
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of metastable phases of 2D transition metal dichalcogenides
Maria S.
Sokolikova
 and
Cecilia
Mattevi
*
and
Cecilia
Mattevi
*
Department of Materials, Imperial College London, London SW7 2AZ, UK. E-mail: c.mattevi@imperial.ac.uk
First published on 26th May 2020
Abstract
The different polymorphic phases of transition metal dichalcogenides (TMDs) have attracted enormous interest in the last decade. The metastable metallic and small band gap phases of group VI TMDs displayed leading performance for electrocatalytic hydrogen evolution, high volumetric capacitance and some of them exhibit large gap quantum spin Hall (QSH) insulating behaviour. Metastable 1T(1T′) phases require higher formation energy, as compared to the thermodynamically stable 2H phase, thus in standard chemical vapour deposition and vapour transport processes the materials normally grow in the 2H phases. Only destabilization of their 2H phase via external means, such as charge transfer or high electric field, allows the conversion of the crystal structure into the 1T(1T′) phase. Bottom-up synthesis of materials in the 1T(1T′) phases in measurable quantities would broaden their prospective applications and practical utilization. There is an emerging evidence that some of these 1T(1T′) phases can be directly synthesized via bottom-up vapour- and liquid-phase methods. This review will provide an overview of the synthesis strategies which have been designed to achieve the crystal phase control in TMDs, and the chemical mechanisms that can drive the synthesis of metastable phases. We will provide a critical comparison between growth pathways in vapour- and liquid-phase synthesis techniques. Morphological and chemical characteristics of synthesized materials will be described along with their ability to act as electrocatalysts for the hydrogen evolution reaction from water. Phase stability and reversibility will be discussed and new potential applications will be introduced. This review aims at providing insights into the fundamental understanding of the favourable synthetic conditions for the stabilization of metastable TMD crystals and at stimulating future advancements in the field of large-scale synthesis of materials with crystal phase control.
 Maria S. Sokolikova | Maria Sokolikova received her MSc degree in Chemistry from Lomonosov Moscow State University in 2013. She then joined Dr Mattevi research group at Imperial College London as a PhD student holding the President's Scholarship. Maria completed her PhD in Materials Science from Imperial College London in 2020. Presently, Maria is a postdoctoral researcher in the Department of Materials at Imperial College London and her research focuses on colloidal growth of 2D materials for electrocatalysis. |
 Cecilia Mattevi | Dr Cecilia Mattevi received her Laurea degree in Materials Science from the University of Padua and a PhD in Materials Science from the University of Padua in 2008. After a postdoctoral appointment at Rutgers University, Dr Mattevi joined the Materials Department at Imperial College London, becoming a Junior Research Fellow in 2010 and then a Lecturer and Royal Society University Research Fellow in 2012. Cecilia is a Fellow of the Royal Society of Chemistry and her research interest includes the liquid phase synthesis of 2D materials with tailored characteristics and the investigation of their properties for energy applications. |
Introduction
The isolation of graphene has sparked widespread interest in layered materials. A large family of those materials is represented by the transition metal dichalcogenides which comprise about 40 compounds with a chemical formula of ME2 (M, transition metal; E, chalcogen).1 While they have been studied for over half a century in their bulk form2,3 for their photoelectrocatalytic activity,4–6 lubricity7 to superconductivity,8,9 it was not until recently that they have been isolated in their elementary building blocks, which are triatomic layers, and several of their properties started to emerge and their potential identified. These emerging 2D materials offer new opportunities for the realization of novel technologies and for the exploration of novel condensed matter phenomena.Interestingly, compounds of group IV, V, VI, VII and X are nearly isostructural with three possible crystallographic orders in the monolayer form,10 and they present several analogies. Group VI TMDs have been studied more extensively than others and in particular the Se and S compounds (MoS2, MoSe2, WS2 and WSe2) owing to the richness and the uniqueness of the properties concerning each of their polymorphs. Additionally, the relative abundance of transition metal compounds, the good chemical stability in air compared to the dichalcogenides of other groups metals, make them more accessible. Group VI TMDs started to attract large interest a decade ago owing to the ability to display an indirect-to-direct bandgap crossover with bright photoluminescence (PL) in the monolayer form at room temperature11 and for the valley degree of freedom associated to the non-centrosymmetric semiconducting phase.12 Additional unique properties to this class of materials include mechanical flexibility, electrostatic coupling, large carrier mobility, gate-tunability, piezoelectricity in monolayers and odd number of layers, as discussed in recent reviews.13–16
It is now emerging that there is a variety of other properties associated to different polymorphs of group VI TMDs; some of those arising from the different crystallographic phases that they can acquire. In addition to the thermodynamically stable 2H polymorph with the trigonal-prismatic coordination of transition metal atoms, there are various polymorphs that are broadly defined as 1T phases (perfect 1T and distorted 1T′, 1T′′ and 1T′′′) and where the metal atoms present an octahedral (or distorted octahedral) coordination. These phases are generally characterized by a metal- (1T) or semimetal-like (1T′) behaviour.
Indeed, the electronic band structure is heavily affected by the coordination of transition metal atoms which leads to a different splitting of the d-orbitals. The key opportunity arising from the TMD polymorphism is to influence the electronic structure of a given material and so to achieve a high density of electronic states at the Fermi level, which can enable efficient electrocatalytic hydrogen evolution from water. This ability was initially attributed to the localized metallic states on molybdenum-terminated edges of 2H MoS2 clusters.17 Soon after, it became clear that the basal planes of the 1T phase MoS2 or WS2 could enable accessibility to a large number of electrocatalytically active sites thus approaching Pt performance.18
Several 1T′ polymorphs have been demonstrated since. The 1T′ phases of single layer WSe2 and WTe2 have been reported to be large gap quantum spin Hall (QSH) insulators suitable for application in spintronic devices and in the case of WSe2 also operable at ambient temperature.19–21 Their chemical stability makes them more advantageous of the currently known large gap QSH insulators such as stanene22 and two-dimensional In–Sb compounds which can be used in an inert atmosphere only.23 On the other hand, it is predicted that a spontaneous symmetry breaking in the undistorted metallic 1T phase would open up a band gap and lead to the emergence of robust ferroelectricity in the novel 1T′′′ phase.24
Despite the rapid progress in investigating the properties of 1T(1T′) phases of group VI TMDs over a small scale (about hundreds of nanometres over the basal plane), it is still quite arduous to obtain those phases as continuous over large areas. The 1T(1T′) phases are metastable, thus their direct synthesis is very challenging because the formation energy of the 1T′ phase is higher than that of the 2H phase;25 therefore, the 2H ME2 is the preferable phase obtained.
The most common way, and historically the first one to be used, for achieving those 1T(1T′) phases is via conversion of the 2H phase. This is generally achieved by increasing the electron density via exposure to alkali metals.26,27 While the design of direct synthesis methods for the 1T(1T′) phases is still at the early stages of investigation. Here, we provide a comprehensive overview of topochemical methods and direct synthesis methods designed so far to obtain the 1T(1T′) phases of group VI TMDs. We will discuss the chemical prerequisites for the formation of the 1T(1T′) phases in vapour phase and liquid phase. We will present the possible reaction pathways that can take place in liquid phase which lead to the nucleation and growth of crystalline metastable phases.
We will then review the intrinsic properties of 1T(1T′) phases and we highlight the differences in the materials morphology and properties between 1T(1T′) phases obtained via destabilization of 2H phase versus direct synthetic approaches. Finally, we will introduce newly emerging properties which are unique to the 1T(1T′) phases as they arise from the lower symmetry. We will conclude the review with an outlook for the future of the different synthetic approaches to metastable phases in view of their applications and possible large-scale production of these materials.
1. Structural polymorphs of group VI TMDs
TMDs exhibit a variety of structural polymorphs. The polymorphs are determined by the very different chalcogen coordination geometry that transition metal atoms can accommodate, while the polytypes are determined by the stacking order of two or more individual triatomic layers with the same symmetry.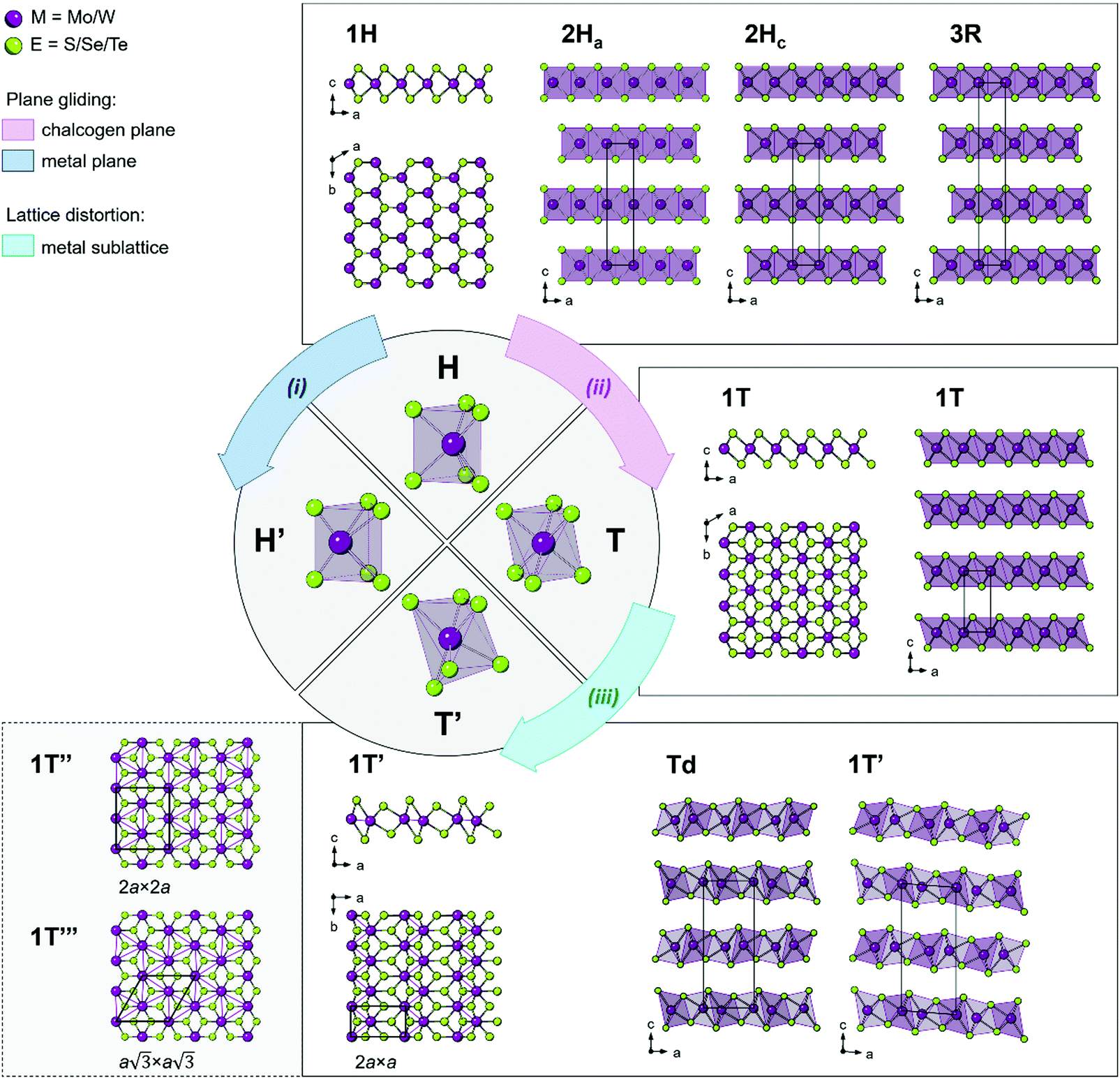 | ||
| Fig. 1 Bulk polymorphs of group VI TMDs. In the schematics, the arrows indicate the pathways for possible structural transformation (i) from the 1H to the 1H′ phase, (ii) from the 1H to the 1T phase, and (iii) from the 1T to the 1T′ phase via either (i) metal plane gliding, (ii) chalcogen plane gliding, or (iii) spontaneous distortion in the metal sublattice. The crystallographic projections of the 1H, 1T, and 1T′ monolayers along the b (side view) and c (top view) axes are shown. The known polytypes (stacking polymorphs) of the 1H, 1T, and 1T′ structures are presented in the corresponding fields. Bottom left panel illustrates two additional clustering patterns – tetramerization (1T′′) and trimerization (1T′′′) – proposed in the distorted 1T TMDs. | ||
Two types of structural transitions in the 1H-type TMD monolayers associated with the atomic planes gliding within one triatomic layer are assumed to give rise to two distinct polymorphs: 1H′ and 1T42 (Fig. 1, paths i and ii). The transversal displacement of the transition metal plane could produce the distorted 2H phase, which is referred to as the 2H′ phase and is considered as a 60° rotational phase of the 2H structure42 (Fig. 1, path i). Experimentally, the formation of a new distorted 2H phase, 2Hd, was observed in the few-layered 2H MoTe2 and WxMo1−xTe2 when an external electric field was applied.43 The phase was found to be unstable and is considered to be a transitional structural state between the semiconducting 2Hc and the semimetallic 1T′ (or Td) phases discussed below. The structure of the mentioned 2Hd state was not inferred at this stage.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1), with the metal centres of two adjacent layers located above each other, is reported in literature.44
m1), with the metal centres of two adjacent layers located above each other, is reported in literature.44
The perfect octahedral 1T structure of group VI disulphides and diselenides is typically higher in energy by 700–900 meV per formula unit than the trigonal-prismatic 1H structure,45 which renders this phase metastable.25,46 It is prone to spontaneous structural distortions, caused by charge density waves (CDW) which lead to clustering of transition metal atoms and buckling of chalcogen planes to accommodate these metal atom displacements47,48 (Fig. 1, path iii). Different distorted 1T structures of chemically exfoliated MoS2 and WS2 have been proposed in literature back in 1990s.47 The most common superstructures include the zigzag chain (2a × a) formation, tetramerization (2a × 2a), and trimerization 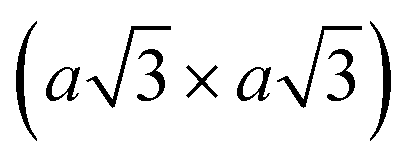 of transition metal atoms in the ab plane as shown in Fig. 1. The former one is widely referred to as the 1T′ polymorph and was established by scanning tunnelling microscopy (STM) studies of chemically exfoliated MoS2 and WS2 single layers47,49–51 (Fig. 1). The three-dimensional arrangement of the 1T′ single layers can lead to the formation of two polytypes: the orthorhombic Td phase and the monoclinic 1T′ phase (Fig. 1). Additionally, tetramerization and trimerization within the transition metal sublattice would result in the 1T′′ and 1T′′′ phases, respectively.52 According to first-principles calculations, the semimetallic 1T′ phase of group VI TMDs monolayers generally possesses the lowest formation energy among the competing distorted octahedral phases.45,53 However, a spontaneous trimerization of transition metal atoms, involving only a slight displacement from the equilibrium positions, is found to be favourable in 1T MoS2 monolayers, leading to the formation of the semiconducting 1T′′′ phase.45,54 Such distinct distortion patterns in 1T group VI TMD monolayers are in good agreement with the early predictions that weak distortions (trimerization) are to be expected in the systems with short M–E bonds; whereas, strong distortions (zigzag chain formation) are characteristic to the systems with long M–E bonds.55 It has also been predicted that the spontaneous symmetry breaking in 1T group VI TMD monolayers would lead to the emergence of robust ferroelectricity in the 1T′′′ phase, with the electrical dipoles perpendicular to the ab plane.24,54 Recently, the preparation of 1T′′′ MoS2 single crystals (space group P31m) by deintercalation of KMoS2 crystals has been reported.56 Unlike the metallic 1T and semimetallic 1T′ phases, bulk 1T′′′ MoS2 was reported to be a semiconductor with an indirect band gap of 0.65 eV. Although the 1T′′′ phase was predicted to be formed only in MoS2 monolayers, the small energy difference between the 1T′ and 1T′′′ monolayers of WS2 and MoSe2 (36.5 and 57.8 meV per formula unit, respectively) suggests that these metastable phases might be obtained under favourable synthesis conditions.54
of transition metal atoms in the ab plane as shown in Fig. 1. The former one is widely referred to as the 1T′ polymorph and was established by scanning tunnelling microscopy (STM) studies of chemically exfoliated MoS2 and WS2 single layers47,49–51 (Fig. 1). The three-dimensional arrangement of the 1T′ single layers can lead to the formation of two polytypes: the orthorhombic Td phase and the monoclinic 1T′ phase (Fig. 1). Additionally, tetramerization and trimerization within the transition metal sublattice would result in the 1T′′ and 1T′′′ phases, respectively.52 According to first-principles calculations, the semimetallic 1T′ phase of group VI TMDs monolayers generally possesses the lowest formation energy among the competing distorted octahedral phases.45,53 However, a spontaneous trimerization of transition metal atoms, involving only a slight displacement from the equilibrium positions, is found to be favourable in 1T MoS2 monolayers, leading to the formation of the semiconducting 1T′′′ phase.45,54 Such distinct distortion patterns in 1T group VI TMD monolayers are in good agreement with the early predictions that weak distortions (trimerization) are to be expected in the systems with short M–E bonds; whereas, strong distortions (zigzag chain formation) are characteristic to the systems with long M–E bonds.55 It has also been predicted that the spontaneous symmetry breaking in 1T group VI TMD monolayers would lead to the emergence of robust ferroelectricity in the 1T′′′ phase, with the electrical dipoles perpendicular to the ab plane.24,54 Recently, the preparation of 1T′′′ MoS2 single crystals (space group P31m) by deintercalation of KMoS2 crystals has been reported.56 Unlike the metallic 1T and semimetallic 1T′ phases, bulk 1T′′′ MoS2 was reported to be a semiconductor with an indirect band gap of 0.65 eV. Although the 1T′′′ phase was predicted to be formed only in MoS2 monolayers, the small energy difference between the 1T′ and 1T′′′ monolayers of WS2 and MoSe2 (36.5 and 57.8 meV per formula unit, respectively) suggests that these metastable phases might be obtained under favourable synthesis conditions.54
It is important to note that for group VI disulphides and diselenides, the 1T′ polymorph is metastable under ambient conditions and can only be obtained under specific experimental conditions. On the contrary, bulk WTe2 forms the orthorhombic Td polymorph (space group Pnm21).57 Bulk MoTe2 is normally obtained in the 2H polymorph (α-phase); however, the monoclinic 1T′ MoTe2 polymorph (space group P21/m) exists at temperatures above 900 °C (β-phase).57,58 Due to a very small energy difference between the 2H and 1T′ polymorphs of MoTe2 (40 meV per formula unit25), both polymorphs are reported almost equally probable depending on the synthesis conditions.59,60 The orthorhombic Td polymorph (γ-phase) of MoTe2 can be obtained by cooling the monoclinic 1T′ (β-) phase down to cryogenic temperatures61–64 or it can be stabilized at room temperature in the presence of substitutional doping with tungsten.65 On the other hand, the Td polymorph of WTe2 can be transformed into the lower symmetry 1T′ phase at pressure over 15.5 GPa.66,67 Due to a large energy difference between the 2H and 1T′ phases, the metastable group VI diselenides and disulphides are challenging to obtain in a pure form and the reports on their successful synthesis are limited.21,68–71 The crystal structure of the monoclinic 1T′ phase of group VI diselenides and disulphides is still debated with the possible space groups being either P21/m or C2/m; the latter has only been reported recently and is referred to as the 2M phase, where M stands for the monoclinic lattice system.72 Unlike the semiconducting 2H counterparts, the corresponding 1T′ and Td phases of group VI TMDs are semimetallic.61 Furthermore, the non-centrosymmetric Td polymorph exhibits topologically nontrivial surface electronic states,73,74 and bulk Td phases of WTe2 and MoTe2 have been extensively investigated as potential candidates for the observation of 3D Weyl fermions.64,75–77 Recently, single layers of group VI TMDs with the 1T′ symmetry have been predicted to host topologically protected edge states,46,78 and large QSH gaps have been verified experimentally in monolayer 1T′ WTe2,19 1T′ WSe2,20,21 and 1T′ MoS2.79
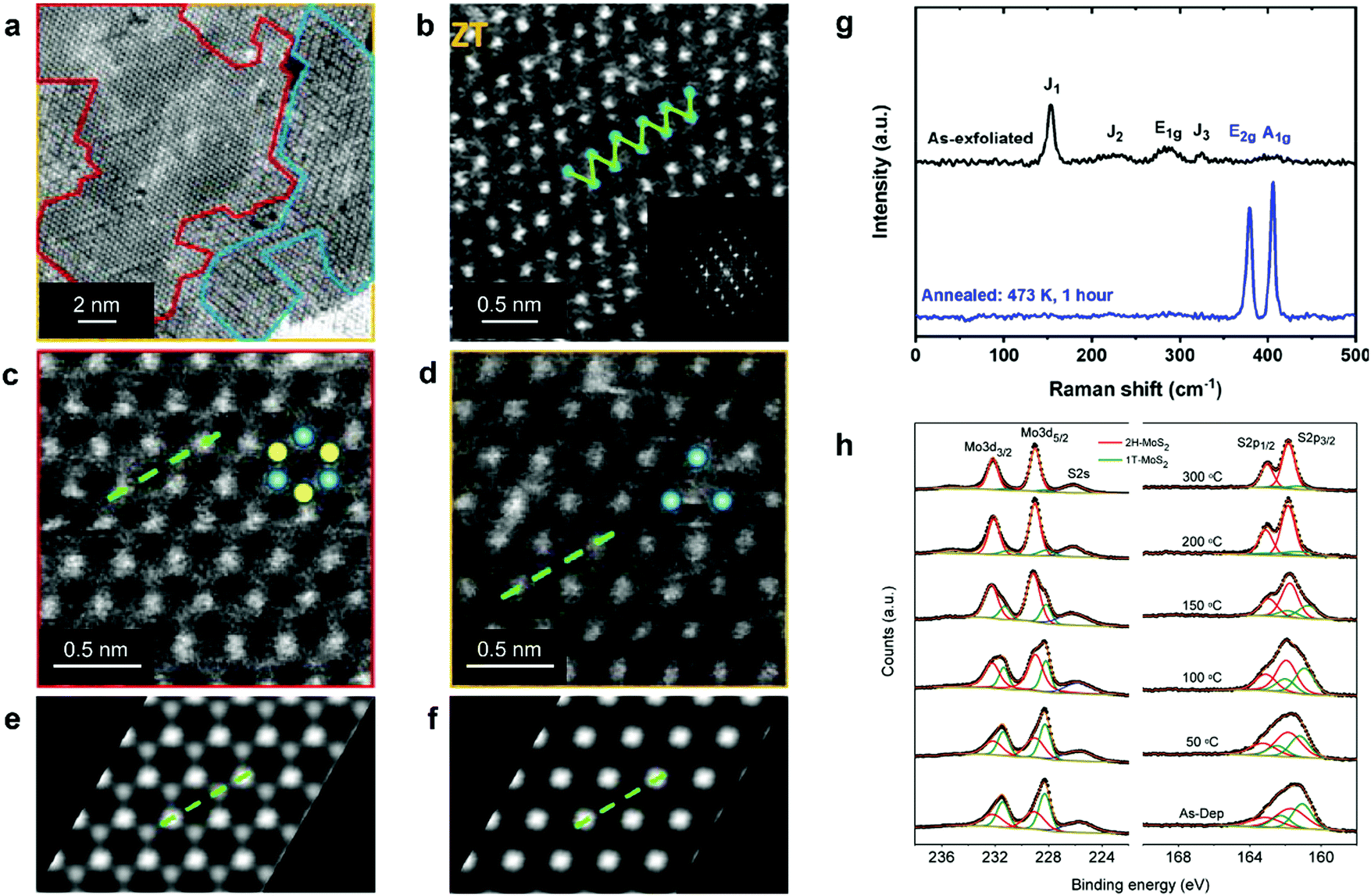 | ||
| Fig. 2 Distinguishing the 1H, 1T, and 1T′ polymorphs. (a) STEM image of chemically exfoliated MoS2 displaying co-existing 1H, 1T, and 1T′ phases; the corresponding areas are enclosed by red, yellow, and blue curves, respectively. (b–d) Representative HAADF STEM images of the 1T′, 1H, and 1T MoS2 lattice, respectively. (e and f) Simulated HAADF STEM images of the ideal 1H and 1T MoS2 monolayers, respectively. (g) Raman spectra of chemically exfoliated 1T′ (top) and annealed 2H (bottom) MoS2 flakes, illustrating the characteristic vibrational modes of these two phases. (h) XPS spectra of the Mo 3d, S 2s and S 2p core level electrons of mixed-phase 1T/2H MoS2 nanosheets, demonstrating the gradual recovery of the 2H phase during thermal annealing. The 1T and 2H phase contributions are given in green and red, respectively. Separate panels are reproduced with permission: (a and c–f) from ref. 85, copyright 2012, American Chemical Society; (b) from ref. 51, copyright 2016, American Chemical Society; (g) from ref. 263, copyright 2019, American Chemical Society; and (h) from ref. 264, copyright 2011, American Chemical Society. | ||
Spectroscopic techniques, such as Raman spectroscopy and X-ray photoemission spectroscopy (XPS), offer a relatively accurate way of detecting co-existing polymorphs in TMD nanostructures over micron-sized areas. In the original work by J. Sandoval et al., it has been reported that the octahedral 1T phase of chemically exfoliated MoS2 displayed two Raman-active modes, Eg and A1g, at 287 and 408 cm−1, respectively, while the E12g mode of the 2H phase (at 383 cm−1) was absent.87 In addition to those, three previously unassigned modes, J1, J2, and J3, at 156, 226, and 333 cm−1, respectively, were found (Fig. 2g). These modes were attributed to the 2a × a superstructure in the octahedral phase. The wider set of Raman-active modes of the 1T′ and Td phases is indicative of their lower symmetry as compared to the 2H phase and is usually reported for WTe2 and MoTe2.62,88–90 The 1T′ signature J1, J2, and J3, modes were also observed in the 1T′ MoS2, WS2, MoSe2, and WSe2 single crystals68,91 and few-layered nanosheets.71,86,92,93 It should be noted that the J1 mode consistently exhibits the highest intensity in Raman spectra of nearly phase-pure 1T′ group VI disulphides.86,92,93 Further, the overall Raman signal intensity is significantly lower in the 1T(1T′) phase TMDs than in the 2H samples of equivalent thickness as has been experimentally demonstrated in the case of MoS294–96 and WSe2 nanosheets.71,97
In XPS spectra, both the 1T and 1T′ phases are reported to present peaks systematically shifted to lower binding energies as compared to the 2H phase, however, XPS is not sensitive to the differences between the 1T and 1T′ phases98 (Fig. 2h). In chemically exfoliated TMDs, the shift to lower binding energies is typically ascribed to partial reduction of transition metal centres during alkali metal intercalation.91
2. Access to the metastable crystal phases of group VI TMDs
The metastable crystal phases can be either accessed through destabilisation of the thermodynamically stable polymorphs, for instance, due to electron transfer, or synthesised directly, when the growth conditions are optimised in such a way that the metastable product is kinetically preferred.2.1.1. Chemical treatment. The first reports on the metallic 1T(1T′) group VI TMD polymorphs date back to the pioneering works by R. Frindt et al. on chemical exfoliation of these materials27,50 (Fig. 3a). Chemical exfoliation of layered materials typically proceeds in two steps.47,50,99 At first, bulk TMD powders are intercalated with alkali metals. The most commonly used intercalants include n-BuLi,83,100,101t-BuLi,102 and LiBH4.50,103 The subsequent hydrolysis of the intercalated alkali metal cations leads to the generation of hydrogen gas bubbles that split the individual layers apart. The exfoliation can be facilitated by gentle shaking or sonication. It has been demonstrated that upon intercalation of alkali metal species between the layers of bulk crystals of group VI TMDs, the crystal phase would change from the 2H to 1T. It has been suggested that the mechanism of this structural transition should be imputed to the electron transfer from the intercalated alkali metal species leading to an increased electron count on the transition metal d-orbitals.47 Specifically, for the case of MoS2 monolayers, it has been shown that the injection of 4 electrons per unit cell allows lowering the 2H-to-1T transition barrier from 1.59 to only 0.27 eV.48 Thus, upon this electron transfer (chemical reduction), the octahedral coordination of transition metal becomes favoured over the trigonal-prismatic and thus the 2H-to-1T structural phase transition occurs.104 In the absence of intercalated species, the 2H-to-1T phase transition due to accumulation of negative charges was also observed in Re doped MoS2 monolayers under continuous electron beam scanning during STEM imaging.42 The transition occurs through the initial formation of numerous non-parallel molybdenum zigzag chains which is followed by chalcogen plane gliding, reducing mechanical strain and forming a domain of the trigonal 1T phase.42
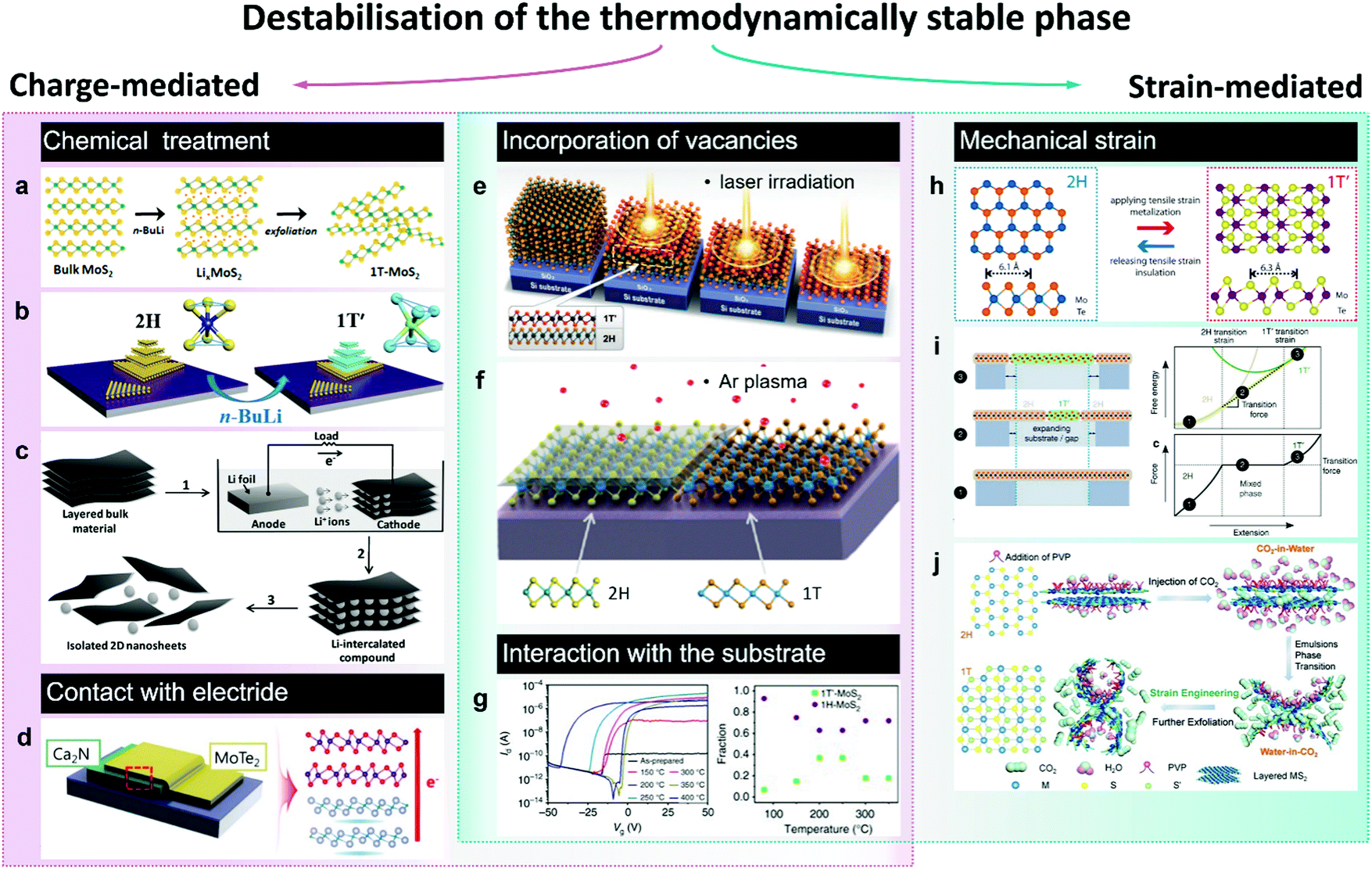 | ||
| Fig. 3 Accessing the metastable 1T(1T′) phase via destabilisation of the thermodynamically stable (2H) phase. The approaches to induce the 2H-to-1T′ transition in group VI TMDs are divided in two categories: charge-mediated, as chemical treatment (panels a–c) and contact with an electride (panel d); and strain-mediated, such as tensile strain-induced transition between the 2H and 1T′(1T) polymorphs (panels h–j). Panels e–g represent the phase transitions caused by the combination of charge and strain as in the case of chalcogen vacancies (panels e and f) or due to the interaction with the substrate (panel g). Separate panels are adapted with permission: (a) from ref. 100, copyright 2017, American Chemical Society; (b) from ref. 114, copyright 2018, American Chemical Society; (c) from ref. 108, copyright 2011, John Wiley & Sons; (d) from ref. 123, copyright 2017, American Chemical Society; (e) from ref. 155, copyright 2015, American Association for the Advancement of Science; (f) from ref. 138, copyright 2017, American Chemical Society; (g) from ref. 128, copyright 2017, Springer Nature; (h) from ref. 130, copyright 2016, American Chemical Society; (i) from ref. 25, copyright 2014, Springer Nature; and (j) from ref. 134, copyright 2017, John Wiley & Sons. | ||
Generally, the 1T(1T′) phases produced via chemical exfoliation approach are attained as small crystalline (within the range 1–10 nm, Fig. 4) domains often embedded into the 2H matrix85 (Fig. 2a and 4c). Patches of both 1T and 1T′ phases are found in chemically exfoliated MoS2 nanosheets.49,85,101 Whereas a characteristic distortion in the ab plane of chemically exfoliated WS2 indicates the crystal phase to be 1T′,47,51 although the resulting crystal phase is commonly denoted as 1T.83 The ratio between the converted 1T(1T′) and pristine 2H phases strongly depends on the type and concentration of reducing agent (alkali metal) and the TMD material.102 The maximum amount of 1T(1T′) phases achievable via chemical exfoliation approach is reported to be ∼70–80% of the overall material92,101 (typically, estimated from by XPS data, Fig. 4). The metastable 1T(1T′) phases obtained by chemical exfoliation are reported to gradually decay into the 2H polymorph over time; this process is considerably promoted in the restacked films once the stabilising ions have been removed during drying.49,87,105 A full conversion to the 2H phase occurs if the material is subjected to high temperatures, such as ∼100 °C for MoS227 and ∼200 °C for WS2.50
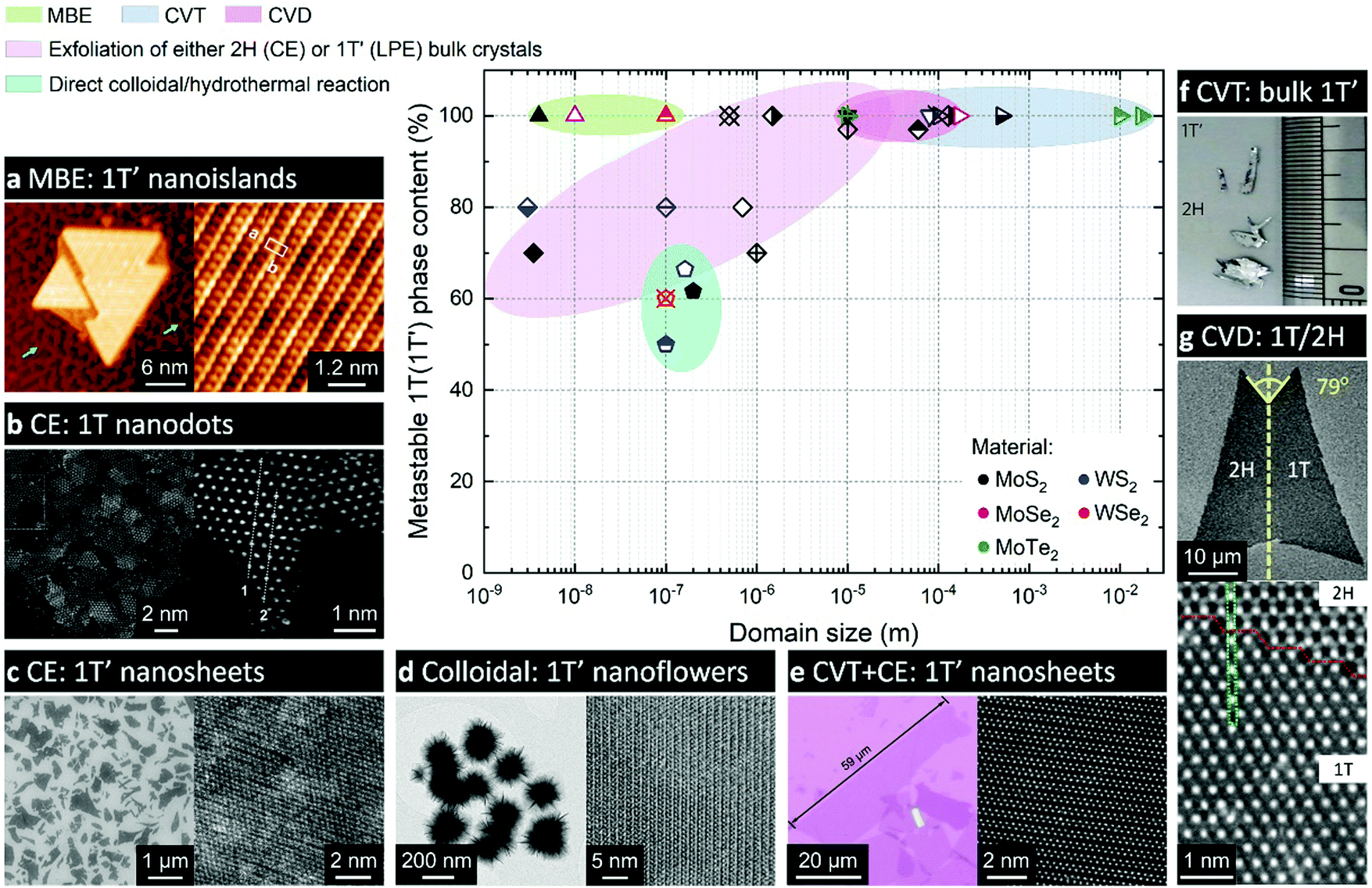 | ||
| Fig. 4 Comparison of various experimental approaches to produce the metastable 1T(1T′) phases of group VI TMDs. The approaches are grouped into the following categories: molecular beam epitaxy (MBE); chemical vapour transport (CVT) growth; chemical vapour deposition (CVD); chemical (CE) or liquid-phase (LPE) exfoliation of bulk crystals; and direct synthesis approaches, such as colloidal and hydrothermal growth. The experimental data are taken from the following articles: ref. 21, 56, 58, 68–71, 79–81, 83, 93, 101, 103, 112, 144, 147, 148, 153, 162 and 164. Separate panels are adapted with permission: (a) from ref. 70, copyright 2019, American Chemical Society; (b) from ref. 112, copyright 2018, John Wiley & Sons; (c) from ref. 103, copyright 2013, American Chemical Society; (d) from ref. 71, copyright 2019, Springer Nature; (e) from ref. 69, copyright 2019, John Wiley & Sons; (f) from ref. 153, copyright 2015, Springer Nature; and (g) from ref. 164, copyright 2018, American Chemical Society. | ||
Recently, it has been demonstrated that the lifetime of the 1T(1T′) phases in intercalated few-layered MoS2 flakes can be considerably extended via the hydrogenation of intercalated lithium species.86 Results of DFT calculations suggest that LiH is a good electron donor that stabilises the 1T′ phase in multi-layered flakes for over 3 months. Although LiH, encapsulated between the TMD layers, is stable, the surface bound lithium hydride can easily convert into Li2CO3 and Li3N, what makes this approach unsuitable for stabilisation of the 1T′ phase in chemically exfoliated single layers.
A morphological aspect should be also taken into consideration as the non-uniform distribution of intercalated lithium hinders the control over the dimensions and distribution of the converted 1T(1T′) patches. The intercalation kinetics affects the conversion efficiency and the lateral dimensions of the patches of metastable phase.106,107 A uniform lithiation has been reported by an electrochemical intercalation process where Li foil serves as an anode and bulk TMD material as a cathode108 (Fig. 3c). After lithium insertion, lithiated powders are exfoliated by mild sonication in water producing predominantly monolayered nanosheets with lateral dimensions of up to 1 μm. Moreover, it has been demonstrated that the lithium migration within the van der Waals (vdW) gaps of lithiated few-layered material can be controlled by an external electric field so that a reversible 2H-to-1T′ phase transformation can be uniformly extended over micron-sized areas.109 Such controllable phase switching has been demonstrated in lithiated MoS2 thin films, which have been used as neuromorphic memory devices,109 and in ultra-thin (∼2–5 nm) MoS2 nanosheets upon a reversible electrochemical lithiation.110 Another method to achieve a uniform intercalation in group VI TMDs has been recently reported by J. Zheng et al.111 Performed prior to intercalation, the expansion of TMD powders by reacting with hydrazine under hydrothermal conditions has facilitated the alkali metal naphthalenide insertion. This allowed producing single layer (80%) MoS2 sheets of up to 400 μm2 in size; the metastable 1T(1T′) phase content, however, was not specified.111
It is interesting to note that combined with ball-milling of 2H-phase bulk crystals, chemical (organolithium) treatment and exfoliation approach has recently been shown to produce MoS2, MoSe2, WS2, Mo0.5W0.5S2 and MoSSe nanodots with lateral sizes of 2–5 nm and a high (70%) content of the metastable 1T phase112 (Fig. 4b).
Phase conversion can also be achieved via a post-growth treatment of CVD grown 2H TMD few-layered flakes and films with solutions of n-BuLi in nonpolar solvents113,114 (Fig. 3b). Considering that CVD grown TMD flakes present much larger (tens of microns) lateral sizes compared to the ones of chemically exfoliated flakes (submicron for monolayers), this could allow for the production of larger continuous domains of the metastable 1T(1T′) phases. The n-BuLi treatment of polycrystalline films of 2H–MoS2 and 2H–MoSe2 grown on Si/SiO2 wafers led to a partial conversion into the metallic 1T phase, with an overall conversion efficiency of ∼50%.113 Further, the lithium-induced structural transformations in the CVD grown 2H–MoS2 flakes appear to be thickness-dependent.114 The increased energy barrier of the 2H-to-1T phase transition in MoS2 monolayers implies much longer treatment time in order to achieve the phase conversion compared to the multi-layered counterparts.114 This, therefore, paves the way to the 2D metal–semiconductor junctions in CVD grown TMD devices by controlling the exposure to lithiating agent (n-BuLi). In the case of 2H-WSe2 monolayers, a complete conversion into the metallic 1T phase has been reported and the deposition of a PMMA mask has allowed for the patterning of 2H/1T heterojunctions.97 While a complete conversion to the 1T(1T′) phases via lithium intercalation of bulk MoSe2 and WSe2 powders have not been achieved.102,115 Finally, the 2H-to-1T phase conversion has been demonstrated to be induced also by exposure to the vapours of electron donating compounds, such as butylamine and trimethylamine, as was shown in the cases of MoS2 and MoSe2.116 It has been proposed that this approach can be potentially employed to fabricate new passive sensing devices based on detecting the abrupt changes in material conductivity in the presence of strong donor analytes, such as nerve gas.
2.1.2. Electrostatic gating. The dynamical control of the structural phase transitions in TMD monolayers by an applied electric field can be potentially interesting for application in light-weight flexible electronic devices, including non-volatile memory devices.117 Bias-voltage driven T-to-H crystal phase transformations have been previously demonstrated in TaS2 crystals.118 Y. Li et al. have proposed that the semiconductor-to-semimetal phase transition can be achieved in some group VI TMDs using an electrostatic gating.119 It has been found that the transition between the 2H and 1T′ phases can be driven by excess electric charge in the monolayer. Thus, the excess charge densities less than −0.04e or greater than 0.09e per formula unit would suffice to induce the 2H-to-1T′ transformation in an undoped MoTe2 monolayer; however, to achieve this transition in lighter TMD monolayers, such as MoS2 and WS2, much greater excess charge densities are needed. Alternatively, a constant voltage of −1.6 or 4.4 V is required to induce the semiconductor-to-semimetal transition in monolayer MoTe2 on HfO2 in the electrostatic gating device configuration.119 A reversible 2H-to-1T′ phase transition was achieved in MoTe2 by ionic liquid gating in a field-effect transistor configuration.120 The required gate voltages could be substantially reduced by varying the chemical composition, such as by alloying119,121 or forming Janus monolayers.122
2.1.3. Electron doping by contacting with an electron reservoir. Long-range crystal lattice modulation can be achieved by contacting the 2H TMD (MoTe2) with a single crystalline 2D electride as has been demonstrated in ref. 123 (Fig. 3d). Electrides are a class of inorganic compounds in which electrons serve as anions.124 Layered electrides containing two-dimensionally confined anionic electrons are generally characterised by exceptionally high charge carrier concentrations (1.4 × 1022 cm−3) and high electron mobilities at room temperature (160 cm2 V−1 s−1).125 Similarly to van der Waals solids, layered electrides can be exfoliated into 2D sheets.126 Unlike the chemical treatment discussed earlier, electron transfer from the electride allows for achieving higher levels of doping (about 1014 cm−2). Such high doping densities can also be achieved by ionic modulation, however, contacting with electride is effective over larger distances from the contact interface. For instance, forming a vertical heterostructure between [Ca2N]+·e− and few-layered MoTe2 has led to a uniform 2H-to-1T′ phase transformation over a micron sized area even as far away as 100 nm from the electride.123
A reversible 2H-to-1T phase transition has been demonstrated in MoS2 monolayers by an injection of hot electrons generated by plasmon excitation in Au nanospheres.127 Beyond that, electron injection from gold substrate was reported to cause the 1H-to-1T′ phase transition in MoS2 single layers during thermal annealing leading to the formation of co-existing domains of 1H and 1T′ phases; this electron-driven transition was facilitated by the interfacial strain128 (Fig. 3g). The 1T′ phase content reached its maximum (37%) at the annealing temperature about 200–250 °C.
2.1.4. Applying mechanical strain. According to density functional calculations, group VI TMDs are predicted to undergo a transition from the 2H to the 1T′ phase under tensile strain.25 It is identified that a 0.3–3% uniaxial strain applied along the b (armchair) direction of 2H MoTe2 monolayers allows for the transition into the 1T′ phase at room temperature25 (Fig. 3i). The strain-induced 2H-to-1T′ phase transition in MoTe2 monolayers is presumed to occur due to simultaneous transition metal and tellurium atoms displacement and does not proceed through an intermediate 1T state.129 The process involves a distortion of the transition metal sublattice accompanied by a simultaneous gliding and rippling of chalcogen plane.129 Experimentally, a reversible strain-induced 2H-to-1T′ phase transition has been demonstrated in 20 nm thick MoTe2 films by applying a tensile strain using an AFM tip130 (Fig. 3h). Notably, such a phase transition was realised even at room temperature if a small tensile strain of 0.2% was applied.
Among the emerging approaches to induce the 2H-to-1T phase transformation in group VI TMDs, we would like also to mention the microwave plasma treatment131,132 and the supercritical CO2 treatment.133,134 These less conventional approaches allow for a controllable phase engineering also in the liquid phase. Recently, X. Tong et al. have discovered that during the supercritical CO2 treatment, aqueous suspensions of TMD nanosheets transform into water-in-CO2 emulsions, causing a strain-induced 2H-to-1T reconstruction in WS2 and MoS2 flakes confined at the convex water/CO2 interface134 (Fig. 3j). The metastable 1T phase content achieved by this approach is reported to vary from 70%133 to 90%.134 Similar to supercritical CO2 treatment, mild microwave plasma treatment has been reported to introduce the 1T phase domains in the exfoliated 2H MoS2 nanosheets.132 C. Sharma et al. have used forming gas microwave plasma to convert exfoliated 2H MoS2 flakes into the 1T phase with a 70% efficiency. The phase transition is assumed to proceed due to the momentum transfer from the plasma ions to the MoS2 lattice, causing the chalcogen plane gliding.131
2.1.5. The effect of chalcogen vacancies. Another type of electron donors are the chalcogen vacancies formed in TMD nanosheets. The presence of chalcogen vacancies plays a key role in promoting the 2H-to-1T phase transition by a dual mechanism: generation of extra charges and weakening of the metal–chalcogen bonds. Both facilitate the structural transformation.135 Specifically, in MoS2, the difference in the formation energy between the 2H and 1T phase decreases almost to zero when the vacancy concentration reaches 8 at%.136 Acting as point defects, the vacancies also serve as nucleation sites of the 1T′ phase being formed in the 2H matrix.
Local 2H-to-1T′ transition occurring in mechanically exfoliated few-layered MoTe2 flakes under laser irradiation is commonly attributed to the formation of Te vacancies due to local heating137 (Fig. 3e). This transition is irreversible; however, the 2H phase can be restored by high temperature annealing in tellurium atmosphere.137
In the case of MoS2, it has been shown that the chalcogen vacancies can be introduced into the material by gentle Ar plasma treatment138 (Fig. 3f). The phase transformation was demonstrated to occur in the vicinity of single point defects and the lateral sizes of the 1T′ MoS2 domains were typically about a few nm.138 Combined with shadow mask technique, lateral 2H/1T′ homojunctions, including 1D conducting 1T′ channels, were patterned by Ar plasma treatment in MoS2 monolayers.139 A controlled laser beam 2H/1T′ patterning has also been performed in few-layered MoS2 flakes.140 However, Ar plasma treatment might be preferable for the phase patterning on TMD monolayers since this method is considered less disruptive compared to laser beam irradiation.139 Other methods of introducing chalcogen vacancies in MoS2, as for example harsh electrochemical etching, to cause the 2H-to-1T structural transformation are just emerging.135 In their recent work, X. Gan et al. have demonstrated that the 1T phase content in multi-layered MoS2 can be varied by either increasing the etching potential or the reaction time, although the spatial distribution of the metastable phase is not possible to control.135
2.2.1. The role of a substrate in a crystal phase-selective growth. The use of engineered substrates has been found beneficial for growing the metastable phases of materials with large energy difference between the stable and metastable polymorphs (550 and 330 meV per formula unit in MoS2 and MoSe2, respectively25). Nanometre-sized 1T′ MoSe2 islands were selectively formed on Au(111) substrates which were treated with selenium prior to the deposition, whereas only the thermodynamically stable 1H polymorph was obtained if the layer of selenium was not pre-deposited70 (Fig. 4a). Such a difference in the resulting crystal phase was attributed to the formation of an electron donating Mo layer between the Au surface and the pre-sputtered Se overlayer that facilitate the growth of the 1T′ phase. MBE deposition of 1T′ MoS2 nanoclusters (30–50 atoms) on Au(111) substrates has also been reported.79 It has been also hypothesized that the gold substrate itself serves as an electron reservoir that stabilised the nucleating 1T′ phase.
Thermal expansion coefficients of various substrates might be another important growth parameter to consider when the formation of a metastable phase is targeted. Typically, the formation of metastable polymorphs of MoS2 does not occur in a direct chemical vapour deposition (CVD) reaction due to the huge energy barrier for the 1H-to-1T phase transition. Recently, Z. Wang et al. reported the direct growth of the 1T–1H bi-phase TMD monolayers, in which the formation of the metastable phase is associated with the in-plane thermal strain imposed by the substrate during the cooling step.141
2.2.2. Synthesis from electron donating precursors. High phase-purity mm-sized 1T and 1T′ phase MoS2 single crystals have been demonstrated through a delicate deintercalation of Li or K ions from LiMoS2 and KMoS2 compounds.27,69,142,143 Ternary KMoS2 is typically produced by reacting the corresponding alkali metal molybdate, K2MoO4, with dry H2S that is then followed by a mild reduction in H2/N2 atmosphere27,142 (Fig. 5, reactions i and ii). Alternatively, LiMoS2 and KMoS2 can be obtained by fusing MoS2 with either Li2S or K2S under vacuum in the presence of metallic Mo56,143,144 (Fig. 5, reaction iii). The deintercalation of these alkali metal thiomolybdate precursors is reported to proceed in two stages. At first, the ternary compounds are exposed to water to hydrate the alkali metal ions intercalated between MoS2 layers143 (Fig. 5, reaction iv):
| LiMoS2 + H2O = Li1−x(H2O)yMoS2 + xLiOH + x/2H2. |
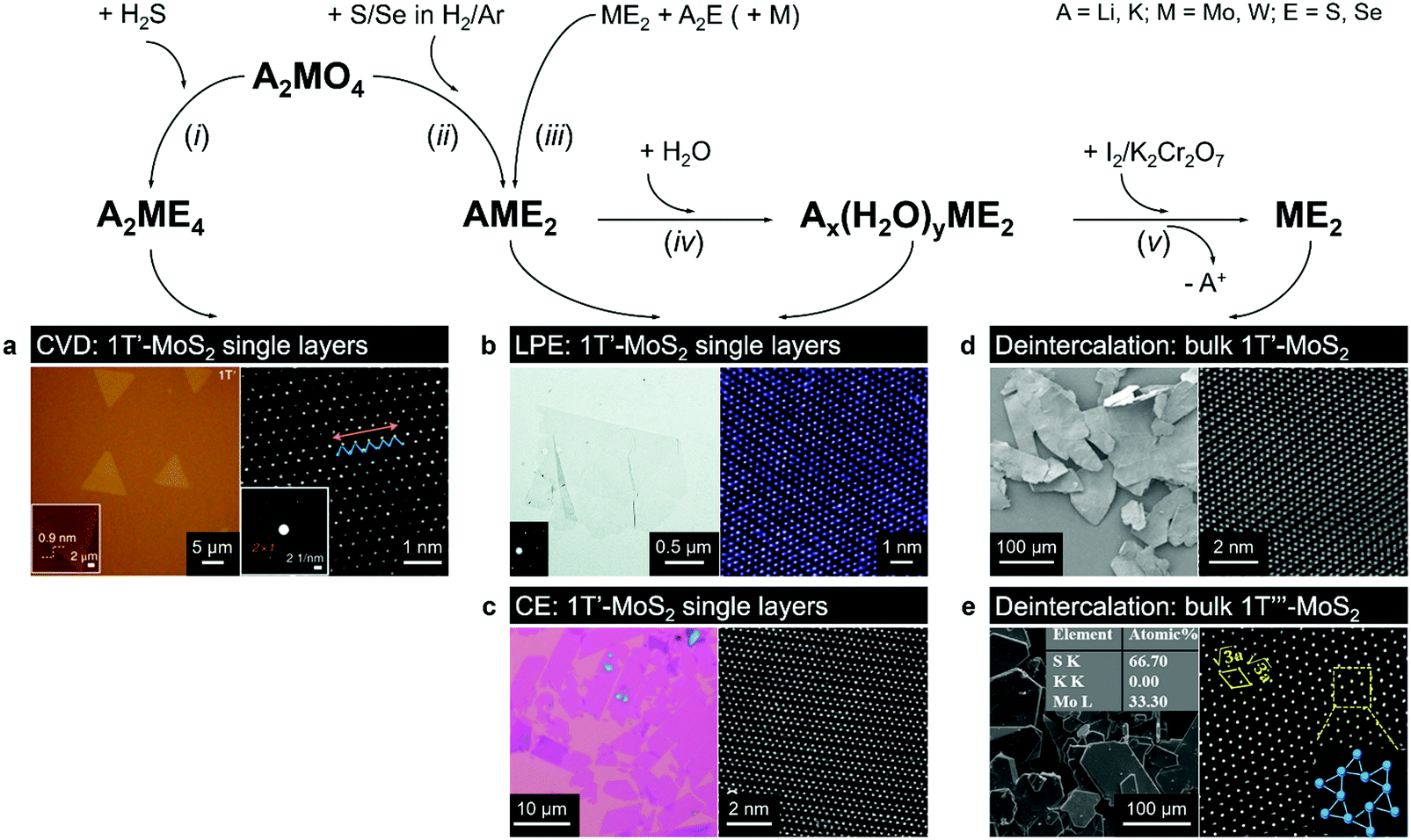 | ||
| Fig. 5 Direct vapour-phase (CVT, CVD) growth of the 1T(1T′) polymorphs from alkali metal containing precursors. Bulk 1T′ crystals can be produced by gentle deintercalation of bulk intercalation compounds Ax(H2O)yME2 (panels d and e); 1T′ monolayers can be obtained by chemical (CE, panel c) or liquid-phase (LPE, panel b) exfoliation of bulk intercalation compounds Ax(H2O)yME2 and AME2; high-quality 1T′ monolayers can be grown by CVD method from A2ME4 precursor (panel a). Separate panels are adapted with permission: (a) from ref. 147, copyright 2018, Springer Nature; (b) from ref. 144, copyright 2017, The Royal Society of Chemistry; (c) from ref. 69, copyright 2019, John Wiley & Sons; (d) from ref. 68, copyright 2018, Springer Nature; and (e) from ref. 56, copyright 2019, American Chemical Society. | ||
This leads to the lattice expansion in the c direction. It is assumed that the increased interlayer distance facilitates the alkali metal ions removal owing to the reduced interaction with the chalcogen atoms of basal planes.143 The alkali metal is removed by a treatment with a mild oxidiser, such as I227,69 (Fig. 5, reaction v):
| Li1−x(H2O)yMoS2 + (1 − x)/2I2 = MoS2 + (1 − x)LiI + H2O. |
Using lithiated compounds might be preferable since smaller ionic radius of lithium leads to its more effective hydration and complete removal from the interlayer gaps, while the deintercalation of larger potassium ions is believed to cause numerous lattice defects.142,145 It has been reported that the structural disorder in the intercalation compounds of MoS2 is externally stabilised by hydrated ions trapped between the layers.146 Thus, K0.7MoS2, similarly to LixMoS2 (x ≤ 1), typically demonstrates the 2a × 2a superstructure,145 whereas the structure of the hydrated Kx(H2O)yMoS2, refined by diffraction methods (XRD, SAED) and STM imaging, is assigned to the monoclinic symmetry, similar to the one reported for WTe2 (2a × a).142,146 Depending on the starting composition of the intercalation compound, different distorted phases of MoS2 were demonstrated.146 Gentle deintercalation of Lix(H2O)yMoS2 with iodine solution was shown to result the high-quality 1T MoS2 single crystals.143 Distorted MoS2 structures produced by oxidation of Kx(H2O)yMoS2 strongly depend on the amount of intercalated potassium.146 An incomplete oxidation of Kx(H2O)yMoS2 (x ≈ 0.3) leads to the formation of MoS2 crystals with the 2a × a (1T′) structure, whereas the final product of a prolonged (complete) oxidation displays the 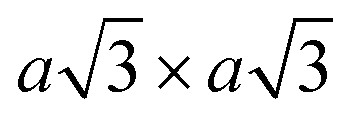 (1T′′′) superstructure in the ab plane, where, according to STM, Mo atoms are slightly displaced from their ideal positions forming triangles (trimerization).58,146 A similar 2a × a (1T′) structure was obtained by oxidation of Kx(H2O)yMoS2 (x < 0.3)146 implying a composition-dependant difference in the symmetry of these intercalation compounds. Additionally, the strength of the oxidiser is also reported to affect the crystal symmetry of deintercalated metastable MoS2. By using a mild oxidiser, like I2, to treat hydrated KMoS2 crystals, the 1T′ phase MoS2 crystals were synthesised.68 While by using strong oxidisers, K2Cr2O7 and Br2, it was possible to attain the 1T′′′ polymorph of MoS227,56,146 (Fig. 5e). It is also reported that intercalation compounds containing other alkali metals, such as sodium and rubidium, can be used as precursors in the synthesis of 1T′′′ MoS2 single crystals.56
(1T′′′) superstructure in the ab plane, where, according to STM, Mo atoms are slightly displaced from their ideal positions forming triangles (trimerization).58,146 A similar 2a × a (1T′) structure was obtained by oxidation of Kx(H2O)yMoS2 (x < 0.3)146 implying a composition-dependant difference in the symmetry of these intercalation compounds. Additionally, the strength of the oxidiser is also reported to affect the crystal symmetry of deintercalated metastable MoS2. By using a mild oxidiser, like I2, to treat hydrated KMoS2 crystals, the 1T′ phase MoS2 crystals were synthesised.68 While by using strong oxidisers, K2Cr2O7 and Br2, it was possible to attain the 1T′′′ polymorph of MoS227,56,146 (Fig. 5e). It is also reported that intercalation compounds containing other alkali metals, such as sodium and rubidium, can be used as precursors in the synthesis of 1T′′′ MoS2 single crystals.56
Bulk crystals of the 1T′ MoSe2 with lateral size ∼100 μm have been demonstrated through a similar deintercalation (with I2 solution) of a precursor obtained through a chemical reaction between alkali metal containing molybdenum precursors, such as potassium or sodium molybdate, and elemental selenium annealed in an inert atmosphere in an ampoule69 or in an open system in a reducing H2/Ar atmosphere68 (Fig. 5d). The role of alkali metal cations (Na, K) is similar to that of lithium in chemical exfoliation process; namely, being electron donors, these cations presumably stabilise the 1T′ phase over the 2H phase during the solid-state reaction of the precursor formation. To the best of our knowledge, a direct growth of bulk 1T′ WS2 and 1T′ WSe2 crystals has not been reported so far.
High-purity 1T′ monolayers can then be isolated from the parent bulk crystals either mechanically68 or via chemical exfoliation in solution.69 In the case of chemical exfoliation, an additional lithium intercalation stage of bulk 1T′ crystals is introduced followed by exfoliation in water. With such approach, the 1T′ phase content in the final product reaches 97%69 (Fig. 4e and 5c). A successful exfoliation of 1T′ MoS2 monolayers by gentle sonication and even shaking of the hydrated NaxMoS2 or LiMoS2 crystals immersed in water has also been demonstrated69,93,144 (Fig. 5b). As shown by C. Guo et al., the alkali metal cations can then be removed from the colloids of MoS2 monolayers by repeated dialysis.144 The reported sizes of thus exfoliated 1T′ MoS2 monolayers vary from a few μm144 to a few tens of μm69 in the case of agitation-assisted exfoliation and chemical exfoliation, respectively. Direct exfoliation of bulk 1T′ TMD crystals can effectively avoid an incomplete phase transition that is typically manifested as co-existing 2H/1T/1T′ phases in chemically exfoliated material.85 Moreover, this approach also allows protecting the in-plane framework of the 1T′ phase in TMD monolayers from fragmentation due to lithium intercalation thus potentially producing 1T′ domains with larger lateral dimensions.
A phase-selective CVD growth of MoS2 monolayers on mica based on using a similar type of precursor – potassium thiomolybdate, K2MoS4 – has been proposed recently147 (Fig. 5a). The authors found out that the crystallisation of the 1T′ phase of MoS2 becomes favourable in potassium-enriched CVD growth due to the stabilisation of the 1T′ phase in the intermediate KxMoS2 compound. The crystal phase control was achieved by changing the atmosphere from inert to reducing by adding H2 into the carrier gas. The MoS2 flakes produced by decomposition of K2MoS4 precursor in an inert (Ar) atmosphere display the 2H phase, whereas high-purity 1T′ MoS2 flakes were produced if H2 gas was introduced. Thus, 1T′ MoS2 monolayers with over 90% phase purity can be selectively grown on mica in the temperature range 650–750 °C and H2 concertation of approximately 5–12% in the carrier gas mixture (Fig. 4). The choice of a suitable substrate may also play an important role since mica may assist in trapping potassium cations produced during the precursor decomposition facilitating the formation of the 1T′ phase. Interestingly, 1T′/2H heterobilayers could be selectively produced by simply changing pure Ar to Ar/H2 mixture during the crystal growth. The authors have also demonstrated that Cs2MoS4 and Rb2MoS4 could be suitable for a phase-selective synthesis of 1T′ MoS2.147 Additionally, the growth of 1T′ WS2 under the same conditions was achieved if K2WS4 was used as an alkali metal containing precursor.147
Similarly to vapour deposition approaches, the reported protocols of hydro- or solvothermal syntheses of the metastable 1T(1T′) group VI TMD polymorphs utilise a reaction involving ammonium or alkali metal containing precursors. Typically, 1T(1T′) WS2 and MoS2 nanostructures are obtained by reacting ammonium containing salts, like (NH4)2WO4,148 (NH4)10W12O41·H2O,82 and (NH4)6Mo7O24,81 with the chalcogen precursor, such as thiourea or thioacetamide, or by thermal decomposition of ammonium thiomolybdate149 in solution phase (Fig. 6a). Thus produced material exhibits the morphology of ultra-thin nanosheets or nanoribbons composed of nm-sized co-existing 2H and 1T(1T′) single-crystalline domains. The 1T(1T′) phase content in these nanostructures reaches ∼61.6%81 (Fig. 4). The metastable phases are likely to be stabilised by charged precursor residues trapped between the layers of few-layered nanosheets and nanoribbons. Specifically, presence of the intercalated ammonium uniformly distributed across the nanosheets has been repeatedly verified by elemental mapping and XPS analysis.81,82,150 Apart from cationic species, it is also assumed that the 1T′ phase in MoS2 produced by a solvothermal reaction from (HN4)2MoS4 can be stabilised by intercalated anionic thiomolybdate moieties.149 To verify that, the complete removal of salt residues was achieved by prolonged dialysis and the absence of intercalated ammonium in the synthesised material has been demonstrated by XPS analysis. The solvothermal strategy has been further adapted to form arrays of the 1T′-rich (60%) nanosheets (MoS2) on conductive support (single wall carbon nanotubes) for the application in electrochemical devices.150
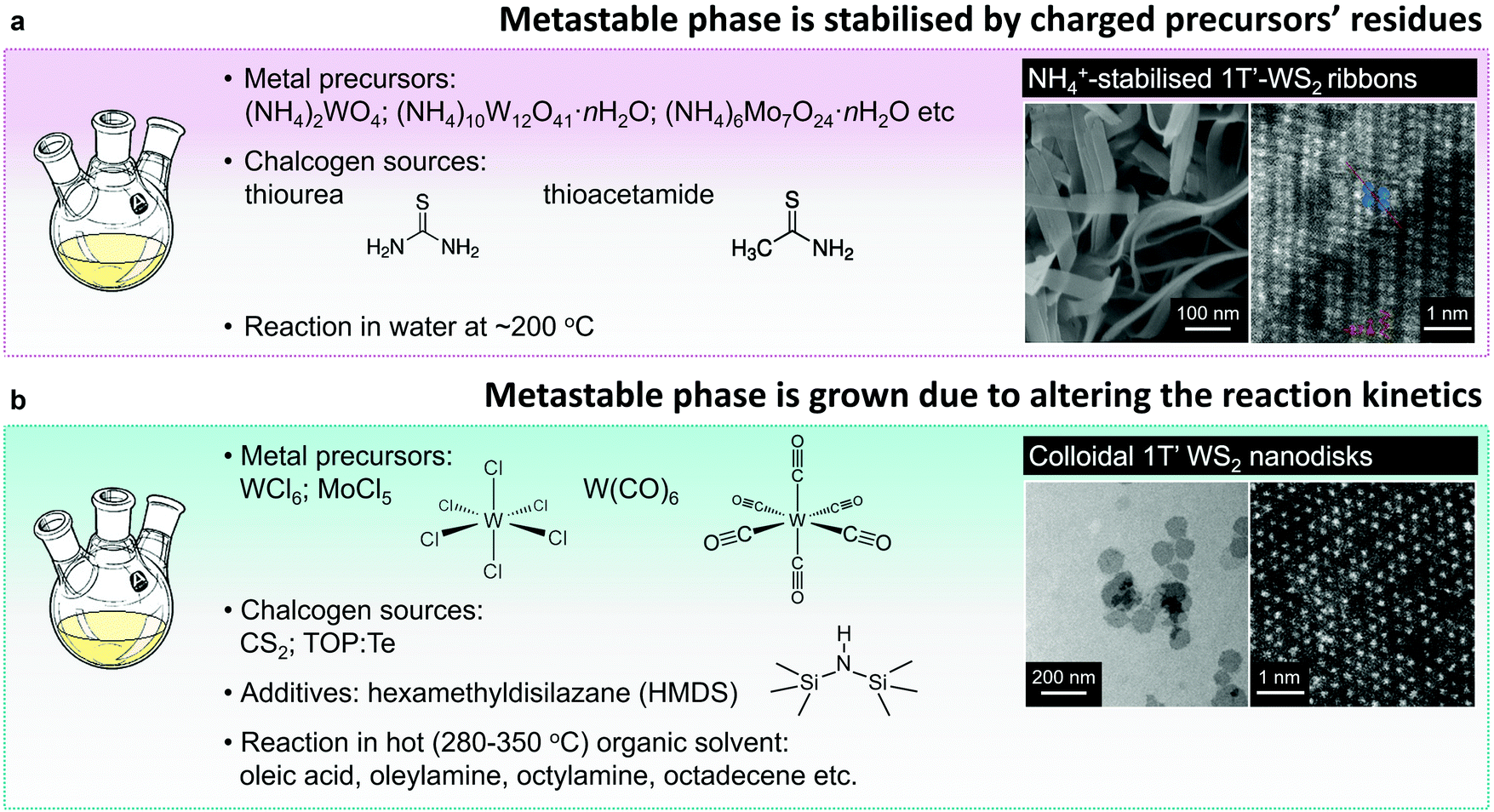 | ||
| Fig. 6 Direct solution-phase growth of the 1T′ polymorphs. (a) Hydro-/solvothermal reactions between the ammonium containing precursors produce the metastable 1T′ phase stabilised by precursors’ residues. (b) Colloidal synthesis of the metastable 1T′ phase achieved due to the fine control over the reaction kinetics. Figures in panels (a) and (b) are adapted with permission from ref. 82, copyright 2015, John Wiley & Sons, and ref. 80, copyright 2014, American Chemical Society. | ||
An interesting chemical approach to produce the 1T phase of TMDs that rely on the conversion of non-van der Waals solids, such as MAX and MXene phases, has been reported recently.151,152 In this approach, the transition metal atom coordination might be templated by the symmetry of a parent MAX phase. J. Tang et al. have demonstrated that high quality TiS2 sheets can be synthesised by a high temperature chalcogenation of Ti3C2Tx in H2S atmosphere.151 This approach has been further extended to produce group IV and VI disulphides and diselenides, and 1T phases of MoS2 and WS2, in particular.152 Interestingly, the metastable 1T phases have only been obtained in the presence of phosphorus vapour in the chalcogen atmosphere, leading to the formation of P-doped 1T/2H mixed-phase material.152
2.2.3. Reaction kinetics-driven direct synthesis of the metastable phase. Phase-selective synthesis through a fine tuning of the growth conditions is achievable in TMDs with a small energy difference between the thermodynamically stable and metastable polymorphs. MoTe2 displays the difference of only about 35–40 meV per formula unit between the 2H and 1T′ polymorphs,25,153 which is markedly different from the rest of group VI TMDs, and therefore is the most suitable candidate for developing the phase-selective growth strategies. The second candidate could be WTe2 with the difference between stable 1T′ and metastable 2H being only 90 meV per formula unit.25 However, the synthesis of the metastable 2H WTe2 has not been demonstrated so far.
In vapour phase deposition approaches, the crystal phase tunability has been demonstrated by finely controlling the reaction parameters. For instance, islands of 1T′ WSe2 extended over ∼100 nm have been selectively synthesised on bilayer graphene on 6H-SiC(0001) substrates by molecular beam epitaxy (MBE) by keeping the deposition temperature not higher than 280 °C20,21 (Fig. 4). In CVD approach, the crystal phase selectivity is often achieved by controlling the precursor flux. Thus, polycrystalline films of 1T′ MoTe2 with single crystalline domains of ∼10 nm are commonly reported through tellurisation of Mo films or Mo islands in a tellurium-deficient atmosphere.60,154 In this case, the formation of the metastable phase is attributed to tellurium deficiency in the produced MoTex material that lowers the free energy of 1T′ polymorph.154 In particular, Te deficiency greater than 2 at% suffices the stabilisation of the 1T′ phase over the 2H in MoTe2.25,155 The lateral dimensions of such phase-uniform polycrystalline 1T′ MoTe2 films are reported to reach a few cm2.59 Interestingly, prolonging the tellurisation time has facilitated a complete solid-to-solid 1T′-to-2H phase transformation; moreover, the formation of single crystalline 2H MoTe2 domains as large as 2.34 mm in diameter has been achieved by annealing in a tellurium-enriched atmosphere.154 Additionally, the formation of the metastable 1T′ phase by tellurisation of Mo films was attributed to a mechanical strain accumulated between the tellurised and pristine Mo regions.59 In such a case, during a prolonged tellurisation, gradual relaxation of the strain triggers structural transformation into the 2H phase.59 It has also been reported that either Mo or MoO3 films can be used as a precursor for the synthesis of few-layered 1T′ MoTe2.156 Regardless of the precursor choice, the 1T′ polymorph of MoTe2 was obtained if Te supply was insufficient; however, using MoO3 as precursor led to the formation of morphologically uniform, ∼3 nm thick films (film roughness ≈1 nm), whereas the roughness of the films produced from Mo precursor under the same conditions was ≈5 nm.156 The choice of molybdenum precursor may, however, plays an important role in the CVD synthesis under high tellurium flux. Thus, the same research group have found that MoO3 reacts more easily with Te vapour forming the 2H phase, while Mo or MoO3−x produce the 1T′ phase under the same conditions.157 Recently, L. Zhou et al. have demonstrated that planar 1T′/2H MoTe2 heterostructures can be produced by a one-step tellurisation of MoO3 films using a partially overlapped geometry.158 The formation of 2H phase is attributed to the trapping of Te gas in the gap between the substrate and the cover and therefore to higher concentration of Te residing over the surface of MoO3 film that led to more complete tellurisation; whereas uniform 1T′ phase is obtained in the exposed areas of the substrate. L. Yang et al. have demonstrated that the rate of MoO3 film tellurisation can be controlled by adjusting both the carrier gas flow rate and the reaction temperature; this has allowed the authors to identify the conditions for the selective growth of 1T′ and 2H MoTe2 few-layered films.159 Alternatively, lateral 1T′/2H homojunctions can be formed in few-layered MoTe2 films by optimising the precursor flux in such a way that the incomplete 1T′-to-2H transition is arrested leading to isolated 2H domains being formed in continuous 1T′ matrix.60,160 However attractive as a lever to direct the crystallisation of MoTe2 in the metastable crystal phase, tellurium deficiency of the 1T′ phase produced via metal film tellurisation can significantly hamper its potential use in electronics where high structural perfection is required.161 High-quality 1T′ MoTe2 single crystals can be grown directly from elemental Mo and Te by chemical vapour transport (CVT) reaction.58,153,162 In this method, a stoichiometric mixture of elemental precursors is sealed in an evacuated ampoule and then maintained at elevated temperature, typically 800–1000 °C, to produce polycrystalline MoTe2 product, which is further recrystallised by CVT method using either TeCl4,63,77 TeBr4,162 or I258 as transport agent. The monoclinic 1T′ phase of MoTe2 is thermodynamically stable at the temperatures reported for the CVT processes (900–1100 °C),27,153 and can be preserved in the final product if rapidly quenched down to room temperature.58,77 The dimensions of thus produced 1T′ MoTe2 single crystals are reported to be as large as 17 × 10 × 2 mm3 (162) (Fig. 4f). Moreover, D. Keum et al. have shown that stoichiometric 1T′ MoTe2 single crystals can be grown from Mo and Te powders in a molten salt (NaCl).153 The use of salt was recognised essential since this allows dissolving Te and thus prevents its sublimation and formation of Te deficient product. Similar to ref. 58, 1T′ MoTe2 single crystals were attained if the product was quenched, whereas the 2H phase is a product of slow cooling to room temperature. Atomically-thin 1T′ MoTe2 flakes with the lateral size exceeding 10 μm and exhibiting high structural perfection can then be mechanically exfoliated from the bulk crystals onto supports (Si/SiO2) for further examination.58,153
By optimising the quenching regime, all three, 1T, 1T′, and 2H, phases of MoTe2 can be selectively produced starting from MoO3 and Te powder precursors.163 The fact that all three phases were obtained through the same growth procedure with the only variation being the cooling rate led the authors to conclude that during the direct CVD reaction, MoTe2 nucleates in the 1T phase. The 1T phase is obtained if after the growth stage the final product is rapidly cooled in the presence of CO2 and H2 gases. Trapping of the metastable 1T phase in this case might be caused by the interplay of CO2 and H2 adsorption and interaction with the substrate. If the temperature is reduced more gradually then the 1T′ phase is found to be dominating in the final product. Finally, natural cooling allows enough time for the crystal lattice to transform into the 2H phase.119
A different approach to obtain the metastable phases on monolayered TMDs via CVD has been demonstrated for the synthesis of the 1T polymorph of WS2164 and it is based on the use of a catalyst. A mixture of Fe3O4 and NaCl, serving as a catalyst and a growth promoter, was added into the CVD-tubular furnace (WO3 and elemental S as material precursors) leading to the formation of monolayered 1T/2H WS2 flakes with a butterfly-like shapes, where one of the laterally stitched wings adopted the 1T and the other – the 2H phase. Single crystalline domains in these butterfly-like WS2 monolayers reached 80 μm (Fig. 4g). It has also been shown that these 1T/2H WS2 structures could serve as seeds for the laterally expanded 1T/1T and 2H/2H WS2/MoS2 planar heterostructures.164
Owing to the non-equilibrium nature of liquid-phase crystallisation, the reaction pathway in the systems with diverse polymorphism greatly depends on subtle variations in growth conditions and may often lead to the formation of metastable reaction products.165–167 First, the crystal growth conditions may be optimised in such a way that the metastable reaction product is kinetically preferred over the equilibrium product.168 In fact, it is known that in the systems with numerous polymorphs present, the crystallisation often proceeds through the formation of thermodynamically unstable polymorphs that successively transform into the equilibrium phase; this is known as Ostwald's step rule (Fig. 7b).169 The formation of the metastable phase can dominate the crystallisation kinetics owing to a lower nucleation barrier than that of the equilibrium phase170 (Fig. 7a) and so one can assume that the metastable polymorph can be isolated if the crystallisation is arrested before the next, more stable, polymorph nucleates and grows. It is assumed that high supersaturations might be prerequisite for such multi-step crystallisation.171,172 Also, the free energy of different polymorphs may be altered due to the interaction of the growing crystal with its environment, such as solvent and, especially in the case of colloidal growth, capping ligands, thus lowering the energy barrier for the preferential formation of metastable polymorph and potentially preventing its transformation into the thermodynamically stable one.80 In TMD systems, the effects of nanocrystal nucleation and growth kinetics on the preferential formation of one polymorph over the other are still at very early stages of investigation.
 | ||
| Fig. 7 Homogenous nucleation of the metastable phase in a system with diverse polymorphism. (a) Nucleation barriers of the metastable (purple) and the stable (green) phase in a supersaturated solution. (b) Schematic diagram illustrating the crystallisation pathway of the stable phase proceeding through a series of intermediate metastable phases A, B, and C; ΔG* denotes the energy barriers of the respective phase transitions. Separate panels are adapted with permission: (a) from ref. 170, copyright 2017, The Royal Society of Chemistry; (b) from ref. [Nature Physics volume 5, pages 68–73 (2009); 10.1038/nphys1148], copyright 2008, Springer Nature. | ||
Y. Sun et al. have reported that the metastable 1T′ polymorph of MoTe2 is directly accessible in colloidal synthesis.173 In their work, flower-like 1T′ MoTe2 nanostructures having diameters of 100–150 nm were grown in solution phase by continuously injecting molybdenum precursor into a hot mixture (300 °C) containing trioctylphosphine telluride (TOP:Te) in trioctylphosphine (TOP) and oleylamine (OlAm). Molybdenum precursor was prepared by dissolving MoCl5 in oleic acid (OlAc). Adding hexamethyldisilazane (HMDS) into tellurium precursor prior to the molybdenum complex injection is required for the formation of crystalline, uniform MoTe2 nanostructures. The authors hypothesised that the interaction with organic ligands can effectively modify the surface energy of MoTe2 nanocrystals and so further lower the energy barrier for forming the metastable structure. Once formed, the metastable 1T′ phase is kinetically trapped due to the pinning at grain boundaries in the petals of MoTe2 nanoflowers. Following a similar synthesis protocol, Y. Sun et al. have obtained high-quality WTe2 nanoflowers using WCl6 complex in OlAc as transition metal precursor.174 Interestingly, colloidal WTe2 nanoflowers are attained in the orthorhombic Td phase. Additionally, the authors have demonstrated that by using mixtures of MoCl5 and WCl6 as transition metal source, nanostructured WxMo1−xTe2 solid solutions with composition-dependant Td/1T′ crystal phase heterogeneity spanning the Td WTe2 and 1T′ MoTe2 polymorphs can be accessed.174
On the other hand, colloidal synthesis of the metastable polymorphs of group VI disulphides and diselenides remains majorly unexplored. Generally, the metastable polymorphs of these materials are considerably less accessible due to the greater energy barriers of their formation.46 Consequently, the solution-phase growth of group VI disulphides and diselenides, whether by a thermolytic decomposition of a single source precursor, like [Mo2O2S2(S2COEt)2],175 or by a direct reaction between molecular precursors, such as MoCl5 and CS2,176 commonly results in the thermodynamically stable 2H polymorph. To date, the reported protocols include the use of tungsten and molybdenum hexacarbonyls177 and chlorides,178–180 molybdenyl acetylacetonate,181 sodium molybdate179,182 and sodium tungstate,183 as a source of transition metal. Chalcogen precursors include carbon disulphide,176,180 diphenyl diselenide;177,180,181 complexes of elemental sulphur178 or selenium179,182,183 in organic solvents, like oleylamine or octadecene. Despite such a variety of precursors available for a direct solution-phase growth of group VI TMDs, only the semiconducting 2H phase was synthesised. However, a slow sulphidation of colloidal W18O49 nanorods, serving as sacrificial seeds, by in situ generated H2S has led to the formation of irregularly-shaped ultrathin WS2 disks demonstrating co-existing 2H and 1T′ crystal phases.184
B. Mahler et al. have reported on a phase-selective colloidal synthesis of WS2 nanosheets80 (Fig. 6b). Disk-like WS2 nanostructures with the distorted octahedral 1T′ crystal structure were synthesised through a reaction between WCl6 complex in oleic acid and CS2 in hot (320 °C) oleylamine, whereas the 2H WS2 nanosheets were grown from the same molecular precursors in the presence of an additive, hexamethyldisilazane (HMDS). HMDS is an essential chemical in colloidal synthesis of another class of layered materials: narrow-gap AIV–BVI semiconductors, such as GeS and SnS.185,186 It has been noted that the growth from the respective transition metal halides in hot oleylamine in the absence of HMDS leads to amorphous product, whereas in the presence of HMDS, highly-uniform, single crystalline plates of SnS and GeS are being formed.185–187 In the case group VI TMDs, Y. Sun et al. have reported that the presence of HMDS is required for the formation of highly crystalline MoTe2,173 WTe2174 and WxMo1−xTe2174 colloidal nanosheets. M. Kobylinski et al. have suggested that in general, the role of HMDS in colloidal synthesis from transition metal chlorides is two-fold: first, HMDS may react with the transition metal source, forming the corresponding (trimethylsilyl)amino complex; second, HMDS may bind chloride ions and remove them from the reaction mixture as (trimethylsilyl) chloride. While B. Mahler et al. hypothesised that in the specific case of colloidal synthesis of WS2, HMDS may effectively tune the reactivity of tungsten precursor enabling the phase-selective growth.80 Specifically, they presume that HMDS may partially substitute the oleate ligands complexing tungsten ions, leading to the formation of a more active form of tungsten precursor. Their alternative hypothesis suggests that HMDS may act as a capping ligand during the nanocrystal growth, stabilising the 2H polymorph over the 1T′. In order to verify which of the two possible mechanism is more likely to drive the reaction, the authors replace oleic acid (OlAc) with another strongly binding ligand – dodecanethiol (DDT). The authors suggest that unlike OlAc, DDT does not react with HMDS thus the formation of an activated tungsten (trimethylsilyl)amino complex is not possible in the DDT protocol. Indeed, the material grown from WCl6–DDT complex in the presence of HMDS exhibits the 1T′ phase. These findings suggest that the formation of the thermodynamically stable 2H phase of WS2 can be mainly attributed to the activation of tungsten complex by reacting with HMDS, whereas less reactive tungsten oleate may lead to the direct growth of the 1T′ phase. It should be noted that a detailed study of the suggested intermediates in the WCl6–OlAc and WCl6–DDT systems, their activity and possible reactions with HMDS is not provided in the cited work.80
In addition to 1T′ WS2, the direct growth of the metastable 1T′ polymorph of WSe2 has been reported recently71,188 (Fig. 4d). It is worth mentioning that the 1T′ polymorphs of WSe2 and MoSe2 were not previously demonstrated by Li intercalation and were only achieved via direct synthesis.68,71 Uniform 1T′ WSe2 nanoflowers with an average diameter of 200 nm and composed of highly-crystalline atomically-thin petals were grown through a reaction between W(CO)6 and trioctylphosphine selenide (TOP:Se) in hot oleic acid (300 °C).71 Prior to that work, only a few experimental protocols on colloidal synthesis of WSe2 nanostructures were present, but only the formation of thermodynamically stable 2H polymorph was reported.177,180 In order to elucidate the reaction pathway, M. Sokolikova et al. have performed a parametric study of growth conditions. The activity of both precursors was systematically varied by replacing W(CO)6 with WCl6, TOP:Se with elemental Se; additionally, the activity of tungsten precursor was modified by complexing with two additives: hexamethyldisilazane (HMDS) and tetradecylphosphonic acid (TDPA). Interestingly, the metastable 1T′ crystal phase was consistently grown under these conditions; only negligible changes in the morphology were reported. Based on these observations, the authors have proposed that under the described experimental conditions, the formation of metastable 1T′ polymorph might be attributed to the crystallisation in kinetically-driven regime; however, switching between the supposedly kinetically-driven and thermodynamically-controlled regimes has not been unambiguously demonstrated.
Later, J. Geisenhoff et al. have demonstrated a phase-selective synthesis of WSe2 nanoflowers that was achieved in a biligand system of trioctylphosphine oxide (TOPO) and oleic acid (OlAc).188 The metastable 1T′ phase of WSe2 was obtained when the fraction of OlAc in the ligand mixture was high, while keeping the oleic acid fraction to minimum has led to the growth of the thermodynamically stable 2H phase. The authors indicate that by coordinating with oleic acid, a less reactive form of tungsten precursor is formed; this leads to nucleation of fewer seeds and to larger nanocrystals compared to the synthesis in TOPO-rich mixtures. However, the effect of ligand mixture composition on the preferential nucleation of one of the other crystal phases remains unclear. Regardless of the ligand ratio, colloidal WSe2 was found to nucleate in the metastable 1T′ phase; however, in the systems, containing low fraction of OlAc, it converts into the 2H phase over time.188 Whereas in the case of ligand mixtures with high OlAc content or of syntheses in OlAc, the final product seemingly retains the metastable 1T′ phase.188
Recently, it has been proposed that the energy barrier for the homogeneous nucleation of the metastable 1T phase could be lowered in a magneto-hydrothermal process.136 The preferential nucleation of the 1T polymorph in high magnetic fields is facilitated by the higher magnetic susceptibility of the 1T structure, compared to the 2H phase. Using this approach, W. Ding et al. have selectively synthesised 1T MoS2 and WS2 nanosheets through a hydrothermal reaction between either ammonium heptamolybdate ((NH4)6Mo7O24·4H2O) and thiourea or tungsten hexachloride (WCl6), and thioacetamide under an external magnetic field of ∼9 T.136 The nucleation of the 2H phase from the same precursors in the absence of magnetic field (control) has also been demonstrated.
Considering the present evidence, we suggest that the understanding of crystallisation pathways leading to the formation of metastable polymorphs of group VI TMDs in colloidal solution is at early stages. A thorough investigation of the nucleation mechanism, including the nature of reaction intermediates formed from molecular precursors, possible prenucleation clusters, energetics of the nanocrystal–solution interface, and surface dynamics of capping ligands on the nanocrystal facets, is needed in order to gain control over the phase-selective growth of various polymorphs of group VI TMDs.
It is interesting to note that unlike other van der Waals solids, such as colloidal SnS186 and SnSe,189 GeS and GeSe,185 InSe,190 and group IV191,192 and V192 disulphides and diselenides, that are acquired in a form of two-dimensional platelets and disks, group VI TMDs obtained through a solution-phase (hydro-/solvothermal81,193 and colloidal71,173,174,179,180) reaction between similar type of precursors typically exhibit branched morphology of flower-like structures composed of ultra-thin petals. The overall anisotropic morphology of van der Waals compounds is expected since the lateral extension in the ab plane is energetically preferred due to surface energy difference between the undercoordinated edge facets and the basal planes, where dangling bonds are absent.192,194 In some cases, like layered SnSe189 and Bi2Te3,195 and non-layered PbS196 systems, the growth of large regularly-shaped single-crystalline nanostructures does not proceed through the extension of primary nucleated seeds but rather occurs via the two-dimensional oriented attachment of the seeds. The self-assembly process is driven by the minimisation of surface energy associated with the unsaturated bonds, for example, in the lateral surface of SnSe seeds, and can be effectively controlled by varying the precursors’ concentrations.189 However, neither of these crystallisation mechanisms can explain the formation of branched flower-like structures of group VI TMDs produced via a solution-phase reaction. The formation of 3D flower-like nanostructures composed of individual 2D nanosheets might be caused by a nucleation from a dense amorphous state formed in the supersaturated solution as proposed for TiS2 nanoflowers.197 S. Prabakar et al. have found out that TiS2 flower-like structures evolve in colloidal solution starting with the formation of amorphous globules and proceeding through the appearance of randomly oriented crystalline platelets on the surface of amorphous globules to the growth of these petals originating from the central core so that the amorphous core is no longer apparent.197 Similar formation mechanism was later described by O. Meiron et al.198 and by C. Zhang et al.182 for colloidal MoS2xSe2−2x and MoSe2 nanoflowers, respectively. O Meiron et al. have identified amorphous blobs that are formed at the very early stages of the reaction; these later serve as material reservoirs for the curled and tangled nanosheets that crystallise inside the amorphous clusters.198 Over time, amorphous material is fully consumed and uniform nanoflowers are formed. Despite a considerable progress in controlled synthesis of TMDs, we highlight that a deeper understanding of solution-phase nucleation and crystallisation processes of group VI TMDs is required in order to fully employ their potential in practical applications.
3. Comparison of various techniques to access the metastable 1T(1T′) phases
Fig. 4 provides a comprehensive overview of the reported approaches to access the metastable 1T(1T′) phases of group VI TMDs. The data points were taken from the references, where the lateral dimensions of continuous domains (not necessarily single crystalline) of the metastable 1T(1T′) phase were clearly stated and the metastable phase content was determined from the results of either XPS or Raman spectroscopy. These are listed in Table 1.| Material | Lateral size | % of 1T(1T′) | Ref. |
|---|---|---|---|
| Molecular beam epitaxy | |||
| MoS2 | 4 nm | 100 | 79 |
| MoSe2 | 10 nm | 100 | 70 |
| WSe2 | 100 nm | 100 | 21 |
| Chemical vapour transport | |||
| MoS2 | 100 μm | 100 | 56 |
| 150 μm | 100 | 68 | |
| 500 μm | 100 | 69 | |
| MoSe2 | 170 μm | 100 | 68 |
| MoTe2 | 10 μm | 100 | 58 |
| 1 cm | 100 | 153 | |
| 1.7 cm | 100 | 162 | |
| Chemical vapour deposition | |||
| MoS2 | 10 μm | 100 | 147 |
| WS2 | 80 μm | 100 | 164 |
| Chemical exfoliation (both 2H and 1T′ crystals) | |||
| MoS2 | 3.5 nm | 70 | 112 |
| WS2 | 3 nm | 80 | 112 |
| 100 nm | 80 | 83 | |
| MoS2 | 1 μm | 70 | 101 |
| 700 nm | 80 | 103 | |
| 10 μm | 97 | 69 | |
| 60 μm | 97 | 69 | |
| 500 nm | 100 | 93 | |
| 1.5 μm | 100 | 265 | |
| Direct colloidal/hydrothermal synthesis | |||
| MoS2 | 200 nm | 61.6 | 81 |
| WS2 | 100 nm | 50 | 80 |
| 162 nm | 66.4 | 148 | |
| WSe2 | 100 nm | 60 | 71 |
The most commonly reported approach to synthesise the metastable phase in measurable quantities is chemical exfoliation from bulk TMD powders. In chemically exfoliated MoS2 and WS2, the 1T(1T′) phase is found as nm-scale domains embedded in the 2H matrix, although the total 1T(1T′) phase content reaches ∼70–80%. It should be noted that there is little evidence that this approach is effective in the case of group VI diselenides. On the other hand, CVT growth, that up until recently was employed to synthesise cm-sized single crystals of 1T′ MoTe2 only, has been extended to the case of molybdenum disulphide and diselenide. The growth of the metastable phase is achieved with the assistance of alkali metal containing precursors and 1T′ MoS2 and 1T′ MoSe2 crystals with lateral dimensions of a few hundreds of μm are produced by gentle deintercalation of the intermediate products. Furthermore, chemical exfoliation from bulk 1T′ MoS2 and 1T MoSe2 crystals results in unprecedentedly large nanosheets with the metastable phase content of nearly 100%. CVD growth, that typically leads to the formation of the thermodynamically stable polymorph, can be optimised in such a way that the metastable phase is selectively produced. Thus, in the presence of a catalyst or alkali metal containing precursors, the growth of single crystalline domains of 1T WS2 and 1T′ MoS2 with the sizes of a few tens of μm has been reported. Another deposition approach, molecular beam epitaxy, allows for the growth of islands of 1T′ phase with lateral dimensions varying from a few nm as in the case of MoS2 and MoSe2 to nearly reaching 100 nm in the case of WSe2. It should be noted that the selectivity of MBE process is rather low at present as the formation of separate domains of the 2H phase is often reported.
Solution-phase approaches, such as hydrothermal and colloidal reactions, allow for the synthesis of measurable quantities of the 1T′ phase containing material, in some cases reaching a few grams. The lateral size of 1T′ domains are reported within the range from a few tens to a few hundreds of nm, and the metastable phase content is about 50–60%.
4. How stable the metastable 1T(1T′) phases are?
Although being thermodynamically metastable under ambient conditions, the 1T(1T′) polymorph is kinetically stable over the course of a few days to a few months, depending on the material morphology and environment. Thus, chemically exfoliated 1T′ and 1T′′′ MoS2 monolayers gradually convert back to the 2H phase over 3–5 days in air as demonstrated by Raman87 and STM146 studies. The conversion is attributed to the restacking of exfoliated material in suspension that leads to the van der Waals interactions between the layers being recovered. The transformation into the 2H phase is significantly promoted when the exfoliated 1T′ TMDs are deposited to form thin films owing to the removal of water from the interlayer space of exfoliated sheets during drying.87 Synthesised through a direct hydrothermal reaction, the 1T-phase MoS2 was reported to slowly convert into the 2H phase over 90 days in water. This was accompanied by a gradual loss of the catalytic activity.1995. Property-related applications of the metastable 1T(1T′) phases
The metastable 1T(1T′) polymorphs of group VI TMDs are found to be promising for various applications, including electrocatalysis,83,103 electrochemical energy storage,101,200 sensors,201,202 transparent electrodes for nanoscaled optoelectronics110 and photovoltaics,203 transport layers for solar cells,204 and even microwave absorbers for application in electromagnetic shielding and stealth camouflage.205 The incorporation of domains of the 1T phase in the 2H matrix induces a robust ferromagnetism in the intrinsically nonmagnetic semiconducting TMDs, further expanding the range of potential applications to spintronic devices.206,207 The difficulty in accessing the input of the metastable phase is commonly associated with the fact that in many cases, the 1T(1T′) phase co-exists with the thermodynamically stable 2H phase in these produced materials. Here, we review the state-of-the-art research articles related to the understanding of how the intrinsic properties of the 1T(1T′) phases can enable novel applications.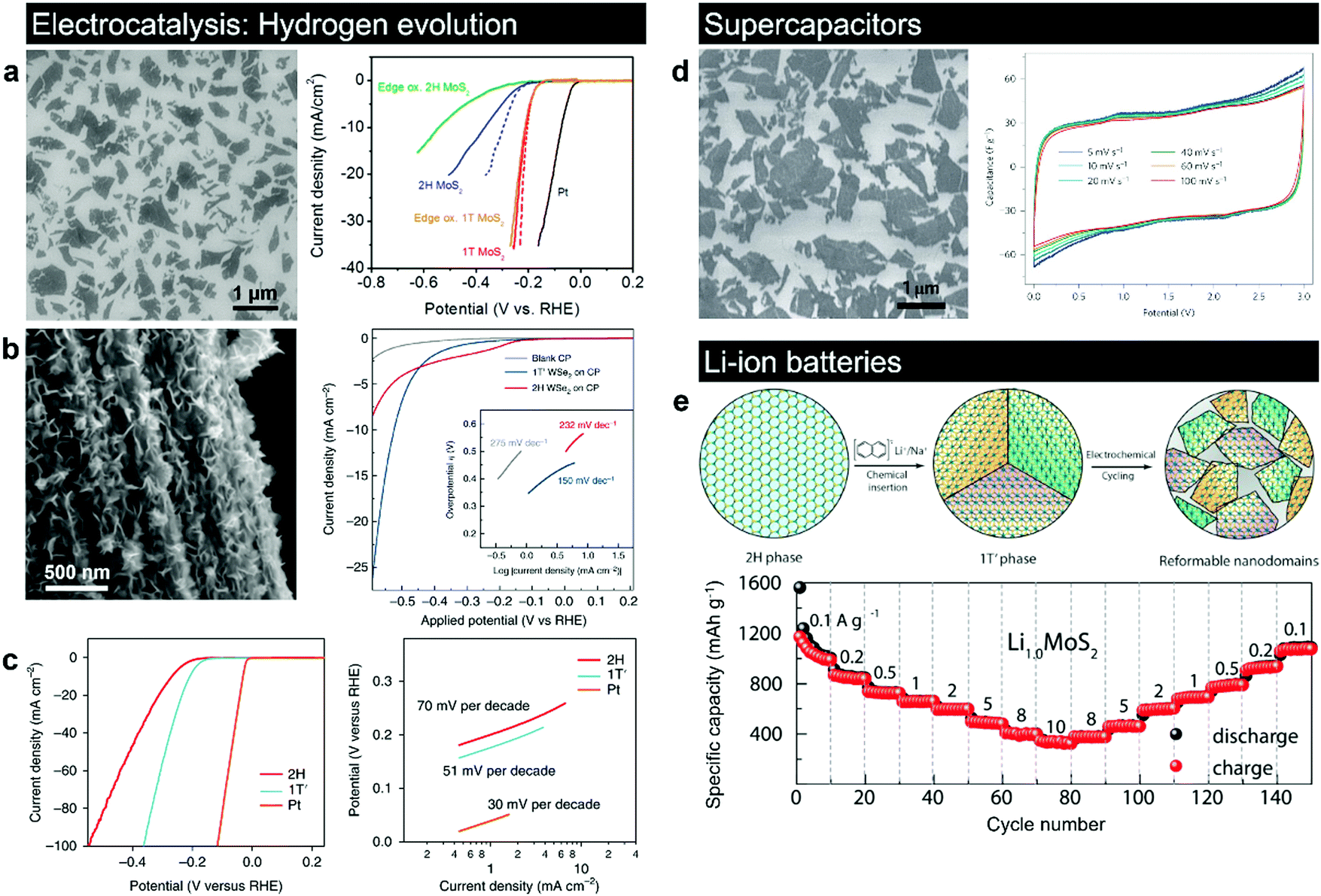 | ||
| Fig. 8 Applications of the metastable 1T(1T′) phases of group VI TMDs in electrochemical energy storage and conversion. (a) SEM image (left) of micron-sized chemically exfoliated 1T′ MoS2 nanosheets and the polarisation curves (right) of 1T′ and 2H MoS2 demonstrating the basal plane activation in the 1T′ polymorph. (b) SEM image (left) of 1T′ WSe2 nanosheets grown directly on carbon fibres demonstrating lower HER overpotentials and lower Tafel slopes (right) compared to the corresponding 2H phase. (c) Polarisation curves (left) and the corresponding Tafel slopes (right) of the CVD grown 1T′ MoS2 flakes. (d) SEM image (left) of micron-sized chemically exfoliated MoS2 nanosheets and CV curves (right) of a symmetric supercapacitor device in an organic electrolyte acquired at various scan rates. (e) A schematic (top) illustrating the domain size reduction during a continuous cycling of lithiated MoS2 nanosheets and charge–discharge plots (bottom) at different rates for the lithiated MoS2-based batteries. Separate panels are adapted with permission: (a) from ref. 103, copyright 2013, American Chemical Society; (b) from ref. 71, copyright 2019, Springer Nature; (c) from ref. 147, copyright 2018, Springer Nature; (d) from ref. 101, copyright 2015, Springer Nature; and (e) from ref. 200, copyright 2016, American Chemical Society. | ||
The enhanced catalytic activity of the metastable polymorphs of group VI TMDs is also associated with higher conductivity of the metallic (semimetallic) 1T(1T′) phases. Recently, it has been suggested that the characteristics of the interface between active material and functional substrate, as well as the electron transport across active material, affect the catalytic performance even more than the thermodynamics of catalytic reaction. Compared to the semiconducting 2H counterparts, the 1T(1T′) phases of WS2, MoS2 and MoSe2 greatly benefit from faster charge transfer in metallic polymorphs.102,199,224 Additionally, in the case of metallic polymorphs, the catalyst–substrate interface becomes essentially metal–metal, which leads to more effective electron injection into the active material.225 It is worth mentioning that the 1T′ phase of WSe2 have shown the enhanced electrocatalytic activity compared to its 2H counterpart (Fig. 8b); however, in this case the material was obtained uniquely via direct synthesis.71
The catalytic activity of metastable 1T and 1T′ polymorphs of group VI TMDs cannot be compared directly as both phases are often reported interchangeably.83 Further, L. Wang et al. have suggested that during electrochemical cycling, the 1T phase of MoS2 irreversibly transforms into the more catalytically active 1T′ phase.226 According to first principle calculations, the 1T phase readily transforms into the 1T′ phase at room temperature and this transition is considerably promoted by hydrogen adsorption during the H2 evolution. Moreover, the authors have experimentally proved that the 1T-to-1T′ transition does occur in chemically exfoliated MoS2 and they suggest that such a transition may explain the self-optimising catalytic behaviour reported for the group VI TMD catalysts.226 Additionally, there is not yet a clear evidence of the effect that a fully extended 1T′ phase and large single crystalline domains of 1T′ phase can have on the HER performance. For instance, the enhanced catalytic activity in mixed-phase 1T′/2H, as well as in polycrystalline single phase, TMD systems is occasionally attributed to the strained crystalline domain boundaries in the basal plane, providing a higher density of catalytically active sites.227,228 Certainly, the bottom-up synthesis approach paves the way to expand the portfolio of 1T′ phases and to control the crystallinity, morphology and crystal phase in multi-layered form (Fig. 8c).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.231
000 cycles.231
Based on first-principle calculations, 1T MoS2 monolayers have been identified as a suitable anode material for lithium-ion batteries due to their ability to absorb Li ions at high concentrations without aggregation.232 T. Xiang et al. have realised hybrid 1T MoS2/graphene electrodes exhibiting a capacity of 666 mA h g−1 at high current density (3500 mA g−1).233 It is interesting to note that the arrays of vertically aligned few-layered MoS2 nanosheets composing the electrodes were grown on graphene via a single-step hydrothermal reaction and the interlayer spacing in thus grown MoS2 reaches 9.8 Å dictated by the graphene lattice; such lattice expansion facilitates a faster Li diffusion essential for a high performance lithium-ion battery. In bulk MoS2 crystals, the repeating lithiation process is reported to form a mosaic-like nanocrystalline domains, which ensure high rate capability and excellent stability in rechargeable lithium-ion batteries.200 This process is schematically shown in Fig. 8e. Such phase restructuring in pre-lithiated MoS2 ensures a faster and more stable charge/discharge rate than in either chemically exfoliated or bulk material.200 The 1T phases have seen recent utilization also in composites for batteries electrodes and we refer the reader to the following reviews for more details on this subject.234–236 Covering these works is beyond the scope of the current review as here, we aim at discussing the applications that directly arise from the intrinsic characteristics of the metallic 1T phases of group VI TMDs.
The metastable 1T(1T′) phases of group VI TMDs have been also proposed to be used in miniaturised biosensors. In particular, chemically exfoliated metallic 1T WS2 has been employed as a conductive platform for an indirect enzymatic detection of fenitrothion; the biosensor expressed good linearity in the a broad concentration range (1–1000 nM) with the detection limit as low as 2.86 nM of fenitrothion.201 On the other hand, the semimetallic 1T′ MoTe2 and WTe2 polymorphs are reported to be ultra-sensitive surface enhanced Raman scattering (SERS) platforms for molecular detection.237 In this case, the enhancement of Raman signal is achieved due to the polarisation of analyte molecules (rhodamine 6G) owing to the coupling with the 1T′ phase SERS platform. The reported detection limits are as low as 40 and 400 fM of rhodamine 6G on pristine few-layered 1T′ WTe2 and 1T′ MoTe2 flakes, respectively; however, these can be further improved when the sensor is integrated on a Bragg reflector.
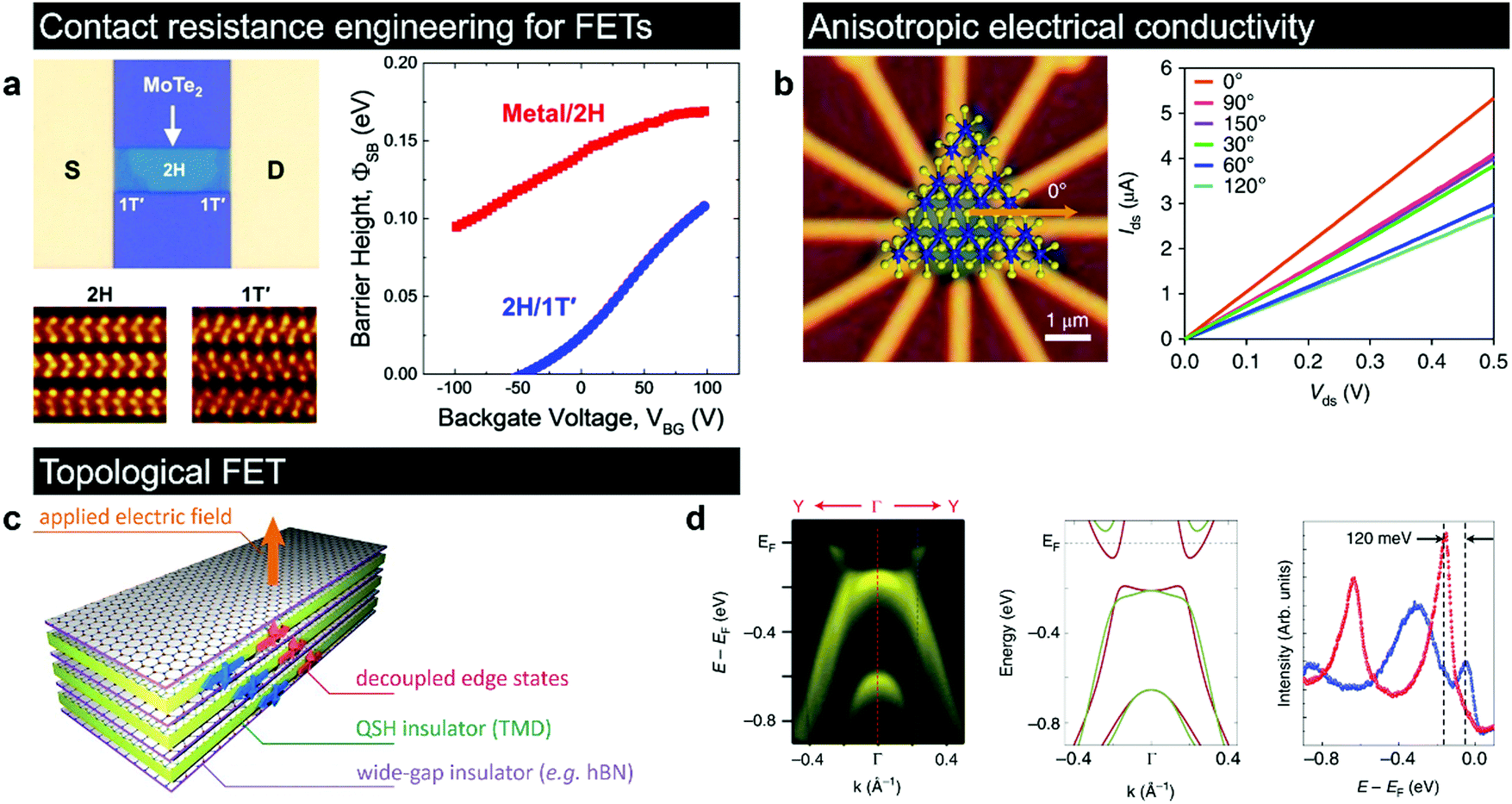 | ||
| Fig. 9 Applications of the metastable 1T(1T′) phases of group VI TMDs in novel electronics. (a) Optical image (left) of an FET device with patterned 1T′/2H MoTe2 contacts and the Schottky barrier height (right) in the 2H MoTe2 only device and in the device with patterned 1T′/2H contacts. (b) Electrical characterisation of a CVD grown 1T′ MoS2 flake (left) demonstrating an anisotropic electrical transport (right). (c) A schematic of a topological FET device based on van der Waals heterostructures of alternating monolayer 1T′ group VI TMD and single-/few-layer wide-gap insulators. (d) Electronic characterisation of 1T′ WSe2 monolayer. High-resolution ARPES band dispersion (left) and the corresponding calculated bands (middle) demonstrate the band inversion in monolayer 1T′ WSe2; an indirect band gap in 1T′ WSe2 calculated from two energy dispersion curves (right) is characteristic to 2D TI. Separate panels are adapted with permission: (a) from ref. 160, copyright 2019, American Chemical Society; (b) from ref. 147, copyright 2018, Springer Nature; (c) from ref. 46, copyright 2014, American Association for the Advancement of Science; and (d) from ref. 20, copyright 2018, Springer Nature. | ||
Due to the structural anisotropy in the ab plane, 1T′ single layers have been predicted to exhibit anisotropic transport characteristics. Anisotropic electrical conductivity of the 1T′ phase have been recently demonstrated in chemically exfoliated250 and CVD grown 1T′ MoS2 flakes147 (Fig. 9b). The conductance reaches its maximum along the a (zigzag) direction and the anisotropic ratio is as high as 2,147 that exceeds the reported anisotropic conductance for black phosphorus.251 This leads to envision application of the 1T′ phases for neuromorphic devices where anisotropic transport is sought in different device components. Black phosphorous, a highly anisotropic 2D material, is being investigated in synaptic devices and higher devices complexities could be enabled by 2D materials with different anisotropic transport.
Bulk nonmagnetic Td WTe2 demonstrates an extremely large magnetoresistance (XMR); the primary source of which is established to originate from the carrier compensation.252 Thus, at 4.5 K, positive magnetoresistance of 452700% was observed in Td WTe2 in a magnetic field of 14.7 T; moreover, the magnetoresistance value reaches 13000000% at 0.53 K in a magnetic field of 60 T and no saturation of magnetoresistance is evidenced even in very high magnetic fields.253 Later, the XMR behaviour was discovered in the Td phase of MoTe2.254,255 Furthermore, the magnetoresistance observed in Td MoTe2 in high magnetic fields can be tuned by an applied tensile strain (0.5%).256 At low temperatures and in high magnetic fields, the magnetoresistance variation approaches 30% under uniaxial tensile strain perpendicular to the zigzag chain direction, while the strain along the zigzag chain direction effectively suppresses the magnetoresistance.256
Conclusions and future outlook
In summary, in the past few years there have been exciting fast developments in the elucidation of the potential of the metastable phases of TMDs, ranging from electrocatalysis83,103 to condensed matter phenomena such as 2D topological insulators.19 Here, we have reviewed the concomitant significant step towards the production of these phases in measurable quantities and the key applications that these phases can uniquely enable. The most widely used method to synthesise metastable phases is based on the intercalation with alkali metals in organic solvent83,114 or in a dry form.50,103 This is a versatile route which can be applied to bulk powders, with an associated exfoliation of the flakes, or on individual flakes, often CVD grown, on a substrate. Thus, it provides with the ability to produce highly concentrate suspensions of the 1T(1T′) phases in water as well as monolayers extended over large areas. The latter is compatible with the integration in planar devices such as field-effect transistors.95,155 The first provides stable colloidal suspensions of the 1T phases in water which can be processed from solution via vacuum filtration103 or dip-coating,257 to generate uniform thin films for different applications from electrocatalysis103 and photoassisted electrocatalysis,257 to energy storage devices.101In the form of thin films, the 1T phase can be converted into the 2H phase upon annealing.27,50 Thus, the accessibility of the 2H phase from the 1T(1T′) phase is advantageous as it provides an alternative approach to the commonly used exfoliation of the 2H phase in organic solvent by applying a shear force,258 allowing for the processability of water-based suspensions which are compatible with large-scale production and deposition techniques, such as ink-jet printing,259 3D printing,260,261 and spray coating,262 to name a few.
While chemical treatment has intrinsic advantages and it has been very effective for initial studies of 1T phases, as the field progresses, the limitations are now emerging. The first one is represented by the use of Li-chemistry which is air sensitive, involving the use of a controlled atmosphere. Although the intercalation of MoS2 and WS2 bulk powders have demonstrated the 2H-to-1T(1T′) phase transformation, the conversion of the corresponding diselenides (MoSe2 and WSe2) have not been reported. While the 1T′ phases of MoSe2 and WSe2 have been obtained via direct synthesis, demonstrating that these metastable phases can actually be achieved.68,71
Furthermore, the 1T′ phase resulting from the Li intercalation is limited to the 75% of the material and it has a patchy distribution across the basal plane of individual flakes with a polycrystalline nature, where individual domains can be limited to very few nm.85 The impact of the polycrystallinity on the electrical, mechanical and electrocatalytic properties is still not known yet. Thus, the search for bottom-up approaches to the synthesis arises from the need of fully controlling the phase morphology, the materials processability and the possibility to establish a wider spectrum of metastable phases and compounds composition.
Progress has been made in the direct synthesis of metastable phases via both vapour-based deposition techniques and in liquid phase from molecular precursors which led to the first demonstrations of the 1T′ phase of MoS2147 and WSe2.71 Most of the reported vapour-phase syntheses are based on chemical vapour transport and have demonstrated the synthesis of bulk crystals of metastable 1T′ MoSe268 and 1T′ MoS2 phases.68,69 The achievement of the 1T′ phase monolayers via CVD approach has been limited to MoTe2, WTe2,161 and MoS2.147 These bottom-up approaches lead to nearly continuous 1T′ phase across large areas (over tens of microns) of over 80% and exhibit a single crystalline nature across hundreds of nanometres. As the atomically thin nature of the flakes is not preserved with CVT synthesis, when bulk crystals are produced, they need to undergo a liquid phase exfoliation step to be used in their 2D form. CVD, which can enable scalability of the synthesis, have the advantage of producing anisotropic monolayers over large areas for application in catalysis, nanoelectronics, neuromorphic systems, quantum technologies and advanced spectroscopies; however, altering the reaction energetics to selectively produce the metastable phase is still challenging.
MBE have been used to demonstrate single crystalline phase of 1T′ WSe220,21 on bilayer graphene terminated SiC substrates, and of 1T′ MoSe270 and 1T′ MoS279 on Au substrates. Although the 1T′ domains are small (less than 100 nm), these are sufficiently extended to demonstrate new condensed-matter phenomena so far just predicted, thanks to the single crystalline nature and of the 1T′ phase.
Direct synthesis in solution is the most recent synthesis method demonstrated and it has the advantage of involving low temperatures (<300 °C), high percentage of the 1T′ phase in the final product and single crystalline nature across flakes sizes of 100 nm.71,80,173 Synthesised through a direct colloidal reaction, the nanosheets can form stable dispersions in non-polar organic solvents. This synthesis route presents features compatible with a large-scale production and the possibility of translating the chemistry of the synthesis into a hydrothermal process.
Branched flower-like nanostructures with atomically thin petals are generally obtained via liquid phase synthesis.71,81,173,199 Such morphology is very advantageous for catalysis and supercapacitors as it maximizes the exposure of active sites versus a 2D nanosheets geometry where flakes are planarly stacked, losing some of the accessible surface area. These nanoflowers can also be directly grown on the electrode materials where they can be strongly anchored enabling a good electrical contact. The possibility of obtaining individualized planar 2D nanosheets could, however, expand the applications of the 1T′ phases produced in liquid phase.
The next key challenge lies in understanding the nucleation stage in solution phase to be able to reliably control the flake morphology (between branched sheets and individual sheets) and the crystal phase. As stated by the Ostwald's step rule, which has been based on empirical observations in zeolite materials, the crystallization from a solution occurs in steps in such a way that often thermodynamically unstable phases occur first, followed by a transformation in the thermodynamically stable phase.169 Thus, more theoretical understanding and experimental studies of the early stages of nucleation of TMDs are needed to predict the course of the growth and thus to design the ways of controlling morphology and to lock the crystal phase. The ability to produce individualized flakes in solution with micrometric lateral sizes can expand the potential applications competing with CVD methods. Accessing pure 1T phases of single crystalline nature across large areas of TMDs is necessary to study the intrinsic physical and chemical properties and so far there is a clear indication that only direct synthesis methods enable to approach 100% of metastable phase with large single crystals. Also, to fully understand the crystallographic structure of the most exotic metastable 1T′ and 1T′′′ phases and their related properties is necessary to have high-quality single crystals.
For practical applications, the metastable phases should be stabilized in the absence of charged species, i.e. precursor residues or intercalated cations, to produce pristine, kinetically stable materials. To this goal, the Ostwald's step rule could be again useful to predict how to preserve a metastable phase beyond the nucleation point.
The recent evidence that the 1T TMDS can also be obtained via chemical transformation of non-van der Waals materials, further expands the potential routes of the synthetic approaches possibly making them numerous.152 Thus, these new synthesis strategies should pave the way to a new class of atomically thin materials which present new physicochemical features promising in electrocatalytic and energy storage applications as well as new fundamental condensed matter phenomena which can enable new technologies. Recent demonstration of 2D topological insulating properties in the 1T′ TMD monolayers could lead to the fabrication of topological field-effect transistors operating at room temperature46 (Fig. 9c and d). The anisotropic transport in 1T′ phases dictated by the anisotropic crystal structure could see applications in artificial synaptic devices to emulate biological systems.
The investigation on the exotic physical properties related to the most rarely observed or predicted metastable crystal phases of group VI TMDs (1T′ and 1T′′′) is progressing very rapidly and there is an incredible potential to exploit these highly anisotropic but stable phases in future nanoelectronics, optoelectronic systems and quantum technology.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the award of funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (ERC Consolidator grant agreement no. [819069]) and CM would like to acknowledge the award of a Royal Society University Research Fellowship (UF160539) by the UK Royal Society.References
- J. A. Wilson and A. D. Yoffe, The transition metal dichalcogenides discussion and interpretation of the observed optical, electrical and structural properties, Adv. Phys., 1969, 18, 193–335 CrossRef CAS.
- R. B. Murray, R. A. Bromley and A. D. Yoffe, The band structures of some transition metal dichalcogenides. II. Group IVA; octahedral coordination, J. Phys. C: Solid State Phys., 1972, 5, 746–758 CrossRef CAS.
- R. A. Bromley, R. B. Murray and A. D. Yoffe, The band structures of some transition metal dichalcogenides. III. Group VIA: trigonal prism materials, J. Phys. C: Solid State Phys., 1972, 5, 759–778 CrossRef CAS.
- H. Tributsch and J. C. Bennett, Electrochemistry and photochemistry of MoS2 layer crystals, J. Electroanal. Chem. Interfacial Electrochem., 1977, 81, 97–111 CrossRef CAS.
- H. Tributsch, Photoelectrochemical behaviour of layer-type transition metal dichalcogenides, Faraday Discuss. Chem. Soc., 1980, 70, 189 RSC.
- J. A. Baglio, Characterization of n-type semiconducting tungsten disulfide photoanodes in aqueous and nonaqueous electrolyte solutions, J. Electrochem. Soc., 1982, 129, 1461 CrossRef CAS.
- J. M. Martin, C. Donnet, T. Le Mogne and T. Epicier, Superlubricity of molybdenum disulphide, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10583–10586 CrossRef CAS PubMed.
- R. B. Somoano and A. Rembaum, Superconductivity in intercalated molybdenum disulfide, Phys. Rev. Lett., 1971, 27, 402–404 CrossRef CAS.
- J. A. Woollam and R. B. Somoano, Superconducting critical fields of alkali and alkaline-earth intercalates of MoS2, Phys. Rev. B: Solid State, 1976, 13, 3843–3853 CrossRef CAS.
- M. Chhowalla, et al., The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- A. Splendiani, et al., Emerging photoluminescence in monolayer MoS2, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS PubMed.
- H. Zeng, J. Dai, W. Yao, D. Xiao and X. Cui, Valley polarization in MoS2 monolayers by optical pumping, Nat. Nanotechnol., 2012, 7, 490–493 CrossRef CAS PubMed.
- W. Zhao, R. M. Ribeiro and G. Eda, Electronic structure and optical signatures of semiconducting transition metal dichalcogenide nanosheets, Acc. Chem. Res., 2015, 48, 91–99 CrossRef CAS PubMed.
- H. Schmidt, F. Giustiniano and G. Eda, Electronic transport properties of transition metal dichalcogenide field-effect devices: surface and interface effects, Chem. Soc. Rev., 2015, 44, 7715–7736 RSC.
- D. L. Duong, S. J. Yun and Y. H. Lee, van der Waals layered materials: Opportunities and challenges, ACS Nano, 2017, 11, 11803–11830 CrossRef CAS PubMed.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, 2D transition metal dichalcogenides, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS.
- T. F. Jaramillo, et al., Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- Q. Tang and D. Jiang, Mechanism of hydrogen evolution reaction on 1T-MoS2 from first principles, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- S. Tang, et al., Quantum spin Hall state in monolayer 1T′-WTe2, Nat. Phys., 2017, 13, 683–687 Search PubMed.
- M. M. Ugeda, et al., Observation of topologically protected states at crystalline phase boundaries in single-layer WSe2, Nat. Commun., 2018, 9, 3401 Search PubMed.
- P. Chen, et al., Large quantum-spin-Hall gap in single-layer 1T′ WSe2, Nat. Commun., 2018, 9, 2003 Search PubMed.
- Y. Xu, et al., Large-gap quantum spin Hall insulators in tin films, Phys. Rev. Lett., 2013, 111, 136804 CrossRef PubMed.
- D. V. Gruznev, et al., Two-dimensional In–Sb compound on silicon as a quantum spin Hall insulator, Nano Lett., 2018, 18, 4338–4345 CrossRef CAS PubMed.
- S. N. Shirodkar and U. V. Waghmare, Emergence of ferroelectricity at a metal-semiconductor transition in a 1T monolayer of MoS2, Phys. Rev. Lett., 2014, 112, 157601 CrossRef PubMed.
- K.-A. N. Duerloo, Y. Li and E. J. Reed, Structural phase transitions in two-dimensional Mo- and W-dichalcogenide monolayers, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- P. Joensen, R. F. Frindt and S. R. Morrison, Single-layer MoS2, Mater. Res. Bull., 1986, 21, 457–461 CrossRef CAS.
- F. Wypych and R. Schöllhorn, 1T-MoS2, a new metallic modification of molybdenum disulfide, J. Chem. Soc., Chem. Commun., 1992, 1386–1388 RSC.
- X. Zhao, S. Ning, W. Fu, S. J. Pennycook and K. P. Loh, Differentiating polymorphs in molybdenum disulfide via electron microscopy, Adv. Mater., 2018, 30, 1802397 CrossRef PubMed.
- D. Puotinen and R. E. Newnham, The crystal structure of MoTe2, Acta Crystallogr., 1961, 14, 691–692 CrossRef CAS.
- P. B. James and M. T. Lavik, The crystal structure of MoSe2, Acta Crystallogr., 1963, 16, 1183 CrossRef CAS.
- R. G. Dickinson and L. Pauling, The crystal structure of molybdenite, J. Am. Chem. Soc., 1923, 45, 1466–1471 CrossRef CAS.
- W. G. Fisher and M. J. Sienko, Stoichiometry, structure, and physical properties of niobium disulfide, Inorg. Chem., 1980, 19, 39–43 CrossRef CAS.
- X. Fan, D. J. Singh, Q. Jiang and W. T. Zheng, Pressure evolution of the potential barriers of phase transition of MoS2, MoSe2 and MoTe2, Phys. Chem. Chem. Phys., 2016, 18, 12080–12085 RSC.
- A. P. Nayak, et al., Pressure-induced semiconducting to metallic transition in multilayered molybdenum disulphide, Nat. Commun., 2014, 5, 3731 CrossRef CAS PubMed.
- Z.-H. Chi, et al., Pressure-induced metallization of molybdenum disulfide, Phys. Rev. Lett., 2014, 113, 036802 CrossRef PubMed.
- D. Xiao, G.-B. Liu, W. Feng, X. Xu and W. Yao, Coupled spin and valley physics in monolayers of MoS2 and other group-VI dichalcogenides, Phys. Rev. Lett., 2012, 108, 196802 CrossRef PubMed.
- K. F. Mak, K. He, J. Shan and T. F. Heinz, Control of valley polarization in monolayer MoS2 by optical helicity, Nat. Nanotechnol., 2012, 7, 494–498 CrossRef CAS PubMed.
- A. M. Jones, et al., Optical generation of excitonic valley coherence in monolayer WSe2, Nat. Nanotechnol., 2013, 8, 634–638 CrossRef CAS PubMed.
- R. Suzuki, et al., Valley-dependent spin polarization in bulk MoS2 with broken inversion symmetry, Nat. Nanotechnol., 2014, 9, 611–617 CrossRef CAS PubMed.
- J. Shi, et al., 3R MoS2 with broken inversion symmetry: A promising ultrathin nonlinear optical device, Adv. Mater., 2017, 29, 1701486 CrossRef PubMed.
- Z. Zeng, et al., Controlled vapor growth and nonlinear optical applications of large-area 3R phase WS2 and WSe2 atomic layers, Adv. Funct. Mater., 2019, 29, 1806874 CrossRef.
- Y.-C. Lin, D. O. Dumcenco, Y.-S. Huang and K. Suenaga, Atomic mechanism of the semiconducting-to-metallic phase transition in single-layered MoS2, Nat. Nanotechnol., 2014, 9, 391–396 CrossRef CAS PubMed.
- F. Zhang, et al., Electric-field induced structural transition in vertical MoTe2- and Mo1–xWxTe2-based resistive memories, Nat. Mater., 2019, 18, 55–61 CrossRef CAS PubMed.
- R. B. Murray and A. D. Yoffe, The band structures of some transition metal dichalcogenides: band structures of the titanium dichalcogenides, J. Phys. C: Solid State Phys., 1972, 5, 3038–3046 CrossRef CAS.
- A. Singh, S. N. Shirodkar and U. V. Waghmare, 1H and 1T polymorphs, structural transitions and anomalous properties of (Mo,W)(S,Se)2 monolayers: first-principles analysis, 2D Mater., 2015, 2, 035013 CrossRef.
- X. Qian, J. Liu, L. Fu and J. Li, Quantum spin Hall effect in two-dimensional transition metal dichalcogenides, Science, 2014, 346, 1344–1347 CrossRef CAS PubMed.
- J. Heising and M. G. Kanatzidis, Structure of restacked MoS2 and WS2 elucidated by electron crystallography, J. Am. Chem. Soc., 1999, 121, 638–643 CrossRef CAS.
- G. Gao, et al., Charge mediated semiconducting-to-metallic phase transition in molybdenum disulfide monolayer and hydrogen evolution reaction in new 1T′ phase, J. Phys. Chem. C, 2015, 119, 13124–13128 CrossRef CAS.
- X. R. Qin, D. Yang, R. F. Frindt and J. C. Irwin, Real-space imaging of single-layer MoS2 by scanning tunneling microscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3490–3493 CrossRef PubMed.
- H. L. Tsai, J. Heising, J. L. Schindler, C. R. Kannewurf and M. G. Kanatzidis, Exfoliated-restacked phase of WS2, Chem. Mater., 1997, 9, 879–882 CrossRef CAS.
- K. K. Amara, et al., Dynamic structural evolution of metal–metal bonding network in monolayer WS2, Chem. Mater., 2016, 28, 2308–2314 CrossRef CAS.
- W. Zhao, et al., Metastable MoS2: Crystal structure, electronic band structure, synthetic approach and intriguing physical properties, Chem. – Eur. J., 2018, 24, 15942–15954 CrossRef CAS PubMed.
- H. L. Zhuang, M. D. Johannes, A. K. Singh and R. G. Hennig, Doping-controlled phase transitions in single-layer MoS2, Phys. Rev. B, 2017, 96, 165305 CrossRef.
- E. Bruyer, et al., Possibility of combining ferroelectricity and Rashba-like spin splitting in monolayers of the 1T-type transition-metal dichalcogenides MX2 (M = Mo, W; X = S, Se, Te), Phys. Rev. B, 2016, 94, 195402 CrossRef.
- C. Rovira and M. H. Whangbo, Factors governing the charge density wave patterns of layered transition-metal compounds of octahedral coordination with d2 and d3 electron counts, Inorg. Chem., 1993, 32, 4094–4097 CrossRef CAS.
- Y. Fang, et al., Structural determination and nonlinear optical properties of new 1T′′′-type MoS2 compound, J. Am. Chem. Soc., 2019, 141, 790–793 CrossRef CAS PubMed.
- B. E. Brown, The crystal structures of WTe2 and high-temperature MoTe2, Acta Crystallogr., 1966, 20, 268–274 CrossRef CAS.
- L. Zhou, et al., Sensitive phonon-based probe for structure identification of 1T′ MoTe2, J. Am. Chem. Soc., 2017, 139, 8396–8399 CrossRef CAS PubMed.
- J. C. Park, et al., Phase-engineered synthesis of centimeter-scale 1T′- and 2H-molybdenum ditelluride thin films, ACS Nano, 2015, 9, 6548–6554 CrossRef CAS PubMed.
- Y. Yoo, Z. P. DeGregorio, Y. Su, S. J. Koester and J. E. Johns, In-plane 2H-1T′ MoTe2 homojunctions synthesized by flux-controlled phase engineering, Adv. Mater., 2017, 29, 1605461 CrossRef PubMed.
- W. G. Dawson and D. W. Bullett, Electronic structure and crystallography of MoTe2 and WTe2, J. Phys. C: Solid State Phys., 1987, 20, 6159–6174 CrossRef CAS.
- S. Chen, T. Goldstein, D. Venkataraman, A. Ramasubramaniam and J. Yan, Activation of new Raman modes by inversion symmetry breaking in type II Weyl semimetal candidate T′-MoTe2, Nano Lett., 2016, 16, 5852–5860 CrossRef CAS PubMed.
- Y. Qi, et al., Superconductivity in Weyl semimetal candidate MoTe2, Nat. Commun., 2016, 7, 11038 CrossRef CAS PubMed.
- J. Jiang, et al., Signature of type-II Weyl semimetal phase in MoTe2, Nat. Commun., 2017, 8, 13973 CrossRef CAS PubMed.
- D. Rhodes, et al., Engineering the structural and electronic phases of MoTe2 through W substitution, Nano Lett., 2017, 17, 1616–1622 CrossRef CAS PubMed.
- Y. Zhou, et al., Pressure-induced Td to 1T′ structural phase transition in WTe2, AIP Adv., 2016, 6, 075008 CrossRef.
- J. Xia, et al., Pressure-induced phase transition in Weyl semimetallic WTe2, Small, 2017, 13, 1701887 CrossRef PubMed.
- Y. Yu, et al., High phase-purity 1T′-MoS2- and 1T′-MoSe2-layered crystals, Nat. Chem., 2018, 10, 638–643 CrossRef CAS PubMed.
- J. Peng, et al., High phase purity of large-sized 1T′-MoS2 monolayers with 2D superconductivity, Adv. Mater., 2019, 31, 1900568 CrossRef PubMed.
- F. Cheng, et al., Interface Engineering of Au(111) for the Growth of 1T′-MoSe2, ACS Nano, 2019, 13, 2316–2323 CAS.
- M. S. Sokolikova, P. C. Sherrell, P. Palczynski, V. L. Bemmer and C. Mattevi, Direct solution-phase synthesis of 1T′ WSe2 nanosheets, Nat. Commun., 2019, 10, 712 CrossRef CAS PubMed.
- Y. Fang, et al., Observation of superconductivity in pressurized 2 M WSe2 crystals, J. Mater. Chem. C, 2019, 7, 8551–8555 RSC.
- Y. Sun, S.-C. Wu, M. N. Ali, C. Felser and B. Yan, Prediction of Weyl semimetal in orthorhombic MoTe2, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 161107 CrossRef.
- Z. Wang, et al., MoTe2: A type-II Weyl topological metal, Phys. Rev. Lett., 2016, 117, 056805 CrossRef PubMed.
- P. Li, et al., Evidence for topological type-II Weyl semimetal WTe2, Nat. Commun., 2017, 8, 2150 CrossRef PubMed.
- Y. Wu, et al., Observation of Fermi arcs in the type-II Weyl semimetal candidate WTe2, Phys. Rev. B, 2016, 94, 121113 CrossRef.
- K. Deng, et al., Experimental observation of topological Fermi arcs in type-II Weyl semimetal MoTe2, Nat. Phys., 2016, 12, 1105–1110 Search PubMed.
- A. Pulkin and O. V. Yazyev, Robustness of the quantum spin Hall insulator phase in monolayer 1T′ transition metal dichalcogenides, J. Electron Spectrosc. Relat. Phenom., 2017, 219, 72–76 CrossRef CAS.
- H. Xu, et al., Observation of gap opening in 1T′ phase MoS2 nanocrystals, Nano Lett., 2018, 18, 5085–5090 CrossRef CAS PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, Colloidal synthesis of 1T-WS2 and 2H-WS2 nanosheets: Applications for photocatalytic hydrogen evolution, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- Q. Liu, et al., Gram-scale aqueous synthesis of stable few-layered 1T-MoS2: Applications for visible-light-driven photocatalytic hydrogen evolution, Small, 2015, 11, 5556–5564 CrossRef CAS PubMed.
- Q. Liu, et al., Stable metallic 1T-WS2 nanoribbons intercalated with ammonia ions: The correlation between structure and electrical/optical properties, Adv. Mater., 2015, 27, 4837–4844 CrossRef CAS PubMed.
- D. Voiry, et al., Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- Y. Guo, et al., Probing the dynamics of the metallic-to-semiconducting structural phase transformation in MoS2 crystals, Nano Lett., 2015, 15, 5081–5088 CrossRef CAS PubMed.
- G. Eda, et al., Coherent atomic and electronic heterostructures of single-layer MoS2, ACS Nano, 2012, 6, 7311–7317 CrossRef CAS PubMed.
- S. J. R. Tan, et al., Chemical stabilization of 1T′ phase transition metal dichalcogenides with giant optical Kerr nonlinearity, J. Am. Chem. Soc., 2017, 139, 2504–2511 CrossRef CAS PubMed.
- S. Jiménez Sandoval, D. Yang, R. F. Frindt and J. C. Irwin, Raman study and lattice dynamics of single molecular layers of MoS2, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3955–3962 CrossRef PubMed.
- X. Ma, et al., Raman scattering in the transition-metal dichalcogenides of 1T′-MoTe2, Td-MoTe2, and Td-WTe2, Phys. Rev. B, 2016, 94, 214105 CrossRef.
- S.-Y. Chen, C. H. Naylor, T. Goldstein, A. T. C. Johnson and J. Yan, Intrinsic Phonon Bands in High-Quality Monolayer T′ Molybdenum Ditelluride, ACS Nano, 2017, 11, 814–820 CrossRef CAS PubMed.
- C.-H. Lee, et al., Tungsten ditelluride: a layered semimetal, Sci. Rep., 2015, 5, 10013 CrossRef CAS PubMed.
- S. J. R. Tan, et al., Temperature- and Phase-Dependent Phonon Renormalization in 1T′-MoS2, ACS Nano, 2018, 12, 5051–5058 CrossRef CAS PubMed.
- D. Pierucci, et al., Evidence for a narrow band gap phase in 1T′ WS2 nanosheet, Appl. Phys. Lett., 2019, 115, 032102 CrossRef.
- C. Guo, et al., Observation of superconductivity in 1T′-MoS2 nanosheets, J. Mater. Chem. C, 2017, 5, 10855–10860 RSC.
- X. Fan, et al., Fast and efficient preparation of exfoliated 2H MoS2 nanosheets by sonication-assisted lithium intercalation and infrared laser-induced 1T to 2H phase reversion, Nano Lett., 2015, 15, 5956–5960 CrossRef CAS PubMed.
- A. D. Mohite and M. Chhowalla, Phase-engineered low-resistance contacts for ultrathin MoS2 transistors, Nat. Mater., 2014, 13, 1128–1134 CrossRef PubMed.
- R. Kappera, et al., Metallic 1T phase source/drain electrodes for field effect transistors from chemical vapor deposited MoS2 transistors from chemical vapor deposited MoS2, APL Mater., 2014, 2, 092516 CrossRef.
- Y. Ma, et al., Reversible semiconducting-to-metallic phase transition in chemical vapor deposition grown monolayer WSe2 and applications for devices, ACS Nano, 2015, 9, 7383–7391 CrossRef CAS PubMed.
- D. Voiry, A. Mohite and M. Chhowalla, Phase engineering of transition metal dichalcogenides, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC.
- L. Niu, et al., Production of Two-Dimensional Nanomaterials via Liquid-Based Direct Exfoliation, Small, 2016, 12, 272–293 CrossRef CAS PubMed.
- D. M. Sim, et al., Long-term stable 2H-MoS2 dispersion: Critical role of solvent for simultaneous phase restoration and surface functionalization of liquid-exfoliated MoS2, ACS Omega, 2017, 2, 4678–4687 CrossRef CAS PubMed.
- M. Acerce, D. Voiry and M. Chhowalla, Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- A. Ambrosi, Z. Sofer and M. Pumera, 2H → 1T phase transition and hydrogen evolution activity of MoS2, MoSe2, WS2 and WSe2 strongly depends on the MX2 composition, Chem. Commun., 2015, 51, 8450–8453 RSC.
- D. Voiry, et al., Conducting MoS2 nanosheets as catalysts for hydrogen evolution reaction, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- L. Wang, Z. Xu, W. Wang and X. Bai, Atomic mechanism of dynamic electrochemical lithiation processes of MoS2 nanosheets, J. Am. Chem. Soc., 2014, 136, 6693–6697 CrossRef CAS PubMed.
- D. Yang, S. J. Sandoval, W. M. R. Divigalpitiya, J. C. Irwin and R. F. Frindt, Structure of single-molecular-layer MoS2, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 12053–12056 CrossRef CAS PubMed.
- K. K. Bardhan, J. C. Wu, J. S. Culik, S. H. Anderson and D. D. L. Chung, Kinetics of intercalation and desorption in graphite, Synth. Met., 1980, 2, 57–84 CrossRef CAS.
- M. Azhagurajan, T. Kajita, T. Itoh, Y.-G. Kim and K. Itaya, In situ visualization of lithium ion intercalation into MoS2 single crystals using differential optical microscopy with atomic layer resolution, J. Am. Chem. Soc., 2016, 138, 3355–3361 CrossRef CAS PubMed.
- Z. Zeng, et al., Single-layer semiconducting nanosheets: High-yield preparation and device fabrication, Angew. Chem., Int. Ed., 2011, 50, 11093–11097 CrossRef CAS PubMed.
- X. Zhu, D. Li, X. Liang and W. D. Lu, Ionic modulation and ionic coupling effects in MoS2 devices for neuromorphic computing, Nat. Mater., 2019, 18, 141–148 CrossRef CAS PubMed.
- F. Xiong, et al., Li intercalation in MoS2: In situ observation of its dynamics and tuning optical and electrical properties, Nano Lett., 2015, 15, 6777–6784 CrossRef CAS PubMed.
- J. Zheng, et al., High yield exfoliation of two-dimensional chalcogenides using sodium naphthalenide, Nat. Commun., 2014, 5, 2995 CrossRef.
- C. Tan, et al., Preparation of high-percentage 1T-phase transition metal dichalcogenide nanodots for electrochemical hydrogen evolution, Adv. Mater., 2018, 30, 1705509 CrossRef PubMed.
- M. Naz, et al., A new 2H-2H′/1T cophase in polycrystalline MoS2 and MoSe2 thin films, ACS Appl. Mater. Interfaces, 2016, 8, 31442–31448 CrossRef CAS PubMed.
- L. Sun, et al., Layer-dependent chemically induced phase transition of two-dimensional MoS2, Nano Lett., 2018, 18, 3435–3440 CrossRef CAS PubMed.
- A. Y. S. Eng, A. Ambrosi, Z. Sofer, P. Šimek and M. Pumera, Electrochemistry of transition metal dichalcogenides: Strong dependence on the metal-to-chalcogen composition and exfoliation method, ACS Nano, 2014, 8, 12185–12198 CrossRef CAS PubMed.
- A. L. Friedman, et al., Evidence for chemical vapor induced 2H to 1T phase transition in MoX2 (X = Se, S) transition metal dichalcogenide films, Sci. Rep., 2017, 7, 3836 CrossRef PubMed.
- M. Wuttig and N. Yamada, Phase-change materials for rewriteable data storage, Nat. Mater., 2007, 6, 824–832 CrossRef CAS PubMed.
- J.-J. Kim, et al., Observation of a phase transition from the T phase to the H phase induced by a STM tip in 1T-TaS2, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, R15573–R15576 CrossRef CAS.
- Y. Li, K.-A. N. Duerloo, K. Wauson and E. J. Reed, Structural semiconductor-to-semimetal phase transition in two-dimensional materials induced by electrostatic gating, Nat. Commun., 2016, 7, 10671 CrossRef CAS PubMed.
- Y. Wang, et al., Structural phase transition in monolayer MoTe2 driven by electrostatic doping, Nature, 2017, 550, 487–491 CrossRef CAS PubMed.
- C. Zhang, et al., Charge mediated reversible metal–insulator transition in monolayer MoTe2 and WxMo1–xTe2 alloy, ACS Nano, 2016, 10, 7370–7375 CrossRef CAS PubMed.
- Y. Sun, Z. Shuai and D. Wang, Janus monolayer of WSeTe, a new structural phase transition material driven by electrostatic gating, Nanoscale, 2018, 10, 21629–21633 RSC.
- S. Kim, et al., Long-range lattice engineering of MoTe2 by a 2D electride, Nano Lett., 2017, 17, 3363–3368 CrossRef CAS.
- J. L. Dye, Electrons as Anions, Nature, 2003, 301, 607–609 CAS.
- K. Lee, S. W. Kim, Y. Toda, S. Matsuishi and H. Hosono, Dicalcium nitride as a two-dimensional electride with an anionic electron layer, Nature, 2013, 494, 336–340 CrossRef CAS.
- D. L. Dru, et al., Experimental demonstration of an electride as a 2D material, J. Am. Chem. Soc., 2016, 138, 16089–16094 CrossRef.
- Y. Kang, et al., Plasmonic hot electron induced structural phase transition in a MoS2 monolayer, Adv. Mater., 2014, 26, 6467–6471 CrossRef CAS PubMed.
- X. Yin, et al., Tunable inverted gap in monolayer quasi-metallic MoS2 induced by strong charge-lattice coupling, Nat. Commun., 2017, 8, 486 CrossRef PubMed.
- H. H. Huang, et al., Controlling phase transition for single-layer MTe2 (M = Mo and W): modulation of the potential barrier under strain, Phys. Chem. Chem. Phys., 2016, 18, 4086–4094 RSC.
- S. Song, et al., Room temperature semiconductor-metal transition of MoTe2 thin films engineered by strain, Nano Lett., 2015, 16, 188–193 CrossRef.
- C. H. Sharma, A. P. Surendran, A. Varghese and M. Thalakulam, Stable and scalable 1T MoS2 with low temperature-coefficient of resistance, Sci. Rep., 2018, 8, 12463 CrossRef PubMed.
- S. Reshmi, M. V. Akshaya, B. Satpati, P. K. Basu and K. Bhattacharjee, Structural stability of coplanar 1T-2H superlattice MoS2 under high energy electron beam, Nanotechnology, 2018, 29, 205604 CrossRef CAS PubMed.
- Y. Qi, et al., CO2-Induced Phase Engineering: Protocol for Enhanced Photoelectrocatalytic Performance of 2D MoS2 Nanosheets, ACS Nano, 2016, 10, 2903–2909 CrossRef CAS PubMed.
- X. Tong, Y. Qi, J. Chen, N. Wang and Q. Xu, Supercritical CO2-Assisted Reverse-Micelle-Induced Solution-Phase Fabrication of Two-Dimensional Metallic 1T-MoS2 and 1T-WS2, ChemNanoMat, 2017, 3, 466–471 CrossRef CAS.
- X. Gan, et al., 2H/1T phase transition of multilayer MoS2 by electrochemical incorporation of S vacancies, ACS Appl. Energy Mater., 2018, 1, 4754–4765 CrossRef CAS.
- W. Ding, et al., Highly Ambient-Stable 1T-MoS2 and 1T-WS2 by Hydrothermal Synthesis under High Magnetic Fields, ACS Nano, 2019, 13, 1694–1702 CAS.
- Y. Tan, et al., Controllable 2H-to-1T′ phase transition in few-layer MoTe2, Nanoscale, 2018, 10, 19964–19971 RSC.
- J. Zhu, et al., Argon plasma induced phase transition in monolayer MoS2, J. Am. Chem. Soc., 2017, 139, 10216–10219 CrossRef CAS PubMed.
- Z. Wang, et al., Local engineering of topological phase in monolayer MoS2, Sci. Bull., 2019, 64, 1750–1756 CrossRef CAS.
- H. Mine, et al., Laser-beam-patterned topological insulating states on thin semiconducting MoS2, Phys. Rev. Lett., 2019, 123, 146803 CrossRef CAS PubMed.
- Z. Wang, et al., Synthesizing 1T–1H two-phase Mo1–xWxS2 monolayers by chemical vapor deposition, ACS Nano, 2018, 12, 1571–1579 CrossRef CAS PubMed.
- F. Wypych, T. Weber and R. Prins, Scanning tunneling microscopic investigation of Kx(H2O)yMoS2, Surf. Sci., 1997, 380, L474–L478 CrossRef.
- Y. Fang, et al., Structure re-determination and superconductivity observation of bulk 1T MoS2, Angew. Chem., 2018, 130, 1246–1249 CrossRef.
- C. Guo, et al., High-quality single-layer nanosheets of MS2 (M = Mo, Nb, Ta, Ti) directly exfoliated from AMS2 (A = Li, Na, K) Crystals, J. Mater. Chem. C, 2017, 5, 5977–5983 RSC.
- F. Wypych, C. Solenthaler, R. Prins and T. Weber, Electron diffraction study of intercalation compounds derived from 1T-MoS2, J. Solid State Chem., 1999, 436, 430–436 CrossRef.
- F. Wypych, T. Weber and R. Prins, Scanning tunneling microscopic investigation of 1T-MoS2, Surf. Sci., 1998, 380, L474–L478 CrossRef.
- L. Liu, et al., Phase-selective synthesis of 1T′ MoS2 monolayers and heterophase bilayers, Nat. Mater., 2018, 17, 1108–1114 CrossRef CAS PubMed.
- Z. Liu, et al., Colloidal synthesis of 1T′ phase dominated WS2 towards endurable electrocatalysis, Nano Energy, 2018, 50, 176–181 CrossRef CAS.
- J. Ekspong, et al., Stable sulfur-intercalated 1T′ MoS2 on graphitic nanoribbons as hydrogen evolution electrocatalyst, Adv. Funct. Mater., 2018, 28, 1802744 CrossRef.
- Q. Liu, et al., Electron-doped 1T-MoS2 via interface engineering for enhanced electrocatalytic hydrogen evolution, Chem. Mater., 2017, 29, 4738–4744 CrossRef CAS.
- J. Tang, et al., MXene derived TiS2 nanosheets for high-rate and long-life sodium-ion capacitors, Energy Storage Mater., 2020, 26, 550–559 CrossRef.
- Z. Du, et al., Conversion of non-van der Waals solids to 2D transition-metal chalcogenides, Nature, 2020, 577, 492–496 CrossRef CAS PubMed.
- D. H. Keum, et al., Bandgap opening in few-layered monoclinic MoTe2, Nat. Phys., 2015, 11, 482–486 Search PubMed.
- X. Xu, et al., Millimeter-Scale Single-Crystalline Semiconducting MoTe2 via Solid-to-Solid Phase Transformation, J. Am. Chem. Soc., 2019, 141, 2128–2134 CrossRef CAS PubMed.
- S. Cho, et al., Phase patterning for ohmic homojunction contact in MoTe2, Science, 2015, 349, 625–628 CrossRef CAS PubMed.
- L. Zhou, et al., Synthesis of high-quality large-area homogenous 1T′ MoTe2 from chemical vapor deposition, Adv. Mater., 2016, 28, 9526–9531 CrossRef CAS PubMed.
- L. Zhou, et al., Large-area synthesis of high-quality uniform few-layer MoTe2, J. Am. Chem. Soc., 2015, 137, 11892–11895 CrossRef CAS.
- L. Zhou, et al., Role of molecular sieves in the CVD synthesis of large-area 2D MoTe2, Adv. Funct. Mater., 2017, 27, 1603491 CrossRef.
- L. Yang, et al., Tellurization velocity-dependent metallic–semiconducting–metallic phase evolution in chemical vapor deposition growth of large-area, few-layer MoTe2, ACS Nano, 2017, 11, 1964–1972 CrossRef CAS PubMed.
- R. Ma, et al., MoTe2 lateral homojunction field-effect transistors fabricated using flux-controlled phase engineering, ACS Nano, 2019, 13, 8035–8046 CrossRef CAS PubMed.
- J. Zhou, et al., Large-Area and High-Quality 2D Transition Metal Telluride, Adv. Mater., 2017, 29, 1603471 CrossRef PubMed.
- R. Sankar, et al., Polymorphic layered MoTe2 from semiconductor, topological insulator, to Weyl semimetal, Chem. Mater., 2017, 29, 699–707 CrossRef CAS.
- T. A. Empante, et al., Chemical vapor deposition growth of few-layer MoTe2 in the 2H, 1T′, and 1T phases: Tunable properties of MoTe2 films, ACS Nano, 2017, 11, 900–905 CrossRef CAS PubMed.
- Y.-C. Lin, et al., Stable 1T tungsten disulfide monolayer and its junctions: Growth and atomic structures, ACS Nano, 2018, 12, 12080–12088 CrossRef CAS.
- B.-R. Chen, et al., Understanding crystallization pathways leading to manganese oxide polymorph formation, Nat. Commun., 2018, 9, 2553 CrossRef.
- S. Lee, et al., Multiple pathways of crystal nucleation in an extremely supersaturated aqueous potassium dihydrogen phosphate (KDP) solution droplet, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13618–13623 CrossRef CAS.
- H. Zhang and J. F. Banfield, Understanding polymorphic phase transformation behavior during growth of nanocrystalline aggregates: Insights from TiO2, J. Phys. Chem. B, 2000, 104, 3481–3487 CrossRef CAS.
- J. D. Dunitz and J. Bernstein, Disappearing polymorphs, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- R. A. Van Santen, The Ostwald step rule, J. Phys. Chem., 1984, 88, 5768–5769 CrossRef CAS.
- W. Sun and G. Ceder, Induction time of a polymorphic transformation, CrystEngComm, 2017, 19, 4576–4585 RSC.
- D. Bonn and N. Shahidzadeh, Multistep crystallization processes: How not to make perfect single crystals, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13551–13553 CrossRef CAS.
- D.-K. Bučar, R. W. Lancaster and J. Bernstein, Disappearing polymorphs revisited, Angew. Chem., Int. Ed., 2015, 54, 6972–6993 CrossRef.
- Y. Sun, et al., Low-temperature solution synthesis of few-layer 1T′-MoTe2 nanostructures exhibiting lattice compression, Angew. Chem., Int. Ed., 2016, 55, 2830–2834 CrossRef CAS.
- Y. Sun, K. Fujisawa, M. Terrones and R. E. Schaak, Solution synthesis of few-layer WTe2 and Mo(x)W(1−x)Te2 nanostructures, J. Mater. Chem. C, 2017, 5, 11317–11323 RSC.
- N. Savjani, et al., Synthesis of lateral size-controlled monolayer 1H-MoS2@oleylamine as supercapacitor electrodes, Chem. Mater., 2016, 28, 657–664 CrossRef CAS.
- D. Son, et al., Colloidal synthesis of uniform-sized molybdenum disulfide nanosheets for wafer-scale flexible nonvolatile memory, Adv. Mater., 2016, 28, 9326–9332 CrossRef CAS PubMed.
- W. Jung, et al., Colloidal synthesis of single-layer MSe2 (M = Mo, W) nanosheets via anisotropic solution-phase growth approach, J. Am. Chem. Soc., 2015, 137, 7266–7269 CrossRef CAS PubMed.
- L. Cheng, et al., Ultrathin WS2 nanoflakes as a high-performance electrocatalyst for the hydrogen evolution reaction, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed.
- D. Sun, S. Feng, M. Terrones and R. E. Schaak, Formation and interlayer decoupling of colloidal MoSe2 nanoflowers, Chem. Mater., 2015, 27, 3167–3175 CrossRef CAS.
- Y. Sun, et al., Low-temperature solution synthesis of transition metal dichalcogenide alloys with tunable optical properties, J. Am. Chem. Soc., 2017, 139, 11096–11105 CrossRef CAS.
- X. Zhou, et al., Fast colloidal synthesis of scalable Mo-rich hierarchical ultrathin MoSe(2−x) nanosheets for high-performance hydrogen evolution, Nanoscale, 2014, 6, 11046–11051 RSC.
- C. Zhang, et al., Phosphine-free synthesis and shape evolution of MoSe2 nanoflowers for electrocatalytic hydrogen evolution reactions, CrystEngComm, 2018, 20, 2491–2498 RSC.
- O. E. Meiron, V. Kuraganti, I. Hod, R. Bar-Ziv and M. Bar-Sadan, Improved catalytic activity of Mo(1−x)W(x)Se2 alloy nanoflowers promotes efficient hydrogen evolution reaction in both acidic and alkaline aqueous solutions, Nanoscale, 2017, 9, 13998–14005 RSC.
- R. Mastria, et al., In-plane aligned colloidal 2D WS2 nanoflakes for solution-processable thin films with high planar conductivity, Sci. Rep., 2019, 9, 9002 CrossRef.
- D. D. Vaughn, R. J. Patel, M. A. Hickner and R. E. Schaak, Single-Crystal Colloidal Nanosheets of GeS and GeSe, J. Am. Chem. Soc., 2010, 132, 15170–15172 CrossRef CAS PubMed.
- M. M. Kobylinski, C. Ruhmlieb, A. Kornowski and A. Mews, Hexagonally shaped two-dimensional tin(II) sulfide nanosheets: Growth model and controlled structure formation, J. Phys. Chem. C, 2018, 122, 5784–5795 CrossRef CAS.
- Z. Deng, et al., Solution synthesis of ultrathin single-crystalline SnS nanoribbons for photodetectors via phase transition and surface processing, ACS Nano, 2012, 6, 6197–6207 CrossRef CAS PubMed.
- J. Q. Geisenhoff, A. K. Tamura and A. M. Schimpf, Using ligands to control reactivity, size and phase in the colloidal synthesis of WSe2 nanocrystals, Chem. Commun., 2019, 55, 8856–8859 RSC.
- D. D. Vaughn, S. In Il and R. E. Schaak, A precursor-limited nanoparticle coalescence pathway for tuning the thickness of laterally-uniform colloidal nanosheets: The case of SnSe, ACS Nano, 2011, 5, 8852–8860 CrossRef CAS PubMed.
- K. H. Park, K. Jang, S. Kim, H. J. Kim and S. U. Son, Phase-controlled one-dimensional shape evolution of InSe nanocrystals, J. Am. Chem. Soc., 2006, 128, 14780–14781 CrossRef CAS.
- D. Yoo, M. Kim, S. Jeong, J. Han and J. Cheon, Chemical synthetic strategy for single-layer transition-metal chalcogenides, J. Am. Chem. Soc., 2014, 136, 14670–14673 CrossRef CAS PubMed.
- S. Jeong, D. Yoo, J. Jang, M. Kim and J. Cheon, Well-defined colloidal 2-D layered transition-metal chalcogenide nanocrystals via generalized synthetic protocols, J. Am. Chem. Soc., 2012, 134, 18233–18236 CrossRef CAS PubMed.
- W. Li, et al., Flowerlike WSe2 and WS2 microspheres – one-pot synthesis, formation mechanism and application in heavy metal ion sequestration, Chem. Commun., 2016, 52, 4481–4484 RSC.
- Y. Xie, et al., Metallic-like stoichiometric copper sulfide nanocrystals: Phase- and shape-selective synthesis, near-infrared surface plasmon resonance properties, and their modeling, ACS Nano, 2013, 7, 7352–7369 CrossRef CAS PubMed.
- J. Song, et al., Solvothermal growth of bismuth chalcogenide nanoplatelets by the oriented attachment mechanism – an in situ PXRD study, Chem. Mater., 2015, 27, 3471–3482 CrossRef CAS.
- C. Schliehe, et al., Ultrathin PbS sheets by two-dimensional oriented attachment, Science, 2010, 329, 550–553 CrossRef CAS PubMed.
- S. Prabakar, C. W. Bumby and R. D. Tilley, Liquid-phase synthesis of flower-like and flake-like titanium disulfide nanostructures, Chem. Mater., 2009, 21, 1725–1730 CrossRef CAS.
- O. E. Meiron, L. Houben and M. Bar-Sadan, Understanding the formation mechanism and the 3D structure of Mo(SxSe1−x)2 nanoflower, RSC Adv., 2015, 5, 88108–88114 RSC.
- X. Geng, et al., Pure and stable metallic phase molybdenum disulfide nanosheets for hydrogen evolution reaction, Nat. Commun., 2016, 7, 10672 CrossRef CAS PubMed.
- K. Leng, et al., Phase restructuring in transition metal dichalcogenides for highly stable energy storage, ACS Nano, 2016, 10, 9208–9215 CrossRef CAS PubMed.
- M. Z. M. Nasir, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, Two-dimensional 1T-phase transition metal dichalcogenides as nanocarriers to enhance and stabilize enzyme activity for electrochemical pesticide detection, ACS Nano, 2017, 11, 5774–5784 CrossRef CAS PubMed.
- E. Rahmanian, et al., 1T-phase tungsten chalcogenides (WS2, WSe2, WTe2) decorated with TiO2 nanoplatelets with enhanced electron transfer activity for biosensing applications, ACS Appl. Nano Mater., 2018, 1, 7006–7015 CrossRef CAS.
- W. Wei, K. Sun and Y. H. Hu, An efficient counter electrode material for dye-sensitized solar cells—flower-structured 1T metallic phase MoS2, J. Mater. Chem. A, 2016, 4, 12398–12401 RSC.
- P. Huang, et al., Water-Soluble 2D Transition Metal Dichalcogenides as the Hole-Transport Layer for Highly Efficient and Stable p–i–n Perovskite Solar Cells, ACS Appl. Mater. Interfaces, 2017, 9, 25323–25331 CrossRef CAS PubMed.
- M. Piao, et al., Crystal phase control synthesis of metallic 1T-WS2 nanosheets incorporating single walled carbon nanotubes to construct superior microwave absorber, J. Alloys Compd., 2020, 815, 152335 CrossRef CAS.
- L. Cai, et al., Vacancy-induced ferromagnetism of MoS2 nanosheets, J. Am. Chem. Soc., 2015, 137, 2622–2627 CrossRef CAS PubMed.
- C. Yang, et al., Phase-driven magneto-electrical characteristics of single-layer MoS2, Nanoscale, 2016, 8, 5627–5633 RSC.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, Catalyzing the hydrogen evolution reaction (HER) with molybdenum sulfide nanomaterials, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- Z. Gholamvand, et al., Comparison of liquid exfoliated transition metal dichalcogenides reveals MoSe2 to be the most effective hydrogen evolution catalyst, Nanoscale, 2016, 8, 5737–5749 RSC.
- H. Li, et al., Activating and optimizing MoS2 basal planes for hydrogen evolution through the formation of strained sulphur vacancies, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- D. Voiry, et al., The role of electronic coupling between substrate and 2D MoS2 nanosheets in electrocatalytic production of hydrogen, Nat. Mater., 2016, 15, 1003–1009 CrossRef CAS PubMed.
- Y. Yan, et al., Vertically oriented MoS2 and WS2 nanosheets directly grown on carbon cloth as efficient and stable 3-dimensional hydrogen-evolving cathodes, J. Mater. Chem. A, 2015, 3, 131–135 RSC.
- M. Zou, et al., WSe2 and W(SexS1−x)2 nanoflakes grown on carbon nanofibers for the electrocatalytic hydrogen evolution reaction, J. Mater. Chem. A, 2015, 3, 18090–18097 RSC.
- L. Lin, et al., Engineered 2D Transition Metal Dichalcogenides—A Vision of Viable Hydrogen Evolution Reaction Catalysis, Adv. Energy Mater., 2020, 10, 1903870 CrossRef CAS.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, 2D Transition-Metal-Dichalcogenide-Nanosheet-Based Composites for Photocatalytic and Electrocatalytic Hydrogen Evolution Reactions, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- J. K. Nørskov, et al., Trends in the exchange current for hydrogen evolution, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- B. Hinnemann, et al., Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- M. V. Bollinger, et al., One-dimensional metallic edge states in MoS2, Phys. Rev. Lett., 2001, 87, 196803 CrossRef CAS PubMed.
- A. Parija, et al., Mapping catalytically relevant edge electronic states of MoS2, ACS Cent. Sci., 2018, 4, 493–503 CrossRef CAS PubMed.
- C. Tsai, K. Chan, F. Abild-Pedersen and J. K. Nørskov, Active edge sites in MoSe2 and WSe2 catalysts for the hydrogen evolution reaction: a density functional study, Phys. Chem. Chem. Phys., 2014, 16, 13156–13164 RSC.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Theoretical insights into the hydrogen evolution activity of layered transition metal dichalcogenides, Surf. Sci., 2015, 640, 133–140 CrossRef CAS.
- M. A. Lukowski, et al., Enhanced hydrogen evolution catalysis from chemically exfoliated metallic MoS2 nanosheets, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- S. S. Chou, et al., Understanding catalysis in a multiphasic two-dimensional transition metal dichalcogenide, Nat. Commun., 2015, 6, 8311 CrossRef CAS PubMed.
- Y. Zhou, et al., Revealing the contribution of individual factors to hydrogen evolution reaction catalytic activity, Adv. Mater., 2018, 30, 1706076 CrossRef PubMed.
- L. Wang, et al., Self-optimization of the active site of molybdenum disulfide by an irreversible phase transition during photocatalytic hydrogen evolution, Angew. Chem., Int. Ed., 2017, 56, 7610–7614 CrossRef CAS PubMed.
- J. Zhu, et al., Boundary activated hydrogen evolution reaction on monolayer MoS2, Nat. Commun., 2019, 10, 1348 CrossRef PubMed.
- Y. Ouyang, et al., Activating Inert Basal Planes of MoS2 for Hydrogen Evolution Reaction through the Formation of Different Intrinsic Defects, Chem. Mater., 2016, 28, 4390–4396 CrossRef CAS.
- Q. Ji, et al., Metallic vanadium disulfide nanosheets as a platform material for multifunctional electrode applications, Nano Lett., 2017, 17, 4908–4916 CrossRef CAS PubMed.
- R. Naz, et al., Highly defective 1T-MoS2 nanosheets on 3D reduced graphene oxide networks for supercapacitors, Carbon, 2019, 152, 697–703 CrossRef CAS.
- X. Wang, et al., Unveiling highly ambient-stable multilayered 1T-MoS2 towards all-solid-state flexible supercapacitors, J. Mater. Chem. A, 2019, 7, 19152–19160 RSC.
- B. Xu, et al., Adsorption and diffusion of lithium on 1T-MoS2 monolayer, Comput. Mater. Sci., 2014, 93, 86–90 CrossRef CAS.
- T. Xiang, et al., Vertical 1T-MoS2 nanosheets with expanded interlayer spacing edged on a graphene frame for high rate lithium-ion batteries, Nanoscale, 2017, 9, 6975–6983 RSC.
- X. Cao, C. Tan, X. Zhang, W. Zhao and H. Zhang, Solution-Processed Two-Dimensional Metal Dichalcogenide-Based Nanomaterials for Energy Storage and Conversion, Adv. Mater., 2016, 28, 6167–6196 CrossRef CAS PubMed.
- S. Shi, Z. Sun and Y. H. Hu, Synthesis, stabilization and applications of 2-dimensional 1T metallic MoS2, J. Mater. Chem. A, 2018, 6, 23932–23977 RSC.
- Q. Yun, et al., Layered Transition Metal Dichalcogenide-Based Nanomaterials for Electrochemical Energy Storage, Adv. Mater., 2020, 32, 1903826 CrossRef CAS PubMed.
- L. Tao, et al., 1T′ transition metal telluride atomic layers for plasmon-free SERS at femtomolar levels, J. Am. Chem. Soc., 2018, 140, 8696–8704 CrossRef CAS PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Single-layer MoS2 transistors, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed.
- Z. Yin, et al., Single-layer MoS2 phototransistors, ACS Nano, 2012, 6, 74–80 CrossRef CAS PubMed.
- O. Lopez-Sanchez, D. Lembke, M. Kayci, A. Radenovic and A. Kis, Ultrasensitive photodetectors based on monolayer MoS2, Nat. Nanotechnol., 2013, 8, 497–501 CrossRef CAS PubMed.
- B. W. H. Baugher, H. O. H. Churchill, Y. Yang and P. Jarillo-Herrero, Optoelectronic devices based on electrically tunable p–n diodes in a monolayer dichalcogenide, Nat. Nanotechnol., 2014, 9, 262–267 CrossRef CAS PubMed.
- S. Das, H.-Y. Chen, A. V. Penumatcha and J. Appenzeller, High performance multilayer MoS2 transistors with scandium contacts, Nano Lett., 2013, 13, 100–105 CrossRef CAS PubMed.
- J. H. Sung, et al., Coplanar semiconductor-metal circuitry defined on few-layer MoTe2 via polymorphic heteroepitaxy, Nat. Nanotechnol., 2017, 12, 1064–1070 CrossRef CAS PubMed.
- X. Xu, et al., Scaling-up Atomically Thin Coplanar Semiconductor–Metal Circuitry via Phase Engineered Chemical Assembly, Nano Lett., 2019, 19, 6845–6852 CrossRef CAS PubMed.
- W. Zhang, R. Mazzarello, M. Wuttig and E. Ma, Designing crystallization in phase-change materials for universal memory and neuro-inspired computing, Nat. Rev. Mater., 2019, 4, 150–168 CrossRef CAS.
- R. H. Friend and A. D. Yoffe, Electronic properties of intercalation complexes of the transition metal dichalcogenides, Adv. Phys., 1987, 36, 1–94 CrossRef CAS.
- R. B. Somoano, V. Hadek and A. Rembaum, Alkali metal intercalates of molybdenum disulfide, J. Chem. Phys., 1973, 58, 697–701 CrossRef CAS.
- R. Zhang, et al., Superconductivity in potassium-doped metallic polymorphs of MoS2, Nano Lett., 2016, 16, 629–636 CrossRef CAS PubMed.
- X.-C. Pan, et al., Pressure-driven dome-shaped superconductivity and electronic structural evolution in tungsten ditelluride, Nat. Commun., 2015, 6, 7805 CrossRef PubMed.
- G. Nam, et al., In-plane anisotropic properties of 1T′-MoS2 layers, Adv. Mater., 2019, 31, 1807764 CrossRef PubMed.
- F. Xia, H. Wang and Y. Jia, Rediscovering black phosphorus as an anisotropic layered material for optoelectronics and electronics, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- I. Pletikosić, M. N. Ali, A. V. Fedorov, R. J. Cava and T. Valla, Electronic structure basis for the extraordinary magnetoresistance in WTe2, Phys. Rev. Lett., 2014, 113, 216601 CrossRef PubMed.
- M. N. Ali, et al., Large, non-saturating magnetoresistance in WTe2, Nature, 2014, 514, 205–208 CrossRef CAS PubMed.
- F. C. Chen, et al., Extremely large magnetoresistance in the type-II Weyl semimetal MoTe2, Phys. Rev. B, 2016, 94, 235154 CrossRef.
- S. Lee, et al., Origin of extremely large magnetoresistance in the candidate type-II Weyl semimetal MoTe2−x, Sci. Rep., 2018, 8, 13937 CrossRef PubMed.
- J. Yang, et al., Elastic and electronic tuning of magnetoresistance in MoTe2, Sci. Adv., 2017, 3, eaao4949 CrossRef PubMed.
- F. M. Pesci, et al., MoS2/WS2 heterojunction for photoelectrochemical water oxidation, ACS Catal., 2017, 7, 4990–4998 CrossRef CAS.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Liquid exfoliation of layered materials, Science, 2013, 340, 1226419 CrossRef.
- D. McManus, et al., Water-based and biocompatible 2D crystal inks for all-inkjet-printed heterostructures, Nat. Nanotechnol., 2017, 12, 343–350 CrossRef CAS PubMed.
- J. Orangi, F. Hamade, V. A. Davis and M. Beidaghi, 3D Printing of additive-free 2D Ti3C2Tx (MXene) ink for fabrication of micro-supercapacitors with ultra-high energy densities, ACS Nano, 2020, 14, 640–650 CrossRef CAS PubMed.
- W. Yang, et al., 3D printing of freestanding MXene architectures for current-collector-free supercapacitors, Adv. Mater., 2019, 31, 1902725 CrossRef PubMed.
- Y.-Y. Peng, et al., All-MXene (2D titanium carbide) solid-state microsupercapacitors for on-chip energy storage, Energy Environ. Sci., 2016, 9, 2847–2854 RSC.
- H. K. Ng, et al., Effects of structural phase transition on thermoelectric performance in lithium-intercalated molybdenum disulfide (LixMoS2), ACS Appl. Mater. Interfaces, 2019, 11, 12184–12189 CrossRef CAS PubMed.
- G. Eda, et al., Photoluminescence from chemically exfoliated MoS2, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- C. Guo, et al., High-quality single-layer nanosheets of MS2 (M = Mo, Nb, Ta, Ti) directly exfoliated from AMS2 (A = Li, Na, K) crystals, J. Mater. Chem. C, 2017, 5, 5977–5983 RSC.
| This journal is © The Royal Society of Chemistry 2020 |