
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
I2-catalyzed intramolecular oxidative amination of C(sp3)–H bond: efficient access to 3-acylimidazo[1,2-a]pyridines under neat condition†
Lilan
Huang‡
a,
Wenqing
Yin‡
 a,
Jian
Wang
b,
Chunfang
Gan
a,
Yanmin
Huang
a,
Chusheng
Huang
*a and
Yimiao
He
*a
a,
Jian
Wang
b,
Chunfang
Gan
a,
Yanmin
Huang
a,
Chusheng
Huang
*a and
Yimiao
He
*a
aGuangxi Key Laboratory of Natural Polymer Chemistry and Physics, Nanning Normal University, Nanning 530001, P. R. China. E-mail: heyimiao@gxtc.edu.cn; huangcs@gxtc.edu.cn
bKey Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, P. R. China
First published on 18th January 2019
Abstract
An efficient and “green” protocol for the synthesis of 3-acylimidazo[1,2-a]pyridines through intramolecular oxidative α-amination of carbonyl compounds has been developed. The reaction proceeds smoothly utilizing I2 as a catalyst and H2O2 as an oxidant under neat condition with broad substrate scope. Several complex nitrogen-containing fused rings are conveniently constructed, which are not easy to access by traditional methods.
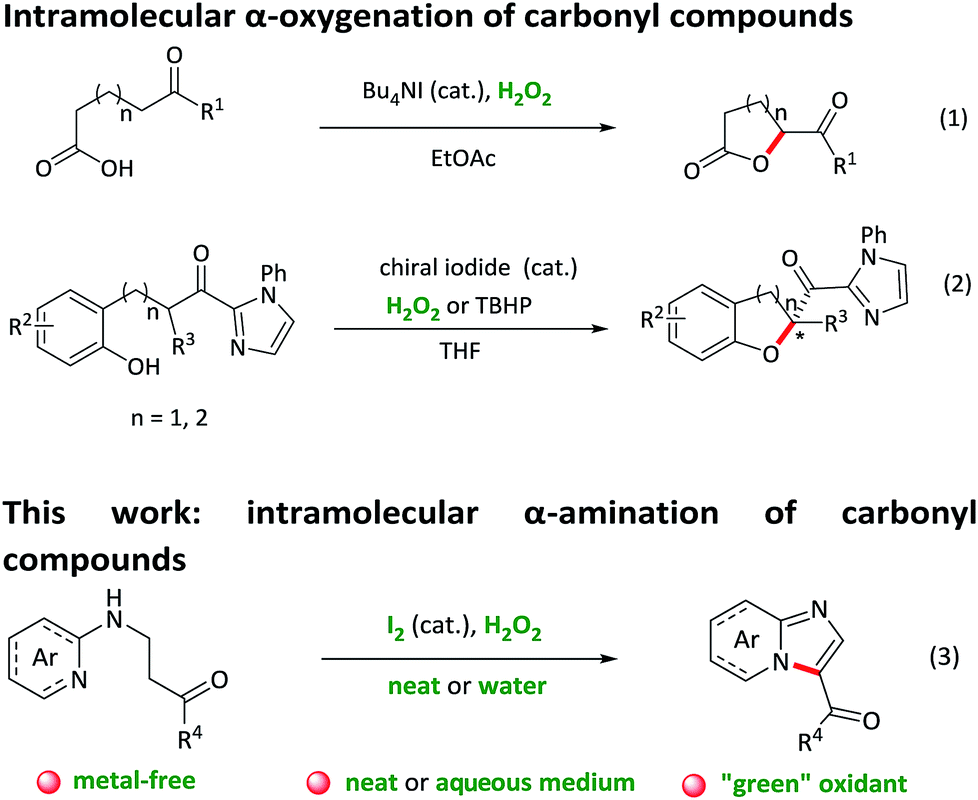
Intramolecular C–H bond activation to build C–X (N/O) bonds is of great significance in the construction of heterocyclic scaffolds, which could afford wide application for the direct synthesis of biologically active molecules and drug candidates.1 In contrast to unsaturated C–H bond activation reactions, transformations of Csp3–H bond to Csp3–X bonds are still limited.2 Over the past decades, several groups have developed transition-metal-catalysed intramolecular aliphatic C–H aminations and oxygenations, affording an atom-economic and efficient access to a series of N/O-containing heterocyclic compounds.3 Nevertheless, most of these methods face the limitations of transition metal residuals, expensive ligands, harsh conditions and narrow substrate scope. Recently, more environmentally-benign iodide-catalysed reactions for the preparation of heterocyclic compounds utilizing inexpensive and readily available TBHP or H2O2 as an oxidant have attracted enormous attention.4 For example, Ishihara and co-workers reported an intramolecular oxidative C–H bond α-oxygenation reaction of carbonyl compounds catalysed by in situ generated tetrabutylammonium (hypo)iodite with either hydrogen peroxide or tert-butyl hydroperoxide as a green oxidant (Scheme 1, eqn (1) and (2)).5 However, the application of this “green” catalytic cycle (iodide as the catalyst and H2O2 as the oxidant) for intramolecular α-amination of the carbonyl compounds, to the best of our knowledge, has been barely documented,6 although which allows a facile and “green” formation of a great bioactive N-heterocycles.
 | ||
| Scheme 1 Intramolecular α-oxygenation and α-amination of carbonyl compounds. | ||
Imidazo[1,2-a]pyridine represents an important heterocyclic scaffold with broad ranges of biological activities.7 Many commercially available drugs such as zolpidem, alpidem, zolimidine, olprinone, saripidem, and necopidem contain the core structure of imidazo[1,2-a]pyridine.8 Therefore, a variety of synthetic methods, including intramolecular aminooxygenation of alkenes or alkynes,9 tetrabutylammonium iodide catalyzed oxidative coupling reactions,10 copper-catalyzed aromatic aminations,11 three component couplings,12 and silver-catalyzed oxidative cross-coupling reactions,13 have been developed for the preparation of imidazo[1,2-a]pyridine derivatives. However, there are still some drawbacks which require to be solved in the traditional synthetic methods such as the use of complex starting materials, metal vestigial, narrow substrate scopes and harsh conditions. Therefore, the development of a metal-free, “green” and efficient method for the construction of imidazo[1,2-a]pyridine scaffold is still necessary.
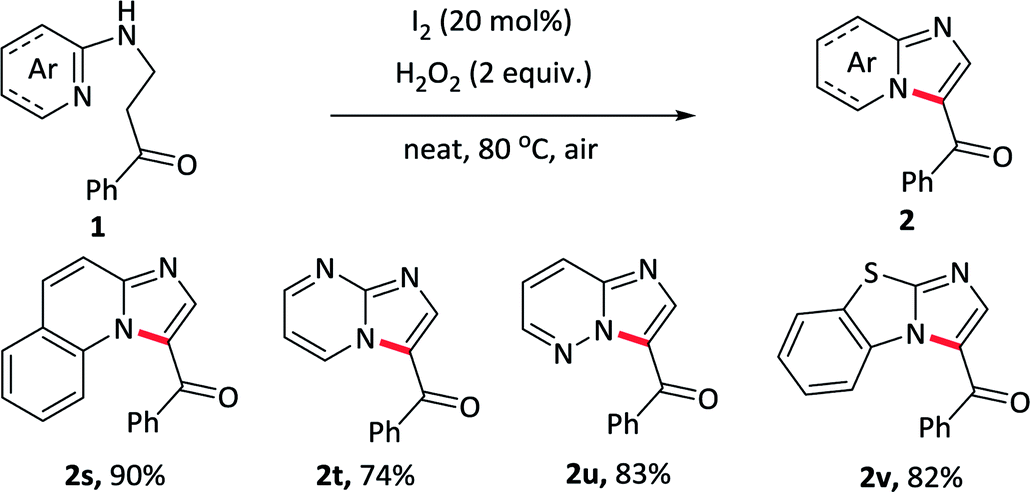
Herein, we develop a convenient, efficient and “green” approach for the construction of 3-acylimidazo[1,2-a]pyridines through intramolecular oxidative α-amination of carbonyl compounds, which utilizing I2 as a catalyst and H2O2 as the only oxidant. To point out, the reaction proceeds under neat condition and the use of H2O2 as an oxidant generates H2O as the only by-product, which fully meet the requirement of green chemistry. Besides pyridine as a nitrogen source, the reaction undergoes equally efficiently with other N-heterocycles such as quinoline, pyrimidine, pyridazine and benzo[d]thiazole, thus providing a facile and environmentally sustainable pathway to prepare more complex nitrogen-containing fused rings.
Initially, the reaction was carried out by using 1-phenyl-3-(pyridin-2-ylamino)propan-1-one 1a as a model substrate in toluene with I2 as a catalyst and TBHP as an oxidant at 50 °C under air condition (Table 1, entry 1). Pleasingly, the reaction of 1a in the presence of 20 mol% catalyst with 2 equiv. of the oxidants gave the desired 2a in 71% yield. After screening various oxidants, H2O2 was found to be the most effective and provided 2a in 75% yield (entries 1–4). Replacing the catalyst I2 with other iodides such as TBAI, NaI did not improve the reaction (entry 5 and –6). Subsequently, various solvents were also evaluated and toluene revealed the best (entries 7–10). A further improvement was achieved upon elevating the temperature to 80 °C, allowing the reaction yield up to 87% (entry 11). The more important thing was that, the reaction could even undergo in water or under neat condition, giving a similar yield of 85% and 88% (entry 12–13). We also investigated the influence of the loadings of the catalyst on the reaction, and it revealed that increasing the amount of catalysts to 30 mol% did not improve the reaction, however, decreasing the amount of catalysts to 10 mol% resulted in a lower yield (entry 14 and 15). The blank experiments demonstrated that both catalyst and oxidant were essential for this reaction (entry 16 and 17). Consequently, the optimum reaction conditions were determined to be 1-phenyl-3-(pyridin-2-ylamino)propan-1-one 1a in the presence of I2 (20 mol%) as well as H2O2 (2 equiv.) under neat condition or in water at 80 °C.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Oxidant | Solvent | T [°C] | Yieldb [%] |
| a Reaction conditions: 1a (0.2 mmol), catalyst (20 mol%), oxidant (0.4 mmol) in solvent (1 mL), air. TBHP (70 wt% in H2O), H2O2 (30 wt% in H2O). b Isolated yields. c TBPB = t-butylperoxybenzoate. d DTBP = di-tert-butyl peroxide. e Catalyst (30 mol%). f Catalyst (10 mol%). g Without oxidant. h Without catalyst. | |||||
| 1 | I2 | TBHP | Toluene | 50 | 71 |
| 2 | I2 | H2O2 | Toluene | 50 | 75 |
| 3 | I2 | TBPBc | Toluene | 50 | 15 |
| 4 | I2 | DTBPd | Toluene | 50 | 11 |
| 5 | TBAI | H2O2 | Toluene | 50 | 56 |
| 6 | NaI | H2O2 | Toluene | 50 | 54 |
| 7 | I2 | H2O2 | THF | 50 | 61 |
| 8 | I2 | H2O2 | DCM | 50 | 63 |
| 9 | I2 | H2O2 | CH3CN | 50 | 52 |
| 10 | I2 | H2O2 | DMF | 50 | Trace |
| 11 | I2 | H2O2 | Toluene | 80 | 87 |
| 12 | I 2 | H 2 O 2 | H 2 O | 80 | 85 |
| 13 | I 2 | H 2 O 2 | Neat | 80 | 88 |
| 14e | I2 | H2O2 | H2O | 80 | 86 |
| 15f | I2 | H2O2 | H2O | 80 | 67 |
| 16g | I2 | H2O | 80 | trace | |
| 17h | H2O2 | H2O | 80 | trace | |
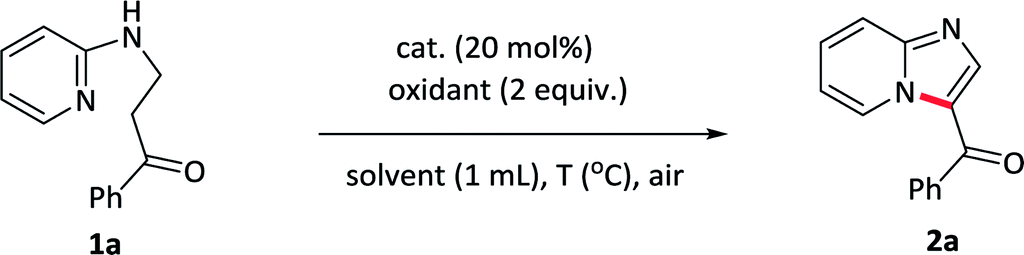
With optimized conditions in hand, various substituents at pyridine component of 1-phenyl-3-(pyridin-2-ylamino)propan-1-ones 1b–1i were first tested under standard conditions to form the corresponding imidazo[1,2-a]pyridin-3-yl(phenyl)methanone 2b–2i. As showed in Table 2, pyridines bearing electron-rich groups (2b, 2h–2i) such as methyl, methoxy at the meta- or para-position underwent an oxidative cycloamination process smoothly to afford the desired products in good yields (81–93%). Slightly lower yields were achieved for weak electron-withdrawing groups at pyridine component such as fluoro, chloro, bromo substituents, maybe due to the electronic effect (2c–2e). Several strong electron-withdrawing groups, such as trifluoromethyl (2f) and ester (2g) substituents, the yield sharply decreased to 48% and 67%, respectively. These results indicated that electronic effect exerted by the substituents in pyridine component had an important influence on this reaction. In addition, further transformations could be made for halogen-substituent in pyridine component through the cross-coupling reactions.
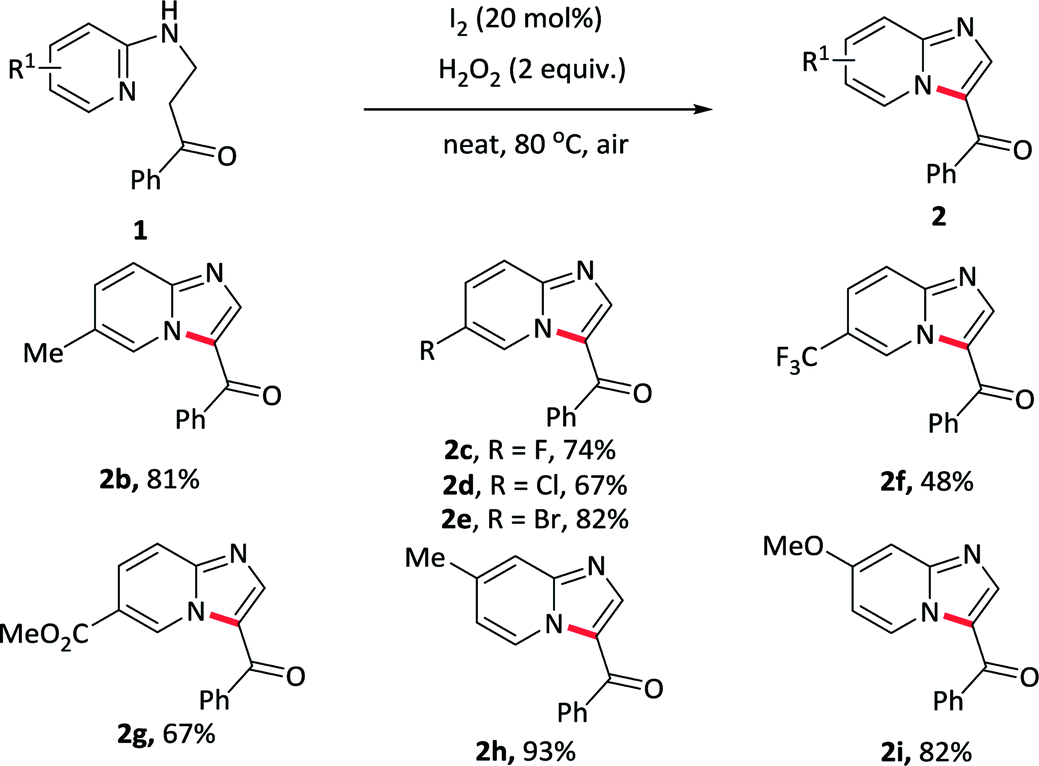
The substrates bearing various substituents at the carbonyl terminal position were subsequently evaluated (Table 3). Groups such as electron-rich methyl, electron-withdrawing fluoro, chloro, bromo substituents on the arene ring were tolerant well and afforded the desired products in similar yields (2j–2n), suggesting that electronic effect exerted by the substituents at the terminal of the carbonyl component had almost no effect on this reaction. Exchanging the phenyl ring to other aromatic rings, such as thiophene, gave the product 2o in 83% yield. Furthermore, alkyl substituents were also compatible with this reaction and received the target products in moderate to excellent yields (2p–2q). However, the reaction did not proceed when the substrate carried with the ester substituent at the terminal (2r).
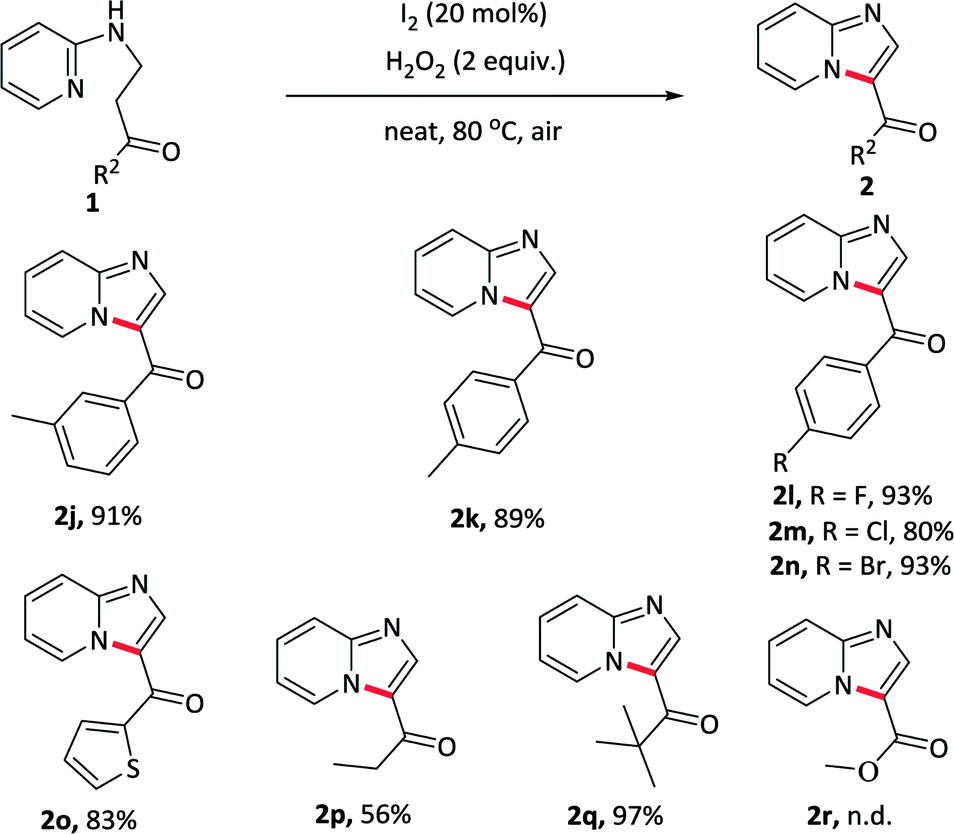
To elaborate the substrate universalities of this strategy for the intramolecular oxidative amination reaction, substrates bearing other azaheterocyclic arenes other than the pyridine were also prepared and the results were displayed in Table 4. The substrate carrying with quinoline ring 2s gave the desired product in excellent yield under standard conditions. Other nitrogen-containing hexatomic rings, such as pyrimidine and pyridazine, delivered the target outcomes 2t and 2u in 74% and 83% yields, respectively. In addition, the five-membered azaheterocyclic arene such as benzo[d]thiazole, also was tolerant well and obtained the desired products 2v in 82% yields. The above-mentioned nitrogen-containing fused ring derivatives 2s–2v were not easy to prepare in conventional methods.
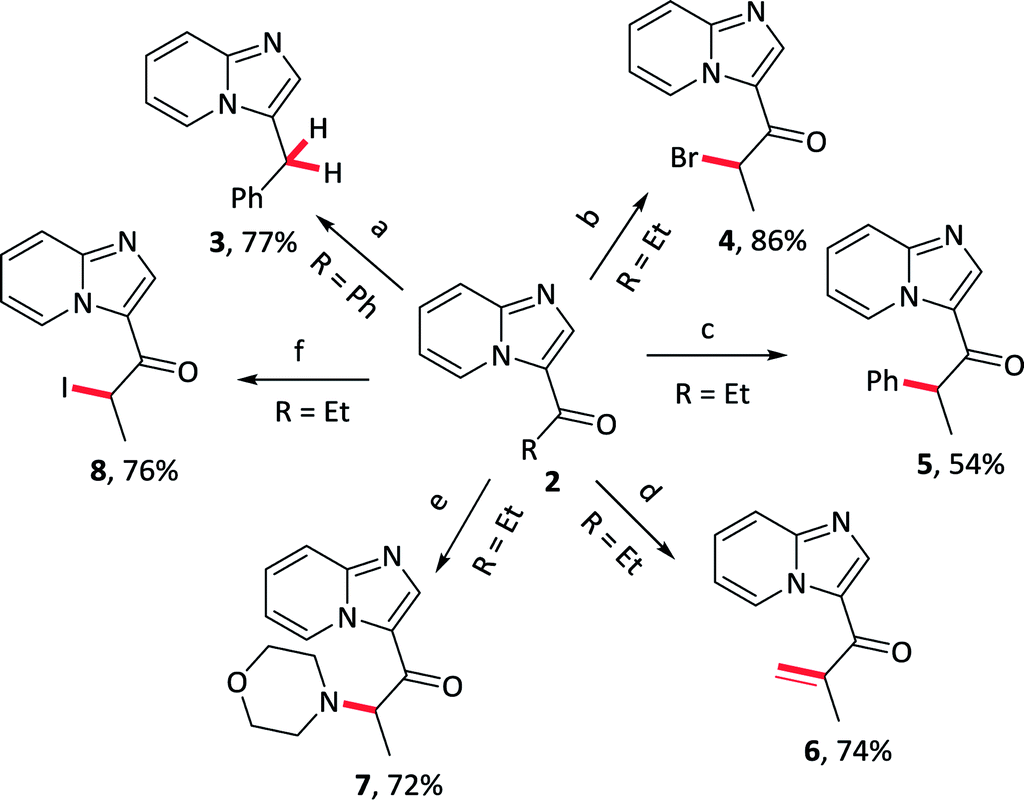
Further elaborations of 3-acylimidazo[1,2-a]pyridines were subsequently conducted; several representative examples were provided in Table 5. The acyl group could be transformed to the methylene, as well as several functionalization were effectively received in α position of carbonyl groups, such as bromination, arylation, alkenylation, amination and iodization.
| a Reaction conditions: 2a (0.2 mmol), N2H4 (2 equiv.), toluene (1 mL), μW; then KOH (7 equiv.), μW. b 2p (0.2 mmol), NBS (1.2 equiv.), p-TsOH·H2O (0.2 equiv.), CH3CN (1 mL), 60 °C. c 2p (2 equiv.), PhI (0.1 mmol), t-BuOK (5 equiv.), DMF (1 mL), 60 °C. d 2p (0.1 mmol), DABCO (0.5 equiv.), K2S2O8 (2 equiv.), DMSO (1 mL), 120 °C. e 2p (0.2 mmol), NBS (1.4 equiv.), PTSA (1 equiv.), CH3CN (1 mL), 60 °C; then morpholine (3 equiv.), K2CO3 (2.5 equiv.), CH3CN (1 mL), rt. f 2p (0.2 mmol), NBS (1.4 equiv.), PTSA (1 equiv.), CH3CN (1 mL), 60 °C; then NaI (1.1 equiv.), acetone (1 mL), rt. |
|---|
|
The scalability of this approach was verified by running the reaction of 1a on both 4 and 10 mmol scales (Scheme 2). 2a was isolated in acceptable yields in both of the cases.
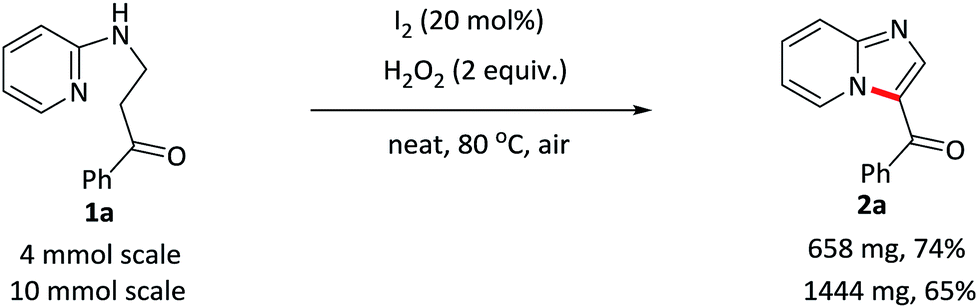 | ||
| Scheme 2 Large-scale synthesis of 2a. | ||
The mechanism for this intramolecular oxidative amination reaction was subsequently studied. Firstly, model substrate 1a was subjected to the hypervalent iodide reagents PhI(OAc)2 or Bu4NIO3 or Bu4NIO4, and the targeted molecule 2a was not detected along with recovery of 1a in 91% yield (page S24,† eqn (1)). Subsequently, increasing the loadings of I2 from 0.2 equiv. to 2 equiv. in the absence of H2O2, the reaction turned out to be messy (page S24,† eqn (2)). Furthermore, when TEMPO, a radical scavenger, was added into the system, the reaction was not suppressed and the targeted product 2a was delivered in 73% yield (S5,† eqn (3)), which indicated that the reaction probably didn't involve a radical process.
Based on the above results and previous reports,14 a plausible mechanism pathway for I2/H2O2-catalyzed synthesis of 3-acylimidazo[1,2-a]pyridines 2a starting from 1-phenyl-3-(pyridin-2-ylamino)propan-1-one 1a is depicted in Scheme 3. In the first step, I2 or the released HI is oxidized by H2O2 to form HOI. In the second step, 1a reacts with HOI to produce a possible intermediate A or B, which then experiences a similar SN2 process to receive a cyclic intermediate C with release of HI. The intermediate C is oxidized by HOI and finally obtain the target product 2a.
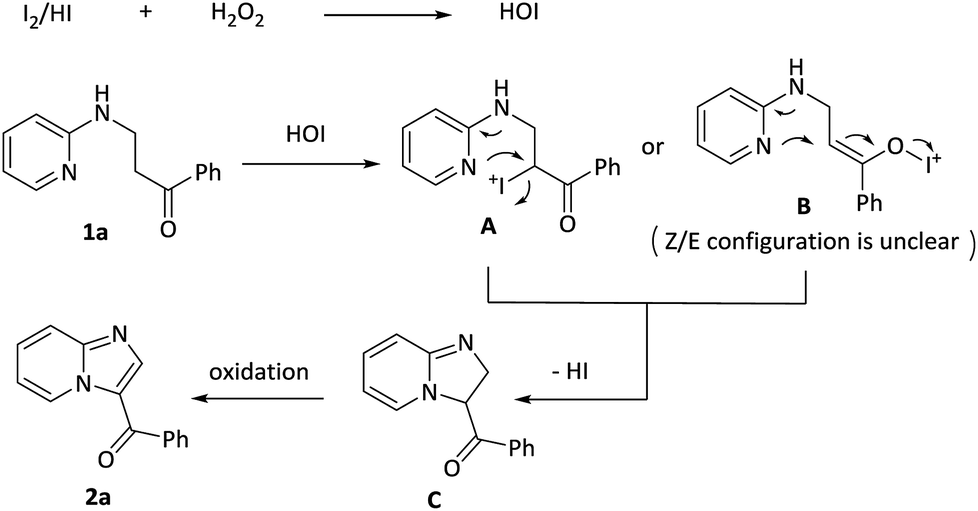 | ||
| Scheme 3 Proposed mechanism. | ||
In summary, we have developed a general and efficient method for the construction of 3-acylimidazo[1,2-a]pyridines through intramolecular oxidative α-amination of carbonyl compounds under metal-free conditions. The reaction proceeded smoothly with I2 as a catalyst and H2O2 as an oxidant under neat condition. This protocol exhibits general substrate scope and several complex nitrogen-containing fused rings could be conveniently accessed via this approach.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21702034, 21861007, 21762008), Natural Science Foundation of Guangxi Province (2017GXNSFBA198089).References
- (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS; (b) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS.
- (a) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635 CrossRef CAS; (b) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS.
- (a) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS; (b) A. P. Smalley and M. J. Gaunt, J. Am. Chem. Soc., 2015, 137, 10632 CrossRef CAS; (c) R. A. Leal, C. Bischof, Y. V. Lee, S. Sawano, C. C. McAtee, L. N. Latimer, Z. N. Russ, J. E. Dueber, J.-Q. Yu and R. Sarpong, Angew. Chem., Int. Ed., 2016, 55, 11824 CrossRef CAS.
- (a) L. J. Ma, X. P. Wang, W. Yu and B. Han, Chem. Commun., 2011, 47, 11333 RSC; (b) J. S. Tian, K. W. J. Ng, J. R. Wong and T. P. Loh, Angew. Chem., Int. Ed., 2012, 51, 9105 CrossRef CAS; (c) X. B. Zhang and L. Wang, Green Chem., 2012, 14, 2141 RSC; (d) M. Lamani and K. R. Prabhu, Chem. - Eur. J., 2012, 18, 14638 CrossRef CAS; (e) J. Zhang, J. Jiang, Y. Li, Y. Zhao and X. Wan, Org. Lett., 2013, 15, 3222 CrossRef CAS; (f) Y. H. Lv, Y. Li, T. Xiong, Y. Lu, Q. Liu and Q. Zhang, Chem. Commun., 2014, 50, 2367 RSC; (g) Q. Jiang, B. Xu, A. Zhao, J. Jia, T. Liu and C. C. Guo, J. Org. Chem., 2014, 79, 8750 CrossRef CAS; (h) H. Huang, W. Chen, Y. Xu and J. Li, Green Chem., 2015, 17, 4715 RSC; (i) J. Q. Han, H. H. Zhang, P. F. Xu and Y. C. Luo, Org. Lett., 2016, 18, 5212 CrossRef CAS.
- (a) M. Uyanik, D. Suzuki, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2011, 50, 5331 CrossRef CAS; (b) M. Uyanik, H. Okamoto, T. Yasui and K. Ishihara, Science, 2010, 328, 1376 CrossRef CAS; (c) M. Uyanik, H. Hayashi and K. Ishihara, Science, 2014, 345, 291 CrossRef CAS.
- (a) W. C. Gao, S. Jiang, R. L. Wang and C. Zhang, Chem. Commun., 2013, 49, 4890 RSC; (b) G. Satish, P. Ashok, R. Thangeswaran and A. Ilangovan, J. Org. Chem., 2015, 80, 5167 CrossRef CAS.
- (a) S. M. Hanson, E. V. Morlock, K. A. Satyshur and C. Czajkowski, J. Med. Chem., 2008, 51, 7243 CrossRef CAS; (b) S. Kang, R. Y. Kim, M. J. Seo, S. Lee, Y. M. Kim, M. Seo, J. J. Seo, Y. Ko, I. Choi, J. Jang, J. Nam, S. Park, H. Kang, H. J. Kim, J. Kim, S. Ahn, K. Pethe, K. Nam, Z. No and J. Kim, J. Med. Chem., 2014, 57, 5293 CrossRef CAS.
- L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305 CrossRef CAS.
- (a) H. G. Wang, Y. Wang, D. D. Liang, L. Y. Liu, J. C. Zhang and Q. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5677 Search PubMed; (b) D. Chandra Mohan, S. N. Rao and S. Adimurthy, J. Org. Chem., 2013, 78, 1266 CrossRef CAS; (c) H. Cao, X. Liu, J. Liao, J. Huang, H. Qiu, Q. Chen and Y. Chen, J. Org. Chem., 2014, 79, 11209 CrossRef CAS; (d) H. Zhan, L. Zhao, J. Liao, N. Li, Q. Chen, S. Qiu and H. Cao, Adv. Synth. Catal., 2015, 357, 46 CrossRef CAS.
- L. J. Ma, X. P. Wang, W. Yu and B. Han, Chem. Commun., 2011, 47, 11333 RSC.
- R. L. Yan, H. Yan, C. Ma, Z. Y. Ren, X. A. Gao, G. S. Huang and Y. M. Liang, J. Org. Chem., 2012, 77, 2024 CrossRef CAS.
- N. Chernyak and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2743 CrossRef CAS PubMed.
- C. He, J. Hao, H. Xu, Y. Mo, H. Liu, J. Han and A. Lei, Chem. Commun., 2012, 48, 11073 RSC.
- (a) J. Barluenga, M. Marco-Arias, F. Gonzalez-Bobes, A. Ballesteros and J. M. Gonzalez, Chem. Commun., 2004, 2616 RSC; (b) J. Barluenga, M. MarcoArias, F. Gonzalez Bobes, A. Ballesteros and J. M. Gonzalez, Chem.–Eur. J., 2004, 10, 1677 CrossRef CAS; (c) J.-S. Tian, K. W. J. Ng, J.-R. Wong and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 9105 CrossRef CAS; (d) T. Nobuta, N. Tada, A. Fujiya, A. Kariya, T. Miura and A. Itoh, Org. Lett., 2013, 15, 574 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10118c |
| ‡ L. L. Huang and W. Q. Yin contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |