
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear-infrared fluorescein dyes containing a tricoordinate boron atom†
Naoki
Ando
 a,
Hiroki
Soutome
a and
Shigehiro
Yamaguchi
*ab
a,
Hiroki
Soutome
a and
Shigehiro
Yamaguchi
*ab
aDepartment of Chemistry, Graduate School of Science, Integrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo, Chikusa, Nagoya 464-8602, Japan. E-mail: yamaguchi@chem.nagoya-u.ac.jp
bInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo, Chikusa, Nagoya 464-8602, Japan
First published on 3rd July 2019
Abstract
Bora-fluoresceins (BFs), fluorescein analogues containing a tricoordinate boron atom instead of an oxygen atom at the 10-position of the fluorescein skeleton, were synthesized as a new family of fluorescein analogues. The deprotonated BFs exhibited absorption and fluorescence in the near-infrared region, which were significantly red-shifted relative to those of hitherto-known heteroatom-substituted fluorescein analogues on account of the orbital interaction between the tricoordinate boron atom and the fluorescein skeleton. BFs also showed multi-stage changes resulting from a Lewis acid–base equilibrium at the boron center in combination with a Brønsted acid–base equilibrium at the phenolic hydroxy group.
Introduction
Xanthene is one of the most representative π-conjugated scaffolds for fluorescent dyes, exemplified by fluoresceins and rhodamines. A variety of xanthene-based fluorescence probes have so far been developed and applied to the visualization of various biological events by fluorescence imaging.1–9 These dyes typically exhibit absorption and fluorescence in the visible region. To increase their practical utility in current biological applications, xanthene dyes exhibiting long-wavelength absorption and emission, particularly in the near-infrared (NIR) region, remain attractive target compounds. The use of NIR light entails several advantages, e.g., deep tissue penetration, photo-toxicity mitigation, suppression of auto-fluorescence interference, and evasion of cross-talk with conventional fluorescence probes emitting in the visible region in multi-color imaging.10–15In the development of NIR-emissive xanthene dyes, a variety of structural modifications have been explored extensively. A typical approach is the π-expansion of the xanthene skeleton. For instance, naphthofluoresceins, where the fluorescein skeleton is extended with two naphthalene moieties, present absorption and emission in the red-to-NIR region (Fig. 1a).16–20 Alternatively, the replacement of the endocyclic oxygen atom at the 10-position of the xanthene skeleton with other heteroatoms represents an effective approach to achieve longer-wavelength absorption and emission. A series of xanthene dyes containing various main-group elements, including group 14,21–29 15,30–34 and 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35–42 elements, have been synthesized and their properties have been investigated. Nevertheless, boron-containing xanthene dyes have not received much attention to date. Recently, borinate-substituted xanthene dyes have been reported independently by Egawa and Stains (Fig. 1b).43,44 However, these studies remain limited to tetracoordinate boron-containing derivatives. Our motivation was to prepare the missing part of the heteroatom-substituted xanthene dyes, i.e., tricoordinate boron-containing fluoresceins.
35–42 elements, have been synthesized and their properties have been investigated. Nevertheless, boron-containing xanthene dyes have not received much attention to date. Recently, borinate-substituted xanthene dyes have been reported independently by Egawa and Stains (Fig. 1b).43,44 However, these studies remain limited to tetracoordinate boron-containing derivatives. Our motivation was to prepare the missing part of the heteroatom-substituted xanthene dyes, i.e., tricoordinate boron-containing fluoresceins.
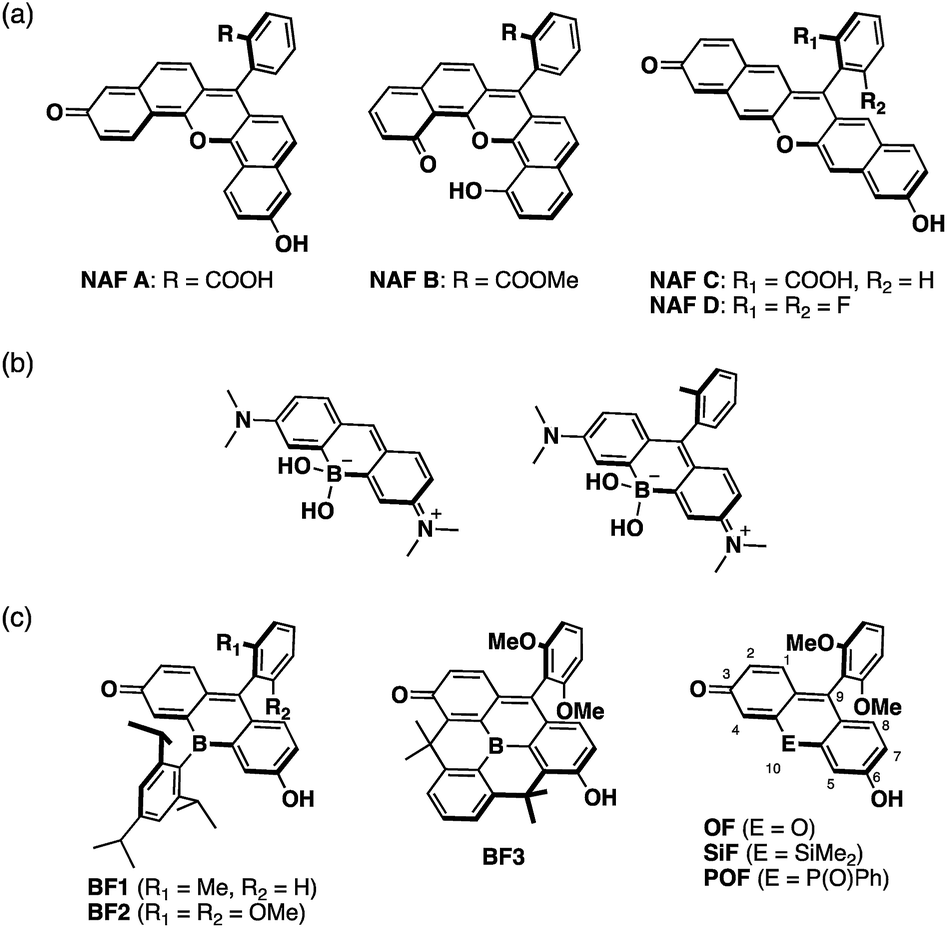 | ||
| Fig. 1 Molecular structures of (a) naphthofluoresceins, (b) previously reported borinate-substituted xanthene dyes, and (c) bora-fluoresceins and their analogues studied in this work. An overview of the numbering of the fluorescein skeleton is also depicted. | ||
A tricoordinate boron atom introduced into the π-conjugated system should perturb the electronic structure through orbital interaction between the vacant p orbital on the boron atom and the π* orbital of the π-skeleton (p–π* interaction).45–54 As target compounds, we designed bora-fluoresceins BF1–3, which contain a tricoordinate boron atom at the 10-position of the fluorescein skeleton (Fig. 1c). Herein, we disclose that these bora-fluoresceins exhibit significantly red-shifted absorption and emission as well as unusual multi-stage responses to Brønsted and Lewis bases. The impact of the boron atom on these properties will be discussed in this article.
Results and discussion
The key step in the synthetic route to bora-fluoresceins BF1–3 (Scheme 1) is the synthesis of boron-substituted dihydroxyxanthone precursor 4. While other heteroatom-substituted dihydroxyxanthones have been synthesized via the introduction of hydroxy groups by the Sandmeyer reaction,25,30 this strategy may be problematic for the bora-xanthone 4 due to the high reactivity of the triarylborane moiety. Therefore, we decided to prepare 4 from dichlorinated triarylborane 1 by a borylation and a subsequent oxidation of the boronic esters and benzylic moiety in a stepwise manner. Namely, the palladium-catalyzed borylation of 1a with bis(pinacolate)diboron furnished diborylated 2a in 61% yield. The selective oxidation of the two boronic esters in 2a was accomplished by treatment with 2 equiv. of Oxone to yield the corresponding dihydroxy-substituted triarylborane 3a. Further oxidation of 3a with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) in MeOH afforded boron-substituted dihydroxyxanthone 4 in 87% yield. After protecting the phenolic hydroxy groups in 4 with t-butyldimethylchlorosilane (TBDMSCl), the reaction with aryl Grignard reagents, followed by acidic workup with p-toluenesulfonic acid, finally produced bora-fluoresceins BF1 and BF2 as deep purple solids in 27% and 37% yield from 1a, respectively.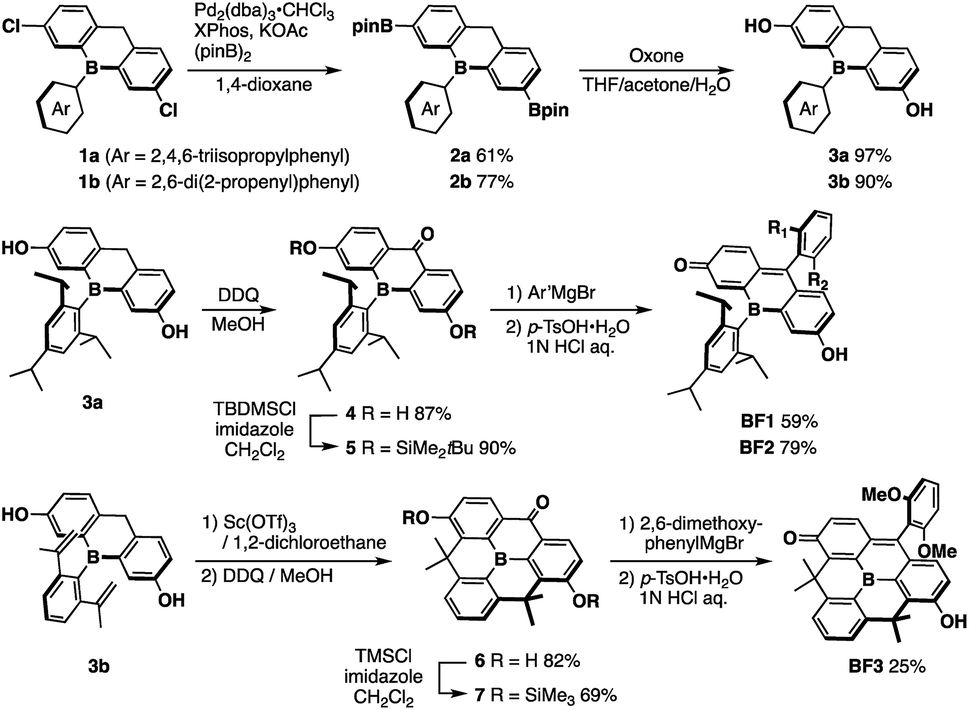 | ||
| Scheme 1 Synthesis of bora-fluoresceins BF1–3. | ||
This synthetic method was also applied for the preparation of B-phenyl-planarized bora-fluorescein BF3. 2,6-Di(2-propenyl)phenyl-substituted precursor 3b, which was obtained by the method described above, was treated with Sc(OTf)3 to promote a two-fold intramolecular Friedel–Crafts cyclization.55 Using a procedure similar to that employed for the synthesis of BF1 and BF2, albeit employing trimethylchlorosilane (TMSCl), B-phenyl-planarized BF3 was obtained as purple solids in 9.8% yield from 1b.
All bora-fluorescein derivatives are sufficiently stable to be handled under ambient conditions and can be purified by column chromatography on silica gel. However, in polar solvents such as acetonitrile, the color of o-tolyl-substituted BF1 immediately changed from purple to yellow, while 2,6-dimethoxyphenyl-substituted BF2 retained its purple color. This difference was confirmed by UV-vis absorption spectroscopy (Fig. S5, ESI†), which suggested that the discoloration of BF1 in acetonitrile is mainly due to the coordination of a solvent molecule at the boron center (vide infra). Consequently, introduction of a proper aryl group at the 9-position of the fluorescein skeleton is essential to ensure the chemical stability of the BFs by suppressing undesired nucleophilic attacks not only at the 9-position but also at the boron center.
The structures of BF2 and BF3 were determined by single crystal X-ray diffraction analysis (Fig. 2). Although the crystal structure of BF2 contains two crystallographically independent molecules in the unit cell, only one is shown in Fig. 2 as both are structurally similar. In both BF2 and BF3, the boron atoms adopt a trigonal planar geometry with a C–B–C angle sum of 360°. The central six-membered rings that contain the boron atoms exhibit nearly planar geometries. The remaining two six-membered rings in the fluorescein skeleton adopt a coplanar conformation with dihedral angles of 4.7° (BF2) and 6.2° (BF3). These results suggest that the boron atom at the 10-position effectively participates in π-conjugation, irrespective of the sterics around the boron center. It is noteworthy that the fluorescein skeleton exhibits an unsymmetrical structure in the crystalline state. The six-membered C1–C6 ring adopts a quinoidal structure with marked bond alternation, while the other six-membered C8–C13 ring adopts a benzene-like structure. The unsymmetrical structures of BF2 and BF3 are probably due to crystal packing effects, i.e., the formation of intermolecular hydrogen bonds between the hydroxy group in the phenol moiety of one molecule and the carbonyl group of the p-quinone methide moiety of an adjacent molecule (Fig. S1 and S3, ESI†).
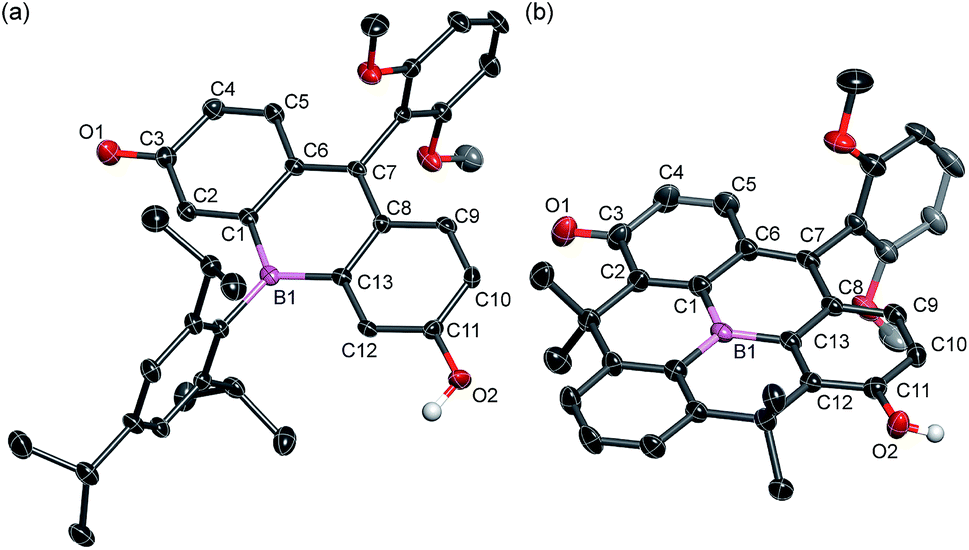 | ||
| Fig. 2 Crystal structures of (a) BF2 and (b) BF3 (50% probability of thermal ellipsoids). Hydrogen atoms, except for those on the phenolic hydroxy groups, are omitted for clarity. | ||
Under basic conditions, bora-fluoresceins showed characteristic absorption and fluorescence in the NIR region (Fig. 3 and Table 1). BF2 exhibited a broad absorption band with the maximum wavelength (λabs) of 538 nm (ε = 0.84 × 104 M−1 cm−1) in acetonitrile. Upon addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the Brønsted base to the acetonitrile solution of BF2, a significantly red-shifted absorption band emerged at λabs = 851 nm (ε = 1.90 × 104 M−1 cm−1). This absorption band in the NIR region was attributed to the deprotonated form of BF2, which stems from the equilibrium at the phenolic hydroxy group. This notion is consistent with the behavior of other heteroatom-substituted fluorescein analogues, for which similar spectral changes have been observed (Fig. S7, ESI†). Under basic conditions, deprotonated BF2 exhibited NIR fluorescence with the maximum at λem = 907 nm, while the fluorescence quantum yield is low (ΦF = 0.003).
 | ||
| Fig. 3 (a) Absorption and (b) fluorescence spectra of heteroatom-substituted fluorescein dyes in the presence of DBU in acetonitrile. | ||
| Dye | λ abs [nm] | ε [104 M−1 cm−1] | λ em [nm] | Stokes shift [cm−1] |
|---|---|---|---|---|
| a Measured after addition of an excess amount of DBU in acetonitrile with a sample concentration of 1.2 to 3.2 × 10−5 M−1. b Ref. 20. c Reported as a Cs salt in acetonitrile. | ||||
| BF2 | 851 | 1.90 | 907 | 730 |
| BF3 | 801 | 3.40 | 843 | 620 |
| OF | 524 | 9.95 | 544 | 700 |
| SiF | 612 | 12.3 | 627 | 390 |
| POF | 646 | 8.43 | 669 | 530 |
| NAF D , | 802 | 9.60 | 839 | 550 |
A comparison of BF2 with other heteroatom-substituted analogues clearly demonstrated the impact of the tricoordinate boron atom on the electronic structure. The λabs of BF2 (851 nm in acetonitrile) under basic conditions is significantly red-shifted relative to those of the corresponding oxygen (OF: 524 nm; Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 7330 cm−1), dimethylsilyl (SiF: 612 nm; Δ = 4560 cm−1), and phosphine oxide (POF: 624 nm; Δ = 3730 cm−1) analogues. Meanwhile, the molar absorption coefficient of BF2 (ε = 1.90 × 104 M−1 cm−1) is about one-fifth that of OF, while their Stokes shifts are comparable. In the fluorescence spectra, deprotonated BF2 exhibits the most red-shifted fluorescence in the NIR region, whereas OF, SiF, and POF only exhibit fluorescence in the visible-to-red region. It is noteworthy that the NIR absorption and fluorescence of BF2 are even further red-shifted relative to those of the naphthofluorescein derivative NAF D (Fig. 1a), where the fluorescein skeleton is extended with two naphthalenes.20 These comparisons clearly show that the tricoordinate boron atom significantly perturbs the electronic structure of the fluorescein skeleton.
= 7330 cm−1), dimethylsilyl (SiF: 612 nm; Δ = 4560 cm−1), and phosphine oxide (POF: 624 nm; Δ = 3730 cm−1) analogues. Meanwhile, the molar absorption coefficient of BF2 (ε = 1.90 × 104 M−1 cm−1) is about one-fifth that of OF, while their Stokes shifts are comparable. In the fluorescence spectra, deprotonated BF2 exhibits the most red-shifted fluorescence in the NIR region, whereas OF, SiF, and POF only exhibit fluorescence in the visible-to-red region. It is noteworthy that the NIR absorption and fluorescence of BF2 are even further red-shifted relative to those of the naphthofluorescein derivative NAF D (Fig. 1a), where the fluorescein skeleton is extended with two naphthalenes.20 These comparisons clearly show that the tricoordinate boron atom significantly perturbs the electronic structure of the fluorescein skeleton.
To elucidate the significant influence of the boron atom on the photophysical properties of the fluorescein dyes, the electronic structure of the deprotonated BF2 was theoretically compared to those of OF, SiF, and POF by DFT calculations at the B3LYP/6-31+G(d) level of theory (Fig. 4). A remarkable difference was observed for their LUMO levels, where the specific orbital interaction between the fluorescein π-skeleton and the substituent at the 10-position plays a crucial role. POF has a low-lying LUMO due to the σ*–π* interaction between the P–Ph bond and the fluorescein skeleton, in addition to the inductive effect of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety. Compared to the LUMO in POF that of BF2 lies even lower by 0.23 eV. The low-lying LUMO level of BF2 was attributed to the effective orbital interaction between the vacant p orbital on the boron atom and the π* orbital of the fluorescein skeleton. In contrast to the significant dependence of the LUMOs on the substituent at the 10-position of the fluorescein skeleton, the HOMOs are delocalized over the fluorescein skeleton with a node at the 10-position, regardless of the substituent. Consequently, BF2 has a comparable HOMO level with those of the other analogues, except for POF, which has a slightly lower-lying HOMO due to the inductive effect of the PO moiety. Overall, BF2 exhibits the narrowest HOMO–LUMO gap in this series of fluorescein analogues.
O moiety. Compared to the LUMO in POF that of BF2 lies even lower by 0.23 eV. The low-lying LUMO level of BF2 was attributed to the effective orbital interaction between the vacant p orbital on the boron atom and the π* orbital of the fluorescein skeleton. In contrast to the significant dependence of the LUMOs on the substituent at the 10-position of the fluorescein skeleton, the HOMOs are delocalized over the fluorescein skeleton with a node at the 10-position, regardless of the substituent. Consequently, BF2 has a comparable HOMO level with those of the other analogues, except for POF, which has a slightly lower-lying HOMO due to the inductive effect of the PO moiety. Overall, BF2 exhibits the narrowest HOMO–LUMO gap in this series of fluorescein analogues.
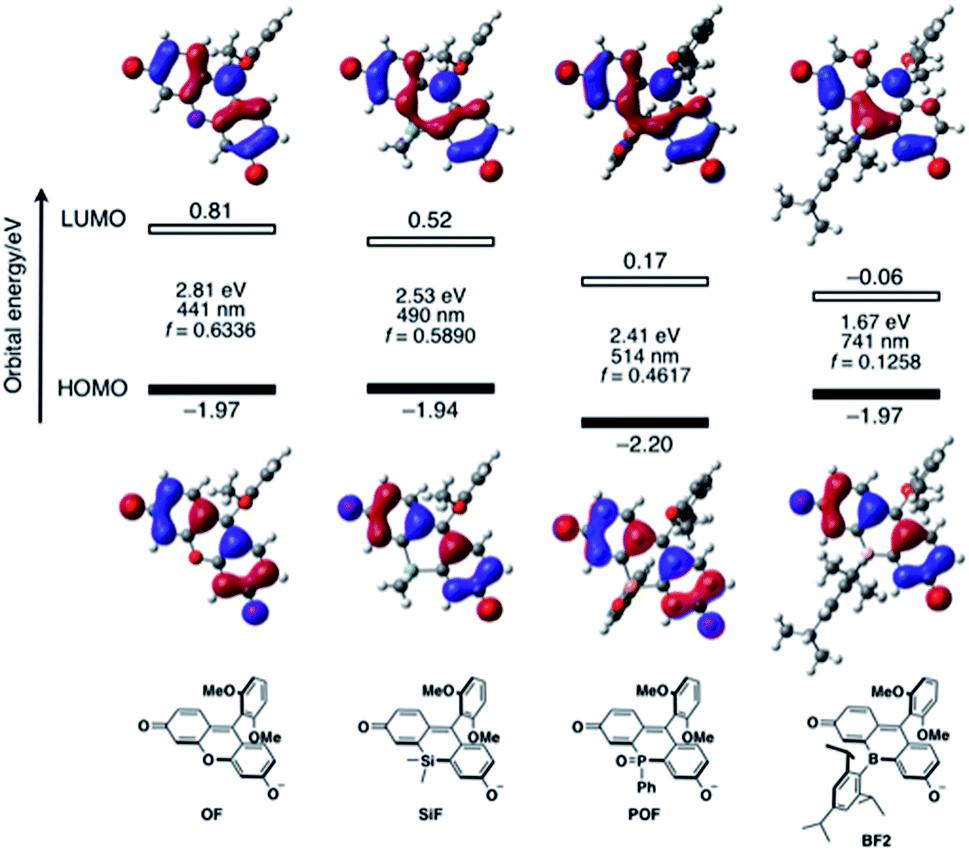 | ||
| Fig. 4 Energy diagrams and Kohn–Sham plots of the HOMOs and LUMOs for the deprotonated forms of the fluorescein dyes OF, SiF, POF, and BF2 calculated at the B3LYP/6-31+G(d) level of theory. The results of TD-DFT calculations at the same level of theory are also shown. | ||
TD-DFT calculations at the same level of theory suggested that deprotonated BF2 exhibits a much red-shifted absorption maximum in the NIR region than the other fluorescein analogues (Fig. 4). The calculated oscillator strength for BF2 is also significantly reduced compared to those of the other derivatives, which is consistent with the experimentally observed smallest molar absorption coefficient of BF2. This difference in the oscillator strength should be related to the contribution of the substituent at the 10-position to the LUMO, i.e., the overlap between the HOMO and LUMO should decrease with increasing contribution of the substituent at the 10-position. Consequently, BF2 has the smallest oscillator strength. Importantly, the narrow HOMO–LUMO gap observed in BF2 is no longer retained once the vacant p orbital on the boron atom is occupied by a Lewis base externally added to form a tetracoordinate species (vide infra) (Fig. S14, ESI†). Therefore, only tricoordinate boron species that ensure the effective orbital interaction through the vacant p orbital on the boron atom can attain NIR-absorption and fluorescence.
The response to Lewis bases, such as fluoride ions, is a characteristic feature of boron-containing π-conjugated compounds, and should endow the bora-fluoresceins with unusual properties. Indeed, BF2 exhibits drastic multi-stage color changes from purple to yellow and blue upon addition of tetra-n-butylammonium fluoride (TBAF). These color changes were monitored by UV-vis absorption spectroscopy. Upon addition of TBAF to an acetonitrile solution of BF2 (2.8 × 10−5 M), the broad absorption band at λabs = 538 nm diminished with the concomitant emergence of a new absorption band at λabs = 436 nm (Fig. 5a). Successive addition of excess TBAF resulted in the appearance of an intense and sharp absorption band at λabs = 585 nm, which is reminiscent of the deprotonation of fluorescein dyes (Fig. 5b). Ultimately, the thus obtained mixture shows intense red fluorescence with the fluorescence maximum at λem = 595 nm with a high quantum yield of ΦF = 0.89.
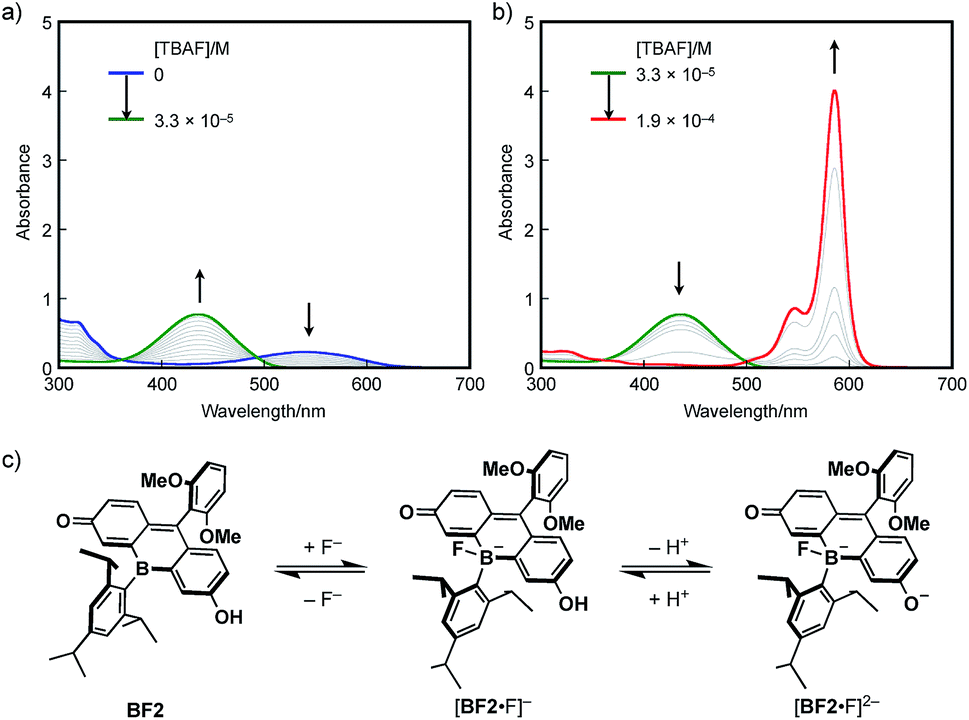 | ||
| Fig. 5 UV-vis absorption spectral changes of BF2 in acetonitrile (2.8 × 10−5 M) upon addition of TBAF: (a) [TBAF] = 0 to 3.3 × 10−5 M−1 and (b) 3.3 × 10−5 to 1.9 × 10−4 M−1. (c) Equilibrium between neutral BF2, borate [BF2·F]−, and borate fluorescein anion [BF2·F]2−. | ||
These changes can be interpreted in terms of a complexation of a fluoride ion at the boron center, followed by deprotonation of the phenolic hydroxy group in the fluorescein skeleton (Fig. 5c). Specifically, BF2 initially incorporates a fluoride ion to form tetracoordinate borate [BF2·F]−. With increasing TBAF concentration, subsequent deprotonation by the basic fluoride ion from [BF2·F]− generates borate fluorescein anion [BF2·F]2−.56–58 For a detailed study on the electronic structure of [BF2·F]− and [BF2·F]2−, TD-DFT calculations were performed at the B3LYP/6-31+G(d) level of theory (Fig. S14, ESI†). The absorption bands observed at 436 nm for [BF2·F]− and 585 nm for [BF2·F]2− should be ascribed to π–π* transitions, which mainly consist of the HOMO–LUMO transitions in both complexes. It should be noted that the LUMOs of these complexes are delocalized over the fluorescein skeleton with a subtle contribution from the boron moiety, which stands in stark contrast to the orbital interaction observed in the LUMO of neutral BF2 bearing a tricoordinate boron atom. Moreover, complexation with a fluoride ion switches the role of the boron moiety in the π-conjugated system from a π-electron-accepting unit to a strong σ-donating unit, giving rise to the red-shifted absorption of [BF2·F]2− compared to that of deprotonated OF. In this sense, the boron moiety in the fluorescein skeleton enables multi-stage electronic structure changes accompanied by drastic color changes in the visible-to-NIR region.
BF2 underwent complexation not only with fluoride ions, but also with weak neutral Lewis bases such as pyridine. It should be noted that in this case Lewis acid–base complexation predominates over the deprotonation of the phenolic hydroxy group (Fig. S11, ESI†). However, the binding constant of BF2 with pyridine in acetonitrile is low (K = 31 M−1) due to the steric congestion around the boron center induced by the bulky triisopropylphenyl group. In this context, we discovered that the Lewis acidity of the boron center was further enhanced by planarization of the B-phenyl group without the loss of chemical stability. Upon treatment of B-phenyl-planarized BF3 with DBU in acetonitrile, deprotonated BF3 showed red-shifted absorption at λabs = 801 nm, which is similar to the behavior of BF2. The hypsochromic shift of 730 cm−1 relative to the absorption of BF2 (851 nm) is mainly due to the electron-donating effect of the methylene bridges in BF3 (Fig. S13, ESI†). Deprotonated BF3 exhibits NIR fluorescence (λem = 843 nm) with an improved fluorescence quantum yield (ΦF = 0.03). In contrast, upon treatment with pyridine, BF3 showed a new hypsochromically shifted absorption band at 445 nm accompanied by a drastic color change from purple to yellow (Fig. S12, ESI†). On the basis of the UV-vis spectral changes, the binding constant of BF3 toward pyridine was determined (K = 2.1 × 103 M−1), which indicated that the complexation ability of BF3 toward pyridine is higher than that of BF2 by two orders of magnitude.
Conclusions
In summary, we have successfully synthesized bora-fluoresceins BF1–3, which contain a tricoordinate boron atom at the 10-position in the fluorescein skeleton. The deprotonated forms of bora-fluoresceins exhibit absorption and fluorescence in the NIR region. In particular, deprotonated BF2 shows absorption and emission maxima at 851 and 907 nm, respectively. These values are the most red-shifted of all hitherto reported heteroatom-substituted fluorescein dyes, demonstrating the significant impact of the boron atom on the electronic structure of fluorescein dye. The Lewis acidity of the boron atom offers a unique switching mechanism for the photophysical properties in addition to the Brønsted acid–base equilibrium on the fluorescein skeleton. Namely, the complexation of a Lewis base at the boron center of the bora-fluoresceins results in a significant hypsochromic shift of the absorption and emission. Consequently, bora-fluoresceins show multi-stage changes in their absorption and fluorescence properties, which is a noticeably different behavior from that of other heteroatom-substituted fluoresceins. The results of this study not only provide insight into the design principles of xanthene dyes, but also demonstrate the utility of tricoordinate boron atoms in the development of NIR dyes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grants 18H03909 and 18H05261. N. A. thanks the JSPS for a Research Fellowship for Young Scientists. The authors also thank Prof. Dr T. Sasamori (Nagoya City Univ.) and Dr M. Hirai (Nagoya Univ.) for their help with the X-ray crystallographic analysis. The synchrotron X-ray crystallography experiment was performed at the BL40XU beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposal 2017B1179).Notes and references
- T. Terai and T. Nagano, Curr. Opin. Chem. Biol., 2008, 12, 515–521 CrossRef CAS.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS.
- H. Zheng, X.-Q. Zhan, Q.-N. Bian and X.-J. Zhang, Chem. Commun., 2013, 49, 429–447 RSC.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2014, 9, 855–866 CrossRef CAS.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS.
- J. Yin, Y. Hu and J. Yoon, Chem. Soc. Rev., 2015, 44, 4619–4644 RSC.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS.
- R. Weissleder and V. Ntziachristos, Nat. Med., 2003, 9, 123–128 CrossRef CAS.
- J. V Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef.
- K. Kiyose, H. Kojima and T. Nagano, Chem.–Asian J., 2008, 3, 506–515 CrossRef CAS.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- L. G. Lee, G. M. Berry and C.-H. Chen, Cytometry, 1898, 10, 151–164 CrossRef.
- K. Xu, B. Tang, H. Huang, G. Yang, Z. Chen, P. Li and L. An, Chem. Commun., 2005, 5974–5976 RSC.
- M. Sibrian-Vazquez, J. O. Escobedo, M. Lowry, F. R. Fronczek and R. M. Strongin, J. Am. Chem. Soc., 2012, 134, 10502–10508 CrossRef CAS.
- E. Azuma, N. Nakamura, K. Kuramochi, T. Sasamori, N. Tokitoh, I. Sagami and K. Tsubaki, J. Org. Chem., 2012, 77, 3492–3500 CrossRef CAS.
- K. Sezukuri, M. Suzuki, H. Hayashi, D. Kuzuhara, N. Aratani and H. Yamada, Chem. Commun., 2016, 52, 4872–4875 RSC.
- J. Arden-Jacob, J. Frantzeskos, N. U. Kemnitzer, A. Zilles and K. H. Drexhage, Spectrochim. Acta, Part A, 2001, 57, 2271–2283 CrossRef CAS.
- J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, ACS Chem. Biol., 2013, 8, 1303–1310 CrossRef CAS PubMed.
- K. Kolmakov, V. N. Belov, C. A. Wurm, B. Harke, M. Leutenegger, C. Eggeling and S. W. Hell, Eur. J. Org. Chem., 2010, 3593–3610 CrossRef CAS.
- M. Fu, Y. Xiao, X. Qian, D. Zhao and Y. Xu, Chem. Commun., 2008, 1780–1782 RSC.
- T. Egawa, Y. Koide, K. Hanaoka, T. Komatsu, T. Terai and T. Nagano, Chem. Commun., 2011, 47, 4162–4164 RSC.
- Y. Kushida, T. Nagano and K. Hanaoka, Analyst, 2015, 140, 685–695 RSC.
- T. Ikeno, T. Nagano and K. Hanaoka, Chem.–Asian J., 2017, 12, 1435–1446 CrossRef CAS.
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, ACS Chem. Biol., 2011, 6, 600–608 CrossRef CAS.
- H. Nie, J. Jing, Y. Tian, W. Yang, R. Zhang and X. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 8991–8997 CrossRef CAS.
- A. Fukazawa, S. Suda, M. Taki, E. Yamaguchi, M. Grzybowski, Y. Sato, T. Higashiyama and S. Yamaguchi, Chem. Commun., 2016, 52, 1120–1123 RSC.
- X. Chai, X. Cui, B. Wang, F. Yang, Y. Cai, Q. Wu and T. Wang, Chem.–Eur. J., 2015, 21, 16754–16758 CrossRef CAS.
- X. Zhou, R. Lai, J. R. Beck, H. Li and C. I. Stains, Chem. Commun., 2016, 52, 12290–12293 RSC.
- X. Chai, J. Xiao, M. Li, C. Wang, H. An, C. Li, Y. Li, D. Zhang, X. Cui and T. Wang, Chem.–Eur. J., 2018, 24, 14506–14512 CrossRef CAS.
- T. Hirayama, A. Mukaimine, K. Nishigaki, H. Tsuboi, S. Hirosawa, K. Okuda, M. Ebihara and H. Nagasawa, Dalton Trans., 2017, 46, 15991–15995 RSC.
- M. R. Detty, P. N. Prasad, D. J. Donnelly, T. Ohulchanskyy, S. L. Gibson and R. Hilf, Bioorg. Med. Chem., 2004, 12, 2537–2544 CrossRef CAS.
- J. Liu, Y.-Q. Sun, H. Zhang, H. Shi, Y. Shi and W. Guo, ACS Appl. Mater. Interfaces, 2016, 8, 22953–22962 CrossRef CAS.
- G. Dejouy, M. Laly, I. E. Valverde and A. Romieu, Dyes Pigm., 2018, 159, 262–274 CrossRef CAS.
- B. Calitree, D. J. Donnelly, J. J. Holt, M. K. Gannon, C. L. Nygren, D. K. Sukumaran, J. Autschbach and M. R. Detty, Organometallics, 2007, 26, 6248–6257 CrossRef CAS.
- Y. Koide, M. Kawaguchi, Y. Urano, K. Hanaoka, T. Komatsu, M. Abo, T. Terai and T. Nagano, Chem. Commun., 2012, 48, 3091–3093 RSC.
- M. W. Kryman, G. A. Schamerhorn, K. Yung, B. Sathyamoorthy, D. K. Sukumaran, T. Y. Ohulchanskyy, J. B. Benedict and M. R. Detty, Organometallics, 2013, 32, 4321–4333 CrossRef CAS.
- M. W. Kryman, G. A. Schamerhorn, J. E. Hill, B. D. Calitree, K. S. Davies, M. K. Linder, T. Y. Ohulchanskyy and M. R. Detty, Organometallics, 2014, 33, 2628–2640 CrossRef CAS.
- L. V. Lutkus, H. E. Irving, K. S. Davies, J. E. Hill, J. E. Lohman, M. W. Eskew, M. R. Detty and T. M. McCormick, Organometallics, 2017, 36, 2588–2596 CrossRef CAS.
- N. Shimomura, Y. Egawa, R. Miki, T. Fujihara, Y. Ishimaru and T. Seki, Org. Biomol. Chem., 2016, 14, 10031–10036 RSC.
- X. Zhou, L. Lesiak, R. Lai, J. R. Beck, J. Zhao, C. G. Elowsky, H. Li and C. I. Stains, Angew. Chem., Int. Ed., 2017, 56, 4197–4200 CrossRef CAS PubMed.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS.
- A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC.
- Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC.
- E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS.
- K. H. Lee, H.-Y. Lee, D. H. Lee and J.-I. Hong, Tetrahedron Lett., 2001, 42, 5447–5449 CrossRef CAS.
- H. Tong, G. Zhou, L. Wang, X. Jing, F. Wang and J. Zhang, Tetrahedron Lett., 2003, 44, 131–134 CrossRef CAS.
- D. A. Jose, P. Kar, D. Koley, B. Ganguly, W. Thiel, H. N. Ghosh and A. Das, Inorg. Chem., 2007, 46, 5576–5584 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data, details of the computational studies, and crystallographic data for BF2 and BF3. CCDC 1913398 and 1913399. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02314c |
| This journal is © The Royal Society of Chemistry 2019 |