
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular triazine-based carborane-containing carboxylic acids – synthesis and characterisation of potential boron neutron capture therapy agents made of readily accessible building blocks†
Martin
Kellert
 a,
Dennis J.
Worm
c,
Paul
Hoppenz
c,
Menyhárt B.
Sárosi
a,
Peter
Lönnecke
a,
Bernd
Riedl
b,
Johannes
Koebberling
b,
Annette G.
Beck-Sickinger
c and
Evamarie
Hey-Hawkins
*a
a,
Dennis J.
Worm
c,
Paul
Hoppenz
c,
Menyhárt B.
Sárosi
a,
Peter
Lönnecke
a,
Bernd
Riedl
b,
Johannes
Koebberling
b,
Annette G.
Beck-Sickinger
c and
Evamarie
Hey-Hawkins
*a
aLeipzig University, Faculty of Chemistry and Mineralogy, Institute of Inorganic Chemistry, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bBayer AG, Aprather Weg 18A, 42113 Wuppertal, Germany
cLeipzig University, Faculty of Life Sciences, Institute of Biochemistry, Brüderstrasse 34, 04103 Leipzig, Germany
First published on 14th June 2019
Abstract
Based on a modular combination of s-triazine, the well-known 9-mercapto-1,7-dicarba-closo-dodecaborane(12) and commercially available carboxylic acids, namely thioglycolic acid, glycine, and Nα-Boc-L-lysine, several carboxylic acid derivatives were synthesised and fully characterised. The thioglycolic acid derivative was introduced into a peptide hormone by solid phase peptide synthesis. High activity and selective internalisation into peptide receptor-expressing cells was observed. With a very high boron content of twenty boron atoms, these derivatives are interesting as selective Boron Neutron Capture Therapy (BNCT) agents.
Introduction
For almost six decades, boron neutron capture therapy (BNCT) has been stated to be a very promising method for cancer treatment.1 It combines two non-toxic components, boron-10 containing reagents and thermal or epithermal neutrons, to produce cytotoxic species, which are able to destroy malignant tissue. Boron-10 containing drugs bearing a tumour-selective moiety to address the infected site are highly advantageous.2,3–5 Ideally, the non-toxic bioconjugate accumulates selectively in the malignant tissues in the required amount of 20–30 μg g−1 tumour tissue and can then be irradiated with thermal or epithermal neutrons.5,6 It depends on the biomolecule if the BNCT drug is just accumulated in the tumour tissue or internalised into the cancer cells; internalisation of course increases the efficacy of this cancer treatment.7,8,9–16 The particles which are generated on neutron capture are lithium and helium nuclei (α particles) with a high linear energy transfer (LET).5,17 These high-energy particles have a mean free path of about 5 to 10 μm, and thus a limited radius of destructive action.3–5 While the combination of suitable boron-rich molecules with tumour-selective biomolecules opens up a very selective tumour therapy which only affects malignant tissue and spares normal tissue,7,9,11–14,18 there are still some major challenges, including, but not limited to, the selectivity of the chosen biomolecules for a specific type of tumour, the required high concentration of boron-10 in the cancer cell, the water solubility of the final bioconjugate, and the neutron beam quality,19 which are the focus of recent studies.10,14–16,20,21Some ortho- and meta-carboranyl s-triazine derivatives were previously synthesised as potential BNCT agents (Chart 1). However, these compounds show only limited water solubility and cellular boron uptake due to unspecific accumulation, caused by the lack of tumour-selective moieties.22–28
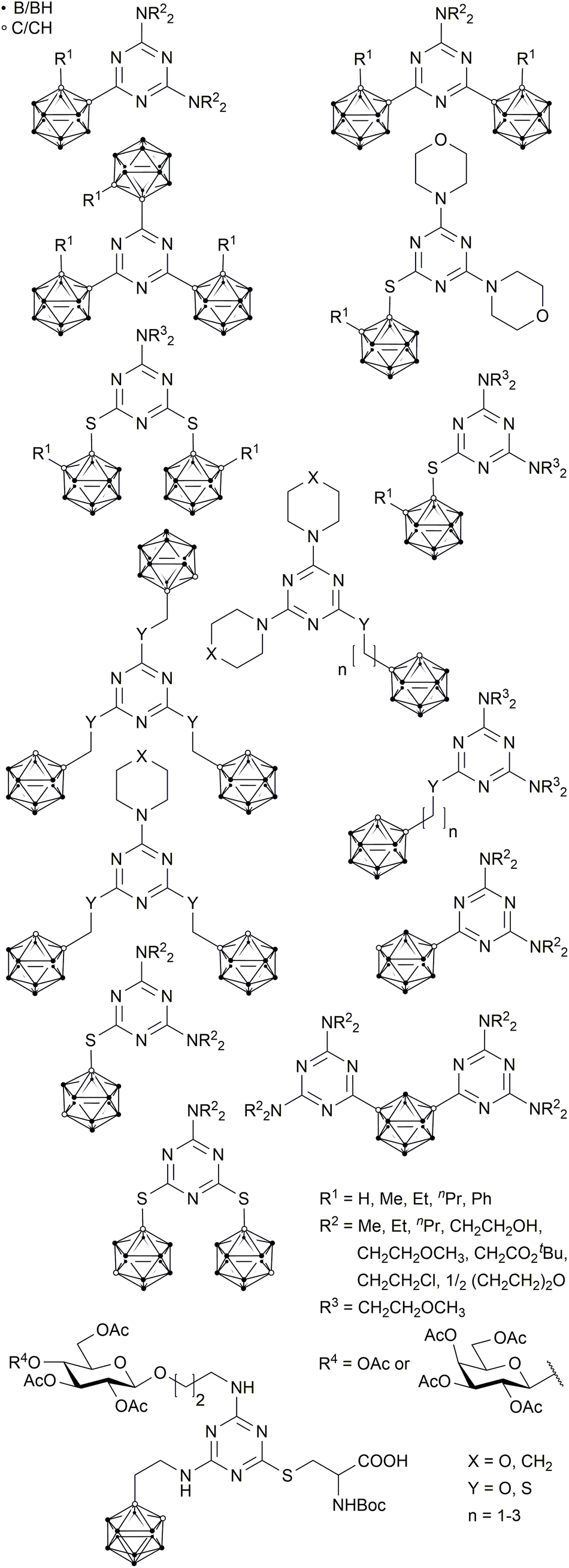 | ||
| Chart 1 Known s-triazine-based carborane derivatives.22–28,31 | ||
Another derivative, reported by Ronchi et al., with a β-D-glucose substituent, an ortho-carborane cluster and a cysteine moiety (Chart 1) is remarkably water-soluble and has an amino acid function suitable for coupling reactions with biomolecules. However, the number of synthetic steps required to prepare this compound is rather high (8 steps); thus, the overall yield tends to be very small.29
Here, we present a modular system based on s-triazine, where readily accessible starting materials are combined with meta-carboranes to boron-rich compounds, which can be easily coupled with suitable biomolecules to generate promising BNCT agents. We used a neuropeptide Y (NPY) analogue that selectively binds to the human Y1 receptor (hY1R), which is highly expressed on breast cancer cells.30,31
Results and discussion
The modular system is based on three components (Fig. 1).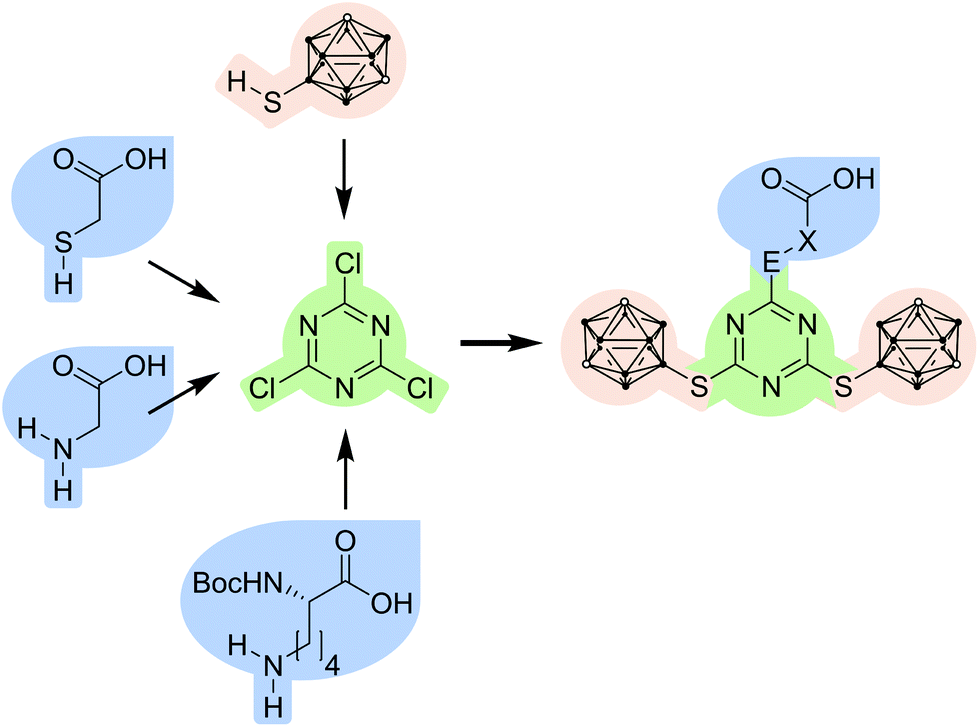 | ||
| Fig. 1 Combination of a carborane-based building block (pale red) and carboxylic acid derivatives (blue) with cyanuric chloride (green) as a central branching unit and scaffold to give s-triazine-based carborane-containing carboxylic acids (E = NH or S; X = CH2 or (CH2)4CHNHBoc (Boc = tert-butoxycarbonyl)). | ||
The first component, 1,3,5-triazine or s-triazine, is widely used, not only in biologically active compounds.32,33–35 It is easily obtained by substitution reactions of cyanuric chloride; the substitution pattern can be controlled by stoichiometry, temperature and the respective nucleophile.33–35,36,37 Several carborane-containing s-triazine derivatives have been prepared, also as potential BNCT agents (Chart 1).22,23,25–29
The second component are meta-carborane (1,7-dicarba-closo-dodecaborane(12)) derivatives. One problem with ortho-carborane derivatives is their facile deboronation under basic conditions.38–40 Therefore, we have employed meta-carborane which is much more stable under basic conditions in aqueous media, with a special focus on 9-substituted meta-carboranes.38,39,41,42 9-Mercapto-1,7-dicarba-closo-dodecaborane(12) has a highly nucleophilic thiol group due to the electron-withdrawing character at the 9-position,14,42–45 and was therefore favoured over 1-mercapto-meta-carborane in this approach. This substitution pattern also allows further functionalisation at the cluster carbon atoms for tuning of specific properties, such as water solubility or improved selectivity towards specific cancer cell types, or even the introduction of transition metal complexes or other moieties which can be used for imaging or localisation of the administered bioconjugate.45 This is an additional advantage in comparison with known carborane-triazine compounds (Chart 1).
The third component are commercially available carboxylic acids with nucleophilic side chains (S, O, N atoms), such as 2-mercaptoacetic acid (thioglycolic acid), 2-hydroxyethanoic acid (glycolic acid), aminoacetic acid (glycine) and Nα-(tert-butoxycarbonyl)-L-lysine (Nα-Boc-L-lysine). As the S, O or N moiety has a higher nucleophilicity as the respective carboxylate, which is produced under basic conditions, the coupling reaction with the s-triazine derivative is straightforward without requiring comprehensive protection strategies; only the α-amino group in lysine needs to be Boc protected to avoid undesired side reactions.
Synthesis and characterisation
9-Mercapto-1,7-dicarba-closo-dodecaborane(12) (2)46 is a suitable nucleophile for substitution reactions with cyanuric chloride.42,43 The reaction was carried out under inert conditions with N,N-diisopropylethylamine (DIPEA) as base (Scheme 1). Attempts to use caesium carbonate and tetra-n-butylammonium chloride in tetrahydrofuran were unsuccessful. The substitution ratio was controlled by the stoichiometry (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and temperature.37 The reaction conditions could be optimised to obtain 2-chloro-4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine (3) in quantitative yield, which is much better than reported for related 1-mercapto-carboranyl substituted s-triazine derivatives.25,26 The disubstituted s-triazine derivative 3 is more stable than cyanuric chloride itself and can even be purified by column chromatography. The next step was the replacement of the remaining chloro substituent in 3 with a functional group that would allow coupling of the carborane-rich molecule with a tumour-selective biomolecule. For this purpose, glycolic acid, thioglycolic acid, glycine and Nα-Boc-L-lysine were used to modify 3. After optimisation of the reaction conditions (Table S2, ESI†), 2-{[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]thio}acetic acid (4), [4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]glycine (5) and N6-[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]-N2-(tert-butoxycarbonyl)-L-lysine (6) could be successfully synthesised in moderate to good yields from 3 and thioglycolic acid, glycine, and Nα-Boc-L-lysine, respectively (Scheme 1). The L-lysine derivative 6 is an artificial amino acid and could even be employed directly in peptide synthesis.20 However, the synthesis of 2-{[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]oxy}acetic acid by reaction of 3 with glycolic acid was not successful. One possible reason could be the lower nucleophilicity of oxygen in comparison to sulfur and nitrogen. Compounds 3–6 were fully characterised by NMR and IR spectroscopy, and mass spectrometry. Furthermore, single crystals suitable for X-ray structure determination could be obtained of compound 3 from acetone solution, 4 from methanol solution and 5 from a mixture of chloroform and acetone.
:1) and temperature.37 The reaction conditions could be optimised to obtain 2-chloro-4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine (3) in quantitative yield, which is much better than reported for related 1-mercapto-carboranyl substituted s-triazine derivatives.25,26 The disubstituted s-triazine derivative 3 is more stable than cyanuric chloride itself and can even be purified by column chromatography. The next step was the replacement of the remaining chloro substituent in 3 with a functional group that would allow coupling of the carborane-rich molecule with a tumour-selective biomolecule. For this purpose, glycolic acid, thioglycolic acid, glycine and Nα-Boc-L-lysine were used to modify 3. After optimisation of the reaction conditions (Table S2, ESI†), 2-{[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]thio}acetic acid (4), [4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]glycine (5) and N6-[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]-N2-(tert-butoxycarbonyl)-L-lysine (6) could be successfully synthesised in moderate to good yields from 3 and thioglycolic acid, glycine, and Nα-Boc-L-lysine, respectively (Scheme 1). The L-lysine derivative 6 is an artificial amino acid and could even be employed directly in peptide synthesis.20 However, the synthesis of 2-{[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]oxy}acetic acid by reaction of 3 with glycolic acid was not successful. One possible reason could be the lower nucleophilicity of oxygen in comparison to sulfur and nitrogen. Compounds 3–6 were fully characterised by NMR and IR spectroscopy, and mass spectrometry. Furthermore, single crystals suitable for X-ray structure determination could be obtained of compound 3 from acetone solution, 4 from methanol solution and 5 from a mixture of chloroform and acetone.
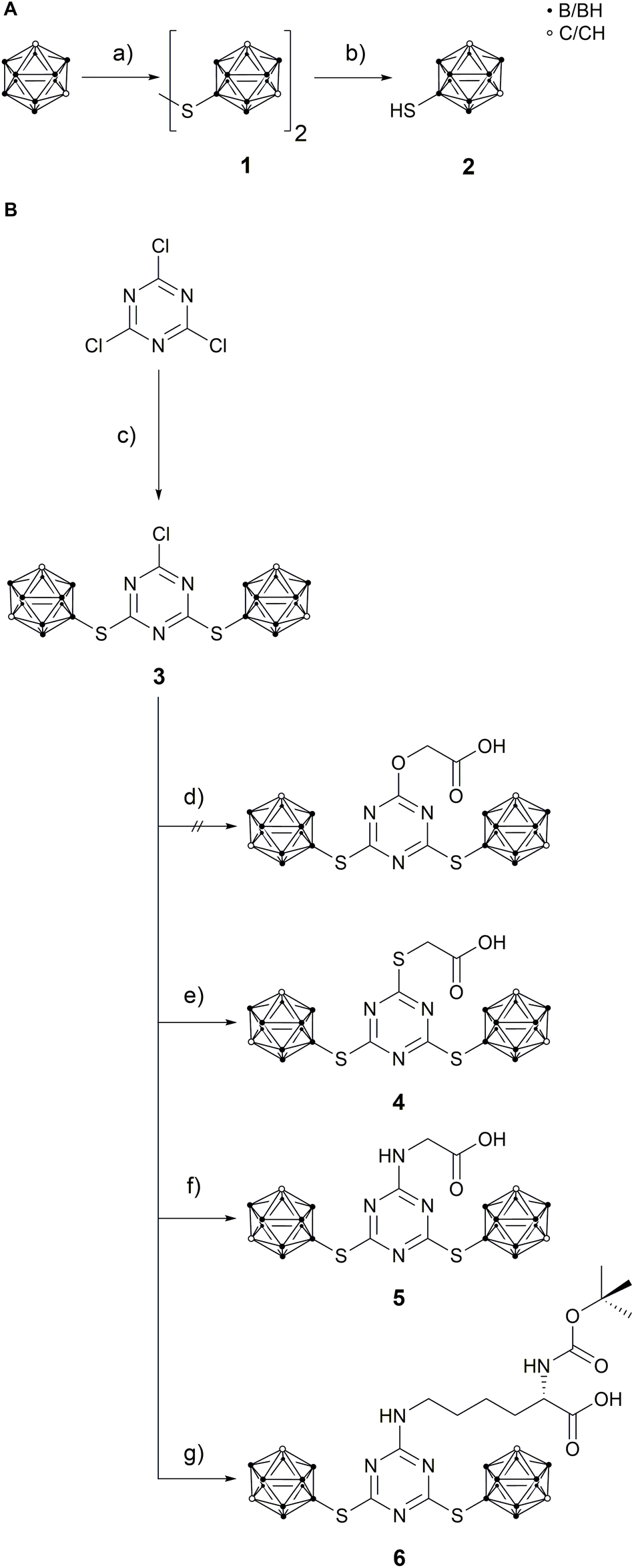 | ||
| Scheme 1 A: Synthesis of 9-mercapto-1,7-dicarba-closo-dodecaborane(12) (2). (a) S2Cl2, AlCl3, DCM, 40 °C, 4 h, not isolated;48 (b) Zn, HCl, HOAc, 120 °C, 4 h, 71%;48 B: synthetic route to compounds 4–6. (c) 2, N,N-diisopropylethylamine (DIPEA), acetonitrile (MeCN), reflux, 5 h, quant.; (d) glycolic acid, NaOH, MeCN/H2O (1:1.25), 85 °C, 18 h, no product obtained; (e) thioglycolic acid, DIPEA, MeCN, reflux, 3 h, 27%; (f) glycine, NaOH, MeCN/H2O/ethyl acetate (EtOAc) (3.33:2:1), 95 °C, 6 h, 60%; (g) Nα-Boc-L-lysine, NaOH, MeCN/H2O (1:1), 85 °C, 6 h, 69%. | ||
In the molecular structure of 3 (Fig. 2), one carborane cage is slightly moved out of the ring plane caused by packing effects and intramolecular repulsion between the carborane clusters. The triazine ring is rather distorted with CNC and NCN bond angles varying from 112.7(1)° to 114.2(1)° and 125.6(1)° to 128.6(2)°, respectively. This is not unusual and was already described for other s-triazine derivatives.47
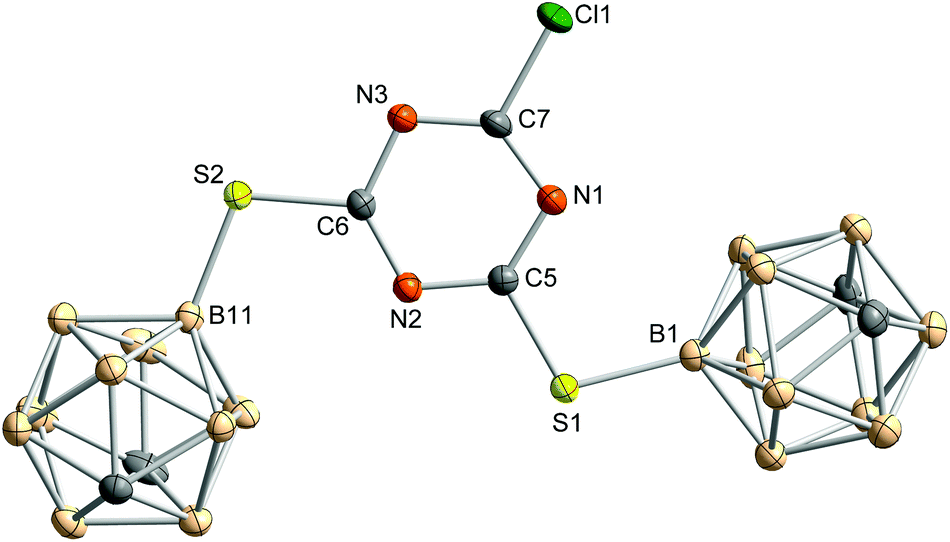 | ||
| Fig. 2 Molecular structure of 2-chloro-4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine (3). Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 50% level of probability. Selected bond lengths, distances (pm) and bond angles (°): Cl1–C7 172.7(2), S1–C5 174.2(2), S2–C6 173.4(2), S1–B1 187.2(2), S2–B11 185.9(2), ◆⋯N1 0.4, ◆⋯C5 1.5, ◆⋯N2 1.2, ◆⋯C6 0.0, ◆⋯N3 1.0, ◆⋯C7 0.9, ◆⋯S1 4.9, ◆⋯S2 4.3, ◆⋯Cl1 3.8, ◆⋯B1 41.0, ◆⋯B11 1.2; C5–S1–B1 106.1(1), C6–S2–B11 107.6(1); ◆ represents the mean plane of the triazine core, formed by N1, C5, N2, C6, N3 and C7. | ||
In the molecular structure of 4 (Fig. 3), the carborane clusters are slightly moved out of the plane of the triazine ring, as was already observed for 3. In crystals of 4, a mixture of carboxylic acid derivative and methyl ester (ca. 68:32) is present. Apparently, on recrystallisation from methanol, formation of the methyl ester occurred which co-crystallised with the free carboxylic acid.
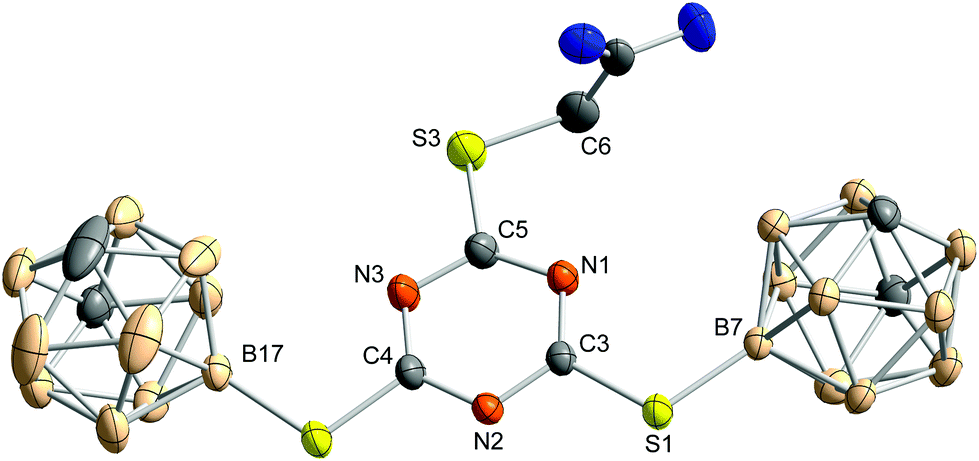 | ||
| Fig. 3 Molecular structure of 2-{[4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazin-2-yl]thio}acetic acid (4) obtained from methanol. Thermal ellipsoids are drawn at the 50% level of probability. The acid and the methyl ester co-crystallise in a ratio of 0.678(7):0.322(7). Here, only the acid is shown. Hydrogen atoms and co-crystallised methanol molecules are omitted for clarity. Selected bond lengths, distances (pm) and bond angles (°): S1–C3 174.4(2), S1–B7 186.2(2), S2–C4 174.2(2), S2–B17 186.3(3), S3–C5 173.8(2), S3–C6 177.7(3), ◆⋯N1 0.8, ◆⋯C3 0.1, ◆⋯N2 0.6, ◆⋯C4 0.7, ◆⋯N3 0.0, ◆⋯C5 0.8, ◆⋯S1 2.0, ◆⋯S2 2.2, ◆⋯S3 8.5, ◆⋯B7 13.0, ◆⋯B17 16.7, ◆⋯C6 27.9, C3–S1–B7 109.9(1), C4–S2–B17 107.0(1), C5–S3–C6 103.5(1); ◆ represents the mean plane of the triazine core, formed by N1, C3, N2, C4, N3 and C5. | ||
In the molecular structure of the glycine derivative 5, the formation of trimers from three symmetry-independent molecules is observed via hydrogen bonding between the secondary amine group of the glycine moiety and the carbonyl oxygen of the free acid and between the hydroxy group of the free acid and one nitrogen atom of the triazine ring (Fig. 4). Such a trimeric hydrogen-bonded arrangement is unprecedented for triazine derivatives.49
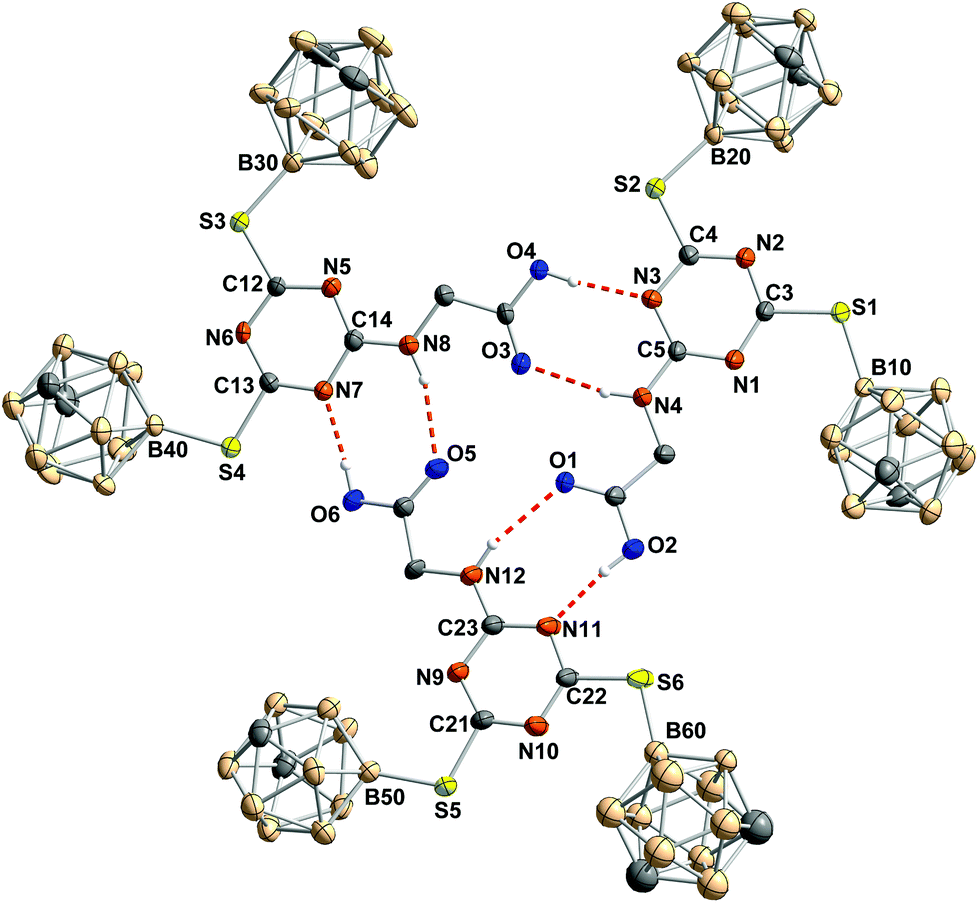 | ||
| Fig. 4 Arrangement of the three symmetry-independent molecules in 5 viewed along the a axis. Hydrogen atoms, not involved in hydrogen bonding, and solvent molecules are omitted for clarity. Thermal ellipsoids of all non-hydrogen atoms are drawn at the 50% level of probability; hydrogen atoms have a fixed atom radius of 13.5 pm. Selected bond lengths, distances (pm) and bond angles (°): A: S1–C3 175.8(6), S1–B10 187.3(7), S2–C4 175.5(6), S2–B20 185.7(6), N4–C5 133.9(7), ◆⋯N1 0.2, ◆⋯C3 0.1, ◆⋯N2 0.6, ◆⋯C4 0.7, ◆⋯N3 0.2, ◆⋯C5 0.2, ◆⋯S1 0.6, ◆⋯S2 1.2, ◆⋯N4 4.4, ◆⋯B10 18.1, ◆⋯B20 9.0, C3–S1–B10 109.2(3), C4–S2–B20 106.7(3), C5–N4–C6 122.0(5); B: S3–C12 174.3(5), S3–B30 185.7(7), S4–C13 175.2(5), S4–B40 186.5(6), N8–C14 132.7(7), ▲⋯N5 1.8, ▲⋯C12 0.9, ▲⋯N6 0.4, ▲⋯C13 0.7, ▲⋯N7 0.3, ▲⋯C14 1.5, ▲⋯S3 3.1, ▲⋯S4 5.5, ▲⋯N8 5.8, ▲⋯B30 25.2, ▲⋯B40 30.1, C12–S3–B30 107.9(3), C13–S4–B40 105.7(3), C14–N8–C15 122.6(5); C: S5–C21 175.1(6), S5–B50 186.4(7), S6–C22 174.4(6), S6–B60 187(1), N12–C23 131.1(8), ●⋯N9 1.1, ●⋯C21 0.4, ●⋯N10 1.3, ●⋯C22 0.9, ●⋯N11 0.6, ●⋯C23 1.6, ●⋯S5 7.5, ●⋯S6 9.4, ●⋯N12 4.6, ●⋯B50 8.6, ●⋯B60 6.0, C21–S5–B50 107.8(3), C22–S6–B60 106.4(5), C23–N12–C24 123.3(6); ◆ represents the mean plane of one triazine core, formed by N1, C3, N2, C4, N3 and C5; ▲ represents the mean plane of one triazine core, formed by N5, C12, N6, C13, N7 and C14; ● represents the mean plane of one triazine core, formed by N9, C21, N10, C22, N11 and C23. | ||
Due to packing effects in the solid state, the trimer of 5 does not show C3 symmetry. Some atoms are slightly moved out of the mean plane formed by the non-hydrogen atoms which participate in the hydrogen donor–acceptor interactions.
Peptide conjugates and biochemical evaluation
The carboxylic acid in compounds 4–6 facilitates coupling with tumour-selective biomolecules. One prominent example of a breast tumour-selective peptide is the modified neuropeptide Y, [F7,P34]-NPY. It is selective for G protein-coupled human Y1 receptors, which are overexpressed in breast cancer cells. Importantly, normal breast tissue contains predominantly Y2 receptors.30,31 Studies were already carried out for ortho-carborane derivatives which were coupled to [F7,P34]-NPY via the primary amino group of lysine-4.20,48 The formation of a highly stable peptide bond between the carboxylic acid of boron-rich derivatives and an amino group of the biomolecule of interest (e.g. lysine in specific peptides) is a much-favoured coupling strategy20,48,50–53 and was also employed here for the thioglycolic acid derivative 4. Compound 4 could be incorporated up to three times into the breast cancer-selective peptide [F7,P34]-NPY at positions 4, 18 and 22.20 All conjugates were characterised by RP-HPLC and ESI-MS, verifying the correct composition and excellent purity. Analytical data for [K4(4′),F7,K18,22(4′),P34]-NPY are exemplarily shown in Fig. 5.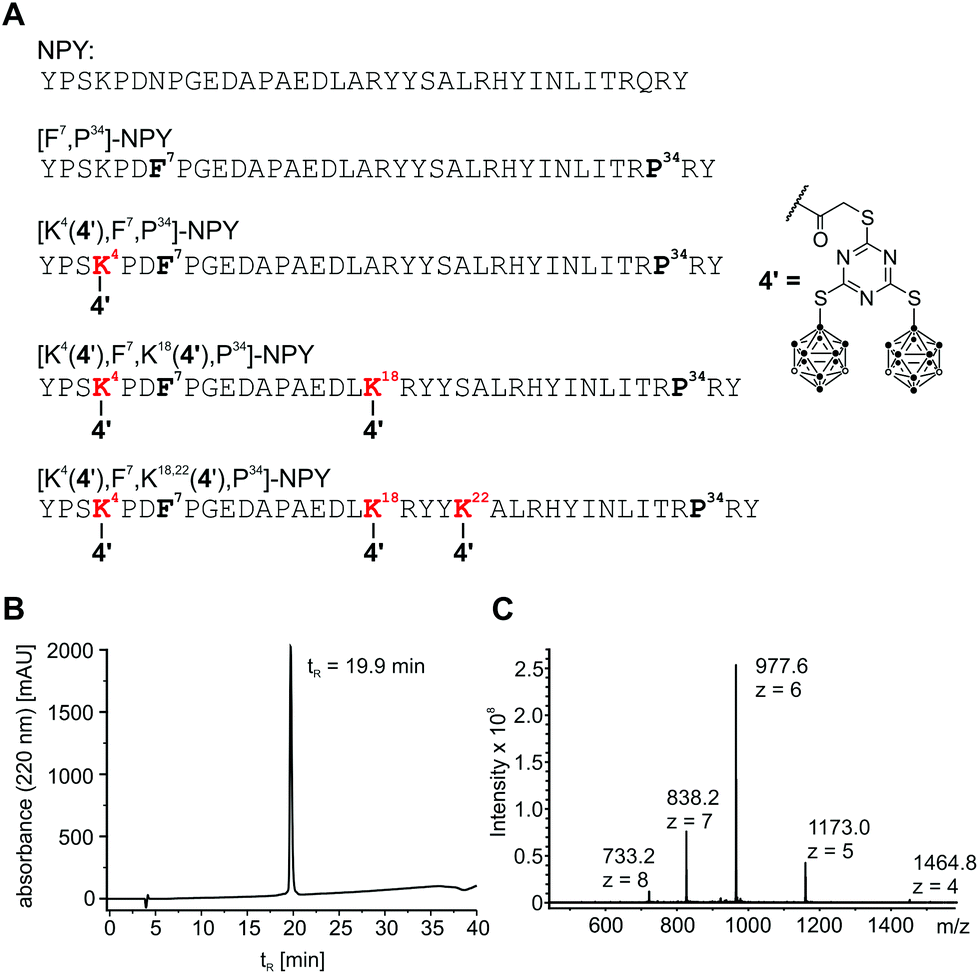 | ||
| Fig. 5 Sequence of synthesised peptide conjugates and analytical data of [K4(4′),F7,K18,22(4′),P34]-NPY. (A) Peptide positions 4, 18 and 22 were used for modification with 4. (B) Analytical RP-HPLC data of [K4(4′),F7,K18,22(4′),P34]-NPY. (C) ESI mass spectrum showing multiple charged ion species of [K4(4′),F7,K18,22(4′),P34]-NPY. MWcalc/avg: 5859.6 Da. | ||
After successful synthesis, all conjugates were tested for their ability to activate and internalise the human Y1 receptor (hY1R). The peptide conjugate with one biscluster ([K4(4′),F7,P34]-NPY) exhibits full agonism with an EC50 value of 8.1 nM, which is only slightly higher than that of the unmodified control peptide [F7,P34]-NPY (Fig. 6A). However, incorporation of more than one derivative 4 in conjugates [K4(4′),F7,K18(4′),P34]-NPY and [K4(4′),F7,K18,22(4′),P34]-NPY led to a significant reduction of activity of the peptides at the hY1 receptor. Thus, [K4(4′),F7,K18(4′),P34]-NPY can partially activate the hY1R with an EC50 > 1 μM, while [K4(4′),F7,K18,22(4′),P34]-NPY is inactive. Furthermore, the peptide conjugates were also investigated for their hY1R internalisation profile by fluorescence microscopy studies. Without peptide stimulation, the fluorescently labelled hY1R (green) was predominantly localised in the cell membrane (Fig. 6B). Stimulation with 100 nM of the native ligand NPY or [F7,P34]-NPY for 1 h led to strong internalisation as observable by the green fluorescence in intracellular vesicles. The mono-substituted conjugate [K4(4′),F7,P34]-NPY was also able to internalise the hY1R while the conjugates with more than one thioglycolic acid derivative 4 were not able to induce internalisation. This is in agreement with their activity.
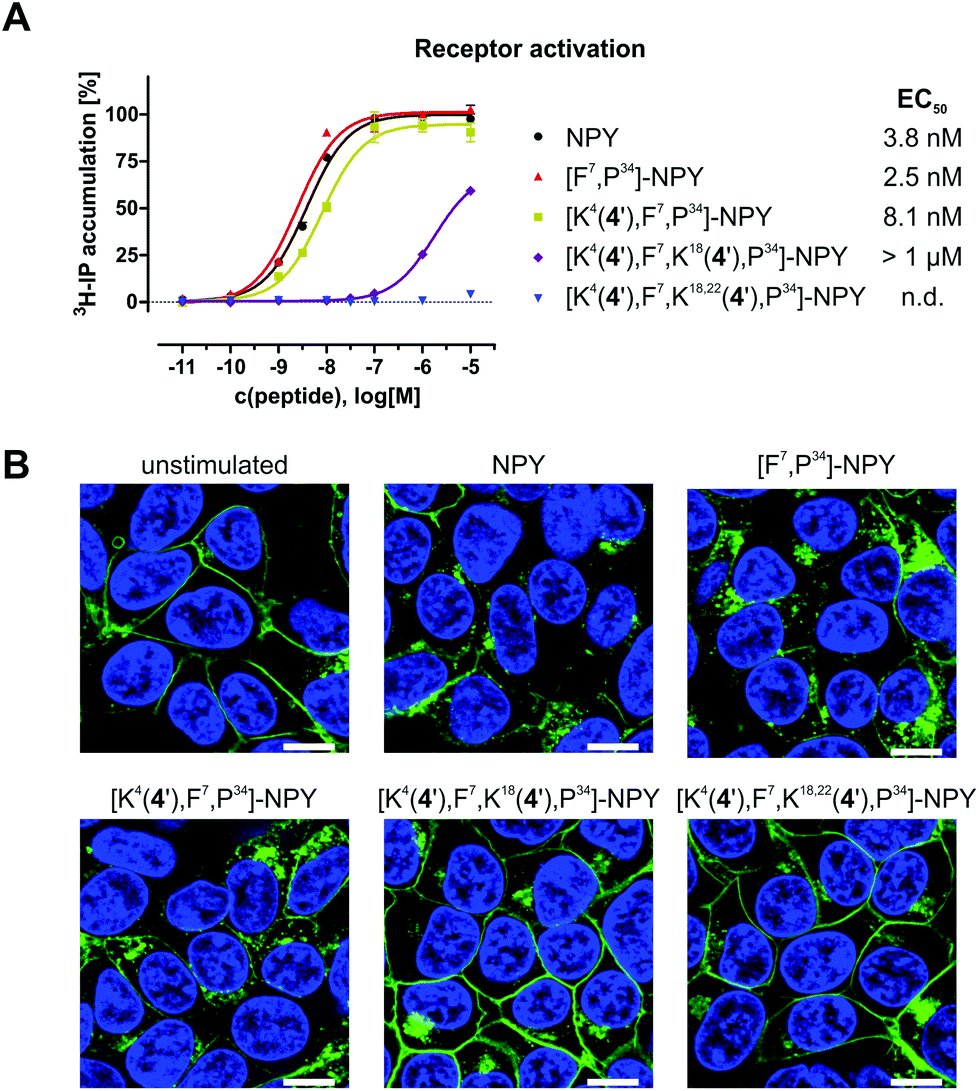 | ||
| Fig. 6 Receptor activation and internalisation data of modified [F7,P34]-NPY. (A) hY1 receptor activation data of synthesised peptide conjugates. Sigmoidal concentration–response curves were determined from at least two independent inositol phosphate (IP) accumulation experiments, each performed in duplicates. (B) Receptor internalisation studies of HEK293 cells stably expressing the hY1R fused to eYFP (green). Cells were stimulated with 100 nM peptide for 1 h at 37 °C and cell nuclei were stained with Hoechst 33342 (blue). Scale bar: 10 μm. | ||
Experimental section
Materials and methods
All reactions were carried out under nitrogen atmosphere using Schlenk techniques, if not reported otherwise. Anhydrous diethyl ether, dichloromethane and tetrahydrofuran were obtained with an MBRAUN solvent purification system MB SPS-800. Acetonitrile was dried over CaH2, distilled and stored over molecular sieve (3 Å) under nitrogen atmosphere. All other solvents were purchased and used as received. 1,2-Di(1,7-dicarba-closo-dodecaboran-9-yl)disulfane (1) and 9-mercapto-1,7-dicarba-closo-dodecaborane(12) (2) were prepared according to the literature procedure.46 All other compounds are commercially available.Thin-layer chromatography (TLC) with silica gel 60 F254 on glass available from Merck KGaA was used for monitoring the reactions. Carborane-containing spots were visualised with a 5–10% solution of PdCl2 in methanol. For chromatography, silica gel (60 Å) with a particle diameter in the range of 0.035 to 0.070 mm or a Biotage Isolera 1 or a Biotage Isolera 4 automatic purification system with SNAP (particle diameter: 0.040 to 0.065 mm) and SNAP Ultra (spherical particle, diameter: 0.025 mm) cartridges were used. The triazine and carborane species were detected by an integrated UV/vis detector (Isolera 1) or evaporative light scattering detector (ELSD) A-120 (Isolera 4). For chromatography, solvents were distilled before use.
NMR measurements were carried out on a Bruker AVANCE III HD spectrometer with an Ascend™ 400 magnet at room temperature. Tetramethylsilane was used as internal standard for 1H and 13C{1H} NMR spectra, and 11B and 11B{1H} NMR spectra were referenced to the Ξ scale.54 NMR spectra were recorded at the following frequencies: 1H: 400.16 MHz, 13C: 100.63 MHz, 11B: 128.38 MHz. All chemical shifts are reported in ppm. Assignment of the 1H and 13C signals was based on 2D NMR spectra (H,H-COSY, HMQC, HSQC, HMBC) or selective TOCSY experiments for the lysine derivative. Identification of the boron atom attached to sulfur was possible by comparison of the proton-coupled and -decoupled 11B NMR spectra. NMR data were interpreted with MestReNova.55 NMR signals that appear as broad overlapping signals with the shape of a multiplet in either 1H, 11B{1H} or 11B NMR spectra are described as bra. IR data were obtained with a PerkinElmer FT-IR spectrometer Spectrum 2000 as KBr pellets and with a Thermo Scientific Nicolet iS5 with an ATR unit in the range from 4000 to 400 cm−1. Electrospray ionisation mass spectrometry was performed with an ESI ESQUIRE 3000 PLUS spectrometer with an IonTrap analyser from Bruker Daltonics or a MicroTOF spectrometer from Bruker Daltonics with a ToF analyser in negative or positive mode. As solvents for the measurements, dichloromethane, acetonitrile, methanol or mixtures of these solvents were used. Elemental analysis was conducted with a VARIO EL elemental analyser from Heraeus. Melting points were determined with a Gallenkamp MPD350·BM2.5 melting point device.
X-ray diffraction experiments
Measurements were performed with a Gemini diffractometer (Rigaku Oxford Diffraction) with Mo-Kα radiation (λ = 71.073 pm), ω-scan rotation. Data reduction was performed with CrysAlis Pro56 including the program SCALE3 ABSPACK57 for empirical absorption correction. The structures were solved by dual space methods (SHELXT-2014)58 and the refinement of all non-hydrogen atoms was performed with SHELXL-2018.59 H atoms were calculated on idealised positions for 1, 5 and for disordered parts of a structure. In all other cases, H atoms were located on difference Fourier maps calculated at the final stage of the structure refinement. Structure figures were generated with ORTEP,60 Diamond61 or Mercury 3.9.62 CCDC 1898709 (1), 1898710 (3), 1898711 (4), 1898712 (5), 1898713 (7), 1898714 (8-acetone) and 1898715 (8-methanol)† contain the supplementary crystallographic data for this paper.:2, v/v) showed that the product was of sufficient purity. Compound 3 was isolated as 6.59 g (14.2 mmol, quantitative yield, Rf = 0.63) of a slightly yellow solid. Tm: 245–256 °C (ethyl acetate/acetone; decomposition). Elemental analysis: C7H22B20ClN3S2, calculated (%): C 18.12, H 4.78, N 9.05; found (%): C 18.16, H 4.64, N 8.75. IR (KBr): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3446 (m), 3072 (m), 3060 (m), 3050 (m), 2962 (w), 2617 (s), 2390 (w), 2091 (w), 1988 (w), 1718 (w), 1624 (w), 1562 (w), 1501 (s), 1477 (s), 1456 (s), 1432 (m), 1312 (m), 1274 (s), 1252 (s), 1166 (m), 1150 (m), 1105 (w), 1067 (m), 1036 (w), 992 (m), 954 (m), 920 (w), 863 (s), 846 (s), 806 (m), 790 (m), 773 (m), 760 (m), 732 (m), 676 (w), 624 (w), 576 (w), 507 (w), 376 (w) cm−1. 1H NMR (acetone-d6): δ = 1.52–3.54 (bra, 18H, B10H9), 3.82 (br, s, 4H, CH) ppm. 13C{1H} NMR (acetone-d6): δ = 56.1 (br, s, CH), 168.5 (s, CqCl), 182.6 (s, CqS) ppm. 11B{1H} NMR (acetone-d6): δ = −18.1 (br, s, 2B), −16.8 (s, 2B), −13.8 (s, 4B), −12.8 (bra, 4B), −10.4 (s, 2B), −5.9 (br, s, 4B), −4.0 (s, 2B, BS) ppm. 11B NMR (acetone-d6): δ = −17.5 (bra, m, 4B), −13.2 (bra, m, 8B), −10.4 (d, 1JBH = 152 Hz, 2B), −5.9 (d, 1JBH = 165 Hz, 4B), −4.0 (s, 2B, BS) ppm. ESI MS: positive mode, C7H23B20ClN3S2, m/z calculated: 465.1; found: 465.4 (51%, [M + H]+); LiC14H44B40Cl2N6S4, m/z calculated: 935.1; found: 935.6 (100%, [2M + Li]+); negative mode, C7H22B20Cl2N3S2, m/z calculated: 499.5; found: 499.3 (38%, [M + Cl]−).
:1 to 100% ethyl acetate, v/v) and 600 mg (1.15 mmol, 27%, Rf = 0.18, 100% ethyl acetate) of 4 were obtained as a slightly yellow solid. Tm: 175–177 °C (acetone). IR (KBr): = 3431 (w), 3054 (m), 2968 (m), 2926 (m), 2604 (s), 2092 (w), 1713 (s), 1471 (s), 1245(s), 1156 (m), 1063 (w), 990 (w), 951 (m), 864 (m), 844 (s), 788 (m), 756 (m), 727 (m), 675 (w), 625 (w), 460 (w) cm−1. 1H NMR (acetone-d6): δ = 1.44–3.50 (bra, 18H, B10H9), 3.80 (br, s, 4H, 4CH), 4.17 (s, 2H, CH2), 11.28 (br, s, 1H, COOH) ppm. 13C{1H} NMR (acetone-d6): δ = 32.6 (s, CH2), 56.1 (br, s, CH), 169.7 (s, CqS), 179.4 (s, CqOOH), 179.6 (s, CqS) ppm. 11B{1H} NMR (acetone-d6): δ = −18.2 (s, 2B), −16.9 (s, 2B), −13.9 (s, 4B), −12.7 (s, 4B), −10.4 (s, 2B), −5.9 (s, 4B), −3.8 (s, 2B, BS) ppm. 11B NMR (acetone-d6): δ = −17.6 (bra, 4B), −13.3 (bra, 8B), −10.4 (d, 1JBH = 150 Hz, 2B), −5.9 (d, 1JBH = 162 Hz, 4B), −3.8 (s, 2B, BS) ppm. ESI MS: positive mode, C9H26B20N3O2S3, m/z calculated: 521.31608, found: 521.31583 (100%, [M + H]+).
:1:0.05, v/v/v). After one day, the reaction was stopped by adding 2 M hydrochloric acid to receive a pH < 7. All organic solvents were removed under reduced pressure. The remaining aqueous layer was extracted four times with 50 mL diethyl ether. The combined organic layers were dried over magnesium sulfate, filtered and the solvent was removed under reduced pressure. After column chromatography (ethyl acetate/n-hexane, 1:1, v/v, 2.5% glacial acetic acid) 176 mg (0.35 mmol, 60%, Rf = 0.14) of 5 were isolated as a slightly yellow solid. Tm: 241–243 °C (ethyl acetate). IR (KBr): = 3290 (m), 3056 (s), 2927 (m), 2606 (s), 1714 (s), 1562 (s), 1521 (s), 1494 (s), 1415 (s), 1243 (s), 1176 (s), 1064 (m), 991 (m), 950 (m), 850 (s), 801 (m), 757 (w), 730 (m), 668 (w), 624 (w), 450 (w) cm−1. 1H NMR (acetone-d6): δ = 1.40–3.60 (bra, 18H, B10H9), 3.71 (br, s, 4H, CH), 4.30 (d, 3JHH = 6.3 Hz, 2H, CH2), 6.94 (t, 3JHH = 6.0 Hz, 1H, NH), 10.8 (br, s, 1H, COOH) ppm. 13C{1H} NMR (acetone-d6): δ = 42.7 (s, CH2), 55.7 (br, s, CH), 55.8 (br, s, CH), 164.7 (s, CqNH), 171.5 (s, CqS), 179.9 (s, CqOOH) ppm. 11B{1H} NMR (acetone-d6): δ = −18.6 (s, 2B), −17.0 (s, 2B), −14.0 (s, 4B), −12.8 (s, 4B), −10.5 (s, 2B), −5.9 (s, 4B), −3.3 (s, 2B, BS) ppm. 11B NMR (acetone-d6): δ = −18.6 (bra, 2B), −17.0 (overlapping signals, 2B), −14.0 (bra, 4B), −12.8 (bra, 4B), −10.5 (d, 1JBH = 151 Hz, 2B), −5.9 (d, 1JBH = 166 Hz, 4B), −3.3 (s, 2B, BS) ppm. ESI MS: positive mode, C9H27B20N4O2S2, m/z calculated: 504.35497, found: 504.35463 (100%, [M + H]+).
:1, v/v). After the reaction was finished the mixture was acidified with 1 M hydrochloric acid and the resulting precipitate was immediately filtered off and washed with water until pH 7 was reached. The precipitate was purified by column chromatography (acetone/n-hexane, 1:1, v/v, 2.5% glacial acetic acid). 138 mg (0.20 mmol, 46.5%, Rf = 0.65) of 6 were isolated as a colourless solid. Tm: 178–180 °C (acetone). IR (KBr): = 3856 (w), 3455 (m), 3258 (m), 3138 (m), 3058 (m), 2980 (m), 2934 (m), 2864 (m), 2603 (s), 1698 (s), 1614 (s), 1569 (m), 1520 (s), 1500 (s), 1452 (m), 1435 (m), 1410 (s), 1369 (s), 1307 (m), 12477 (s), 1218 (m), 1201 (m), 1166 (s), 1117 (m), 1059 (m), 1022 (w), 994 (w), 954 (m), 863 (s), 850 (s), 801 (m), 760 (m), 731 (m), 668 (w), 642 (w), 590 (w), 418 (w) cm−1. 1H NMR (acetonitrile-d3): δ = 1.40 (s, 9H, C(CH3)3), 1.50–3.30 (bra, 18H, B10H9), 1.59 (br, m, 4H, CH2), 1.77 (br, m, 2H, CH2), 3.38 (br, s, 4H, CH), 3.41 (m, 2H, CH2), 4.04 (m, 1H, CH), 5.54 (d, 3JHH = 7.8 Hz, 1H, NH), 6.20 (m, 1H, NH), 9.50 (s, 1H, COOH) ppm. 13C{1H} NMR (acetonitrile-d3): δ = 23.9 (s, CH2), 28.6 (s, C(CH3)3), 30.2 (s, CH2), 32.1 (s, CH2), 41.2 (s, CH2), 54.4 (s, CH), 55.9 (br, s, CH), 56.0 (br, s, CH), 79.9 (s, Cq(CH3)3), 156.7 (s, CqS), 174.5 (s, Cq), 178.5 (s, CqO), 179.6 (s, CqOOH) ppm. 11B{1H} NMR (acetonitrile-d3): δ = −18.4 (s, 2B), −17.0 (s, 2B), −14.1 (s, 4B), −12.9 (s, 4B), −10.6 (s, 2B), −6.0 (s, 4B), −3.5 (s, 2B, BS) ppm. 11B NMR (acetonitrile-d3): δ = −18.4 (bra, 2B), −17.0 (br, d, 1JBH = 192 Hz, 2B), −13.5 (bra, 8B), −10.6 (d, 1JBH = 154 Hz, 2B), −6.0 (d, 1JBH = 166 Hz, 4B), −3.5 (s, 2B, BS) ppm. Detailed assignment of all spectroscopic data is given in the ESI.† ESI MS: Negative mode, C18H42B20N5O4S2, m/z calculated: 673.46669, found: 673.46701 (100%, [M − H]−).
= 3446 (m), 3072 (m), 3060 (m), 3050 (m), 2962 (w), 2617 (s), 2390 (w), 2091 (w), 1988 (w), 1718 (w), 1624 (w), 1562 (w), 1501 (s), 1477 (s), 1456 (s), 1432 (m), 1312 (m), 1274 (s), 1252 (s), 1166 (m), 1150 (m), 1105 (w), 1067 (m), 1036 (w), 992 (m), 954 (m), 920 (w), 863 (s), 846 (s), 806 (m), 790 (m), 773 (m), 760 (m), 732 (m), 676 (w), 624 (w), 576 (w), 507 (w), 376 (w) cm−1. 1H NMR (acetone-d6): δ = 1.52–3.54 (bra, 18H, B10H9), 3.82 (br, s, 4H, CH) ppm. 13C{1H} NMR (acetone-d6): δ = 56.1 (br, s, CH), 168.5 (s, CqCl), 182.6 (s, CqS) ppm. 11B{1H} NMR (acetone-d6): δ = −18.1 (br, s, 2B), −16.8 (s, 2B), −13.8 (s, 4B), −12.8 (bra, 4B), −10.4 (s, 2B), −5.9 (br, s, 4B), −4.0 (s, 2B, BS) ppm. 11B NMR (acetone-d6): δ = −17.5 (bra, m, 4B), −13.2 (bra, m, 8B), −10.4 (d, 1JBH = 152 Hz, 2B), −5.9 (d, 1JBH = 165 Hz, 4B), −4.0 (s, 2B, BS) ppm. ESI MS: positive mode, C7H23B20ClN3S2, m/z calculated: 465.1; found: 465.4 (51%, [M + H]+); LiC14H44B40Cl2N6S4, m/z calculated: 935.1; found: 935.6 (100%, [2M + Li]+); negative mode, C7H22B20Cl2N3S2, m/z calculated: 499.5; found: 499.3 (38%, [M + Cl]−).
:1 to 100% ethyl acetate, v/v) and 600 mg (1.15 mmol, 27%, Rf = 0.18, 100% ethyl acetate) of 4 were obtained as a slightly yellow solid. Tm: 175–177 °C (acetone). IR (KBr): = 3431 (w), 3054 (m), 2968 (m), 2926 (m), 2604 (s), 2092 (w), 1713 (s), 1471 (s), 1245(s), 1156 (m), 1063 (w), 990 (w), 951 (m), 864 (m), 844 (s), 788 (m), 756 (m), 727 (m), 675 (w), 625 (w), 460 (w) cm−1. 1H NMR (acetone-d6): δ = 1.44–3.50 (bra, 18H, B10H9), 3.80 (br, s, 4H, 4CH), 4.17 (s, 2H, CH2), 11.28 (br, s, 1H, COOH) ppm. 13C{1H} NMR (acetone-d6): δ = 32.6 (s, CH2), 56.1 (br, s, CH), 169.7 (s, CqS), 179.4 (s, CqOOH), 179.6 (s, CqS) ppm. 11B{1H} NMR (acetone-d6): δ = −18.2 (s, 2B), −16.9 (s, 2B), −13.9 (s, 4B), −12.7 (s, 4B), −10.4 (s, 2B), −5.9 (s, 4B), −3.8 (s, 2B, BS) ppm. 11B NMR (acetone-d6): δ = −17.6 (bra, 4B), −13.3 (bra, 8B), −10.4 (d, 1JBH = 150 Hz, 2B), −5.9 (d, 1JBH = 162 Hz, 4B), −3.8 (s, 2B, BS) ppm. ESI MS: positive mode, C9H26B20N3O2S3, m/z calculated: 521.31608, found: 521.31583 (100%, [M + H]+).
:1:0.05, v/v/v). After one day, the reaction was stopped by adding 2 M hydrochloric acid to receive a pH < 7. All organic solvents were removed under reduced pressure. The remaining aqueous layer was extracted four times with 50 mL diethyl ether. The combined organic layers were dried over magnesium sulfate, filtered and the solvent was removed under reduced pressure. After column chromatography (ethyl acetate/n-hexane, 1:1, v/v, 2.5% glacial acetic acid) 176 mg (0.35 mmol, 60%, Rf = 0.14) of 5 were isolated as a slightly yellow solid. Tm: 241–243 °C (ethyl acetate). IR (KBr): = 3290 (m), 3056 (s), 2927 (m), 2606 (s), 1714 (s), 1562 (s), 1521 (s), 1494 (s), 1415 (s), 1243 (s), 1176 (s), 1064 (m), 991 (m), 950 (m), 850 (s), 801 (m), 757 (w), 730 (m), 668 (w), 624 (w), 450 (w) cm−1. 1H NMR (acetone-d6): δ = 1.40–3.60 (bra, 18H, B10H9), 3.71 (br, s, 4H, CH), 4.30 (d, 3JHH = 6.3 Hz, 2H, CH2), 6.94 (t, 3JHH = 6.0 Hz, 1H, NH), 10.8 (br, s, 1H, COOH) ppm. 13C{1H} NMR (acetone-d6): δ = 42.7 (s, CH2), 55.7 (br, s, CH), 55.8 (br, s, CH), 164.7 (s, CqNH), 171.5 (s, CqS), 179.9 (s, CqOOH) ppm. 11B{1H} NMR (acetone-d6): δ = −18.6 (s, 2B), −17.0 (s, 2B), −14.0 (s, 4B), −12.8 (s, 4B), −10.5 (s, 2B), −5.9 (s, 4B), −3.3 (s, 2B, BS) ppm. 11B NMR (acetone-d6): δ = −18.6 (bra, 2B), −17.0 (overlapping signals, 2B), −14.0 (bra, 4B), −12.8 (bra, 4B), −10.5 (d, 1JBH = 151 Hz, 2B), −5.9 (d, 1JBH = 166 Hz, 4B), −3.3 (s, 2B, BS) ppm. ESI MS: positive mode, C9H27B20N4O2S2, m/z calculated: 504.35497, found: 504.35463 (100%, [M + H]+).
:1, v/v). After the reaction was finished the mixture was acidified with 1 M hydrochloric acid and the resulting precipitate was immediately filtered off and washed with water until pH 7 was reached. The precipitate was purified by column chromatography (acetone/n-hexane, 1:1, v/v, 2.5% glacial acetic acid). 138 mg (0.20 mmol, 46.5%, Rf = 0.65) of 6 were isolated as a colourless solid. Tm: 178–180 °C (acetone). IR (KBr): = 3856 (w), 3455 (m), 3258 (m), 3138 (m), 3058 (m), 2980 (m), 2934 (m), 2864 (m), 2603 (s), 1698 (s), 1614 (s), 1569 (m), 1520 (s), 1500 (s), 1452 (m), 1435 (m), 1410 (s), 1369 (s), 1307 (m), 12477 (s), 1218 (m), 1201 (m), 1166 (s), 1117 (m), 1059 (m), 1022 (w), 994 (w), 954 (m), 863 (s), 850 (s), 801 (m), 760 (m), 731 (m), 668 (w), 642 (w), 590 (w), 418 (w) cm−1. 1H NMR (acetonitrile-d3): δ = 1.40 (s, 9H, C(CH3)3), 1.50–3.30 (bra, 18H, B10H9), 1.59 (br, m, 4H, CH2), 1.77 (br, m, 2H, CH2), 3.38 (br, s, 4H, CH), 3.41 (m, 2H, CH2), 4.04 (m, 1H, CH), 5.54 (d, 3JHH = 7.8 Hz, 1H, NH), 6.20 (m, 1H, NH), 9.50 (s, 1H, COOH) ppm. 13C{1H} NMR (acetonitrile-d3): δ = 23.9 (s, CH2), 28.6 (s, C(CH3)3), 30.2 (s, CH2), 32.1 (s, CH2), 41.2 (s, CH2), 54.4 (s, CH), 55.9 (br, s, CH), 56.0 (br, s, CH), 79.9 (s, Cq(CH3)3), 156.7 (s, CqS), 174.5 (s, Cq), 178.5 (s, CqO), 179.6 (s, CqOOH) ppm. 11B{1H} NMR (acetonitrile-d3): δ = −18.4 (s, 2B), −17.0 (s, 2B), −14.1 (s, 4B), −12.9 (s, 4B), −10.6 (s, 2B), −6.0 (s, 4B), −3.5 (s, 2B, BS) ppm. 11B NMR (acetonitrile-d3): δ = −18.4 (bra, 2B), −17.0 (br, d, 1JBH = 192 Hz, 2B), −13.5 (bra, 8B), −10.6 (d, 1JBH = 154 Hz, 2B), −6.0 (d, 1JBH = 166 Hz, 4B), −3.5 (s, 2B, BS) ppm. Detailed assignment of all spectroscopic data is given in the ESI.† ESI MS: Negative mode, C18H42B20N5O4S2, m/z calculated: 673.46669, found: 673.46701 (100%, [M − H]−).
2,4,6-Tris(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine ( 7 ) and 4,6-bis(1,7-dicarba-closo-dodecaboran-9-ylthio)-1,3,5-triazine-2(1H)-one ( 8 ) were obtained as by-products in several reactions (Table S2, ESI); details, molecular structures and analytical data are given in the ESI.†
Peptide synthesis
:5:5, v/v) for 2 h and a subsequent precipitation of the crude peptide in ice-cold diethyl ether. The peptide was washed and dissolved in ACN/H2O and subsequently lyophilised. Purification of the crude peptides was achieved by preparative reversed phase (RP)-HPLC using a Phenomenex Aeris® 3.6u PEPTIDE XB-C18 (250 mm × 21.2 mm, 3.6 μm, 100 Å) column with a flow rate of 15 mL min−1, linear gradients of eluent B (0.08% (v/v) TFA in ACN) in eluent A (0.1% (v/v) TFA in water) and detection at λ = 220 nm. The purity of the peptides was determined by analytical RP-HPLC using a Phenomenex Jupiter® 4u Proteo C12 90 Å (250 mm × 4.6 mm, 4 μm, 90 Å, 0.6 mL min−1) column. MALDI-ToF mass spectrometry (UltraflexIII, Bruker, Bremen, Germany) and ESI Ion-trap (HCT, Bruker Daltonics) or ESI Orbitrap (Orbitrap Elite, Thermo Scientific) mass spectrometry were used to verify the identity of the pure peptides. The observed masses were in agreement with the calculated masses and a purity of >95% was obtained for all peptides in the analytical RP-HPLC.
Biological methods
:1, v/v) supplemented with 15% (v/v) FCS and hygromycin (100 μg mL−1). COS-7 cells, stably co-expressing the hY1R, C-terminally fused to eYFP, and the chimeric G protein GαΔ6qi4myr, were cultured in DMEM high glucose supplemented with 10% (v/v) FCS, hygromycin B (133 μg mL−1) and G418-sulfate (1.5 mg mL−1).
000 cells per well) and cultured overnight. Cells were labeled with [2-3H]-myo-inositol (2 μCi mL−1) in medium without penicillin and streptomycin overnight and stimulated with different peptide concentrations in DMEM supplemented with 10 mM LiCl and 0.1% (w/v) BSA for 1 h at 37 °C. Compounds were typically tested in a concentration range from 10−5 M to 10−11 M in duplicates. Radioactive IP species were isolated by anion-exchange chromatography and measured by scintillation counting. Data analysis was performed with GraphPad Prism 5.03. Obtained raw dpm values were normalised to NPY and EC50 and pEC50 values were calculated from sigmoidal concentration–response curves. Each peptide was tested at least two times independently.
000 cells per well and cultured for 24 h until the cells reached confluency. The medium was aspirated and the cells were starved in 200 μL Opti-MEM® with 1 μL nuclear stain Hoechst 33342 (0.5 mg mL−1) for 30 min under standard incubation conditions. For non-stimulated cells, the solution was aspirated and 200 μL Opti-MEM® were added. For testing the receptor internalisation of the NPY analogues, the starving medium was aspirated and 200 μL Opti-MEM® containing 100 nM peptide conjugate were added to the cells. After 1 h of stimulation at 37 °C, the cells were washed once and maintained in Opti-MEM®. A Zeiss Axio Observer microscope with an ApoTome.2 Imaging System and a 63× immersion oil objective was used for fluorescence image acquisition. The nuclear stain Hoechst 33342 was visualised by the DAPI filter (excitation 335–383 nm; emission 420–470 nm) and the eYFP-tag on the receptor by the YFP filter (excitation 488–512 nm; emission 520–550 nm). Image processing was performed with Zeiss ZEN 2 software.
Conclusions
A modular approach was used for the synthesis of meta-carboranyl-substituted s-triazines with carboxylic acid functions, namely the thioglycolic acid derivative 4, the glycine derivative 5, and the Nα-Boc-L-lysine derivative 6, which can be coupled with suitable biomolecules for selective tumour targeting as was demonstrated here for compound 4. The peptide conjugate of one thioglycolic acid derivative 4 with breast-tumour selective [F7,P34]-NPY, [K4(4′),F7,P34]-NPY, exhibits full agonism, while incorporation of more than one derivative 4 leads to a significant loss of activity of the peptide conjugates at the human Y1 receptor. It can be speculated that the hydrophobicity of the conjugates with more than one derivative 4 is too high and leads thereby to a hydrophobic collapse of the conjugate. More hydrophilic derivatives could potentially result in higher activity of the peptide conjugates. Further potential is offered by the L-lysine derivative 6, which might be directly employed as an artificial amino acid in the preparation of peptides, proteins or even antibodies. Furthermore, deprotection of the tert-butoxycarbonyl group in 6 will allow further derivatisation, for example for increasing the water solubility or the introduction of an imaging agent.9,64Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ramona Oehme, Dr Susan Billig, Dr Claudia Birkemeyer and Regina Reppich for measuring the mass spectra, Manuela Roßberg for elemental analysis, Stefanie Märcker-Recklies and Ines Rein for recording the infrared spectra, Ronny Müller for support in peptide synthesis and Kristin Löbner for cell culture support.Notes and references
- G. L. Locher, Am. J. Roentgenol. Radium Ther., 1936, 36, 1–18 CAS.
- (a) R. F. Barth, P. Mi and W. Yang, Cancer Commun., 2018, 38, 1–15 CrossRef PubMed; (b) G. Calabrese, A. Daou, E. Barbu and J. Tsibouklis, Drug Discovery Today, 2018, 23, 63–75 CrossRef CAS PubMed; (c) I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 11, 1433–1450 CrossRef; (d) G. W. Kabalka, Expert Opin. Ther. Pat., 1998, 8, 545–551 CrossRef CAS; (e) H. Hatanaka, J. Neurol., 1975, 209, 81–94 CrossRef CAS PubMed.
- R. F. Barth, A. H. Soloway and R. G. Fairchild, Cancer Res., 1990, 50, 1061–1070 CAS.
- A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed.
- M. F. Hawthorne, Angew. Chem., Int. Ed., 1993, 32, 950–984 ( Angew. Chem. , 1993 , 105 , 997–1033 ) CrossRef.
- (a) V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef; (b) W. Petry and J. Neuhaus, Phys. J., 2007, 6, 31–37 CAS.
- X. Q. Pan, H. Wang, S. Shukla, M. Sekido, D. M. Adams, W. Tjarks, R. F. Barth and R. J. Lee, Bioconjugate Chem., 2002, 13, 435–442 CrossRef CAS PubMed.
- (a) S. Shukla, G. Wu, M. Chatterjee, W. Yang, M. Sekido, L. A. Diop, R. Müller, J. J. Sudimack, R. J. Lee, R. F. Barth and W. Tjarks, Bioconjugate Chem., 2003, 14, 158–167 CrossRef CAS PubMed; (b) Y. Iguchi, H. Michiue, M. Kitamatsu, Y. Hayashi, F. Takenaka, T.-i. Nishiki and H. Matsui, Biomaterials, 2015, 56, 10–17 CrossRef CAS PubMed; (c) A. Doi, S. Kawabata, K. Iida, K. Yokoyama, Y. Kajimoto, T. Kuroiwa, T. Shirakawa, M. Kirihata, S. Kasaoka, K. Maruyama, H. Kumada, Y. Sakurai, S.-i. Masunaga, K. Ono and S.-I. Miyatake, J. Neurooncol., 2008, 87, 287–294 CrossRef CAS PubMed; (d) I. Romero-Canelón, B. Phoenix, A. Pitto-Barry, J. Tran, J. J. Soldevila-Barreda, N. Kirby, S. Green, P. J. Sadler and N. P. E. Barry, J. Organomet. Chem., 2015, 796, 17–25 CrossRef; (e) K. Kettenbach, H. Schieferstein, C. Grunewald, D. Iffland, L. M. Reffert, G. Hampel, C. L. Schütz, N. H. Bings and T. L. Ross, Radiochim. Acta, 2015, 103, 799–809 CAS; (f) W. Mier, D. Gabel, U. Haberkorn and M. Eisenhut, Z. Anorg. Allg. Chem., 2004, 630, 1258–1262 CrossRef CAS.
- R. Dubey, S. Kushal, A. Mollard, L. Vojtovich, P. Oh, M. D. Levin, J. E. Schnitzer, I. Zharov and B. Z. Olenyuk, Bioconjugate Chem., 2015, 26, 78–89 CrossRef CAS PubMed.
- B. Feng, K. Tomizawa, H. Michiue, S.-I. Miyatake, X.-J. Han, A. Fujimura, M. Seno, M. Kirihata and H. Matsui, Biomaterials, 2009, 30, 1746–1755 CrossRef CAS PubMed.
- H. Michiue, Y. Sakurai, N. Kondo, M. Kitamatsu, F. Bin, K. Nakajima, Y. Hirota, S. Kawabata, T.-i. Nishiki, I. Ohmori, K. Tomizawa, S.-I. Miyatake, K. Ono and H. Matsui, Biomaterials, 2014, 35, 3396–3405 CrossRef CAS PubMed.
- C.-H. Lai, Y.-C. Lin, F.-I. Chou, C.-F. Liang, E.-W. Lin, Y.-J. Chuang and C.-C. Lin, Chem. Commun., 2012, 48, 612–614 RSC.
- R. Otero, S. Seoane, R. Sigüeiro, A. Y. Belorusova, M. A. Maestro, R. Pérez-Fernández, N. Rochel and A. Mouriño, Chem. Sci., 2016, 7, 1033–1037 RSC.
- D. J. Worm, S. Els-Heindl, M. Kellert, R. Kuhnert, S. Saretz, J. Koebberling, B. Riedl, E. Hey-Hawkins and A. G. Beck-Sickinger, J. Pept. Sci., 2018, 32, e3119 CrossRef PubMed.
- G. Ciofani, V. Raffa, A. Menciassi and A. Cuschieri, Nanoscale Res. Lett., 2008, 4, 113–121 CrossRef PubMed.
- P. J. Kueffer, C. A. Maitz, A. A. Khan, S. A. Schuster, N. I. Shlyakhtina, S. S. Jalisatgi, J. D. Brockman, D. W. Nigg and M. F. Hawthorne, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6512–6517 CrossRef CAS PubMed.
- (a) J. Chadwick and M. Goldhaber, Nature, 1935, 135, 65–65 CrossRef CAS; (b) H. J. Taylor and M. Goldhaber, Nature, 1935, 135, 341–341 CrossRef CAS.
- Y. Hattori, S. Kusaka, M. Mukumoto, K. Uehara, T. Asano, M. Suzuki, S.-i. Masunaga, K. Ono, S. Tanimori and M. Kirihata, J. Med. Chem., 2012, 55, 6980–6984 CrossRef CAS PubMed.
- T. Hartman and J. Carlsson, Radiother. Oncol., 1994, 31, 61–75 CrossRef CAS PubMed.
- V. M. Ahrens, R. Frank, S. Stadlbauer, A. G. Beck-Sickinger and E. Hey-Hawkins, J. Med. Chem., 2011, 54, 2368–2377 CrossRef CAS PubMed.
- (a) K. Y. Choi, G. Saravanakumar, J. H. Park and K. Park, Colloid Surf., B, 2012, 99, 82–94 CrossRef CAS PubMed; (b) S. Agosteo, P. Colautti, J. Esposito, A. Fazzi, M. V. Introini and A. Pola, Appl. Radiat. Isot., 2011, 69, 1664–1667 CrossRef CAS PubMed; (c) H. Kumada, A. Matsumura, H. Sakurai, T. Sakae, M. Yoshioka, H. Kobayashi, H. Matsumoto, Y. Kiyanagi, T. Shibata and H. Nakashima, Appl. Radiat. Isot., 2014, 88, 211–215 CrossRef CAS; (d) E. Durisi, K. Alikaniotis, O. Borla, F. Bragato, M. Costa, G. Giannini, V. Monti, L. Visca, G. Vivaldo and A. Zanini, Appl. Radiat. Isot., 2015, 106, 63–67 CrossRef CAS PubMed; (e) T. Onishi, H. Kumada, K. Takada, F. Naito, T. Kurihara and T. Sakae, Appl. Radiat. Isot., 2018, 140, 5–11 CrossRef CAS PubMed.
- C.-H. Lee, H.-G. Lim, J.-D. Lee, Y.-J. Lee, J. Ko, H. Nakamura and S. Ook Kang, Appl. Organomet. Chem., 2003, 17, 539–548 CrossRef CAS.
- Y. Azev, I. Slepukhina and D. Gabel, Appl. Radiat. Isot., 2004, 61, 1107–1110 CrossRef CAS.
- (a) H. Nakamura, A. Shoji, A. Takeuchi, H. S. Ban, J.-D. Lee and S. O. Kang, Aust. J. Chem., 2011, 64, 1430–1437 CrossRef CAS; (b) J.-D. Lee, S.-J. Cho, S.-H. Kim, G.-Y. Chai and C.-H. Lee, Bull. Korean Chem. Soc., 2012, 33, 3136–3138 CrossRef CAS; (c) C.-H. Lee, J. H. Yun, G. F. Jin, Y. J. Jung, S. O. Kang and J.-D. Lee, KR20090016794(A), 2009, 1–16.
- J.-D. Lee, S. Y. Kim and S. O. Kang, Biochem. Biophys. J. Neutron Ther. Cancer Treatments (BBJNTCT), 2014, 2, 8–12 Search PubMed.
- C.-H. Lee, G. F. Jin, H.-S. Kim, H. Nakamura and J.-D. Lee, Bull. Korean Chem. Soc., 2008, 29, 697–700 CrossRef CAS.
- C.-H. Lee, G. Jin, J. Yoon, S. Kim, J.-D. Lee, H. Nakamura and S. O. Kang, Synthesis, 2008, 1193–1200 CrossRef CAS.
- C.-H. Lee, G. F. Jin, J. H. Yoon, Y. J. Jung, J.-D. Lee, S. Cho, H. Nakamura and S. O. Kang, Tetrahedron Lett., 2008, 49, 159–164 CrossRef CAS.
- S. Ronchi, D. Prosperi, F. Compostella and L. Panza, Synlett, 2004, 1007–1010 CAS.
- J. C. Reubi, M. Gugger and B. Waser, Cancer Res., 2001, 61, 4636–4641 CAS.
- I. U. Khan, D. Zwanziger, I. Böhme, M. Javed, H. Naseer, S. W. Hyder and A. G. Beck-Sickinger, Angew. Chem., Int. Ed., 2010, 49, 1155–1158 ( Angew. Chem. , 2010 , 122 , 1174–1177 ) CrossRef CAS PubMed.
- (a) F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 ( Angew. Chem. , 2016 , 128 , 11984–11988 ) CrossRef CAS PubMed; (b) T. Balakrishnan, E. Murugan and A. Siva, Appl. Catal., A, 2004, 273, 89–97 CrossRef CAS; (c) N. Klee, P. E. Wong, B. Baragaña, F. E. Mazouni, M. A. Phillips, M. P. Barrett and I. H. Gilbert, Bioorg. Med. Chem. Lett., 2010, 20, 4364–4366 CrossRef CAS PubMed; (d) J. Salabert, R. M. Sebastián and A. Vallribera, Chem. Commun., 2015, 51, 14251–14254 RSC; (e) Z. J. Kamiński, B. Kolesińska, J. Kolesińska, G. Sabatino, M. Chelli, P. Rovero, M. Błaszczyk, M. L. Główka and A. M. Papini, J. Am. Chem. Soc., 2005, 127, 16912–16920 CrossRef PubMed; (f) G.-H. Kuo, A. Deangelis, S. Emanuel, A. Wang, Y. Zhang, P. J. Connolly, X. Chen, R. H. Gruninger, C. Rugg, A. Fuentes-Pesquera, S. A. Middleton, L. Jolliffe and W. V. Murray, J. Med. Chem., 2005, 48, 4535–4546 CrossRef CAS PubMed; (g) A. Olczak, M. Blaszczyk, M. L. Glówka, B. Kolesińska and Z. J. Kamiński, Pol. J. Chem., 2008, 82, 1413–1423 CAS; (h) C. Zhu, C. Deng, J.-Y. Cao and Y.-Z. Wang, Polym. Degrad. Stab., 2015, 121, 42–50 CrossRef CAS; (i) K. Venkataraman and D. R. Wagle, Tetrahedron Lett., 1979, 32, 3037–3040 CrossRef; (j) J. K. Srivastava, P. Dubey, S. Singh, H. R. Bhat, M. K. Kumawat and U. P. Singh, RSC Adv., 2015, 5, 14095–14102 RSC.
- K. Arya and A. Dandia, Bioorg. Med. Chem. Lett., 2007, 17, 3298–3304 CrossRef CAS PubMed.
- C. Shen, T. Sheng, Q. Zhu, S. Hu and X. Wu, CrystEngComm, 2012, 14, 3189–3189 RSC.
- J. R. Dudley, J. T. Thurston, F. C. Schaefer, D. Holm-Hansen, C. J. Hull and P. Adams, J. Am. Chem. Soc., 1951, 73, 2986–2990 CrossRef CAS.
- (a) B. Bitter and H. Zollinger, Angew. Chem., 1958, 70, 246–247 CrossRef CAS; (b) D. M. Tigelaar, A. E. Palker, C. M. Jackson, K. M. Anderson, J. Wainright and R. F. Savinell, Macromolecules, 2009, 42, 1888–1896 CrossRef CAS.
- C. Afonso, N. Lourenco and A. Rosatella, Molecules, 2006, 11, 81–102 CrossRef CAS PubMed.
- V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS.
- D. Grafstein and J. Dvorak, Inorg. Chem., 1963, 2, 1128–1133 CrossRef CAS.
- (a) M. A. Fox, W. R. Gill, P. L. Herbertson, J. A. H. MacBride and K. Wade, Polyhedron, 1996, 15, 565–571 CrossRef CAS; (b) J. Yoo, J.-W. Hwang and Y. Do, Inorg. Chem., 2001, 40, 568–570 CrossRef CAS.
- (a) P. Kaszynski, Collect. Czech. Chem. Commun., 1999, 64, 895–926 CrossRef CAS; (b) Z. Juan, H. Pengcheng, C. Gong and Z. Maosheng, Prog. Chem., 2012, 24, 556–567 Search PubMed; (c) L. I. Zakharkin and V. N. Kalinin, Izv. Akad. Nauk. SSSR, 1965, 7, 1287 Search PubMed; (d) J. A. Potenza, W. N. Lipscomb, G. D. Vickers and H. Schröder, J. Am. Chem. Soc., 1966, 88, 628–629 CrossRef CAS.
- V. N. Kalinin and V. A. Ol'shevskaya, Russ. Chem. Bull. Int. Ed., 2008, 57, 815–836 CrossRef CAS ; also published in Russian in Izvestiya Akademii Nauk. Seriya Khimicheskaya, 2008, 3, 801–822.
- A. M. Spokoyny, C. D. Lewis, G. Teverovskiy and S. L. Buchwald, Organometallics, 2012, 31, 8478–8481 CrossRef CAS PubMed.
- (a) J. Plešek, Z. Janoušek and S. Heřmánek, Collect. Czech. Chem. Commun., 1978, 43, 1332–1338 CrossRef; (b) R. Frank, S. Boehnke, A. Aliev and E. Hey-Hawkins, Polyhedron, 2012, 39, 9–13 CrossRef CAS; (c) J. M. Oliva and C. Viñas, J. Mol. Struct., 2000, 556, 33–42 CrossRef CAS; (d) J. Plešek and S. Heřmánek, Collect. Czech. Chem. Commun., 1981, 46, 687–693 CrossRef.
- S. Boehnke, S. Saretz, M. Kellert and E. Hey-Hawkins, Biochem. Biophys. J. Neutron Ther. Cancer Treatments (BBJNTCT), 2013, 1, 22–27 Search PubMed.
- L. I. Zakharkin and I. V. Pisareva, Phosphorus Sulfur, 1984, 20, 357–370 CrossRef CAS.
- P. Tsitsa, E. Antoniadou-Vyza, S. J. Hamodrakas, E. E. Eliopoulos, A. Tsantili-Kakoulidou, E. Lada-Hytiroglou, C. Roussakis, I. Chinou, A. Hempel, N. Camerman, F. P. Ottensmeyer and D. A. Vanden Berghe, Eur. J. Med. Chem., 1993, 28, 149–158 CrossRef CAS.
- V. M. Ahrens, R. Frank, S. Boehnke, C. L. Schütz, G. Hampel, D. S. Iffland, N. H. Bings, E. Hey-Hawkins and A. G. Beck-Sickinger, ChemMedChem, 2015, 10, 164–172 CrossRef CAS PubMed.
- S. M. F. Vilela, D. Ananias, P. Silva, M. Nolasco, L. D. Carlos, V. de Zea Bermudez, J. Rocha, J. P. C. Tomé and F. A. Almeida Paz, CrystEngComm, 2014, 16, 8119–8137 RSC.
- R. Frank, V. Ahrens, S. Boehnke, S. Hofmann, M. Kellert, S. Saretz, S. Pandey, M. Sárosi, Á. Bartók, A. G. Beck-Sickinger and E. Hey-Hawkins, Pure Appl. Chem., 2015, 87, 163–171 CAS.
- R. Frank, V. M. Ahrens, S. Boehnke, A. G. Beck-Sickinger and E. Hey-Hawkins, ChemBioChem, 2016, 17, 308–317 CrossRef CAS PubMed.
- S. Stadlbauer, R. Frank, M. Scholz, S. Boehnke, V. M. Ahrens, A. G. Beck-Sickinger and E. Hey-Hawkins, Pure Appl. Chem., 2012, 84, 2289–2298 CAS.
- R. Frank and E. Hey-Hawkins, J. Organomet. Chem., 2015, 798, 46–50 CrossRef CAS.
- (a) R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CAS; (b) R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS PubMed.
- MestReNova, v12.0 0-20080, Mestrelab Research S.L., 2017 Search PubMed.
- CrysAlis Pro: Data Collection and Data Reduction Software Package, Rigaku Oxford Diffraction Search PubMed.
- SCALE3 ABSPACK: Empirical Absorption Correction using Sperical Harmonics Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–565 CrossRef CAS.
- K. Brandenburg, Diamond, v4.5.3, Crystal Impact GbR, Bonn, Germany Search PubMed.
- Mercury, v4.1.0, The Cambridge Crystallographic Data Centre, 2019 Search PubMed.
- S. Hofmann, R. Frank, E. Hey-Hawkins, A. G. Beck-Sickinger and P. Schmidt, Neuropeptides, 2013, 47, 59–66 CrossRef CAS PubMed.
- (a) A. Toppino, M. E. Bova, S. Geninatti Crich, D. Alberti, E. Diana, A. Barge, S. Aime, P. Venturello and A. Deagostino, Chem. – Eur. J., 2013, 19, 721–728 CrossRef CAS PubMed; (b) A. R. Genady, J. A. Ioppolo, M. M. Azaam and M. E. El-Zaria, Eur. J. Med. Chem., 2015, 93, 574–583 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data of compounds 1, 3, 4, 5, 7 and 8 (CCDC 1898709–1898715), synthetic procedures and characterisation of 1 and 2 as well as by-products 7 and 8; theoretical calculations for formation of 7 and 8. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02130b |
| This journal is © The Royal Society of Chemistry 2019 |