
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA H-bond stabilized quinone electrode material for Li–organic batteries: the strength of weak bonds†
Louis
Sieuw
a,
Alia
Jouhara
b,
Éric
Quarez
 b,
Chloé
Auger
b,
Jean-François
Gohy
a,
Philippe
Poizot
*b and
Alexandru
Vlad
*a
b,
Chloé
Auger
b,
Jean-François
Gohy
a,
Philippe
Poizot
*b and
Alexandru
Vlad
*a
aInstitut de la Matière Condense et des Nanosciences (IMCN), Université Catholique de Louvain, Place L. Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: alexandru.vlad@uclouvain.be
bInstitut des Matériaux Jean Rouxel (IMN), UMR CNRS 6502, Université de Nantes, 2 rue de la Houssinière, B.P. 32229, 44322 Nantes Cedex 3, France. E-mail: philippe.poizot@cnrs-imn.fr
First published on 9th October 2018
Abstract
Small organic materials are generally plagued by their high solubility in battery electrolytes. Finding approaches to suppress solubilization while not penalizing gravimetric capacity remains a challenge. Here we propose the concept of a hydrogen bond stabilized organic battery framework as a viable solution. This is illustrated for 2,5-diamino-1,4-benzoquinone (DABQ), an electrically neutral and low mass organic chemical, yet with unusual thermal stability and low solubility in battery electrolytes. These properties are shown to arise from hydrogen bond molecular crystal stabilization, confirmed by a suite of techniques including X-ray diffraction and infrared spectroscopy. We also establish a quantitative correlation between the electrolyte solvent polarity, molecular structure of the electrolyte and DABQ solubility – then correlate these to the cycling stability. Notably, DABQ displays a highly reversible (above 99%) sequential 2-electron electrochemical activity in the solid phase, a process rarely observed for similar small molecular battery chemistries. Taken together, these results reveal a potential new strategy towards stable and practical organic battery chemistries through intramolecular hydrogen-bonding crystal stabilization.
Introduction
From an economic point of view, the future of battery materials research is bright, as it is crucial to the continuous development of energy-consuming electronic devices such as smartphones and laptops.1,2 On another account, the emergence of new, higher performing energy storage technologies is also required to keep up with the transition towards renewable energy sources, which presents the downside of being intermittent.Inorganic compounds have long thrived both as research and commercial battery materials.3–6 However, the redox chemistry of organic molecules is increasingly attracting interest as it is rich and has the potential to produce electrode material candidates combining high capacities and practical redox potentials.7–12 Moreover, organic materials allow for transition-metal free battery compositions making possible the fabrication of all-organic batteries. Several families of organic redox compounds have been investigated so far including carbonyls, amines, sulfur compounds and nitroxide radicals.9,13–15 Belonging to the carbonyl family, quinone-based compounds have been extensively explored and a rich library for quinone-battery chemistries is already available.11,16 However, limiting the solubility of these molecules in battery electrolytes while maintaining high gravimetric capacities remains a persistent challenge.
Until now, three main strategies have been explored to address this issue:9,11,16 polymerization,17–23 introduction of ionic groups7,10,24–32 and grafting on mesoporous solid supports (e.g. carbon or silica nanoparticles).33–36 While successfully reducing the solubility of the quinone materials, these strategies add to synthesis complexity, manufacturing costs and molecular weight that is inversely proportional to the gravimetric storage capacity. This calls for new paradigms in choice, design and synthesis of new organic battery materials. For instance, taking inspiration from nature, where hydrogen bonding (H-bonding) creates strong interactions and stabilizes complementary bases on opposite strands of DNA,37 H-bonding could also be explored to create stable molecular crystal materials and electrodes for battery applications. Few attempts in this direction exist and H-bonding seems indeed to be suitable to organic battery materials as well. For example, Luo et al., have combined di-sodium rhodizonate with a hydroxyl-rich sodium alginate binder and detailed a self-healing mechanism of the electrode during electrochemical cycling.38 However, rhodizonate is an intrinsically insoluble battery chemical, so that the impact of H-bonding was on the macroscopic structural stability of the electrode rather than on the molecular level solubility effect. Thus, our study goes a step further by demonstrating that the solubility of electrically neutral small organic compounds can be controlled through hydrogen intermolecular bonding, and that consequently the electrochemical cycling improved, without the need of a binder in the electrode formulation.
Herein we prove that the unusual thermal stability and low solubility in battery electrolytes of 2,5-diamino-1,4-benzoquinone (DABQ) arise from hydrogen bond molecular crystal stabilization. Electronic delocalization polarizes the amino and carbonyl groups, which are at the origin of the strong H-bond network that stabilizes the crystal. DABQ solubility is found to be strongly dependent on the nature of the electrolyte solvent and we also observe a clear correlation between the dipolar momentum of the solvent and the solubility. Consequently, stable cycling with high coulombic efficiency is achieved in the solid state. By combining the H-bonding properties of this molecule with the use of low polarity battery electrolytes, we intend to initiate a new approach towards the insolubilization of quinone materials, a concept that could be generalized to other organic battery materials as well.
Furthermore, the electrochemistry of diamino-quinone and -anthraquinone derivatives has so far been investigated only as part of a larger molecular system, through covalent fixation within a covalent organic framework.39 The stable electrochemical cycling was assigned to the rigid fixation at the expense of lower gravimetric capacity since a larger molecular mass COF unit cell was used. It is also worth mentioning that the introduction of amine groups has been shown to affect the cycling stability of phenazine derivatives,40 however the origin of this observation was not detailed and there was no specific mention of any particular intra-/inter-molecular interaction.
Results and discussion
DABQ was obtained through the oxidation of 2,5-diaminohydroquinone dihydrochloride in aqueous medium, following the procedure published by Das et al.41 A pure purple-red powder was obtained (Fig. S1†).DABQ was first studied by powder X-ray diffraction (PXRD) using synchrotron radiation at the European Synchrotron Radiation Facility (ESRF, Grenoble) with a wavelength of 0.74580 Å making the ab initio structure determination possible. The Rietveld refinement of the diffractogram is displayed in Fig. 1A, and allowed us to determine the structure of DABQ (Fig. 1B) in the P21/n space group. The unit cell has a volume of 299.88 (4) Å3 and the following parameters: a = 5.2532 (4) Å, b = 5.3210 (3) Å, c = 10.9671 (7) Å, β = 101.9740 (17)° (ESI Tables 1–4†). The individual molecules assemble into parallel layers stacked along the b-axis with adjacent layers being perpendicular to each other (Fig. S2†). Intermolecular H-bonding is observed between the polarized carbonyl and amino groups. However, these bonds only form between perpendicular molecules and between molecules of nearest parallel layers, not between those in the same layer. This results in particles with lamellar structures, as evidenced by SEM analysis (Fig. S3†).
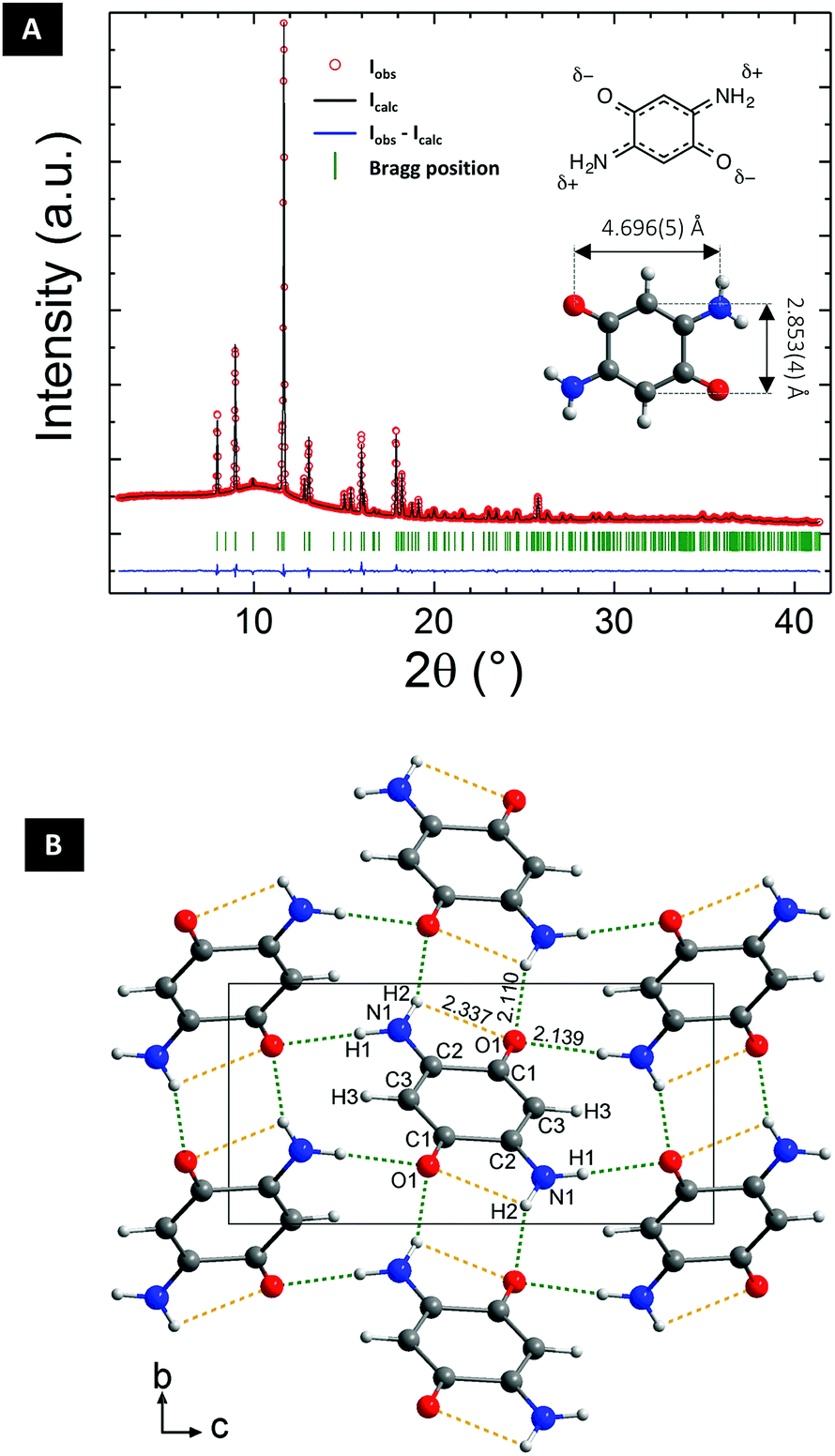 | ||
| Fig. 1 Ab initio structure determination of DABQ. (A) X-ray powder diffraction pattern (final Rietveld refinement) of crystalline DABQ with the molecular structure depicted in the inset. (B) View of the solved DABQ crystal structure (yellow dotted line: intramolecular H-bonds; green dotted line: intermolecular H-bonds). | ||
DABQ can be regarded as a molecular hybrid of 1,4-benzoquinone (BQ) and para-phenylenediamine (p-PDA) (Fig. 2A). The Fourier-transform infrared (FTIR) spectrum of DABQ contains the stretching peak of the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in BQ, as well as the stretching peaks of C–N and bending peaks of N–H in p-PDA (Fig. 2B). However, the CO stretching peak interestingly undergoes a bathochromic shift (about 100 cm−1) in the case of DABQ as compared to BQ. This shift can be explained by the weakening of the CO bond, which further supports the aforementioned intermolecular H-bond formation, reinforced by the electronic density delocalization.
O) in BQ, as well as the stretching peaks of C–N and bending peaks of N–H in p-PDA (Fig. 2B). However, the CO stretching peak interestingly undergoes a bathochromic shift (about 100 cm−1) in the case of DABQ as compared to BQ. This shift can be explained by the weakening of the CO bond, which further supports the aforementioned intermolecular H-bond formation, reinforced by the electronic density delocalization.
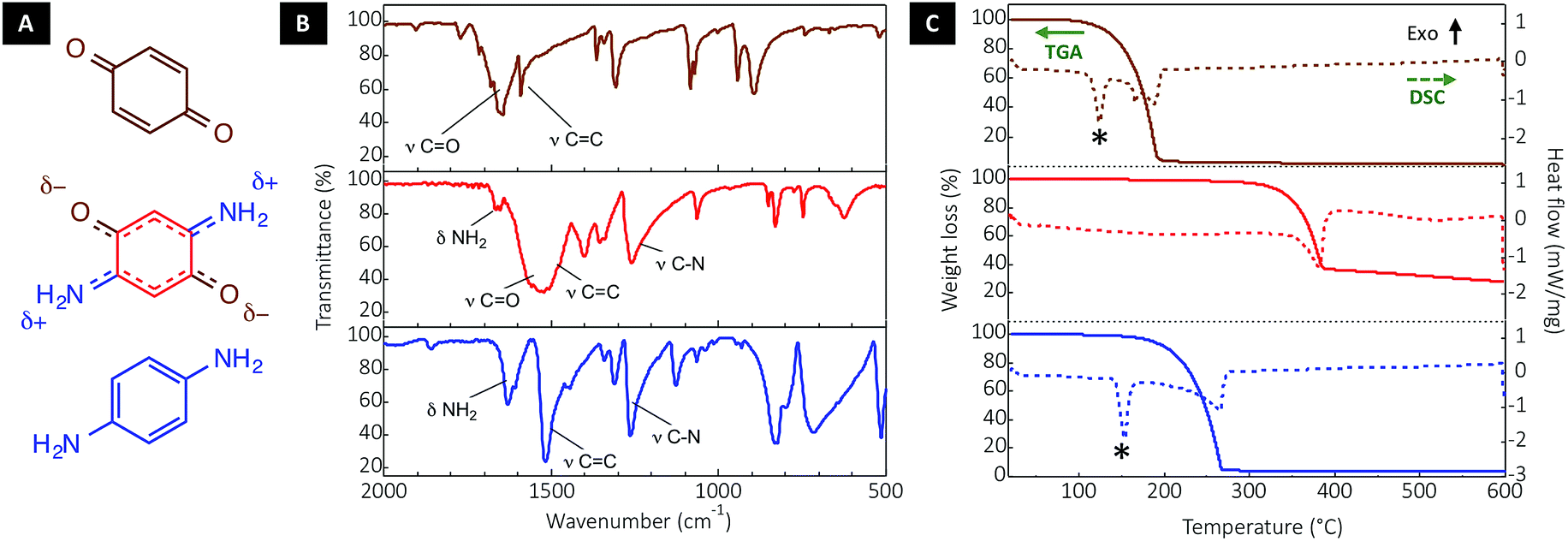 | ||
| Fig. 2 Stabilization of the DABQ molecular crystal through H-bonding. (A) Molecular structure of DABQ and of the reference compounds (i.e., BQ and p-PDA) and their corresponding (B) FTIR spectra as well as (C) thermogravimetry (TG) and differential scanning calorimetry (DSC) plots. The stars mark the melting point in the DSC plots. | ||
The thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) further confirm that the intermolecular interactions translate into a considerable gain of stability for DABQ. Indeed, DABQ shows no melting behavior with direct thermal decomposition at a temperature above 300 °C. This is unusually high for a neutral organic low molecular mass compound. For instance, BQ and p-PDA both display a decomposition onset around 120 °C and 170 °C respectively; and for both compounds, melting precedes vaporization.
According to the above-mentioned analyses, the crystalline phase of DABQ is determined to have intermolecular hydrogen bonding involving the N–Hδ+ and δ−O–C end groups. Any chemical modification or steric hindering is expected to weaken the H-bond strength and thus affect the crystal stability, as discussed above. Indeed, we synthesized two N-substituted alkyl derivatives of DABQ (N-methyl and N-ethyl DABQ) and their solubility was increased, as also evidenced by faster capacity decay during galvanostatic cycling (Fig. S4†). Since the cycling performances of these derivatives were considerably diminished, we did not proceed to further extended analysis.
When considering the application of DABQ as a battery electrode material, it is important to note that a solvent which can interfere with the molecule’s crystal structure through H-bonding or electrostatic interactions could lead to a different degree of solvation and consequently to partial or total dissolution of DABQ. The polarity of the electrolyte solvent is thus expected to play an important role and control the saturation solubility of DABQ. Overall, we observed limited solubility of DABQ in various organic and inorganic solvents, already indicative of stronger interactions between DABQ molecules than between DABQ and solvent molecules. The synthesis protocol of DABQ is based on its intrinsic poor solubility in water (DABQ precipitates instantaneously) but also on methanol and ethers (used for purification). Since our interest is in the applicability of DABQ as a battery electrode material, we next focused our attention on the quantification of DABQ solubility in conventional battery electrolytes and subsequent correlation with cycling stability and efficiency.
The solubility of DABQ in a series of electrolytes with varying dipolar moments was determined through UV-vis titration following an experimental approach recently reported42 (refer to Fig. S5† and associated content for experimental details). Given the concentration of the solute being directly proportional to the absorbance of the solution, the solubility of DABQ in the electrolytes can be calculated with relative precision, using the DABQ strong absorption band at 330 nm.
Different electrolyte formulations were tested for solubility and galvanostatic cycling was measured. These formulations are based on lithium bis(trifluoromethylsulfonyl)imide (LiTFSI) and lithium hexafluorophosphate (LiPF6) supporting salts in various mixtures of carbonates or glymes in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio (1:1 v/v): ethylene carbonate (EC)/dimethyl carbonate (DMC), 1,2-dimethoxyethane (DME)/1,3-dioxolane (DOL) and tetraethylene glycol dimethyl ether (TEGDME)/1,3-dioxolane (DOL). The effect of salt concentration as well as the associated impact on electrochemical cycling efficiency were also assessed. The results are summarized in Fig. 3 and discussed in the following.
:1 volume ratio (1:1 v/v): ethylene carbonate (EC)/dimethyl carbonate (DMC), 1,2-dimethoxyethane (DME)/1,3-dioxolane (DOL) and tetraethylene glycol dimethyl ether (TEGDME)/1,3-dioxolane (DOL). The effect of salt concentration as well as the associated impact on electrochemical cycling efficiency were also assessed. The results are summarized in Fig. 3 and discussed in the following.
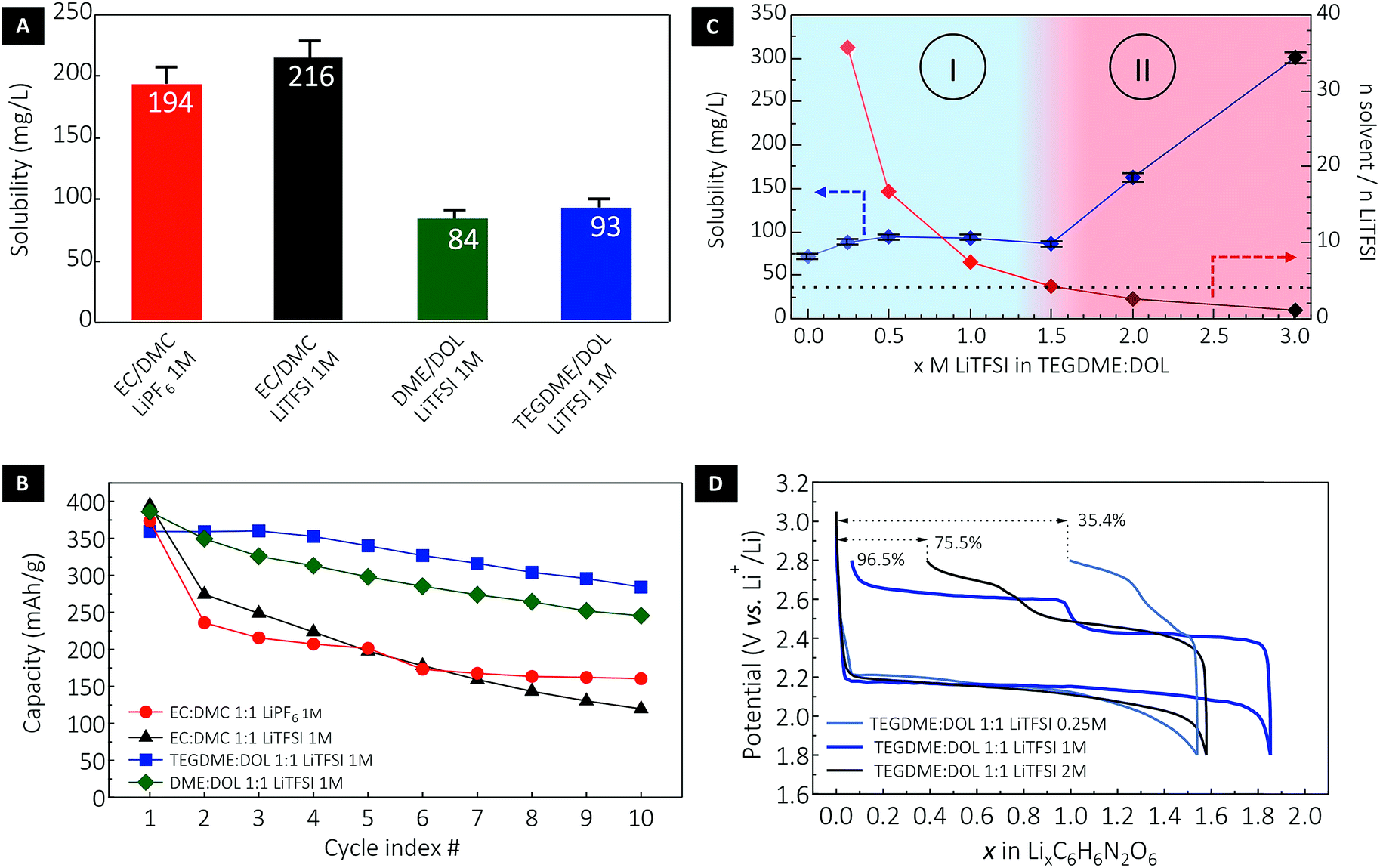 | ||
| Fig. 3 Solubility of DABQ in different electrolyte formulations and consequences for cycling reversibility. (A) Saturation concentration of DABQ in a series of representative electrolytes and (B) the corresponding half-cell cycling stability at a lithium insertion/extraction rate of 1Li+/1e− in 10 h (i.e. equivalent to 40 hours per one full cycle). (C) Solubility of DABQ in TEGDME/DOL electrolyte as a function of LiTFSI supporting salt concentration and (D) the associated potential–composition traces for the 1st cycle. | ||
Considering electrolytes with a 1 M concentration in a carrier salt, the solubility of DABQ was found to be higher in carbonates when compared to glyme based electrolytes (Fig. 3A): approximately 200 mg L−1 in EC/DMC (1:1 v/v) for both LiPF6 and LiTFSI salts, and in the range of 90 mg L−1 for DME/DOL (1:1 v/v) and TEGDME/DOL (1:1 v/v) electrolytes at 1 M LiTFSI. To place these values in perspective, BQ displays a solubility of about 200 g L−1 in TEGDME/DOL (1:1 v/v), and is thus 2.000 times more soluble than DABQ in the same electrolyte formulation.
The measured solubility of DABQ correlates with the dipole moments of the aprotic electrolyte solvents used for these experiments. The ethers, with low values for dipole moments of 1.25 D for DOL, 1.15 D for DME and 2.45 D for TEGDME, disrupt the H-bond based crystallinity of DABQ to a lesser extent than EC-based electrolytes (μ = 4.61 D for EC).43 EC is considerably more polar than the other electrolyte constituents, enhancing the formation of H-bonds between its carbonyl group and the amine groups of DABQ, thus leading to increased solubility. Interesting to note is that DMC has the lowest dipole momentum amongst all tested electrolyte solvents, of 0.76D and correspondingly, DABQ also displays the lowest solubility in this solvent, of about 65 mg L−1 (Fig. S6†).
The impact of DABQ solubility on the electrochemical cycling stability is clearly observed in Fig. 3B. It is to be stressed that low rate cycling (1Li+ or 1e− exchanged per 1 molecule of DABQ in 10 hours) was deliberately chosen here to emphasize the impact of solubility on cycling stability over a long time interval. Fast cycling, although leading to an apparently greater cycle number stability, implies also shorter test times (a lower measurement time per cycle) which can mask the slow solubilisation of the materials as well as underlying redox-reactions between reactive lithium and dissolved material. Cycling stability analysis revealed that while all electrolyte formulations yielded an initial capacity close to the theoretical value of 388 mA h g−1 (corresponding to a two-electron redox process), the ether electrolytes showed enhanced stability with much lower capacity loss over cycling, with similar degradation tendencies (as displayed by the slope of the capacity decay curves, Fig. 3B) for DME/DOL and TEGDME/DOL. This is consistent with the close DABQ solubility values in these electrolytes. Regardless of the nature of the carrier salt, galvanostatic cycling of DABQ in carbonate electrolytes (except for the DMC LiTFSI 1 M) systematically resulted in important capacity loss, assigned to a higher solubility of DABQ in the EC containing electrolytes. Clear evidence of active material dissolution and electrode morphology change are also provided by post-cycling cell inspection and SEM analysis of the electrode (Fig. S7†).
In an effort to further understand and control the dissolution process of DABQ in glyme electrolytes and thus reduce capacity loss during cycling, we assessed the variation of the solubility with changing carrier salt (LiTFSI) concentrations. Indeed, electrolytes with high salt concentrations – ‘solvent-in-salt electrolytes’ – are known to decrease the solubility of electroactive species; notably, these have been extensively used to suppress the polysulfide shuttle effect in lithium–sulfur cells.44 However, a progressively increasing carrier salt concentration had the opposite effect on DABQ solubility, as a non-linear upward trend was observed. Starting from a value of 72 mg L−1 for the pure TEGDME/DOL solvent, the DABQ solubility remained constant – around 90 mg L−1 – for LiTFSI concentrations ranging from 0.25 M to 1.5 M (Fig. 3C). Interestingly, an abrupt increase in solubility was observed for salt concentrations higher than 1.5 M, leading to two distinct regions/regimes in the solubility diagram (Fig. 3C, regions I and II).
Whereas the common ion effect could account for the lower solubility of polysulfides in high molarity electrolytes, clearly another mechanism should be considered to explain the increase in DABQ solubility with the increase in salt concentration. For instance, the solubility can be correlated to the solvent (TEGDME/DOL) to salt (LiTFSI) molar ratio (MR) in the electrolyte (Fig. 3C) and thus to the molecular structure of the electrolyte. As shown in Fig. 3C, the solubility remains nearly constant for LiTFSI concentrations ranging from 0 to 1.5 M. This corresponds to electrolytes with the salt-to-solvent MR above 4.25. Below this ratio, a rapid increase in solubility is observed – notably, the solubility of DABQ has about doubled for a 2.68 MR in the 2 M LiTFSI electrolyte.
Through the study of isolated Li+ solvates, Henderson et al. determined that the coordination number of Li+ in glyme–Li salt binary mixtures was in the range of 4 to 6.45–48 Although the TFSI− counter anions also play a role in the occupation of the free Li+ coordination sites,49 it is reasonable to assume that, at a specific threshold of MR – close to 4 – the coordination environment will not be saturated and the respective free sites may interact with the DABQ molecules. In the process, this can facilitate the solubilization of DABQ in the electrolyte, thus explaining the increasing solubility values of DABQ for LiTFSI concentrations beyond approximately 1.5 M.
The correlation between the electrolyte salt concentration and cycling efficiency is also found to be non-linear, as other factors influence the extent of material utilization and cycling stability. Increasing the LiTFSI content from 1 M to 2 M leads to a considerable solubility increase that is indeed translated into a lower first cycle efficiency and consequently quicker loss of capacity with cycling (Fig. 3D and S7†). Conversely, while an electrolyte with only 0.25 M LiTFSI slightly reduces the solubility, the electrochemical cycling stability and efficiency are considerably affected, with poor material utilization at the first cycle and high cell polarization detected. This is ascribed to the ionic conductivity of the electrolyte, rendered insufficient by the decrease in electrolyte salt concentration.
Having established a correlation between the solubility of DABQ and the cycling stability, we undertook a mechanism study to confirm and understand the solid-phase electrochemical process occurring with DABQ in TEGDME/DOL 1:1 LiTFSI 1 M, as this electrolyte formulation delivered the best results. Given the noticeable polarization values observed during the regular galvanostatic cycling (up to 450 mV for the first electron and 300 mV for the second at a rate of 1Li+/10 h, Fig. 3D), a cell was cycled using a galvanostatic intermittent titration technique (GITT). Experimentally, a constant current equivalent to a cycling rate of 1Li+/10 h was applied over 2 hours (i.e., Δx = 0.2), followed by a relaxation period of 2 hours at zero current (Fig. 4A). Basically, this measurement confirms that the overall electrochemical process occurs according to two successive phase transitions (two separated plateaus), each involving one electron per molecule. The first phase transition process (1st plateau) appears significantly more sluggish than the second one (2nd plateau). Indeed, the quasi-equilibrium state of this biphasic system is not reached, which is not the case for the second phase transition. However, by extrapolation we can estimate the quasi-equilibrium potential values for the two biphasic systems at ∼2.4 V and 2.35 V vs. Li+/Li, respectively. The lower (as compared to BQ) electrochemical reduction potential of DABQ is the inevitable result of the richer electronic environment of the carbonyl double bond due to the electron delocalization in DABQ, as also confirmed by the ν(CO) vibration band shift in FTIR analysis (Fig. 2).
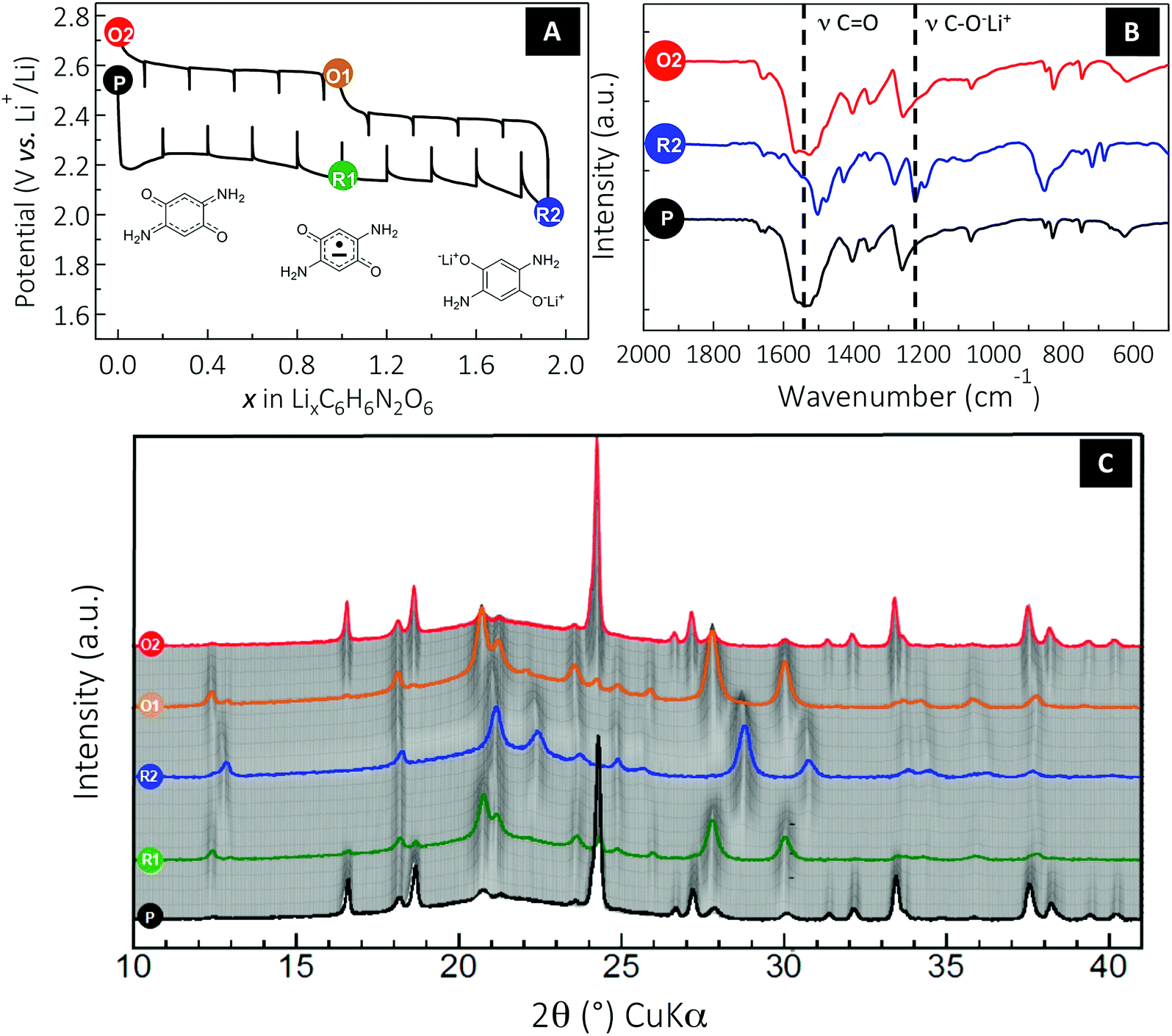 | ||
| Fig. 4 Monitoring the reversible solid-state electrochemical Li insertion/deinsertion process occurring in DABQ using TEGDME:DOL 1:1 LiTFSI 1 M as the electrolyte. (A) A typical potential–composition curve for a Li half-cell using DABQ as the positive electrode cycled in GITT mode at a rate of 1Li+/10 h for Δx = 0.2 followed by an OCV period of 2 hours. (B) Ex situ FTIR of the pristine (P), two-electron reduced (R2) and two-electron oxidized (O2) DABQ. (C) In situ X-ray diffraction powder patterns of an electrode made of DABQ collected during the first cycle. The cell was cycled in the 2.0–2.8 V potential range using an intermittent galvanostatic mode. More specifically, a cycling rate of 1Li+/10 h was applied for a period of 2 h, separated by resting periods of 1 h during which the XRD patterns were collected. Note that XRD patterns labelled (P) and (O2) formally correspond to the DABQ crystallized form, (R1) and (O1) to the monolithiated semiquinone radical form Li1C6H6N2O2, and (R2) to the fully lithiated Li2C6H6N2O2 phase. | ||
To get further insight into the electrochemical mechanism and the reversibility of the DABQ redox process, ex situ FTIR was carried out (Fig. 4B). Therefore, three samples of the cycled DABQ electrode material were retrieved and denoted as follows (Fig. 4): pristine electrode (P), after a 2-electron reduction (R2), then after a full reversible 2-electron oxidation (O2). Consistent with the carbonyl group reduction mechanism, the CO stretching band disappeared upon the 2-electron reduction of DABQ (R2), accompanied by the appearance of a phenolate stretching band at 1220 cm−1. This reaction was found to be perfectly reversible, as evidenced by the spectrum of the re-oxidized DABQ (O2) being identical to that of the pristine material (P).
Finally, in situ XRD patterns were collected to monitor the structural changes during the lithium insertion/deinsertion process in the DABQ electrodes. The gradual fading of most of the reflections accompanied by the appearance of new peaks indicates a biphasic reaction for each sequentially exchanged electron. In agreement with the GITT measurement (Fig. 4a), the 2-electron reduction thus proceeds through two distinct phase transitions with three related phases: pristine C6H6N2O2 (DABQ), monolithiated semiquinone radical form Li1C6H6N2O2, and then the fully lithiated Li2C6H6N2O2.
These major structural changes are the result of H-bond disruption in DABQ driven by reduction of the carbonyl groups. Since the electronic as well as the coordination environment of the O atoms is changing during carbonyl bond reduction accompanied by lithium insertion (as also confirmed by ex situ FTIR measurements, Fig. 4b), the intermolecular H-bonds are disrupted. The stabilization of the newly formed crystalline phase takes place through coordination of the Li+ cations in the vicinity of newly formed phenolate groups. Remarkably, this two-step reduction is fully reversible, the diffraction patterns of Li1C6H6N2O2 (O1) and C6H6N2O2 (O2) in the re-oxidation route matching those of Li1–C6H6N2O2 (R1) and C6H6N2O2 (P) obtained during the reduction, respectively. Furthermore, no broadening of the peaks is observed, which means no amorphization of the material. The structure is thus flexible and strong enough to allow for subsequent lithium insertions (deinsertions) and H-bond regeneration. The stable intermediate Li1C6H6N2O2 structure (R1 or O1) probably contributes to the material’s strength.
An important aspect to be highlighted here is the peculiar properties of H-bond stabilized DABQ in that the electrochemistry of this solid material proceeds exclusively via intrinsically insoluble phases (DABQ–C6H6N2O2via H-bonding and Li2C6H6N2O2). By contrast, whereas the reduced form of benzoquinone becomes poorly soluble in organic electrolytes due to the formation of a delocalized organic polyanion (Li2–C6H4O2),7,42 the oxidized form (benzoquinone, BQ, C6H4O2) is highly soluble due to the weaker van der Waals intermolecular interactions. In fact, the solubility of BQ is so high (measured in excess of 200 g L−1, Fig. S8†) that the solid-state electrochemical cycling is impossible to carry out. As such, the exceptional H-bonding feature grants DABQ far superior cycling performances.
Conclusion
In this work, a new conceptual strategy for small quinone battery material insolubilization is presented. The intermolecular hydrogen bonding in 2,5-diamino-1,4-benzoquinone (DABQ), reinforced by the strongly polarized amino and carbonyl groups, is found to be robust enough to result in an unusually low solubility in organic battery electrolytes. We further provide a correlation between the electrolyte solvent polar momentum, the solvation state of the electrolyte and the solubility of DABQ, directly influencing the cycling stability of the solid electrode cells. With an optimized electrolyte formulation we achieved full material utilization under galvanostatic cycling (theoretical capacity of 388 mA h g−1 for a reversible 2-electron process) and remarkable cycling stability when compared to organic battery materials similar in composition (e.g., 1,4-benzoquinone). Furthermore, through ex situ FTIR and in situ PXRD, the chemical and structural changes undergone by DABQ upon electrochemical cycling are shown to be perfectly reversible, attesting the stability of the electrochemical process and the unusual robustness of this peculiar molecular crystal. Our work paves the way to new designs of H-bond stabilized organic molecular compounds for battery applications. If combined with non-polar electrolyte solvents, H-bond interacting binders and/or conductive carbon frameworks, this could lead to a new strategy towards insoluble small organic battery materials with practical impact.Experimental methods
Synthesis of 2,5-diaminobenzoquinone (DABQ)
Commercially available 2,5-diaminohydroquinone dihydrochloride (>90%, Sigma Aldrich) (213 mg, 1 mmol) was dissolved in 20 mL of Milli-Q water and an excess of triethylamine (Sigma Aldrich) (1.4 mL, 10 mmol) was added as a proton scavenger while stirring the mixture in air at room temperature. The addition was followed by a change of color from violet to dark red and formation of a dark purple precipitate. The suspension was stirred for 30 minutes and the precipitate was filtered off and washed with water, methanol and finally with ether. After drying under vacuum, the reaction afforded an 80% yield of pure DABQ (110 mg). 1H NMR (300 MHz, DMSO-d6, TMS): δ = 7.76 (br, 2H; NHax), δ = 7.02 (br, 2H; NHeq), δ = 5.30 (s, 2H; aromatic CH) (Fig. S1†). FTIR: 3348 cm−1, 3158 cm−1 (νN–H); 1663 cm−1, 1653 cm−1 (δN–H); 1563 cm−1 (νCO); 1522 cm−1, 1402 cm−1, 1356 cm−1 (νCC); 1259 cm−1 (νC–N) 1065 cm−1 (νC–O) (Fig. 2 and S1†).
Characterization techniques
The crystal structure of DABQ was solved from PXRD data collected using the ESRF synchrotron (Grenoble) with a wavelength λ = 0.74580 Å. FTIR spectroscopy was carried out on pristine DABQ powders using a Bruker Alpha P spectrometer with a single reflection ATR module. Thermogravimetry (TG) and differential scanning calorimetry (DSC) experiments were carried out under argon with a SENSYSevo instrument from Setaram using a heating rate of 5 °C min−1 between 25 and 600 °C. Scanning electron microscopy (SEM) images were collected using a JEOL JSM-7600F microscope. UV-visible spectra were recorded using a Shimadzu UV-1700 PharmaSpec. Samples were analyzed in 1 cm length quartz cuvettes. Saturated solutions of DABQ in the studied electrolyte systems were prepared by adding 2 mg of the powder to 3 mL of the electrolyte. After equilibrium was reached, the resulting supernatant was carefully filtered through Millipore Millex-HV PVDF 0.45 μm filters to remove excess insolubilized DABQ. The saturated solution was subsequently diluted by a factor of 100. Standard solutions were prepared with four for each electrolyte with DABQ concentrations of 2, 5, 10 and 20 μM. Absorbance was measured in the 200–800 nm range for all solutions. The calibration curve was obtained by linking the maximum absorbance (λmax = 330 nm) of the standard solutions to their concentration in DABQ using the common Beer–Lambert law: A = εlC.Electrochemical study
Pristine DABQ powder was ground with Ketjenblack EC600JD carbon black (AkzoNobel) in a 67:33 mass ratio (mtot. = 100 mg) using a planetary ball mill (1 hour; 400 rpm; five 5 mm steel balls and stainless steel container). The mixed powders were dried in a vacuum oven at 70 °C for 12 hours and kept under argon atmosphere afterwards. The electrodes were tested in half-cell configuration using Li–metal foil (Alfa Aesar) as both reference and counter electrode. CR2032 (SS316) coin-cells were assembled in an argon-filled glovebox. One sheet of Whatman Grade GF/D Glass Microfiber Filters was used as a separator between the working electrode and the lithium disk. The following electrolytes (Solvionic) were used in a 1:1 (v/v) ratio: EC/DMC LiPF6 1 M; EC/DMC LiTFSI 1 M; DME/DOL LiTFSI 1 M and TEGDME/DOL LiTFSI 1 M. Other electrolyte formulations (TEGDME/DOL 1:1 (v/v) LiTFSI 0.25 M/0.5 M/1.5 M/2 M/3 M) were homemade using TEGDME (>99%, Sigma Aldrich), DOL (99.8%, Sigma Aldrich) and high-purity LiTFSI (3 M). The galvanostatic cycling experiments were performed using an Arbin BT-2043 multichannel potentiostat battery tester.
Ex situ FTIR measurements
Swagelok-type cells were used for the sake of simplicity using a Li metal disc as the negative electrode and a fiberglass separator soaked with TEGDME/DOL 1:1 (v/v) LiTFSI 1 M as the electrolyte. The composite electrode material made of DABQ powder ground with Ketjenblack EC600JD carbon black (67:33 in mass ratio) was collected after cell disassembly in an argon-filled glovebox before cycling, after the first reduction and after the first full cycle (first discharge/charge). The collected samples were gently washed with DMC, centrifuged to remove any trace of LiTFSI then placed in a drying glass oven (Buchi B-585 glass-oven Kugelrohr) and heated at 120 °C for 3 hours. Finally, the FTIR analyses were carried out on the resulting dried powders using a Bruker Alpha P spectrometer with a Single reflection ATR module installed in the glovebox.
In situ powder XRD
Diffractograms were recorded using a PANanalytical X’Pert Pro diffractometer operated in Bragg–Brentano reflection geometry with a long line focus Cu-anode X-ray source, and a X’Celerator RTMS detector. Data were collected in the 10–40° 2θ range with a step of 0.017° and an acquisition time of 1.9 s per step. Measurements were performed with a Multi Purpose Sample Stage (MPSS) able to accommodate the in situ Swagelok-type cell described elsewhere.50 A Li metal disk was used as the negative electrode whereas the positive electrode composition was 70 wt% of DABQ, 25 wt% Ketjenblack EC-600JD, and 5 wt% PTFE. This mixture was pressed at 1 ton on a stainless steel (AISI 316L) grid current collector.Conflicts of interest
There are no conflict of interest to declare.Acknowledgements
LS and AV acknowledge FSR UCL and F.R.S.-FNRS through grant No. J.0111.16 ‘Equinox’ for financial support, and wish to thank Y. Filinchuk and F. Morelle (UCL) for help with XRD data analysis. LS and JFG are grateful to CfB in the frame of the ARC BATTAB 14/19-057. AJ and PP wish to thank J. Gaubicher, O. Crosnier and S. Grolleau (IMN) for their assistance.References
- M. Armand and J.-M. Tarascon, Issues and challenges facing rechargeable lithium batteries, Nature, 2001, 414, 359–367 CrossRef PubMed.
- M. Armand and J.-M. Tarascon, Building better batteries, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- H. Li, Z. Wang, L. Chen and X. Huang, Research on advanced materials for Li-ion batteries, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Positive electrode materials for Li-ion and Li-batteries, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- L. Croguennec and M. R. Palacin, Recent achievements on inorganic electrode materials for lithium-ion batteries, J. Am. Chem. Soc., 2015, 137, 3140–3156 CrossRef CAS PubMed.
- L. Shi and T. Zhao, Recent advances in inorganic 2D materials and their applications in lithium and sodium batteries, J. Mater. Chem. A, 2017, 5, 3735–3758 RSC.
- H. Chen, M. Armand, G. Demailly, F. Dolhem and P. Poizot, From Biomass to a Renewable LixC6O6 Organic Electrode for Sustainable Li-Ion Batteries, ChemSusChem, 2008, 1, 348–355 CrossRef CAS PubMed.
- M. Armand, et al., Conjugated dicarboxylate anodes for Li-ion batteries, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed.
- Y. Liang, Z. Tao and J. Chen, Organic electrode materials for rechargeable lithium batteries, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- S. Renault, et al., A green Li–organic battery working as a fuel cell in case of emergency, Energy Environ. Sci., 2013, 6, 2124–2133 RSC.
- B. Häupler, A. Wild and U. S. Schubert, Carbonyls: Powerful Organic Materials for Secondary Batteries, Adv. Energy Mater., 2015, 5(1–34), 1402034 CrossRef.
- P. Poizot, F. Dolhem and J. Gaubicher, Progress in all-organic rechargeable batteries using cationic and anionic configurations: Toward low-cost and greener storage solutions?, Curr. Opin. Electrochem., 2018, 9, 70–80 CrossRef CAS.
- É. Deunf, et al., Solvation, exchange and electrochemical intercalation properties of disodium 2,5-(dianilino)terephthalate, CrystEngComm, 2016, 18, 6076–6082 RSC.
- H. Zhang, C. Mao, J. Li and R. Chen, Advances in electrode materials for Li-based rechargeable batteries, RSC Adv., 2017, 7, 33789–33811 RSC.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, The rise of organic electrode materials for energy storage, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- Q. Zhao, Z. Zhu and J. Chen, Molecular Engineering with Organic Carbonyl Electrode Materials for Advanced Stationary and Redox Flow Rechargeable Batteries, Adv. Mater., 2017, 29(1–25), 1607007 CrossRef PubMed.
- D. Häringer, P. Novák, O. Haas, B. Piro and M. Pham, Poly (5-amino-1,4-naphthoquinone), a Novel Lithium-Inserting Electroactive Polymer with High Specific Charge, J. Electrochem. Soc., 1999, 146, 2393–2396 CrossRef.
- T. L. Gall, K. H. Reiman, M. C. Grossel and J. R. Owen, Poly(2,5-dihydroxy-1,4-benzoquinone-3,6-methylene): a new organic polymer as positive electrode material for rechargeable lithium batteries, J. Power Sources, 2003, 119, 316–320 CrossRef.
- L. Kai, Z. Jianming, Z. Guiming and Y. Yang, Poly(2,5-dihydroxy-1,4-benzoquinonyl sulfide) (PDBS) as a cathode material for lithium ion batteries, J. Mater. Chem., 2011, 21, 4125–4131 RSC.
- Z. Song, et al., A quinone-based oligomeric lithium salt for superior Li–organic batteries, Energy Environ. Sci., 2014, 7, 4077–4086 RSC.
- B. Häupler, T. Hagemann, C. Friebe, A. Wild and U. S. Schubert, Dithiophenedione-containing polymers for battery application, ACS Appl. Mater. Interfaces, 2015, 7, 3473–3479 CrossRef PubMed.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Poly(benzoquinonyl sulfide) as a High-Energy Organic Cathode for Rechargeable Li and Na Batteries, Adv. Sci., 2015, 2(1–9), 1500124 CrossRef PubMed.
- A. Vlad, et al., Exploring the potential of polymer battery cathodes with electrically conductive molecular backbone, J. Mater. Chem. A, 2015, 3, 11189–11193 RSC.
- Z. Zhu, H. Li, J. Liang, Z. Tao and J. Chen, The disodium salt of 2,5-dihydroxy-1,4-benzoquinone as anode material for rechargeable sodium ion batteries, Chem. Commun., 2015, 51, 1446–1448 RSC.
- X. Jiangfeng, et al., A Novel Coordination Polymer as Positive Electrode Material for Lithium Ion Battery, Cryst. Growth Des., 2008, 8, 280–282 CrossRef.
- H. Chen, et al., Electrochemical Reactivity of Lithium Chloranilate vs. Li and Crystal Structures of the Hydrated Phases, Electrochem. Solid-State Lett., 2009, 12, 102–106 CrossRef.
- R. H. Zeng, et al., Synthesis and properties of a lithium–organic coordination compound as lithium-inserted material for lithium ion batteries, Electrochem. Commun., 2010, 12, 1253–1256 CrossRef CAS.
- S. Wang, et al., Organic Li4C8H2O6 Nanosheets for Lithium-Ion Batteries, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS PubMed.
- S. Gottis, A.-L. Barrès, F. Dolhem and P. Poizot, Voltage Gain in Lithiated Enolate-Based Organic Cathode Materials by Isomeric Effect, ACS Appl. Mater. Interfaces, 2014, 6, 10870–10876 CrossRef CAS PubMed.
- S. Wang, et al., All organic sodium-ion batteries with Na4C8H 2O6, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef CAS PubMed.
- J. Geng and P. Poizot, Evaluation of polyketones with N-cyclic structure as electrode material for electrochemical energy storage: case of pyromellitic diimide dilithium salt, Chem. Commun., 2011, 47, 2414–2416 RSC.
- Y. Lu, et al., Flexible and Free-Standing Organic/Carbon Nanotubes Hybrid Films as Cathode for Rechargeable Lithium-Ion Batteries, J. Phys. Chem. C, 2017, 121, 14498–14506 CrossRef CAS.
- B. Genorio, K. Pirnat, R. Cerc-Korosec, R. Dominko and M. Gaberscek, Electroactive organic molecules immobilized onto solid nanoparticles as a cathode material for lithium-ion batteries, Angew. Chem., Int. Ed., 2010, 49, 7222–7224 CrossRef CAS PubMed.
- L. Zhao, et al., A MC/AQ Parasitic Composite as Cathode Material for Lithium Battery, J. Electrochem. Soc., 2011, 158, A991–A996 CrossRef CAS.
- K. Pirnat, R. Dominko, R. Cerc-korosec, B. Genorio and M. Gaberscek, Electrochemically stabilised quinone based electrode composites for Li-ion batteries, J. Power Sources, 2012, 199, 308–314 CrossRef CAS.
- A. Jaffe, A. Saldivar Valdes and H. I. Karunadasa, Quinone-functionalized carbon black cathodes for lithium batteries with high power densities, Chem. Mater., 2015, 27, 3568–3571 CrossRef CAS.
- J. D. Watson and F. H. C. Crick, Molecular structure of nucleic aids: A structure for deoxyribose nucleic acid, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- C. Luo, et al., Self-Healing Chemistry between Organic Material and Binder for Stable Sodium-Ion Batteries, Chem, 2017, 3, 1050–1062 CAS.
- S. Wang, Exfoliation of Covalent Organic Frameworks into Few-Layer Redox-Active Nanosheets as Cathode Materials for Lithium-Ion Batteries, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- B. Tian, et al., Amino group enhanced phenazine derivatives as electrode materials for lithium storage, Chem. Commun., 2017, 53, 2914–2917 RSC.
- H. S. Das, et al., Straightforward synthesis of substituted p-Quinones: Isolation of a key intermediate and use as a bridging ligand in a diruthenium complex, Chem.–Eur. J., 2010, 16, 2977–2981 CrossRef CAS PubMed.
- P. Jezowski, et al., Safe and recyclable lithium-ion capacitors using sacrificial organic lithium salt, Nat. Mater., 2018, 17, 167–173 CrossRef CAS PubMed.
- T. L. Bucholz and Y. L. Loo, Polar aprotic solvents disrupt interblock hydrogen bonding and induce microphase separation in double hydrophilic block copolymers of PEGMA and PAAMPSA, Macromolecules, 2008, 41, 4069–4070 CrossRef CAS.
- L. Suo, Y. Hu, H. Li, M. Armand and L. Chen, A new class of Solvent-in-Salt electrolyte for high-energy rechargeable metallic lithium batteries, Nat. Commun., 2013, 4(1–9), 1481 CrossRef PubMed.
- C. Zhang, et al., Chelate Effects in Glyme/Lithium Bis(trifluoromethanesulfonyl)amide Solvate Ionic Liquids, Part 2: Importance of Solvate-Structure Stability for Electrolytes of Lithium Batteries, J. Phys. Chem. B, 2014, 118, 17362–17373 CAS.
- W. A. Henderson, N. R. Brooks, W. W. Brennessel and V. G. Young, Tetraglyme-Li+ Cation Solvate Structures: Models for Amorphous Concentrated Liquid and Polymer Electrolytes (II), Chem. Mater., 2003, 15, 4685–4690 CrossRef CAS.
- W. A. Henderson, et al., Glyme–Lithium Bis(trifluoromethanesulfonyl)imide and Glyme–Lithium Bis(perfluoroethanesulfonyl)imide Phase Behavior and Solvate Structures, Chem. Mater., 2005, 17, 2284–2289 CrossRef CAS.
- W. A. Henderson, Glyme–lithium salt phase behavior, J. Phys. Chem. B, 2006, 110, 13177–13183 CrossRef CAS PubMed.
- R. Raccichini, J. W. Dibden, A. Brew, J. R. Owen and N. García-Aráez, Ion Speciation and Transport Properties of LiTFSI in 1,3-Dioxolane Solutions: A Case Study for Li–S Battery Applications, J. Phys. Chem. B, 2018, 122, 267–274 CrossRef CAS PubMed.
- M. Morcrette, et al., In situ X-ray diffraction techniques as a powerful tool to study battery electrode materials, Electrochim. Acta, 2002, 47, 3137–3149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and protocols. CCDC 1854000. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02995d |
| This journal is © The Royal Society of Chemistry 2019 |