Applications in catalysis, photochemistry, and photodetection: general discussion
Javier
Aizpurua
 ,
Jeremy
Baumberg
,
Valérie
Caps
,
Emiliano
Cortes
,
Bart
de Nijs
,
Yuri
Diaz Fernandez
,
Laura
Fabris
,
Simon
Freakley
,
Sylwester
Gawinkowski
,
Daniel
Glass
,
Junyang
Huang
,
Bartlomiej
Jankiewicz
,
Jacob
Khurgin
,
Priyank Vijaya
Kumar
,
Reinhard J.
Maurer
,
Peter
McBreen
,
Niclas Sven
Mueller
,
Jeong Y.
Park
,
Jhon
Quiroz
,
Sebastian
Rejman
,
Rosa Mayela
Romero Gómez
,
Jorge
Salmon-Gamboa
,
Sebastian
Schlücker
,
Zachary
Schultz
,
Amaresh
Shukla
,
Yonatan
Sivan
,
Madasamy
Thangamuthu
,
Laura
Torrente-Murciano
,
Xiaofei
Xiao
,
Hongxing
Xu
and
Chao
Zhan
,
Jeremy
Baumberg
,
Valérie
Caps
,
Emiliano
Cortes
,
Bart
de Nijs
,
Yuri
Diaz Fernandez
,
Laura
Fabris
,
Simon
Freakley
,
Sylwester
Gawinkowski
,
Daniel
Glass
,
Junyang
Huang
,
Bartlomiej
Jankiewicz
,
Jacob
Khurgin
,
Priyank Vijaya
Kumar
,
Reinhard J.
Maurer
,
Peter
McBreen
,
Niclas Sven
Mueller
,
Jeong Y.
Park
,
Jhon
Quiroz
,
Sebastian
Rejman
,
Rosa Mayela
Romero Gómez
,
Jorge
Salmon-Gamboa
,
Sebastian
Schlücker
,
Zachary
Schultz
,
Amaresh
Shukla
,
Yonatan
Sivan
,
Madasamy
Thangamuthu
,
Laura
Torrente-Murciano
,
Xiaofei
Xiao
,
Hongxing
Xu
and
Chao
Zhan
First published on 15th May 2019
Sylwester Gawinkowski opened a general discussion of the paper by Jorge Salmon-Gamboa: Why you are using SiO2 nanoparticles? Do they have any function or are they only the substrate to attach other active nanoparticles to? You have shown that only gold nanoparticles attached to silica nanoparticles do not influence the rate of reaction significantly. You have also demonstrated that adding platinum decorations on the gold nanoparticles causes a strong increase in the reaction rate. The significance of the gold nanoparticles would be more clearly shown with a simple experiment in which you have no gold nanoparticles but still have platinum decorations.Jorge Salmon-Gamboa replied: Silica nanoparticles were chosen as a nano-sized inert substrate for Au nanoparticles. This choice of substrate provides a larger surface area covered with the active Au-Pt nanoparticles, in contrast to the situation when the active particles are placed directly onto a flat substrate. Aiming for applications, the SiO2–Au–Pt nanoparticles can then in turn be attached onto a flat surface, forming a solid device that can be submerged into water, avoiding the problem of water contamination by nanoparticles. The role of the Au nanoparticles was investigated. Under illumination in the LSP spectral band (556-566 nm), SiO2–Pt did not enhance the reaction rate. In contrast, under the same conditions, SiO2–Au–Pt showed a considerably enhanced rate, proving that hot carriers were generated in Au (see Fig. 1 below).
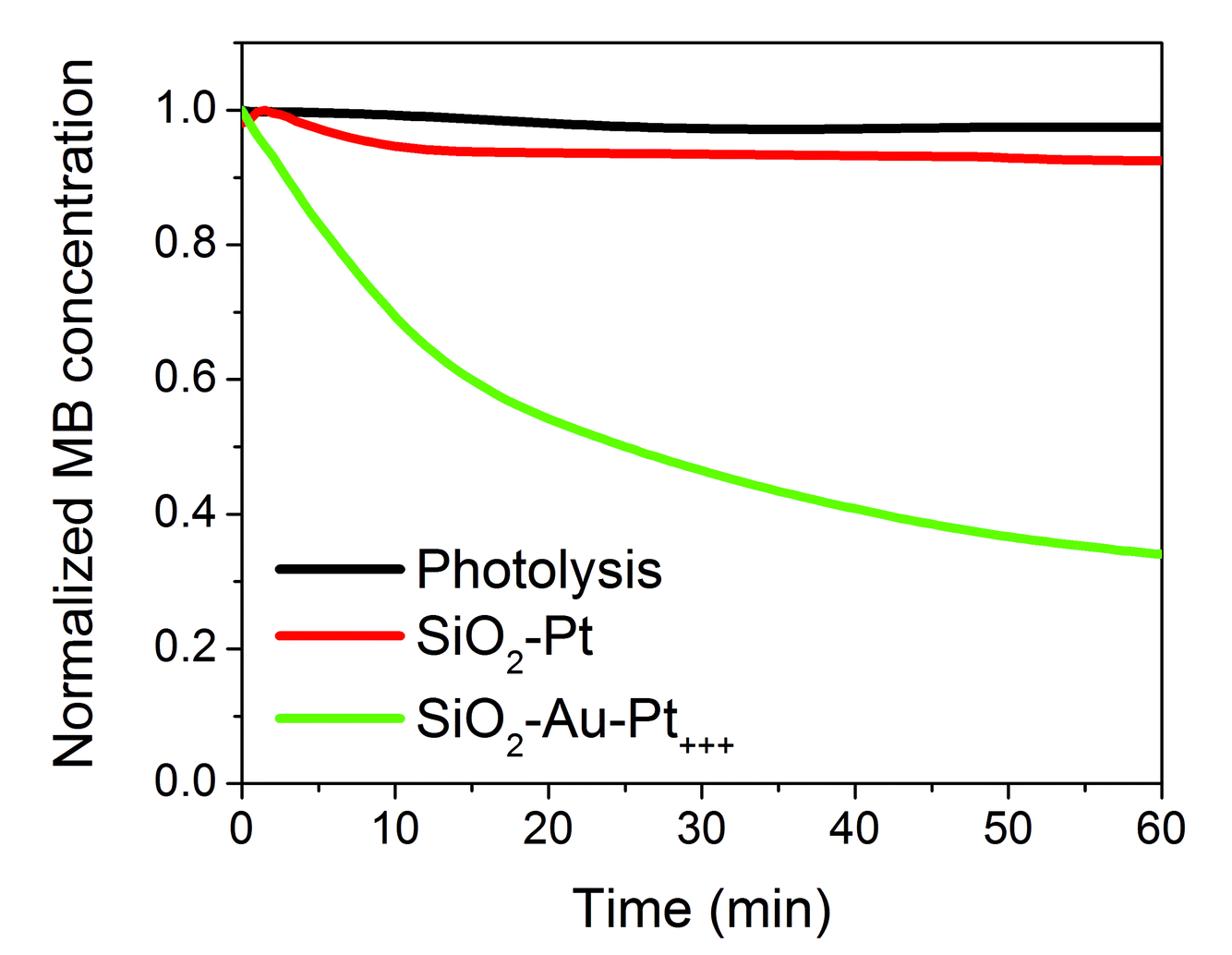 | ||
| Fig. 1 Degradation of methylene blue over time by catalysts with Au (SiO2–Au–Pt+++) and without Au (SiO2–Pt). | ||
Yuri Diaz Fernandez continued the discussion: I have two questions: (1) Can you comment on the dispersity of the size distribution of metal and SiO2 particles and how well controlled are these in your system? (2) In the paper it was not clearly stated which normalisation criteria were used. Was the data normalised considering the extinction efficiency of gold, or considering the metal content in the samples? Did you correct you data for changes in the nanoparticle concentration due to material lost during synthesis/purification? If you consider this dilution effect, your efficiency may be higher.
Jorge Salmon-Gamboa answered: (1) The size distribution of silica nanoparticles had a good uniformity, confirmed by TEM. They were fabricated following the Stöber synthesis method, which is known for its well-controlled growth and size uniformity. On the other hand, the size distribution of Au nanoparticles was broader (9-15 nm in diameter). As the LSP resonances are relatively broad, the size distribution of the nanoparticles does not have a detrimental effect for the studied processes from an optical point of view. (2) The data were normalised considering the maximum optical absorption of the colloidal samples, so that the effect of nanoparticle concentration is taken into account.
Bartlomiej Jankiewicz commented: As we have heard from Professor Halas platinum is considered an endangered element and so it could be reasonable to recover the catalyst you have discussed. Thus, did you consider making core–shell structures with magnetic cores, i.e. Fe3O4@SiO2@Au–Pt, so that you can recover them?
Jorge Salmon-Gamboa replied: Using magnetic cores was not considered at the beginning of the project, but it seems to be a very interesting approach for recovery.
Priyank Vijaya Kumar asked: Your schematic shows a nice core–shell structure. However, is this really the case in experiments? Do you see a nice shell of Pt formed on the core or do you see some mixing with the core, like an alloy? Is there any structural characterisation or other experiments you could do to confirm this?
Jorge Salmon-Gamboa responded: Pump–probe experiments were carried out on heterostructures without Pt and with different Pt loadings. It was found that the electron transfer occurred from Au to Pt and it depends on the amount of Pt. This effect would not appear in the case of alloy formation. Also, the localised surface plasmon resonance is sensitive to the material, and optical simulations suggest that alloy formation was not the case. The HAADF-STEM elemental maps presented in the paper also confirm this.
Sebastian Schlücker requested: Could you please comment on the mechanism of this photodegradation reaction on the atomic scale? What analytical techniques did you use or do you plan to use (e.g. UV/vis absorption, NMR, MS)?
Jorge Salmon-Gamboa replied: Photogenerated hot carriers interact with the surrounding aqueous medium, producing reactive oxygen species. Hot electrons can ionise adsorbed oxygen molecules forming superoxide anion radicals (˙O2−). These highly reactive species can degrade organic molecules.
Optical spectroscopy was used to monitor methylene blue absorbance during degradation experiments. We plan to use complementary analytical techniques in the future to determine the products of the reaction. This was beyond the scope of the present work.
Jhon Jhon Quiroz Torres queried: It has been demonstrated in core–shell multimetallic architectures, comprised of plasmonic and catalytic metals, that the energy concentrated via LSPR excitation can be dissipated through absorption processes in the catalytic metal. Have you estimated the contributions from absorption and scattering to the calculated extinction spectra of the different Au@Pt core–shell architecture models that you presented? How does it change?
Jorge Salmon-Gamboa answered: An estimation of the contribution from absorption and scattering to the calculated extinction was determined by numerical simulations. The total absorption, scattering and extinction cross sections of Au and Au–Pt nanoparticles deposited on SiO2 nanoparticles are presented in the Fig. 2 below. An increase in absorption and scattering cross sections is observed as the quantity of Pt increases on the surface of Au. This effect was also noted experimentally.
 | ||
| Fig. 2 Numerical simulations of extinction, absorption and scattering cross sections for (a) 15 nm diameter Au nanoparticles on SiO2 and (b–d) SiO2–Au–Pt heterostructures with (b) a 0.5 nm thickness of Pt shell, (c) a 2 nm radius of Pt seeds and (d) a 0.5 nm Pt shell and 2 nm radius Pt seeds. Au nanoparticle size is the same for all simulations. | ||
Xiaofei Xiao asked: Thanks, I have two questions. Firstly, how did you estimate the concentration of the small particles – did you consider different concentrations, and what is the effect on the final spectrum? Secondly, I am curious whether a method exists to add another layer of silicon or silica on the surface of the finial structure?
Jorge Salmon-Gamboa replied: The concentration of small Au nanoparticles can be estimated considering the average size of a gold nanoparticle obtained by TEM, the concentration of K-Au used (number of Au+ ions), and the concentration of the SiO2 cores. Then it is assumed that all Au+ ions reduce to form small Au nanoparticles, distributed over all the silica cores. Ultimately, comparison of the absorption spectra allows one to normalise different solutions to the nanoparticle concentrations. Different concentrations of Au were considered. The LSPR spectrum red-shifts as the size of the Au seeds increases. When the Au nanoparticles are big enough they merge together to form a complete Au shell, and finally, for thicker Au layers, the spectrum blue shifts.
As for multilayered coatings, it is possible to add more silica layers as described by P. Wang.1
1 P. Wang, A. V. Krasavin, F. N. Viscomi, A. M. Adawi, J.-S. G. Bouillard, L. Zhang, D. J. Roth, L. Tong and A. V. Zayats, Laser Photonics Rev., 2018, 12, 1800179.
Sebastian Rejman enquired: Have you conducted any kinetic studies of the reaction, and determined the turnover number and frequencies?
Is it possible to do SERS on these nanoparticles? This could provide valuable information about the mechanism of the reaction you studied and possibly other reactions you could run on this system.
Jorge Salmon-Gamboa responded: Turnover number and frequencies were not determined, as it was not within the scope of this work. In principle, they can be calculated. In general, Turnover Frequency is TOF = (number of molecules reacted)/(number of active sites)(time). This measure does not account for the number of incident photons and becomes irrelevant for reporting efficiencies of plasmonic photocatalysts, as pointed out by E. Cortes during this Faraday Discussion.
Performing SERS measurements on these nanoparticles seems to be very interesting, for this and other reactions. We will attempt this in the future.
Simon Freakley remarked: The particle density is very high on the SiO2 particles – in thermal catalysis a major deactivation pathway would be particle sintering or evolution of the nanostructure to produce alloys or loss of shell material via agglomeration. Do you see any signs of plasmon-induced evolution of the materials?
Jorge Salmon-Gamboa replied: We did not see any signs of material evolution and degradation. The absorption spectra were checked before and after the experiments and also, in order to check the behaviour and stability of the heterostructures, under high power laser illumination. The extinction spectra of the samples were monitored and did not change over time. Since the localised surface plasmon resonance is very sensitive to shape, material and agglomeration, we would detect these effects if present. Therefore, we can rule out nanoparticle material evolution.
Javier Aizpurua questioned: In the optical spectroscopy of your structure in Fig. 2 in your paper, there seems to be some response in the long wavelength part of the spectrum, which is not fully captured in the simulations. Have you played with the concentration of metallic particles on the SiO2 core to reproduce this better? Is there any chance that magnetic modes are induced in these structures? How important is it to faithfully reproduce the concentration and the percolation to properly address the near field?
Rosa Mayela Romero Gómez responded: Indeed, this is very complex system with random particle distributions which may have many collective resonant effects. Potentially, uncontrolled aggregation of nanoparticles in the experiment may result in collective modes with additional long wavelength resonances. This sort of resonance is difficult to reproduce in the simulations due to random realisation of particle configurations. The change of nanoparticle concentration influences the resonance, leading to its broadening.
SiO2 nanoparticles, with the same dimensions as investigated in this work, do not exhibit resonant modes in the range of 400-800 nm due to their low relative refractive index. Decoration of silica particles with Au and Au–Pt nanoparticles may or may not lead to additional magnetic modes due to a collective interaction1 but we did not observe these effects.
1 V. Ponsinet, P. Barois, S. M. Gali, P. Richetti, J. B. Salmon, A. Vallecchi, M. Albani, A. Le Beulze, S. Gomez-Grana, E. Duguet, S. Mornet and M. Treguer-Delapierre, Phys. Rev. B, 2015, 92, 220414(R).
Jorge Salmon-Gamboa added: The near-field is of paramount importance for the hot-electron excitation and related processes. Unfortunately, as for the effects discussed above, random realisation of the nanoparticle configurations makes it difficult to simulate and compare the near-field effects. The only conclusion that can be made is that the field is enhanced at the Au nanoparticles, and in the case of their agglomerates, additional near-field features may exist. All these near-field enhancements contribute to hot-electron generation; at the same time, they are different for different random realisations of nanoparticle assemblies. All these effects, combined together, provide the observed averaged effect.
Jeong Y. Park queried: In the paper, it is stated that “SiO2-core based nanoparticles were chosen instead due to the dielectric properties of SiO2, so that a Schottky barrier between metal and semiconductor is avoided, and thus more hot-carriers are available for reduction processes.” I am not quite sure about this statement. I am wondering if fewer hot electron carriers would be available if the authors use a more reactive support such as TiO2. In addition, photocatalytic systems using SiO2 are not effective photocatalysts because SiO2 doesn’t contribute any photocatalytic activity and absorption on metal nanoparticles is quite low. Could the authors comment on this?
Rosa Mayela Romero Gómez replied: The SiO2 nanoparticles do not contribute catalytically – they were used as a nanosize inert support for Au nanoparticles to increase the surface area. On the other hand, if a semiconductor was used instead (TiO2 for example), some hot electrons generated from Au would migrate to the semiconductor through the Schottky barrier and fewer electrons would be available on the surface of Pt, thus the overall photocatalytic activity of the heterostructure might be less. This effect was previously noted.1
1 S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67.
Chao Zhan commented: Surface area is very important for catalysts. Are the Pt surface areas of the Pt NPs and Pt-SiO2–Au the same? You use the term “Pt shell” in the paper, but usually it is small Pt nanoparticles.
Jorge Salmon-Gamboa responded: Surface area is of paramount importance for catalysts. In our experiment, comparison is done between SiO2–Pt nanoparticles and SiO2–Au–Pt nanoparticles with different amounts of Pt. So the Pt nanoparticles cover Au differently for different Pt concentrations and evolve from individual Pt nanoparticles to a complete “rough” shell around the Au nanoparticles. Therefore, we did not ensure precisely the same surface areas of the active particles, but, since the Pt nanoparticles on SiO2–Pt are very small (∼ 3 nm in diameter), they deliver a higher surface area than the Au–Pt particles (∼12 nm). Despite having a higher surface area, the reaction rate was not enhanced in the case of SiO2–Pt, whereas SiO2–Au–Pt showed a significant reaction rate enhancement. The goal of the experiment was to show the role of the combination of the plasmonic-Au and catalyst-Pt in the heterostructures. The term “Pt shell” is used for those heterostructures with a higher Pt loading (where Pt shells formed, as confirmed by the HAADF-STEM elemental maps), which showed higher reactions rates.
Peter McBreen opened a general discussion of the paper by Madasamy Thangamuthu: Thank you for your interesting paper. You describe a minor hot electron driven pathway in Fig. 7B in your paper. Have you considered a pathway other than nitrogen splitting through electron injection into the LUMO? For example, a pathway that first involves N–H bond formation. The adsorption energy for nitrogen on TiO2 or on oxidized aluminum is still quite small. Do you believe that N2 needs to be strongly coupled to TiO2 (strongly adsorbed) or to the aluminum triangles to facilitate such electron transfer into the LUMO?
Madasamy Thangamuthu answered: Electron injection into the LUMO of N2 can potentially break the bonds either directly in one step or in three steps: i) NH, ii) NH2 and iii) NH3. We expect that the three step mechanism is more likely to occur in our configuration since the experimental conditions are mild. Currently we are studying the mechanism by analyzing the different by-products of the reaction. A strong adsorption of N2 on catalytic surfaces is very important to reduce the bond dissociation energy. The TiO2 surface offers suitable active sites for N2 adsorption. Moreover, in our configuration aluminium covers less than 10% of the overall electrode surface. It would be very interesting to study the adsorption mechanism using first principles calculations. We are very much interested to collaborate with theoretical chemists to understand and optimize the N2 adsorption.
Yuri Diaz Fernandez remarked: In your paper the efficiency is calculated considering the total incident photon flux. Is there any particular reason to do this taking into account that with the relatively low extinction values of your samples most of the light will be not be absorbed or scattered?
Madasamy Thangamuthu responded: The interesting point is how much ammonia can be produced per incident flux.
Bart de Nijs asked: Based on the TEM images the structures seem to have a relatively large separation, which means that most of the field enhancement is coming from just the structure. If the structures or different types of structures were closer together a coupling between these structures would occur and provide a coupled plasmon mode. The coupled mode will have the advantage of a much higher field but the optical resonance will be at a longer wavelength (i.e. less energy per photon). Could you say something about this tradeoff? Do you think a high field would benefit this reaction or does the change to a longer wavelength make this light catalyzed process less effective?
Madasamy Thangamuthu answered: A coupled mode could definitely improve the efficiency due to the higher field in the gap (hot spot) but smaller gaps also lead to an increased confinement which lowers the volume of action. Energetically it is also possible to induce the reaction at a longer wavelength.
Jeremy Baumberg commented: I am interested in your UV spectroscopy, because the simulations show that 80% extinction is expected, and you measure (in the small peak) about 5%. Understanding this difference is crucial to get a connection to the photocatalysis. Including a thin oxide layer should not make so much difference, and coupling is weak. This seems rather surprising.
Madasamy Thangamuthu replied: Both the simulations and experimental spectra showing the plasmonic resonance wavelength of the aluminium nanotriangles agree well. The simulations have been done by taking the average refractive index of the substrate and air. The variation might be due to the plasmonic interaction with the substrate.
Yonatan Sivan enquired: Can I ask you to elaborate on the thermal control experiments you have done? Did you measure the temperature rise in your sample?
I am asking because the increase in absorption you report can lead either to the generation of non-thermal (i.e., “hot”) electrons, or to a purely thermal effect (heating), both of which can lead to faster reactions. In that respect, you claim that you added illumination at 650 nm which did not make a difference. However, your Fig. 1D indicates that the AlNTs absorb rather weakly at that wavelength. In fact, it is not clear what is measured (extinction upon illuminating the whole layer (i.e., extinction = 1 − transmission) or a single AlNT), and whether the simulation is of the exact same configuration? Otherwise, how can you explain the very different spectra of the experiment and simulation? They are similar just in terms of the resonance position, but the latter is pronounced only in the simulation.
Could it be that the absorption due to the 650 nm illumination is just small with respect to the 365 nm illumination? If so, I would not say that this test ruled out the possibility of thermal effects.
Madasamy Thangamuthu responded: No, we didn’t measure the temperature changes but we observed that there is no temperature influence on photocurrent measurement by irradiating the electrodes far from the resonance wavelength. The conditions we used in our experiment, 5 mW cm−2 CW narrow band light sources, are very mild so we expect that they don’t raise the temperature significantly. We measured extinction (1−T) for the whole layer not for a single AlNT. AlNTs absorb about 7% of the light (experimental absorption spectra, Fig. 4E in our paper) at 650 nm illumination. A comparison of measurements performed under dark and illuminated conditions revealed no measurable difference in the photocurrent. We conclude that under these conditions there is no thermal contribution.
Bartlomiej Jankiewicz commented: During the presentation of Professor Laura Fabris we have heard that in order to somehow influence TiO2 reactivity by plasmonics the shell thickness in the case of nanoparticles should be around 4–5 nm to avoid electron quenching. Thus, did you try to reduce the thickness of the TiO2 layer over the Al nanostructures and how did it affect the efficiency of the process you were investigating?
Madasamy Thangamuthu answered: We didn’t reduce the thickness of the TiO2 layer. All our experiments were performed with 100 nm TiO2 on top of AlNTs. Thus, we didn’t study the electron quenching phenomenon.
Jacob Khurgin remarked: As far as I know there is no shortage of TiO2 in the earth’s crust. So why do we need plasmons to absorb UV light? It will get eventually absorbed in TiO2 anyway provided we have enough of it. And making the TiO2 particles small will ensure that absorption takes place near the surface.
Madasamy Thangamuthu replied: UV plasmonics is highly useful for chemical reactions that are require higher energy to break strong chemical bonds. Although TiO2 is present in sufficiently large amounts in the earth’s crust, it fails as a photocatalyst in a number of ways such as by having a fast charge-carrier recombination, poor charge extraction, etc. Ammonia is one of the most highly produced chemicals worldwide. To compete against the strongly polluting Haber–Bosch process a large ammonia yield per volume is desirable. Hence, using plasmonic enhancement has the potential to increase the efficiency and, therefore, the yield per volume.
Laura Torrente-Murciano questioned: You are using ethanol as a hole scavenger in these reaction systems. Have you tried to remove it? If no reaction (or very little) takes place in the absence of ethanol, it would confirm that although thermal effects might have a role in the resulting activity, the generation of hot electrons is the main pathway for the reaction to take place.
Madasamy Thangamuthu replied: Yes, we performed experiments in the absence of ethanol and observed that after some time the original photocurrent response as well as ammonia production efficiency is significantly reduced. This confirms that the chemical reaction takes place by electron injection (either by hot electron injection or near-field coupling where TiO2 generates electrons) into the N2 molecule rather than from a thermal contribution.
Reinhard J. Maurer said: The Haber-Bosch process is costly in terms of reaction conditions, but existing plants follow highly optimised processes. The main business-critical cost is the cost of methane. There is also the additional aspect of transitioning a heterogeneous catalytic reaction to an electrochemical setup. In this context, I wonder if nitrogen dissociation is the best reaction for which to develop hot-electron-enhancement procedures rather than finding new (potentially plasmonic) routes to deliver hydrogen for this reaction.
Madasamy Thangamuthu answered: Thank you for the comment. I agree that new plasmonic routes to produce hydrogen are as important as the nitrogen reduction reaction.
Amaresh Shukla asked: Regarding the Al nanotriangle structures, usually Al has an oxide layer. You haven’t shown this in the band gap or simulations. Does this not affect your predictions?
Madasamy Thangamuthu answered: We agree that the Al nanostructure has a 2-3 nm native oxide layer. We checked with SIE simulation that this 3 nm native oxide does not change the spectral features of the system.
Chao Zhan commented: The experimental conditions are a nitrogen-saturated solution (pH=4) containing 10% ethanol under illumination by a narrowband 365 nm beam with a 5 mW cm−2 power. The catalysts are TiO2 and Al–TiO2. All of these are as same as the reaction conditions for hydrogen evolution. In the plenty of reported papers of photocatalysis, the product is only hydrogen. And under acidic conditions, hydrogen evolution is more likely to happen. Nitrogen reduction is much more difficult than hydrogen evolution, and for synthetic ammonia isotope experiments are needed.
Madasamy Thangamuthu replied: Thank you for the very nice feedback. Isotopic experiments are definitely needed to support synthetic ammonia production. We agree that the electrons generated in our electrodes can also produce hydrogen gas as it needs less energy than ammonia production. Currently we are investigating it by measuring ammonia as well as hydrogen gas from the reaction chamber.
Sebastian Rejman enquired: What is your opinion about moving your system to the gas phase? In solution, the concentration of nitrogen is limited by its solubility and the proton concentration is limited by pH. Reactions in the gas phase allow for way higher concentrations.
Madasamy Thangamuthu responded: Indeed, nitrogen has a limited solubility in water and also, as mentioned, the proton concentration in solution is an issue. Our long-term goal is to produce ammonia in the gas phase.
Bartlomiej Jankiewicz opened a general discussion of the papers by Valérie Caps: The fabrication process that you describe in your paper seems to be quite random because it involves simply mixing nanoparticles with TiO2 particles. Did you observe any variation of the CH4 generation efficiency between various batches? Did you expose your photocatalyst to sunlight to see if you can produce methane?
Valérie Caps answered: The preparation of Au/TiO2 composites was achieved by colloidal deposition. In this method, a colloidal solution of gold is prepared by chemical reduction (using sodium borohydride as a reducing agent) before deposition on titania. Titania is then introduced, upon vigorous stirring, in this perfectly stable, i.e. homogeneous, solution of NaBH4/NaBR4 (R = O, OH)-protected Au NPs exhibiting a narrow size distribution (3.0 ± 0.7 nm as evidenced by TEM). In about 30 min, the burgundy colored gold solution becomes colorless and the white titania powder turns pink, highlighting adsorption of Au NPs onto titania. Adsorption is complete, 100% of Au NPs are adsorbed, as determined by ICP. The composite is washed with water to remove the NaBH4-derived protecting agent (Na, B < 100 ppm in the final composite as determined by ICP). The limited increase in Au NP size after deposition (to 3.6 ± 1.5 nm) indicates that Au NPs are well adsorbed and stabilized by the titania support. So the Au NP size is well controlled within the given standard deviation. Only their distribution over the support is random, adsorption occurring preferentially on titania surface defects. The standard deviation of CH4 production rates did not exceed 15% between batches. The reaction has not been carried out using direct sunlight as the energy source, only with the lamp mimicking the solar spectrum which contains less UV irradiation (1% vs. 4%).
Laura Torrente-Murciano commented: I really enjoyed this systematic study.
You claim that there is not a size effect of the gold nanoparticles in the 3 and 15 nm range, however, it is possible that there are competing effects as you increase the particle size that results in overall similar reactivities. Have you tried to normalise the activity per surface area? Such a calculation might reveal the real size effect on the catalytic activity.
Valérie Caps replied: Au/TiO2 composites containing 3 nm or 15 nm Au NPs on average exhibit similar mass-normalized CH4 production rates, both under visible irradiation and under solar irradiation. The increase in Au NP average size results in a decrease in metallic surface, i.e. the dispersion or fraction of surface atoms vs. total number of atoms in the particle, from about 37% to 10%, based on geometrical calculations for cubo-octahedral NPs, as described in the paper. This means that surface Au atoms are about 4 times more active in Au15nm/TiO2. Au15nm/TiO2 also exhibits the highest internal quantum yield of the study (0.012% under simulated solar irradiation). The subsequent decrease in CH4 production rate observed with Au50nm/TiO2 of about 80% correlates with the loss of gold dispersion (10% to 3%, i.e. 70% loss). The variation of the solar CH4 production rates normalized by the gold surface as a function of Au NP size thus consists of an increase from 3 nm to 15 nm and stabilization (plateau) from 15 nm to 50 nm. It seems that the co-catalytic effect may not be limiting and that the effect of metallic dispersion may be counterbalanced by the change in Au NP density (from about 90 NP/μm2 in Au3nm/TiO2 to 1 and ≪0.1 NP/μm2 in Au15nm/TiO2 and Au50nm/TiO2, respectively (Fig. 8 in our paper)), which affects competitive adsorption of the reactants (CO2vs. H2O), as suggested by changes in selectivity. It is noted that the average CH4 production rate observed over Au50nm/TiO2 is similar to that observed over bare TiO2. Hence the primary electron trap function (density effect) of the Au NPs appears inhibited in the solar process while the co-catalyst function is limited by the low Au dispersion (size effect). In the visible process, the variation of the solar CH4 production rates normalized by the gold surface as a function of Au NP size consists of an increase from 3 nm to 15 nm and an even more drastic increase from 15 nm to 50 nm, which clearly indicates that the plasmonic effect dominates over both electron trap and co-catalytic effects.
Jeremy Baumberg asked: How is the Au NP surface and interface changed after calcination? Does the particle change shape – are there sharp asperities on the nanoparticles? Does it matter if the calcination is above the melting point of the NPs (well below the bulk melting point)?
Valérie Caps responded: In the post-calcined series, the whole Au/TiO2 composite is submitted to calcination. Au NPs drastically increase in size from 3.6 nm in the uncalcined composite to 5.1 nm, 10.0 nm and 42 nm when calcination is performed at 450°C, 700°C and 900°C, respectively (Table 1 in our paper). This corresponds to a decrease in gold dispersion from 37% to 27%, 15% and 4%, respectively, based on geometrical calculations using cubo-octahedral shapes for the particles. The surface of the Au NPs thus dramatically decreases: only about 10% of the surface atoms remain on the surface after calcination at 900°C. Interaction of Au NPs with titania also dramatically increases with increasing calcination temperature, as shown by the significant red shift (up to 90 nm, Fig. 5a in our paper) in the position of the surface plasmon resonance (LSPR) maximum, which is much more moderate in the pre-calcined series (Fig. 5b in our paper), and by HRTEM images (Fig. 7 left in our paper), showing inclusion of Au atoms inside the titania lattice at 900°C and the presence of titania layers around the Au NPs. This strong metal–support interaction is key in stabilizing the supported Au NPs and in retaining discrete NPs despite the fact that calcination is performed at a much higher temperature than the melting temperature of the Au NPs (Tammann). The initially pseudo-round-shaped Au NPs become more and more facetted with increasing temperature, but the Au NPs retain a general cubo-octahedral geometry.
In the pre-calcined series, calcination of titania is performed in the absence of Au NPs, before their deposition. The impact of calcination on Au NP size, shape and interaction with titania is thus much more limited. Au NP size varies from 3.6 to 9 nm (Table 1 in our paper), the LSPR maximum is between 530 and 560 nm (Fig. 5b in our paper), and no shape change is detected by HRTEM (cubo-octahedral particles). The more modest, yet effective, variations are due to the change of chemical composition and structure of the titania surface which controls adsorption of the gold sol. In particular, longer adsorption times are needed to adsorb Au NPs onto the more perfect, more crystallized surfaces, which increases the probability for aggregation (cf. the larger standard deviations observed at higher calcination temperatures in the pre-calcined vs. post-calcined series, Table 1 in our paper).
Daniel Glass enquired: Your paper discusses the treatment of TiO2 films by calcination at high temperatures, as a result seeing different properties of the films. Can you comment on how the calcination effects possible phase changes of the TiO2?
Additionally, heating to high temperatures can introduce or change bulk and surface defects – can you comment on what effect this could have on the reactions described?
Valérie Caps answered: The uncalcined titania semi-conductor (SC) used in our Au/TiO2 composites exhibits 100% anatase phase. It remains 100% anatase upon calcination up to 700°C. Diffraction peaks related to the rutile phase, namely rutile (101), are observed only after calcination at 900°C (Fig. 4 in our paper). TEM (Fig. 7 right in our paper) and TRMC results (Fig. 12-14 in our paper) suggest that the anatase-to-rutile transition in both pre- and post-calcined Au/TiO2 composites is initiated from 700°C, leading to the elongation of anatase crystallites, but the crystallised rutile phase appears only at 900°C in the XRD spectra.
Concerning bulk and surface defects, increasing calcination temperature generally induces an increase in the crystallisation degree of TiO2, as shown by the narrowing of the diffraction peaks related to the anatase phase (Fig. 4 in our paper). So the density of bulk and surface defects is expected to decrease. This should positively impact the photocatalytic performances in the SC-driven solar process, as defects are known to act as recombination centers and to lower charge carrier lifetime. The surface-normalized solar activities of the composites indeed increase with increasing calcination temperature up to 700°C. However, narrowing of XRD peaks with increasing calcination temperature also implies an increase in the crystallite size, which can be estimated from the Debye-Scherrer law. The size of anatase crystallites increases from around 9 nm in the uncalcined sample to 15 nm, 45 nm and 80 nm in composites calcined at 450°C, 700°C and 900°C, respectively. The lower (surface-normalized) solar activities of the composites calcined at 900°C indicates that the beneficial effect expected from the increased crystallinity of the sample and decreased number of defects is counterbalanced by the particularly large crystallite size. The latter indeed significantly lengthens the path for the charge carriers generated within the SC bulk to reach the surface, which increases the probability of charge recombination before charge carriers can react at the surface and carry out any reaction (water oxidation to generate the active reducing agent, CO2 reduction). After calcination at 900°C, this effect becomes predominant also over the potential increase in charge carrier lifetime expected from the appearance of the anatase-rutile heterojunction.
Hongxing Xu addressed Valérie Caps and Madasamy Thangamuthu: How close is your study to industrialization in terms of both the efficiency and the cost?
Valérie Caps replied: The best internal quantum yield observed in our study under simulated solar irradiation is 0.012%. It is likely to increase to a certain extent under real solar irradiation, due to the higher amount of UV photons (4% of the full solar spectrum vs. only 1% in the lamp emission spectrum). This value is however at least two orders of magnitude lower than what would be required for industrialization.
Madasamy Thangamuthu answered: The efficiency of the industrial process is 10-15% and we are at 0.06% and hence our study is far from industrialisation. Furthermore, in terms of cost, it is too early to compare. More optimization is required to move towards industrialisation.
Javier Aizpurua questioned: I am wondering about the spatial resolution of these chemical effects reported in this session. We all assume an improvement of product generation connected to the near field induced at some spot, but we mostly trace the far field. Couldn’t we put more emphasis in spatially resolved techniques that tell us something about where particularly designed chemistry is happening in space? Can we identify chemical hotspots using these techniques?
Valérie Caps responded: We can get indirect information about the location of the chemical reaction on e.g. Au/TiO2 composites by using an approach such as the one described in our paper. Indeed, by controlling the size of metal nanoparticles (via size-controlled synthesis such as colloidal deposition which leads to narrow size distributions) and surface area of the support (by calcination), the average density of plasmonic objects may be controlled. Distribution of the particles over the support remains random. Nevertheless, the way in which Au NP density impacts the photocatalytic properties (activity + selectivity) of the composite under visible vs. solar (UV + visible) irradiation gives us information on the role of Au–TiO2 perimeter sites (here in the adsorption of CO2 in the solar process for example) vs. the role of longer range chemical processes (such as hydrogen spillover from the titania surface to Au NPs) and titania surface (ensuring water activation). Besides, state of the art developments of EELS-STEM techniques now allow the mapping of a surface plasmon on single metal nanoparticles using sub-nm resolved EELS spectrum-imaging. We intend to perform such a study within the French Microscopy Network METSA in order to complete our experimental study with more direct local information on where reactions take place.
Emiliano Cortes contributed: There are some techniques that could identify the places where the reaction is taking place within a single nanostructure. This has been for a long time an important goal for the whole heterogeneous catalysis community (see for example ref. 1). However, the ultimate resolution may need to reach the atomic scale to disentangle the role of step-edges, surface-defects and other (low coordinated) atoms in the surface, which are usually claimed to be the most reactive places on the surface of the material.2 We have used a series of different approaches (including chemical tracking of reactive hot-spots, super-resolution microscopy, opto-electrochemistry, etc.) to try to resolve in energy and space these transformations.
1 C.-Y. Wu, W. J. Wolf, Y. Levartovsky, H. A. Bechtel, M. C. Martin, F. D. Toste and E. Gross, Nature, 2017, 541, 511–515.
2 J.-H. Zhong, X. Jin, L. Meng, X. Wang, H. S. Su, Z. L. Yang, C. T. Williams and B. Ren, Nat. Nanotechnol., 2017, 12, 132–136.
Sebastian Schlücker added: I agree that we need experimental techniques probing reactivity with nanoscale resolution. Nanoscale IR spectroscopy, tip-enhanced Raman scattering, and super-resolution fluorescence microscopy are a few examples of experimental approaches which can achieve this.
Jeremy Baumberg noted: This is a very difficult challenge, and electrochemical TERS is perhaps one technique that will be able to access such information, but it is not high throughput and not easy to extract useful chemistry of hot electrons. The use of the Nanoparticle-on-Mirror (NPoM) system allows creation of precise nm-scale hotspots and also may deliver better defined spatial resolution.
Niclas Sven Mueller opened a general discussion of the paper by Junyang Huang: In your paper you estimated the local plasmonic heating efficiency to be 1.5 times higher in the gap region than in the area 10 nm around the aggregate. How did you get to this estimate? Did you consider the effect of plasmonic hot spots between the nanoparticles?
Junyang Huang replied: Thank you. This estimation is based on the steady-state Coulomb temperature profile from the metal surface, assuming the metal has a uniform temperature. The concentrated optical field in the hot-spot is likely to contribute additional heating but is not included in this estimation.
Hongxing Xu questioned: Is this a dimer or an aggregate? How easy is it to get the dimer together?
Junyang Huang responded: The SERS substrates are based on AuNP aggregates bridged by CB[n] molecular spacers. The resultant aggregates generate 400-fold near-field enhancement (|E|/|E0|) by trapping incident light in the hot-spot at resonant frequencies and are consequently utilized as SERS substrates for their 1010-fold Raman enhancement.1 Fabricating dimeric colloidal structures through self-assembly approaches is possible but known to be challenging, especially if an open interface is desired.
1 C. Carnegie, R. Chikkaraddy, F. Benz, B. de Nijs, W. M. Deacon, M. Horton, W. Wang, C. Readman, S. J. Barrow, O. A. Scherman and J. J. Baumberg, ACS Photonics, 2017, 4, 2681–2686.
Sebastian Schlücker queried: In your supramolecular chemistry CB-based approach to make dimers what is the approximate size range (min to max) of molecules that fit into the hot spot and still yield single-molecule SERS?
Junyang Huang answered: In this presented work, the aggregates are used to probe an ensemble of molecules in a large number of hot-spots, however, CB[n]s have been shown to facilitate single-molecule SERS.1 There have been extensive studies carried out on the topic of encapsulation and association of various sized guest molecules in a range of CB[n] cavities (i.e. n = 5,6,7,8,10).2 However, it has been reported that molecules which cannot fit into CB[n] cavities can also be incorporated interstitially in the hotspot between nanoparticles and yield SERS detection.3
1 B. de Nijs, F. Benz, S. J. Barrow, D. O. Sigle, R. Chikkaraddy, A. Palma, C. Carnegie, M. Kamp, R. Sundararaman, P. Narang, O. A. Scherman and J. J. Baumberg, Nat. Commun., 2017, 8, 994.
2 S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406.
3 B. de Nijs, M. Kamp, I. Szabó, S. J. Barrow, F. Benz, G. Wu, C. Carnegie, R. Chikkaraddy, W. Wang, W. M. Deacon, E. Rosta, J. J. Baumberg and O. A. Scherman, Faraday Discuss., 2017, 205, 505–515.
Laura Torrente-Murciano commented: It is very impressive the way that you control and manipulate the space between the Au NPs in the agglomerates. Do you think that the molecular spacer might have additional roles beyond keeping the Au nanoparticles at specific distances? Can they accept hot carriers as demonstrated previously in the case of adsorbed molecules?
Junyang Huang replied: Thank you for the comments. We show that the presented optically controlled redox chemistry can be achieved with both CB[n]-linked aggregates and salt-induced aggregates. However, there are differences between the two types of aggregates which we take into consideration. It is found that CB[n]-induced aggregates exhibit an extended fractal morphology while using salt results in more compact clusters of nanoparticles. This could influence the in-coupling and out-coupling of light into and from the aggregates, which could, in turn, change the heating efficiency and Raman response. Using CB[n] also allows us to obtain nanoparticle aggregates at a lower salt concentration. The ionic concentration of the suspension can play an important role as the dithionite ions are negatively charged and how easily they can approach the nanoparticle surfaces depends on the Debye length. In addition, the host–guest chemistry between CB[7] and methyl viologen could also influence the redox dynamics. It has been demonstrated in previous work that using CB[n] to capture methyl viologen molecules in plasmonic junctions allows us to observe hot electrons hopping through the junction through the molecule and the subsequent redox chemistry on a single molecule can be tracked by SERS.1
1 B. de Nijs, F. Benz, S. J. Barrow, D. O. Sigle, R. Chikkaraddy, A. Palma, C. Carnegie, M. Kamp, R. Sundararaman, P. Narang, O. A. Scherman and J. J. Baumberg, Nat. Commun., 2017, 8, 994.
Hongxing Xu asked: What is the advantage of using SERS to study hot electrons compared to other technologies?
Junyang Huang responded: Most analytical techniques probe chemical changes in the bulk of the sample or reactor, whereas SERS allows for the probing of chemical properties at or near metal interfaces where charge carrier transfer and heterogeneous catalysis occurs. The immense signal enhancement obtainable using SERS for these interface signals means that they far exceed any Raman signal from the bulk environment. In addition, the large amount of signal allows for real-time observation of chemical reactions,1,2 and it allows for small sample volumes to be probed enabling rapid screening of substrates.
1 B. de Nijs, F. Benz, S. J. Barrow, D. O. Sigle, R. Chikkaraddy, A. Palma, C. Carnegie, M. Kamp, R. Sundararaman, P. Narang, O. A. Scherman and J. J. Baumberg, Nat. Commun., 2017, 8, 994.
2 E. A. Sprague-Klein, B. Negru, L. R. Madison, S. C. Coste, B. K. Rugg, A. M. Felts, M. O. McAnally, M. Banik, V. A. Apkarian, M. R. Wasielewski, M. A. Ratner, T. Seideman, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2018, 140, 10583–10592.
Zachary Schultz said: In your nanoparticle aggregates, the chromophores all undergo redox. Can you comment on the nature of the SERS signal, whether this is resonantly enhanced by either an electronic state or a charge-transfer interaction with the nanoparticles? Also, at what wavelength are your studies performed?
Junyang Huang answered: Thank you for addressing this point. The reduction of the methyl viologen is a result of charge transfer from the chemical fuel dithionite. The Raman intensity of the methyl viologen radical cation (MV+˙) indeed experiences an electronic resonance enhancement in the Raman scattering. The MV+˙ radical has a broad absorption from 500 nm to 700 nm which is close to the excitation wavelength used in the experiments (785 nm). However, the methyl viologen dication (MV2+) does not absorb in the visible range and therefore does not experience the same enhancement.
Jeremy Baumberg opened a general discussion of the paper by Zachary Schultz: This is really interesting work, although here the Ag nanostructures are rather complex to understand. My question is how it might be possible to distinguish surface rectification compared to light-induced charging at the interface? I am trying to understand what might be clear evidence for the χ(2) effect you are discussing. Also, you use a linear fit on your Stark shifts, while it definitely looks much more like a quadratic dependence here. You noted it is different on Au, and it might be nice to include this also, together with a comment about why these are not linear.
Zachary Schultz replied: We have looked at the Stark shift from nitrile (-CN) groups on a number of surfaces. The shift in the frequency corresponds to the electric field experienced by the CN bond. The work on Au was published last year.1 The behavior on Au is more linear; however, the linearity has been attributed to chemistry that occurs on the surface associated with double layer phenomena. As chemical reactions alter the surface chemistry, for example oxidation of the Ag, the linearity of the shift vs. applied potential decreases. Au, a chemically more inert surface, shows better linearity in the double layer region. At potentials where electron transfer reactions are observed on Au, the linearity also goes away. The connection to χ(2) originates from further back, in our previous work;2 where plotting SERS intensity against the CN stretch frequency showed a dependence that resembled electric field induced second harmonic generation trends (see ref. 3). We are currently working to correlate the second harmonic intensity to the Stark Spectroscopy results we report. We believe this will address the correlation to χ(2) more directly.
1 D. A. Nelson and Z. D. Schultz, J. Phys. Chem. C, 2018, 122, 8581–8588.
2 D. T. Kwasnieski , H. Wang and Z. D. Schultz , Chem. Sci., 2015, 6 , 4484–4494.
3 R. M. Corn, M. Romagnoli, M. D. Levenson and M. R. Philpott, Chem. Phys. Lett., 1984, 106, 30–35.
Hongxing Xu asked: Can you explain what you mean by ‘rectify the field’? Is it a general effect? What is the efficiency?
Zachary Schultz responded: ‘Rectify the field’ means convert the AC field into a DC field. In this case the AC plasmon resonance, and associated field, drives a DC potential on the surface. Optical rectification is a 2nd order nonlinear process and has been previously reported,1 however, it has not been widely appreciated in the plasmonic community. Our group and a few others have reported this effect in plasmonic materials. As commented on in this discussion by Professor Baumberg, there are still some remaining questions about the origin of the DC potential, but optical rectification is consistent with multiple observations. It is a general effect, though the exact conditions that optimize the process in plasmonic materials is still an open question. In regards to efficiency, this is difficult to address. I think we would have to agree on a definition of efficiency first.
1 Y.R. Shen, The Principles of Nonlinear Optics, Wiley, 1984.
Sebastian Schlücker enquired: Have you thought about mapping out the Stark effect at the nanoscale using molecular rulers, e.g., by changing the distance between the nitrile group and the metal surface?
Zachary Schultz replied: This is something we have considered, and others have tried previously. We have used different length thiolate molecules to change the gap distance between surfaces, but the nitrile is always at a fixed distance. Moving the nitrile within the gap may show interesting behavior. The challenge is the availability of molecules, but this is something that could be overcome.
Bart de Nijs queried: In your article you provide a brief description on how the self-assembled monolayers (SAMs) are made. Have you done any characterization on the formed SAMs, could you say something about for example the grafting density of the SAM angle, or are these parameters of less importance for this work?
Zachary Schultz answered: We have not done extensive characterization of the self-assembled monolayers. We use a high concentration of thiols to promote a dense, or complete layer. The roughness of the surface makes detailed characterization complicated. In general, we use replicate measurements for verification as defects should be random and more difficult to reproduce.
Javier Aizpurua commented: Your work involves non-linearities which are very interesting. We have recently reported theoretically the non-linear response in plasmonic gaps, where we have identified that the tunnelling current is a key source of non-linearity of the response, for instance in high-harmonic generation.1 Since rectification is connected with these non-linearities, of type χ(2), I think it would be very interesting to understand also the optical rectification originating from the tunneling current. On another issue, could we use some specific molecule that is fluorescent, where this kind of rectifying effect could change the electronic structure, and this change manifested in the fluorescence?
1 G. Aguirregabiria, D. C. Marinica, R. Esteban, A. K. Kazansky, J. Aizpurua and A. G. Borisov, Phys. Rev. B, 2018, 97, 115430.
Zachary Schultz responded: One could look at changes in fluorescence intensity that originate from the rectified fields on the surface. A voltage sensitive dye might provide signal, provided quenching at the metal surface can be avoided. Rather than fluorescence we have reported on changes in the Raman signal that originate from photoreactions on the surface, specifically the conversion of aminothiophenol (or nitrothiophenol) to dimercaptoazobenzene. These reactions are believed to result from hot-electrons, but our work shows that the rectified current alters the rate at which they occur. I would expect the same to be true for the fluorescence indicators of electron transfer. These chemical reactions require electron transfer, which generally implicates hot electrons. The Stark shift from the nitrile is a measure of the surface charge, which provides insight into the rectified field. As we noted in response to the earlier question about χ(2), we are working to better correlate these effects.
Hongxing Xu asked: Since SERS is a very sensitive technology, is it also good for the study of catalysis? What is your opinion on SERS for general sensing applications so far?
Junyang Huang answered: In order to facilitate further understanding of the fundamental mechanism and catalytic reactions on the active sites during catalysis processes, it is crucial that structural and dynamic insights are acquired on a molecular level. A range of operando methodologies have been developed to track catalysis processes under realistic working conditions, from which SERS is one of the most informative ones, as it provides dynamic chemical information down to a single molecular level and can be integrated with other methods such as electron microscopy and scanning probe techniques to unfold molecular dynamics with a high spatial resolution. Recently a few demonstrations have shown that SERS is highly promising for understanding catalysis processes.1-3
1 J.-H. Zhong, X. Jin, L. Meng, X. Wang, H. S. Su, Z. L. Yang, C. T. Williams and B. Ren, Nat. Nanotechnol., 2017, 12, 132–136.
2 H. Zhang, C. Wang, H.-L. Sun, G. Fu, S. Chen, Y.-J. Zhang, B.-H. Chen, J. R. Anema, Z.-L. Yang, J.-F. Li and Z.-Q. Tian, Nat. Commun., 2017, 8, 15447.
3 J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. Miguel Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2019, 4, 60–67.
Zachary Schultz answered: SERS is something I am very interested in and believe it is one tool for studying catalysis. In SERS the importance of junctions and chemical enhancements for some molecules may limit the utility. For example, the surface science community has studied the dependence of crystal planes for adsorption and reactivity, but this is difficult to study in nanometer sized junctions. Some molecules have very poor cross-sections and, even with SERS, are hard to detect at low concentrations. For other molecules and reactions, I think SERS is an invaluable tool for monitoring chemical transformations. I also think the work being done with tip-enhanced Raman imaging, though a more complex experiment, has real potential to elucidate the connections between chemical reactions and surface sites.
Chao Zhan added: PEMS which has been widely investigated over the past four decades can enlighten our understanding and development of PMCR. In its earliest incarnations PMCR was often studied using PERS. In such experiments the nanostructure-based plasmonic enhancement was exploited in two ways: enabling a strong photochemical response, and allowing the time-dependent concentration of the products, often produced in meagre quantities, to be reliably measured.
Nevertheless, PEMS and PMCR are different in crucial ways. Usually, there are more challenges in PMCR compared with PEMS because of the molecular transformation. For example, PEMS measurements obtain the better sensitivity when molecules bond to the plasmonic nanostructure, while the over-strong bonding/adsorption causes problems for PMCR, like the blocking of actives sites. Thus, PERS is useful but careful design is needed.
Bartlomiej Jankiewicz remarked: A very interesting example of problems with applications of SERS is the history of Klarite substrate, which I am sure has been used by many researchers gathered at this Faraday Discussion. It has been withdrawn from the market because the main customers of the company producing Klarite were researchers who were just using it to compare the commercially available substrate with their home-made SERS substrates. Because of that production of Klarite was unprofitable and has been stopped. But I would like to mention a very recent initiative, the NATO Exploratory Team SET-ET-109 “Surface-enhanced Raman Scattering for Defense Applications”. This team, which was proposed by colleagues from the Edgewood Chemical Biological Center (ECBC) and which includes scientist from several countries, will attempt to answer questions needed for SERS to be applicable to defense applications.
Zachary Schultz replied: The interesting thing about Klarite is that it was originally designed to be a reproducible substrate. I think our understanding of SERS has advanced and a higher signal and reproducible substrate could be produced. I think the outstanding question for SERS applications is understanding why some molecules show enhancement and others do not. For example, rhodamine 6G gives an excellent signal but sugars are difficult to detect. Perhaps a better understanding of chemical effects is what needs to be studied.
Jeremy Baumberg continued: To add to this, as the inventor and initial commercialiser of Klarite, the real challenge was to create a SERS substrate that was completely reproducible, which is why it was of real benefit to the community. In fact it was purchased in large quantity also by bioresearchers working in this area.
One of the main challenges is not just to create a quantitative and reproducible substrate. We note that the Au:CB colloidal aggregate system we now use is a similar low-cost but bottom-up version, see various papers at https://www.np.phy.cam.ac.uk/publications, including most recently ref. 1. For Klarite a major challenge was to package such a high-fidelity sensor, so that it could be stored and deployed when needed, without the sensor surface showing contamination already. Packaging is to be ignored at peril when developing sensors! We managed to achieve this (first in Mesophotonics, and then transferred to Renishaw), but often people complained about contamination when they opened their sensor, but it merely picked up whatever they had at trace concentrations in their own labs! The advantage of the new colloidal systems is that the sensor is only self-assembled when it is needed (though remaining quantitative and reproducible), and then the component nanoparticles and chemicals can be kept clean much more easily. Our forthcoming publication will detail this aspect.
1 B. de Nijs, M. Kamp, I. Szabó, S. J. Barrow, F. Benz, G. Wu, C. Carnegie, R. Chikkaraddy, W. Wang, W. M. Deacon, E. Rosta, J. J. Baumberg and O. A. Scherman, Faraday Discuss., 2017, 205, 505–515.
Laura Fabris commented: If one reads the literature on SERS, one realizes that the majority of publications cover the application of SERS to detect some kind of analyte, while fundamental studies have recently become extremely scarce. We all agree that SERS can be quite effective for qualitative detection even by simply adding salts and inducing nanoparticle aggregation around the analyte. However, this approach has led to inconsistencies in the enhancement factors and detection platforms, drastically hindering the applicability of SERS beyond the research lab. It is therefore important to modify our approach so that we can produce results that lead to (at least) some semiquantitative results. I believe that identifying materials design rules to obtain materials that produce homogeneous and reproducible enhancements is mandatory. In addition, designing sensing platforms that are simple to set up would be the way to go to extend the applicability of the technique. If nothing changes, we will be still here in twenty years discussing why SERS has not reached the levels of applicability that it had promised many years ago. We need to start learning from our mistakes and address them systematically with quantifiable metrics that can confirm our success.
Zachary Schultz noted: I think this echoes the earlier comments in the meeting to define standards. We discussed efficiencies for catalysis, but for SERS based sensing, an agreed upon standard for enhancement would also be useful.
Conflicts of interest
There are no conflicts to declare.| This journal is © The Royal Society of Chemistry 2019 |