
Synthesis of imides via palladium-catalyzed three-component coupling of aryl halides, isocyanides and carboxylic acids†
Bo
Wang‡
,
Dan
He‡
,
Beige
Ren
and
Tuanli
Yao
 *
*
Shaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi’an, 710021, China. E-mail: yaotuanli@sust.edu.cn
First published on 11th December 2019
Abstract
A palladium-catalyzed three-component synthesis of acyclic imides from feedstock aryl halides, carboxylic acids and isocyanides through the intermediacy of isoimides has been developed. The key to the success of this approach was controlled isocyanide slow addition and organic/aqueous biphasic conditions. This transition-metal-catalyzed approach features readily available starting materials, atom- and step-economy, good functional group compatibility and gram-scale synthetic capability. Utilization of this new method is illustrated in the late-stage functionalization of drugs Carprofen, Loxoprofen and Flurbiprofen. This strategy has also been successfully applied in the synthesis of cyclic imides including phthalimide, homophthalimide, and 2H-2-benzazepine-1,3-dione derivatives.
Imides are present as a core structural motif in natural products and pharmaceutical agents. For example, natural products penimide A,1a berkeleyamide C,1b and pestalamides A,1c and approved drugs thalidomide, anircetam,1d and ethosuximide1e all contain an imide moiety (Scheme 1a). Imides also have a wide range of applications in agrochemicals2a and materials science.2b In addition, imides are versatile synthetic building blocks.3 The wide occurrence and high value of imides have driven the continuous development of new synthetic methods.
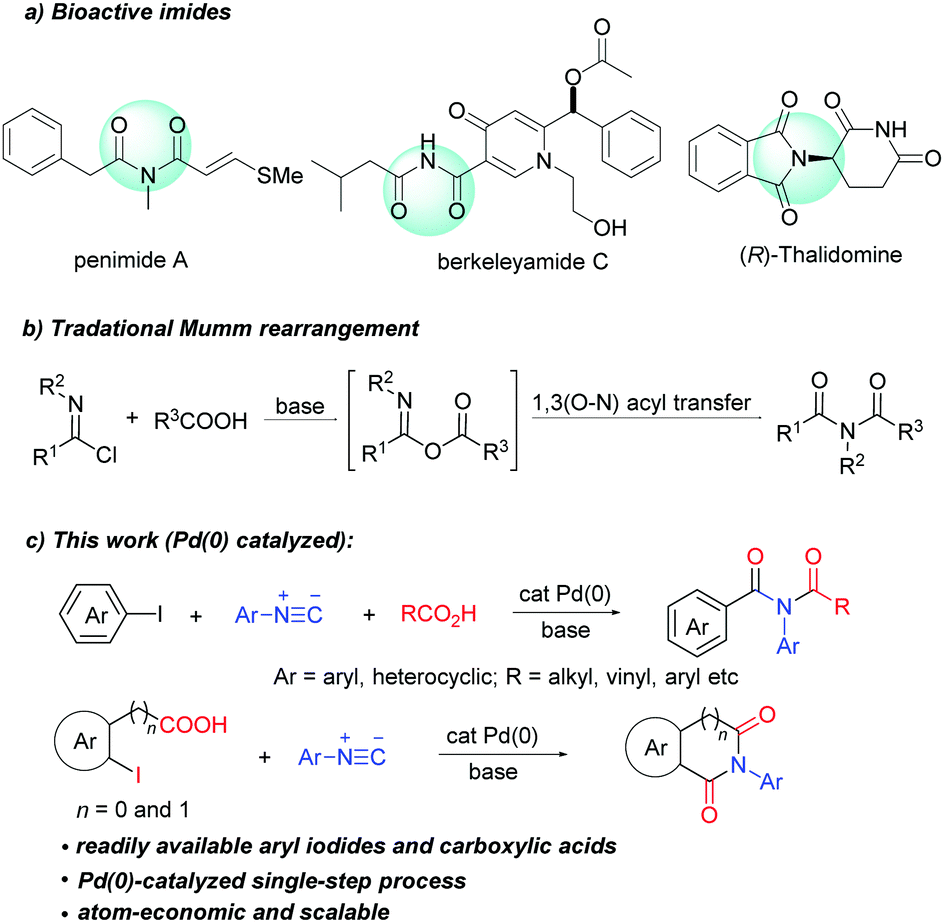 | ||
| Scheme 1 Examples of biologically relevant imides, traditional Mumm rearrangement and palladium-catalyzed three-component synthesis of imides. | ||
Mumm rearrangement of isoimides (Scheme 1b) is a traditional method for the synthesis of imides,4 in which the use of reactive reagents (POCl3 and SOCl2) leads to limited functional group compatibility and creates significant chemical waste. Transition-metal catalysed Mumm rearrangement has been developed recently. Unfortunately, these methods are limited to the synthesis of only specific imides such as N-acyl propiolamides,5N-acetyl-indole-3-carboxamides6 and α-amio imides.7 Furthermore, these methods require the use of pre-prepared carboxylic salts, which greatly limits the scope of applicable carboxylic acids. An ideal solution to overcome these limitations would be the development of a general and practical transition-metal-catalyzed process that utilizes feedstock chemicals such as aryl halides and carboxylic acids to arrive at acyclic imides. To the best of our knowledge, there is no example of catalytic imide synthesis employing aryl halides and carboxylic acids. Herein, we disclose the first palladium(0) catalyzed three-component coupling of aryl halides, isocyanides and carboxylic acids, which enables straightforward synthesis of a broad range of acyclic imides. This strategy has also been successfully applied in the synthesis of cyclic imides such as phthalimide, homophthalimide and 2-benzazepine-1,3-dione derivatives.
Our investigation of this Pd-catalyzed tandem process was commenced by surveying reaction conditions using isocyanide 1a, aryl iodide 2a and cesium pivalate (3a) as the model. In the presence of Pd(OAc)2 (10 mol%) and PPh3 (20 mol%), no detectable imide 4a was produced (Table 1, entry 1). To our delight, changing the palladium precatalyst to Pd2(dba)3 or Pd(dba)2 afforded 4a in 7% and 14% isolated yields, respectively (Table 1, entries 2 and 3). The previously known indole product from isocyanide insertion/benzylic C(sp3)–H activation was not observed.8 More sterically hindered P(o-tol)3 and sterically demanding and electron-rich phosphine Ad2PnBu are detrimental to the reaction (Table 1, entries 4 and 5). On the other hand, bidentate phosphines were more efficient and DEPhos afforded a 43% yield of 4a (Table 1, entries 6–9). Switching the solvent to more polar DMF was inferior (Table 1, entry 10). Since CsOPiv is very hydroscopic, an organic/aqueous biphasic condition (toluene/water) using pivalic acid, Cs2CO3 and phase transfer reagent n-Bu4NCl was developed to facilitate operation and extend the reaction to more complex carboxylic acids (Table 1, entries 11–13). Remarkably, the isolated yield of 4a was increased to 70% upon using BINAP as the ligand (Table 1, entry 13) and no corresponding amide from reacting with water was observed. Decreasing the reaction temperature to 80 °C did not affect the result (Table 1, entry 14). Further decreasing the reaction temperature (Table 1, entry 15) or reducing the loading of the Pd catalyst (Table 1, entry 16) all led to lower yields of 4a. It should be noted that slow addition of isocyanide 1a is the key to the success of the reaction. When adding 1a in one portion, no 4a was produced at all under otherwise identical conditions (compare Table 1, entries 14 and 17). We reasoned that slow addition of isocyanides would restrict the highly coordinating nature of isocyanides,9 which facilitated their insertion by the phenylpalladium intermediate and protected the catalyst from poisoning at the same time (see Scheme 3).10
|
|
||||
|---|---|---|---|---|
| Entry | Pd catalyst | Ligand | Solvent | 4aaa (yield) (%) |
| a Reaction conditions: isocyanide 1a (0.2 mmol) in solvent (1 mL) was added by syringe pump to a solution of aryl iodide 2a (0.3 mmol, 1.5 equiv.), CsPiv (0.24 mmol, 1.2 equiv.), Pd catalyst (10 mol%), and ligand (20 mol%) in solvent (1 mL) over 1 h at 100 °C and was further stirred for 0.5 h after addition. b Isolated yield. c PivOH (0.24 mmol, 1.2 equiv.), Cs2CO3 (0.12 mmol, 0.6 equiv.) and n-Bu4NCl (1.0 equiv.) in H2O (0.4 ml) were used. d 80 °C. e 60 °C. f Pd(dba)2 (5 mol%) and BINAP (10 mol%). g 1a was added in one portion. | ||||
| 1 | Pd(OAc)2 | PPh3 | Toluene | 0 |
| 2 | Pd2(dba)3 | PPh3 | Toluene | 7 |
| 3 | Pd(dba)2 | PPh3 | Toluene | 14 |
| 4 | Pd(dba)2 | P(o-tol)3 | Toluene | 3 |
| 5 | Pd(dba)2 | Ad2PnBu | Toluene | 0 |
| 6 | Pd(dba)2 | BINAP | Toluene | 29 |
| 7 | Pd(dba)2 | dppm | Toluene | 36 |
| 8 | Pd(dba)2 | dppe | Toluene | 12 |
| 9 | Pd(dba)2 | DEPhos | Toluene | 43 |
| 10 | Pd(dba)2 | DEPhos | DMF | 0 |
| 11c | Pd(dba)2 | DEPhos | Toluene/H2O | 24 |
| 12c | Pd(dba)2 | dppm | Toluene/H2O | 45 |
| 13c | Pd(dba)2 | BINAP | Toluene/H2O | 70 |
| 14c,d | Pd(dba)2 | BINAP | Toluene/H2O | 73 |
| 15c,e | Pd(dba)2 | BINAP | Toluene/H2O | 21 |
| 16c,d,f | Pd(dba)2 | BINAP | Toluene/H2O | 57 |
| 17c,d,g | Pd(dba)2 | BINAP | Toluene/H2O | 0 |
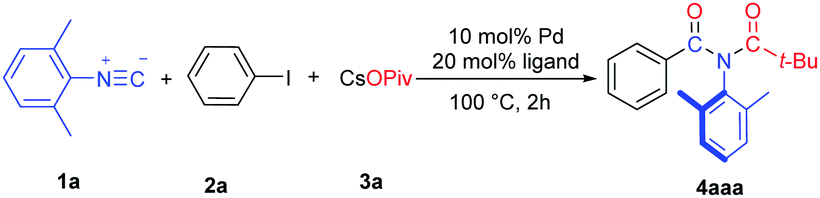
We first examined the substrate scope with respect to the aryl halides under the optimized reaction conditions (Table 1, entry 13), and the results are shown in Table 2. Aryl iodides with either an electron-withdrawing or an electron–donating group worked smoothly in this chemistry (4aba–4aha). The structure of 4ada was confirmed by single crystal X-ray diffraction.11 It should be noted that 2-iodobiphenyl reacted chemoselectively, affording imide 4aha in 70% yield without the formation of the previously reported fluorenone imine product.12 1-Iodonaphthalene was also a suitable substrate, providing imide 4aia in 71% yield. We were pleased to find that hetero aromatic halides were compatible with the reaction conditions. Thus, 4-bromoquinoline and 3-iodo-2-methoxy pyridine provided the corresponding imide products 4aja and 4aka in good yields.
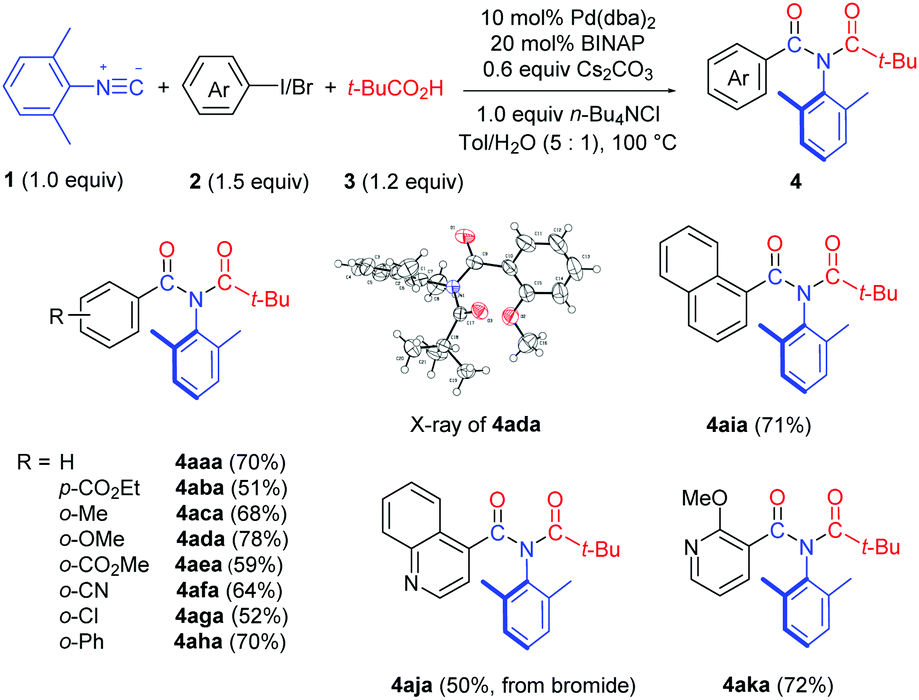
Both bis-ortho-substituted and mono-ortho-substituted isocyanides afforded good yields of imide products and functional groups such as chloride, methoxy group and acrylate are all well tolerated (Table 3). No imide product 4hda was obtained when employing isocyanobenzene. This observation demonstrated the importance of steric hindrance of isocyanides, which might promote the reductive elimination of an aryl imidoyl palladium carboxylate intermediate (see Scheme 3). 1-Isocyanonaphthalene afforded imide 4jda in 79% yield.
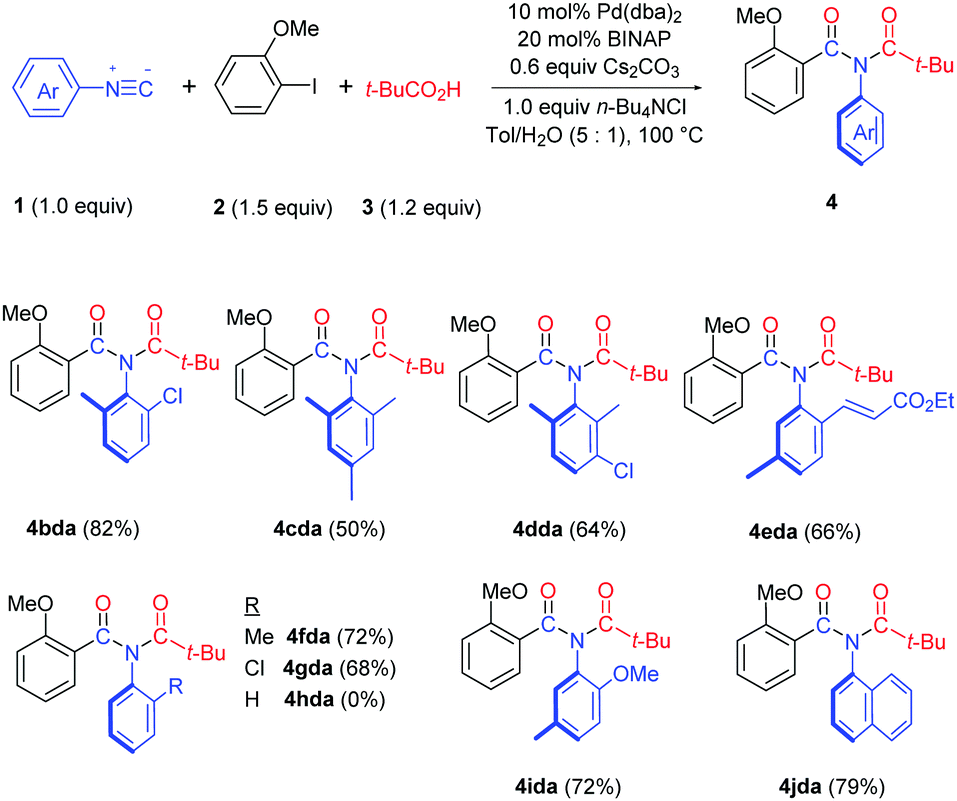
The reaction proceeded very well with various carboxylic acids such as aromatic, vinylic, saturated carbocyclic and benzyl carboxylic acids, providing the corresponding imide products (4adb–adi) in good to excellent yields (Table 4). Even very bulky 9H-xanthene-9-carboxylic acid afforded imide 4adj in a 40% yield. Interestingly, when employing Cl3CCO2H, F3CCO2H or N-protected amino acid Boc-Ala-OH in the reaction, amide 4adk was obtained due to hydrolysis of the corresponding imide products. Gratifyingly, this method has been successfully applied in the late-stage functionalization of drugs Carprofen, Loxoprofen and Flurbiprofen (4adl–adn).
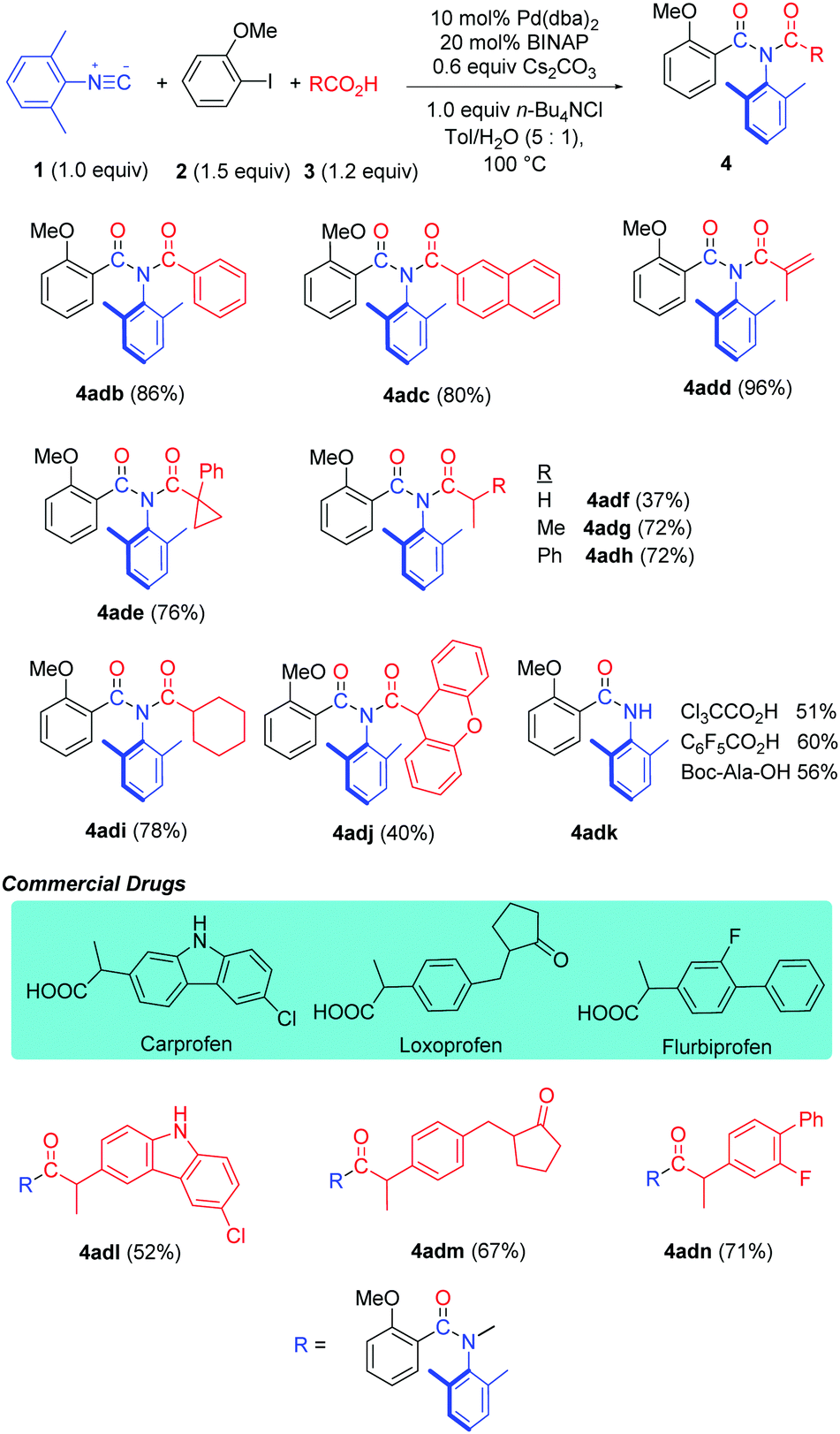
This strategy has also been successfully applied in the synthesis of cyclic imides by using 2-halobenzoic acids (Table 5). Various phthalimides were prepared in moderate to good yields (6a–f). Homophthalimide 6g and 2H-2-benzazepine-1,3-dione 6h can also be prepared in useful yields from 2-(2-iodophenyl)acetic acid and 2-(3-iodobenzo[b]thiophen-2-yl)benzoic acid, respectively.
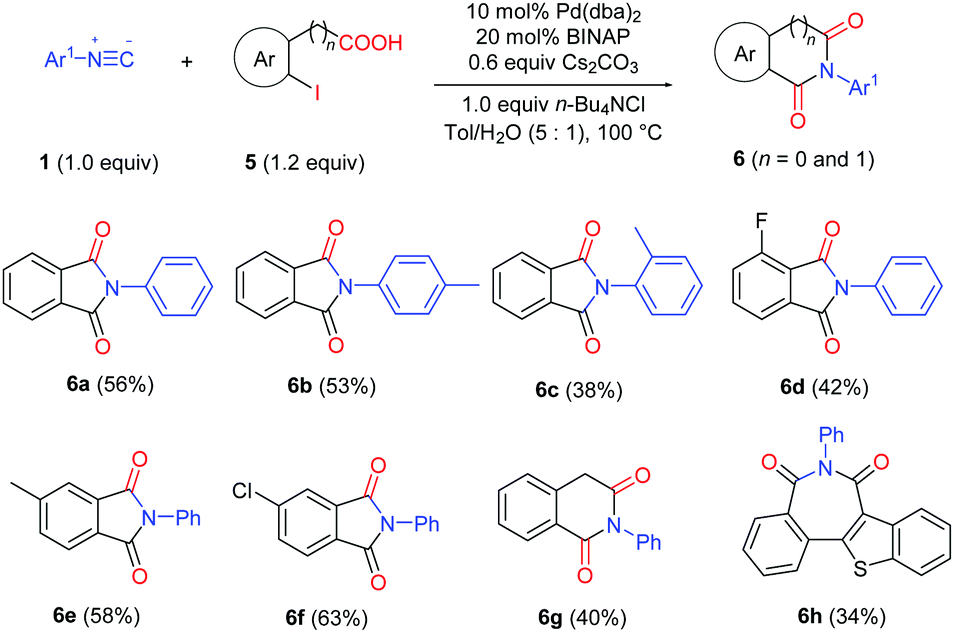
Since the imide products possess N-Ar axial chirality when using asymmetric aryl isocyanides, we performed the reaction using unsymmetrical isocyanides 1f and 1k and chiral carboxylic acid (S)-3h (Scheme 2). Slightly enantioenriched products 4fdh and 4kdh were obtained. The diastereoselectivity was not improved when employing (R)-BINAP as the ligand.13
 | ||
| Scheme 2 Diastereoselectivity of palladium-catalyzed Mumm rearrangement. | ||
We propose a mechanism for this process as illustrated in Scheme 3 by employing iodobenzene (2a) as an example. Pd(0) adds oxidatively to iodobenzene to generate the phenylpalladium(II) intermediate I. Isocyanide insertion of this intermediate followed by halide/carboxylate exchange affords imidoylpalladium(II)-carboxylate complex III, whose reductive elimination leads to isoimide IV and regenerates the Pd(0) catalyst. 1,3(O–N) acyl transfer of isoimide IV (Mumm rearrangement) produces imide product 4.
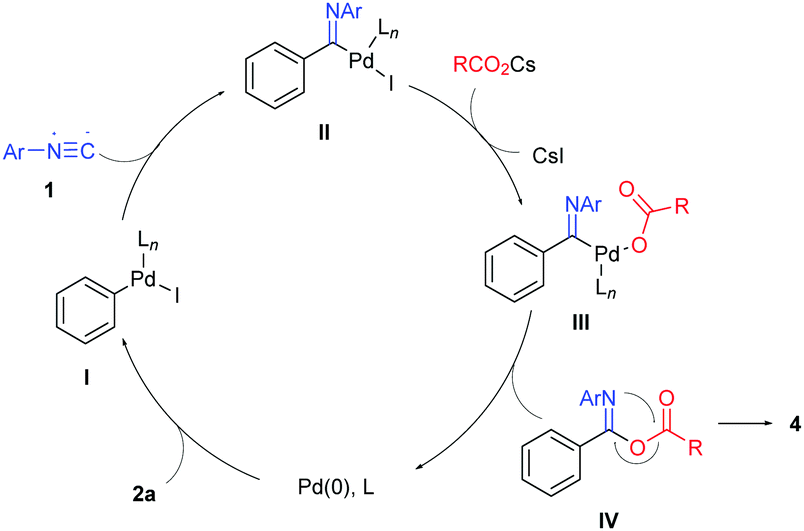 | ||
| Scheme 3 Proposed mechanism. | ||
In summary, we have developed a general and efficient method to prepare acyclic imides by palladium-catalyzed three-component coupling of aryl halides, isocyanides and carboxylic acids. Key to the success of this approach was controlled isocyanide slow addition and organic/aqueous biphasic conditions. This transition-metal-catalyzed approach features readily available starting materials, atom- and step-economy and good functional group compatibility. Additionally, we have also demonstrated expansion of this strategy to the synthesis of cyclic imides.
We are grateful for financial support from Shaanxi University of Science and Technology (BJ15-33).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. Pacher, A. Raninger, E. Lorbeer, L. Brecker, P. P.-H. But and H. Greger, J. Nat. Prod., 2010, 73, 1389 CrossRef CAS PubMed; (b) A. A. Stierle, D. B. Stierle and B. Patacini, J. Nat. Prod., 2008, 71, 856 CrossRef CAS PubMed; (c) G. Ding, L. Jiang, L. Guo, X. Chen, H. Zhang and Y. Che, J. Nat. Prod., 2008, 71, 1861 CrossRef CAS PubMed; (d) K. Nakamura, CNS Drug Rev., 2002, 8, 70 CrossRef CAS PubMed; (e) S. J. Flatters and G. J. Bennett, Pain, 2004, 109, 150 CrossRef CAS PubMed.
- (a) Y. Hisada and A. Fujinami, Agric. Biol. Chem., 1987, 51, 1547 Search PubMed; (b) Z. Cheng, X. Zhu, E. T. Kang and K. G. Neoh, Macromolecules, 2006, 39, 1660 CrossRef CAS.
- (a) C. Patzelt, A. Pöthig and T. Gulder, Org. Lett., 2016, 18, 3466 CrossRef CAS PubMed; (b) S. Takebayashi, J. M. John and S. H. Bergens, J. Am. Chem. Soc., 2010, 132, 12832 CrossRef CAS PubMed.
- (a) O. Mumm, H. Hesse and H. Volquartz, Ber. Dtsch. Chem. Ges., 1915, 48, 379 CrossRef CAS; (b) J. S. P. Schwarz, J. Org. Chem., 1972, 37, 2906 CrossRef CAS.
- Y. He, Y. Wang, X. Liang, B. Huang, H. Wang and Y.-M. Pan, Org. Lett., 2018, 20, 7117 CrossRef CAS PubMed.
- (a) G. Qiu, C. Chen, L. Yao and J. Wu, Adv. Synth. Catal., 2013, 355, 1579 CrossRef CAS; (b) Z. Hu, D. Liang, J. Zhao, J. Huang and Q. Zhu, Chem. Commun., 2012, 48, 7371 RSC; (c) S. Chen, W.-X. Wei, J. Wang, Y. Xia, Y. Shen, X.-X. Wu, H. Jing and Y.-M. Liang, Adv. Synth. Catal., 2017, 359, 3538 CrossRef CAS; (d) J. Peng, L. Liu, Z. Hu, J. Huang and Q. Zhu, Chem. Commun., 2012, 48, 3772 RSC.
- (a) M. Rueping and C. Vila, Org. Lett., 2013, 15, 2092 CrossRef CAS PubMed; (b) X. Ye, C. Xie, Y. Pan, L. Han and T. Xie, Org. Lett., 2010, 12, 4240 CrossRef CAS PubMed; (c) I. Guerrero, S. S. Marcos and A. Correa, Chem. Commun., 2018, 54, 1627 RSC.
- T. Nanjo, C. Tsukano and Y. Takemoto, Org. Lett., 2012, 14, 4270 CrossRef CAS PubMed.
- Y. Yamamoto, Coord. Chem. Rev., 1980, 32, 193 CrossRef CAS.
- For selected references using controlled slow addition in Pd-catalyzed reactions, see: (a) R. J. Sullivan, G. P. R. Freure and S. G. Newman, ACS Catal., 2019, 9, 5623 CrossRef CAS; (b) X. Hua, J. Masson-Makdissi, R. J. Sullivan and S. G. Newman, Org. Lett., 2016, 18, 5312 CrossRef CAS PubMed; (c) M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667 CrossRef CAS PubMed; (d) V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Org. Lett., 2013, 15, 5114 CrossRef CAS PubMed.
- CCDC 1962679† contains the supplementary crystallographic data for 4ada.
- M. Tobisu, S. Imoto, S. Ito and N. Chatani, J. Org. Chem., 2010, 75, 4835 CrossRef CAS PubMed.
- For racemization of chiral imides via N-Ar bond rotation, see: K. Kondo, T. Iida, H. Fujita, T. Suzuki, K. Yamaguchi and Y. Murakami, Tetrahedron, 2000, 56, 8883 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1962679. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08438j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |