
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceParahydrogen based NMR hyperpolarisation goes micro: an alveolus for small molecule chemosensing†
Lorenzo
Bordonali
 ,
Nurdiana
Nordin
,
Erwin
Fuhrer
,
Neil
MacKinnon
and
Jan G.
Korvink
,
Nurdiana
Nordin
,
Erwin
Fuhrer
,
Neil
MacKinnon
and
Jan G.
Korvink
Institute for Microtechnology, Karlsruhe Institute for Technology, Hermann-von-Helmholtz Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 7th January 2019
Abstract
Complex mixtures, commonly encountered in metabolomics and food analytics, are now routinely measured by nuclear magnetic resonance (NMR) spectroscopy. Since many samples must be measured, one-dimensional proton (1D 1H) spectroscopy is the experiment of choice. A common challenge in complex mixture 1H NMR spectroscopy is spectral crowding, which limits the assignment of molecular components to those molecules in relatively high abundance. This limitation is exacerbated when the sample quantity itself is limited and concentrations are reduced even further during sample preparation for routine measurement. To address these challenges, we report a novel microfluidic NMR platform integrating signal enhancement via parahydrogen induced hyperpolarisation. The platform simultaneously addresses the challenges of handling small sample quantities through microfluidics, the associated decrease in signal given the reduced sample quantity by Signal Amplification by Reversible Exchange (SABRE), and overcoming spectral crowding by taking advantage of the chemosensing aspect of the SABRE effect. SABRE at the microscale is enabled by an integrated PDMS membrane alveolus, which provides bubble-free hydrogen gas contact with the sample solution. With this platform, we demonstrate high field NMR chemosensing of microliter sample volumes, nanoliter detection volumes, and micromolar concentrations corresponding to picomole molecular sensitivity.
1 Introduction
Identification of the molecular constituency of complex mixtures at low concentrations continues to be an important challenge. Nuclear magnetic resonance (NMR) is one of the preferred analytical methods since it is able to handle the large concentration range, the complexity of the physico-chemical properties and structural diversity of the molecules, and is quantitative. Where NMR must improve is in sensitivity, to reach a concentration limit of detection on the order of low μM for reasonable measurement times, and spectral crowding, with all 1H signals appearing in the relatively small chemical shift dispersion of 12 ppm.NMR detector miniaturisation is considered an effective design strategy to significantly decrease the limit of detection for NMR. The limit of detection is inversely proportional to the NMR sensitivity; for common geometries like solenoidal, saddle or Helmholtz coils, sensitivity itself scales inversely with the coil radius1,2 so that there usually is a net gain in sensitivity by reducing the size of the detector. NMR micro-coils permit the detection of NMR signals from very limited amounts of matter in the nanoliter range, and they lend themselves to be integrated in microfluidic handling systems, thus enabling on-chip NMR experiments.3–5
Micro-detectors have improved mass sensitivity, but still suffer in terms of concentration sensitivity and thus many molecules of interest escape detection. In this case, one way to further increase sensitivity is to resort to signal enhancement techniques such as dynamic nuclear polarisation (DNP),6 chemically induced DNP (CIDNP),7 optically hyperpolarised 129Xe (ref. 8) or para-hydrogen induced polarisation (PHIP).9 These methods provide access to levels of polarisation far beyond what is usually achievable with magnets alone.
para-Hydrogen (p-H2) induced polarisation in particular has revived applications in magnetic fields comparable to the Earth's magnetic field and lower (down to zero-field), enabling high resolution NMR spectroscopy without the need for polarising magnets.10 Hydrogenative PHIP methods rely on the pairwise nuclear spin transfer to an unsaturated molecule during a catalytic hydrogenation reaction where parahydrogen is used. The two protons formerly in the p-H2 molecule provide the spin order reservoir to build up a huge non-equilibrium nuclear spin population in the hydrogenated molecule. A non-hydrogenative sub-class of PHIP has been introduced during the past decade,11 the so-called signal amplification by reversible exchange effect (SABRE). SABRE relies on polarisation transfer during the momentary and reversible contact of a p-H2 molecule (the source of spin alignment), an iridium-containing catalyst, and a molecule of interest or substrate (here also referred to as analyte). This adds the most welcome opportunity for continuous hyperpolarisation to NMR measurements.12,13
Additionally, SABRE can be exploited as a selective chemosensing tool. During the SABRE sequence, enhanced di-hydride signals form in the far-shielded regions of the spectrum (∼−21 ppm). The chemical shifts of the hydride peaks are intimately related to the unique long-range scalar coupling constants between the two protons from p-H2 and the protons of the substrate bound in the catalytic complex. The nature of the scalar coupling network is strongly dependent on the molecular structure; therefore, it is possible to quantitatively discriminate the components of a complex mixture by observing enhanced chemically-shifted di-hydritic doublets instead of the full, non- or weakly-enhanced and often overcrowded NMR spectrum. Importantly, the spectral complexity is simplified since signals are only observed for molecules capable of interaction with the SABRE catalyst. SABRE's chemosensing abilities have already been demonstrated while profiling aromatic species in complex artificial mixtures and natural extracts (e.g. coffee) at low micromolar concentrations.14,15
While SABRE specifically has so far been applied in the realm of high-volume NMR spectroscopy and imaging, other forms of NMR hyperpolarisation have already been proven beneficial in micro-scale contexts. For instance, Jiménez-Martínez et al. designed an on-chip hyperpolarised Xenon NMR system,16 Causier et al. presented a 3D-printed polariser to address hyperpolarisation with different gas species using a hand-wound microcoil,17 Bouchard et al. exploited NMR to visualize chemical processes in catalytic microreactors.18 Recently, Lehmkuhl et al. demonstrated an in-line flow setup for SABRE on a bench-top NMR spectrometer based on a 3D printed membrane reactor19 while Mompeán et al. presented a photo-CIDNP NMR probe for limited-volume samples by leveraging the advantages of microcoils.20
The design of a SABRE platform that integrates the hyperpolarisation step, the fluidics handling system and the NMR detection hardware is constrained mainly by physical factors, such as the magnetic environment required to facilitate the polarisation transfer, the gas pressure needed for high enhancement factors, and the generally fast relaxation times of the nuclear magnetisation. With a few exceptions,21–23 the polarisation step is traditionally carried out at low fields, e.g. in the stray field region of the NMR magnet, or in a dedicated low-field polariser.24–26 When implementing a compact hyperpolarisation unit, one is compelled to carry out the hyper-polarisation step inside the magnet itself, in or close to the magnetic iso-center where the magnetic field is most homogeneous. PHIP is theoretically able to yield nuclear polarisations of 100%, i.e. an enhancement factor ε of the order of ε ≃ 105 within a strong static magnetic field and under ideal conditions;27,28 on the contrary, the SABRE experiment has been known to be much less effective at high field (ε ≃ 10 or less),29 although recent advances have pushed this limit to much higher values (ε > 103).22,23,30
In this contribution we present an NMR platform for in-field SABRE hyperpolarisation featuring a straightforward p-H2 micro-polariser. The system is based on a micro coil chip fabricated with MEMS-technology5 and a PDMS membrane liquid–gas contactor, or ‘alveolus’. We provide proof-of-concept evidence that the system is able to detect the hydritic signals necessary for chemosensing, achieving performances comparable to the bulk experiments. Additionally, we demonstrate chemosensing of strongly diluted solutions of SABRE-active analytes using the presented system, with picomole material quantities detected.
2 Results
In the following section, we describe the technical details behind the design and use of the experimental setup, as well as the result of the test SABRE experiments carried out to evaluate the efficacy of the setup.A detailed drawing of the NMR probe head is displayed in Fig. 1. The polariser probe head consists of three functional elements: 1) a PDMS membrane liquid–gas contacting element (polariser) to deliver the enriched p-H2 gas mixture to the sample; 2) a copper printed circuit board supporting both the NMR micro coil chip and the pre-tuning and matching circuit; 3) a sample collecting chamber valleyside of the NMR detector. The frame supporting the NMR sensor and the polariser was machined from acrylic polymer. The complete probe head design had to satisfy two major constraints: the NMR coil needed to reside in the high-homogeneity region of the static magnetic field, and the support frame would have to fit inside a micro-gradient sleeve.
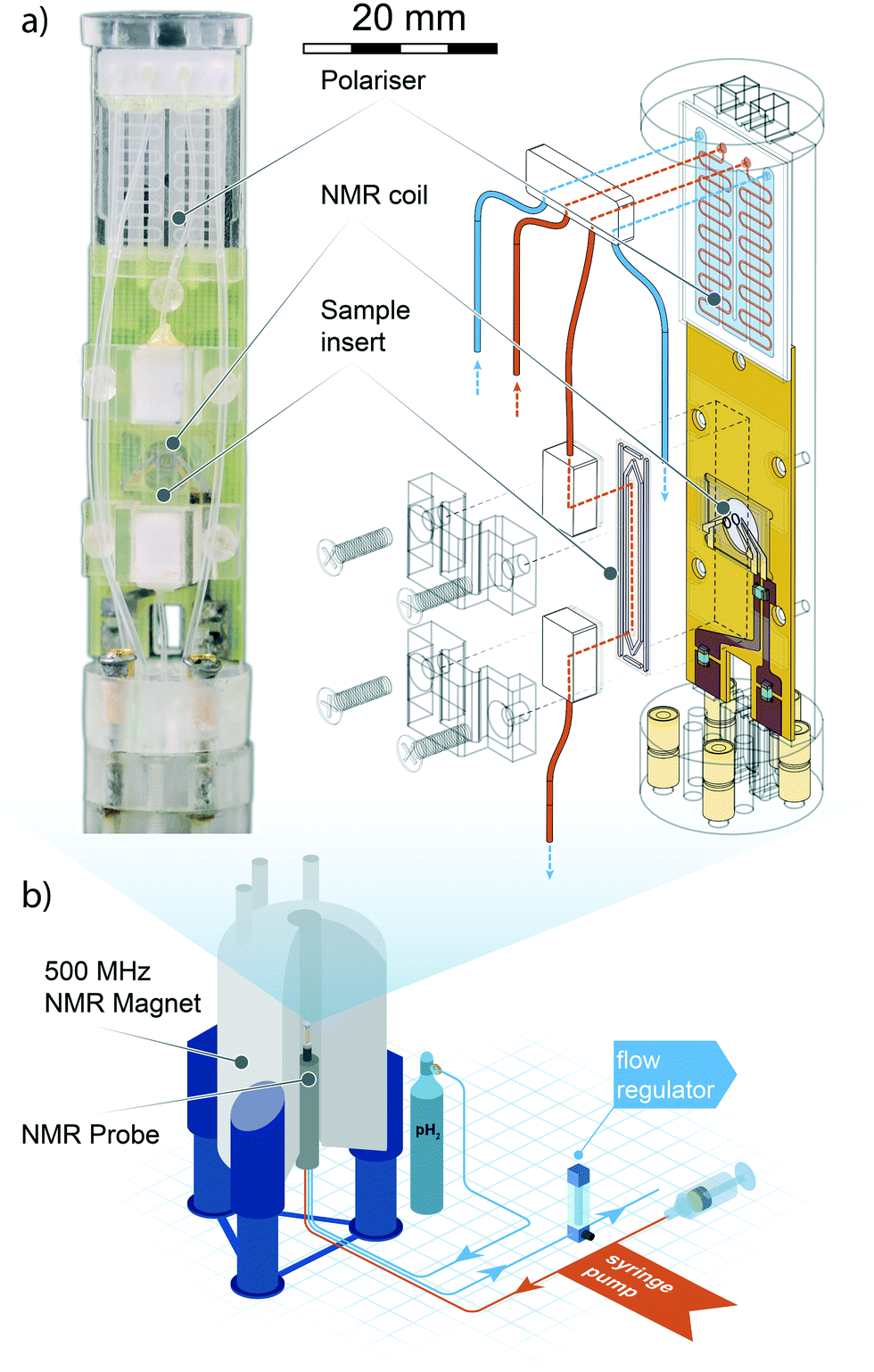 | ||
| Fig. 1 Top left: Photograph of the micro-SABRE platform mounted on the head of the NMR probe. Top right: Schematic drawing displaying the main components of the micro-NMR platform. Red lines indicate gas flow paths, blue lines are fluidic flow paths. The meandering channel gas–liquid contactor visible at the upper extremity of the platform is described in Fig. 2. Bottom: An overview of the main components of the experimental layout. The gas and solution are pneumatically transported through the NMR probe mounted in an 11.74 T NMR magnet, where the custom probe head insert performs the SABRE experiment. Gas flow is regulated valley-side of the NMR probe to ensure proper pressure equilibration throughout the system. The sample solutions are injected with a standard syringe pump; any excess fluid running out of the detection area is collected in a spill-out chamber inside the NMR probe body. The area of the helmholtz pair is 1.13 mm2, the detection volume enclosed by the coil is 0.56 μL. For a close-up view of the NMR detector refer to section 3 of ESI.† | ||
The electronic circuit consists of a symmetric series C–(L‖C)–C configuration where the capacitor in the L‖C circuit is used to tune the probehead to the 1H NMR frequency, while matching to 50 Ω is achieved with fixed capacitors connected to the 1H channel connector and ground. The NMR coil used in this work is a microfabricated 1.2 mm diameter Helmoltz coil pair encased in a glass chip. Fabrication details of have been reported elsewhere.5 In section 5 we provide a brief summary of the fabrication process.
2.1 PDMS membrane gas–liquid contactor
The active core of the setup consists of a two-chamber micro-contactor (in the following also referred to as polariser) composed of two meandering channels carved in a glass substrate and separated by a polydimetilsiloxane (PDMS) membrane. The gas side of the contactor features a 180 nm long channel whose width was maximised to the value of 210 μm to achieve wide surface coverage of the flexible and permeable membrane. A meandering geometry was chosen to ensure an homogeneous flow of p-H2 gas. On the liquid side, a l = 180 mm long meandering channel with a (w × d) = 0.3 mm × 0.1 mm cross section, arranged orthogonal to the gas channel, guides the sample through the polariser. The total area available for the exchange of gaseous species is 38 mm2. Fabrication details are provided in section 5. A Teflon connector was tailor-cut to couple to the contactor inlets and outlets, secured in place by an adhesive layer, and further sealed along its perimeter with epoxy resin. The polariser is able to withstand an hydrodynamic pressure of about 5 bar, although above 2 bar the flexible PDMS membrane starts to bend and obstruct the flow of the sample solution. To ensure the structural integrity of the micro coil chip, the maximum working gas pressure was constrained to a safe value of 2 bar. The low working pressure relaxes the requirements for the tight-fit of connections between the various modules of the system; fluidic and pneumatic connections rely on thin polyethylene tubings, secured to the inlets and outlets of each module with epoxy resin. On all connections, the fittings were found to be able to withstand pressures of up to 3 bar without any gas or liquid leakage.2.2 Fluidic control
The part of the setup that resides outside of the NMR magnet is illustrated in Fig. 1b: the NMR probe is connected to the p-H2 gas cylinder by 1 mm inner diameter PE tubings that plug directly into the inlet gas connectors on the bottom of the probe head's base. An additional pair of tubings connects a syringe pump to the polariser fluidic inlet and the outlet of the spillover chamber to the outside of the magnet. p-H2 gas pressure can be applied to the venting outlet of the spillover chamber to greatly reduce the absolute pressure differential between the liquid phase and the gas phase in the polariser. This operation prevents the soft PDMS membrane from overly flexing and blocking the microchannel. Sample flow was regulated between 20 and 300 μL min−1.2.3 Micro-SABRE
High-field SABRE experiments were performed on a fully deuterated methanol (Me-d4) mixture containing two common benchmark substrates (pyridine and nicotinamide) and a third component referred to as ‘co-substrate’, 1-methyl-1,2,3-triazole (mtz). The SABRE process is schematically depicted in Fig. 2: a nitrogen-containing molecule and a p-H2 molecule reversibly bind to the metal center of an Ir-containing catalyst. During the short coordination time, singlet spin order of the para-hydrogen molecule is converted into detectable nuclear magnetic polarisation due to symmetry breaking. In a favourable magnetic environment, usually at static field strengths of about 5 mT, polarisation flows through the temporary spin network formed by exchange-coupled spins in the catalytic complex. The end result is a hyperpolarised substrate molecule that eventually detaches from the complex. The process is thus reversible and can be repeated indefinitely, on the condition that the catalyst remains in its active form.31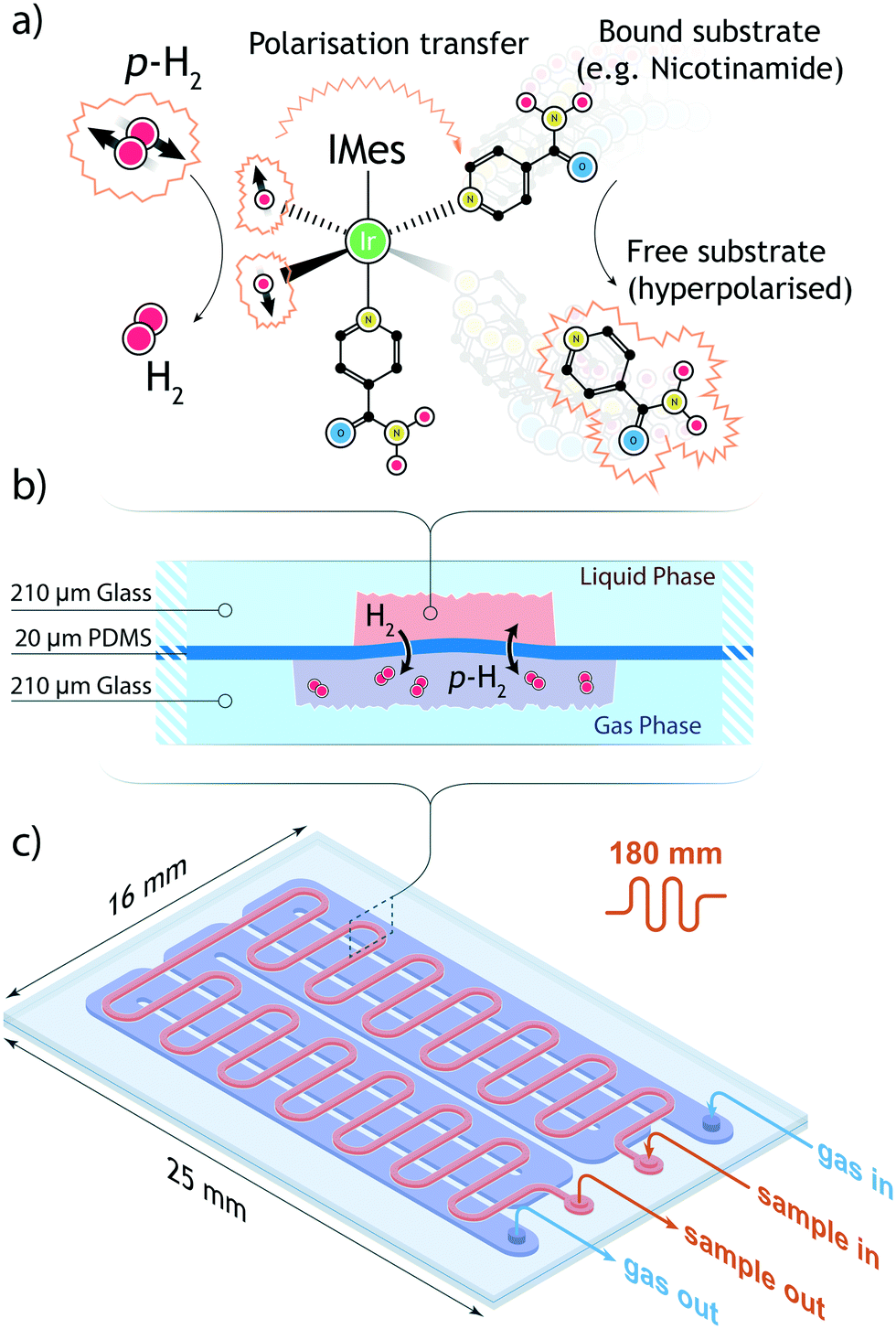 | ||
| Fig. 2 (a) Schematic of the SABRE process. A p-H2 molecule coordinates to an iridium-centered catalyst. Each hydride then has a distinct, enhanced signal in the 1H NMR spectrum. In the presence of a coordinated substrate, the chemical shift of the hydride is slightly modified. Simultaneously, spin-order is transferred through the coupling network from the p-H2 to the target molecule. A hyperpolarised substrate and an o-H2 molecule are released from the complex. (b) Schematic of the gas–liquid contact channel (c) schematic of the meandering channel for improved contact area. On the gas side, the total channel length is 180 mm while the enclosed volume is 4.8 μL. On the gas side, the fluidic path is 120 mm long and the channel volume is 20.2 μL. The total area available for gas exchange is 45 mm2. | ||
In a high magnetic field the nuclear spins do not satisfy the strong coupling condition as experienced in a low field environment. In this case, SABRE happens via spin cross-relaxation and is much less efficient than its low-field counterpart. The feasibility of SABRE at high magnetic field has been demonstrated and a maximum enhancement of ∼3.09 was found for one of the three pyridine peaks (at 8.5 ppm), where the effect is more pronounced.29
Therefore, rather than focusing on the weak enhancements of substrate proton signals, we also monitored the formation of the dihydritic signals at far-removed positions in the proton NMR spectrum. The emergence of strong dihydride signals reveals that a buildup of usable polarisation forms inside the polariser chamber, a hallmark of the high-field SABRE phenomenon, under the condition that an excess of co-substrate mtz is maintained in the solution.32 The presence of co-substrate at much higher concentrations with respect to both the catalyst and the analytes has the function of moderating the association–deassociation rates of the analytes to the iridium centre, to the point where the free-to-bound substrate ratio is determined by the concentration of co-substrate only. Therefore, a linear dependence develops between enhanced signal amplitudes and substrate concentration.
Optimal transfer of polarisation to nuclear species of the substrate is possible but requires implementation of advanced techniques and lies outside the focus of this paper. Nonetheless, an interesting feature of the hydride signal is its close relationship to the particular substrate that coordinates to the catalytic complex, whereby each molecule generates a dihydride peak at a distinct chemical shift14 in the negative chemical shift region [−21, −24] ppm.
The SABRE experiment was first carried out on a Micro-5 Bruker NMR probe head, by preparing a high concentration test solution inside a 5 NMR tube. Capillaries were inserted through the mounted PTFE cap, to provide an inlet and outlet for p-H2 gas. The assembled probe head was placed inside a wide-bore 11.74 Tesla magnet (500 MHz 1H frequency). The sample solution was thoroughly bubbled in situ with fresh p-H2 at a flow rate of 0.5 L min−1 for 20 s prior to acquisition. The NMR measurement was set up as a standard π/2-pulse FID reading sequence; an optimal flip angle was found by running a nutation experiment at fixed power (35 W for a Bruker 5 mm saddle coil and 1 W for the micro-Helmholtz setup) and varying pulse lengths.
The NMR spectrum of the sample is shown in Fig. 3a. The maximum observed enhancement factors for the analyte 1H signals, calculated as the ratio between the peak integrals of the hyperpolarised signal over the thermal equilibrium signal, are: ∼4-fold for nicotinamide peaks, ∼1.2-fold for pyridine and ∼4.6-fold for mtz. On the negative side of the frequency axis, sharp non-overlapping di-hydride peaks appeared at distinct positions: pyridine at −22.2 ppm (pyX) and −22.9 ppm (pyA), nicotinamide at −21.9 ppm (NAX) and −23.1 ppm (NAA). These signals only appear after the solution is loaded with fresh p-H2 and have a longitudinal relaxation time (T1) of about 20 s. It is important to note that at the concentrations tested in this experiment the relative intensities of the recorded peaks do not reflect the molar fractions of the associated molecules in the test solution.
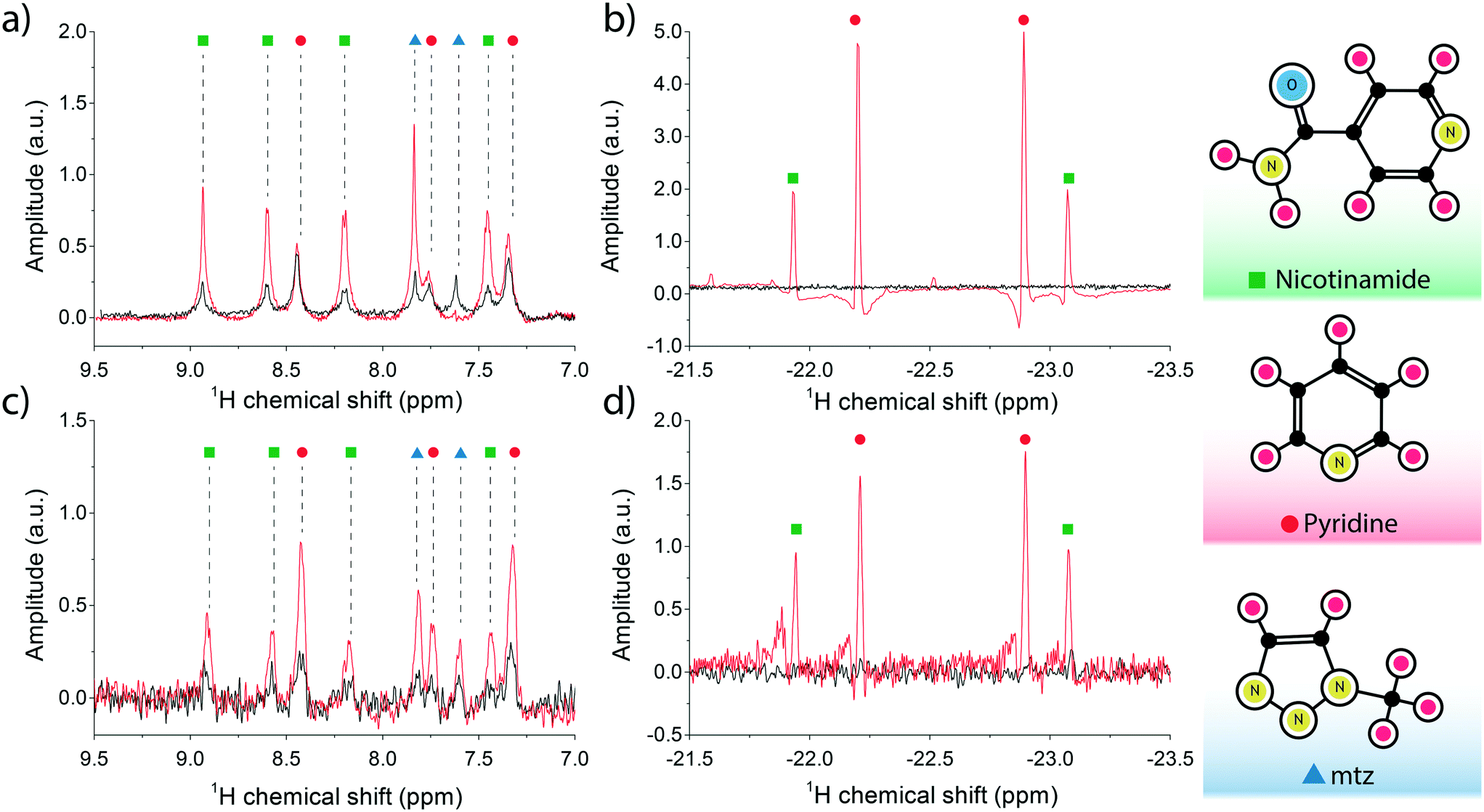 | ||
| Fig. 3 (a and b) Single acquisition 1H spectra of the SABRE mixture (25 mM pyridine, 25 mM nicotinamide, 26 mM mtz, 2.4 mM Ir-IMes catalyst) performed at B = 11.7 T and T = 30 °C. The SABRE measurement (in red) was recorded immediately after thoroughly bubbling the sample solution with a 90% p-H2 enriched mixture for 20 s. The reference measurement at thermal equilibrium is plotted in black. The analyte signal peaks are shown on the left in (a), and the di-hydritic region to the right in (b). (c and d) 16-fold averaged 1H spectra for the high-field SABRE experiment performed at B = 11.7 T and T = 30 °C and under active microflow conditions. The flow rate was set at the optimal value of 60 μL min−1. The reference measurement, in black, was recorded at rest. Peak assignment follows the colour scheme reported on the right edge of the figure, i.e. nicotinamide peaks are labelled with green dots, pyridine with red, and mtz with blue in plots (a) through (d). | ||
The SABRE experiment was then replicated on an identically prepared solution using the micro-coil and micro-polariser system under active flow conditions, at a constant flow rate of 60 μL min−1. Fig. 3b reports the results of the high field SABRE experiment compared to the signal at thermal equilibrium. Both thermal and hyperpolarised signals were averaged 16-fold. In this case we observed a ∼2.5-fold maximum 1H signal enhancement for nicotinamide, ∼3.8-fold for pyridine, and ∼4.3-fold for mtz. Di-hydritic peaks associated to the three SABRE-active molecules emerged in the far negative frequency region of the spectrum at the same positions as the bulk SABRE experiments. Importantly, the relative intensities of the signals in this region perfectly match the experiment in bulk, whereas the same correlation cannot be drawn for signals from analyte protons of hyperpolarised non-bound molecules when comparing bulk measurements (Fig. 3a) and micro-NMR measurements (Fig. 3b).
2.4 SABRE-driven chemosensing
To bring the system into the chemosensing regime, the catalyst and co-substrate must be present in solution at much higher concentrations than the target analytes. Therefore, a concentration series was prepared for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pyridine:nicotinamine mixtures in the range 50–250 μM with steps of 50 μL. SABRE experiments on the bulk solutions were first implemented on the Bruker probe by bubbling p-H2 at atmospheric pressure for 20 s, recording for each sample a single scan immediately after arresting the p-H2 gas flow. The five solutions were successively flowed through the micro-polariser at 1 bar p-H2 pressure and constant flow rate of 40 μL min−1, to ensure both maximum signal enhancement and maximum available time for signal averaging. For each sample solution, a total of 128 averages at 1 s intervals were found sufficient to bring the signal above the background noise to a level comparable to the single scan bulk measurements.
:1 pyridine:nicotinamine mixtures in the range 50–250 μM with steps of 50 μL. SABRE experiments on the bulk solutions were first implemented on the Bruker probe by bubbling p-H2 at atmospheric pressure for 20 s, recording for each sample a single scan immediately after arresting the p-H2 gas flow. The five solutions were successively flowed through the micro-polariser at 1 bar p-H2 pressure and constant flow rate of 40 μL min−1, to ensure both maximum signal enhancement and maximum available time for signal averaging. For each sample solution, a total of 128 averages at 1 s intervals were found sufficient to bring the signal above the background noise to a level comparable to the single scan bulk measurements.
Fig. 4 reports the hydritic region of the proton NMR spectrum for chemosensing experiments in bulk (Fig. 4a) and on the micro-system (Fig. 4c). Three clearly distinguishable hydride peaks in the spectra have been integrated and their value plotted against the known molecular concentration in Fig. 4b) and d) to highlight the linear relationship between the two quantities. The leftmost peak associated with nicotinamide (NAX) was not quantified due to an overlap with an antiphase peak, possibly originating from the equivalent hydrides in the symmetric complex [Ir(IMes)(H)2(mtz)3]Cl, which is the most abundant configuration at very low analytes concentrations.
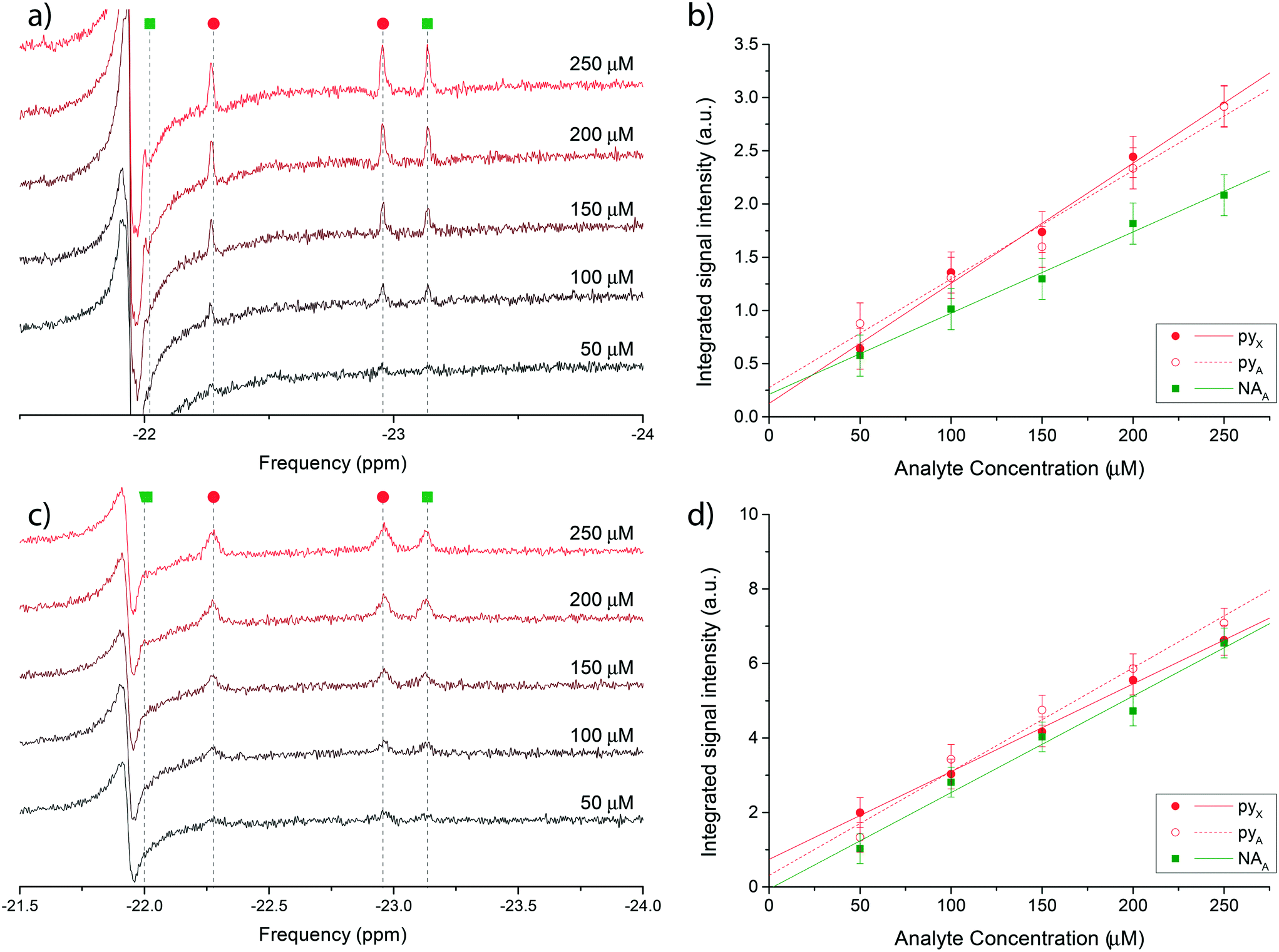 | ||
| Fig. 4 a) Single scan 1H @500 MHz NMR spectra of a nicotinamide and pyridine concentration series ranging from 50 μM to 250 μM, performed in bulk on a Bruker Micro-5 NMR probe. Only the di-hydritic region is displayed. c) 128-averaged spectra of the concentration series recorded on the micro-NMR setup. Peak assignment in (a) and (c) follows the colour scheme reported in Fig. 3. (b and d) Corresponding signal integrals of di-hydride peaks in plots (a) and (c) as a function of nominal nicotinamide and pyridine concentrations. The X hydride for nicotinamide was not quantified due to the overlap with a constant antiphase signal in the same region. The mass sensitivity range corresponding to the concentration range covered by the experiments is 15–75 pmol. | ||
Uncertainties on the integrated signal intensity were assigned based on the background noise level, propagating the error through the integration routine. Linear regressions of the signal integrals are also shown in Fig. 4b) and d) to check for the zero crossing at null concentration. The intercept at null concentration was found to be within the experimental error.
3 Discussion
The presented system was specifically designed to enable repolarisation of the sample, which is the most useful characteristic of the SABRE experiment and a net advantage over hydrogenative hyperpolarisation techniques. By virtue of this property, the NMR sequence can be run for an indefinitely long time, provided that re-circulation of the sample is implemented, for instance by using a peristaltic pump on a closed fluidic circuit.The avoidance of strong turbulent gas flows, a common characteristic of bubbling polarisers, solves the issue of evaporation of the volatile solvents, reduces the delay between complete polarisation of the sample and NMR acquisition, and eliminates the magnetic susceptibility mismatch problem at gas–liquid interfaces. Control over the stoichiometry of the sample solution is preserved over many iterations of the experiment, improving reproducibility of measurements.33
Currently, the main source of signal loss is caused by the finite amount of time required for the sample to reach the NMR detector once leaving the polariser chamber. The necessity for an optimized time of flight has been recently highlighted for hyperpolarised gases undergoing a parahydrogen-driven enhancement under constant flow conditions in tightly packed catalytic beds.34 The same phenomenon is clearly visible in the experiments performed with the presented setup and, as discussed next, it is largely due to the interplay between polarisation buildup inside the contactor unit and T1 relaxation during transport to the detector. Fig. 5 reports polarisation levels as a function of sample flow through the system and illustrates the issue: if the flow rate is lower than a certain threshold, much of the polarisation generated in the contactor module is lost via nuclear-lattice T1 relaxation while sample volume elements flow towards the detector area. Conversely, if the flow rate is much higher than the threshold, polarisation buildup is hindered by the low residence time of the sample in the meandering channel. An optimal polarisation level is achieved by restricting the flow rate between these two regimes. In our specific case, this condition corresponds to flow rates in the range 40–80 μL min−1.
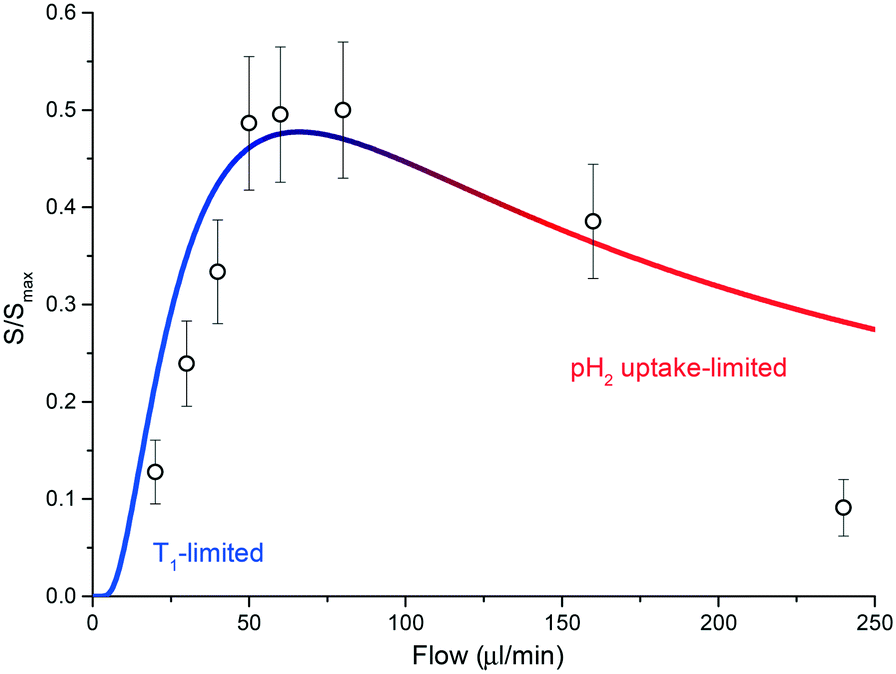 | ||
| Fig. 5 A plot of eqn (1) for T1 = 50 s and tb = 3.5 s. Geometric parameters entering the equation are as declared in the main text. The T1 limited and gas-uptake limited regimes are highlighted in blue and red, respectively. The integral of the signal for the full di-hydritic spectral region, as a function of sample flow, is also reported. Values have been uniformly scaled so that the maximum value (at 60 μL min−1) matches the ratio between the referenced hydride signal integrals from the bulk sample SABRE measurement (Fig. 3a) and the alveolus SABRE measurement (Fig. 3b), which is about 50%. | ||
Diffusion of hydrogen gas through a 20 μm PDMS porous membrane is enhanced by an applied pressure of 2 bar. The diffusion process continues inside the methanol solution, having a diffusion coefficient DH/Me-d4 = 17.18 × 10−5 cm2 s−1 for hydrogen molecules.35 Under these conditions, the mean transverse time necessary for the gas molecules to completely permeate the solution is td ≃ d2/(2DH/Me-d4) ∼ 300 ms, for a channel height of d = 100 μm;36 therefore, the maximum volumetric flow rate that allows for saturation of a fluidic element in the contactor can be roughly estimated as fmax = ldw/td ≃ 1.1 mL min−1. As reported in Fig. 5, SABRE efficiency starts to decrease at much smaller values than fmax, signifying that td is not the limiting time scale for the process. In other words, SABRE in methanol is not diffusion-limited. This hypothesis is supported by the fact that signal buildup time tb for SABRE, i.e. the time required for saturation of the hyperpolarised signal, is of the order of 1–10 s in high magnetic field environments.37
In order for polarisation to build up, the residence time for the sample solution inside the polariser has to be comparable to tb. Considering the concerted effects of polarisation buildup and relaxation, the available hyperpolarised signal intensity S at the detector position as a function of the flow rate f can be expressed as:
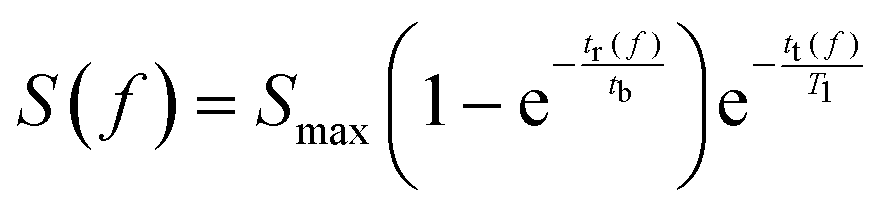 | (1) |
At about 60 μL min−1, the residence time is ∼5.5 s and the transfer time to the detector is ∼8.5 s. We measured a long average T1 time for the di-hydride peaks, of the order of 50 s, while the signal build-up time constant was found to be ∼3.5 s.
Usually, to ensure continuous hyperpolarisation, a dynamic equilibrium has to emerge between the uptake of fresh, highly enriched parahydrogen gas and the removal of hydrogen molecules that participated in SABRE and had lost their singlet character, converting back into ortho-hydrogen. In our case T1 ≫ tb, therefore this otherwise cumbersome requirement is greatly relaxed. Nontheless, the presence of leftover ‘exhausted’ hydrogen molecules in the sample solution can negatively affect the net equilibrium polarisation.
Once inside the sample insert, the fraction of sample that is actually irradiated and detected by the coil depends on the NMR measurement time. The two expansion of the Helmholtz pair, separated by a 550 μm-wide gap, define a total detection volume of about 620 nL. However, since the sample insert channel is 270 μm, the irradiated sample volume is only about 300 nL. Excitation time (the pulse width) is of the order of 10 μs and at this time scale the fluidic element barely moves inside the detection area. The acquisition time for a single scan during an active flow of 60 μL min−1 is about 50 ms. During this time, the excited sample moves about 90 μm along the channel direction. We can then safely assume that the detected signal is coming from a large portion of the irradiated volume, i.e. about 90%.
From eqn (1) we estimated that only about S(60 μL min−1)/Smax ∼ 47% of the polarised signal is available for detection after the sample shuttling step. The calculated value is in quantitative agreement with the decrease in signal when the SABRE routine is run through the alveolus with respect to the sample bulk measurement. A relative decrease of about 50% in the hydride signal was observed.
Fast transport times of the order of a few milliseconds can be achieved by minimising the sample transfer distance, at the cost of requiring a longer path for thorough saturation of the hyperpolarised signal. Realistically, the dead time between polarisation and detection could be completely eliminated by performing the p-H2 diffusion directly inside the detection region. However, additional design elements in or around the sample space would likely deteriorate the quality of the NMR signal because of magnetic susceptibility discontinuities, as confirmed by preliminary tests. Large in situ polarisers are not affected by this inconvenience because the detection volume is large compared to that of the active element of the polariser (e.g., a capillary tubing).12,33 For this reason, we believe that integration of on-chip polarisers will mostly rely on in-line, rather than stacked, designs.
The correct choice of the materials used, as well as the appropriate measures to prevent de-gassing of the solution and the formation of bubbles within the NMR-detector window, are essential prerequisites to avoid magnetic susceptibility mismatch artifacts in the NMR signal. A further source of issues comes from chemical stability of the materials when exposed to harsh chemicals such as methanol and other aggressive organic solvents. The combination of glass and PDMS as the main building materials for fabrication solves both problems at once. The use of photoresists (such as Ordyl) and adhesives that are not as chemically resistant as glass and PDMS did not seem to compromise the stability of the fabricated sample inserts and micro-polariser. The use of relatively large amounts of polymeric materials came at the cost of additional broad background signals in the NMR spectra. However, the solid contributions to the NMR signal are constant and can be simply stored and subtracted to achieve clean spectra.
A further advantage of our system is the possibility of using the sample inserts as consumables, which is useful in those cases where sample purity is of paramount importance, e.g., when small concentrations of the target molecule need to be recorded. Sample inserts can be mass produced at reduced costs while the storage and collecting chambers can be easily cleaned and reused. This modularity allows one to experiment with variations on the polarisation schemes without compromising the complete setup.
The test measurements presented here constitute an attempt to reproduce the results of ref. 29 and 14 in an NMR micro system. Despite the low enhancement observed for the analyte NMR signals, we were able to replicate the results of the classical high field experiments while only sampling a fraction of 0.0001 of the volume with respect to the bulk solution measurement, namely ∼300 nL instead of 1 mL. In addition, chemosensing capabilities have been demonstrated by achieving sensing linearities at micromolar concentrations, a regime at which both the bulk measurement devices and micro-NMR hardware often struggle to provide sufficient signal intensities to overcome background noise. Given the 300 nL detection volume and 50 μM analyte concentration, the platform proved capable of detecting 15 pmol of material.
4 Conclusions
The positive results reported in our work encourages further developments to integrate PHIP polarisers with NMR micro-detectors.Improvements over the current results can be obtained by implementing a modification of the SABRE experiment with polarisation transfer NMR sequences, if necessary.
The complexity of the fluidic network could also be increased to automate on-demand titration of analytes for chemosensing purposes and to further optimize p-H2 gas delivery. We envision a future version of the probe head where micro-reactor technology will be exploited to design an efficient p-H2-liquid contactor by accounting for high-pressure microfluidics, whereby higher amounts of p-H2 gas can be dissolved in the target solution.
Compared to other hyperpolarisation strategies, parahydrogen-based signal enhancement techniques such as SABRE are well-suited for lab-on-a-chip applications. Compared to DNP, for example, the problem of dealing with the generation and transport of microwaves is avoided. No thermal cycle is necessary to hyperpolarize the sample, unlike in dissolution DNP, which greatly reduces the technical challenges associated with refrigeration and dissolution.
As the classes of SABRE-active molecules increase in number, due to significant contributions such as LIGHT-SABRE,30 SABRE-SHEATH,38 and SABRE-Relay,39 and with the introduction of water-soluble SABRE catalysts,40,41 the implementation of continuous hyperpolarisaton methodologies becomes increasingly attractive for applications in chemical analysis.
We therefore anticipate that the integration of a hyperpolarised NMR micro detector in organ-on-a-chip and body-on-a-chip systems42,43 will open new horizons in the field of systems biology and metabolomics.13
5 Experimental
5.1 Sample preparation
Prior to the experiments, p-H2 with a purity of >95% was prepared from commercial H2 with a custom parahydrogen generator (described elsewhere31) and stored in a 0.5 L gas bottle at 35 bar. A batch of pre-catalyst solution was prepared by dissolving 15.4 mg of Ir-IMes in 5 mL of fully deuterated methanol (Me-d4, Sigma Aldrich 151947, CAS 811-98-3). 30 mg of 1-methyl-1,2,3-triazole (mtz, ChemPur GmbH BD59300, 99.999%, CAS 16681-65-5) were added to achieve a 15:1 mtz:catalyst concentration ratio (72 mM mtz, 4.8 mM Ir-IMes). 100 mM and 1 mM stock solutions of pyridine (Sigma Aldrich 270970, CAS 110-86-1) and nicotinamide (Sigma Aldrich 72340, CAS 98-92-0) were prepared separately. For the highly-enhanced SABRE experiment of Fig. 3, 500 μL of catalytic solution was transferred into an NMR pressure glass tube (Wilmad quick pressure valve 5 mm, 500 MHz) and flushed with p-H2 gas pressurised at 5 bar, shaken vigorously and left to rest for about one hour to allow for activation of the Ir-IMes pre-catalyst. 250 μL of 100 mM pyridine solution and 250 μL 100 mM nicotinamide solution were then added to the mixture. Molar concentrations for the prepared test solution are as follows: 25 mM pyridine, 25 mM nicotinamide, 36 mM mtz, 2.4 mM activated Ir-IMes catalyst. To test the SABRE micro-polariser under chemosensing conditions, a series of five solutions with progressively increasing analyte concentrations were prepared by mixing 250 μL of activated catalyst solution with a 1:1 mixture of 1 mM pyridine and 1 mM nicotinamide in Me-d4, adding solvent to the target volume of 1 mL. 1:15:X catalyst:mtz:analyte mixtures with X = 0.003, 0.006, 0.009, 0.01, 0.013 were then obtained, for a corresponding analyte concentration series in the range 50–250 μM.
5.2 NMR micro-detector
Fabrication details of the MEMS NMR detector used in this work were reported in ref. 5. Briefly, the detector was of a 3 layer-stack design: the top and bottom layers featured Au traces on borosilicate glass substrates, connecting to a copper wire coil wound around a photoresist post of 1.2 mm diameter. Each half of the Helmholtz pair was encapsulated in resin. The third, middle layer (550 μm thick) was fabricated from glass chips and served as a spacer for the coil pair. Additionally, the spacer defined the sample insert cross-section (5 mm × 0.55 mm). The final chip size was 10 mm × 8 mm × 2 mm (W × L × H), and the sample insert size was 5 mm × 25 mm × 0.5 mm.5.3 Sample insert fabrication
A 100 mm diameter, 100 μm thick D263-T ECO Schott glass wafer substrate was cleaned using a sequence of acetone, isopropanol (IPA), and deionised water (DI) baths. The wafer was prepared for dry resist lamination by exposing the clean surface to oxygen plasma at 200 W RF power for 10 minutes. Three fresh layers of 90 μm-thick Ordyl dry resist were laminated onto the silicon substrate using a standard lamination device (Mylam 12, GMP) at 85 °C and roll speed 1 cm s−1 for a total nominal resist thickness of 270 μm. A 60 s post-lamination bake was performed at 85 °C on a hotplate. The Ordyl stack was then exposed to UV light for a total dose of 180 mJ cm−2. The Ordyl patterns were developed for 7 minutes in Ordyl developer under ultrasonic agitation. The developed structures were rinsed in IPA and DI water and dried under nitrogen flow. Finally, thermal bonding to a clean top glass wafer was performed with a hot embossing device (EVG 510HE) with an applied pressure of ∼6 bar, at a temperature of 95 °C for 30 minutes. Inlet and outlet holes were drilled into the top glass wafers before bonding, by machining the thin glass with a nanosecond UV laser (Trumpf TruMark 5000) pulsed at 30 kHz. The final thickness of the glass-Ordyl-glass sandwich was 460 μm, yielding an inner fluidic channel height of 260 μm. The diced chips were then further reinforced against inner pressure and solvent leakage by injecting polyurethane based resin through openings in the outer ordyl walls and between the glass slides to fully seal the space around the central fluidic channel (Figure in ESI†).5.4 PDMS membrane polariser fabrication
Patterns for the two meandering channels (gas and liquid sides of the polariser) were engraved on a 4-inch 210 μm thick square 263D-Eco Shott glass wafer using a nano-second UV laser beam (Trumpf TruMark 5000) pulsed at 30 kHz. Inlet and outlet holes were machined by letting the laser cut through the full glass layer. Dicing was also performed by laser-engraving along the perimeter of the polariser halves. The prepared halves were then cleaned in an acetone–IPA–DI water ultrasonic bath sequence, dried under pressurised nitrogen flow and baked at 200 °C for 2 hours.Meanwhile, a liquid PDMS elastomer solution (Dow Corning Sylgard 184) was prepared using the standard 10:1 elastomer-to-binding agent ratio. A 125 μm Kapton film sheet was cut into a 4-inch round shape, cleaned with isopropyl alcohol and dried under nitrogen gas flow. The Kapton film was vacuum-clamped on a spin-coater and 2.5 mL of PDMS solution was statically dispensed and spun at 2000 revolutions minute−1 for 5 minutes to achieve a uniform layer of 20 μm PDMS. The Kapton handling film was transferred to a hot plate and baked at 120 °C for 5 min to cure the silicon film.
The bottom half of the polariser (gas side) was prepared for bonding to the PDMS membrane by activating the contact glass surface in an oxygen plasma cleaner at 200 W RF power for 10 min. The PDMS was similarly activated by driving the plasma cleaner at 50 W RF power for 20 s. The Kapton-supported PDMS film was then immediately put in contact with the activated surface of the polariser half under a standard laminator (Mylam 12, GMP) at room temperature and roll speed of 1 cm s−1. After a waiting time of 10 min the handling polyimide layer was peeled off the bonded PDMS film. The exposed PDMS and the second glass half of the polariser (top half, liquid side) were then treated with oxygen plasma under the same conditions for PDMS and glass surface activation, respectively.
Finally, the top half was aligned to the bottom half under a microscope and brought into contact with the activated PDMS layer. The completed stack was placed on a hot plate set at 80 °C for 2 h under mechanical pressure, to further increase bond strength at the glass-PDMS interfaces.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
LB, NM, and JGK acknowledge initial support for this work by the ERC Project NMCEL 290586. LB acknowledges subsequent funding from the Carl Zeiss Stiftung. EF and JGK acknowledge additional support by the European Union's Future and Emerging Technologies Framework (H2020-FETOPEN-1-2016-2017-737043-TISuMR). NN and NM acknowledge funding from the Deutsche Forschungsgemeinschaft through the project Bio-PRICE (DFG MA 6653/1-1). N. N. wishes to thank the Ministry of Higher Education of Malaysia for the scholarship (SLAB Scheme) and the BioInterfaces International Graduate School (BIF-IGS, http://www.bif-igs.kit.edu). We thank the Institute of Biology (IBG) at KIT for access to their NMR instrument.References
- D. I. Hoult and R. E. Richards, J. Magn. Reson., 1969, 1976(24), 71–85 Search PubMed.
- T. L. Peck, R. L. Magin and P. C. Lauterbur, J. Magn. Reson., Ser. B, 1995, 108, 114–124 CrossRef CAS PubMed.
- K. Ehrmann, N. Saillen, F. Vincent, M. Stettler, M. Jordan, F. M. Wurm, P.-A. Besse and R. Popovic, Lab Chip, 2007, 7, 373–380 RSC.
- R. C. Meier, J. Höfflin, V. Badilita, U. Wallrabe and J. G. Korvink, J. Micromech. Microeng., 2014, 24, 45021 CrossRef.
- N. Spengler, J. Höfflin, A. Moazenzadeh, D. Mager, N. MacKinnon, V. Badilita, U. Wallrabe and J. G. Korvink, PLoS One, 2016, 11, 1–16 CrossRef PubMed.
- K. Hausser and D. Stehlik, Adv. Magn. Reson., 1968, 3, 79–139 CrossRef CAS.
- R. Kaptein, K. Dijkstra and K. Nicolay, Nature, 1978, 293–294 CrossRef CAS.
- A.-M. Oros and N. J. Shah, Phys. Med. Biol., 2004, 49, R105 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645 CrossRef CAS PubMed.
- T. Theis, P. Ganssle, G. Kervern, S. Knappe, J. Kitching, M. P. Ledbetter, D. Budker and A. Pines, Nat. Phys., 2011, 7, 571–575 Search PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1705–1708 CrossRef PubMed.
- J.-B. Hövener, N. Schwaderlapp, T. Lickert, S. B. Duckett, R. E. Mewis, L. A. R. Highton, S. M. Kenny, G. G. R. Green, D. Leibfritz and J. G. Korvink, Nat. Commun., 2013, 4, 2946 CrossRef PubMed.
- J.-B. Hövener, S. Knecht, N. Schwaderlapp, J. Hennig and D. von Elverfeldt, ChemPhysChem, 2014, 15, 2451–2457 CrossRef PubMed.
- N. K. J. Hermkens, N. Eshuis, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, Anal. Chem., 2016, 88(6), 3406–3412 CrossRef CAS PubMed.
- N. K. J. Hermkens, R. L. E. G. Aspers, M. C. Feiters, F. P. J. T. Rutjes and M. Tessari, Magn. Reson. Chem., 2017, 56, 633–640 CrossRef PubMed.
- R. Jiménez-Martínez, D. J. Kennedy, M. Rosenbluh, E. a. Donley, S. Knappe, S. J. Seltzer, H. L. Ring, V. S. Bajaj and J. Kitching, Nat. Commun., 2014, 5, 3908 CrossRef PubMed.
- A. Causier, G. Carret, C. Boutin, T. Berthelot and P. Berthault, Lab Chip, 2015, 15, 2049–2054 RSC.
- L.-S. Bouchard, S. R. Burt, M. S. Anwar, K. V. Kovtunov, I. V. Koptyug and A. Pines, Science, 2008, 319, 442–445 CrossRef CAS PubMed.
- S. Lehmkuhl, M. Wiese, L. Schubert, M. Held, M. Küppers, M. Wessling and B. Blümich, J. Magn. Reson., 2018, 291, 8–13 CrossRef CAS PubMed.
- M. Mompeán, R. M. Sánchez-donoso, A. D. Hoz, V. Saggiomo, A. H. Velders and M. V. Gomez, Nat. Commun., 2018, 9, 108 CrossRef PubMed.
- M. Haake, J. Natterer and J. Bargon, J. Am. Chem. Soc., 1996, 118, 8688–8691 CrossRef CAS.
- A. N. Pravdivtsev, A. V. Yurkovskaya, N. N. Lukzen, H.-M. Vieth and K. L. Ivanov, Phys. Chem. Chem. Phys., 2014, 16, 18707 RSC.
- N. Eshuis, R. L. E. G. Aspers, B. J. A. van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, Angew. Chem., Int. Ed., 2015, 14527–14530 CrossRef CAS PubMed.
- M. Goldman, H. Jóhannesson, O. Axelsson and M. Karlsson, C. R. Chim., 2006, 9, 357–363 CrossRef CAS.
- J.-B. Hövener, E. Y. Chekmenev, K. C. Harris, W. H. Perman, L. W. Robertson, B. D. Ross and P. Bhattacharya, Magn. Reson. Mater. Phys., Biol. Med., 2009, 22, 111–121 CrossRef PubMed.
- K. W. Waddell, A. M. Coffey and E. Y. Chekmenev, J. Am. Chem. Soc., 2011, 133, 97–101 CrossRef CAS PubMed.
- D. Blazina, S. B. Duckett, T. K. Halstead, C. M. Kozak, R. J. K. Taylor, M. S. Anwar, J. a. Jones and H. a. Carteret, Magn. Reson. Chem., 2005, 43, 200–208 CrossRef CAS PubMed.
- S. Bär, T. Lange, D. Leibfritz, J. Hennig, D. von Elverfeldt and J.-B. Hövener, J. Magn. Reson., 2012, 225, 25–35 CrossRef PubMed.
- D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, P. He, K. A. Groome, Q. A. Best, F. Shi, B. M. Goodson, R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, J. Am. Chem. Soc., 2014, 136, 3322–3325 CrossRef CAS PubMed.
- T. Theis, M. Truong, A. M. Coffey, E. Y. Chekmenev and W. S. Warren, J. Magn. Reson., 2014, 248, 23–26 CrossRef CAS PubMed.
- J.-B. Hövener, S. Bär, J. Leupold, K. Jenne, D. Leibfritz, J. Hennig, S. B. Duckett and D. von Elverfeldt, NMR Biomed., 2013, 26, 124–131 CrossRef PubMed.
- N. Eshuis, N. Hermkens, B. J. A. Van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, J. Am. Chem. Soc., 2014, 136, 2695–2698 CrossRef CAS PubMed.
- M. Roth, P. Kindervater, H.-P. Raich, J. Bargon, H. W. Spiess and K. Münnemann, Angew. Chem., Int. Ed., 2010, 49, 8358–8362 CrossRef CAS PubMed.
- D. A. Barskiy, O. G. Salnikov, K. V. Kovtunov and I. V. Koptyug, J. Phys. Chem. A, 2015, 119, 996–1006 CrossRef CAS PubMed.
- K. Sporka, J. Hanika, V. Růžička and M. Halousek, Collect. Czech. Chem. Commun., 1971, 36, 2130–2136 CrossRef CAS.
- P. Atkins and J. De Paula, Atkins' Physical Chemistry, Oxford University Press, 2002 Search PubMed.
- D. A. Barskiy, A. N. Pravdivtsev, K. V. Ivanov, K. L. Kovtunov and I. V. Koptyug, Phys. Chem. Chem. Phys., 2015, 18, 89–93 RSC.
- T. Theis, M. L. Truong, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Am. Chem. Soc., 2015, 1404–1407 CrossRef CAS PubMed.
- S. S. Roy, K. M. Appleby, E. J. Fear and S. B. Duckett, J. Phys. Chem. Lett., 2018, 9, 1112–1117 CrossRef CAS PubMed.
- P. Spannring, I. Reile, M. Emondts, P. P. Schleker, N. K. Hermkens, N. G. vaner Zwaluw, B. J. van Weerdenburg, P. Tinnemans, M. Tessari, B. Blümich, F. P. Rutjes and M. C. Feiters, Chem. – Eur. J., 2016, 22, 9277–9282 CrossRef CAS PubMed.
- W. Iali, A. M. Olaru, G. G. Green and S. B. Duckett, Chem. – Eur. J., 2017, 23, 10491–10495 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- E. W. Esch, A. Bahinski and D. Huh, Nat. Rev. Drug Discovery, 2015, 14, 248–260 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01259h |
| This journal is © The Royal Society of Chemistry 2019 |