
Heterometal incorporation in NH2-MIL-125(Ti) and its participation in the photoinduced charge-separated excited state†
Lauren
Hanna
a,
Conor L.
Long
a,
Xiaoyi
Zhang
 b and
Jenny V.
Lockard
*a
b and
Jenny V.
Lockard
*a
aDepartment of Chemistry, Rutgers University-Newark, Newark, NJ, USA. E-mail: jlockard@newark.rutgers.edu
bAdvanced Photon Source, Argonne National Laboratory, Lemont, IL, USA
First published on 26th August 2020
Abstract
Optical and X-ray spectroscopy studies reveal the location and role of Fe3+ sites incorporated through direct synthesis in NH2-MIL-125(Ti). Fe K-edge XAS analysis confirms its metal–oxo cluster node coordination while time-resolved optical and X-ray transient absorption studies disclose its role as an electron trap site, promoting long-lived photo-induced charge separation in the framework. Notably, XTA measurements show sustained electron reduction of the Fe sites into the microsecond time range. Comparison with an Fe-doped MOF generated through post-synthetic modification indicates that only the direct synthesis approach affords efficient Fe participation in the charge separated excited state.
Efficient photocatalysis for solar energy applications requires materials that exhibit long lived charge separation properties and reversible electron transfer behavior of the catalytic sites. Hybrid materials, such as metal–organic frameworks (MOFs), offer new opportunities in this field through their rich chemical and structural diversity that allows incorporating accessible catalytic sites and tailoring optoelectronic properties. MOFs are porous solid state networks composed of self-assembled metal ions or clusters connected through coordination bonds with organic linkers.1 Photocatalytic behavior in MOFs can be promoted through inclusion of transition metal centers with multiple stable oxidation states and linker sites that can accommodate stable radical entities in the excited state. MIL-125 is a titanium-based MOF, containing octameric Ti–oxo clusters connected in three dimensions by terephthalate linkers.2 The reversible Ti4+/Ti3+ redox conversion at the metal node sites within the framework is accessible through photo-induced linker-to-metal cluster charge transfer (LMCCT) but requires UV-wavelength light irradiation. Borrowing from the heteroatom doping strategies used in traditional inorganic semiconductors like TiO2,3 analogous modifications of MIL-125 and other Ti-based MOFs have been explored to tune the range of usable absorption wavelengths and photoredox properties without simultaneously introducing high rates of charge recombination.4 Introducing an amino group auxochrome to the terephthalate linker precursor for example, red shifts the LMCCT absorption band into the visible region for the modified MOF, NH2-MIL-125(Ti).5 This and other linker substituents tune the overall electronic structure of the MIL-125 framework,4a,c but without significantly impacting the unoccupied orbitals localized on the Ti–oxo nodal cluster and therefore the reductive potential of the photoexcited MOF.4a,6 To modify these unoccupied frontier orbitals, the metal cluster composition should be adjusted as revealed by a recent theoretical investigation of NH2-MIL-125(Ti) upon substitution of different heterometals into the Ti–oxo core.7 Furthermore, possible experimental validation can be found in recent reports of Cu-incorporated NH2-MIL-125(Ti) frameworks that demonstrated enhanced photocatalytic activity for CO2 reduction compared to the parent NH2-MIL-125(Ti) catalyst.4b,d Despite these promising first steps, the local coordination of the heterometal in the MOF cluster has not been experimentally confirmed nor has its direct participation in the photoinduced charge separated excited state tied to the photocatalytic reaction.
In this communication, the coordination environment and excited state contribution of a heterometal incorporated in NH2-MIL-125(Ti) nodal cluster is definitively revealed for the first time. Specifically, we focus on an iron-doped version, NH2-MIL-125(Ti,Fe) with targeted stoichiometry of one Fe3+ center per octameric cluster (Fig. 1a) as a model system. Since doping TiO2 with Fe was found to increase photoactivity by lowering the conduction band, and creating trap states that delays charge recombination,8 we expected that upon population of the LMCCT state of NH2-MIL-125(Ti,Fe), the photo-excited electron would analogously localize onto the Fe3+ sites, generating long-lived transient Fe2+ species.
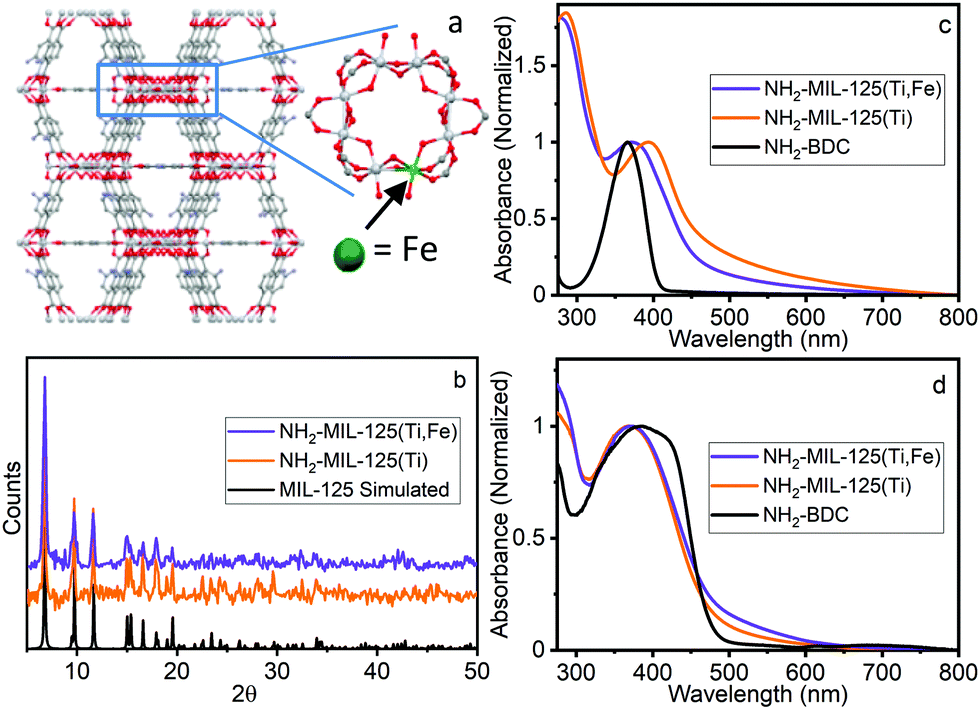 | ||
| Fig. 1 (a) Proposed structure of NH2-MIL-125(Ti,Fe), (b) PXRD, (c) UV-vis absorption and (d) DR spectra of NH2-BDC, NH2-MIL-125(Ti), NH2-MIL-125(Ti,Fe). | ||
X-ray absorption spectroscopy (XAS) is ideally suited for probing the Fe site coordination and electronic structure dynamics associated with the LMCCT state. X-ray transient absorption (XTA) measurements can track electronic structure changes induced by laser excitation and, because of its element specificity, has become an important tool for investigating the photochemistry of both molecular9 and solid-state10 metal-based systems, including other Fe-containing MOFs.11 Here Fe K-edge XAS and XTA are used along with optical transient absorption (OTA) spectroscopy to confirm the location of the Fe sites and investigate the nature and dynamics of the LMCCT and subsequent charge separation.
NH2-MIL-125(Ti) was synthesized following literature procedures.4d NH2-MIL-125(Ti,Fe) was generated using a modified direct synthesis method, where stoichiometric amounts of titanium(IV) tert-butoxide and iron(III) chloride precursors were combined with the 2-aminoterephthalic acid (NH2-BDC) linkers under anhydrous DMF and methanol solvent conditions to achieve a target average doping level of about one Fe site per Ti–oxo cluster as confirmed by atomic absorption spectroscopy (see ESI†). Both MOF syntheses included cetyltrimethylammoniumbromide (CTMAB) surfactant to promote nanoparticle formation and suspension.12 Powder X-ray diffraction (PXRD) patterns compared to that simulated from the MIL-125 crystal structure,2 (Fig. 1b) verified the crystallinity of both frameworks and that the introduction of neither the iron sites nor surfactant altered the overall structure. The optical electronic absorption spectra for the MOF materials were obtained through transmission absorption measurements of the MOF nanoparticle suspensions and diffuse reflectance (DR) measurements of the solid-state powders. As shown in Fig. 1c and d, the NH2-BDC linker, NH2-MIL-125(Ti) and NH2-MIL-125(Ti,Fe) framework spectra all exhibit a peak maximum between 350 and 400 nm, which is attributed to a n → π* transition localized on the aminated linker. For the MOF systems, this band extends into the visible region and gains LMCCT character upon linker coordination with the metal–oxo clusters.6 However, scattering background differences, particularly for transmission measurements of the solid-state MOF suspensions, complicate the comparison of the low intensity, unresolved electronic transitions in this wavelength region, including any contributions from those involving lower energy “trap” states due to introduction of the Fe heterometal sites in the case of NH2-MIL-125(Ti,Fe). Further proof is therefore needed to confirm the participation of the Fe sites in the charge separated excited state and its metal–oxo node location.
To better understand how the iron is incorporated within the framework, steady state XAS measurements were conducted at the Fe K-edge. Both XANES and EXAFS spectra of NH2-MIL-125(Ti,Fe) and several Fe references are depicted in Fig. 2a and b. The XANES pre-edge feature and edge shift is similar to those of Fe2O3 indicating comparable Fe3+ octahedral speciation in the MOF devoid of metallic Fe, or residual FeCl3 impurities. Comparisons with the reference EXAFS spectra further verify the distinct Fe coordination environment in the framework. XAS also reveals how the Fe coordination in the MOF generated using this direct synthesis method diverges from that attempted through a transmetallation approach that involved soaking NH2-MIL-125(Ti) in FeCl3 solution. The XANES pre-edge feature for that system indicates lower symmetry Fe coordination environment and the EXAFS spectrum notably shows longer average first shell scattering path distances in the post synthetically doped MOF. Together, these XAS characteristics signify possible outer-cluster incorporation and/or residual FeCl3 species trapped within the pores.
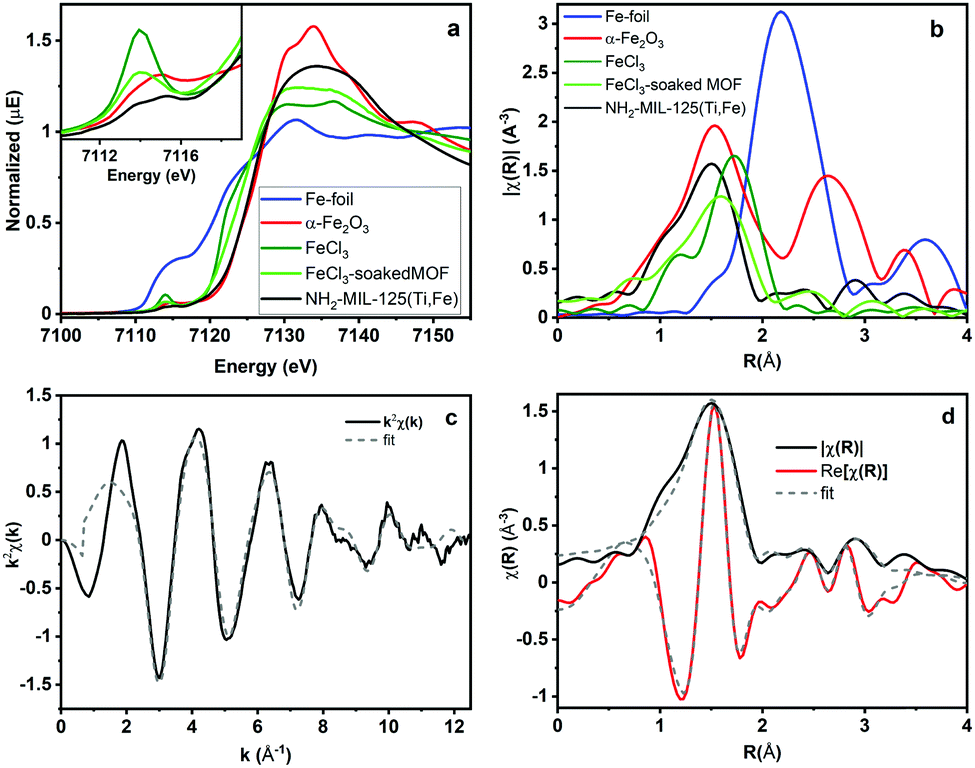 | ||
| Fig. 2 Fe K-Edge (a) XANES, (b) EXAFS spectra and (c) k-space and (d) R-space NH2-MIL-125(Ti,Fe) EXAFS spectrum and corresponding fit. | ||
Quantitative coordination information is provided by fitting the NH2-MIL-125(Ti,Fe) EXAFS spectrum (Fig. 2c) using a model derived from the reported crystal structure of NH2-MIL-125(Ti) in which a titanium site is substituted with iron (Fig. S3, ESI†). As summarized in Table S1 (ESI†), the fit includes Fe–O scattering paths from a distorted octahedral coordination environment, as well as contributions from scattering paths beyond the first shell, including neighboring Ti sites within the node and the closest C sites of the surrounding linkers. The fit reveals longer average first shell Fe–O bond lengths compared to those of the Ti sites found in the parent MOF, which is expected given the typical range reported for these metals in other oxide forms.13 The expanded Fe–O coordination shell appears to be compensated by further changes to the Fe position relative to the neighboring Ti sites as evidenced by the Fe–Ti scattering path distances derived from the fit. To be clear, this EXAFS analysis cannot rule out the possibility that some metal–oxo clusters may contain more than one Fe site (or that some contain no Fe sites), but the fit beyond just the first coordination shell, together with the PXRD results, which show analogous, single phase structure for NH2-MIL-125(Ti,Fe) and NH2-MIL-125(Ti), provides strong evidence that Fe is indeed occupying octahedral sites in the octameric metal–oxo ring cluster.
Optical transient absorption characterization provides our first window into the nature and earliest time dynamics of the initial photoexcited state in these materials. Ultrafast OTA data for NH2-MIL-125(Ti). NH2-MIL-125(Ti,Fe), and the NH2-BDC linker are shown in Fig. 3 and the multiexponential fitting results for the kinetics are summarized in Table S2 (ESI†). The spectra of the linker exhibit a TA band that appears within the instrument response time, at λmax = 585 nm and subsists well beyond the experimentally available delay range with more than 80% of the signal remaining at 1 ns delay time. The spectra of NH2-MIL-125(Ti,Fe) show a similar transient absorption feature that overlaps with an additional broad, unstructured TA band that extends throughout the visible and NIR wavelength range while those of the all-Ti version of this MOF are dominated by this broad redshifted band only. Based on the spectral signatures identified through reported spectro-electrochemical characterization of aminoterephthalate dimethyl ester and a Ti8O8tert-butoxide molecular cluster complex,14 the sharper transient absorption band around 585 nm is assigned to the formation of the NH2-BDC radical cation with the hole localized on the amine and the broad absorption band observed throughout the visible and NIR range is attributed to the radical anion of the metal–oxo cluster node. Comparing the OTA kinetics reveals that both NH2-MIL-125(Ti) and NH2-MIL-125(Ti,Fe) exhibit shorter sub-ns lifetime components. However, while the signal appears to have completely decayed for NH2-MIL-125(Ti) by 1 ns, nearly half of the TA signal remained at this delay time for NH2-MIL-125(Ti,Fe), indicating contribution from substantially longer lifetime components. Notably, our findings for NH2-MIL-125(Ti) diverge from previous OTA characterization of this MOF,14 which reported nearly identical OTA data (spectra and kinetics) to those we found for the NH2-BDC linker. While we observed similar results for NH2-MIL-125(Ti) samples after initial washing, we found that further purification was needed to fully remove residual linker that was presumably trapped within the porous structure. Extensive washing and careful isolation of suspended MOF nanoparticles consistently yielded the OTA data presented here. The appearance of the TA feature around 585 nm in NH2-MIL-125(Ti,Fe), that is mostly absent from the NH2-MIL-125(Ti) TA spectra may indicate a more localized hole on the linker associated with a charge separated state and the larger contribution of longer lifetime components may be attributed to the Fe dopant acting as a “trap” site and serving to delay electron–hole recombination.
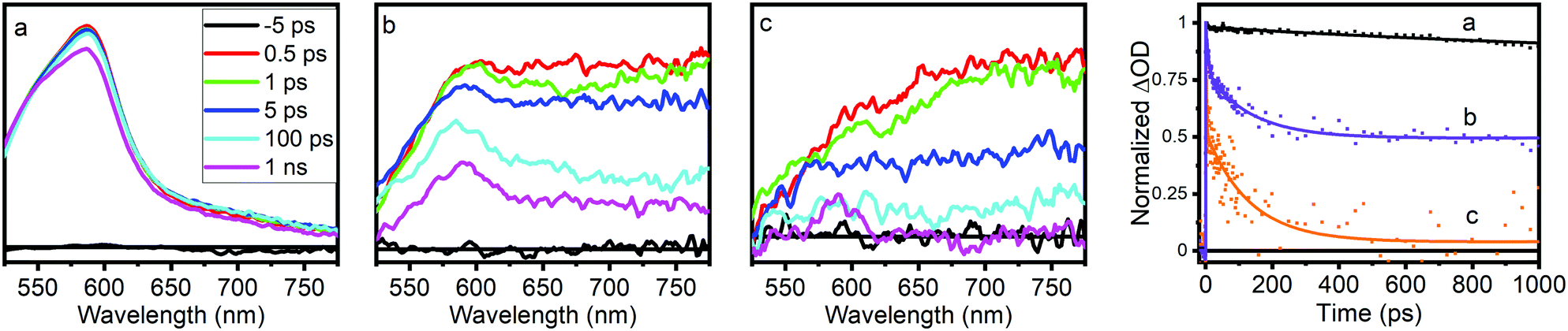 | ||
| Fig. 3 Optical transient absorption spectra and kinetics (collected at 585 nm) of (a) NH2-BDC, (b) NH2-MIL-125(Ti,Fe), and (c) NH2-MIL-125(Ti). | ||
Fe K-edge XTA was used to further elucidate the nature and dynamics of the charge separated state, particularly the role of the Fe dopant, following LMCCT excitation in NH2-MIL-125(Ti,Fe). Fig. 4a highlights the changes observed between the XTA spectra collected using the X-ray bunch synchronized with the laser pump pulse at nominal time zero delay (“laser on”) and those collected using pre-time zero X-ray bunches (“laser off”). A significant edge shift to lower energy is observed for the “laser on” spectrum, as emphasized by the derivative like feature in the corresponding difference spectrum with peak maximum aligned with the rising edge. This edge shift clearly indicates transient reduction of the iron sites upon population of the LMCCT excited state. Notably, no signal changes were observed in XTA spectra collected for the Fe-doped MOF generated using the post-synthetic modification approach. This comparison further supports the novelty of the direct synthesis approach for incorporating the Fe sites within the metal–oxo nodes and suggests that this method of heterometal doping is required for efficient Fe participation in the CS excited state.
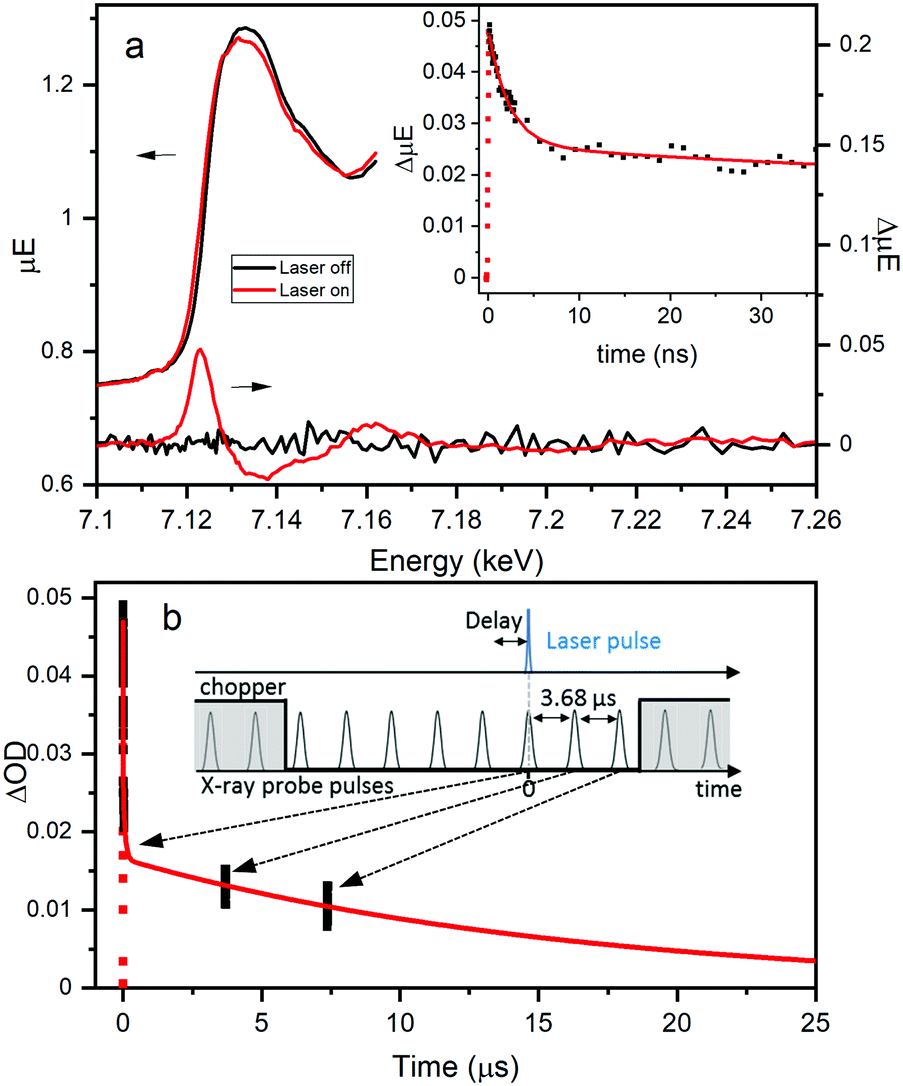 | ||
| Fig. 4 Fe K-Edge XTA (a) spectra and corresponding difference spectra for NH2-MIL-125(Ti,Fe). Inset: Kinetics monitored at 7.123 keV and (b) extended kinetics associated with synchronized and subsequent two X-ray probe pulses. Inset: Schematic representation of XTA timing structure. | ||
Time scans collected by monitoring the XTA difference signal maximum at 7.123 keV for NH2-MIL-125(Ti,Fe) produce the kinetic trace, shown in the inset of Fig. 4a. Successive X-ray bunches, after the synchronized one, were simultaneously collected during these time scans to produce the extended kinetics depicted in Fig. 4b. Kinetic fitting, as summarized in Table S3 (ESI†), reveals a faster 2.3 ns lifetime component, and at least two longer-lived components. While too few data points on the microsecond time scale are available to obtain these longer lifetimes, the signal clearly extends into the 10's of microseconds time range. This exceptionally long lifetime indicates that excitation into the LMCCT band of NH2-MIL-125(Ti,Fe) promotes localization of an electron that remains on the Fe site, preventing charge recombination. The different lifetime components may be associated with the probability of the charge recombination event, as it depends on the position of the electron-trapping Fe site relative to the linker containing the hole. Future studies aim to test this theory by assessing the dependence of XTA kinetics on the doping level (i.e. average number of heteroatoms per metal–oxo cluster). Here, we revealed the coordination and specific role of the Fe sites within the photoexcited framework, illustrating that heterometal-doped Ti-based MOFs produced through direct-synthesis are promising candidates for photocatalysis with ordered and controllable composition.
JVL acknowledges support by the National Science Foundation (DMR-1455127 and 2003910). This research used resources of the Advanced Photon Source and the National Synchrotron Light Source II, U.S. Department of Energy (DOE) Office of Science User Facilities operated for the DOE Office of Science respectively by Argonne National Laboratory under Contract No. DE-AC02-06CH11357 and Brookhaven National Laboratory under Contract No. DE-SC0012704.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef; (b) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755 RSC; (c) M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310 CrossRef CAS; (d) C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222 CrossRef CAS; (e) S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857 CrossRef CAS.
- (a) Y. Cong, J. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976 CrossRef CAS; (b) A. Di Paola, E. García-López, S. Ikeda, G. Marcì, B. Ohtani and L. Palmisano, Catal. Today, 2002, 75, 87 CrossRef CAS; (c) S. N. R. Inturi, T. Boningari, M. Suidan and P. G. Smirniotis, Appl. Catal., B, 2014, 144, 333 CrossRef CAS; (d) Y. Matsumoto, M. Murakami, T. Shono, T. Hasegawa, T. Fukumura, M. Kawasaki, P. Ahmet, T. Chikyow, S.-Y. Koshihara and H. Koinuma, Science, 2001, 291, 854 CrossRef CAS.
- (a) C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D’arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942 CrossRef CAS; (b) Y. Fu, H. Yang, R. Du, G. Tu, C. Xu, F. Zhang, M. Fan and W. Zhu, RSC Adv., 2017, 7, 42819 RSC; (c) M. W. Logan, S. Ayad, J. D. Adamson, T. Dilbeck, K. Hanson and F. J. Uribe-Romo, J. Mater. Chem. A, 2017, 5, 11854 RSC; (d) D. Ao, J. Zhang and H. Liu, J. Photochem. Photobiol., A, 2018, 364, 524 CrossRef CAS; (e) R. M. Abdelhameed, M. M. Q. Simões, A. M. S. Silva and J. Rocha, Chem. – Eur. J., 2015, 21, 11072 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364 CrossRef CAS.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS.
- M. A. Syzgantseva, C. P. Ireland, F. M. Ebrahim, B. Smit and O. A. Syzgantseva, J. Am. Chem. Soc., 2019, 141, 6271 CrossRef CAS.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, J. Phys. Chem. Solids, 2002, 63, 1909 CrossRef CAS.
- (a) C. Bressler, C. Milne, V. T. Pham, A. ElNahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Science, 2009, 323, 489 CrossRef CAS; (b) A. Cannizzo, C. J. Milne, C. Consani, W. Gawelda, C. Bressler, F. van Mourik and M. Chergui, Coord. Chem. Rev., 2010, 254, 2677 CrossRef CAS; (c) L. X. Chen, W. J. H. Jager, G. Jennings, D. J. Gosztola, A. Munkholm and J. P. Hessler, Science, 2001, 292, 262 CrossRef CAS; (d) L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022 CrossRef CAS; (e) H. Cho, M. L. Strader, K. Hong, L. Jamula, E. M. Gullikson, T. K. Kim, F. M. F. de Groot, J. K. McCusker, R. W. Schoenlein and N. Huse, Faraday Discuss., 2012, 157, 463 RSC; (f) W. Gawelda, V.-T. Pham, M. Benfatto, Y. Zaushitsyn, M. Kaiser, D. Grolimund, S. L. Johnson, R. Abela, A. Hauser, C. Bressler and M. Chergui, Phys. Rev. Lett., 2007, 98, 057401 CrossRef; (g) J. V. Lockard, A. A. Rachford, G. Smolentsev, A. B. Stickrath, X. Wang, X. Zhang, K. Atenkoffer, G. Jennings, A. Soldatov, A. L. Rheingold, F. N. Castellano and L. X. Chen, J. Phys. Chem. A, 2010, 114, 12780 CrossRef CAS; (h) M. L. Shelby, M. W. Mara and L. X. Chen,, Coord. Chem. Rev., 2014, 277–278, 291 CrossRef; (i) G. Smolentsev and V. Sundström, Coord. Chem. Rev., 2015, 304–305, 117 CrossRef CAS; (j) R. M. van der Veen, C. J. Milne, A. El Nahhas, F. A. Lima, V.-T. Pham, J. Best, J. A. Weinstein, C. N. Borca, R. Abela, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2009, 48, 2711 CrossRef CAS; (k) L. X. Chen,, Angew. Chem., Int. Ed., 2004, 43, 2886 CrossRef.
- (a) J. Huang, O. Buyukcakir, M. W. Mara, A. Coskun, N. M. Dimitrijevic, G. Barin, O. Kokhan, A. B. Stickrath, R. Ruppert, D. M. Tiede, J. F. Stoddart, J.-P. Sauvage and L. X. Chen, Angew. Chem., Int. Ed., 2012, 51, 12711 CrossRef CAS; (b) B. Pattengale, S. Yang, J. Ludwig, Z. Huang, X. Zhang and J. Huang, J. Am. Chem. Soc., 2016, 138, 8072 CrossRef CAS; (c) F. Zamponi, T. J. Penfold, M. Nachtegaal, A. Lubcke, J. Rittmann, C. J. Milne, M. Chergui and J. A. van Bokhoven,, Phys. Chem. Chem. Phys., 2014, 16, 23157 RSC; (d) X. Zhang, G. Smolentsev, J. Guo, K. Attenkofer, C. Kurtz, G. Jennings, J. V. Lockard, A. B. Stickrath and L. X. Chen,, J. Phys. Chem. Lett., 2011, 2, 628 CrossRef CAS.
- L. Hanna, P. Kucheryavy, C. Liu, X. Zhang and J. V. Lockard, J. Phys. Chem. C, 2017, 121, 13570 CrossRef CAS.
- S. Abedi and A. Morsali, New J. Chem., 2015, 39, 931 RSC.
- (a) K. S. Tanwar, S. C. Petitto, S. K. Ghose, P. J. Eng and T. P. Trainor, Geochim. Cosmochim. Acta, 2009, 73, 4346 CrossRef CAS; (b) P. K. Naicker, P. T. Cummings, H. Zhang and J. F. Banfield, J. Phys. Chem. B, 2005, 109, 15243 CrossRef CAS.
- J. G. Santaclara, M. A. Nasalevich, S. Castellanos, W. H. Evers, F. C. Spoor, K. Rock, L. D. Siebbeles, F. Kapteijn, F. Grozema, A. Houtepen, J. Gascon, J. Hunger and M. A. van der Veen, ChemSusChem, 2016, 9, 388 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05339b |
| This journal is © The Royal Society of Chemistry 2020 |