
Tuning of the flexibility in metal–organic frameworks based on pendant arm macrocycles†
Sungeun
Jeoung
 a,
Songho
Lee
a,
Jae Hwa
Lee
a,
Soochan
Lee
b,
Wonyoung
Choe
b,
Dohyun
Moon
*c and
Hoi Ri
Moon
*a
a,
Songho
Lee
a,
Jae Hwa
Lee
a,
Soochan
Lee
b,
Wonyoung
Choe
b,
Dohyun
Moon
*c and
Hoi Ri
Moon
*a
aDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: hoirimoon@unist.ac.kr
bDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: choe@unist.ac.kr
cBeamline Division, Pohang Accelerator Laboratory, 80 Jigok-ro, 127beon-gil, Nam-gu, Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: dmoon@postech.ac.kr
First published on 24th May 2019
Abstract
An isostructural series of flexible metal–organic frameworks based on macrocycles having diverse pendant arms was developed to tune flexibility depending on functional groups. The pendant arms directing into the pores were found to play a key role in imparting different gate-opening behaviours in the threshold pressure and sorption capacity upon interaction with guest molecules.
The design and syntheses of metal–organic frameworks (MOFs) begin with the selection of the metal nodes and bridging organic ligands.1 Types of metal and organic building blocks and the nature of their assembly determine the geometry and structure of the MOFs. Often, they allow a high degree of flexibility, resulting in so-called ‘flexible MOFs’ or ‘soft porous crystals’.2 Flexible MOFs can reversibly change their form upon the action of external stimuli, owing to the intermolecular degrees of freedom in their components and thus, have tremendous potentials in applications such as adsorption, separation, sensing, catalysis, and biomedical usage.3 Accordingly, researchers studying MOFs have been devoting considerable efforts to establish the fundamental principles that facilitate the syntheses of flexible MOFs. For instance, the O–O axis of a carboxylate group endows significant breathing effect in MIL-53 and MIL-88 by acting as a kneecap.4 Farha et al. recently reported a flexible MOF, NU-1400, based on four connected Zr6 nodes, which afforded degrees of freedom by reducing the connectivity, compared with rigid NU-1000 and NU-901 having eight connected Zr6 nodes.5
However, strategies for the incorporation of selective switching in flexible MOFs, as demanded by the applications based on the flexibility, are still elusive and serendipitous. Hence, designing and controlling the flexibility in these materials are essential and yet, a challenging issue. Several strategies have been demonstrated in order to tune the structural dynamics and responsiveness of the MOFs such as crystal size adjustment, metal substitution, and linker functionalization with various functional groups or flexible side chains.6
Macrocycle-based MOFs have been reported as a subclass of MOFs; in particular, aza-macrocyclic complexes with square planar geometry can act as a linear linker, providing axial sites for carboxylate ligands.7 Furthermore, various types of functional groups can be easily introduced into the pores during the synthesis, by using macrocyclic moieties such as the 1,3,6,8,10,13-hexaazacyclotetradecane Ni(II) complex with pendant arms at 1 and 8 positions.8 Previously, we reported a flexible MOF based on the Ni(II) macrocycle having two propyl arms that behaved as a molecular gate.9 Experimental and theoretical studies found that CO2 molecules mainly interacted with the propyl pendant arms to induce the gate-opening and breathing phenomena. Based on this, we postulated that such a MOF can be an excellent platform for easily securing different functional groups in the pendant arm macrocycles, which would render them different structural dynamics. Thus, we introduced nitrile, hydroxyl, and allyl pendant arms in Ni(II) macrocycles in this study and successfully synthesised three isostructural flexible MOFs, flexMOF(CN), flexMOF(OH), and flexMOF(CH2) (Fig. 1). Upon types of the guest molecules such as CO2 and water, these MOFs showed distinguishable flexible behaviours, which were examined by sorption isotherms, in situ X-ray powder diffraction (XRPD), and single-crystal diffraction (SCD).
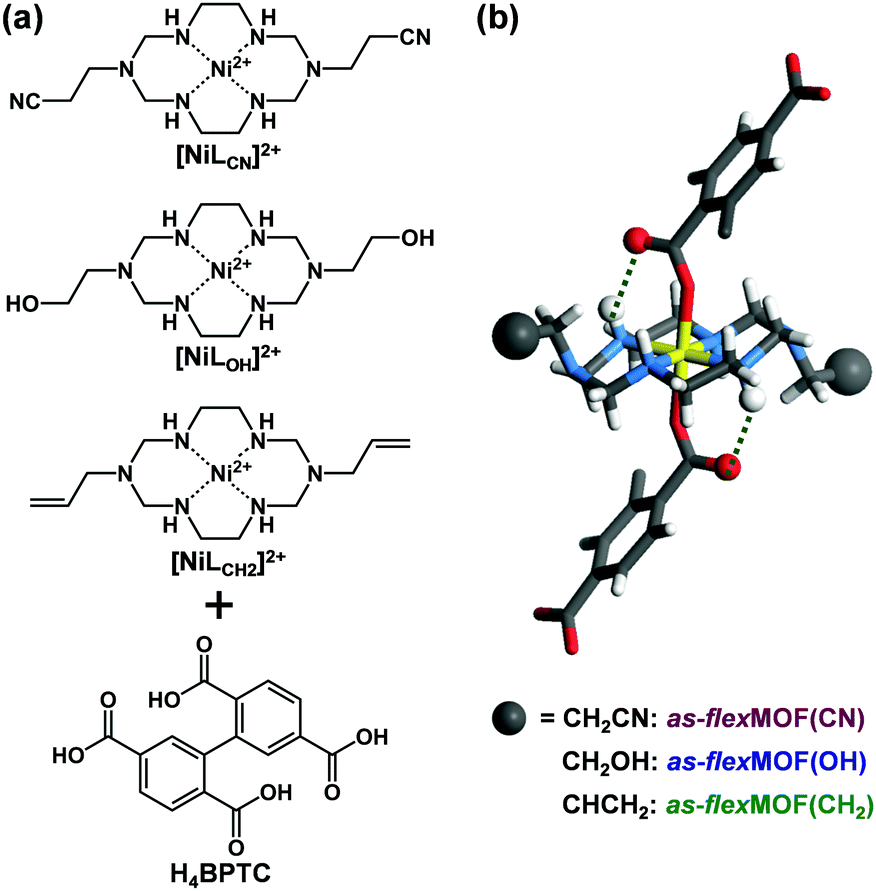 | ||
| Fig. 1 (a) Square planar Ni(II) macrocycles with different pendant arms as metal building blocks and H4BPTC as an organic building block. (b) Representation of the linear connectivity between two building units via coordinate and hydrogen bonds. Colour scheme: Ni, yellow; C, grey; O, red; N, blue; H, white. | ||
To synthesise an isostructural series of flexible MOFs decorated with different functional groups, we induced the self-assembly of a tetradentate ligand, 2,2′,5,5′-biphenyltetracarboxylic acid (H4BPTC), with [NiLR]2+ as a metal building block (R = CN, OH, and CH2 for nitrile, hydroxyl, and allyl substitutes, respectively, Fig. 1a), yielding {[(NiLCN)2(BPTC)]·4DMF·2H2O} (as-flexMOF(CN); DMF = N,N-dimethylformamide), {[(NiLOH)2(BPTC)]·1DMF·3H2O} (as-flexMOF(OH)), and {[(NiLCH2)2(BPTC)]·1CH3CN·5H2O} (as-flexMOF(CH2)). For all the compounds, the square planar Ni(II) macrocycles were coordinated by two BPTC4− ligands in the axial sites, resulting in a six-coordinated octahedral geometry with hydrogen bonds between the uncoordinated oxygen atoms of the carboxylate and secondary amines from the aza-macrocyclic complex (dotted lines in Fig. 1b). Since the BPTC4− moiety in the MOFs is not planar (dihedral angles between two phenyl rings are 52.780° in as-flexMOF(CN), 50.318° in as-flexMOF(OH), and 57.267° in as-flexMOF(CH2)) and coordinates four macrocycles, 3D networks having interconnected pores are formed (top of Fig. S4–S6, ESI†), in which the void space is 52.5% for as-flexMOF(CN), 49.6% for as-flexMOF(OH), and 41.2% for as-flexMOF(CH2) per unit cell volume, as obtained by PLATON calculations.10 Upon drying the MOFs, the 3D structures drastically changed and exhibited structural flexibility, as evidenced by the XRPD patterns (Fig. S7–S9, and details of activation are provided in ESI;† hereafter, dried compounds are designated as cp-flexMOF (cp = closed-pore)). The void volume is also greatly reduced to 6.1%, 6.6%, and 3.9% per unit cell volume for cp-flexMOF(CN), cp-flexMOF(OH), and cp-flexMOF(CH2), respectively. Despite such tremendous changes in the cell volume, all the samples maintained single crystallinity, which was sufficient to obtain SCD data. Fig. 2 shows that the rectangular dimension of as phases in all the three compounds considerably shrank in cp phases (42.6%, 34.1%, and 23.8% decreases for flexMOF(CN), flexMOF(OH), and flexMOF(CH2), respectively). During the dynamic movement, the dihedral angle between the two phenyl rings and those between the carboxylate planes and benzene rings of the ligand changed, and the pendant arms of the macrocycles blocked the pores (Fig. S10–S12, ESI†).
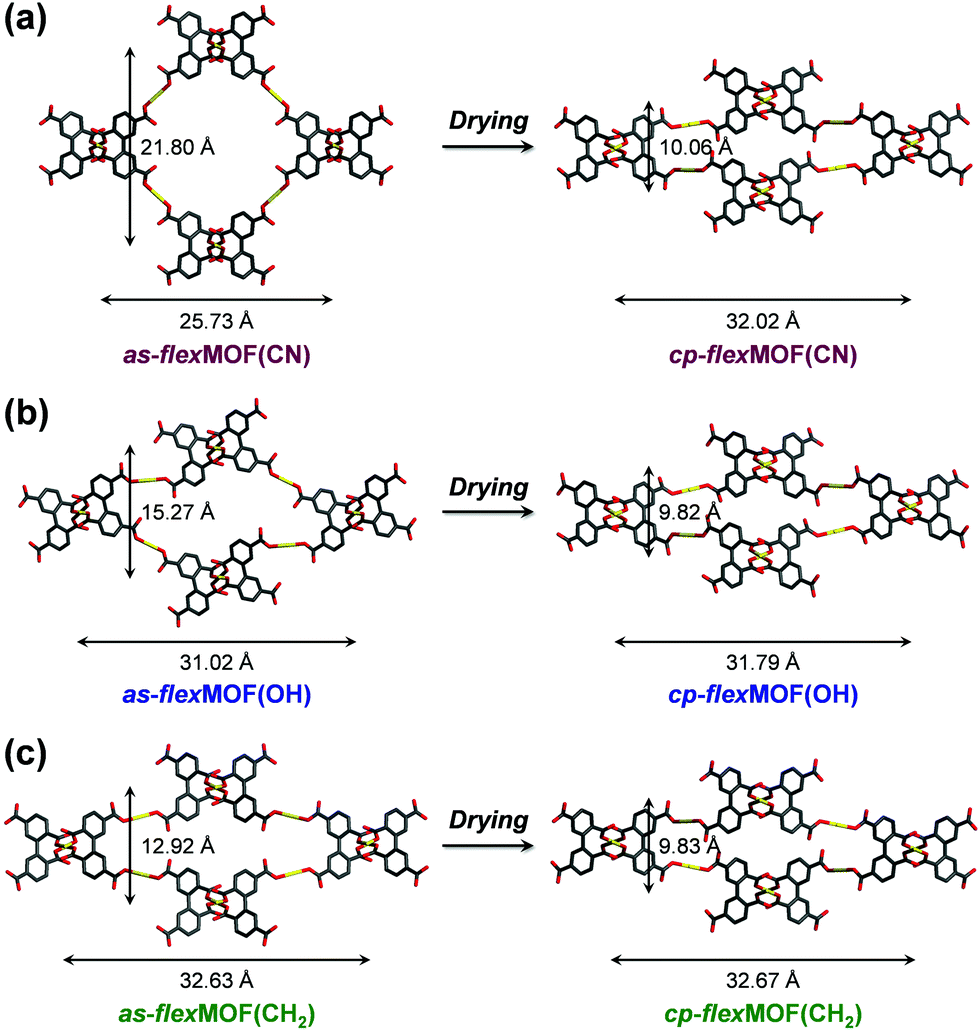 | ||
| Fig. 2 Comparison between the as-synthesised (as) and closed-pore (cp) phases of (a) flexMOF(CN), (b) flexMOF(OH), and (c) flexMOF(CH2) in rectangular dimension. The macrocycles and hydrogen atoms are omitted for clarity. Colour scheme: Ni, yellow; C, grey; O, red. | ||
In order to explore the guest-dependent flexibility of these MOFs, N2 and CO2 gas sorption isotherms were obtained at 77 and 196 K, respectively. All cp phases showed typical type II N2 sorption isotherms, indicating a nonporous nature (Fig. S13, ESI†). However, the characteristic of the CO2 sorption isotherms was interesting (Fig. 3). cp-flexMOF(CN) exhibited gate-opening for CO2 with an abrupt uptake at a threshold pressure of 0.42 bar, and its total adsorption capacity approached 40.1 wt% (204.8 cc g−1 or 9.14 mmol g−1 of the host) at 1 bar. When the allyl pendant arms are introduced, the resulting MOF, cp-flexMOF(CH2) displayed a significantly different threshold pressure of ∼0.70 bar, which was higher than that in cp-flexMOF(CN). This was also accompanied by a decrease in the CO2 uptake capacity (16.7 wt%, 85.5 cc g−1, or 3.82 mmol g−1). The different response of cp-flexMOF(CN) and cp-flexMOF(CH2) toward N2 and CO2 can be explained by the high quadruple moment and polarizability of CO2, which is in contrast to the inert nature of N2.11 It is also noteworthy that as phases of flexMOF(CN) and flexMOF(CH2) were estimated to potentially adsorb ca. 13.9 and 5.4 mmol g−1 of CO2, respectively (greater than the experimental value; Table S11, ESI†), as calculated by grand canonical Monte Carlo (GCMC) adsorption simulation performed with the molecular software package, RASPA 2.0 (details of simulation in ESI†).12 This implies that those cp phases are not fully transformed into as phases under this condition (1 bar of CO2 at 196 K), but are open up to form new phases, lp-flexMOF (Fig. 3; lp stands for large pore phase). Also, both the flexMOF(CN) and flexMOF(CH2) desorbed in a hysteretic and stepwise manner, which adsorption and desorption curves do not merge at the end even under vacuum (<10−5 mbar) at 196 K. In contrast, hydroxyl group-containing cp-flexMOF(OH) showed no stepwise adsorption profile with negligible uptake.
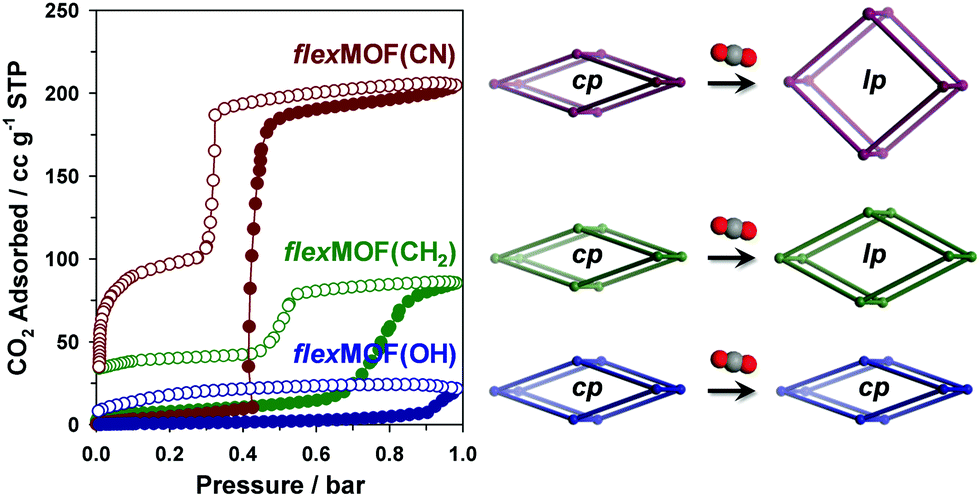 | ||
| Fig. 3 CO2 adsorption/desorption isotherms of flexMOF(CN), flexMOF(OH), and flexMOF(CH2) at 196 K (left) and schematic of their flexible behaviours upon CO2 adsorption (right). Filled and empty circles represent adsorption and desorption curves, respectively. | ||
The forementioned different responsive behaviours were studied by in situ XRPD under the synchronized condition of CO2 sorption. The XRPD patterns of cp-flexMOF(CN) did not change up to 0.4 bar, and a drastic structural change was observed at 0.6 bar, at which the CO2 uptake increased sharply (Fig. S14, ESI†), retaining up to 1 bar. As mentioned previously, this is not a same structure with a as phase, but a lp phase. During desorption, the XRPD patterns were retained up to 0.4 bar, but it changed into a new structure below this pressure, which had neither cp nor lp structure and thereby denoted as np-flexMOF(CN) (Fig. 4a; np stands for narrow pore phase). Such a discovery of a new intermediate phase derived upon desorption has been very rarely reported in flexible MOFs.13
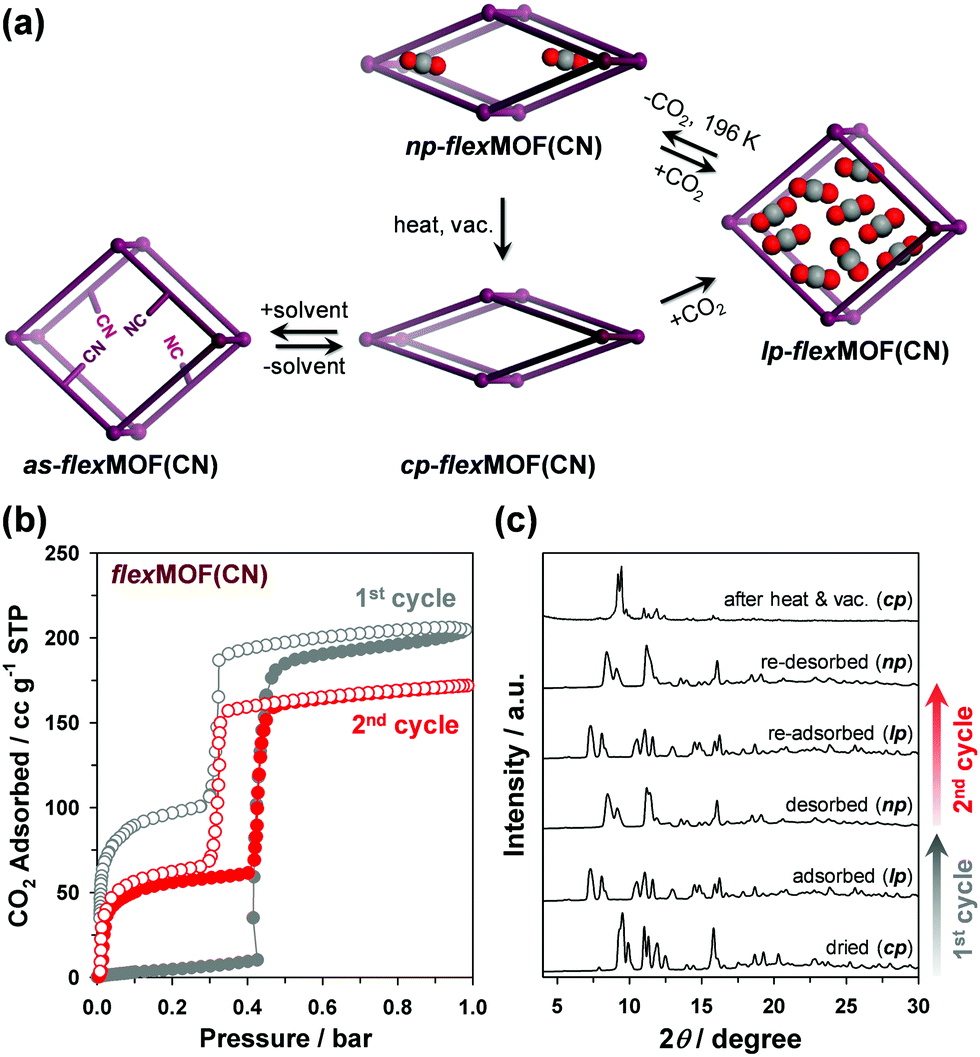 | ||
| Fig. 4 (a) Schematic illustration of the phase transitions of flexMOF(CN) upon guest removal/reintroduction and CO2 adsorption/desorption. (b) Consecutive cycles of CO2 adsorption/desorption isotherms in flexMOF(CN) at 196 K, and (c) corresponding XRPD data measured before CO2 adsorption, after adsorption, desorption (vacuum < 10−5 mbar), re-adsorption, and re-desorption, and after re-activation by heating under vacuum. Filled and empty circles represent adsorption and desorption curves, respectively. | ||
The intermediate structure, np-flexMOF(CN) has its own permanent porosity. After one adsorption–desorption experiment, the np phase is stabilised even under vacuum at 196 K. Interestingly, the successive measurement of the CO2 uptake beginning from np-flexMOF(CN) at 196 K (Fig. 4b) showed type IV adsorption isotherm, which was clearly different from the adsorption isotherm of the first cycle. In this profile, even at the low pressure region CO2 uptake was occurred up to 12.0 wt%, yet the threshold pressure was observed at the same position of the first cycle. The total CO2 uptake decreased from 40.1 to 33.7 wt%, in which the uptake difference is equal to the trapped amount of CO2 after the first cycle, and the desorption curve retraced to the adsorption curve. As revealed by the in situ XRPD (Fig. 4c), the np transformed to the lp phase during the CO2 re-adsorption, and then reverted to its np phase after re-desorption. The further consecutive cycle of CO2 adsorption/desorption for np-flexMOF(CN) fully reproduced this trend (Fig. S15, ESI†), indicating the reversible transition between the np and lp phases (Fig. 4a). Furthermore, heating at 110 °C under vacuum resulted in the transformation into the cp phase (Fig. 4c and Fig. S16, ESI†). Remarkably, at higher temperature (298 K), the np phase can be still identified upon CO2 desorption (Fig. S19, ESI†). The gate-opening pressure of flexMOF(CN) dramatically shifts to a higher pressure upon the temperature increase to room temperature, while flexMOF(CH2) still opens the pores at a very low pressure.
It should be noted that the discrepancy of the flexibility in flexMOF(OH) is attributed to the increased number of hydrogen bonds from as-flexMOF(OH) to cp-flexMOF(OH), as revealed by the SCD analysis, owing to the single-crystal-to-single-crystal transformation (Fig. 5b and Fig. S20 in ESI†). In cp-flexMOF(OH), the hydroxyl groups participate in hydrogen bonding with the carboxylate oxygens and the secondary amines from the macrocycles as well as the hydroxyl groups from other pendant arms to stabilize the cp structure by strong host-host interactions. Considering this aspect, while CO2 molecules failed to open up the structure (Fig. S18, ESI†), certain guest molecules, which can make stronger host–guest interaction such as water, is expected to lead a structural flexibility, and water vapour sorption isotherm (Fig. 5c) shows that the introduction of polar water molecules can open the pores of cp-flexMOF(OH); it adsorbs 144 cc g−1 of water vapour at 298 K in two steps, at 0.08 and 0.65 relative humidity (P/P0), and desorbs with a large hysteresis. The corresponding XRPD results, measured after water adsorption and re-activation by heating at 110 °C under vacuum, also verifies the significant and reversible structural transitions of flexMOF(OH) (Fig. S21, ESI†). Likely, flexMOF(CN) and flexMOF(CH2) also shows the stepwise adsorption profile for water vapour, which was similarly observed in the CO2 sorption (Fig. S22, ESI†). Furthermore, methanol and benzene vapour isotherms of flexMOF(OH) were measured to prove the role of polar protic solvents in controlling the host–guest interaction and opening the closed pores. Resultingly, the pores of flexMOF(OH) was opened by methanol, but there was no adsorption by the non-polar benzene vapour (Fig. S25, ESI†).
 | ||
| Fig. 5 (a) Schematic illustration of the phase transitions of flexMOF(OH) upon guest removal/reintroduction and adsorption/desorption of CO2 and H2O molecules. (b) Hydrogen bonds of cp-flexMOF(CN) as represented by dotted lines, and (c) its water vapour isotherm at 298 K. Filled and empty circles represent adsorption and desorption curves, respectively. | ||
In conclusion, we employed the Ni(II) macrocycle moiety with different pendant arms containing the nitrile, hydroxyl, or allyl groups, to demonstrate that such a MOF platform can readily grant and tune the flexibility based on the functional groups in the pores. Three isostructural MOFs exhibited distinct flexibility, including the gate-opening threshold pressure, sorption capacities, step steepness, and hysteresis. For flexMOF(CN) and flexMOF(CH2), an intermediate new form was discovered from the CO2-trapped np phase. Moreover, the hydroxyl group in flexMOF(OH) induced the strong host–host interaction via numerous hydrogen bonds in the cp phase, which could be attributed to the structural stabilisation, and flexMOF(OH) showed the responsive flexibility upon water over CO2. Therefore, we envision that this pendant arm azamacrocycle-based MOF would not only provide insights for the rational design and fine-tuning of flexible MOFs but also enable their uses in a variety of practical applications.
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Ministry of Science and ICT (No. NRF-2016R1A5A1009405, NRF-2017R1A2B4008757). X-ray crystallography analysis performed at 2D-SMC beamline (2018-3rd-2D-036) in PLS-II were supported by MSIT and POSTECH.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS; (b) J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (c) H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167 CrossRef CAS PubMed.
- (a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed; (b) Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867 RSC.
- (a) J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161 CrossRef CAS; (b) A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC; (c) Z. Chang, D.-H. Yang, J. Xu, T.-L. Hu and X.-H. Bu, Adv. Mater., 2015, 27, 5432 CrossRef CAS; (d) M. Shivanna, Q.-Y. Yang, A. Bajpai, S. Sen, N. Hosono, S. Kusaka, T. Pham, K. A. Forrest, B. Space, S. Kitagawa and M. J. Zaworotko, Sci. Adv., 2018, 4, eaaq1636 CrossRef PubMed; (e) P. Kanoo, R. Haldar, S. K. Reddy, A. Hazra, S. Bonakala, R. Matsuda, S. Kitagawa, S. Balasubramanian and T. K. Maji, Chem. – Eur. J., 2016, 22, 15864 CrossRef CAS; (f) Y.-X. Shi, W.-X. Li, W.-H. Zhang and J.-P. Lang, Inorg. Chem., 2018, 57, 8627 CrossRef CAS PubMed.
- (a) C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828 CrossRef CAS PubMed; (b) G. Férey, Z. Anorg. Allg. Chem., 2012, 638, 1897 CrossRef.
- (a) Y. Zhang, X. Zhang, J. Lyn, K.-I. Otaka, X. Wang, L. R. Redfern, C. D. Malliakas, Z. Li, T. Islamoglu, B. Wang and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 11179 CrossRef CAS PubMed; (b) A. W. Peters, K. Otake, A. E. Platero-Prats, Z. Li, M. R. DeStefano, K. W. Chapman, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2018, 10, 15073 CrossRef CAS PubMed; (c) H. Noh, C.-W. Kung, T. Islamoglu, A. W. Peters, Y. Liao, P. Li, S. J. Garibay, X. Zhang, M. R. DeStefano, J. T. Hupp and O. K. Farha, Chem. Mater., 2018, 30, 2193 CrossRef CAS.
- (a) Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193 CrossRef CAS; (b) A. Schneemann, P. Vervoorts, I. Hante, M. Tu, S. Wannapaiboon, C. Sternemann, M. Paulus, D. C. F. Wieland, S. Henke and R. A. Fischer, Chem. Mater., 2018, 30, 1667 CrossRef CAS; (c) C. M. McGuirk, T. Runčevski, J. Oktawiec, A. Turkiewicz, M. K. Taylor and J. R. Long, J. Am. Chem. Soc., 2018, 140, 15924 CrossRef CAS PubMed; (d) P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839 CrossRef CAS PubMed; (e) S. Henke, A. Schneemann, A. Wütscher and R. A. Fischer, J. Am. Chem. Soc., 2012, 134, 9464 CrossRef CAS PubMed.
- (a) S.-M. Hyun, T. K. Kim, Y. K. Kim, D. Moon and H. R. Moon, Inorg. Chem. Commun., 2013, 33, 52 CrossRef CAS; (b) J. H. Lee and H. R. Moon, J. Inclusion Phenom. Macrocyclic Chem., 2018, 92, 237 CrossRef CAS.
- M. P. Suh, B. Y. Shim and T.-S. Yoon, Inorg. Chem., 1994, 33, 5509 CrossRef CAS.
- S.-M. Hyun, J. H. Lee, G. Y. Jung, Y. K. Kim, T. K. Kim, S. Jeoung, S. K. Kwak, D. Moon and H. R. Moon, Inorg. Chem., 2016, 55, 1920 CrossRef CAS PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- (a) A. Boutin, F.-X. Coudert, M.-A. Springuel-Huet, A. V. Neimark, G. Férey and A. H. Fuchs, J. Phys. Chem. C, 2010, 114, 22237 CrossRef CAS; (b) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791 CrossRef CAS.
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, Mol. Simul., 2015, 41, 1 CrossRef.
- A.-X. Zhu, Q.-Y. Yang, A. Kumar, C. Crowley, S. Mukherjee, K.-J. Chen, S.-Q. Wang, D. O’Nolan, M. Shivanna and M. J. Zaworotko, J. Am. Chem. Soc., 2018, 140, 15572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, crystal structures, details of characterisation results. CCDC 1898504–1898507, 1908259 and 1908261. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc02819f |
| This journal is © The Royal Society of Chemistry 2019 |