
Electropolymerization in a confined nanospace: synthesis of PEDOT nanoparticles in emulsion droplet reactors†
Myoung Won
Lee
a,
Deok-Jin
Kwon
b,
Junmo
Park
a,
Jae-Chul
Pyun
 c,
Young-Joo
Kim
b and
Hyun S.
Ahn
*a
c,
Young-Joo
Kim
b and
Hyun S.
Ahn
*a
aDepartment of Chemistry, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. E-mail: ahnhs@yonsei.ac.kr
bDepartment of Mechanical Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea
cDepartment of Materials Science and Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea
First published on 17th July 2020
Abstract
Poly(3,4-ethylenedioxythiophene) (PEDOT) is an important material widely used in electronics for its hole conducting property. A novel strategy for the synthesis of nanoparticulate PEDOT was developed by emulsion droplet electrochemistry. Taking advantage of the space confinement in emulsions, PEDOT nanoparticles were size controllable without use of a separate template. Potential applications were investigated by implementing the PEDOT nanoparticle decorated electrodes as a supercapacitor and a hole transport layer in an organic light emitting diode.
Detection of stochastic collisions of nano-objects on an electrode has been a research topic of interest in recent years.1–3 Since the first observation of hard nanoparticle collisions by implementation of ultramicroelectrodes (UMEs),4 detection and statistical analysis of the electrode collisions of many types of nano-objects were studied, including those of hard nanoparticles,5–8 soft nanoparticles,9–11 and biologically relevant entities.12–15 More recent adaptations of the electrochemical collision research empowered scientists to utilize these stochastic collisions as a synthetic tool.16–18 Particularly, electrochemistry of emulsions (both oil-in-water and water-in-oil) yielded metal19–23 and metal oxide24 nanoparticles while the synthesis processes were monitored real-time by amperometry. This electrochemical synthetic strategy is particularly significant due to the precise size and composition control it provides and the surfactant-free nature of the resulting nanoparticles.19,20,24 In the present communication, we expand the capabilities of this emulsion-collision based synthetic method to polymeric nanomaterials (Scheme 1). A confined nanospace provided by an emulsion droplet served as a synthesis template, electropolymerization in which resulted in polymeric nanoparticles of size and shape of the droplet. In the current study, oxidative electropolymerization of 3,4-ethylenedioxythiophene (EDOT) was performed to yield nanoparticulate PEDOT (poly-EDOT). The synthesized particles were bound to the electrode relatively strongly for further electrochemical use, and were also detachable and dispersible in a solvent upon ultrasonic stimulation.
 | ||
| Scheme 1 Schematic representation of PEDOT electrosynthetic process by emulsion-collision method and potential applications of the PEDOT NP decorated electrodes. | ||
Polyethylenedioxythiophene is widely implemented as a hole transport layer in various electronic devices including solar cells25–27 and light-emitting diodes.28,29 Common preparation methods for PEDOT (including commercially available PEDOT:PSS) involve rather harsh chemical oxidants and templates.30–32 Electro-oxidative polymerization is another viable and milder synthetic method;33–36 however, electropolymerized PEDOT generally results in thin films rather than colloidal nanoparticles (NPs). Preparation of PEDOT in the form of NPs is generally more preferred due to solution processability and superior hole conductivity.37,38 A previous account reported nanoparticulate PEDOT by an electrochemical method;39 however, to the best of our knowledge, this work is the first case where discrete droplet reactors were used to electrochemically synthesize PEDOT NPs with relatively narrow size distribution.
As shown in Scheme 1, the reactors employed in this work were dichloromethane emulsion droplets dispersed in aqueous continuous phase. To determine the optimum potential for the electrosynthesis experiment in emulsion droplets, EDOT electropolymerization was performed by cyclic voltammetry in dichloromethane (Fig. 1a). In the potential scale adopted in this work (vs. Ag/AgCl), we observed no PEDOT formation at potentials less oxidizing than 1.15 V, and oxidative degradation of solvent occurred at potentials more positive than 1.2 V. Therefore, 1.15 V was chosen as the optimum electrode potential for the collision experiments and all of our data were collected at this potential unless otherwise stated. As commonly reported in the literature, PEDOT synthesized by cyclic voltammetry method resulted in films covering the entire electrode (Fig. 1 and Fig. S1, ESI†).33–36,40
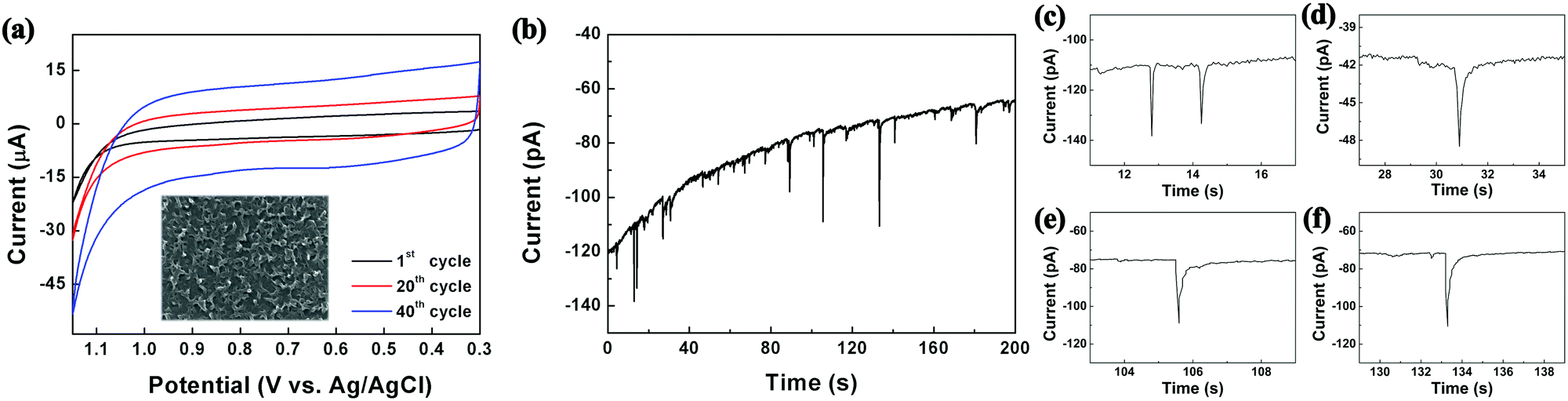 | ||
| Fig. 1 (a) Electropolymerization of EDOT by cyclic voltammetry (0.1 M EDOT, 0.2 M TBAP in DCM at 50 mV s−1 scan rate; inset displays resultant film by SEM). (b) Chronoamperogram exhibiting collision signals of 200 nm droplets onto a 10 μm carbon electrode, with each oxidation spike leading to a synthesized PEDOT NP. (c)–(f) Typical single droplet electrolysis signals. See also Fig. S2 and S3 (ESI†). | ||
The size of the droplet reactors was controllable by varying the droplet synthesis condition (see ESI†), and each droplet contained 25 mM EDOT precursor. Oxidative electropolymerization initiated inside the droplets as they collided on the working electrode, resulting in PEDOT NPs of size concomitant to the precursor droplet, which also serves as a synthesis template (Scheme 1). The electrosynthetic process can be monitored in situ by amperometry, with each of the spike-like signals in Fig. 1 (also see Fig. S2 and S3, ESI†) corresponding to electrochemical synthesis of a PEDOT NP. Due to the radical-based mechanism of the EDOT polymerization process,41 the charge under the spike-like signals does not correlate to an overall one electron oxidation of EDOT monomers contained in the droplet. The integrated charge under the spike signals (e.g. 0.42 pC for 350 nm diameter droplets) was approximately ten-fold greater than that calculated for one electron oxidation per EDOT molecule (0.054 pC for a 350 nm droplet). The excess amount of charge presumably is due to oxidative degradation of the dichloromethane solvent. Considering the proximity in the potential scale of the two oxidation reactions (EDOT oxidation at 1.15 V and dichloromethane oxidation at 1.2 V), slight oxidation of solvent was inevitable during PEDOT NP synthesis. The degree of solvent intervention was small, however, accounting for ca. 1% of total solvent molecules being oxidized: dichloromethane oxidized during PEDOT synthesis was ca. 0.38 pC, whereas the amount of charge required to oxidize all solvent molecules in the droplet was 34 pC (for a 350 nm diameter droplet).
The frequency of these droplet collision events can be estimated from the Stokes–Einstein equation based on the Brownian motion of the droplets in the aqueous continuous phase (see ESI† for detailed calculations).1 Experimentally observed frequency of events agreed well with the calculated results and are summarized in Table S1 (ESI†). After several hundreds of the synthetic collision events, the electrodes were isolated and analyzed by SEM. Fig. 2 displays SEM images of the electrosynthesized PEDOT NPs obtained from droplets of various sizes: 200 nm, 350 nm, 450 nm, and 600 nm in diameter. Correlation between the size of the synthesized PEDOT NPs and that of the precursor emulsion droplets was reasonable based on the statistical analysis of the NP size (Fig. 2), suggesting that the emulsion droplets not only serve as EDOT precursor vessels but also as NP synthesis templates. Morphology of the PEDOT NPs was in general spherical (see Fig. 2 and Fig. S4, ESI†), with kinks and wrinkles typical of polymer NPs. Nanoparticles with smaller than 200 nm diameter were also synthesizable by this method: ca. 40 and 70 nm NPs were obtained by employing 60 nm and 100 nm droplets, respectively (see Fig. S5–S7, ESI†). Particles of smaller size exhibited larger uncertainty in the measurement of their diameters in SEM due to electron beam charging at high magnifications. Adhesion of electrosynthesized PEDOT NPs onto the electrode surface was strong such that direct implementation of NP attached electrodes were feasible (vide infra; see Fig. S13 and S14, ESI†). The particles were also detachable upon ultrasonic stimulation and dispersed in solvent for further processing (Fig. S8, ESI†).
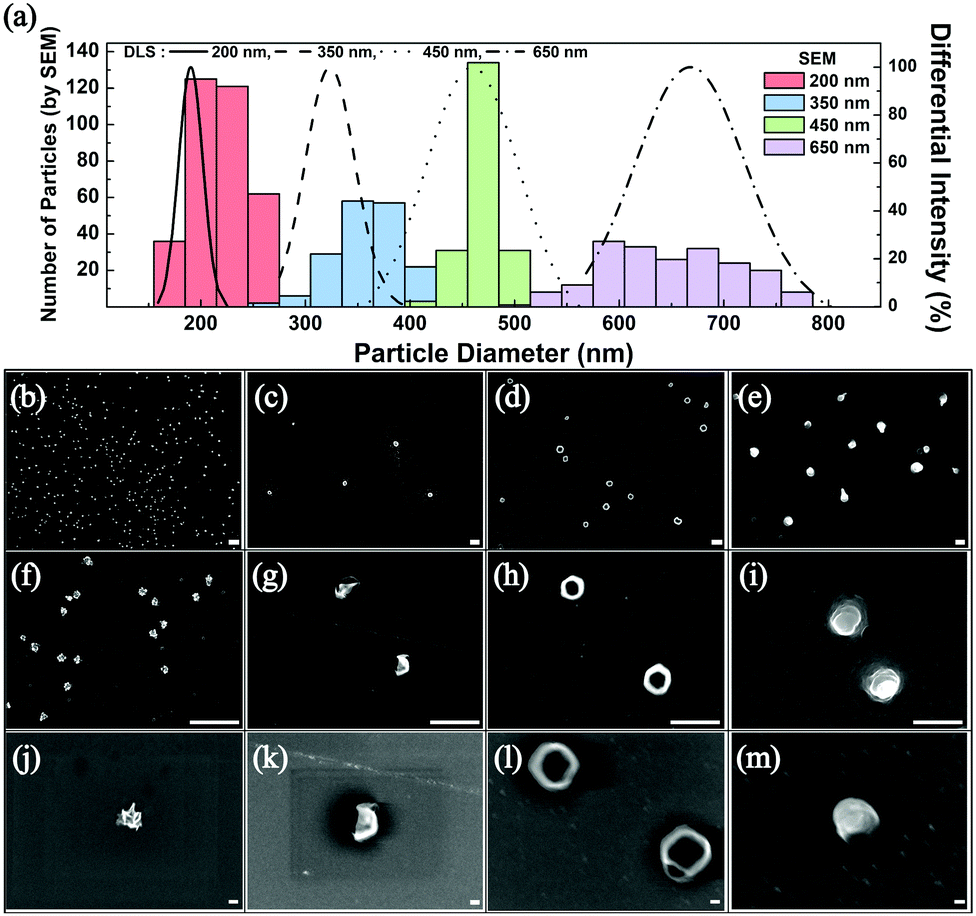 | ||
| Fig. 2 (a) Histograms of PEDOT NP size measured by SEM (200 nm-red, 350 nm-blue, 450 nm-green, and 650 nm-purple) and the size of the precursor droplet determined from dynamic light scattering (DLS; 200 nm-black solid line, 350 nm-dashed line, 450 nm-dotted line, and 650 nm-dash-dotted line). (b)–(m) SEM images of PEDOT NPs ((b), (f) and (j) 200, (c), (g) and (k) 350, (d), (h) and (l) 450, and (e), (i) and (m) 600 nm NPs, respectively on a same magnification scale; scale bars are 1 μm in length for images in (b)–(i) and 100 nm in images (j)–(m)). | ||
Oxidative polymerization process of EDOT is prevalent in the literature, and typically results exclusively in PEDOT formation without significant side reactions.42,43 In order to confirm the chemical identity of the NPs synthesized in this work, spectroscopic analyses were performed. Inspection of the NP attached electrodes by UV-vis and Raman spectroscopy revealed distinctive features corresponding to PEDOT (Fig. S9, S10 and Table S2, ESI†), namely a broad absorption centered around 580 nm44,45 and vibrations corresponding to PEDOT structural motifs.44,45 Because electrosynthesized PEDOT NPs are positively charged, electrolyte anions incorporate into the NPs for charge compensation.46–48 In the case of this work, PF6 anions in the electrolyte incorporated to form PEDOT:PF6 NPs (see also Fig. S4, ESI†). Potential applications of the electrosynthesized PEDOT NPs were investigated by subjecting the NP attached electrodes as (1) a capacitor and (2) a hole transport layer in an organic light emitting diode (OLED). For capacitance measurements PEDOT NPs were synthesized on a 10 μm diameter carbon UME.
As shown in Fig. 3, geometric capacitances of a carbon UME decorated with PEDOT NPs were measured. Deposition times ranging from 15 minutes to 60 minutes were chosen, corresponding to good electrode coverage of PEDOT NPs (ranging in 100 to 2500 NPs depending on size; see ESI† for detailed calculations). Geometric capacitances of 100, 160, and 215 mF cm−1 were observed for 200 nm, 350 nm, 450 nm NP decorated electrodes, respectively. The obtained values compare favorably to literature reported capacitances of PEDOT based materials.49,50 Interestingly, UMEs decorated with larger PEDOT NPs exhibited superior capacitances compared to those with smaller NPs, which is counter intuitive considering larger surface to volume ratio of smaller NPs. We presume that larger NPs in this study (350 and 450 nm; see Fig. S11, ESI†) result in partially hollow structure, which leads to higher surface area and superior capacitor performance compared to 200 nm compact NPs. Capacitance values greater than 200 mF cm−1 is on par or better than that of many polymer-based capacitors in the literature,51,52 and suggests that the PEDOT in this work is a promising candidate material for supercapacitor application. Supercapacitor electrodes based on PEDOT NPs exhibited little fade in performance even after one hundred continuous cycles and in excess of 2 h of continued use (Fig. S14, ESI†).
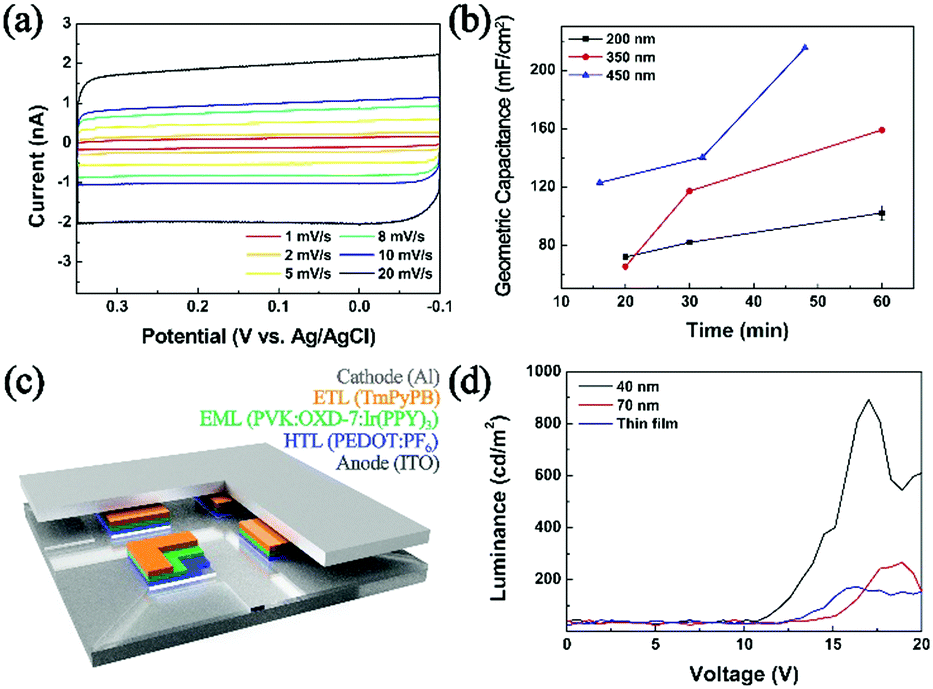 | ||
| Fig. 3 (a) Cyclic voltammograms of 450 nm PEDOT:PF6 (146 NPs) decorated carbon UME at different scan rates. (b) Geometric capacitance (mF cm−2) of PEDOT NPs decorated electrodes (200 nm-black, 350 nm-red, and 450 nm-blue). (c) Schematic representation of an OLED device investigated in this work. (d) Luminescence measurements from OLED devices employing PEDOT:PF6 NPs (40 and 70 nm) and a PEDOT:PF6 thin film (deposited by cyclic voltammetry) as hole transport layers. | ||
Electrosynthesized NPs were also implemented as a hole transport layer in OLED devices (Fig. 3c. For detailed device preparation, see ESI;† configuration similar to that in ref. 53 and 54 was adopted). Devices fabricated with NPs larger than 200 nm did not luminesce, presumably due to circuit shortage caused by a thick hole transport layer. Typical OLED devices in the literature employ commercial PEDOT:PSS (PSS; polystyrene sulfonate) cast films of ca. 40 nm thickness.53,54 When we implemented PEDOT:PF6 NPs of 40 and 70 nm, the device luminesced, with superior performance compared to that with a thin film PEDOT:PF6 hole transport layer (Fig. 3d). Compared to a benchmark device adopting a spin-coated commercial PEDOT:PSS hole transport layer, that with PEDOT:PF6 NPs from this work exhibited approximately 10% luminance (Fig. S12, ESI†). It is well known that PEDOT hole conductivity varies depending on the anion, and that PEDOT:PSS greatly outperforms PEDOT:PF6.55–57 It is encouraging that the PEDOT NPs prepared in this work can actively function as a hole transport layer in a working OLED device, and that it can match 10% luminance of a device with a commercial PEDOT:PSS layer. Although in the current state, the PEDOT NP materials synthesized by the method introduced in this work are not competitive with established commercial analogue, we hope to further improve the methodology such that tuning of the counter anion may be feasible for the conductivity engineering and ultimately an optimized performance of the resulting material as a hole conducting layer in an OLED device.
In summary, we established a novel synthetic strategy towards size-controlled NPs of PEDOT by adaptation of droplet reactor collision electrochemistry. The synthesized particles adhere to the electrode strongly for direct electrochemical use and were also detachable and solvent dispersible upon suitable stimulation. Possible applications of PEDOT NPs were investigated in forms of supercapacitor and hole transport layer in OLED. The synthetic protocol was made possible by extension of soft nano-object collision research, and we hope to extend this strategy further towards various other polymeric nanostructures.
This work was financially supported by the National Research Foundation of Korea under grant NRF-2020R1C1C1007409.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B.-K. Kim, A. Boika, J. Kim, J. E. Dick and A. J. Bard, J. Am. Chem. Soc., 2014, 136, 4849–4852 CrossRef CAS PubMed.
- S. E. Fosdick, M. J. Anderson, E. G. Nettleton and R. M. Crooks, J. Am. Chem. Soc., 2013, 135, 5994–5997 CrossRef CAS PubMed.
- X.-W. Zhang, A. Hatamie and A. G. Ewing, J. Am. Chem. Soc., 2020, 142, 4093–4097 CrossRef CAS PubMed.
- X. Xiao, F.-R. F. Fan, J. Zhou and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 16669–16677 CrossRef CAS PubMed.
- H. Zhou, F.-R. F. Fan and A. J. Bard, J. Phys. Chem. Lett., 2010, 1, 2671–2674 CrossRef CAS.
- Y.-G. Zhou, N. V. Rees and R. G. Compton, ChemPhysChem, 2011, 12, 2085–2087 CrossRef CAS PubMed.
- X. Xiao and A. J. Bard, J. Am. Chem. Soc., 2007, 129, 9610–9612 CrossRef CAS PubMed.
- N. Daryanavard and H. R. Zare, Anal. Chem., 2017, 89, 8901–8907 CrossRef CAS PubMed.
- H. Deng, J. E. Dick, S. Kummer, U. Kragl, S. H. Strauss and A. J. Bard, Anal. Chem., 2016, 88, 7754–7761 CrossRef CAS PubMed.
- Y. Li, H. Deng, J. E. Dick and A. J. Bard, Anal. Chem., 2015, 87, 11013–11021 CrossRef CAS PubMed.
- B.-K. Kim, J. Kim and A. J. Bard, J. Am. Chem. Soc., 2015, 137, 2343–2349 CrossRef CAS PubMed.
- J. E. Dick, C. Renault and A. J. Bard, J. Am. Chem. Soc., 2015, 137, 8376–8379 CrossRef CAS PubMed.
- A. N. Sekretaryova, M. Y. Vagin, A. P. F. Turner and M. Eriksson, J. Am. Chem. Soc., 2016, 138, 2504–2507 CrossRef CAS PubMed.
- J. Y. Lee, B.-K. Kim, M. Kang and J. H. Park, Sci. Rep., 2016, 6, 30022–30027 CrossRef CAS PubMed.
- W. Cheng and R. G. Compton, Angew. Chem., Int. Ed., 2014, 53, 13928–13930 CrossRef CAS PubMed.
- D. A. Robinson and H. S. White, Nano Lett., 2019, 19, 5612–5619 CrossRef CAS PubMed.
- M. Zhou, D. Wang and M. V. Mirkin, Anal. Chem., 2018, 90, 8285–8289 CrossRef CAS PubMed.
- J. Dunevall, H. Fathali, N. Najafinobar, J. Lovric, J. Wigström, A.-S. Cans and A. G. Ewing, J. Am. Chem. Soc., 2015, 137, 4344–4346 CrossRef CAS PubMed.
- Y. E. Jeun, B. Baek, M. W. Lee and H. S. Ahn, Chem. Commun., 2018, 54, 10052–10055 RSC.
- J. H. Park and H. S. Ahn, Appl. Surf. Sci., 2020, 504, 144517 CrossRef.
- M. W. Glasscott, A. D. Pendergast and J. E. Dick, ACS Appl. Nano Mater., 2018, 1, 5702–5711 CrossRef CAS.
- A. D. Pendergast, M. W. Glasscott, C. Renault and J. E. Dick, Electrochem. Commun., 2019, 98, 1–5 CrossRef CAS.
- M. W. Glasscott, A. D. Pendergast, S. Goines, A. R. Bishop, A. T. Hoang, C. Renault and J. E. Dick, Nat. Commun., 2019, 10, 2650 CrossRef PubMed.
- Y. E. Jeun, J. H. Park, J. Y. Kim and H. S. Ahn, Chem. – Eur. J., 2020, 26, 4039–4043 CAS.
- J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Am. Chem. Soc., 2008, 130, 1258–1263 CrossRef CAS PubMed.
- B.-w. Park, M. Pazoki, K. Aitola, S. Jeong, E. M. J. Johansson, A. Hagfeldt and G. Boschloo, ACS Appl. Mater. Interfaces, 2014, 6, 2074–2079 CrossRef CAS PubMed.
- C. Yuan, S. Guo, S. Wang, L. Liu, W. Chen and E. Wang, Ind. Eng. Chem. Res., 2013, 52, 6694–6703 CrossRef CAS.
- G.-F. Wang, X.-M. Tao and R.-X. Wang, Compos. Sci. Technol., 2008, 68, 2837–2841 CrossRef CAS.
- J. H. Kim, C. W. Joo, J. Lee, Y. K. Seo, J. W. Han, J. Y. Oh, J. S. Kim, S. Yu, J. H. Lee, J.-I. Lee, C. Yun, B. H. Choi and Y. H. Kim, Macromol. Rapid Commun., 2016, 37, 1427–1433 CrossRef CAS PubMed.
- Y. Li, D. Wang, X. Pei, Z. Shi, L. Wang, W. Zhang and J. Jin, Soft Matter, 2011, 7, 2682–2686 RSC.
- Y. Zhang and K. S. Suslick, Chem. Mater., 2015, 27, 7559–7563 CrossRef CAS.
- Y. Liu, A. P. F. Turner, M. Zhao and W. C. Mak, Eur. Polym. J., 2018, 99, 332–339 CrossRef CAS.
- R. Asami, M. Atobe and T. Fuchigami, J. Am. Chem. Soc., 2005, 127, 13160–13161 CrossRef CAS PubMed.
- K. Nakabayashi, F. Amemiya, T. Fuchigami, K. Machida, S. Takeda, K. Tamamitsu and M. Atobe, Chem. Commun., 2011, 47, 5765–5767 RSC.
- E. Poverenov, M. Li, A. Bitler and M. Bendikov, Chem. Mater., 2010, 22, 4019–4025 CrossRef CAS.
- T. Lindfors, Z. A. Boeva and R.-M. Latonen, RSC Adv., 2014, 4, 25279–25286 RSC.
- Y. Lu, C. Kacica, S. Bansal, L. M. Santino, S. Acharya, J. Hu, C. Izima, K. Chrulski, Y. Diao, H. Wang, H. Yang, P. Biswas, J. Schaefer and J. M. D’Arcy, ACS Appl. Mater. Interfaces, 2019, 11, 47320–47329 CrossRef CAS PubMed.
- K. Muro, M. Watanabe, T. Tamai, K. Yazawa and K. Matsukawa, RSC Adv., 2016, 6, 87147–87152 RSC.
- Y.-M. Xiao, J.-Y. Lin, J.-H. Wu, S.-Y. Tai and G.-T. Yue, Electrochim. Acta, 2012, 83, 221–226 CrossRef CAS.
- L. Groenendaal, G. Zotti, P.-H. Aubert, S. M. Waybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855–879 CrossRef CAS.
- B. R. Pistillo, K. Menguelti, N. Desbenoit, D. Arl, R. Leturcq, O. M. Ishchenko, M. Kunat, P. K. Baumann and D. Lenoble, J. Mater. Chem. C, 2016, 4, 5617–5625 RSC.
- J. Li and Y. Ma, Synth. Met., 2016, 217, 185–188 CrossRef CAS.
- J. Metsik, K. Saal, U. Mäeorg, R. Lõhmus, S. Leinberg, H. Mändar, M. Kodu and M. Timusk, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 561–571 CrossRef CAS.
- M. Culebras, C. M. Gómez and A. Cantarero, J. Mater. Chem. A, 2014, 2, 10109–10115 RSC.
- W. Fan, C.-Y. Guo and G. Chen, J. Mater. Chem. A, 2018, 6, 12275–12280 RSC.
- A. Savva, S. Wustoni and S. Inal, J. Mater. Chem. C, 2018, 6, 12023–12030 RSC.
- S. Fabiano, N. Sani, J. Kawahara, L. Kergoat, J. Nissa, I. Engquist, X. Crispin and M. Berggren, Sci. Adv., 2017, 3, e1700345 CrossRef PubMed.
- K. Tybrandt, I. V. Zozoulenko and M. Berggren, Sci. Adv., 2017, 3, eaao3659 CrossRef PubMed.
- H. Zhou, G. Han, Y. Chang, D. Fu and Y. Xiao, J. Power Sources, 2015, 274, 229–236 CrossRef CAS.
- H. Zhou, X. Zhi and H.-J. Zhai, Org. Electron., 2018, 63, 149–158 CrossRef CAS.
- D. Ni, Y. Chen, H. Song, C. Liu, X. Yang and K. Cai, J. Mater. Chem. A, 2019, 7, 1323–1333 RSC.
- X. Zhang and P. Samorì, ChemNanoMat, 2017, 3, 362–372 CrossRef CAS.
- T. Earmme and S. A. Jenekhe, J. Mater. Chem., 2012, 22, 4660–4668 RSC.
- L. Qian, Y. Zheng, J. Xue and P. H. Holloway, Nat. Photonics, 2011, 5, 543–548 CrossRef CAS.
- A. N. Aleshin, S. R. Williams and A. J. Heeger, Synth. Met., 1998, 94, 173–177 CrossRef CAS.
- A. N. Aleshin, R. Kiebooms and A. J. Heeger, Synth. Met., 1999, 101, 369–370 CrossRef CAS.
- M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J.-P. Simonato, Prog. Mater. Sci., 2020, 108, 100616 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, relevant calculations, and spectral characterizations of materials. See DOI: 10.1039/d0cc03834b |
| This journal is © The Royal Society of Chemistry 2020 |