominating the acid catalyst.Since this finding, the field has expanded, though the proximity effect for zeolite-based catalysts is still under debate, and difficult to disentangle from the strong diffusion limitation inherent in the small pore zeolite structure. Latest results tend to show that the effects of intimacy are reaction-dependent as well as zeolite-dependent.5,11–16
In the case of amorphous silica–alumina (ASA), which are widely used to enable the conversion of longer alkanes, such strong diffusion limitations are absent as result of wider pores.17 Therefore, one expects that in that case the intimacy effect is less significant. Additionally, due to the lower acidity and wide pores, Pt/ASA allows to operate the refining of feedstock such as FTS waxes via the hydro-isomerization and hydro-cracking of high molecular weight alkanes.18–20 Using Pt deposited on high surface area commercial mesoporous amorphous silica–alumina, Samad et al. reported that tuning the distance between the acid site and the Pt metallic site surprisingly resulted in variations in selectivity in the hydro-isomerization of n-heptane. Pt deposited onto the silica domains of ASA was more selective towards n-heptane isomers compared to Pt deposited onto the alumina domains.21,22 They attributed this difference to the influence of residual chloride present in the samples prepared from H2PtCl6·6H2O, increasing the number of the acid sites on alumina and therefore increasing the cracking yield of the catalyst prepared using this precursor.
Among mesoporous materials, Al-SBA-15 prepared by alumination of SBA-15, appears as a promising model solid acid support for hydro-isomerization, as the textural properties of the SBA-15 are retained upon Al grafting. It exhibits a high specific surface area (800 m2 g−1), high pore volume and a tuneable acidity controlled by the targeted Si/Al ratio.23–25 In addition, the relatively thick walls of the SBA-15 ensure a good thermal and hydrothermal stability.26,27 Post-synthesis alumination of SBA-15 by Al isopropoxide precursors has been shown to result in an Al incorporation mainly in tetrahedral positions and no extra framework Al.28,29 Furthermore, Al grafted model mesoporous silica aluminas has been identified as suitable mesoporous acid components in alkane conversion, with Pt–Al-MCM-41 giving high isomerization yield for n-heptane conversion and with Pt–Al-SBA-15 for n-C12 hydrocracking.30–33 Computational studies of the free energy of the transition state in various zeolites and mesoporous Al-MCM-41 showed that in the case of mesoporous materials, the weaker confinement leads to less reactive acid sites and that the facile molecular diffusion of n-heptane in the pores results in a minimized intra-crystalline gradient of iso-alkenes.34
In order to gain further insight into the intimacy effect and its relation with diffusion limitations, we conducted a study on the influence of the metal–acid sites distance on both a model, structured mesoporous acidic model support, Al-SBA-15 and an amorphous commercial support, Siralox40hpv (Sasol). Pt deposition was performed in a controlled manner either on a γ-Al2O3 binder or on the solid acid. The Pt location on the catalyst and its distance to the acid sites were characterized by HAADF-STEM. The Pt catalysts were then studied in both hydro-isomerization of n-heptane, and hydro-cracking of n-hexadecane.
2. Experimental
2.1 Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEOS:HCl:H2O = 0.017:1.0:5.9:192.35 SBA-15 rods of 0.2 × 0.5–2 μm were obtained. The grafting of Al on SBA-15 (aimed Si/Al = 20 at/at or 10 at/at) was performed following a reported procedure.36 Briefly, Al isopropoxide, Al(O-iPr)3 (Aldrich) (0.83 mmol or 1.66 mmol) was dissolved in anhydrous cyclohexane (Fluka) under N2 atmosphere. The SBA-15 support (1 g), dried at 250 °C for 2 h prior to the reaction, was quickly transferred to the Al solution. The suspension was stirred overnight before filtration and washing with cyclohexane. The solid was then dried overnight at 120 °C and calcined under static air at 500 °C for 4 h (1 °C min−1 ramp). Al content in the final material was determined by ICP/AES and EDX. Low angle XRD measurements confirmed that the hexagonal order of the SBA-15 was not modified by the grafting of Al (ESI† Fig. SI.1).
:TEOS:HCl:H2O = 0.017:1.0:5.9:192.35 SBA-15 rods of 0.2 × 0.5–2 μm were obtained. The grafting of Al on SBA-15 (aimed Si/Al = 20 at/at or 10 at/at) was performed following a reported procedure.36 Briefly, Al isopropoxide, Al(O-iPr)3 (Aldrich) (0.83 mmol or 1.66 mmol) was dissolved in anhydrous cyclohexane (Fluka) under N2 atmosphere. The SBA-15 support (1 g), dried at 250 °C for 2 h prior to the reaction, was quickly transferred to the Al solution. The suspension was stirred overnight before filtration and washing with cyclohexane. The solid was then dried overnight at 120 °C and calcined under static air at 500 °C for 4 h (1 °C min−1 ramp). Al content in the final material was determined by ICP/AES and EDX. Low angle XRD measurements confirmed that the hexagonal order of the SBA-15 was not modified by the grafting of Al (ESI† Fig. SI.1).
Pt–Al-SBA-15(20) and Pt–Al-SBA-15(10) were prepared following the same procedure as for Pt–Al-SBA-15(20)/γ-Al2O3.
2.2 Catalyst characterization
N2 physisorption was performed at −196 °C on a Micromeritics TriStar 3000 system. Prior to analysis, the samples were outgassed in vacuum at 400 °C for 20 h. The total pore volume was reported at p/p0 = 0.995.Pt content was determined by inductively coupled plasma-optical emission spectroscopy (ICP-AES) at Mikroanalytisches Laboratorium Kolbe. The analyses were carried out in an ICP-AES Perkin-Elmer Analyst 200 spectrometer, after a digestion procedure (company process).
H2-Chemisorption was measured on a Micromeritics ASAP 2020 instrument. Prior to the measurement the sample was reduced at 350 °C in H2 for 2 h (heating ramp of 1 °C min−1). The chemisorption was then conducted at 35 °C.
Temperature-programmed desorption (TPD) of ammonia was carried out on a Micromeritics AutoChem II equipped with a thermal conductivity detector (TCD). Typically, 100 mg of catalyst was dried in a flow of He for 15 min at 600 °C, with a heating ramp of 10 °C min−1. The temperature was then decreased to 100 °C and 10 vol% ammonia in He was adsorbed in a pulse-wise manner (10 ml min−1) until oversaturation. Thereafter the physisorbed ammonia was removed by flowing He for 2 h at 100 °C. The ammonia desorption was monitored up to 600 °C with a ramp of 10 °C min−1.
Pyridine adsorption/desorption followed by infrared measurements were conducted on a ThermoFisher Nicolet i5 spectrometer instrument (32 scans, 4 cm−1 resolution, DTGS detector). The pellet was dried by heating in the cell at 200 °C (ramp of 10 °C min−1) for 2 h, and then cooled down to 50 °C. At that temperature, pyridine (99.9%, Sigma-Aldrich) vapor was introduced into the cell to an equilibrium pressure set to 10 mbar. Pyridine was then desorbed by increasing temperature at a pressure of 1.10−5 bar.
Transmission electron microscopy (TEM) measurements, high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) measurements, combined with energy dispersive X-ray spectroscopy (EDX) measurements, were performed on a FEI Talos™ F200X transmission electron microscope, using an X-FEG electron source and a Super-X™ EDX detector. Prior to the measurements the catalysts were embedded in Epofix resin and cut into microtomed slices (70 nm) using a Reichert-Jung Ultracut E ultramicrotome with a Diatome Ultra 35° diamond knife. These sections were deposited on a carbon-coated copper TEM grid.
2.3 Catalysis
3. Results and discussion
3.1 Mesoporous support synthesis and characterisation
The grafting of Al was performed on rod-shaped SBA-15 (0.2 × 0.5–2 μm) with targeted Si/Al ratio of 20 and 10 at/at (hereafter referred to as Al-SBA-15(20) and Al-SBA-15(10) respectively). A slight decrease of the BET surface area from 726 to 657 and 643 m2 g−1 was observed (see Table 1). The hexagonal ordered structure of the SBA-15 was retained after the grafting as confirmed by the presence of the (100) and (110) diffraction peaks in small angle XRD (P6mm hexagonal) (Fig. SI.1†). STEM/EDX mapping showed a uniform Al dispersion on the support with no areas where alumina was more abundant. As opposed to the commercial ASA that contains Al domains, Al-SBA-15 exhibits a homogeneous dispersion of the Al over the SBA-15, allowing a good homogeneity of the acid sites in the support (Fig. SI.2†). The acidity profile as determined by NH3-TPD showed that the grafting of Al resulted in the introduction of acid sites of medium and low strength. An increase in the grafted Al content, and corresponding decrease of the Si/Al ratio, resulted in an increase of the total number of acid sites (Fig. SI.3†). Pyridine desorption followed by infrared spectroscopy allowed us to quantify the ratio of Brønsted and Lewis acid sites contained in these materials. Al-SBA-15 contained both Brønsted and Lewis acid sites. Al-SBA-15(10) exhibited a higher content of Brønsted sites than did Al-SBA-15(20) increasing from 20 to 30% with increasing Al content, as observed in the NH3-TPD desorption profile (Fig. SI.4†). The overall characteristics of the mesoporous supports used are summarized in Table 1. In order to enable the selective deposition of the Pt NPs, a composite of Al-SBA-15(20)/γ-Al2O3 was prepared mixing Al-SBA-15 with a γ-Al2O3 binder. TEM images show a good dispersion of the Al-SBA-15 particles within the binder (see Fig. SI.5†). NH3-TPD of the composite is plotted in Fig. SI.6.† As comparison, amorphous silica–alumina (ASA) Siralox from Sasol was studied. Siralox ASA is obtained by grafting of a silica (solution of orthosilicic acid) on alumina resulting in a non-homogenous dispersion of patches of SiO2 on the alumina. As reported by Daniell et al., Siralox 40 is a silica rich support exhibiting the highest concentration of acid sites of the Siral family.37| Sample | Si/Ala (at/at) | BET surface area (m2 g−1) | Pore diameterb (nm) | Pore volumec (cm3 g−1) | Acid sitesd (μmol g−1) | B/Le |
|---|---|---|---|---|---|---|
| a Determined by ICP-AES. b Pore diameter determined by the BJH method for the desorption branch. c Total pore volume reported at p/p0 = 0.995. d Determined by deconvolution and integration of high-temperature (≥300 °C) peak of NH3-TPD profiles. e Determined by pyridine desorption followed by Infrared spectroscopy. | ||||||
| SBA-15 | — | 726 | 6.4 | 1.7 | — | — |
| Al-SBA-15(20) | 12.7 | 657 | 6.3 | 1.5 | 188 | 20/80 |
| Al-SBA-15(10) | 4.2 | 643 | 6.1 | 1.0 | 257 | 30/70 |
| Composite Al-SBA-15(20)/γ-Al2O3 | — | 420 | 6.0 | 0.6 | 115 | — |
| SiO2–Al2O3 Siralox40hpv | 0.66 | 480 | 13.9 | 1.6 | 183 | 25/75 |
| Composite Siralox40hpv | — | — | — | — | 95 | — |
3.2 Pt catalyst preparation and characterisation
Pt catalysts were prepared using two different Pt precursors [Pt(NH3)4](NO3)2 and H2PtCl6·6H2O, that should result respectively in either deposition of Pt by ion exchange on the solid acid or by strong electrostatic adsorption onto the alumina binder.38,39 The properties of the resulting catalysts are given in Table 2. For comparison, the same Pt deposition procedure was conducted on a composite Siralox40hpv/γ-Al2O3. Based on a commercial ASA, Siralox40hpv. After Pt deposition, the obtained bi-functional catalysts have a Nmetal/Nacid ratio well above 0.03 which is the minimum required value a for optimal bi-functionality (reported in the case of zeolites solid acid).6| Pt sample code | Pt precursora | Ptb (% wt) | Pt particle diameter (TEM) | Pt particle size (H2 chemisorption) | N metal/Nacidc (mol/mol) |
|---|---|---|---|---|---|
| a PTA stands for [Pt(NH3)4](NO3)2 and CPA stands for H2PtCl6·6H2O. b Determined by ICP-OES. c Where Nmetal is the number of Pt surface atoms in mol g−1 determined by H2 chemisorption. | |||||
| Pt/SBA-15 | PTA | 0.49 | nd | 1.6 | — |
| Pt/Al-SBA-15(10) | PTA | 0.44 | 1.9 ± 0.3 | 0.77 | 0.12 |
| Pt/Al-SBA-15(20) | PTA | 0.49 | 1.5 ± 0.4 | 0.85 | 0.16 |
| Pt–Al-SBA-15(20)/γ-Al2O3 | PTA | 0.34 | 1.5 ± 0.2 | 0.70 | 0.28 |
| Pt-γ-Al2O3/Al-SBA-15(20) | CPA | 0.48 | 1.2 ± 0.2 | 0.75 | 0.36 |
| Pt/Siralox | PTA | 0.50 | 1.8 ± 0.3 | 0.95 | 0.16 |
| Pt-γ-Al2O3/Siralox | CPA | 0.44 | 7.1 ± 2.0 | 2.20 | 0.13 |
| Pt–Siralox/γ-Al2O3 | PTA | 0.60 | 9.2 ± 3.0 | 4.80 | 0.08 |
HAADF-STEM images of 70 nm thick microtomy slices of Pt–Al-SBA-15(20) and Pt-γ-Al2O3/Al-SBA-15(20) catalysts are displayed in Fig. 1. In the case of the Pt–Al-SBA-15(20) catalysts (Fig. 1a and b), the images shows the hexagonal order of the SBA-15 regular pore structure with 1.5 ± 0.4 nm Pt NPs (nanometer-sized white dots) located within the mesopores of Al-SBA-15(20). As the acid sites are dispersed along the SBA-15 wall, the Pt–acid site distance is estimated in the range of ∼6 nm or below, hence the two functions are in close proximity.
 | ||
| Fig. 1 HAADF-STEM images of a) and b) Pt–Al-SBA-15(20) showing the SBA-15 domains c) and d) Pt-γ-Al2O3/Al-SBA-15(20) showing mainly the γ-Al2O3 domains. The arrows point to Pt NPs. | ||
In the case of the Pt-γ-Al2O3/Al-SBA-15(20) catalysts (Fig. 1c and d), the images show that 1.2 ± 0.2 nm Pt NPs are located on the γ-alumina binder (identified by its platelet-like structure), component of the composite (Al-SBA-15/γ-Al2O3). In this case, the acid sites on the Al-SBA-15 are further away from the Pt NPs estimated to be at least 100 nm apart. This is significantly different to the metal–acid site distance observed in the Pt–Al-SBA-15. Otherwise, these two catalysts have similar properties and Pt particle size. HAADF-STEM images of Pt–Al-SBA-15(20)/γ-Al2O3 catalysts, obtained by deposition of [Pt(NH3)4](NO3)2 on the composite (Fig. SI.8†), resulted in Pt NPs not only located in the Al-SBA-15 but also partially on the binder. This can be explained by the weak acidity of the Al-SBA-15, therefore leading to a weak ion exchange capacity of the material. This catalyst exhibits therefore an average Pt–acid site distance somewhere between the other two. All the catalysts exhibited a narrow Pt particle size distribution around 1.3 nm.
3.3 n-Heptane hydro-isomerization catalytic studies
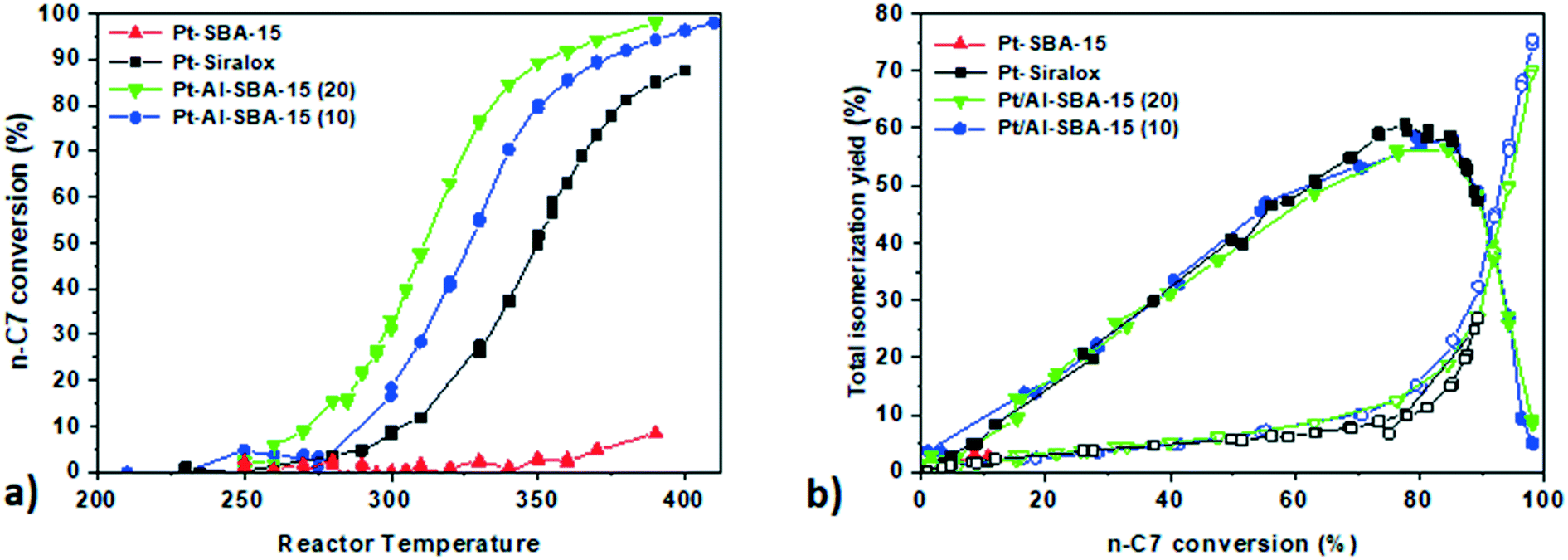 | ||
| Fig. 2 a. Conversion profile of n-heptane against temperature for Pt-SBA-15 (red triangle); Pt–Al-SBA-15(20) (green triangle); Pt–Al-SBA-15(10) (blue circle) and Pt-ASA (black square) b. yield of isomers against n-C7 conversion for the corresponding catalysts (full symbols) and cracking products (open symbols) catalysis performed at 10 bar; H2/n-C7 = 10/1; WHSV = 1.15 gn-C7 gcatalyst h−1. | ||
Pt deposited on non-aluminated SBA-15 (red triangles) did not show any isomerization activity as expected for a monofunctional catalyst only containing metallic sites but no acidity.
Pt–Al-SBA-15(20) showed a higher n-heptane conversion at lower temperature (50% conversion at 310 °C) than to Pt/Siralox (50% conversion at 350 °C). The higher activity of Pt on the Al-SBA-15(20) support compared to Siralox can be explained by its higher content of strong acid sites. Pt/Al-SBA-15(20) exhibited n-heptane conversion at lower temperature (20 °C difference) than Pt/Al-SBA-15(10) suggesting an optimum around this Si/Al ratio for n-heptane hydro-isomerization. The isomer yield is the sum of the mono-branched and di-branched C7 isomers products. The acid sites present in the Al-SBA-15 allowed n-heptane conversion as well as high isomers yield (60% of maximum total isomers yield) similar to the yield of Pt/Siralox (Fig. 2b).
Under these conditions, with an operating pressure 10 bar, no deactivation of the catalysts was observed over the 200 h run, as it was verified by checking the conversion at the end of the run at a lower temperature.
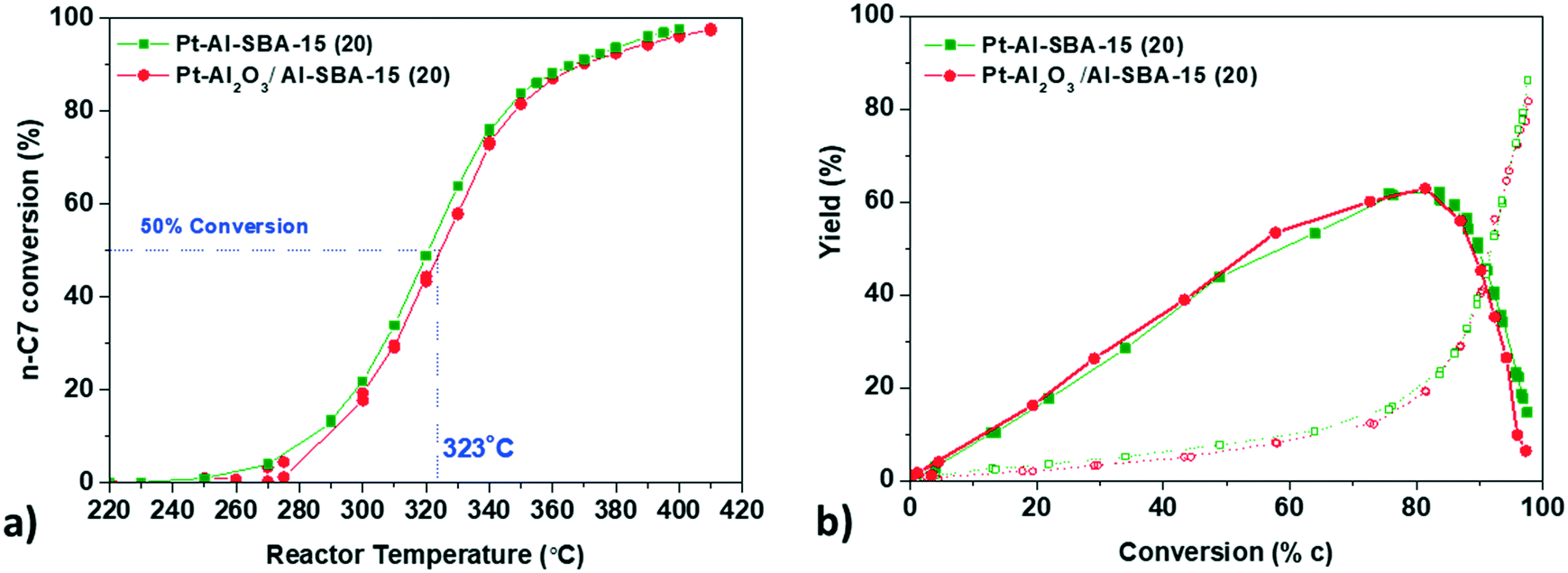 | ||
| Fig. 3 a. Conversion profile of n-heptane against temperature for Pt–Al-SBA-15 (green squares), Pt-γ-Al2O3/Al-SBA-15 (red circles) b. yield of C7 isomers (full symbols) and cracking products (open symbols). Catalysis performed at 10 bar; H2/n-C7 = 10/1; WHSV = 2.3 gn-C7 gsolid acid h−1. | ||
| Sample | WHSV (gC7 gsolid acid−1 h−1) | Temperature (°C) | n-C7 conversion | Maximum total isomers yield (%) | Mono-branched isomers yield (%) | Multi-branched isomers yield (%) | Ratio 2MC6/3MC6a |
|---|---|---|---|---|---|---|---|
| Pt/Al-SBA-15(20) | 2.3 | 345 | 80.5 | 62.2 | 54.2 | 8.0 | 0.82 |
| Pt γ-Al2O3/Al-SBA-15(20) | 2.3 | 345 | 81.2 | 62.5 | 54.5 | 8.0 | 0.82 |
| Sample | WHSV (gC16 gsolid acid−1 h−1) | Temperature (°C) | n-C16 conversion | Maximum total isomers yield (%) | Cracked products (%) | C4/C12b | C5/C11b |
|---|---|---|---|---|---|---|---|
| a Ratio between the yield of 2-methyl-hexane and 3-methyl-hexane. b C4/C12 and C5/C11 molar ratio of cracking products. | |||||||
| Pt/Al-SBA-15(20) | 3.4 | 300 | 88.4 | 71.1 | 17.3 | 1.3 | 1.4 |
| Pt γ-Al2O3/Al-SBA-15(20) | 3.4 | 300 | 85.4 | 75.6 | 13.6 | 1.5 | 1.4 |
Pt–Al-SBA-15(20) and Pt-γ-Al2O3/Al-SBA-15(20) show very similar activity and maximum selectivity towards C7 isomers, with a maximum of isomer yield around 60% at 340 °C and a similar cracking product yield. The catalyst Pt–Al-SBA-15(20)/γ-Al2O3, with a non-selective deposition of Pt both on the Al-SBA-15 and on the binder exhibits the same isomer yield as well (Fig. SI.9a†).
The intimacy effect was explored again using the Siralox based catalysts, with both Pt/Siralox and Pt-γ-Al2O3/Siralox showing similar catalytic performance (see Fig. SI.10†). No influence of the intimacy could be observed, with no effect of the variation of the metal–acid distance between nanoscale and closest intimacy. As the number of strong acid sites of Siralox is lower, a higher temperature is needed to reach similar conversion level of n-heptane (40 °C higher at 50% conversion than for Al-SBA-15(20)).
Locating Pt either on a γ-Al2O3 binder or inside the pores of a model mesoporous solid acid hence does not influence the performance of catalysts for the hydro-conversion of n-heptane. So far, only a few studies have been devoted to the specific topic of the effect of intimacy for n-heptane hydro-isomerization. Interestingly, our findings differ from the results reported earlier by Samad et al.22 They observed differences in activity and selectivity between Pt/Siralox catalysts when the Pt–acid site intimacy was manipulated by directing Pt deposition onto either the alumina-rich or silica-rich domains of the support (note that the two Pt precursors they used to achieve this selective deposition were the same as those used in this paper). The first possible explanation for the difference between these two findings is the difference in reaction conditions for the two studies: they used atmospheric pressure in their catalytic tests, while our assessment was performed at 10 bar, which is closer to relevant industrial conditions, and can influence the reactivity and selectivity of the catalysts. It has been demonstrated that for n-heptane isomerization, an increased operating and H2 pressure can promote the removal of olefins species as well as limit the coke formation.40,41 Therefore, higher overall isomer yield as well as higher stability of the catalysts can be expected under our operating pressure conditions, as it was indeed observed. The second possible explanation is that our approach employing an alumina binder, effectively and systematically located the Pt further than 100 nm from the acid sites, allowing us to study the intimacy effect at a nanometric scale with more certainty. In the case of Samad et al., deposition of Pt by the two precursors towards silica rich or alumina rich domains of the non-homogeneous Siralox does not guarantee a systematic variation of the Pt–acid distance.
4. n-Hexadecane hydro-hydrocracking studies catalytic studies
4.1 Pt–Silica–alumina catalysts
In order to gain understanding of the influence of diffusion limitations of intermediate species within mesoporous solid acids on the final performances of the catalyst, studies with a larger linear alkane with slower diffusion, n-hexadecane, were then conducted. The catalysts showed similar activity trends, albeit at considerably lower temperatures, as for the n-heptane conversion with a higher activity for Pt–Al-SBA-15(20), followed closely by Pt–Al-SBA-15(10) and Pt–Siralox (see Fig. 4). The yield towards iso-hexadecane (i-C16) over the catalysts depicted in Fig. 4b was high for each catalyst with a maximum total C16 isomer yield of 67.5% for Pt–Al-SBA-15(10), 71.1% for Pt–Al-SBA-15(20) and 80% for Pt–Siralox.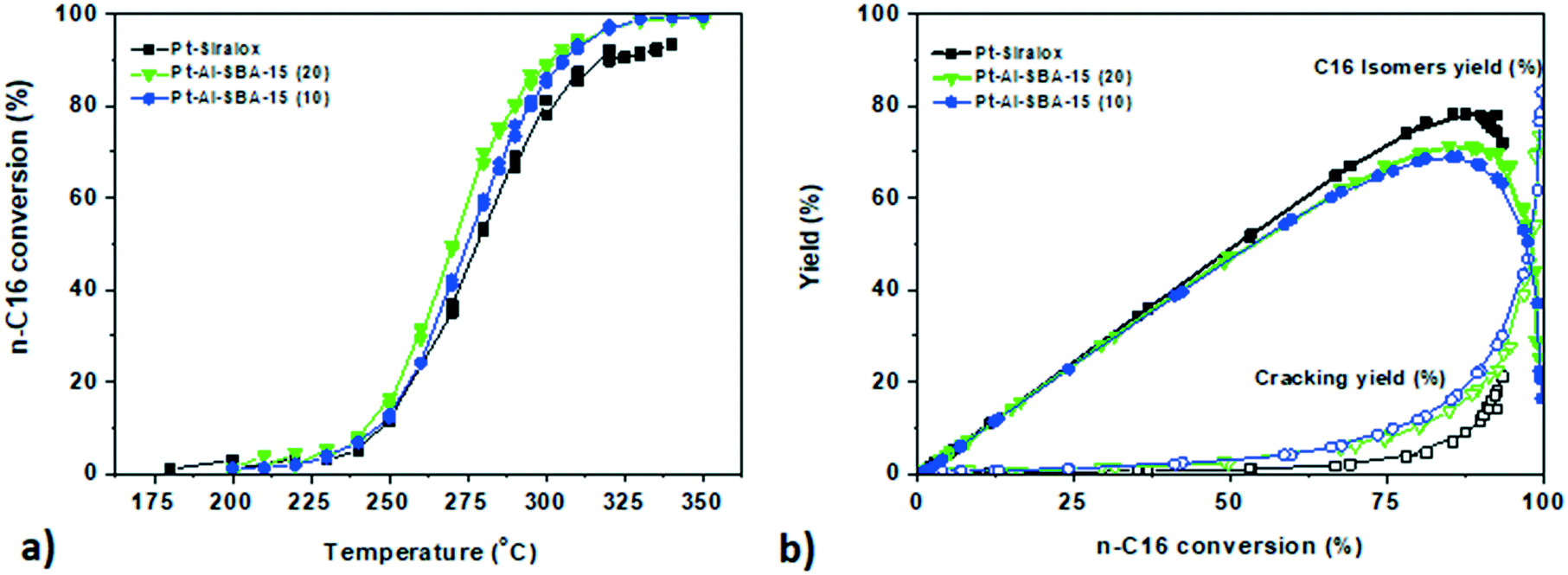 | ||
| Fig. 4 a. Conversion profile of n-hexadecane against temperature for Pt–Al-SBA-15(20) (green triangle); Pt–Al-SBA-15(10) (blue circle) and Pt-ASA (black square) b. yield of isomers of C16 against n-C16 conversion for the corresponding catalysts (full symbols) and cracking products (open symbols). Catalysis performed at 10 bar; H2/n-C16 = 10/1 mol/mol; WHSV = 1.7 gn-C16 gcatalyst h−1. | ||
4.2 Intimacy studies with Pt–Al-SBA-15 catalysts
The Pt catalysts with different degrees of intimacy between the metallic Pt sites and the acid sites were then assessed for n-hexadecane hydro-cracking. Once again, no significant difference on the activity and selectivity of the catalyst was observed. Both catalysts exhibit a steep conversion curve with at 50% conversion occurring at 270 °C and a maximum C16 isomer yield of 75% for Pt-γ-Al2O3/Siralox and 71% for Pt–Al-SBA-15(20) (see Fig. 5 and Table 3). The cracking products distribution for both catalyst is comparable as well with a molar ratio of C4/C12 and C5/C11 close to 1, is pointing to limited secondary cracking5 (see Fig. SI.12†). In passing, we note that these results support that the Pt precursor used (PTA versus CPA) does not affect the results, in other words, residual chlorine does not induce noticeable effects neither for n-heptane nor for n-hexadecane conversion (see Fig. SI.9†).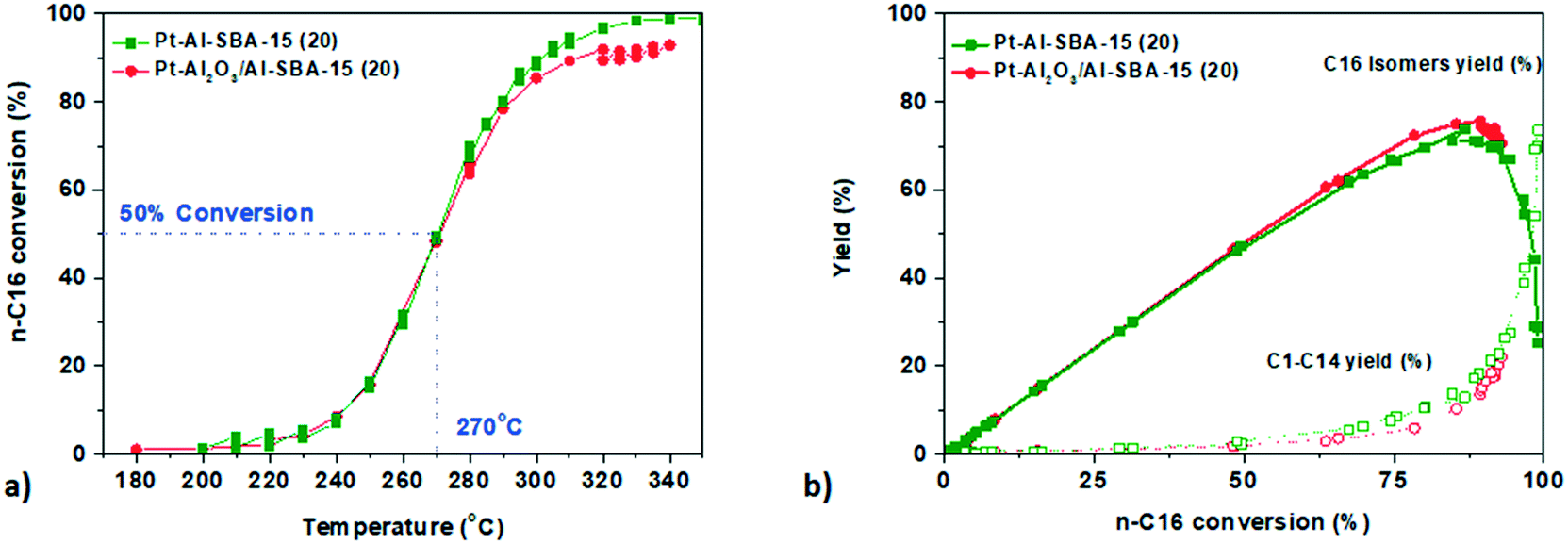 | ||
| Fig. 5 a. Conversion profile of n-hexadecane against temperature for Pt–Al-SBA-15 (green squares), Pt-γ-Al2O3/Al-SBA-15 (red circles) b. yield of C16 isomers (full line) and cracking products (dotted line). Catalysis performed at 10 bar; H2/n-C16 = 10/1 mol/mol; WHSV = 1.7 gn-C16 gsolid acid h−1. | ||
The temperature for 50% conversion of n-hexadecane is lower than for n-heptane (270 °C compared to 323 °C). This can be explained by the higher heat of adsorption of n-hexadecane. The calculated apparent activation energy (see Fig. SI.13†) was decreasing from Eaapp = 178.0 kJ mol−1 for n-heptane conversion to Eaapp = 157.1 kJ mol−1 for n-hexadecane conversion for the same Pt–Al-SBA-15(20) catalyst, in line with literature values.42,43
5. Conclusions
Catalysts with Pt deposited either inside the pores of a mesoporous solid acid (Al-SBA-15 or Siralox) or on a γ-alumina binder were prepared. The distance between the metal and acid function in those catalysts varied from closest (≤10 nm) to nanoscale (≥100 nm) intimacy. These catalysts were studied for the hydro-isomerization of n-heptane and hydro-cracking of n-hexadecane. For these mesoporous acid catalysts intimacy from closest to nanoscale did not affect the performance of the catalyst for the hydro-conversion of alkanes, as the conversion of both n-heptane and of larger and bulkier n-hexadecane led to similar maximum isomer yield. In other words, the Pt location on amorphous silica–alumina acid exhibited no significant influence on catalysis. Clearly, Pt on mesoporous silica–alumina differs from Pt on or in zeolites for which Pt in nanoscale intimacy with the acid sites improved isomer selectivity. In the case of microporous solid acid, when Pt is located inside the zeolite micropores, the reactants can be constrained inside of the porosity of the zeolite and undergo secondary cracking reactions. Therefore, it may be argued that diffusion limitations in the micropores of the zeolites occur and that pore-mouth catalysis on the zeolites surface might be crucial, while that is not the case for mesoporous supports. Finally, we note that most likely the nature of the Pt precursor used (PTA versus CPA) and thus residual chlorine did not affect the catalysis and, therefore, the conclusions from this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Remco Dalebout and Miguel Rivera-Torrente are acknowledged for BET measurements. BP plc is acknowledged for funding, and Prof. Glenn Sunley and Dr. Tegan Roberts (both BP plc) are thanked for fruitful discussions.Notes and references
- J. Weitkamp, ChemCatChem, 2012, 4, 292–306 CrossRef CAS.
- Y. Bi, G. Xia, W. Huang and H. Nie, RSC Adv., 2015, 5, 99201–99206 RSC.
- M. Guisnet, Catal. Today, 2013, 218–219, 123–134 CrossRef CAS.
- L. B. Galperin, S. A. Bradley and T. M. Mezza, Appl. Catal., A, 2001, 209, 257–268 CrossRef CAS.
- N. Batalha, L. Pinard, C. Bouchy, E. Guillon and M. Guisnet, J. Catal., 2013, 307, 122–131 CrossRef CAS.
- M. Guisnet, F. Alvarez, G. Giannetto and G. Perot, Catal. Today, 1987, 1, 415–433 CrossRef CAS.
- J. W. Thybaut, C. S. Laxmi Narasimhan, J. F. Denayer, G. V. Baron, P. A. Jacobs, J. A. Martens and G. B. Marin, Ind. Eng. Chem. Res., 2005, 44, 5159–5169 CrossRef CAS.
- J. Zečević, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 CrossRef PubMed.
- P. B. Weisz and E. W. Swegler, Science, 1957, 126, 31–32 CrossRef CAS PubMed.
- P. B. Weisz, Adv. Catal., 1962, 13, 137–190 CAS.
- P. S. F. Mendes, J. M. Silva, M. F. Ribeiro, A. Daudin and C. Bouchy, J. Ind. Eng. Chem., 2018, 72–83 CrossRef CAS.
- P. S. F. Mendes, A. F. C. Gregório, A. Daudin, C. Bouchy, J. M. Silva and M. F. Ribeiro, Catal. Commun., 2017, 89, 152–155 CrossRef CAS.
- O. Ben Moussa, L. Tinat, X. Jin, W. Baaziz, O. Durupthy, C. Sayag and J. Blanchard, ACS Catal., 2018, 8, 6071–6078 CrossRef CAS.
- G. Lv, C. Wang, K. Chi, H. Liu, P. Wang, H. Ma, W. Qu and Z. Tian, Catal. Today, 2018, 316, 43–50 CrossRef CAS.
- E. G. Acebo, C. Leroux, C. Chizallet, Y. Schuurman and C. Bouchy, ACS Catal., 2018, 8, 6035–6046 CrossRef.
- H. Du, S. Chen, H. Wang and J. Lu, Chin. J. Catal., 2017, 38, 1237–1244 CrossRef CAS.
- J. Kärger, M. Goepel and R. Gläser, Nanotechnology in catalysis Diffusion in nanocatalysis, 2017 Search PubMed.
- A. de Klerk and E. Furimsky, Catalysis in the Refining of Fischer – Tropsch Syncrude, 2010 Search PubMed.
- V. Akhmedov and S. Al-Khowaiter, Catal. Rev.: Sci. Eng., 2007, 49, 33–139 CrossRef CAS.
- V. Calemma, S. Peratello and C. Perego, Appl. Catal., A, 2000, 190, 207–218 CrossRef CAS.
- J. E. Samad, J. Blanchard, C. Sayag, C. Louis and J. R. Regalbuto, J. Catal., 2016, 342, 213–225 CrossRef CAS.
- J. E. Samad, J. Blanchard, C. Sayag, C. Louis and J. R. Regalbuto, J. Catal., 2016, 342, 203–212 CrossRef CAS.
- S. Zeng, J. Blanchard, M. Breysse, Y. Shi, X. Shu, H. Nie and D. Li, Microporous Mesoporous Mater., 2005, 85, 297–304 CrossRef CAS.
- E. de la Rochefoucauld, X. Carrier, J. M. Krafft and J. Blanchard, Stud. Surf. Sci. Catal., 2006, 162, 13–20 CrossRef CAS.
- S. Singh, R. Kumar, H. D. Setiabudi, S. Nanda and D. N. Vo, Appl. Catal., A, 2018, 559, 57–74 CrossRef CAS.
- S. Handjani, S. Dzwigaj, J. Blanchard, E. Marceau, J. M. Krafft and M. Che, Top. Catal., 2009, 52, 334–343 CrossRef CAS.
- S. Handjani, E. Marceau, J. Blanchard, J. M. Krafft, M. Che, P. Mäki-Arvela, N. Kumar, J. Wärn and D. Y. Murzin, J. Catal., 2011, 282, 228–236 CrossRef CAS.
- F. Leydier, C. Chizallet, A. Chaumonnot, M. Digne, E. Soyer, A. A. Quoineaud, D. Costa and P. Raybaud, J. Catal., 2011, 284, 215–229 CrossRef CAS.
- F. Leydier, C. Chizallet, D. Costa and P. Raybaud, J. Catal., 2015, 325, 35–47 CrossRef CAS.
- X. Chen, M. Jia, G. Liu, X. Zhang, L. Wang and Z. Mi, Appl. Surf. Sci., 2010, 256, 5856–5861 CrossRef CAS.
- K. Jaroszewska, A. Masalska, D. Czycz and J. Grzechowiak, Fuel Process. Technol., 2017, 167, 1–10 CrossRef CAS.
- C. Nie, L. Huang, D. Zhao and Q. Li, Catal. Lett., 2001, 71, 117 CrossRef CAS.
- H. W. Lee, J.-K. Jeon, K.-E. Jeong, S.-Y. Jeong, J. Han, B. Seo, S. H. Joo and Y.-K. Park, J. Nanosci. Nanotechnol., 2013, 13, 6074–6078 CrossRef CAS PubMed.
- G. Noh, Z. Shi, S. I. Zones and E. Iglesia, J. Catal., 2018, 368, 389–410 CrossRef CAS.
- H. I. Lee, J. H. M. Kim, G. D. Stucky, Y. Shi, C. Pak and J. H. M. Kim, J. Mater. Chem., 2010, 20, 8483–8487 RSC.
- M. Baca, E. de la Rochefoucauld, E. Ambroise, J. M. Krafft, R. Hajjar, P. P. Man, X. Carrier and J. Blanchard, Microporous Mesoporous Mater., 2008, 110, 232–241 CrossRef CAS.
- W. Daniell, U. Schubert, R. Glöckler, A. Meyer, K. Noweck and H. Knözinger, Appl. Catal., A, 2000, 196, 247–260 CrossRef CAS.
- M. Schreier and J. R. Regalbuto, J. Catal., 2004, 225, 190–202 CrossRef CAS.
- L. Jiao and J. R. Regalbuto, J. Catal., 2008, 260, 342–350 CrossRef CAS.
- K. J. Chao, C. C. Lin, C. H. Lin, H. C. Wu, C. W. Tseng and S. H. Chen, Appl. Catal., A, 2000, 203, 211–220 CrossRef CAS.
- J. M. Campelo, F. Lafont and J. M. Marinas, J. Catal., 1995, 156, 11–18 CrossRef CAS.
- F. Regali, M. Boutonnet and S. Järås, Catal. Today, 2013, 214, 12–18 CrossRef CAS.
- G. Kinger, D. Majda and H. Vinek, Appl. Catal., A, 2002, 225, 301–312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy02510c |
| This journal is © The Royal Society of Chemistry 2020 |