
Total synthesis and antimalarial activity of mortiamides A–D†
Christopher
Bérubé
a,
Dominic
Gagnon
b,
Alexandre
Borgia
a,
Dave
Richard
 b and
Normand
Voyer
*a
b and
Normand
Voyer
*a
aDépartement de Chimie and PROTEO, Université Laval, Faculté des sciences et de génie, 1045 avenue de la Médecine, Québec, QC G1V OA6, Canada. E-mail: normand.voyer@chm.ulaval.ca
bDépartement de microbiologie-infectiologie et d’immunologie, Université Laval, Québec, G1V 0A6, Canada
First published on 20th May 2019
Abstract
Mortiamides A–D (1–4) are head-to-tail cyclic heptapeptides that were identified from a novel Mortierella sp. isolate obtained from marine sediments from Northern Canada. Herein we report the first total synthesis of mortiamides A–D (1–4) on a solid support by concomitant cyclization/cleavage without any oligomerization side reactions, and overall yields up to 48%. We also report on the antiplasmodial activity of mortiamides A–D (1–4). We show that three out of the four tested mortiamides (A, B and D) have moderate antiplasmodial activity, while mortiamide D (4) exhibits low micromolar activity.
Malaria is one of the most serious infectious diseases worldwide. In 2016, the World Health Organization estimated about 216 million cases of malaria in 91 countries. There were five million new cases observed in 2015 alone. Among the five Plasmodium parasite species that cause malaria in humans, two of them, P. falciparum and P. vivax, are responsible for about half a million deaths per year. The increasing resistance of Plasmodium species to drug therapies has created an urgent need to develop new antimalarial agents. Natural products have been a source of amazing drugs for efficient treatment of a variety of diseases and illnesses, including malaria. The planet's biodiversity has long been recognized as an abundant drug cabinet, offering medicinal chemists an immense range of molecular structures serving as inspiring sources for drug discovery.1 From very small structures, such as urea, to complex molecules like taxol, natural products originate from a wide range of organisms. Though tropical plants have been long known as a source for medicinal compounds, organisms from extreme ecosystems such as the polar regions, are largely unexplored for the discovery of new metabolites with useful bioactivities.2 Northern ecosystems’ severe environmental conditions force living organisms to adapt to face high UV radiation, low temperatures, low nutrient inputs, and other stresses. In addition, deep sea organisms are subjected to high pressure, low oxygen levels, and the absence of light.3 These factors contribute to gene modifications and to other adaptive processes producing unique secondary metabolites. Notably, arctic fungi have been underexplored so far. Along this line, Kerr's group isolated recently such species from deep sea sediments from Frobisher Bay in Northern Canada.4 BlastN investigation permitted researchers to place this isolate within the genus Mortierella based on a nucleotide sequence that has 96% similarity with Mortierella antarctica. To date, though nearly 100 Mortierelles species are known,5 few natural products have been isolated from them (Fig. 1).4b,6 Interestingly, Kerr's group identified from this Mortierella species four natural cyclic heptapeptides mortiamides A to D (1–4) (Fig. 1).
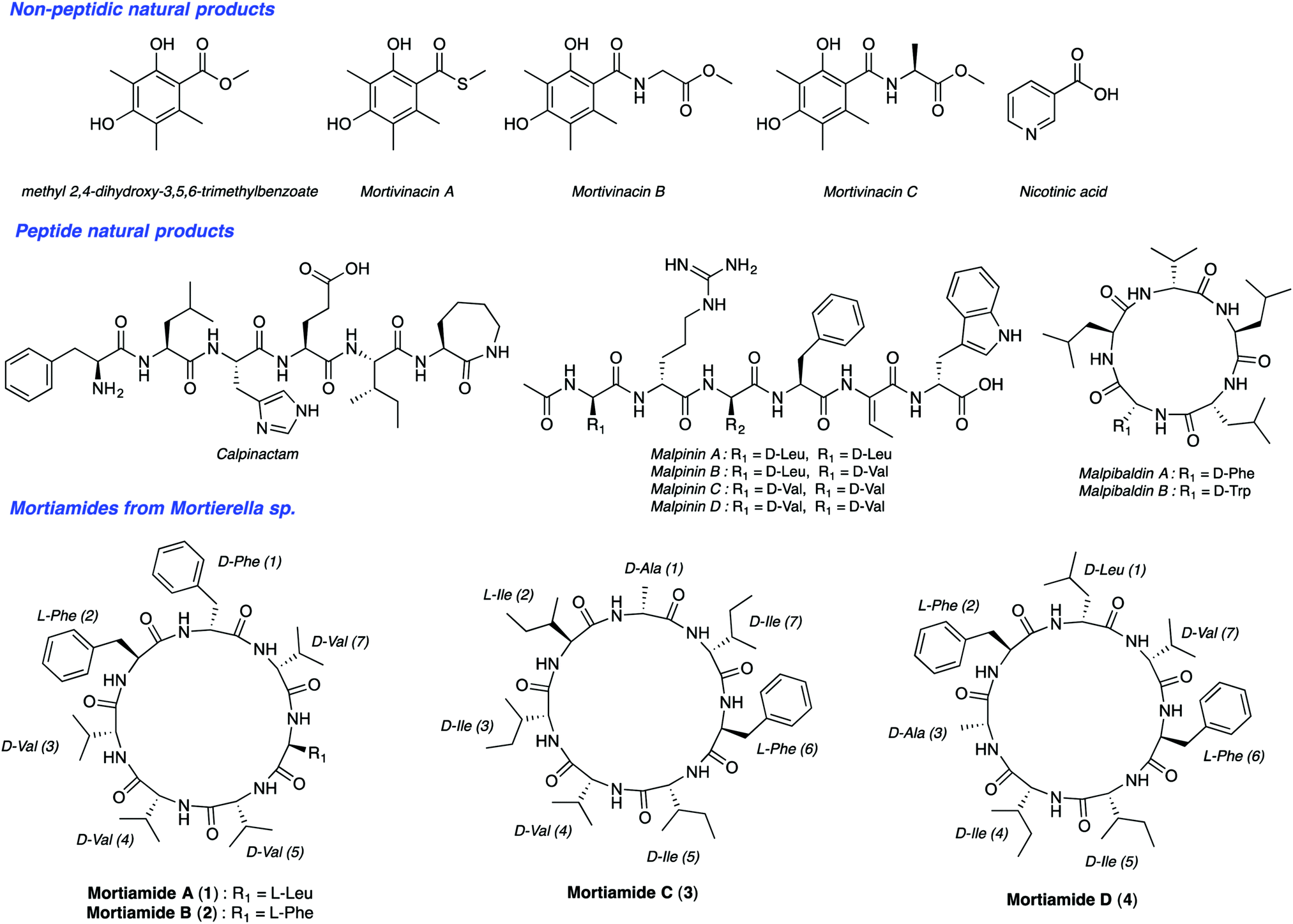 | ||
| Fig. 1 Structure of natural products isolated from Mortierella species. | ||
Analysis of the structures of mortiamides A to D (1–4), composed of D/L hydrophobic amino acids, revealed that they have similarities with natural products having antiplasmodial activity. Indeed, several natural head-to-tail macrocyclic peptides exhibit antiplasmodial activity.7 Among those, several have seven to eight L-α-amino acids, of which only one is polar.8 For example, mahafacyclin B, a hydrophobic cyclic heptapeptide containing four L-phenylalanines, two glycines, and one threonine shows good antimalarial activity (2.2 μM).7c,9
On that basis, we sought to investigate the antiplasmodial potential of mortiamides A to D (1–4). In order to do so, we have developed an efficient synthetic methodology on a solid support which, for the first time, quickly produces the desired cyclic heptapeptides in sufficient quantities for biological investigations.
Since the amino acid sequence influences the oligomerization/cyclization process, we examined the primary sequence of mortiamides A to D (1–4) to identify potential linear precursors.
Those cyclic heptapeptides are characterized by a 21-membered peptide ring containing L- and D-branched and aromatic amino acids. The absence of β-turn-inducing proline and flexible glycine in the sequence makes it more challenging to avoid oligomerization side reactions during cyclization reaction. In addition, mortiamides A to D (1–4) contain five D-amino acids at position 1, 3, 4, 5, and 7, of which three are contiguous. Positions 1 to 4 contain amino acids with alternate configurations, whereas sites 2 and 6 contain L-amino acids. Moreover, the primary sequence of compounds 1–4 contains seven hydrophobic amino acids. Consequently, the on-resin linear precursor could form extended strongly aggregating chains, decreasing the probability of cyclization occurring.10 Inspired by precedents in the literature11 that use oxime resin12 in peptide macrocyclization, we selected a linear precursor to facilitate the peptide folding and cyclization to obtain mortiamides A to D (1–4) by an on-resin acid-catalyzed head-to-tail concomitant cyclization/cleavage. Examples of sequence-dependent cyclization/cleavages that favour the intramolecular process on oxime resin have been described.13 In any macrocyclization process, it is always challenging to avoid undesired oligomerization and epimerization side reactions. However, anchoring the linear peptide on a solid support can reduce the rate of diffusion, thus decreasing the probability of dimerization and oligomerization (pseudodilution effect).14 Hence, we chose to incorporate a D-amino acid (site 1) at the anchor point with the objective to facilitate peptide folding by inserting L-amino acids at positions 2 and 6. Interestingly, incorporating alternating D- and L-amino acids in linear primary sequences causes turn-inducing effects and has been employed to improve the yield of peptide cyclizations.15 With the above considerations in mind, mortiamides A to D (1–4) were synthesized on oxime resin using standard protocols with a 0.43 mmol g−1 loading rate (Fig. 2).16 Briefly, the first amino acid was coupled for 3 h using DIC as a coupling reagent. The N-Boc protecting group was removed using a mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 trifluoroacetic acid (TFA)/dichloromethane, while the second amino acid was activated with hydroxybenzotriazole (6-Cl-HOBt) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HCTU). After completion of the linear precursor on the oxime resin, we proceeded with the key step of the synthesis. The linear peptide was simultaneously cyclized and cleaved from the resin in the presence of diisopropylethylamine (DIEA; 2.5 equiv.) and acetic acid (AcOH; 5 equiv.) in dichloromethane in a respectable 10−2 M cyclization concentration (Fig. 2), leading to very good macrocyclization yields of 1–4 of 35% to 48% without any oligomerization side reactions (>99% of cyclic monomers).
:1 trifluoroacetic acid (TFA)/dichloromethane, while the second amino acid was activated with hydroxybenzotriazole (6-Cl-HOBt) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HCTU). After completion of the linear precursor on the oxime resin, we proceeded with the key step of the synthesis. The linear peptide was simultaneously cyclized and cleaved from the resin in the presence of diisopropylethylamine (DIEA; 2.5 equiv.) and acetic acid (AcOH; 5 equiv.) in dichloromethane in a respectable 10−2 M cyclization concentration (Fig. 2), leading to very good macrocyclization yields of 1–4 of 35% to 48% without any oligomerization side reactions (>99% of cyclic monomers).
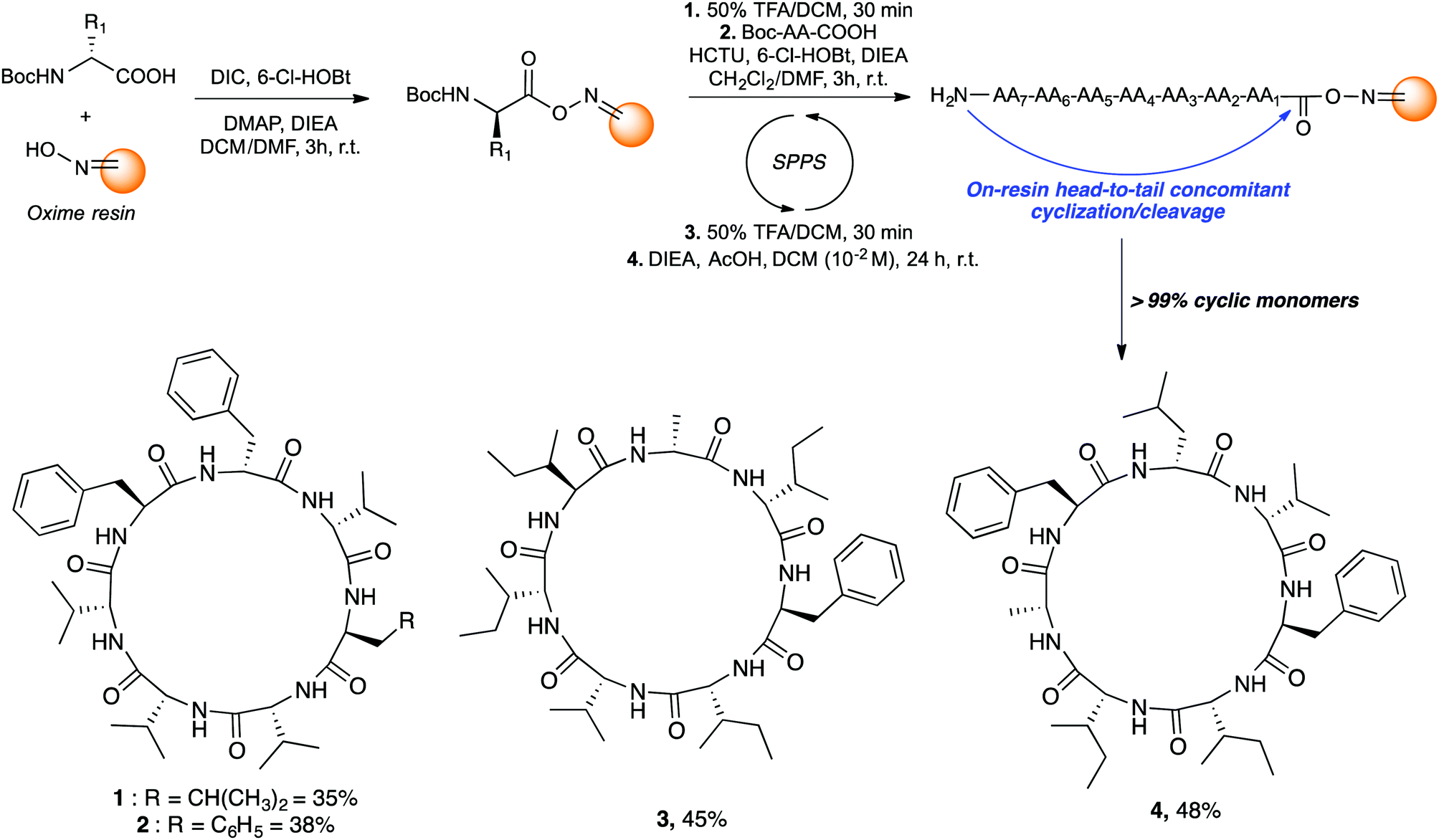 | ||
| Fig. 2 Mortiamides A to D synthetic procedures. | ||
Taking advantage of this powerful synthetic methodology that yields the cyclic monomer almost exclusively, simple triturations with cold ether gave pure mortiamides 1–4. Spectroscopic data of synthetic mortiamides A–D (1–4) matched entirely those reported for the isolated natural product, taking into account the possible conformers equilibrium (see ESI† for details).
In order to investigate whether mortiamides A to D (1–4) exhibit an inhibitory effect on P. falciparum proliferation, in vitro susceptibility assays were conducted and their IC50 were determined (Fig. 3 and 4). Assays were performed on the commonly used lab strain 3D7 and on the chloroquine–mefloquine–pyrimethamine multiresistant strain Dd2.17 The mortiamide A (1) IC50 with 3D7 and Dd2 were 7.85 ± 0.97 μM (n = 4) and 5.31 ± 0.24 μM (n = 5) respectively. The mortiamide B (2) IC50 with 3D7 and Dd2 values were 3.16 ± 0.65 μM (n = 4) and 2.10 ± 0.18 μM (n = 4) respectively. The IC50 value for mortiamide C (3) could not be determined as the molecule was not effective enough at the tested concentrations. The mortiamide D IC50 with 3D7 and Dd2 were 1.31 ± 0.12 μM (n = 11) and 0.94 ± 0.07 μM (n = 13) respectively (Fig. 4). To ensure that the observed growth inhibition effect was not due to red blood cell (RBC) lysis but to a biological effect on the parasite itself, hemolysis assays were performed with mortiamides A, B, and D. The absorbance reading at 405 nm after 72 h of incubation revealed no increase of hemoglobin release in the culture medium supernatant for mortiamides A and B at all tested concentrations (0.78 nM to 25.6 μM) when compared to untreated RBCs. Mortiamide D (4) only showed a mild RBC lysis effect of 19.7% at 25.6 μM and was reduced to 4.4% at 12.8 μM when compared to the untreated sample. Those concentrations are much higher than the IC50 values obtained with mortiamide D (4) proving its activity against the parasite.
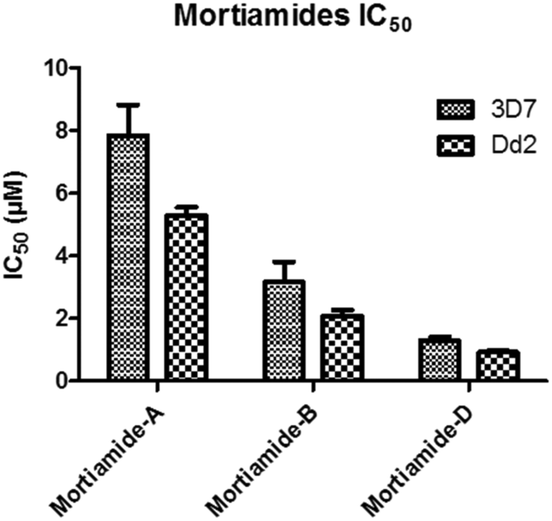 | ||
| Fig. 3 IC50 of mortiamides A, B, and D (1, 2, and 4). | ||
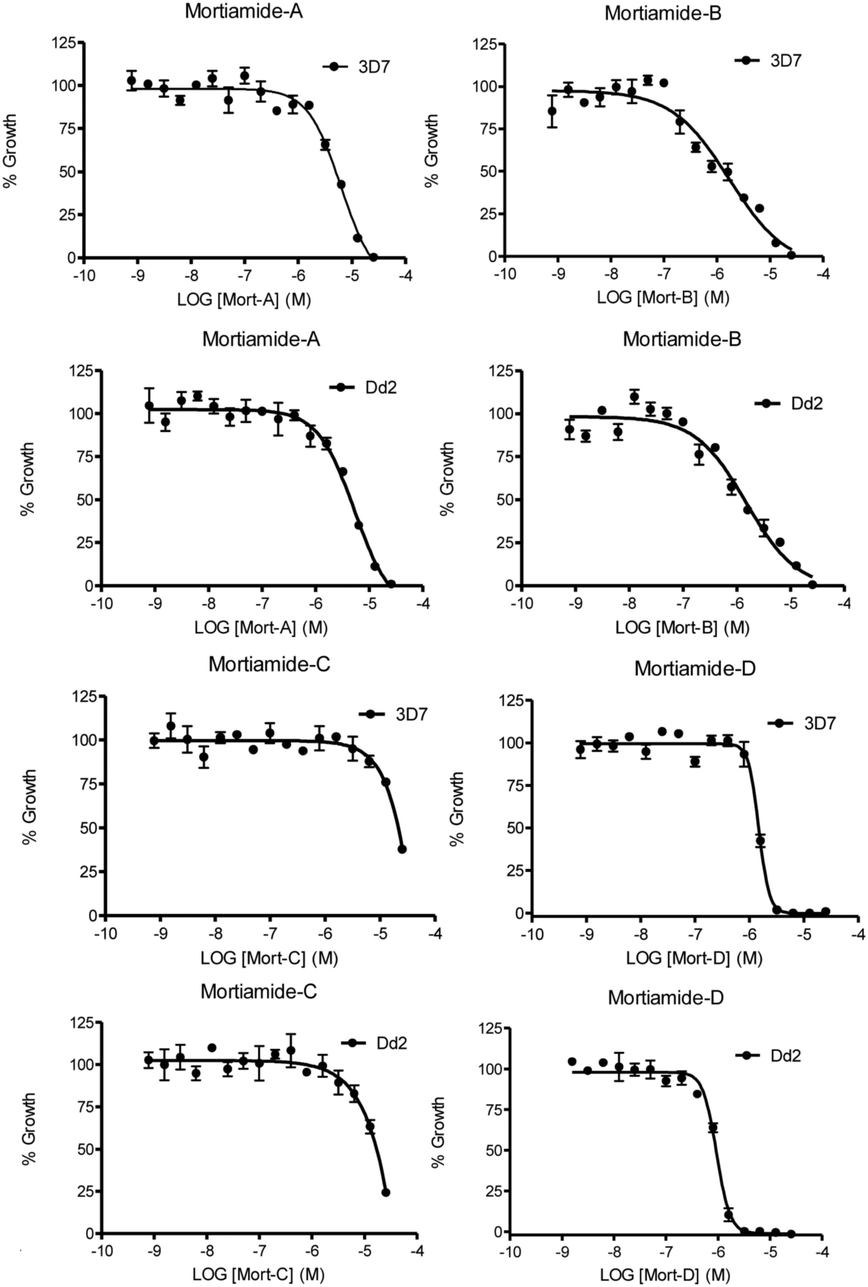 | ||
| Fig. 4 In vitro 72 h viability assays. Shown here are representative assays for both strains treated with each mortiamide. | ||
We achieved the first total synthesis of mortiamides A to D (1–4) on a solid support by concomitant cyclization/cleavage process without any oligomerization side reactions, and with impressive overall yield of 35 to 48%. Access to a sufficient amount of mortiamides allowed us to perform the first antiplasmodial investigation on those natural macrocyclic peptides. Results revealed that mortiamides A and B (1 and 2) have a moderate antiplasmodial activity, while mortiamide D (4) exhibits low micromolar activity. Interestingly, the results demonstrate that small variations on the side chains significantly impact the activity. Although the mechanism of action is not known yet, the results reported can be used to prepare analogs of mortiamide D, to improve its potency against P. falciparum.
This work was supported by the NSERC of Canada, the FRQNT of Quebec, PROTEO, Sentinelle Nord and Université Laval. CB wishes to thank PROTEO, FRQNT and NSERC for postgraduate scholarships. AB thanks the NSERC of Canada for a USRA scholarship. Work in Richard's lab was supported by a Canadian Institutes for Health Research Operating grant (MOP 130359). DR is a Fonds de la recherche du Québec-Santé Junior 2 Fellow. The authors are also thankful to Pierre Audet and François Otis for useful advices in mass spectrometry and HPLC analyses, respectively.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531 CrossRef CAS PubMed; (b) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef CAS PubMed.
- (a) Y. Tian, Y. Li and F. Zhao, Mar. Drugs, 2017, 15, 28 CrossRef PubMed; (b) S. Abbas, M. Kelly, J. Bowling, J. Sims, A. Waters and M. Hamann, Mar. Drugs, 2011, 9, 2423 CrossRef CAS PubMed; (c) Z. E. Wilson and M. A. Brimble, Nat. Prod. Rep., 2009, 26, 44 RSC.
- D. Skropeta and L. Wei, Nat. Prod. Rep., 2014, 31, 999 RSC.
- (a) A. W. Robertson, N. G. McCarville, L. W. MacIntyre, H. Correa, B. Haltli, D. H. Marchbank and R. G. Kerr, J. Nat. Prod., 2018, 81, 858 CrossRef CAS PubMed; (b) A. L. Grunwald, F. Berrue, A. W. Robertson, D. P. Overy and R. G. Kerr, J. Nat. Prod., 2017, 80, 2677 CrossRef CAS PubMed.
- L. Wagner, B. Stielow, K. Hoffmann, T. Petkovits, T. Papp, C. Vagvolgyi, G. de Hoog, G. Verkley and K. Voigt, Persoonia, 2013, 30, 77 CrossRef CAS PubMed.
- (a) K. Nobuhiro, K. Shigenobu, N. Kenichi, M. Rokuro, M. Makoto, Ō. Satoshi and T. Hiroshi, J. Antibiot., 2010, 63, 183 CrossRef PubMed; (b) A. G. Soman, J. Gloer and D. Wicklow, J. Nat. Prod., 1999, 62, 386 CrossRef CAS PubMed; (c) F. Baldeweg, P. Warncke, D. Fischer and M. Gressler, Org. Lett., 2019, 21, 1444 CrossRef CAS PubMed.
- (a) A. Lawer, J. Tai, K. A. Jolliffe, S. Fletcher, V. M. Avery and L. Hunter, Bioorg. Med. Chem. Lett., 2014, 24, 2645 CrossRef CAS PubMed; (b) P. Barbie and U. Kazmaier, Org. Biomol. Chem., 2016, 14, 6036 RSC; (c) C. Baraguey, A. Blond, F. Cavelier, J.-L. Pousset, B. Bodo and C. Auvin-Guette, J. Chem. Soc., Perkin Trans. 1, 2001, 2098, 10.1039/B008538N; (d) V. Rukachaisirikul, S. Chantaruk, C. Tansakul, S. Saithong, L. Chaicharernwimonkoon, C. Pakawatchai, M. Isaka and K. Intereya, J. Nat. Prod., 2006, 69, 305 CrossRef CAS PubMed; (e) C. Auvin-Guette, C. Baraguey, A. Blond, H. S. Xavier, J.-L. Pousset and B. Bodo, Tetrahedron, 1999, 55, 11495 CrossRef CAS; (f) C. Baraguey, C. Auvin-Guette, A. Blond, F. Cavelier, F. Lezenven, J.-L. Pousset and B. Bodo, J. Chem. Soc., Perkin Trans. 1, 1998, 3033, 10.1039/A804003F; (g) C. H. M. Kocken, A. van der Wel, B. Rosenwirth and A. W. Thomas, Exp. Parasitol., 1996, 84, 439 CrossRef CAS PubMed; (h) M. E. F. Pinto, J. M. Batista, J. Koehbach, P. Gaur, A. Sharma, M. Nakabashi, E. M. Cilli, G. M. Giesel, H. Verli, C. W. Gruber, E. W. Blanch, J. F. Tavares, M. S. D. Silva, C. R. S. Garcia and V. S. Bolzani, J. Nat. Prod., 2015, 78, 374 CrossRef CAS PubMed; (i) C. Auvin, C. Baraguey, A. Blond, F. Lezenven, J.-L. Pousset and B. Bodo, Tetrahedron Lett., 1997, 38, 2845 CrossRef CAS; (j) C. Auvin-Guette, C. Baraguey, A. Blond, J.-L. Pousset and B. Bodo, J. Nat. Prod., 1997, 60, 1155 CrossRef CAS; (k) C. Baraguey, A. Blond, I. Correia, J.-L. Pousset, B. Bodo and C. Auvin-Guette, Tetrahedron Lett., 2000, 41, 325 CrossRef CAS.
- S. Ramalho, M. Pinto, D. Ferreira and V. Bolzani, Planta Med., 2018, 84, 558 CrossRef CAS PubMed.
- A. J. J. van den Berg, S. F. A. J. Horsten, J. J. K.-V. Bosch, B. H. Kroes, C. J. Beukelman, B. R. Leeflang and R. P. Labadie, FEBS Lett., 1995, 358, 215 CrossRef CAS.
- E. T. Kaiser, Acc. Chem. Res., 1989, 22, 47 CrossRef CAS.
- (a) A. K. Yudin, Chem. Sci., 2015, 6, 30 RSC; (b) C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509 CrossRef CAS PubMed; (c) J. S. Davies, J. Pept. Sci., 2003, 9, 471 CrossRef CAS PubMed; (d) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef CAS.
- (a) W. F. DeGrado and E. T. Kaiser, J. Org. Chem., 1980, 45, 1295 CrossRef CAS; (b) W. F. DeGrado and E. T. Kaiser, J. Org. Chem., 1982, 47, 3258 CrossRef CAS.
- (a) N. Nishino, M. Xu, H. Mihara and T. Fujimoto, Bull. Chem. Soc. Jpn., 1992, 65, 991 CrossRef CAS; (b) N. Nishino, M. Xu, H. Mihara, T. Fujimoto, Y. Ueno and H. Kumagai, Tetrahedron Lett., 1992, 33, 1479 CrossRef CAS.
- (a) L. T. Scott, J. Rebek, L. Ovsyanko and C. L. Sims, J. Am. Chem. Soc., 1977, 99, 625 CrossRef CAS; (b) S. Mazur and P. Jayalekshmy, J. Am. Chem. Soc., 1979, 101, 677 CrossRef CAS.
- (a) H. Kessler and B. Haase, Int. J. Pept. Protein Res., 1992, 39, 36 CrossRef CAS PubMed; (b) M. Tamaki, S. Akabori and I. Muramatsu, J. Am. Chem. Soc., 1993, 115, 10492 CrossRef CAS.
- (a) C. Bérubé, A. Borgia and N. Voyer, Org. Biomol. Chem., 2018, 16, 9117 RSC; (b) C. Bérubé, A. Borgia and N. Voyer, Tetrahedron Lett., 2018, 59, 4176 CrossRef; (c) C. Bérubé, A. Borgia, G. Simon, D. Grenier and N. Voyer, Phytochem. Lett., 2018, 26, 101 CrossRef.
- (a) G. Françoise, A. D. James, F. Hisashi, B. K. David, M. Olga, C. K. David, A. Masamichi, B. V. Akhil and E. W. Thomas, J. Cell Biol., 1996, 135, 269 CrossRef PubMed; (b) X.-Z. Su, M. T. Ferdig, Y. Huang, C. Q. Huynh, A. Liu, J. You, J. C. Wootton and T. E. Wellems, Science, 1999, 286, 1351 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, mortiamides characterization. See DOI: 10.1039/c9cc02864a |
| This journal is © The Royal Society of Chemistry 2019 |