
Multi-collector ICP-mass spectrometry reveals changes in the serum Mg isotopic composition in diabetes type I patients
Rosa
Grigoryan
 a,
Marta
Costas-Rodríguez
a,
Steven
Van Laecke
b,
Marijn
Speeckaert
c,
Bruno
Lapauw
d and
Frank
Vanhaecke
*a
a,
Marta
Costas-Rodríguez
a,
Steven
Van Laecke
b,
Marijn
Speeckaert
c,
Bruno
Lapauw
d and
Frank
Vanhaecke
*a
aGhent University, Department of Chemistry, Atomic & Mass Spectrometry – A&MS Research Unit, Campus Sterre, Krijgslaan 281-S12, 9000 Ghent, Belgium. E-mail: frank.vanhaecke@ugent.be
bGhent University Hospital, Department of Internal Medicine, C. Heymanslaan 10, 9000 Ghent, Belgium
cGhent University Hospital, Department of Nephrology, C. Heymanslaan 10, 9000 Ghent, Belgium
dGhent University Hospital, Department of Endocrinology, C. Heymanslaan 10, 9000 Ghent, Belgium
First published on 30th May 2019
Abstract
Magnesium is an essential mineral element in the human body, playing a crucial role in the carbohydrate metabolism and insulin action. Mg deficiency has been shown to be associated with diabetes type 1 (T1D) and type 2 (T2D). However, the total serum Mg concentration does not adequately reflect the individual Mg status as a reduced intracellular or ionized serum Mg concentration, i.e. the physiologically active serum fraction, can occur despite a normal Mg serum concentration. Therefore, we explored the isotopic composition of serum Mg as an alternative parameter. A method for Mg isotopic analysis of serum via multi-collector inductively coupled plasma-mass spectrometry (MC-ICP-MS) after acid digestion and isolation of Mg from the concomitant matrix using AG50W-X8 strong cation exchange resin was optimized. Several reference materials of biological and geological origin were included for validation purposes. Mg isotope ratios were expressed relative to both the DSM3 isotopic reference material and ERM-AE143, as an alternative/new isotopic standard. Subsequently, the serum Mg isotopic composition was investigated in patients with T1D and compared to that in healthy individuals (reference population). Patients were re-evaluated after one year. The Mg isotopic composition was significantly lighter for the T1D patients than for the reference population and after one year, a similar shift in the average δMg value was observed. However, for some of the T1D patients, a statistically significant difference was established between the δMg values corresponding to the two sampling events. This variability could be related to the effect of administered insulin on the transcellular kinetics of Mg, inherent characteristics of the T1D patients, such as variable glycemic control, and/or differences in the Mg isotope fractionation accompanying intestinal uptake and/or renal excretion and/or in the distribution of Mg isotopes across body compartments. Further research is required to identify the governing factor(s).
1. Introduction
Magnesium is an essential element involved in bone and teeth mineralization, muscle contraction, nerve impulse transmission and blood clotting. Over 600 enzymes depend on this cation for activation, while also ATP (adenosine triphosphate), the main source of energy in cells, must bind to a Mg2+ ion in order to be biologically active.1 It is the second most abundant intracellular cation and the fourth most abundant mineral element in the body.1,2 The Mg content is on average 486 mg per kg body weight, 99% of which is present in bone and soft tissues, thus leaving 1% of the Mg in the extracellular space.3 The normal serum Mg concentration range is 17.01–24.31 mg L−1, 55% of which is ionized or free Mg (iMg2+), 32% is bound to proteins (especially albumin) and 13% is in the form of a complex with anions such as phosphate, citrate, bicarbonate or sulphate.4,5 The iMg2+ content represents the physiologically active fraction of blood serum.6 The total serum Mg concentration does not adequately reflect the individual Mg status, as a reduced intracellular or ionized serum Mg concentration can occur despite a normal total serum concentration.3In mammalian cells, Mg homeostasis is tightly regulated by mechanisms at the level of Mg2+ entry and efflux across the cell membrane, of intracellular Mg2+ buffering and of organelle compartmentation, following hormonal stimuli.7 Enhanced levels of hormones like insulin and taurine have been associated with increased levels of iMg2+, while adrenalin exerts an opposite effect.8 The hormone insulin, produced in the pancreatic β-cells, plays a critical role in the glucose and lipid metabolism and may modulate Mg transport from the extracellular to the intracellular space.9–11
Diabetes mellitus is a metabolic disorder affecting insulin secretion and/or action and is frequently associated to Mg deficiency (hypomagnesemia).12 Diabetes type 1 (T1D) primarily results from the autoimmune destruction of the pancreatic β-cells, leading to an absolute insulin deficiency and causing hyperglycaemia. T1D patients require injectable insulin therapy, regular carbohydrate counting and glucose monitoring.13,14 T1D is frequently accompanied by alterations in lipid metabolism, inflammatory status, endothelial cell dysfunction and apoptosis.15 T1D patients with worse glycemic control often show lower serum Mg than T1D patients with better glycemic control.16,17 However, no differences were observed in the iMg2+ concentration between healthy controls and young or recently diagnosed T1D patients.18,19 Also, urinary Mg excretion has been associated with elevated fasting blood glucose and HbA1c (glycated hemoglobin) concentrations, and it was found to be augmented or similar in diabetic patients compared to controls.20–22 Tracer experiments with an orally administered isotopically enriched stable isotope of Mg, demonstrated that Mg deficiency was not caused by a reduced dietary Mg absorption, impaired retention or excretion in subjects with T2D.23 The reasons and the clinical consequences of the disrupted Mg homeostasis in diabetic patients remains uncertain although it was hypothesized that a reduced cellular ATP content, a defective tyrosine-kinase activity of the insulin receptor, impairment in the insulin action and worsening of insulin resistance may be involved.10,24 Other possible causes of hypomagnesemia in T2D may be poor gastrointestinal absorption and/or enhanced renal Mg excretion.25 As insulin has anti-magnesiuric effects in the thick ascending limb of the loop of Henle (TAL) and distal convoluted tubule (DCT), insulin deficiency or resistance may exacerbate renal Mg wasting. It has been shown that Mg deficiency reduces insulin sensitivity and thus, glycemic control.25
High-precision isotopic analysis of Cu, Fe, Zn and Ca via multi-collector ICP-mass spectrometry (MC-ICP-MS) has previously been shown a promising approach in a biomedical/clinical context. Some disease conditions were shown to affect the isotope fractionation accompanying biochemical processes, thus altering the isotopic composition of these essential mineral elements in body fluids and/or tissues.26–28 However, to the best of our knowledge, serum Mg isotopic analysis has not been explored for such purpose yet.
Magnesium has three isotopes with the following average natural relative isotopic abundances: 24Mg (79%), 25Mg (10%) and 26Mg (11%).29 Studies on bacteria, fungi and plants revealed Mg isotope fractionation induced by metabolic processes, although the exact mechanisms have not been elucidated yet.30–32 A preferential incorporation of the heavier Mg isotopes in cells and plants with low Mg content and an apparent absence of isotope fractionation in those high in Mg content has been reported.32 Pokharel et al. inferred that the intracellular Mg isotopic composition is governed by the rate of Mg bonding in intracellular compartments relative to the rate of Mg transport into and its efflux from these cells.32 In mammals, faeces are the material most depleted in the heavier Mg isotopes, leaving the body enriched in the heavier isotopes relative to the diet as was shown using a box model by Martin et al.33 Also, an enrichment in the heavier Mg isotopes from herbivores to omnivores in trophic chains has been recently shown.33
The aim of this work was to explore whether the serum Mg isotopic composition can be used for studying the disrupted Mg metabolism in T1D patients. The serum Mg isotopic composition was investigated in patients suffering from T1D and compared to that in assumed healthy individuals (reference population). The T1D patients were re-evaluated after one year. Prior to analysis of these clinical samples, the chromatographic isolation of Mg and the measurement protocol for Mg isotopic analysis by multi-collector inductively coupled plasma-mass spectrometry (MC-ICP-MS) were evaluated for their application in clinical research. Several commercially available biomaterials were analysed and the Mg isotope ratios were expressed relative to both the DSM3 isotopic reference material and to ERM-AE143, as an alternative new isotopic standard.
2. Experimental
2.1. Reagents and materials
Ultrapure water (resistivity > 18.2 MΩ cm) was obtained from a Milli-Q Element water purification system (Merck Millipore, Bedford, MA, USA). Trace metal analysis grade PrimarPlus 14 M nitric acid and 12 M hydrochloric acid (Fisher Chemicals, Leicestershire, UK) were further purified by sub-boiling distillation in a Savillex DST-4000 acid purification system (Savillex Corporation, Eden Prairie, MN, USA). Ultrapure TraceSELECT® 9.8 M hydrogen peroxide and ACS grade acetone were purchased from Sigma Aldrich (Overijse, Belgium).Single-element stock solutions (1000 mg mL−1) used for element quantification were acquired from Merck (Darmstadt, Germany), PlasmaCAL (Quebec, Canada), Inorganic Ventures (Christiansburg, Virginia, USA) and Alfa Aesar GmbH (Karlsruhe, Germany). A standard solution of 1000 μg mL−1 Mg (Inorganic Ventures, USA, lot K2-MG650434) was used as in-house isotopic standard for checking the validity of the isotope ratios.
The international δ-zero reference material DSM3 was used as an internal standard in a standard-sample bracketing approach (SSB) and the ERM-AE143 reference material acquired from BAM (Berlin, Germany) was isotopically characterized relative to the DSM3 reference material.
Two-mL polypropylene chromatographic columns (Eichrom Technologies, Saint Jacques de la Lande, France) and AG50W-X8 strong cation exchange resin (hydrogen form, 8% crosslinkage, 100–200 mesh size) purchased from Bio-Rad (Temse, Belgium) were used for Mg purification.
2.2. Samples
Serum samples from males (18–50 years old) were collected at the Ghent University Hospital. 14 serum samples were taken from assumed healthy individuals (an age-matched reference population) and overall, 26 serum samples were taken from 15 patients with T1D. The follow-up serum sampling for the diabetic patients was performed one year after the first sampling (11 patients, 4 dropped out during this study). Clinical parameters were determined as routine practice using standardized protocols. All T1D patients showed elevated levels of HbA1c (range 6.2–13.3%), indicative of elevated blood glucose levels over the preceding 60 days.34 For diagnosing diabetes, a cut-off value of 6.5% of HbA1c is recommended.35 The glomerular filtration rate (GFR) was within the normal value (>90 mL min−1), except for 4 patients.Serum samples were stored at −20 °C until further sample preparation in a class-10 clean lab (PicoTrace™, Göttingen, Germany) at the Department of Chemistry of Ghent University.
This research was approved by an independent commission for medical ethics connected to the Ghent University hospital and all subjects signed an informed consent concerning this study.
For method development and validation, the reference materials Seronorm™ Trace Elements Serum L-1 (lot 1309438, SERO AS, Billingstad, Norway) and Seronorm™ Trace Elements Whole blood L-1 reference material (lot 1406263) were used.
2.3. Sample preparation
An aliquot of 0.5 mL of blood serum or whole blood was digested in a closed Savillex® PFA beaker using 2 mL of 14 M HNO3 and 0.5 mL of 9.8 M H2O2 at 110 °C for 18 hours. After cooling down, the vessels were opened and the digests were evaporated to dryness at 90 °C. The residues were dissolved in 1 mL of 0.4 M HCl for the subsequent Mg isolation using cation exchange chromatography. The chromatographic separation of Wombacher et al.,36 was re-visited for isolation of Mg from serum and whole blood samples. 1 mL of AG50W-X8 resin (100–200 mesh, hydrogen form) was loaded into a 2 mL polypropylene column with an inner diameter of 0.8 cm. A piece of cotton was placed as bed support. The resin was pre-cleaned with 10 mL of MQ-water, 30 mL of 7 M HCl, followed by another 20 mL of MQ-water. After conditioning of the resin with 10 mL of 0.4 M HCl, 1 mL of the sample digest was loaded onto the column. The matrix was eluted using 34 mL of 0.4 M HCl, 10 mL of the mixture 0.5 M HCl + 95% acetone and 1 mL of 0.8 M HCl. Then, the Mg fraction was eluted using 23 mL of 0.8 M HCl and collected in a Teflon Savillex® beaker. The fraction thus obtained was evaporated to dryness, re-dissolved in 2 mL of 14 M HNO3 + 0.5 mL of 9.8 M H2O2 and heated on a hot plate at 110 °C overnight for the removal of organic compounds (predominantly acetone, in addition to resin material). After that, the pure Mg fraction was evaporated to dryness at 90 °C and the residue subsequently re-dissolved in 1 mL of 2% (v/v) HNO3 for element determinations and isotope ratio measurements. Two procedural blanks were included in each batch (∼25 columns) of samples.Two geological reference materials – Basalt, Hawaiian Volcanic Observatory BHVO-1 and Icelandic Basalt BIR-1 (both from the US Geological Survey, Reston, VA, USA) were also analysed for evaluation of the measurement protocol. About 150 mg of the basalt reference materials were weighed and dissolved in a mixture of 2.5 mL of 27.6 M HF and 5 mL of 14 M HNO3. Closed beakers were heated on hot plate at 90 °C for 72 hours. Samples were evaporated to dryness (at 90 °C), dissolved in freshly prepared 4 mL of aqua regia (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 14 M HNO3:10.6 M HCl, as obtained after subboiling distillation) and heated on a hot plate at 90 °C for 72 hours. Samples were evaporated to dryness and the residue re-dissolved in 1.2 mL of 0.4 M HCl for further Mg isolation as described above.
:3, 14 M HNO3:10.6 M HCl, as obtained after subboiling distillation) and heated on a hot plate at 90 °C for 72 hours. Samples were evaporated to dryness and the residue re-dissolved in 1.2 mL of 0.4 M HCl for further Mg isolation as described above.
2.4 Instrumentation and measurements
A Thermo Scientific Neptune MC-ICP-MS instrument (Bremen, Germany) was used for Mg isotope ratio measurements. The samples were introduced into the plasma using a 100 μL min−1 PFA concentric nebulizer, mounted onto a double spray chamber with cyclonic and Scott-type sub-units. Mg isotope ratio measurements were performed at medium (pseudo) mass resolution on the left side of the peak centres and in static collection mode, involving three Faraday collectors (L3, C, H3) connected to 1011 Ω resistors. Instrument settings and data acquisition parameters are summarized in Table 1. An acid blank (0.28 M HNO3) and procedural blanks were measured at the beginning of each measurement sequence to evaluate their contribution to the Mg2+ signal intensity. The in-house Mg isotopic standard (Inorganic Ventures IV) was measured every five samples for checking the validity of the measurements. The Mg isotope ratio measurements were carried out using approximately 150 μg L−1 of Mg, and samples and standards were concentration-adjusted to within ±5% of the DSM3 isotopic reference material. External correction in a sample-standard bracketing (SSB) approach was applied for mass bias correction. The 26Mg/24Mg and 25Mg/24Mg ratios were expressed in delta notation (δ26Mg and δ25Mg, per mil, ‰) relative to the DSM3 reference material, and calculated according to eqn (1), where x is 25 or 26. | (1) |
| Instrumental settings | ||
|---|---|---|
| RF power, W | 1200 | |
| Gas flow rates, L min−1 | Cool | 15 |
| Auxiliary | 0.70–0.95 | |
| Nebulizer | ∼1 | |
| Interface | ||
| Sampler cone | Ni, Jet-type: 1.1 mm orifice diameter | |
| Skimmer cone | Ni, X-type: 0.8 mm orifice diameter | |
| Sample uptake | 0.1 mL min−1 | |
| Mass resolution mode | (Pseudo) medium | |
| Data acquisition | |
|---|---|
| Mode | Static, multi-collection |
| Integration time, s | 4.194 |
| Number of integrations | 3 |
| Number of blocks | 10 |
| Number of cycles per block | 5 |
| Cup configuration | |||
|---|---|---|---|
| Cup | L3: 24Mg | C: 25Mg | H3: 26Mg |
| Resistor | 1011Ω | 1011Ω | 1011Ω |
A Thermo Scientific Element XR sector field ICP-MS instrument was used for element determination. The sample introduction system consisted of a 200 μL min−1 quartz nebulizer, mounted onto a cyclonic spray chamber. Plasma torch position, gas flow rates and lens settings were optimized in terms of signal intensity (Li, In and U) and oxide levels (UO+/U+). Samples and calibration standards were prepared in 0.28 M HNO3 and Ga (final concentration: 10 μg L−1) was added as internal standard. Concentrations of the target element and some minor and trace elements that can potentially give rise to spectral interference were determined.
3. Results and discussion
3.1 Mg isolation procedure
The procedures used to isolate Mg from geological and biological materials previously reported in the literature mostly rely on ion exchange chromatography using either AG 50W-X8, AG 50W-X12 cationic resins or AG 1-X8, AG 1-X4 and AG MP-1M anionic resins.30,36–40 In this work, the cation exchange resin AG 50W-X8 (100–200 mesh, hydrogen form) was opted for with the aim of obtaining efficient Mg isolation with a sufficiently high sample throughput, as analysis of a high number of samples is often demanded for clinical applications.36 The distribution coefficient of each element varies according to resin and acid type/concentration used.41 A procedure described by Wombacher et al. was re-visited and optimized for Mg isolation from serum and whole blood samples, as the ratio of Mg to the matrix elements differs from that typical in geological samples.36,41 Their protocol relied on a two-stage chemical separation procedure using AG50-X8 resin of 200–400 mesh size. However, in the current work, efficient separation of Mg from the Ca present in the biofluid matrix was achieved using 0.8 M HCl in a one-stage separation procedure. The elution profile for the Seronorm™ Trace Elements Serum L-1 reference material is presented in Fig. 1 and shows that Mg is fully separated from the relevant concomitant elements. Whole blood shows a substantially higher Fe concentration (∼350 mg L−1, most of the Fe is present in the red blood cells) than other biofluids. Therefore, in the case of whole blood, Fe partially co-elutes with Mg. However at the Fe/Mg ratio obtained after the separation (1/10), no significant effect on the Mg isotope ratio results was observed.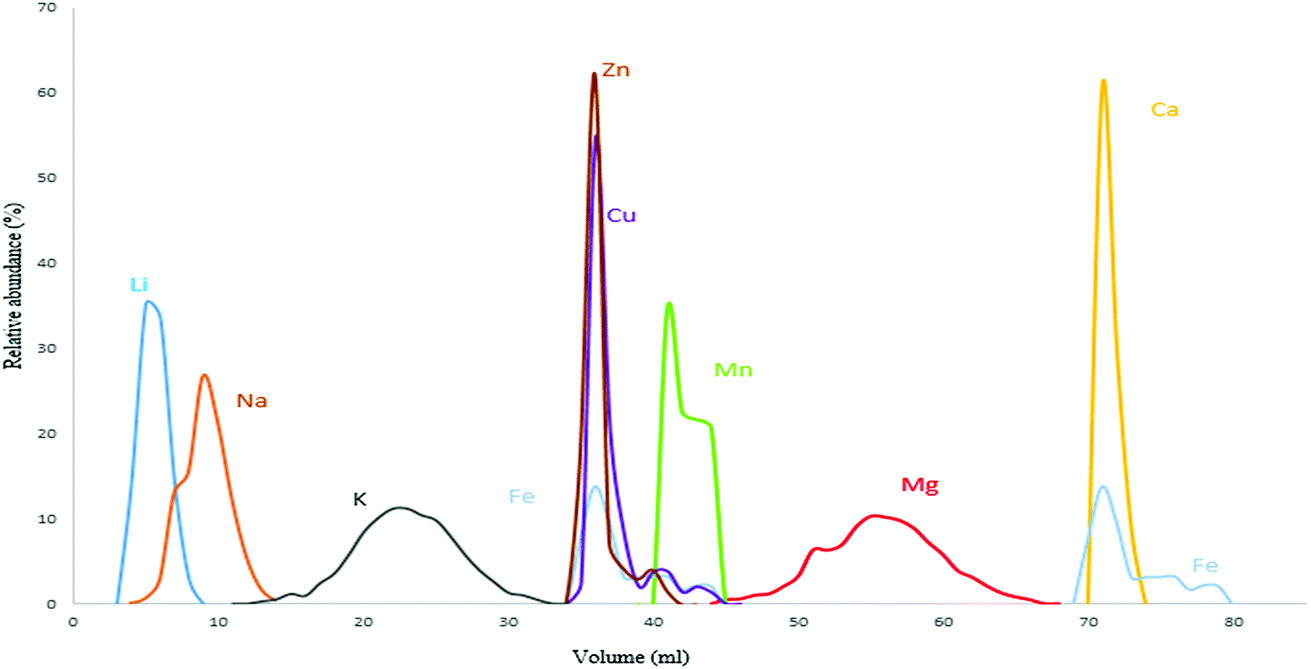 | ||
| Fig. 1 Elution profile for the Seronorm™ Trace Elements Serum L-1. | ||
The Mg yield was 102 ± 2% for the Seronorm serum reference material and 101 ± 1% for the Seronorm whole blood reference material, such that artificial (on-column) Mg isotope fractionation could not affect the Mg isotope ratio results. The Mg yields obtained for the real samples ranged between 95 and 104%, with an average recovery of 99 ± 4% for the controls, 101 ± 2% for the T1D patients year 1 and 101 ± 1% for the T1D patients year 2.
The method was first evaluated using a synthetic serum sample containing the Mg in-house isotopic standard and matrix elements at concentration levels representative for human serum. The δ26Mg value obtained for the synthetic serum solution was −0.61 ± 0.05‰ (N = 3), a value that agrees within the experimental uncertainty with that for the pure in-house standard (δ26Mg = −0.65 ± 0.09‰, N = 167), thus illustrating the validity of the approach.
3.2 Evaluation of the analytical protocol
Mg isotope ratio measurements performed at (pseudo) medium mass resolution on the left side of the peak centres allow the Mg analyte signals to be resolved from these of otherwise interfering polyatomic ions, such as C2+, C2H+, C2H2+, CN+ and NaH+. The isotope ratio measurements were performed at 150 μg L−1 of Mg and the signal intensity achieved was typically ≈ 8.5 V for the 24Mg nuclide.The Mg in-house standard and the ERM-AE143 reference material were used to evaluate the precision and accuracy of the Mg isotope ratios. Additionally, two basalt reference materials (BHVO-1 and BIR-1) were used for comparison purposes, as to date, Mg isotope ratios in biological fluids like serum or whole blood have not been reported yet. Each sample was analysed in triplicate (including the sample preparation and Mg isolation) and measurements were carried out in duplicate. The Mg isotopic composition of the biological and geological materials as well as that of the isotopic standards is presented in Table 2. As can be seen, the δ26Mg values for the geological reference materials, used for assessment of the measurement protocol, were in agreement with reported data. Moreover, the slope of the fractionation line (Fig. 2) was within the range defined by kinetically (slope = 0.5110) and thermodynamically (slope = 0.5210) governed mass-dependent fractionation, respectively.42 As the DSM3 isotopic standard is no longer available, the actual serum samples were measured relative to both, the DSM3 and the ERM-AE143 isotopic reference materials (Table 2). The relationship between δ26MgERM-AE143 and δ26MgDSM3 values obtained for the serum samples (N = 39) is shown in eqn (2) and (3). The intercepts of the linear relations were in concordance with the δ26,25Mg values of the ERM-AE143 directly measured relative to the DSM3 (Table 2).
| δ26MgERM-AE143 = (0.93 ± 0.03) × δ26MgDSM3 + (3.27 ± 0.02), with R2 = 0.96 | (2) |
| δ25MgERM-AE143 = (0.96 ± 0.04) × δ25MgDSM3 + (1.68 ± 0.01), with R2 = 0.96 | (3) |
| Sample | δ 26Mg ± 2SD | δ 25Mg ± 2SD | N | Reported, δ26Mg ± 2SD (ref.) |
|---|---|---|---|---|
| a 2SD: two times the standard deviation. b N: the number of replicates. | ||||
| Mg in-house standard | −0.65 ± 0.09 | −0.34 ± 0.05 | 167 | — |
| Seronorm™ serum L-1 | 0.08 ± 0.06 | 0.04 ± 0.05 | 23 | — |
| Seronorm™ whole blood L-1 | −0.42 ± 0.13 | −0.21 ± 0.07 | 6 | — |
| BIR-1 | −0.36 ± 0.09 | −0.19 ± 0.05 | 6 | −0.30 ± 0.04 (ref. 45) |
| −0.28 ± 0.03 (ref. 46) | ||||
| −0.23 ± 0.23 (ref. 36) | ||||
| BHVO-1 | −0.38 ± 0.12 | −0.21 ± 0.06 | 6 | −0.31 ± 0.08 (ref. 47) |
| −0.22 ± 0.14 (ref. 48) | ||||
| −0.52 ± 0.05 (ref. 49) | ||||
| ERM-AE 143 | −3.28 ± 0.09 | −1.68 ± 0.07 | 48 | −3.30 ± 0.06 (ref. 50) |
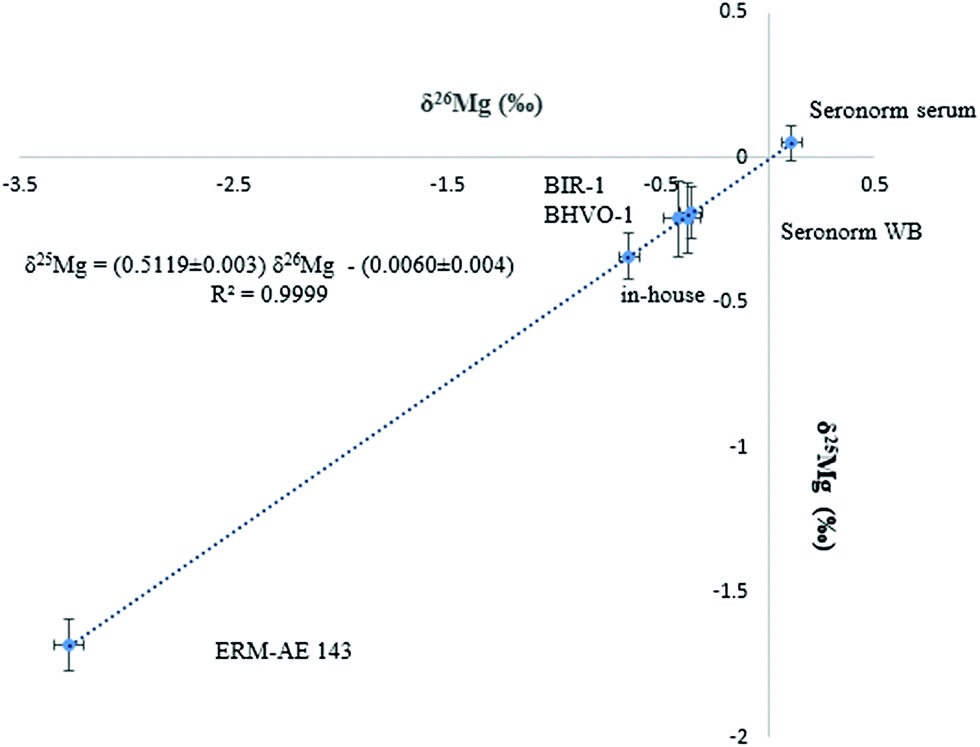 | ||
| Fig. 2 Bivariate Mg isotope ratio plot (or 3-isotope plot) for standards, biological and geological reference materials. δMg values are expressed relative to the DSM3 isotopic reference material. | ||
The internal precision for the δ26Mg value was 0.03‰ (2se). The repeatability for Seronorm™ Trace Elements Whole blood L-1 (N = 3) and Seronorm™ Trace Elements Serum L-1 (N = 6), expressed as 2SD for δ26Mg were 0.13‰ and 0.06‰, respectively. The reproducibility, expressed as long-term precision (2SD), for the δ26Mg value of the in-house isotopic standard was 0.09‰ (Table 2).
The contribution of the procedural blanks ranged between 1 and 3% of the total Mg signal intensity. The maximum bias observed in the δMg values with and without blank correction was <0.01‰, indicating that this contribution is negligible.
3.3 Serum Mg isotopic composition: effect of diabetes type 1
The serum Mg isotopic composition was explored in T1D male individuals and compared to that of healthy controls. The δMg values, expressed relative to the DSM3 and relative to the ERM-E143 isotopic reference materials, are presented in Table 3. The serum Mg isotopic composition for the reference population ranged between −0.26 and 0.06‰ (expressed relative to DSM3), which is within the range reported for bioapatites (tooth enamel and bones) of small carnivore mammals and heavier than that for plants and faeces.33,43 The serum Mg isotopic composition did not show any relation with the age (Spearman's rank correlation coefficient ρ = 0.107, p = 0.608), suggesting that age does not exert a relevant effect on the serum Mg isotopic composition.| Sample ID-age | Reference population | Sample ID-age | Diabetes type 1 population | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Year 1 | Year 2 | ||||||||||||
| DSM3 | ERM-AE143 | DSM3 | ERM-AE143 | DSM3 | ERM-AE143 | ||||||||
| δ 26Mg | δ 25Mg | δ 26Mg | δ 25Mg | δ 26Mg | δ 25Mg | δ 26Mg | δ 25Mg | δ 26Mg | δ 25Mg | δ 26Mg | δ 25Mg | ||
| a Sample ID corresponds to the patient number followed by the age. b IQR is interquartile range. c Sample not available. d Haemolysed sample. e Patients with low GFR. f The internal precision, expressed as 2 times the standard error, ranged between 0.02 and 0.04‰ and the external precision, expressed as the standard deviation of 3–4 replicates of measurements, ranged between 0.02 and 0.20‰. | |||||||||||||
| 1-36 | −0.06 | −0.02 | 3.18 | 1.62 | 15-18 | −1.80 | −0.94 | 1.55 | 0.74 | ||||
| 2-40 | −0.12 | −0.05 | 3.23 | 1.68 | 16-21 | −0.49 | −0.25 | 2.86 | 1.46 | ||||
| 3-30 | −0.39d | −0.21d | 2.84d | 1.44d | 17-47 | −1.31 | −0.68 | 2.09 | 1.05 | −0.27 | −0.14 | 3.04 | 1.55 |
| 4-30 | −0.14 | −0.08 | 3.15 | 1.60 | 18-19 | −0.71 | −0.37 | 2.52 | 1.31 | −1.27 | −0.66 | 2.05 | 1.07 |
| 5-31 | −0.22 | −0.12 | 3.09 | 1.56 | 19-39 | −0.50 | −0.24 | 2.52 | 1.27 | −0.37 | −0.14 | 2.97 | 1.52 |
| 6-47 | −0.26 | −0.13 | 2.92 | 1.51 | 20-40 | −1.33 | −0.69 | 2.04 | 1.02 | −1.54 | −0.80 | 1.76 | 0.88 |
| 7-24 | −0.15 | −0.08 | 3.02 | 1.58 | 21-23 | −0.47 | −0.25 | 2.88 | 1.46 | −1.51 | −0.76 | 1.81 | 0.92 |
| 8-27 | −0.05 | −0.03 | 3.19 | 1.63 | 22-20 | −0.33 | −0.20 | 3.11 | 1.61 | ||||
| 9-37 | 0.01 | 0.01 | 3.23 | 1.66 | 23-28 | −0.29 | −0.13 | 2.78 | 1.44 | −1.32 | −0.65 | 2.03 | 1.04 |
| 10-30 | −0.31d | −0.15d | 2.93d | 1.54d | 24-39 | −0.42 | −0.20 | 3.05 | 1.56 | −0.32 | −0.15 | 3.02 | 1.53 |
| 11-34 | −0.07 | −0.04 | 3.34 | 1.66 | 25-40 | −0.39 | −0.21 | 2.85 | 1.49 | −0.56 | −0.27 | 2.83 | 1.44 |
| 12-26 | −0.03 | −0.01 | 3.18 | 1.68 | 26-50 | −0.61 | −0.32 | −0.48 | −0.24 | 2.84 | 1.42 | ||
| 13-29 | 0.06 | 0.03 | 3.30 | 1.74 | 27-41 | −1.00e | −0.52e | 2.52e | 1.32e | −0.70e | −0.37e | 2.72e | 1.39e |
| 14-42 | −0.03 | 0.00 | 3.24 | 1.69 | 28-48 | −1.17 | −0.61 | 2.13 | 1.10 | −0.92e | −0.48e | 2.42e | 1.25e |
| 29-44 | −0.22e | −0.15e | 3.15e | 1.61e | |||||||||
| Median | −0.07 | −0.04 | 3.19 | 1.65 | −0.50 | −0.25 | 2.65 | 1.38 | −0.70 | −0.37 | 2.72 | 1.39 | |
| IQR | (0.12) | (0.08) | (0.13) | (0.09) | (0.83) | (0.43) | (0.80) | (0.42) | (1.05) | (0.57) | (1.07) | (0.54) | |
The bivariate Mg isotope ratio plot (or 3-isotope plot) for all individuals is shown in Fig. 3. All data are located on the mass-dependent Mg isotope fractionation line, indicating the absence of any resolvable mass-independent isotope effect or polyatomic interference at the level of precision attained. Two of the serum samples from the reference population were haemolysed and thus, excluded from further data treatment. As can be seen in Table 2 for the Seronorm reference materials, the whole blood Mg isotopic composition is lighter (enriched in the lighter Mg isotopes) than that of the serum. The hemolyzed serum samples therefore showed the lightest Mg isotopic composition among the reference population, illustrating contamination of the serum with red blood cells (Fig. 3). It is clear from Fig. 3 that the data points for the T1D patients are shifted towards the lower left corner, i.e. the serum samples systematically show a lighter isotopic composition.
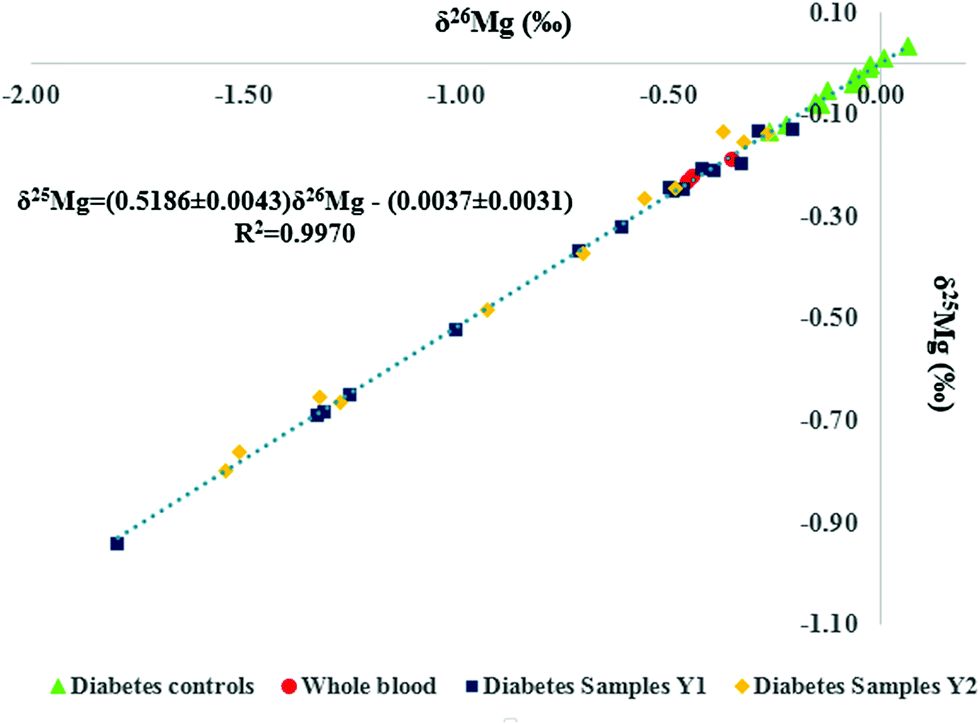 | ||
| Fig. 3 Serum Mg isotopic composition for the reference population (green triangles), T1D patients (Y1, first year of study, blue squares), T1D patients after one year follow-up (Y2, second year of the study, orange diamonds) and the Seronorm™ Trace Elements Whole blood L-1 reference material (red circles). δMg values are expressed relative to the DSM3 isotopic reference material. | ||
Fig. 4 shows box and whisker plots for the Mg concentration (A) and for the Mg isotopic composition (B) of serum. No significant difference was found in terms of serum Mg concentration between the reference and the T1D population, neither among diabetic patients at time = t0 and 1 year later (t = t0 + 1 year) (Kruskal–Wallis test, p > 0.05). Also, in general, the serum Mg concentrations were within the reference range observed in clinical practice (from 17.01–24.31 mg L−1).5 The median Mg concentration for the reference population was 17.9 ± 3.4 mg L−1 (2SD, N = 14), while that for the diabetic patients was 18.9 ± 7.4 mg L−1 (2SD, N = 26). In contrast, the serum Mg isotopic composition was significantly different between the reference population and the T1D population (Fig. 4B). On average, the serum δ26Mg value for diabetic patients was about 0.65‰ lighter than that of the reference population (Table 3) (Kruskal–Wallis test, p < 0.05). For the one year follow-up, the serum Mg isotopic composition of the T1D patients was on average 0.75‰ lighter than that of the healthy controls, similar to the bias previously observed. However, for some T1D individuals, a substantial difference was observed between the serum δ26Mg values for the two sampling events. This variability may be related to the temporary effect of administered insulin on the transcellular kinetics of Mg or to inherent characteristics of the T1D patients, such as a poor glycemic control. Also, differences in the Mg isotope fractionation accompanying intestinal uptake and/or renal excretion and/or in the distribution of Mg isotopes across body compartments could contribute to the light serum Mg isotopic composition of the T1D patients. It has been observed in vitro that insulin affects the iMg2+ concentration in a dose- and time-dependent manner and in absence of insulin, glucose induces the opposite effect.11,44 Also, individual differences can be associated to changes in the response to insulin, e.g., related to its binding to its high-affinity cell surface receptor (receptor tyrosine kinase).
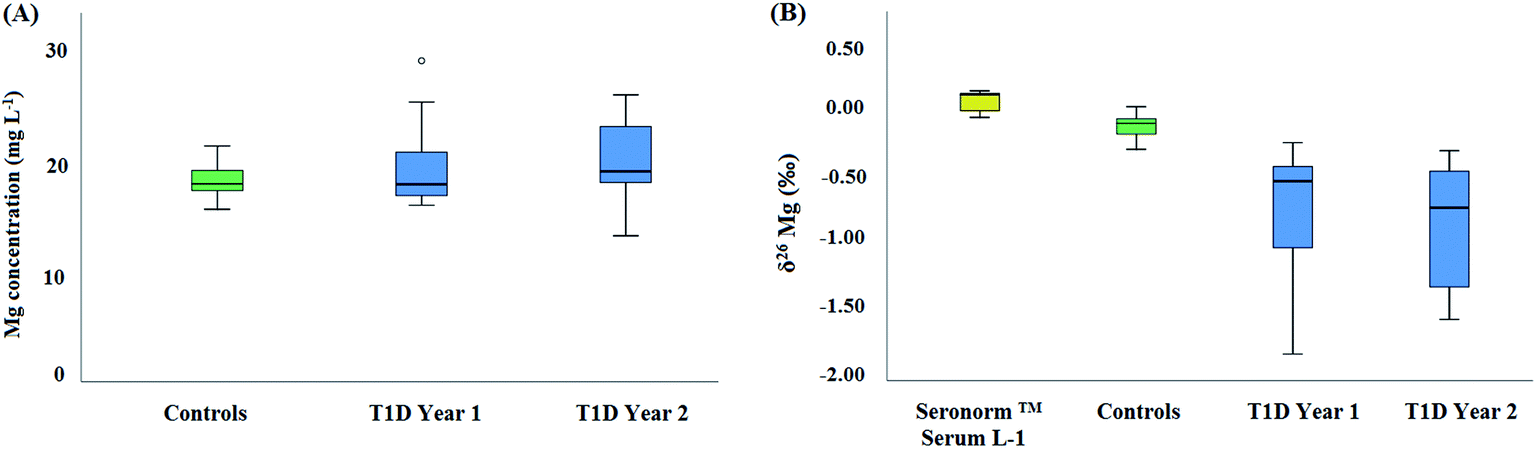 | ||
| Fig. 4 Mg concentrations (A) and δ26Mg values (B) for the reference population and for the T1D patients. Y1 and Y2 correspond to the first and second year of sampling, respectively. The open circle represents an outlier. δ26Mg values are expressed relative to the DSM3 isotopic reference material. | ||
The results obtained in this pilot study indicate that diabetes type 1 induces significant changes in the serum Mg isotopic composition. Further research is required to address the potential effect of insulin administration on the Mg isotope fractionation.
4. Conclusions
A Mg isolation procedure based on the use of the AG 50W-X8 cation exchange resin was shown effective for blood serum and whole blood samples. For whole blood, traces of Fe remain in the purified Mg fraction, but at the Fe/Mg ratio obtained, no effect on the Mg isotope ratio results as determined using MC-ICP-MS was observed. The δ26,25Mg values obtained for geological reference materials and for the alternative/new isotopic reference material ERM-AE143 were in good agreement with those reported in the literature. Mass fractionation lines were within the range of those defined by kinetically governed and thermodynamically governed mass-dependent fractionation. The Mg isotopic composition of diabetes type 1 patients was significantly lighter than that for an age-matched reference population and also showed a similar shift after 1 year follow-up. The Mg isotope fractionation could be affected by administered insulin and its potential effect on the transcellular kinetics of Mg or by inherent characteristics of type 1 diabetes patients, e.g. differences in glucose metabolism, thus inducing a spread within the T1D population. These preliminary results indicate that the disruption of the Mg metabolism in T1D patients is reflected in the serum Mg isotopic composition; further research is required to address the potential effect of insulin administration on the Mg isotope fractionation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Marta Costas-Rodríguez thanks FWO-Vlaanderen for her postdoctoral grant.References
- J. H. F. de Baaij, J. G. J. Hoenderop and R. J. M. Bindels, Physiol. Rev., 2015, 95, 1–46 CrossRef PubMed.
- M. J. Arnaud, Br. J. Nutr., 2018, 99, 24–36 CrossRef PubMed.
- W. Jahnen-Dechent and M. Ketteler, Clin. Kidney J., 2012, 5, 3–14 CrossRef PubMed.
- B. H. Ising, F. Bertschat, G. Theodor, E. Jeremias and A. Jeremias, Eur. J. Clin. Chem. Clin. Biochem., 1995, 33, 365–371 Search PubMed.
- J. J. Dinicolantonio, J. H. O'Keefe and W. Wilson, Open Heart, 2018, 5, e000668 CrossRef PubMed.
- J. D. Wooldridge and C. R. Gregory, Vet. Surg., 1999, 28, 31–37 CrossRef CAS PubMed.
- A. Romani, Miner. Electrolyte Metab., 1993, 19, 282–289 CAS.
- C. Monteiro and M. Bicho, Front Biosci., 2004, 9, 262–276 CrossRef.
- Z. Fu, E. R. Gilbert and D. Liu, Arch. Biochem. Biophys., 2011, 512, 1–23 CrossRef PubMed.
- M. Barbagallo, World J. Diabetes, 2015, 6, 1152–1157 CrossRef PubMed.
- P. Delva, M. Degan, M. Trettene and A. Lechi, J. Endocrinol., 2006, 190, 711–718 CrossRef CAS PubMed.
- R. Swaminathan, Clin. Biochem., 2003, 24, 47–66 CAS.
- B. M. Shields, J. L. Peters, C. Cooper, J. Lowe, B. A. Knight, R. J. Powell, A. Jones, C. J. Hyde and A. T. Hattersley, BMJ Open, 2015, 5, e009088 CrossRef PubMed.
- G. Priya and S. Kalra, Diabetes Ther., 2018, 9, 349–361 CrossRef CAS PubMed.
- J. F. Ndisang, A. Vannacci and S. Rastogi, J. Diabetes Res., 2017, 2017, 10–12 Search PubMed.
- A. Galli-Tsinopoulou, I. Maggana, I. Kyrgios, K. Mouzaki, M. G. Grammatikopoulou, C. Stylianou and K. Karavanaki, J. Diabetes, 2014, 6, 369–377 CrossRef CAS PubMed.
- D. Shahbah, A. Abo, E. Naga, T. Hassan, M. Zakaria, M. Beshir, S. Al Morshedy, M. Abdalhady, E. Kamel, D. A. Rahman, L. Kamel and M. Abdelkader, Medicine, 2016, 95, 1–7 CrossRef PubMed.
- M. Roffi, C. Kanaka, P. E. Mullis, E. Peheim and M. G. Bianchetti, Am. J. Nephrol., 1994, 14, 201–206 CrossRef CAS PubMed.
- G. Matthiesen, K. Olofsson and M. Rudnicki, Diabetes Care, 2004, 27, 1216–1217 CrossRef CAS PubMed.
- I. R. Brown, A. M. Mcbain, J. Chalmers, I. W. Campbell, E. R. Brown and M. J. Lewis, Clin. Chim. Acta, 1999, 283, 119–128 CrossRef CAS.
- Z. Yuan, C. Liu, Y. Tian, X. Zhang, H. Ye, L. Jin, L. Ruan, Z. Sun and Y. Zhu, Ann. Nutr. Metab., 2016, 69, 125–134 CrossRef CAS PubMed.
- P. Mcnair, M. S. Christensen, C. Christiansen and S. Madsbad, Eur. J. Clin. Invest., 1982, 12, 81–85 CrossRef CAS PubMed.
- M. K. Wälti, M. B. Zimmermann, T. Walczyk, G. A. Spinas and R. F. Hurrell, Am. J. Clin. Nutr., 2003, 78, 448–453 CrossRef PubMed.
- S. H. Kwak, K. S. Park, K. Lee and H. K. Lee, J. Diabetes Invest., 2010, 1, 161–169 CrossRef CAS PubMed.
- P. T. Pham, P. T. Pham, S. V Pham, J. M. Miller and P. T. Pham, Clin. J. Am. Soc. Nephrol., 2007, 2, 366–373 CrossRef CAS PubMed.
- M. Costas-Rodríguez, Y. Anoshkina, S. Lauwens, H. Van Vlierberghe, J. Delanghe and F. Vanhaecke, Metallomics, 2015, 7, 491–498 RSC.
- Y. Anoshkina, M. Costas-Rodríguez, M. Speeckaert, W. Van Biesen, J. Delanghe and F. Vanhaecke, Metallomics, 2017, 9, 517–524 RSC.
- M. Costas-Rodríguez, J. Delanghe and F. Vanhaecke, TrAC, Trends Anal. Chem., 2016, 76, 182–193 CrossRef.
- W. A. Brand, T. B. Coplen, J. Vogl, M. Rosner and T. Prohaska, Pure Appl. Chem., 2014, 86, 425–467 CAS.
- E. B. Bolou-Bi, A. Poszwa, C. Leyval and N. Vigier, Geochim. Cosmochim. Acta, 2010, 74, 2523–2537 CrossRef CAS.
- Z. A. Fahad, E. B. Bolou-Bi, R. D. Finlay, J. K. Stephan and V. De Marne, Environ. Microbiol. Rep., 2016, 8, 956–965 CrossRef CAS PubMed.
- R. Pokharel, R. Gerrits, J. A. Schuessler, P. J. Frings, R. Sobotka, A. A. Gorbushina and F. von Blanckenburg, Environ. Sci. Technol., 2018, 52, 12216–12224 CrossRef CAS PubMed.
- J. E. Martin, D. Vance and V. Balter, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 430–435 CrossRef CAS PubMed.
- J. J. Kaneko, Clin. Biochem. Domest. Anim., 2008, 6, 45–80 Search PubMed.
- World Health Organization (WHO), Abbreviated Rep. a WHO Consult., 2011, pp. 1–25 Search PubMed.
- F. Wombacher, A. Eisenhauer, A. Heuser and S. Weyer, J. Anal. At. Spectrom., 2009, 24, 627–636 RSC.
- Y. An and F. Huang, J. Earth Sci., 2014, 25, 822–840 CrossRef CAS.
- V. T.-C. Chang, R. J. P. Williams, A. Makishima, N. S. Belshawl and R. K. O'Nions, Biochem. Biophys. Res. Commun., 2004, 323, 79–85 CrossRef CAS PubMed.
- F. Teng, W.-Y. Li, S. Ke, B. Marty, N. Dauphas, S. Huang, F.-Y. Wu and A. Pourmand, Geochim. Cosmochim. Acta, 2010, 74, 4150–4166 CrossRef CAS.
- M. Schiller, J. A. Baker and M. Bizzarro, Geochim. Cosmochim. Acta, 2010, 74, 4844–4864 CrossRef CAS.
- M. S. Choi, J.-S. Ryu, S.-W. Lee, H. S. Shin and K.-S. Lee, J. Anal. At. Spectrom., 2012, 27, 1955–1959 RSC.
- J. Vogl, B. Brandt, J. Noordmann, O. Rienitz and D. Malinovskiy, J. Anal. At. Spectrom., 2016, 31, 1440–1458 RSC.
- J. E. Martin, D. Vance and V. Balter, Geochim. Cosmochim. Acta, 2014, 130, 12–20 CrossRef CAS.
- M. Barbagallo, R. K. Gupta and L. M. Resnick, Diabetologia, 1993, 36, 146–149 CrossRef CAS PubMed.
- B. Shen, J. Wimpenny, C. A. Lee, D. Tollstrup and Q. Yin, Chem. Geol., 2013, 356, 209–214 CrossRef CAS.
- S. Lee, J. Ryu and K. Lee, Chem. Geol., 2014, 364, 9–19 CrossRef CAS.
- F. Huang, J. Glessner, A. Ianno, C. Lundstrom and Z. Zhang, Chem. Geol., 2009, 268, 15–23 CrossRef CAS.
- X. Liu, F. Teng, R. L. Rudnick, W. F. McDonough and M. L. Cummings, Geochim. Cosmochim. Acta, 2014, 135, 336–349 CrossRef CAS.
- J. Karasinski, E. Bulska, M. Wojciechowski, L. Halicz and A. A. Krata, Talanta, 2017, 165, 64–68 CrossRef CAS PubMed.
- J. Vogl, O. Rienitz, M. Rosner, B. Brandt, S. Kasemann, R. Kraft, D. Malinovskiy, A. Meixner, J. Noordmann, S. Rabb, J. A. Schuessler and R. D. Vocke, in European Winter Conference on Plasma Spectrochemistry, Pau, France, 2019, p. 399 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |