DOI:
10.1039/C9SC02678A
(Edge Article)
Chem. Sci., 2019,
10, 9219-9232
Computational design of syntheses leading to compound libraries or isotopically labelled targets†
Received
2nd June 2019
, Accepted 9th August 2019
First published on 16th August 2019
Abstract
Although computer programs for retrosynthetic planning have shown improved and in some cases quite satisfactory performance in designing routes leading to specific, individual targets, no algorithms capable of planning syntheses of entire target libraries – important in modern drug discovery – have yet been reported. This study describes how network-search routines underlying existing retrosynthetic programs can be adapted and extended to multi-target design operating on one common search graph, benefitting from the use of common intermediates and reducing the overall synthetic cost. Implementation in the Chematica platform illustrates the usefulness of such algorithms in the syntheses of either (i) all members of a user-defined library, or (ii) the most synthetically accessible members of this library. In the latter case, algorithms are also readily adapted to the identification of the most facile syntheses of isotopically labelled targets. These examples are industrially relevant in the context of hit-to-lead optimization and syntheses of isotopomers of various bioactive molecules.
Introduction
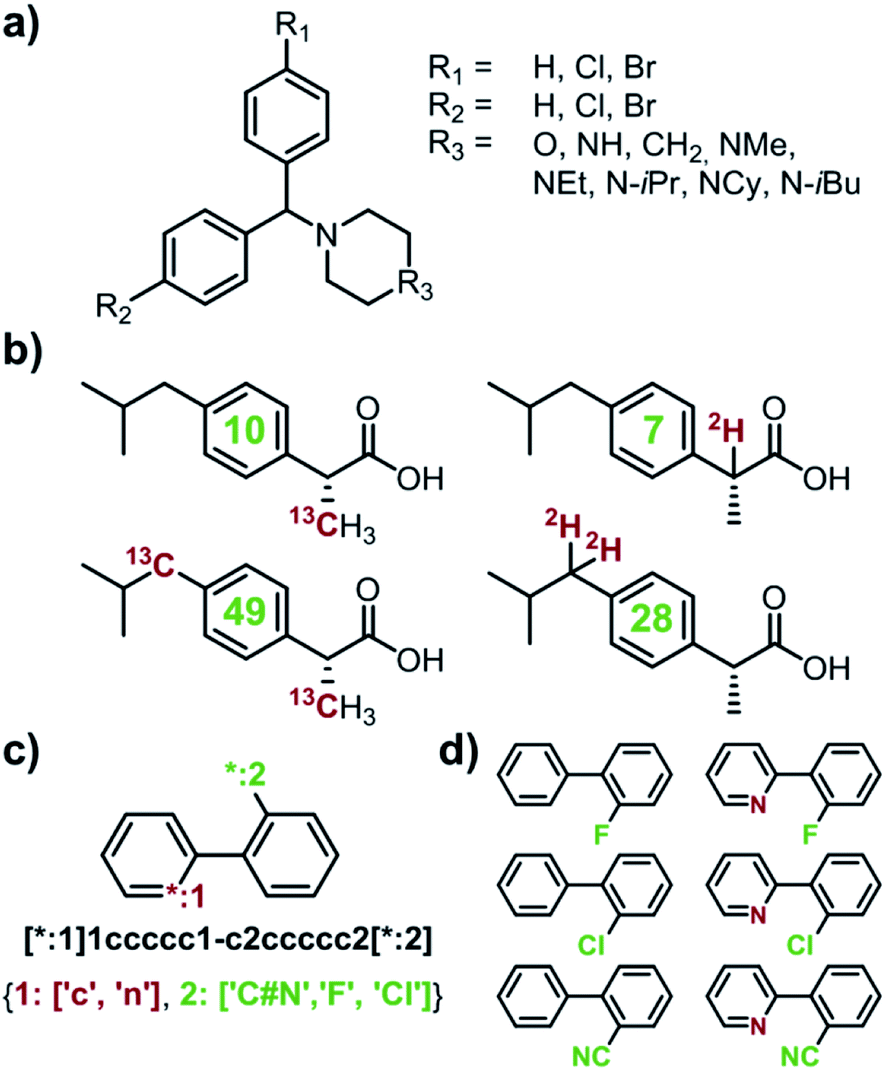
Capitalizing on the advances in artificial intelligence1–4 and constantly increasing computing power, recent years have brought revived interest and significant progress in the decades-old challenge of teaching computers the design of multistep organic syntheses.5–9 Various platforms, differing in the underlying details of search algorithms and reaction-rule formats, have been developed10–21 and one of these platforms, our own Chematica,17–21 has been validated experimentally via successful execution of multiple routes leading to diverse, high-value, medicinally relevant small molecules18 and, more recently, natural products.19 To date, a main effort in this emerging area of chemical research has been on algorithms designing syntheses of one specified target at a time. In several practically/industrially important situations, however, it is desirable to simultaneously design routes to multiple targets. For instance, a medicinal chemist might wish to optimize an existing scaffold and place various substituents in positions of interest (e.g., R1, R2, and R3 in Fig. 1a). Such hit-to-lead or lead optimizations22,23 encompass libraries of multiple synthetic targets, raising some pertinent questions: (1) which of the targets are most readily synthesizable? or (2) how to synthesize all of the targets while making use of some possible common intermediates? Another situation of interest is when one wishes to design syntheses of isotopically labelled compounds that differ from the parent, non-labelled compound by a certain increment of molecular mass – this ability is important to determine drugs' pharmacokinetics,24,25 to study environmental fates of pesticides,26,27 to ascertain food safety,28–30 or to quantify food flavourings,31–33 in many cases using the so-called isotope dilution mass spectroscopy (IDMS) techniques and assays.34 Because isotope labels can be placed in various positions and in various configurations within the molecule leading to isotopomers (Fig. 1b), they, again, constitute a small library of potential targets. In this case, question (1) – which of the labelled compounds offering a desired mass increase are most readily synthesizable – appears most relevant. Currently, there are no retrosynthetic algorithms with which one could address such questions for arbitrary targets. The closest analogue is our earlier work on the so-called Network of Organic Chemistry35–37 (NOC), in which we used Monte-Carlo searches to select (but not design) optimal syntheses leading to multiple targets of interest.37 Unfortunately, NOC is a static network comprising only published literature precedents and so its analyses are limited to already known targets and existing synthetic routes. In addition, Monte Carlo searches are computationally very intensive and typical execution times for the NOC are in days. Here, we describe significantly more efficient and general algorithms for de novo retrosynthetic planning (i.e., planning based on general reaction rules, not existing literature precedents) producing routes to small libraries of arbitrary – that is, both known and unknown – targets, including labelled ones. In our algorithms, retrosynthetic searches for individual targets share the same search graph and can benefit from common intermediates and synthetic strategies. In several cases, these searches utilize a cyclically inspected list of priority-queue-based data structures rather than a single priority-queue; this construction ensures that the overall, multi-target search is not dominated by a sub-search for any individual target. Examples of specific and in all cases viable syntheses we describe evidence that multi-target planning routines make efficient use of common intermediates, reduce the search space significantly (compared to individual, target-by-target searches), and yield complete library-wide plans within minutes. Overall, the methods we describe extend applicability of computational retrosynthetic planning to problems that are ubiquitous and important in pharmaceutical and agrochemical industries, offering savings in terms of planning time and the overall cost of synthesis of compound libraries, as well as minimization of waste (through the maximally efficient use of common intermediates and “root” reactions in the global, library-wide reaction plans). In a broader context, the multi-target design harnesses the computer's ability to store, analyze, and optimize large, interconnected networks of synthetic plans, which may be difficult for human chemists accustomed to planning synthetic solutions to specific targets, one target at a time.
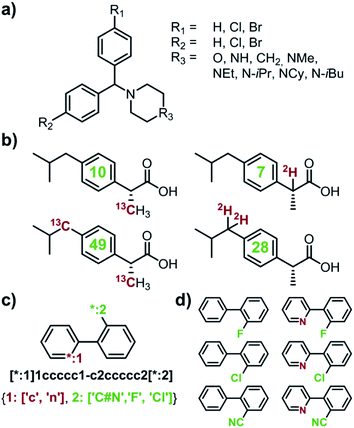 |
| | Fig. 1 Examples of multiple-target libraries. (a) Library of chlorcyclizine derivatives screened for the treatment of hepatitis C virus in ref. 38. (b) Isotopomers of ibuprofen. Top row has two examples of M + 1 isotopomers possible with 13C (left) and 2H (right) labelling. Bottom row has two examples of M+2 isotopomers available via13C (left) and 2H (right) labelling. Green numerals within the molecules give the total numbers of unique (note: some hydrogens and carbons are equivalent) labelled compounds for each case. For the M+2 labelling of this structurally simple target, there are already 49 13C and 28 2H labelling combinations. The number of isotopomers corresponds to sets without 2H labelling of the COOH group which is extremely easily exchangeable. (c) To specify multiple targets of retrosynthetic analyses, it is often convenient to use the so-called Markush structures. Here, a set of molecules is represented as a SMILES string (black) with numbered dummy atoms ([*:1],[*:2]) and dictionary of substituents marked in red and green for positions #1 and #2, respectively. Position [*:1] can correspond to either an aromatic carbon or nitrogen (to yield, respectively, biphenyl and 2-phenylpyridine series), whereas position [*:2] admits either a nitrile, CN, or F, or Cl atoms. In all, the small library thus defined will have six members shown in panel (d). | |
Computational methods
Modern retrosynthetic planners (e.g., MIT's ASKCOS,15 Waller's MCTS,14 or our Chematica17–21) differ in the origin of the underlying reaction rules (machine extracted vs. expert coded) and details of the search routines, but they all rely on the iterative expansion of parent/retron nodes into progeny/synthon nodes and on navigating (with the help of functions scoring individual reaction moves) the thus created bipartite graphs of synthetic options until simple and commercially available substrates are found. This procedure yields pathways that lead to a given target of interest and in which all chemical nodes are “synthesizable” (i.e., they are targets of at least one synthetic pathway tracing back to commercially available substrates) and the reaction nodes are “viable” (i.e., all their substrates are synthesizable). The problem we focus on in the current study is how to extend such generic routines to enable simultaneous synthesis of multiple targets (“a library”).
Algorithm seeking the syntheses of all targets
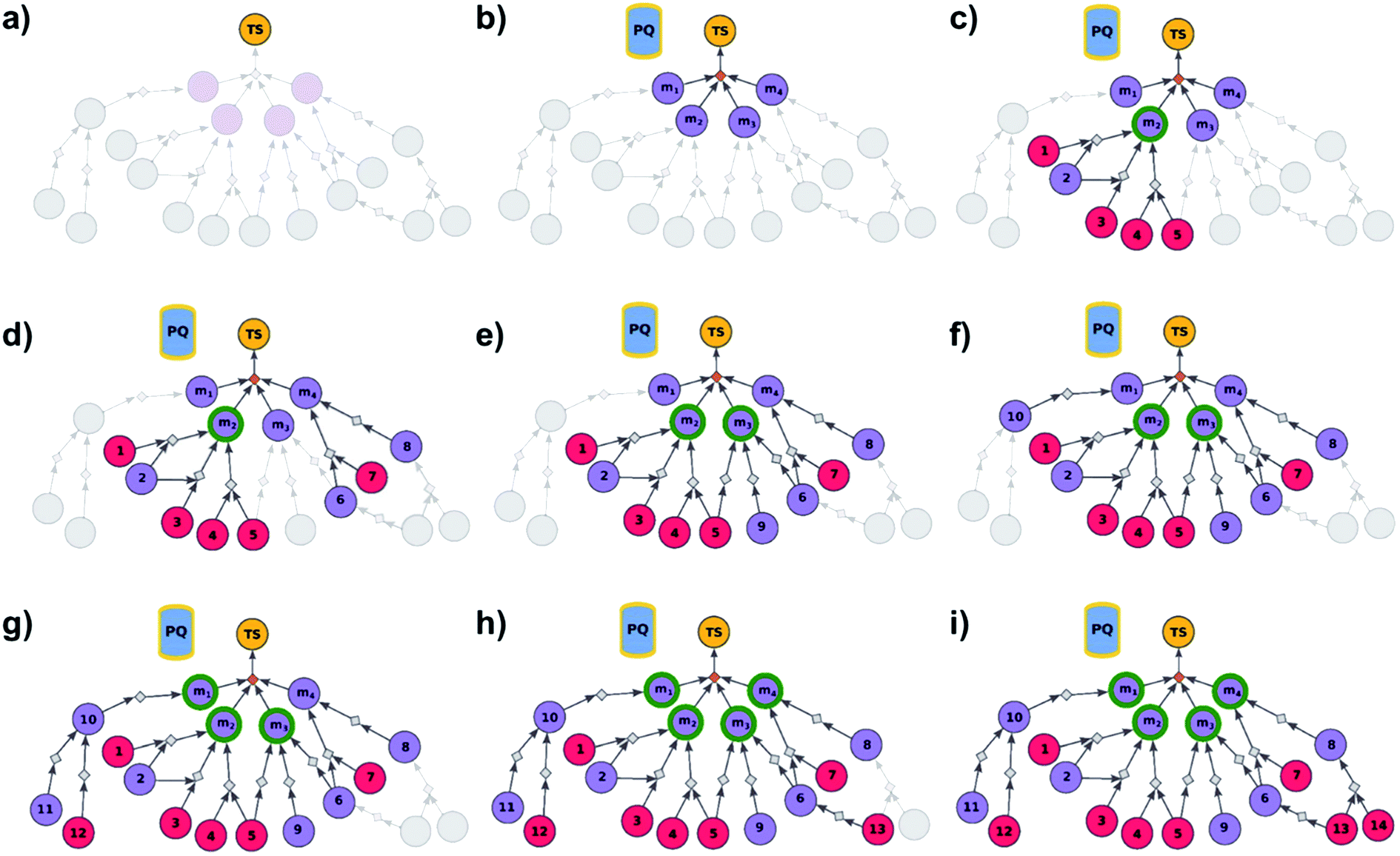
We begin by discussing the simplest problem of identifying syntheses of all members of a library. The algorithm (Fig. 2 and pseudocode in the ESI, Section S1†) initializes by performing a dummy “multicomponent” reaction, rdummy, such that the root node (i.e., the library) is “made” in one step from all molecules in the library, {mi}, i = 1, …, N, serving as its substrates. Chemically, this is a purely fictitious operation but algorithmically, such search-graph construction is important as it serves as an “AND” condition ensuring that any viable syntheses (cf. definition of viability above and in ref. 21) of the root node will also have to make all substrates for the ultimate rdummy reaction – in other words, the search algorithm will not stop until viable syntheses of all mi molecules in the library are identified. Of course, this dummy reaction is not supposed to skew the real searches for mis in any way, and its execution cost is assigned as zero. Starting from the mi nodes, the search graph is iteratively expanded as described before,18 with the already-evaluated sets of synthons stored in a single priority-queue-based data structure (PQ), and with further synthetic navigation guided by desired scoring functions (in Chematica, the synthetic moves are guided by summing the costs of the already-performed reactions and the complexity of synthons created by each reaction;17,18 in programs like ASKCOS or Waller's MCTS, the navigation is based on the scores provided by neural networks trained on large numbers of reaction precedents14,15). The stop points for the search are known and/or commercially available molecules with prices of the latter typically taken from commercial catalogues.
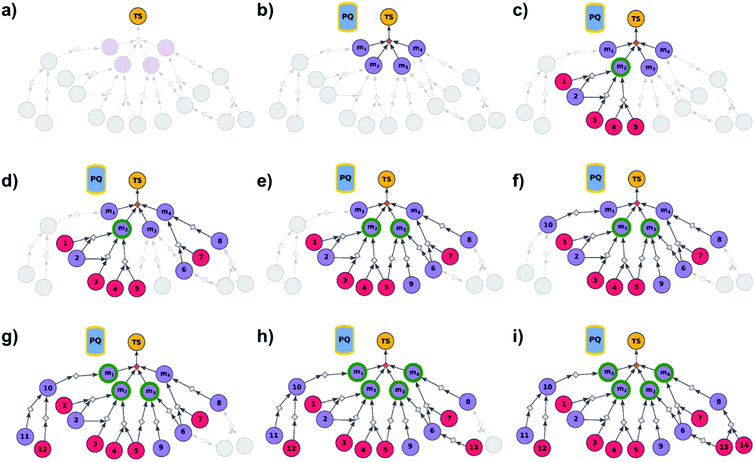 |
| | Fig. 2 Schematic description of the algorithm seeking the syntheses of all targets. (a) The user specifies the target set (here, TS = library of four targets), defining the root node (yellow circle) of the search graph to be explored (shaded parts represent regions of the graph not yet expanded). (b) The search graph is extended by adding four additional substance nodes (violet circles labelled m1, m2, m3, m4), linked with the root node via the same dummy reaction (orange diamond). The search algorithm utilizes a single priority-queue-based data structure, PQ, to navigate the graph expansion according to scores assigned to specific synthetic “options” encountered during the search. For this illustrative example, the algorithm first expands node m2, having a better score than nodes m1, m3, and m4. (c). Three explored reactions (grey diamond-shaped reaction nodes) lead to possibly overlapping substrate sets – i.e., nodes labelled {1, 2}, {2, 3}, and {4, 5}, respectively. Violet circular nodes denote molecules that are unknown in literature and not commercially available; red circular nodes refer to terminal, commercially available, chemicals. Here, one synthesis, i.e., 4, 5 → m2, gives viable synthesis plan for target m2 (green halo denoting that m2 is synthesizable). The search proceeds to (i) find syntheses for the remaining targets m1, m3, m4, and (ii) find alternative synthesis plans for target m2. (d) Search continues expanding node m4, giving reactions with substrates {6, 7}, and {8}, then expanding node m3 (e) resulting in reactions' substrate sets {5}, {6}, {9} (nodes 5 and 9 were already visited in previous expansions; moreover, as node 5 is terminal, path 5 → m3 is a viable synthesis for m3). (f) Furthermore, the region of the search graph related to the synthesis of m1 is visited by exploring node 10, and nodes 11 and 12. (g) The path 12 → 10 → m1 is a viable synthesis, so the target m1 is now also synthesizable. (h) Then, node 6 is expanded, giving terminal node 13. Of note, 6 is a common intermediate for syntheses of targets m3, and m4, and these two targets have new viable syntheses (13 → 6 → m3, and 13 → 6, 7 → m4). Target m4 is now synthesizable, and viable synthesis plans can by retrieved for all targets from initial target set (operation performed by selection algorithm, see main text). (i) The search continues to find more synthesis options, here exploring node 8 to give reaction with substrates 13 and 14 (resulting in alternative synthesis for target m4: 13, 14 → 8 → m4). | |
As the search is allowed to progress, multiple viable syntheses are typically found, forming a solution graph from which most economical plans can be selected (by iteratively propagating the yield-scaled costs from substrates to products and thereby assigning a realistic, monetary cost to each plan). In addition to the selection procedures detailed in our recent publication,21 a unique feature of library-wide design is that we wish to promote convergent synthetic plans that make use not only of common intermediates but also of the smallest number of different synthetic methodologies – to this end, a penalty is added to any new reaction type encountered in the solution graph. Chemically, such penalization ensures that it is more economical to perform the same reaction on a, say, 2 mol scale, rather than two different types of reactions – requiring separate set-ups and likely different reagents – each on a scale of 1 mol.
We observe that if the searches for each library member were performed separately, selection would be made from each individual solution graph and no synergies in terms of common intermediates or reaction types could, in general, be expected. Also, in terms of computational efficiency, the search on a common graph is significantly more compact than the sum of individual searches – as quantified for specific examples we discuss later in the text (cf.Fig. 4, 5 and Table S1†), the number of graph nodes explored before finding the first viable solution is about an order of magnitude smaller for the library-oriented, global search than for separate searches; each ran for a different library member.
Algorithm seeking the easiest syntheses of some targets
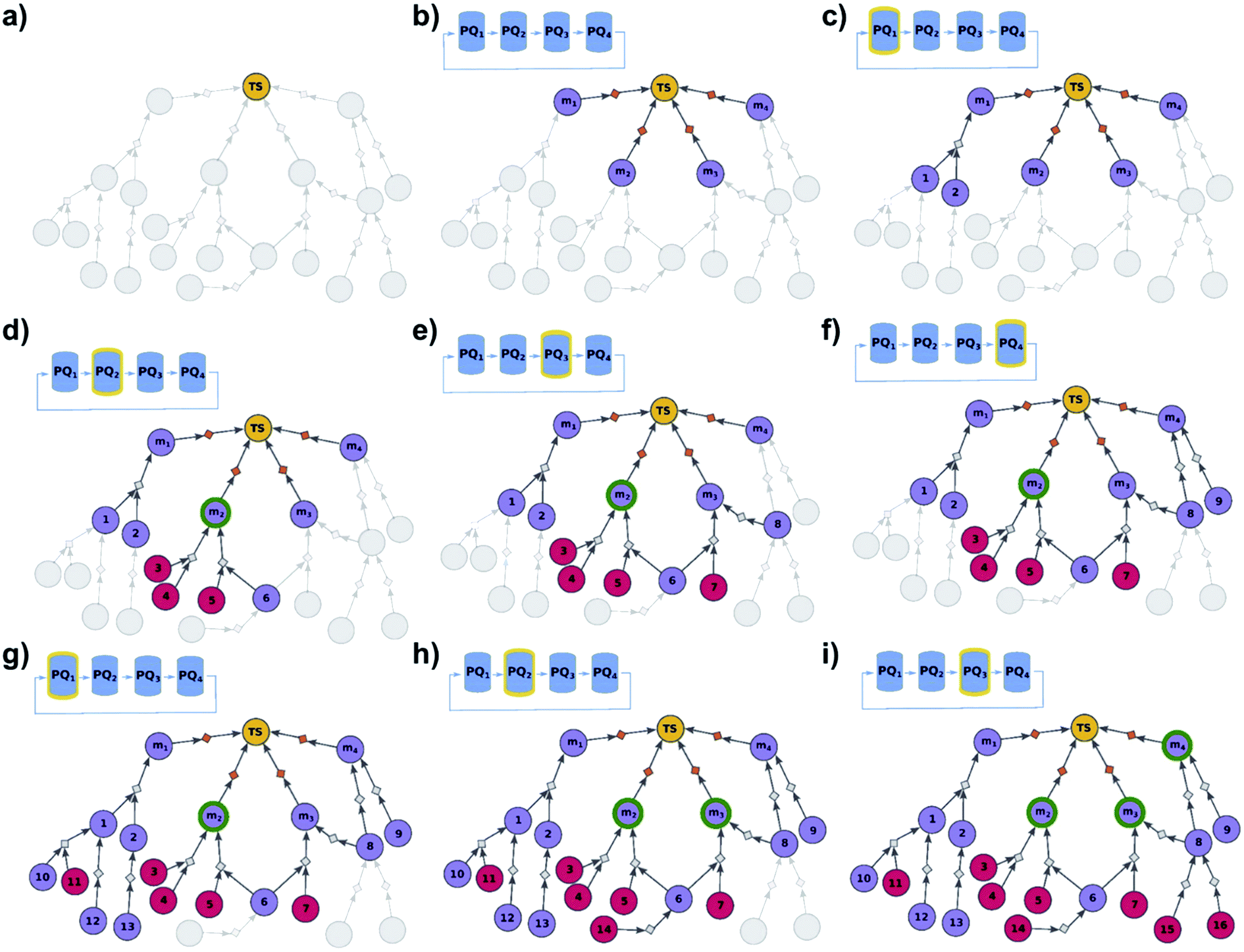
In some cases, not all members of a library are equally difficult to synthesize, and one could wish to perform wet-lab execution of only those that are most readily synthesizable. Algorithmically, this task is a bit more nuanced than finding syntheses of all targets in the library. To illustrate, let us assume that a search for the synthesis of target m1 does not find any solutions after a certain time – if this target is extremely hard to synthesize and yet the algorithm continues to find its synthesis, it might be stuck in this one search while other targets could yield solutions in shorter times. On the other hand, we do not know a priori if these other targets are any simpler. To overcome such problems, we implemented a multi-priority-queue algorithm that allows syntheses of different targets to be explored to comparable levels, while sharing information about common intermediates and chemistries, and returning the best-scoring pathways taking into account the aggregated results for all targets considered. Specifically, the algorithm (Fig. 3 and pseudocode in the ESI, Section S2†) initializes not from a single “dummy” reaction (cf. previous section) but from N such reactions, each linking the terminal root/library node with one specific member of the library {m1, …, mN}. This construction serves as an “OR” condition and ensures that any viable pathway to the root node will also synthesize one of the mi, molecules. Moreover, to ensure that all targets m1, …, mN are being analyzed to comparable degrees, we use the priority-list, PL, rather than a single, global priority queue-based data structure, PQ. This PL (1) has length N; (2) its i-th element, PL[i], is a PQ initialized with a single-element set {m}i; (3) synthon sets from the PL are retrieved in circular order, i.e., PL[1], PL[2], …, PL[N], PL[1], PL[2], …; and (4) when a synthon set S is taken from PL[i], and is further expanded into progeny synthon sets, these progenies are also inserted to PL[i]. In other words, although the search uses one common graph and still benefits from the use of common intermediates/chemistries, each target has its separate PQ which stores the synthetic options for this target and, importantly, is inspected cyclically. As solutions are being found, a selection procedure21 is applied to the entire solution graph (encompassing pathways leading to different targets), to select the best individual syntheses (shortest, most economical, and chemically diverse routes). Unlike the “find all” modality we described earlier, for which the outcome was a global graph encompassing syntheses of all library members, the end result of the “find best” analysis is a list of individual synthetic solutions ranked in descending order of the ease of synthesis.
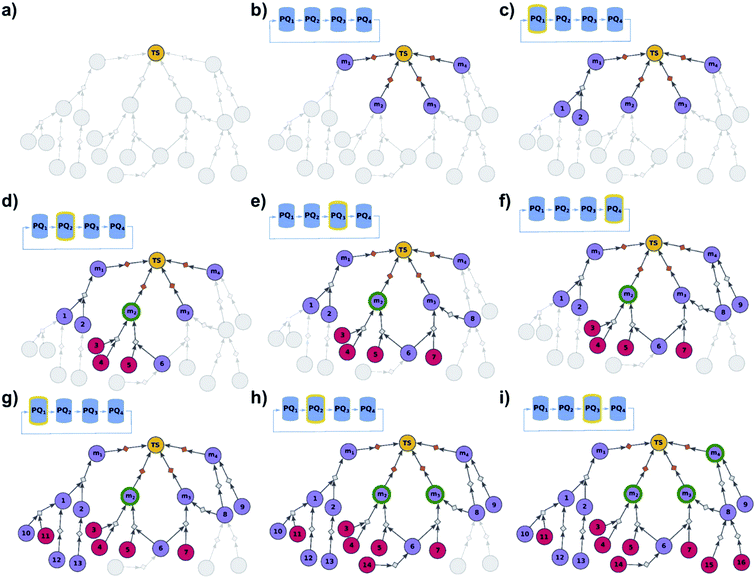 |
| | Fig. 3 Schematic description of the algorithm seeking the syntheses of some, most-synthetically-accessible targets. (a) The root node (yellow circle) corresponds to the target set (here, TS = library of four targets). (b) The graph is extended by creating four substrates (violet circles denoted m1, m2, m3, m4), each linked with the root node via a separate dummy reaction (orange diamonds). Additionally, four priority-queue-based data structures, PQ1, PQ2, PQ3, and PQ4, each corresponding to a separate target node, compose a priority-list, PL, inspected in circular order (first PQ1, then PQ2, PQ3, PQ4, PQ1, etc.). PL is responsible for balanced exploration of syntheses leading to each target. (c) According to PQ1 (orange halo marks this data structure as currently inspected), m1 is expanded, identifying a reaction (grey diamond) from substrates 1 and 2 (violet circle nodes refer to unknown chemicals). (d) Then, as PQ2 is inspected, node m2 is expanded and two reactions are added to the graph (with substrate sets {3, 4} and {5, 6}, respectively; red circular nodes represent terminal, commercially available chemicals). Target m2 is now synthesizable (green halo), with a viable synthesis 3, 4 → m2 already satisfying the condition of finding a synthesis of at least one member of the target library. The search continues to find alternative pathways. (e) According to PQ3, node m3 is expanded, and reactions with substrates {6, 7} and {8} are identified (6 already appeared while searching for syntheses of m2). (f) Then, as PQ4 is inspected, m4 is expanded, giving reaction from node 8 (now, a common intermediate in the synthesis plans leading to m3 and m4) and node 9. (g) Inspection of the priority lists now returns to PQ1, nodes 1 and 2 (previously visited while searching for the synthetic scenario of m1) are explored, giving new nodes 10, 11, 12, and 13. (h) As the search continues, PQ2 is inspected again, discovering viable synthesis (from terminal node 14) leading to a common intermediate of m2 and m3, i.e., node 6. Consequently, m3 becomes synthesizable, and pathways 14 → 5, 6 → m2 and 14 → 6, 7 → m3 become plausible solutions of the initial task. (i) Subsequently, the algorithm inspects PQ3, node 8 is explored by two reactions, each starting at terminal nodes (15 and 16), and m4 becomes synthesizable (newly discovered pathways are 15 → 8 → m3, 16 → 8 → m3, 15 → 8 → m4, and 16 → 8 → m4). All viable pathways identified during the search are retrieved and ranked according to a separate selection algorithm (see ref. 21). | |
Algorithm seeking the most feasible syntheses of multiple isotopomers
For this sub-problem, our aim is to find the most readily synthesizable isotopomer that increases the molecular mass by a user-specified value. The library here is a set of possible isotopomers and the search problem itself is identical to the one described in the previous section with the searches terminating in isotopically labelled (and possibly some non-labelled), commercially available starting materials. The difference lies in the way in which the target library is generated – in particular, we would like to automate the generation of all isotopomers offering the desired mass increase. The procedure to do so begins with specifying the non-labelled target molecule, a desired mass shift, S (positive or negative), and the available_iso list of isotopes one wishes to use. The list should contain isotopes with only positive or only negative mass shifts, corresponding to the sign of S (e.g., if S > 0, 13C but not 11C should be used). This condition precludes generation of several nonsensical isotopomers in which, for example, a mass shift of +1 could be obtained by introducing two 13Cs and one 11C. Additionally, one may wish to specify which atoms should not be labelled (e.g., 2H labelled carboxylic acids, amines, or alcohols are labile, whereas 13C labelling should be avoided in metabolically unstable fragments such as esters or N-methylamines).
With such assumptions, the atoms of the target are arbitrarily ordered, a1, …, aM, and the recursive procedure (for pseudocode, see ESI, Section S3†) is applied to generate a set of desired isotopomers. To explain this procedure, let us consider methanol as the target, a hypothetical available_iso = [2H, 17O, and 18O], and S = 2. Let us define labellings (j, k, S) as a set of labellings of atoms aj, …, ak, giving a mass shift S. Then labellings (1, M, S) refer to the set of isotopomers we ultimately seek (pending de-duplication of possible identical structures). For the specific CH3OH target, let us order atoms a1, a2, a3, a4, a5, a6 = C, H1, H2, H3, O, H4 (upper-right indices are used to distinguish between hydrogen atoms), and commence from a1 = C. As available_iso does not contain any isotopic label for carbon, no isotopic labelling can be applied to a1 and the set of appropriate labellings can be therefore written recursively as equation (*) labellings (1, M, S) = {a1 = 12C} × labellings (2, M, S), where × stands for set multiplication. Moving to the second atom, a2 = H1, it can be labelled deuterium (mass shift of +1) or left unlabelled (no mass shift), and so we have (**) labellings (2, M, S) = {a2 = 2H} × labellings (3, M, S − 1)∪{a2 = 1H} × labellings (3, M, S). By combining (*) and (**) we obtain labellings (1, M, S) = {a1 = 12C, a2 = 2H} × labellings (3, M, S − 1)∪{a1 = 12C, a2 = 1H} × labellings (3, M, S). The procedure is continued until the last atom is reached (a6 for methanol) and all acceptable labelling options are returned. For the methanol example we considered, after de-duplicating the same chemical structures (i.e., removing molecules with the same canonical SMILES representation), there are five viable isotopomers, written here in the SMILES notation that is used as an input to the retrosynthetic search: [2H]CO[2H], C[18O], [2H][17O]C, [2H]C[17O], [2H]C([2H])O.
Results and discussion
Chemical examples implemented in Chematica
The algorithms detailed in the preceding sections can be implemented in various retrosynthetic platforms. Since we have been actively involved in the development of and continue to have access to Sigma-Aldrich’s Chematica, we illustrate how the algorithms function in this particular environment.
As described in several of our publications on Chematica,17–21 this platform is based on the knowledge-base of over 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 expert-coded reaction rules reflecting reaction mechanisms, delineating carefully substituent scope as well as contextual information about potential cross-reactivity conflicts, protection requirements, selectivity issues, etc. (for examples, see ref. 17 and ESI of ref. 18 and 20). The rules have variants applicable to synthesis of isotopically labelled compounds and are augmented by various modules based on quantum-mechanical, molecular-mechanical, machine-learning, or heuristic measures of reactions' electronic and steric requirements.17,20,39 The bipartite synthetic graphs created by the application of reaction rules are navigated with the help of scoring functions assigning costs for each reaction operation performed (with additional costs added to reactions requiring, e.g., protection chemistries) and evaluating structural complexity of the sets of synthon molecules produced in each step. The searches are supplemented by routine checking of the multi-step logic of syntheses – for instance, they prevent dragging highly reactive groups along multiple steps, penalize contraction of certain macrocycles, or allow overcoming local complexity maxima by the use of the so-called tactical combinations. Once feasible routes are found, they are scored according to realistic pricing models21 based on the prices of commercially available starting materials (>200000 chemicals from Sigma-Aldrich including ∼1100 isotopically labelled ones) and approximate yet realistic reaction yields.40,41 The chemical correctness of all these algorithms has been corroborated by successful experimental execution of a number of Chematica-planned, multistep syntheses.18,19
000 expert-coded reaction rules reflecting reaction mechanisms, delineating carefully substituent scope as well as contextual information about potential cross-reactivity conflicts, protection requirements, selectivity issues, etc. (for examples, see ref. 17 and ESI of ref. 18 and 20). The rules have variants applicable to synthesis of isotopically labelled compounds and are augmented by various modules based on quantum-mechanical, molecular-mechanical, machine-learning, or heuristic measures of reactions' electronic and steric requirements.17,20,39 The bipartite synthetic graphs created by the application of reaction rules are navigated with the help of scoring functions assigning costs for each reaction operation performed (with additional costs added to reactions requiring, e.g., protection chemistries) and evaluating structural complexity of the sets of synthon molecules produced in each step. The searches are supplemented by routine checking of the multi-step logic of syntheses – for instance, they prevent dragging highly reactive groups along multiple steps, penalize contraction of certain macrocycles, or allow overcoming local complexity maxima by the use of the so-called tactical combinations. Once feasible routes are found, they are scored according to realistic pricing models21 based on the prices of commercially available starting materials (>200000 chemicals from Sigma-Aldrich including ∼1100 isotopically labelled ones) and approximate yet realistic reaction yields.40,41 The chemical correctness of all these algorithms has been corroborated by successful experimental execution of a number of Chematica-planned, multistep syntheses.18,19
The multi-target design interfaced with Chematica entails specification of the target library, either by drawing all its members in a structure editor or by defining a Markush structure written in the SMILES notation42 (Fig. 1c and d) or, for isotopomers, by specifying the desired mass increase and the isotopes that can be used. The results of the searches are presented to the user as a “global” graph encompassing, depending on the search modality, syntheses of all or some, most synthetically accessible targets. Each substance and reaction node can be expanded to provide, respectively, additional structural and synthetic details (see ESI, Section S5–S11†).
Synthesis of all members of a Prozac-derived library
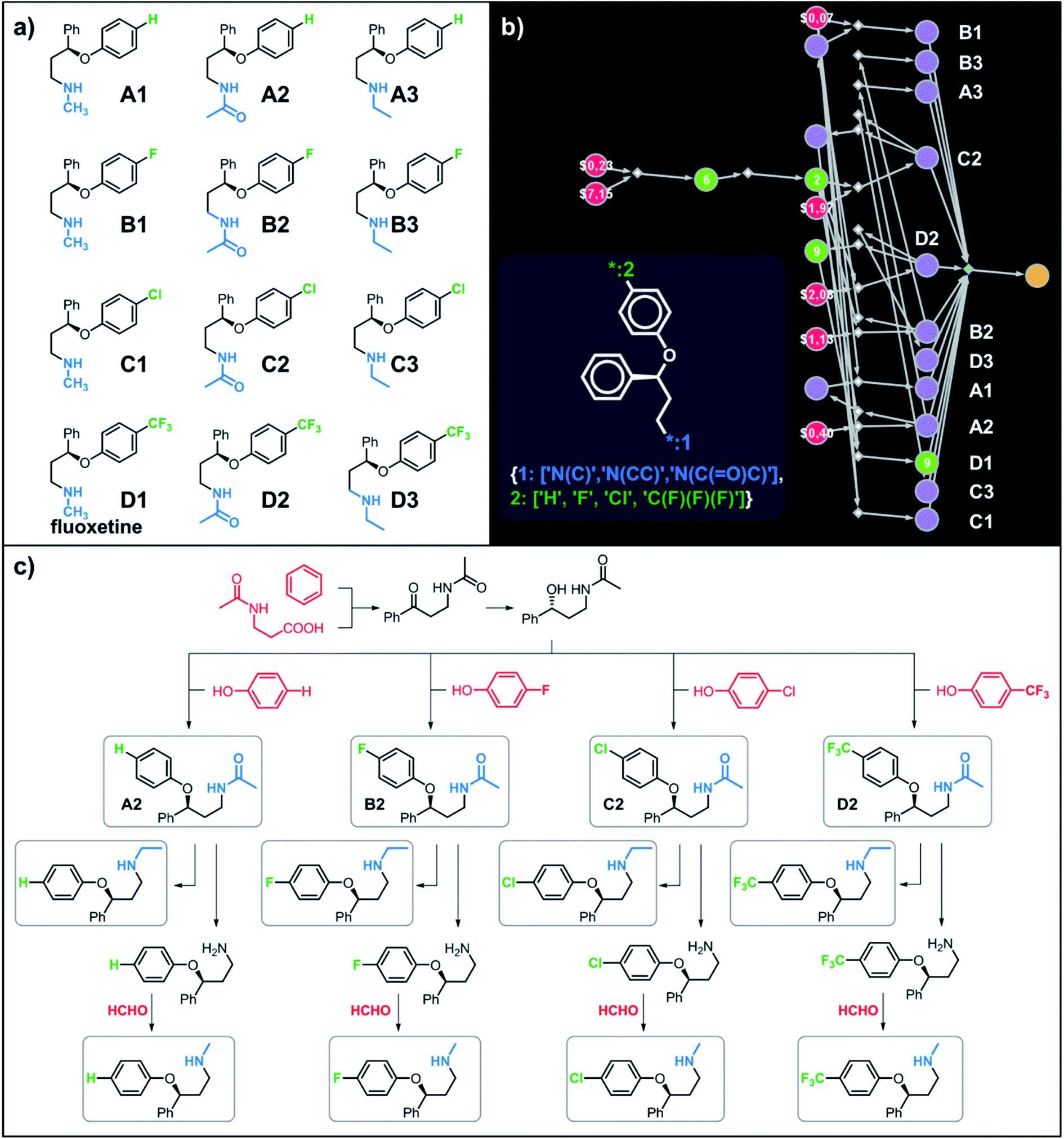
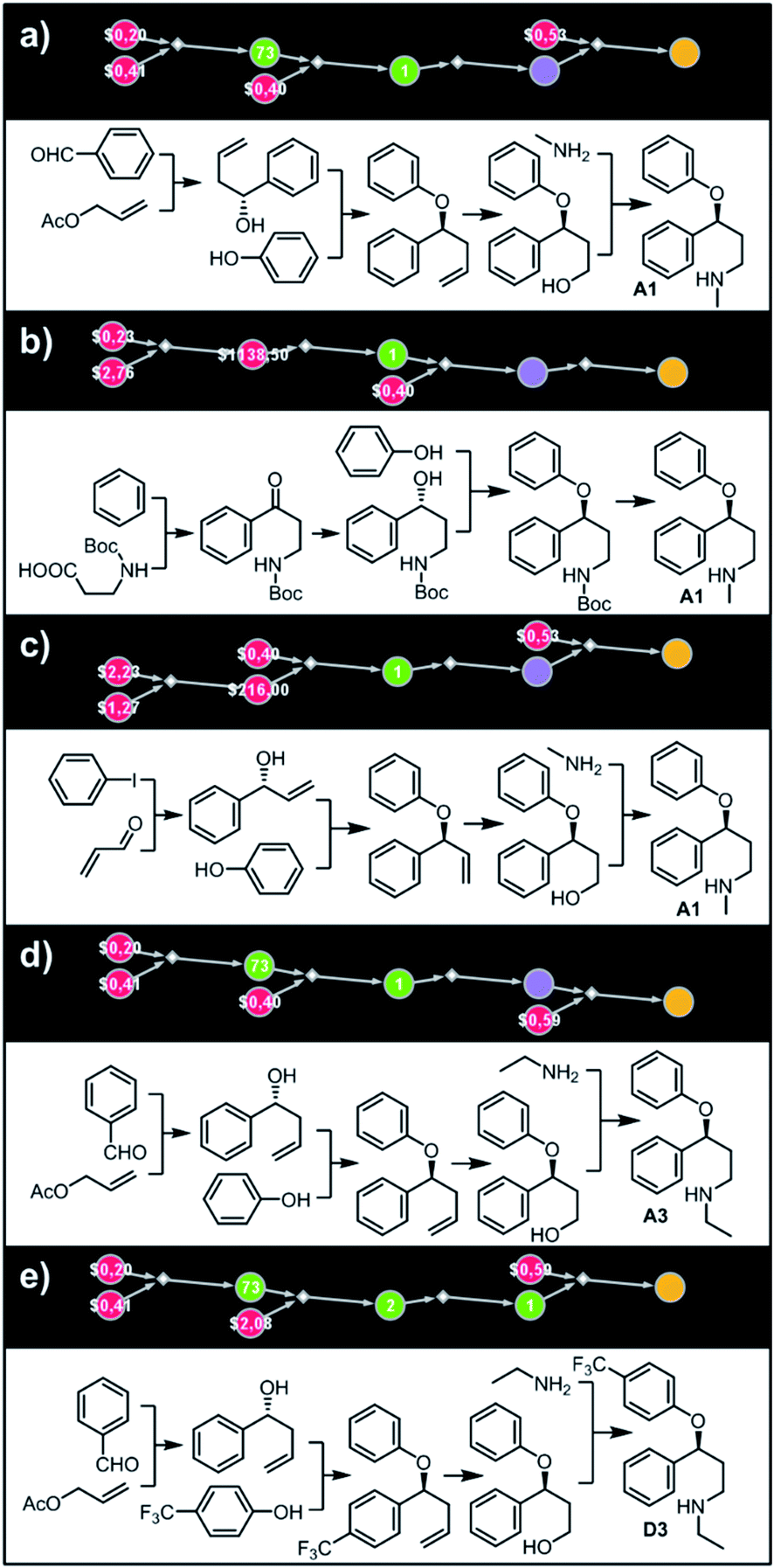
Let us begin with a simple example in which we seek syntheses of all members of a small library around a selective serotonin reuptake inhibitor, fluoxetine (Prozac). The library admits four different substituents (p-F, p-Cl, p-CF3 and H) in the aryl ether part of fluoxetine and three different moieties on the N-terminated side chain (NHMe, NHEt and NHAc), corresponding to 12 compounds in total (Fig. 4a). Within ca. 5 min, Chematica produced tens of global plans for the syntheses of all the library members, with the top-scoring solution graph shown in Fig. 4b and with chemical details elaborated in Fig. 4c (for raw Chematica screenshots and suggested reaction conditions, see Fig. S4†). The common root of all syntheses is the Friedel–Crafts acylation of benzene with acyl chloride derived from N-acetyl β-aminopropionic acid. Subsequent enantioselective reduction (controlled, e.g., by the Corey–Bakshi–Shibata catalyst43 or Noyori's catalyst44) yields an enantioenriched secondary alcohol which is reacted with appropriate phenols under Mitsunobu conditions to give the desired N-acyl series A2–D2. The N-ethyl substituted compounds A3–D3 can then be obtained in one step via reduction of the acetyl moieties while the preparation of N-methyl series A1–D1 requires hydrolysis of acetamide and subsequent reductive amination with formaldehyde. Importantly, the entire scheme to prepare 12 different compounds requires only 18 individual reactions and takes advantages of several common intermediates including interconversions of some library members. We note that the proposed strategy comprising enantioselective reduction of appropriate β-aminoacetophenone44–46 and Mitsunobu displacement with phenol47,48 has already been used in several syntheses of structurally related compounds including our synthesis of hydroxyduloxetine.18 We also emphasize that this “global” synthetic plan is different from the plans that Chematica produces for each target separately – for instance, if the program's task is to make des-trifluoromethyl congener of fluoxetine A1 (Fig. 5a–c and S5†), it uses the enantioselective allylation of aldehyde and ozonolysis mimicking Bracher's approach.49 The same strategy is returned as the top solution if A3 (Fig. 5d and S6†) or D3 (Fig. 5e and S7†) is an individual target. We note that none of the top-three approaches found by Chematica in single-target-oriented searches for A1 (Fig. 5a–c and S5†) – relying on either the Friedel–Crafts acylation with carbamate protected β-aminopropionic acid50 or enantioselective arylation of aldehydes51,52 – are desirable for the design of the entire library. This is so because these syntheses cannot take advantage of late common intermediates and require several additional reactions (e.g., for the optimal individual solution to A1, adaptation to the all-library synthesis would entail a total of 21 distinct reactions (allylation, four displacements with phenols, four hydroborations, and twelve aminations). The software is not using the elegant three-component Mannich reaction (the one we employed previously in ref. 18 for the construction of a structurally similar scaffold of hydroxyduloxetine) because it cannot be adapted for the synthesis of the current library – in particular, the Mannich reaction cannot be performed with acetamide instead of methylamine to yield the N-Ac series A2–D2. We also note that this design example illustrates well typical gains in terms of computational efficiency: the search on a common graph to identify the first viable solution requires exploration of ca. 10 times less nodes compared to the case when the syntheses of the 12 targets are searched separately (cf. Table S1 in the ESI, Section S4†).
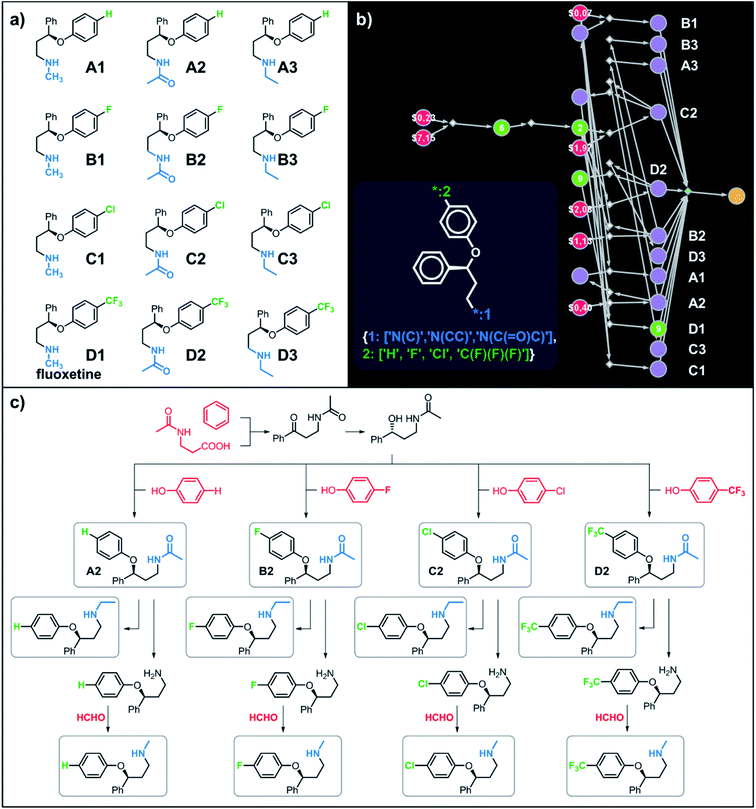 |
| | Fig. 4 Retrosynthetic search to synthesize all members of a library around fluoxetine scaffold. (a) All members of the library. (b) The corresponding Markush structure and the screenshot from Chematica showing the top-scoring solution to synthesize all of the specified targets. In this graph, yellow circular node = Markush structure; small, blue, diamond-shaped node = “dummy” reaction connecting all specific targets A1–D3 to the root node; leftmost column of 12 circular nodes = specific A1–D3 targets, mostly unknown in the NOC repository35,36 (violet nodes) but one known (D1, number 9 inside of the node means that nine syntheses of this compound have been reported in the literature); red nodes = terminal, commercially available chemicals, numbers inside the nodes indicate prices per gram (from Sigma-Aldrich catalog). Note that several substances are common intermediates in multiple pathways and all 12 compounds are made in 18 steps in total. Details of synthetic plans are provided in panel (c). For raw Chematica output and further details, see Fig. S4.† | |
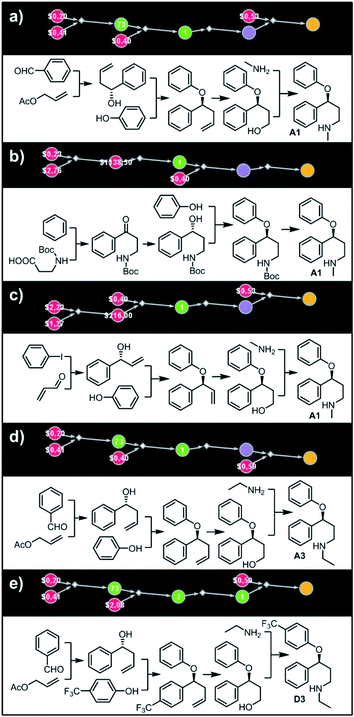 |
| | Fig. 5 Top-scoring solutions proposed by Chematica for the synthesis of individual members of the fluoxetine library: (a–c) target A1, (d) target A3, and (e) target D3. Node coloring scheme is as in Fig. 4. For raw Chematica output and synthetic details, see Fig. S5–S7.† | |
Synthesis of all members of the Almorexant-derived library
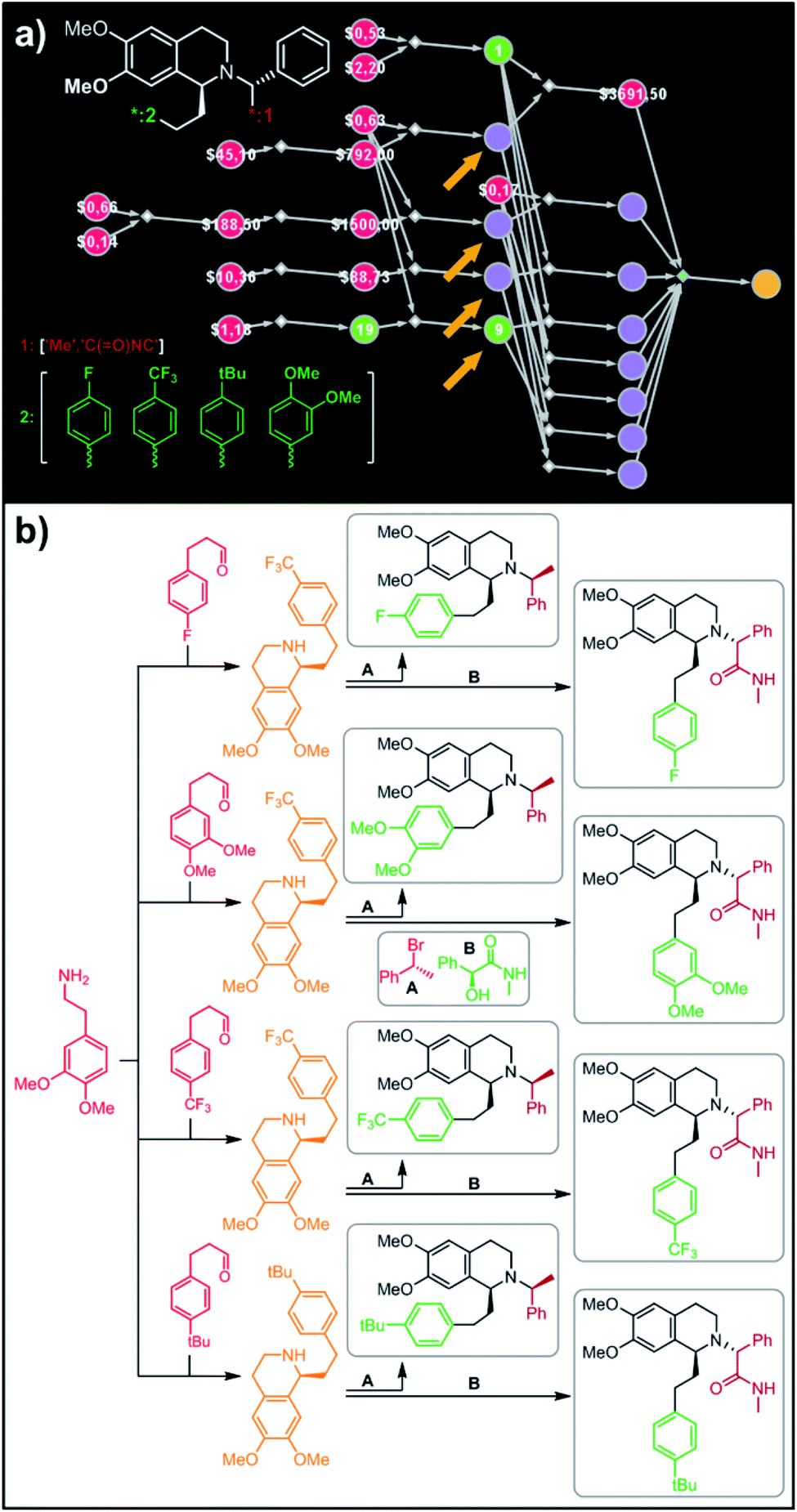
In a more chemically advanced example, we consider synthesis of a library centered around Almorexant, a drug developed by Actelion and GSK for the treatment of insomnia. The library accepts four different substituents (p-F, p-CF3,p-tBu and 3,4-diOMe) in the phenylethyl part of Almorexant and two different N-substituents, corresponding to eight compounds in total (inset in Fig. 6a). Within ca. 30 min, Chematica identified global synthetic plans with the top-scoring solution graph shown in Fig. 6a and with chemical details elaborated in Fig. 6b (for raw Chematica screenshots and suggested reaction conditions, see the ESI, Fig. S8†). The synthesis of each library member commences with the oxidation of appropriate terminal alkenes to aldehydes. The key formation of chiral tetrahydroisoquinolines leading to four common intermediates (marked with arrows in Fig. 6a and colored orange in Fig. 6b) is accomplished via enantioselective Pictet–Spengler cyclisation controlled either by a chiral auxiliary or chiral catalyst.54 Subsequent alkylation with the commercially available secondary benzyl bromide A or condensation with a derivative of mandelic acid B yields the target molecules.
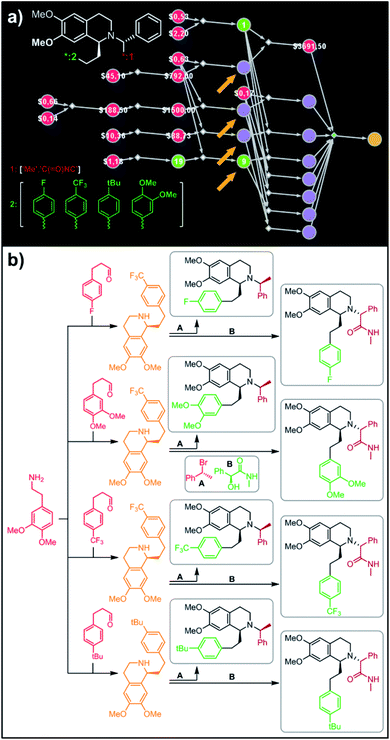 |
| | Fig. 6 Retrosynthetic search to synthesize all members of a library of analogues of Almorexant. (a) Markush structure representing proposed library (white inset, top-left), lists of substituents (bottom left) and graph representation of the top-scoring solution. Chemical details are shown in panel (b). Note that several substances (marked with arrows and colored orange in panel (b)) are common intermediates in multiple pathways while the entire library is prepared from single phenylethylamine. For raw Chematica output and further details, see Fig. S8.† Node coloring scheme is as in Fig. 4. | |
Synthesis of all members of a library of RANKL/RANK inhibitors
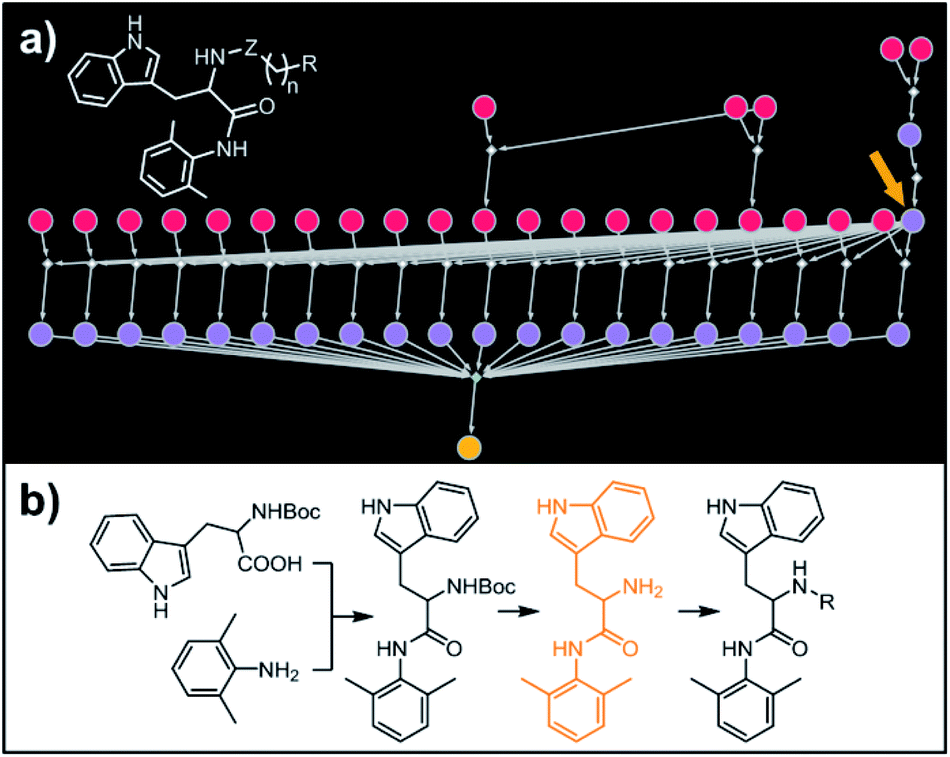
Our last example of all-library design is important in that the computer's autonomous design can be directly compared with and validated against recent experimental work. Specifically, we challenged Chematica with the synthesis of a subset of a library of RANKL/RANK inhibitors reported very recently by Yang and co-workers.53 In this task the library, represented as the Markush structure in the inset of Fig. 7a (for full representation see Fig. S9†), consisted of 20 derivatives of tryptophan with different N-alkyl (Z = CH2) or N-acyl fragments (Z = C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), different linker lengths (n = 1–4) and substituents (R = alkyl, aryl, heteroaryl). The search was allowed to run for ca. 15 min and returned as the top-scoring solution the synthetic plan shown as a graph in Fig. 7a and further detailed in Fig. S10 and S11.† Within this plan, the synthesis of each library member is accomplished in a short (three-four steps) sequence, commencing with the coupling between commercially available N-Boc tryptophan and 2,6-dimethylaniline. Subsequent removal of the protecting group gives a common intermediate (in Fig. 7a, the rightmost node marked with an orange arrow). Finally, coupling with appropriate carboxylic acids or primary mesylates leads to N-acyl or N-alkyl target molecules, respectively. Remarkably, this approach mirrors closely the strategy used in Yang and co-workers' experiments.53
O), different linker lengths (n = 1–4) and substituents (R = alkyl, aryl, heteroaryl). The search was allowed to run for ca. 15 min and returned as the top-scoring solution the synthetic plan shown as a graph in Fig. 7a and further detailed in Fig. S10 and S11.† Within this plan, the synthesis of each library member is accomplished in a short (three-four steps) sequence, commencing with the coupling between commercially available N-Boc tryptophan and 2,6-dimethylaniline. Subsequent removal of the protecting group gives a common intermediate (in Fig. 7a, the rightmost node marked with an orange arrow). Finally, coupling with appropriate carboxylic acids or primary mesylates leads to N-acyl or N-alkyl target molecules, respectively. Remarkably, this approach mirrors closely the strategy used in Yang and co-workers' experiments.53
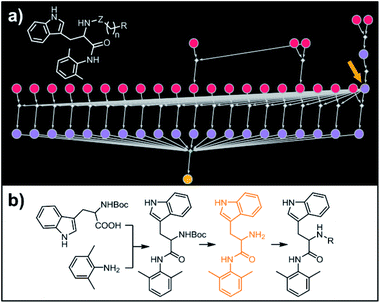 |
| | Fig. 7 Retrosynthetic search to synthesize a subset of a recently reported library of tryptophan derivatives acting as RANKL/RANK inhibitors.53 (a) Markush structure representing proposed library (white inset) and graph representation of the top-scoring solution. All members of the library are prepared from one common intermediate marked with arrow in (a) and colored orange in panel (b) which details synthesis of one of the library members. For Chematica's raw output for the entire library, see Fig. S10 and S11.† | |
Synthesis of the most accessible derivatives of a κ-opioid agonist
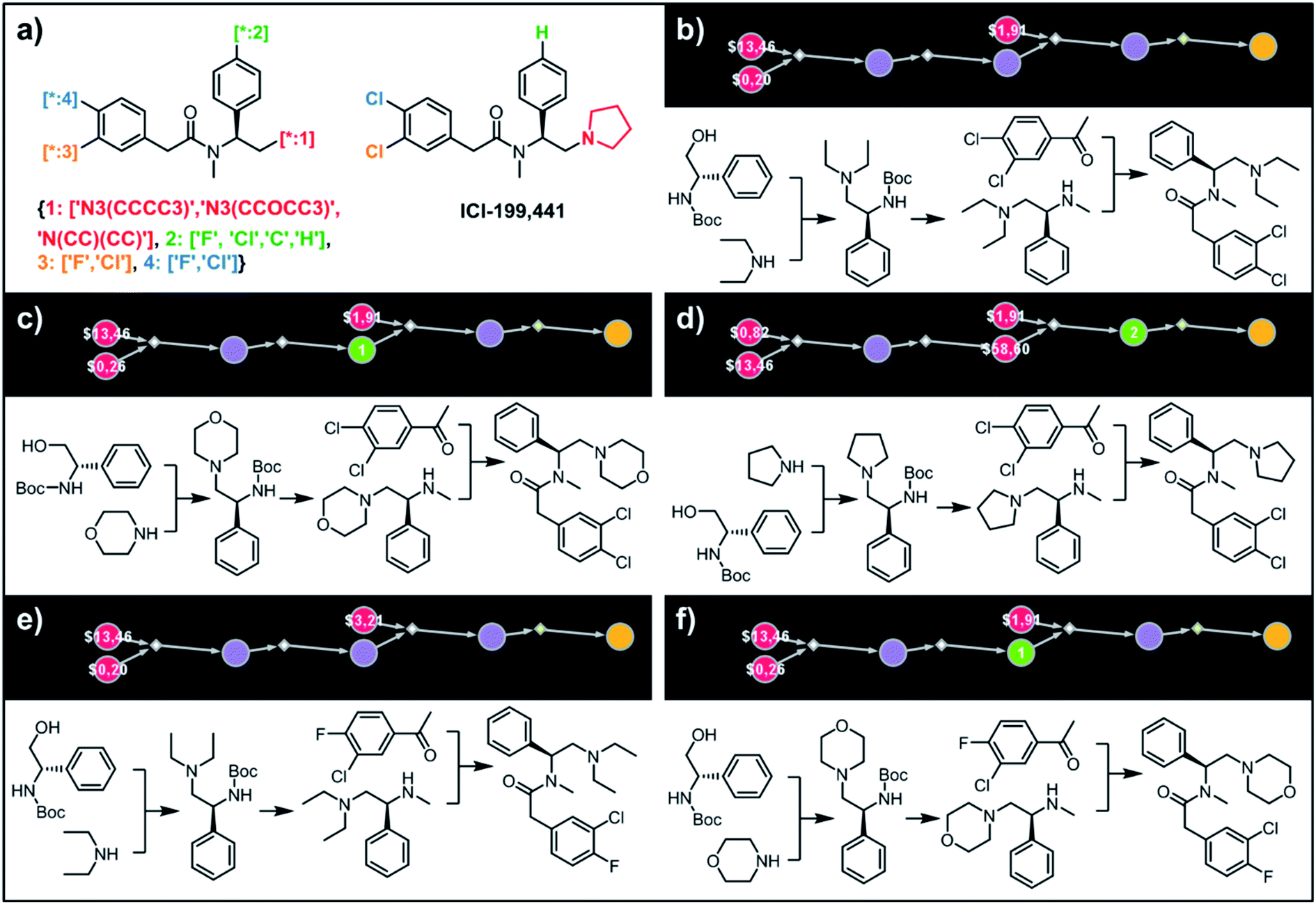
To illustrate the synthetic design of not all but only the most accessible members of a given library, we considered derivatives of a selective κ-opioid agonist55 ICI-199441 (Fig. 8). The ICI-199441 scaffold was decorated with three substituents in the N-terminated side chain, four substituents in the benzylamine part, and four combinations of halogens in the arylacetic acid part, overall corresponding to 48 distinct members of the library (Fig. 8a). The top five of the several hundreds of viable pathways (identified within ∼10 min) are shown in Fig. 8b–f and further detailed in Fig. S12.† In each of these plans, the molecule of interest (one node before the yellow node; compared with the scheme in Fig. 3b) can be obtained in three steps using alkylation of an appropriate secondary amine with a commercially available protected phenylglycinol, removal of the protecting group, and a sulfur catalyzed Willgerodt–Kindler (WK) reaction56 yielding the desired arylacetic acid amides from acetophenones. The application of this last methodology – while chemically correct – might, at first glance, appear counterintuitive given that such amides are usually55,57 formed in reactions of acyl halides or carboxylic acids. These more conventional plans were, indeed, present in the top 100 solutions identified by the software, but the algorithm correctly gave them lower scores based on higher prices of substrates – namely, application of the WK reaction allowed for the use of cheaper acetophenone substrates rather than appropriate arylacetic acids ($1.91 vs. $3.23 per g of dichloroderivative and $3.21 vs. $5.81 per g of the 4-F-3-Cl derivative). Consequently, the diethylamino congeners were found to be more accessible than morpholino- or cyclopentylamino ones. We also note that none of the compounds substituted in the benzylamine part appeared among the most accessible targets as their synthesis requires construction of appropriate chiral aminoalcohols.
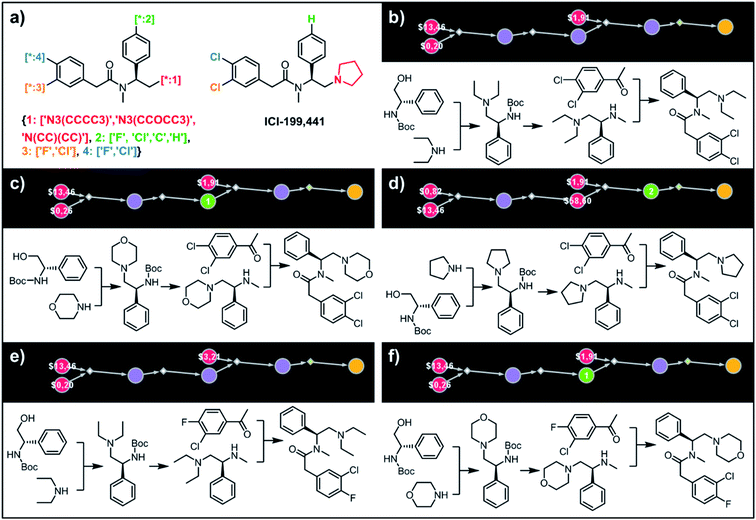 |
| | Fig. 8 Retrosynthetic analysis of ICI199441 derivatives. (a) Left portion shows the Markush structure and dictionary of substituents defining the library of 48 members; right portion shows the structure of the original ICI199441 compound. (b–f) Five top-scoring synthetic plans for library from (a). Note that in plan (d), the algorithm found a commercially available advanced intermediate (red node with price per gram = $58.60) but continued the search until it found less expensive substrates with prices per gram below the user-specified threshold of $50 per g. For further synthetic details, see Fig. S12.† | |
Syntheses of isotopically labelled targets
In our last set of examples, we used the algorithm to determine which isotopically labelled compounds from a given library of isotopomers are synthetically most readily accessible.
(i) Cinacalcet.
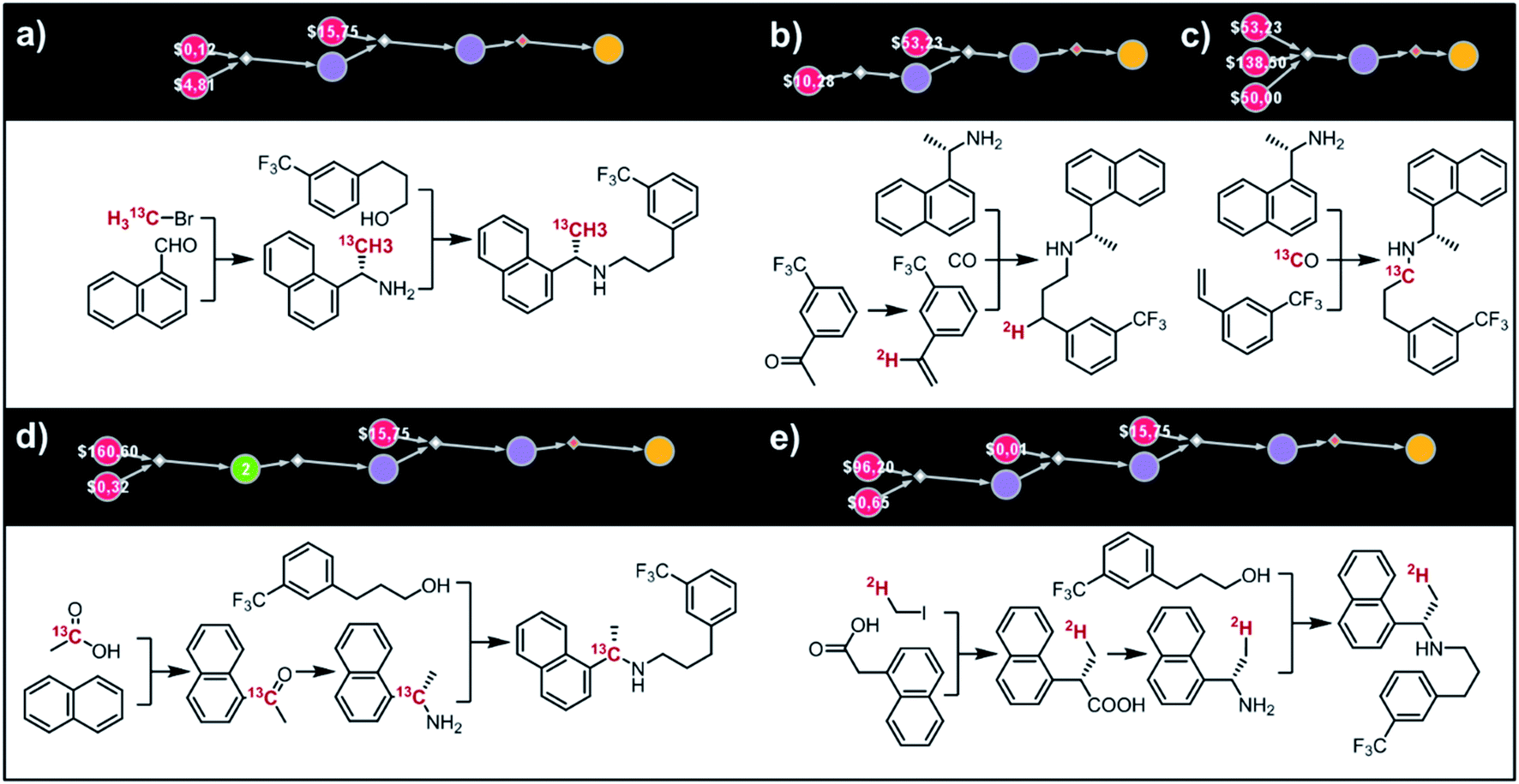
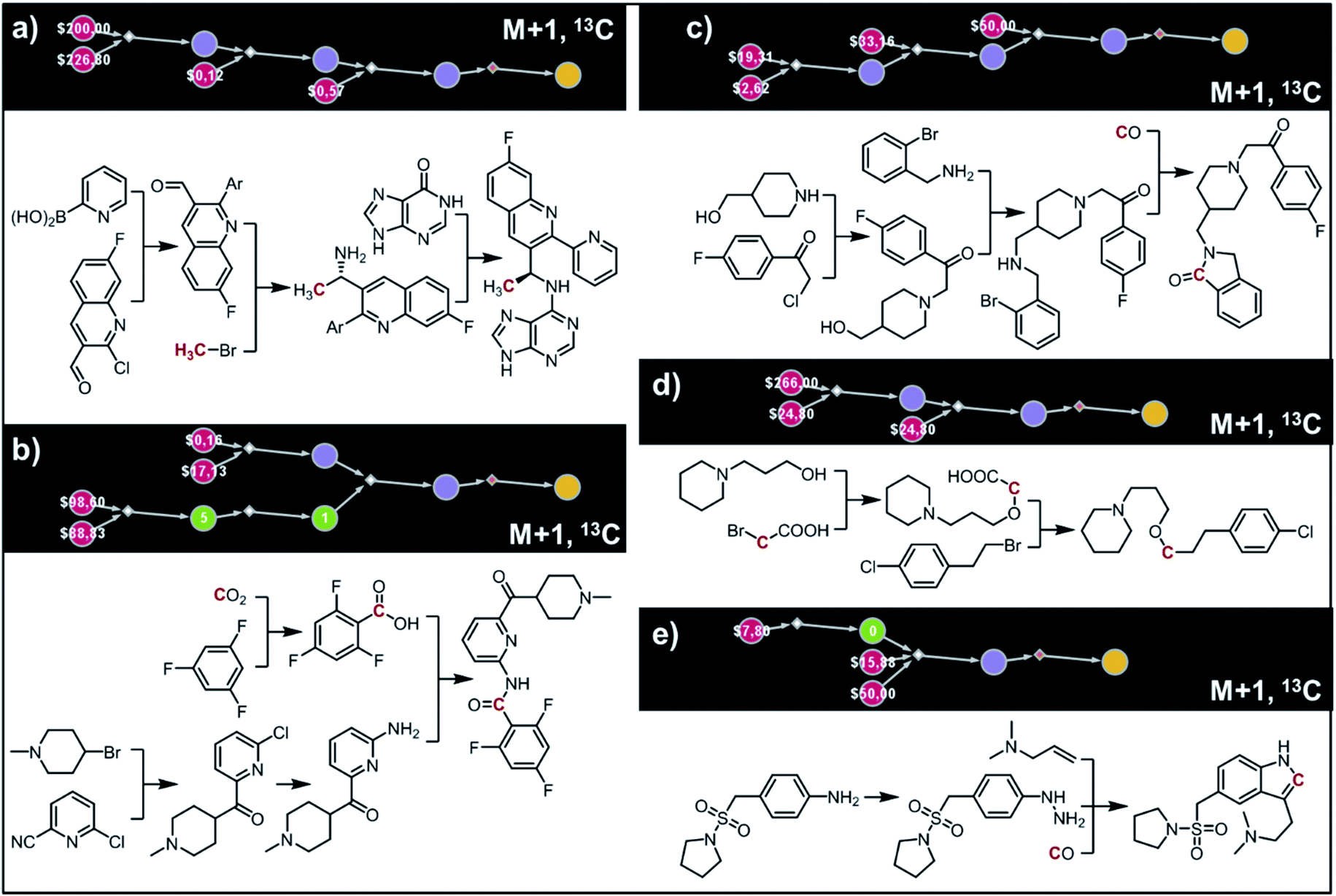
Fig. 9 shows five top-scoring syntheses of Amgen's cinacalcet (Sensipar/Mimpara), whose mass we wish to increase by S =1 by single labelling with either 13C or 2H (there are 39 unique isotopomers). In the first proposed synthesis (Fig. 9a), the 13C isotopic label is located on the methyl group and introduced from bromomethane. In the first step, chiral-auxiliary-directed addition58,59 of an organometallic reagent derived from 13CH3Br yields the enantioenriched secondary amine which is then alkylated with a commercially available alcohol to give the molecule of interest. In the second plan (Fig. 9b), the 2H isotope label is introduced from D2O during the Shapiro reaction proposed as the first step. Subsequent Rh-mediated hydroaminomethylation gives the 2H-cinacalcet also after 2 steps. In the third plan (Fig. 9c), 13C labelled cinacalcet is made in only one step, via three-component hydroaminomethylation utilizing commercially available styrene, enantioenriched secondary amine, and 13C carbon monoxide. We observe that such a carbonylative approach has been already used by Amgen to prepare unlabeled cinacalcet.60 In the fourth plan (Fig. 9d), the labelled 13C atom is located at the chiral carbon and comes from the 13C acetic acid used in the initial Friedel–Crafts acylation of naphthalene.61 Subsequent reductive amination guided by a chiral auxiliary62 or a chiral catalyst63,64 yields the secondary amine transformed into the target molecule following steps from the first plan. Finally, cinacalcet can be labelled with 2H located at the methyl group with a deuterium atom introduced from 2H-methyl iodide participating in direct enantioselective alkylation of naphthylacetic acid65 controlled by a chiral diamine (Fig. 9e). Subsequent transformation of carboxylic acid into secondary amine via the Schmidt reaction66 yields the amine which is subjected to the reaction with alcohol to give the target molecule. We note that the proposed approaches relying on the alkylation of chiral naphthylamine are corroborated by published syntheses67–71 of unlabeled cinacalcet.
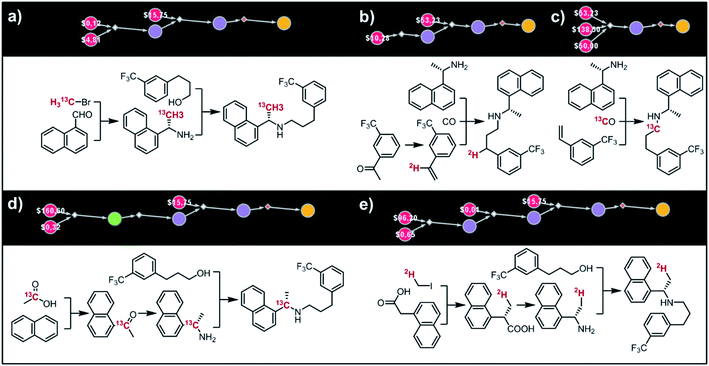 |
| | Fig. 9 Syntheses of cinacalcet singly-labelled with either 13C (a, c and d) or 2H (b and e). Several viable routes were identified within 10 min; five top-scoring routes are shown. For further synthetic details, see Fig. S13.† | |
(ii) AMG-319.
The second example in this section describes synthesis of M + 1 isotopomers of Amgen's AMG-319, the inhibitor targeted for autoimmune diseases72 and head and neck squamous-cell carcinomas.73 The proposed solution (Fig. 10a) commences with the Suzuki coupling of 2-pyridyl boronic acid and 2-chloropyridine. Subsequent conversion to an imine and stereoselective addition58 of the organometallic reagent derived from 13CH3Br yields the enantioenriched benzylic amine which is coupled with hypoxanthine in the last step. We note that this computer-designed synthetic plan resembles Amgen's original route72 to unlabelled AMG-319.
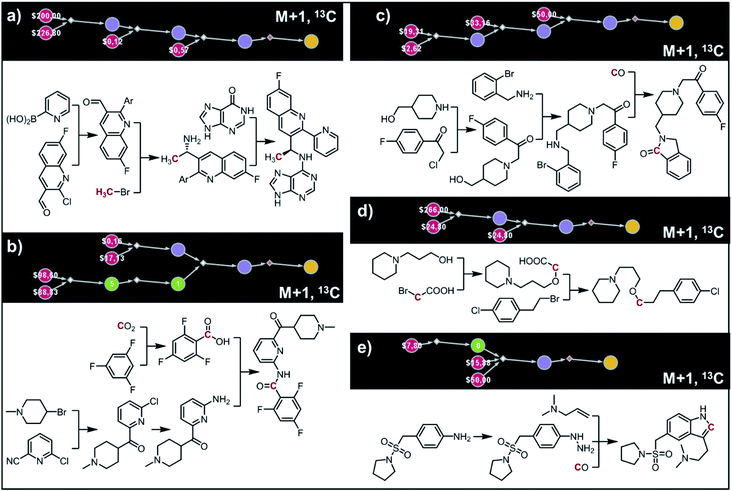 |
| | Fig. 10 Retrosynthetic design of isotopically labeled drug molecules. In all cases, 13C labeling was allowed and the desired mass shift was S = +1. Top-scoring and in all cases chemically viable solutions obtained for (a) a library of 21 isotopomers of AMG-319; (b) a library of 14 isotopomers of lasmiditan; (c) a library of 18 isotopomers of roluperidone; (d) a library of 13 isotopomers of pitolisant; (e) a library of 13 isotopomers of almotriptan. For further details of the pathways, see Fig. S14–S18.† | |
(iii) Lasmiditan.
In the third example, the algorithm is used to design syntheses of M + 1 lasmiditan developed by Eli Lilly for the treatment of acute migraine. After specifying the admissible isotope label (here, 13C), desired mass shift S = 1 (corresponding to 15 isotopomers) and excluding 13C N-methylated isotopomer prone to hepatic cleavage observed previously for N-methyl piperazines,23,74–76 the search was run for ca. 10 min and returned hundreds of viable synthetic plans from which the top-scoring one is shown in Fig. 10b. In the first step, the appropriate 2-chloropyridine is constructed in one step via addition of a Grignard reagent, obtained from the N-methyl-4-bromopiperidine, to 2-cyano-6-chloropyridine. The isotope label is introduced from 13CO2 used for the formation of carboxylic acid from the organolithium reagent derived from trifluorobenzene.77 The following steps resemble the original route to lasmiditan.78 Subsequent amination of 2-chloropyridine and reaction with labelled benzoic acid yield the molecule of interest in a four-step sequence.
(iv) Roluperidone.
In the fourth example, Chematica designs syntheses of M + 1, 13C labelled congener of roluperidone developed by Minerva Neurosciences for the treatment of schizophrenia.79 The top-scoring plan returned after ca. 10 min is shown in Fig. 10c. The proposed short sequence commences with the N-alkylation of hydroxymethylpiperidine with the appropriate chloroacetophenone. Subsequent alkylation of 2-bromobenzylamine with the remaining primary alcohol yields the substrate amenable to intramolecular carbonylative amidation.80 The 13C label is introduced in this step and sourced from 13CO, completing the synthesis in just three steps. The proposed plan resembles Mitsubishi's synthesis of roluperidone utilizing proposed hydroxymethylpiperidine and phenacyl bromide as building blocks and participating in SN2 alkylations of benzolactams and secondary amines;81 moreover, the proposed carbonylative N-alkylation has been used in Astellas' synthesis of N-benzyl benzolactams.82
(v) Pitolisant.
The fifth example illustrates efficient preparation of M + 1, 13C labelled pitolisant (Wakix), developed by Bioprojet for the treatment of hypersomnia.83,84 The search performed for ca. 10 min returned multiple solutions from which the top-scoring one is shown in Fig. 10d. The entire sequence requires only two steps and sources the 13C atom from labelled bromoacetic acid used in the first step to N-alkylate85 hydroxypropylpiperidine. The obtained alkoxyacid is then used in MacMillan's decarboxylative coupling86 with commercially available phenethyl bromide to give the target molecule. The proposed plan resembles Bioprojet's one-step solution87 which also used hydroxypropylpiperidine alkylated with the appropriate alkyl bromide.
(vi) Almotriptan.
In the sixth and last example, we aim to design routes to labelled almotriptan developed by Almirall for the treatment of severe migraine headache. After specifying the plausible isotopes (13C) and mass shift S = 1 and precluding the N-Me labelled isotopomers prone to hepatic cleavage,23,88–90 the search was run for ∼10 min and returned as its top-scoring solution pathway shown in Fig. 10e. Somewhat counterintuitively, the isotope label in the most accessible isotopomer is located inside the indole ring and comes from 13CO. The proposed synthetic plan starts from the commercially available aniline transformed into an appropriate hydrazine. Subsequent Rh-catalyzed tandem hydroformylation/indolisation91 builds the central ring system, introduces the isotope label and attaches the dimethylaminoethyl side chain and yields the target molecule in a two-step sequence. Similar, elegant carbonylative tandem indolisation was already used in Sheldon's one-pot synthesis of unlabeled melatonin.92
Conclusions
In summary, we described how the search routines over large graphs of retrosynthetic scenarios can be adapted to find all or some members of target compound libraries. For the find-all variant, the “global” synthetic plans can benefit from the use of common intermediates and can be significantly different from optimal solutions found for each target separately – that is to say, the global solutions might be counterintuitive for human planners accustomed to optimizing specific synthetic routes rather than interconnected networks of such routes. Another application we consider quite useful is the synthesis of isotopomers. Here, the catalogs of isotopically labelled starting materials are significantly less populous than those of unlabelled building blocks, and many blocks that are considered “basic” are not available in labelled forms. Consequently, synthetic design is often non-intuitive and it is not straightforward to predict which of the potential isotopomers would be the easiest one to make – a question our algorithms can handle rapidly and efficiently. Overall, this work extends the scope of computer-assisted synthetic planning to new problems that are common and important in pharmaceutical and agrochemical industries.
Conflict of interest
While Chematica was originally developed and owned by B. A. G.'s Grzybowski Scientific Inventions LLC, neither he nor the co-authors no longer hold any stock in this company, which is now a property of Merck KGaA, Darmstadt, Germany. The authors continue to collaborate with Merck within the DARPA “Make-It” award. All queries about access options to Chematica (now rebranded as SynthiaTM), including academic collaborations, should be directed to Dr Sarah Trice at E-mail: sarah.trice@sial.com.
Acknowledgements
K. M., P. D. and B. A. G. thank the U.S. DARPA for generous support under the “Make-It” Award, 69461-CH-DRP #W911NF1610384. B. A. G. also gratefully acknowledges personal support from the Institute for Basic Science Korea, Project Code IBS-R020-D1. P. D. thanks Dr Tomasz Badowski for helpful insights.
References
- N. Jones, Nature, 2014, 505, 146–148 CrossRef CAS PubMed.
- M. I. Jordan and T. M. Mitchell, Science, 2015, 349, 255–260 CrossRef CAS PubMed.
- D. Silver, T. Hubert, J. Schrittwieser, I. Antonoglou, M. Lai, A. Guez, M. Lanctot, L. Sifre, D. Kumaran, T. Graepel, T. Lillicrap, K. Simonyan and D. Hassabis, Science, 2018, 362, 1140–1144 CrossRef CAS.
-
I. Sutskever, O. Vinyals and Q. V. Le, Advances in Neural Information Processing Systems, 2014, vol. 27 Search PubMed.
- E. J. Corey and W. T. Wipke, Science, 1969, 166, 178–192 CrossRef CAS.
- H. L. Gelernter, A. F. Sanders, D. L. Larsen, K. K. Agarwal, R. H. Boivie, G. A. Spritzer and J. E. Searleman, Science, 1977, 197, 1041–1049 CrossRef CAS PubMed.
- S. Hanessian, J. Franco and B. Larouche, Pure Appl. Chem., 1990, 62, 1887–1910 CAS.
- J. B. Hendrickson, J. Am. Chem. Soc., 1977, 99, 5439–5450 CrossRef CAS.
- I. Ugi, J. Bauer, K. Bley, A. Dengler, A. Dietz, E. Fontain, B. Gruber, R. Herges, M. Knauer, K. Reitsam and N. Stein, Angew. Chem., Int. Ed. Engl., 1993, 32, 201–227 CrossRef.
- O. Ravitz, Drug Discovery Today: Technol., 2013, 10, e443–e449 CrossRef PubMed.
- M. A. Kayala and P. Baldi, J. Chem. Inf. Model., 2012, 52, 2526–2540 CrossRef CAS PubMed.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein and H. Saller, Org. Process Res. Dev., 2015, 19, 357–368 CrossRef.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS PubMed.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS.
- B. A. Grzybowski, Abstr. Pap. Am. Chem. Soc., 2018, 256, 5 Search PubMed.
- K. Molga, P. Dittwald and B. A. Grzybowski, Chem, 2019, 5, 460–473 CAS.
- T. Badowski, K. Molga and B. A. Grzybowski, Chem. Sci., 2019, 10, 4640–4651 RSC.
-
B. Faller and L. Urban, Hit and Lead Profiling, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed.
-
F. Z. Dörwald, Lead Optimization for Medicinal Chemists, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- A. E. Mutlib, Chem. Res. Toxicol., 2008, 21, 1672–1689 Search PubMed.
- C. J. Unkefer and R. A. Martinez, Drug Test. Anal., 2012, 4, 303–307 Search PubMed.
- C. Planas, A. Puig, J. Rivera and J. Caixach, J. Chromatogr. A, 2006, 1131, 242–252 CrossRef CAS PubMed.
- M. B. Woudneh, M. Sekela, T. Tuominen and M. Gledhill, J. Chromatogr. A, 2006, 1133, 293–299 CrossRef CAS PubMed.
- F. Al-Taher, K. Banaszewski, L. Jackson, J. Zweigenbaum, D. Ryu and J. Cappozzo, J. Agric. Food Chem., 2013, 61, 2378–2384 CrossRef CAS PubMed.
- S. Abu-El-Haj, M. J. Bogusz, Z. Ibrahim, H. Hassan and M. Al Tufail, Food Control, 2007, 18, 81–90 CrossRef CAS.
- S. Chan, M.-F. Kong, Y.-C. Wong, S.-K. Wong and D. W. M. Sin, J. Agric. Food Chem., 2007, 55, 3339–3345 CrossRef CAS PubMed.
- J. Lin, D. H. Welti, F. A. Vera, L. B. Fay and I. Blank, J. Agric. Food Chem., 1999, 47, 2813–2821 CrossRef CAS PubMed.
- M. S. Allen, M. J. Lacey and S. Boyd, J. Agric. Food Chem., 1994, 42, 1734–1738 CrossRef CAS.
- V. Aubry, P. X. Etiévant, C. Giniès and R. Henry, J. Agric. Food Chem., 1997, 45, 2120–2123 CrossRef CAS.
-
M. Berglund, in Handbook of Stable Isotope Analytical Techniques, ed. P. A. de Groot, Elsevier, Introduction to Isotope Dilution Mass Spectrometry (IDMS), 2004, pp. 820–834 Search PubMed.
- M. Fialkowski, K. J. M. Bishop, V. A. Chubukov, C. J. Campbell and B. A. Grzybowski, Angew. Chem., Int. Ed., 2005, 44, 7263–7269 CrossRef CAS PubMed.
- B. A. Grzybowski, K. J. M. Bishop, B. Kowalczyk and C. E. Wilmer, Nat. Chem., 2009, 1, 31–36 CrossRef CAS PubMed.
- M. Kowalik, C. M. Gothard, A. M. Drews, N. A. Gothard, A. Weckiewicz, P. E. Fuller, B. A. Grzybowski and K. J. M. Bishop, Angew. Chem., Int. Ed., 2012, 51, 7928–7932 CrossRef CAS PubMed.
- S. He, J. Xiao, A. E. Dulcey, B. Lin, A. Rolt, Z. Hu, X. Hu, A. Q. Wang, X. Xu, N. Southall, M. Ferrer, W. Zheng, T. J. Liang and J. J. Marugan, J. Med. Chem., 2016, 59, 841–853 CrossRef CAS PubMed.
- W. Beker, E. P. Gajewska, T. Badowski and B. A. Grzybowski, Angew. Chem., Int. Ed., 2019, 58, 4515–4519 CrossRef CAS PubMed.
- G. Skoraczyński, P. Dittwald, B. Miasojedow, S. Szymkuć, E. P. Gajewska, B. A. Grzybowski and A. Gambin, Sci. Rep., 2017, 7, 3582 CrossRef PubMed.
- F. S. Emami, A. Vahid, E. K. Wylie, S. Szymkuć, P. Dittwald, K. Molga and B. A. Grzybowski, Angew. Chem., Int. Ed., 2015, 54, 10797–10801 CrossRef CAS PubMed.
-
http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html, accessed May 27 2019.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS.
- T. Ohkuma, D. Ishii, H. Takeno and R. Noyori, J. Am. Chem. Soc., 2000, 122, 6510–6511 CrossRef CAS.
- N. A. Cortez, G. Aguirre, M. Parra-Hake and R. Somanathan, Tetrahedron: Asymmetry, 2013, 24, 1297–1302 CrossRef CAS.
- J. Wang, D. Liu, Y. Liu and W. Zhang, Org. Biomol. Chem., 2013, 11, 3855–3861 RSC.
- R. K. Rej, T. Das, S. Hazra and S. Nanda, Tetrahedron: Asymmetry, 2013, 24, 913–918 CrossRef CAS.
- D. Liu, W. Gao, C. Wang and X. Zhang, Angew. Chem., Int. Ed., 2005, 44, 1687–1689 CrossRef CAS PubMed.
- F. Bracher and T. Litz, Bioorg. Med. Chem., 1996, 4, 877–880 CrossRef CAS PubMed.
- R. Tang, J. Zhu and Y. Luo, Synth. Commun., 2006, 36, 421–427 CrossRef CAS.
- J. G. Kim and P. J. Walsh, Angew. Chem., Int. Ed., 2006, 45, 4175–4178 CrossRef CAS PubMed.
- C. Nottingham, R. Benson, H. Müller-Bunz and P. J. Guiry, J. Org. Chem., 2015, 80, 10163–10176 CrossRef CAS PubMed.
- M. Jiang, L. Peng, K. Yang, T. Wang, X. Yan, T. Jiang, J. Xu, J. Qi, H. Zhou, N. Qian, Q. Zhou, B. Chen, X. Xu, L. Deng and C. Yang, J. Med. Chem., 2019, 62, 5370–5381 CrossRef CAS PubMed.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef PubMed.
- G. F. Costello, B. G. Main, J. J. Barlow, J. A. Carroll and J. S. Shaw, Eur. J. Pharmacol., 1988, 151, 475–478 CrossRef CAS PubMed.
- E. V. Brown, Synthesis, 1975, 1975, 358–375 CrossRef.
- N. Tsuritani, K. Yamada, N. Yoshikawa and M. Shibasaki, Chem. Lett., 2002, 31, 276–277 CrossRef.
- J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS.
- T. Kohara, Y. Hashimoto and K. Saigo, Tetrahedron, 1999, 55, 6453–6464 CrossRef CAS.
-
O. Thiel, C. Bernard, R. Larsen, M. J. Martinelli and M. T. Raza, WO/2009/002427, June 19, 2008.
- M. Suceveanu, M. Raicopol, R. Enache, A. Finarua and S. I. Rosca, Lett. Org. Chem., 2011, 8, 690–695 CrossRef CAS.
- Ó. Pablo, D. Guijarro, G. Kovács, A. Lledós, G. Ujaque and M. Yus, Chem.–Eur. J., 2012, 18, 1969–1983 CrossRef PubMed.
- C. R. Graves, K. A. Scheidt and S. T. Nguyen, Org. Lett., 2006, 8, 1229–1232 CrossRef CAS PubMed.
- Y. Gao, F. Yang, D. Pu, R. D. Laishram, R. Fan, G. Shen, X. Zhang, J. Chen and B. Fan, Eur. J. Inorg. Chem., 2018, 2018, 6274–6279 CrossRef CAS.
- C. E. Stivala and A. Zakarian, J. Am. Chem. Soc., 2011, 133, 11936–11939 CrossRef CAS PubMed.
- E. Brenna, M. Crotti, F. G. Gatti, A. Manfredi, D. Monti, F. Parmeggiani, S. Santangelo and D. Zampieri, ChemCatChem, 2014, 6, 2425–2431 CrossRef CAS.
- G. B. Shinde, N. C. Niphade, S. P. Deshmukh, R. B. Toche and V. T. Mathad, Org. Process Res. Dev., 2011, 15, 455–461 CrossRef CAS.
-
R. N. Kankan, D. R. Rao and D. R. Birari, WO/2010/100429, March 4, 2010.
-
R. Vlasakova and J. Hajicek, WO/2013/075679, November 21, 2012.
-
T. Szekeres, J. Repasi, A. Szabo, M. Benito Velez and B. Mangion, WO/2008/068625, June 8, 2007.
-
M. Xu, Y. Huang and M. Zhang, US2015/080608, November 28, 2014.
- T. D. Cushing, X. Hao, Y. Shin, K. Andrews, M. Brown, M. Cardozo, Y. Chen, J. Duquette, B. Fisher, F. Gonzalez-Lopez de Turiso, X. He, K. R. Henne, Y.-L. Hu, R. Hungate, M. G. Johnson, R. C. Kelly, B. Lucas, J. D. McCarter, L. R. McGee, J. C. Medina, T. San Miguel, D. Mohn, V. Pattaropong, L. H. Pettus, A. Reichelt, R. M. Rzasa, J. Seganish, A. S. Tasker, R. C. Wahl, S. Wannberg, D. A. Whittington, J. Whoriskey, G. Yu, L. Zalameda, D. Zhang and D. P. Metz, J. Med. Chem., 2015, 58, 480–511 CrossRef CAS PubMed.
-
https://clinicaltrials.gov/ct2/show/NCT02540928, accessed May 27 2019.
- R. Hyland, E. G. H. Roe, B. C. Jones and D. A. Smith, Br. J. Clin. Pharmacol., 2008, 51, 239–248 CrossRef PubMed.
- J. Wójcikowski, Eur. Neuropsychopharmacol., 2004, 14, 199–208 CrossRef.
- N. Nebot, S. Crettol, F. D'Esposito, B. Tattam, D. E. Hibbs and M. Murray, Br. J. Pharmacol., 2010, 161, 1059–1069 CrossRef CAS.
- M. Schlosser, L. Guio and F. Leroux, J. Am. Chem. Soc., 2001, 123, 3822–3823 CrossRef CAS PubMed.
- D. Zhang, M.-J. Blanco, B.-P. Ying, D. Kohlman, S. X. Liang, F. Victor, Q. Chen, J. Krushinski, S. A. Filla, K. J. Hudziak, B. M. Mathes, M. P. Cohen, D. Zacherl, D. L. G. Nelson, D. B. Wainscott, S. E. Nutter, W. H. Gough, J. M. Schaus and Y.-C. Xu, Bioorg. Med. Chem. Lett., 2015, 25, 4337–4341 CrossRef CAS.
-
https://clinicaltrials.gov/ct2/results?term=MIN-101, accessed May 27, 2019.
- M. Mori, K. Chiba and Y. Ban, J. Org. Chem., 1978, 43, 1684–1687 CrossRef CAS.
-
H. Yamabe, M. Okuyama, A. Nakao, M. Ooizumi and K.-I. Saito, US2003212094, February 26, 2001.
-
S. Yoshimura, N. Kawano, T. Kawano, D. Sasuga, T. Koike, H. Watanabe, H. Fukudome, N. Shiraishi, R. Munakata, H. Hoshii and K. Mihara, EP2298747, July 2, 2009.
- J.-C. Schwartz, Br. J. Pharmacol., 2011, 163, 713–721 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2016, 76, 1313–1318 CrossRef CAS PubMed.
- G. Zhao, J. Wu and W.-M. Dai, Synlett, 2012, 23, 2845–2849 CrossRef CAS.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
-
J. Sallares, I. Petschen, X. Camps, W. Schunack, H. Stark and M. Capet, WO/2007/006708, July 5, 2006.
- K. Liu, F. Li, J. Lu, S. Liu, K. Dorko, W. Xie and X. Ma, Drug Metab. Dispos., 2014, 42, 863–866 CrossRef PubMed.
- R. Dixon and A. Warrander, Cephalalgia, 1997, 17, 15–20 CrossRef.
-
P. Anzenbacher and U. M. Zanger, Metabolism of Drugs and Other Xenobiotics, ed. P. Anzenbacher and U. M. Zanger, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- A. M. Schmidt and P. Eilbracht, J. Org. Chem., 2005, 70, 5528–5535 CrossRef CAS PubMed.
- G. Verspui, G. Elbertse, F. A. Sheldon, M. A. P. J. Hacking and R. A. Sheldon, Chem. Commun., 2000, 1363–1364 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02678a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Piotr
Dittwald‡
a and
Bartosz A.
Grzybowski
a,
Piotr
Dittwald‡
a and
Bartosz A.
Grzybowski