
Synthesis of polysubstituted 3-aminoindenes via rhodium-catalysed [3+2] cascade annulations of benzimidates with alkenes†
Binjie
Wu
,
Zi
Yang
,
Hong
Zhang
,
Lianhui
Wang
and
Xiuling
Cui
 *
*
Engineering Research Center of Molecular Medicine of Ministry of Education, Key Laboratory of Fujian Molecular Medicine, Key Laboratory of Xiamen Marine and Gene Drugs, School of Biomedical Sciences, Huaqiao University, Xiamen 361021, P. R. China. E-mail: cuixl@hqu.edu.cn
First published on 13th March 2019
Abstract
A novel Rh-catalysed intermolecular [3+2] cascade cyclization of benzimidates and alkenes has been developed to assemble polysubstituted 3-aminoindenes. The target products are important building blocks for organic synthesis and drug discovery. Cheap and easily available α,β-unsaturated ketones or nitroalkenes are applied as coupling partners. Acyl or nitro groups are easily introduced to 3-aminoindenes at the C-2 position, which makes this reaction particularly attractive for further transformation to synthesize various important building blocks.
Indenamines are key structural frameworks embedded in a large number of biologically active molecules, such as compound A and B, and exhibit a wide range of interesting pharmacological properties (Fig. 1).1 As a result, these molecules provide an attractive platform for structure–activity relationship studies and discovery in drug development.2 Therefore, novel efficient strategies to access such a skeleton are highly desirable.
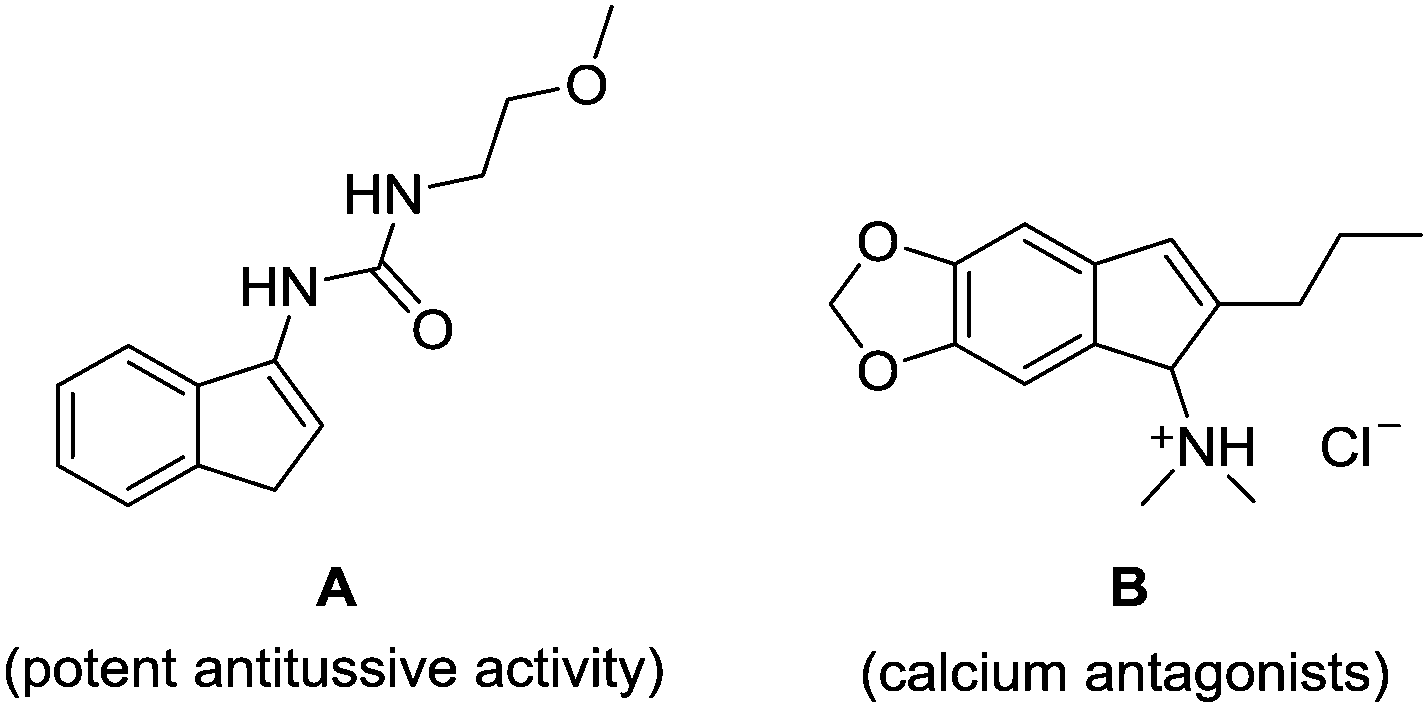 | ||
| Fig. 1 Examples illustrating the importance of indenamines. | ||
Recently, direct transition-metal-catalysed intermolecular cyclization via C–H bond activation3 has been established as a straightforward method to construct such a core.4 For example, Takai and co-workers reported the first catalytic synthesis of 3-aminoindenes via Re catalysed cyclization of aldimines with phenyl acetylenes in 2005 (Scheme 1a).5 Afterwards, Zhao and co-workers reported Rh-catalysed [3+2] annulation of N-unsubstituted ketimines and internal alkynes to afford substituted 1-aminoindene derivatives (Scheme 1b).6 Subsequently, Cramer and co-workers reported an enantioselective synthesis of 1-aminoindenes via rhodium(I)-catalysed C–H functionalization of unsubstituted ketimines with internal alkynes (Scheme 1b).7 Later, Li and co-workers reported a catalytic annulation of N-sulfonyl imines or azomethine ylides with alkynes to form indenamines (Scheme 1b).8 Wang and co-workers reported a rhenium catalysed [3+2] carbocyclization of ketimines and alkynes to approach the unprotected tertiary indenamines in 2016 (Scheme 1b).9 More recently, Yi and co-workers reported an efficient rhodium(III)-catalysed C–H transformation of oximes for the synthesis of indenamine skeletons (Scheme 1b).10 These strategies all focused on the cycloaddition of arylimines with alkynes, which is not beneficial to expand the substrate scope. Herein, we report a protocol to polysubstitute 3-aminoindenes from benzimidates and alkenes through rhodium-catalysed C–H activation/carbocyclization (Scheme 1c). Cheap and easily available α,β-unsaturated ketones11 or nitroalkenes12 are applied as a coupling partner. Acyl or nitro groups could be smoothly introduced to 3-aminoindenes at the C-2 position, which makes the products of this reaction particularly attractive for further transformation to synthesize various important molecules. In addition, this protocol exhibits good functional-group tolerance and excellent regioselectivity.
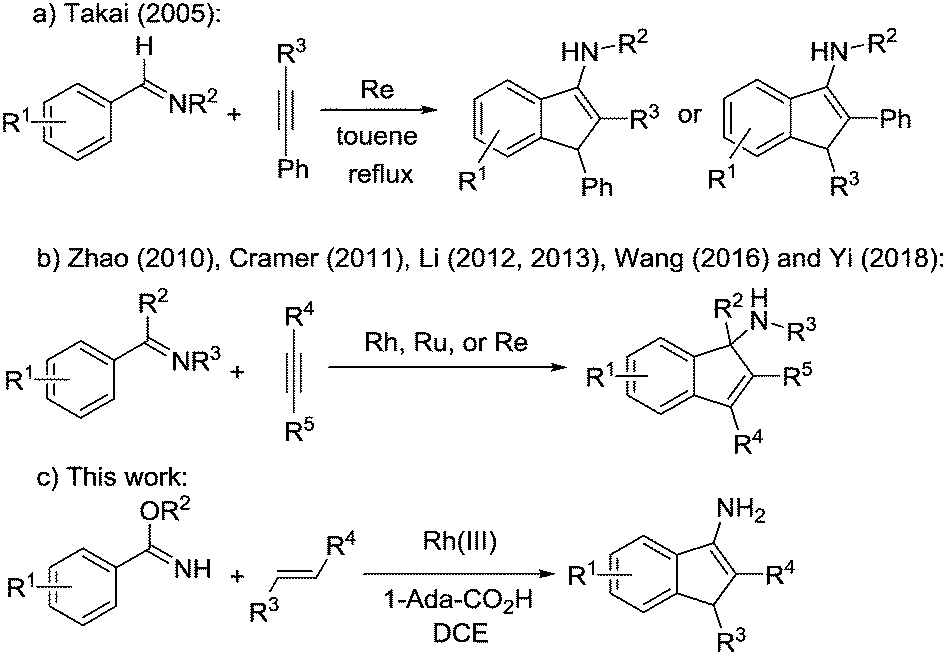 | ||
| Scheme 1 Synthesis of indenamines. | ||
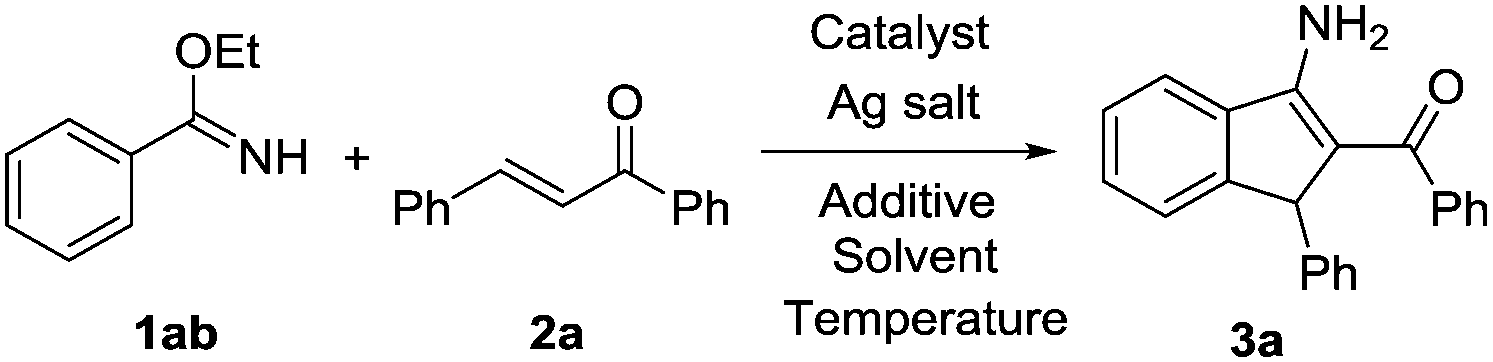
The condensation of ethyl benzimidate (1ab) and 1,3-diphenyl-2-propen-1-one (2a) was initially chosen as a model reaction to optimize the various reaction parameters (Table 1). To our great delight, the desired product 3a was obtained in 39% yield when [Cp*RhCl2]2/AgSbF6 was used as a catalyst and AcOH (1 equiv.) as an additive in DCE at 120 °C under an air atmosphere (entry 1). Other metals, such as Pd(OAc)2, [Ru(p-cymene)Cl2]2, and [Cp*IrCl2]2 complexes, were evaluated as catalysts in our system. Unfortunately, none of them worked for this transformation (entries 2–4). A 42% yield of 3a was obtained when [Cp*RhCl2]2/AgNTf2 was employed as a catalyst (entry 5). Switching the catalyst to [Cp*Rh(MeCN)3](SbF6)2 gave a higher yield of 47% (entry 6). The reaction did not proceed smoothly without an Rh catalyst (entry 7). Then, various additives, including PivOH, 1-Ada-CO2H, NaOAc, and LiOAc, were screened, showing that 1-Ada-CO2H gave the highest yield (entries 8–11). The yield of 3a was decreased to 33% in the absence of an additive (entry 12). Other solvents, such as toluene, 1,4-dioxane, and TFE, were less effective for this catalytic reaction (entries 13–15). The reaction was not significantly affected when carried out under an O2 or N2 atmosphere (entries 16–17). Changing the reaction temperature or decreasing the reaction time to 24 hours did not favor this reaction (entries 18–20).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yieldb (%) |
| a Reaction conditions: 1ab (0.2 mmol), 2a (2.0 equiv.), [Cp*Rh(MeCN)3](SbF6)2 (5.0 mol%), 1-Ada-CO2H (1.0 equiv.), DCE (2 mL), 120 °C, 36 h, under an air atmosphere. b Isolated yields. c Under an N2 atmosphere. d Under an O2 atmosphere. e 110 °C. f 130 °C. g 24 h. N.R. = no reaction. 1-Ada-CO2H = 1-adamantane carboxylic acid. | ||||
| 1 | [Cp*RhCl2]2/AgSbF6 | AcOH | DCE | 39 |
| 2 | Pd(OAc)2 | AcOH | DCE | N.R. |
| 3 | [Ru(p-cymene)Cl2]2 | AcOH | DCE | N.R. |
| 4 | [Cp*IrCl2]2/AgNTf2 | AcOH | DCE | N.R. |
| 5 | [Cp*RhCl2]2/AgNTf2 | AcOH | DCE | 42 |
| 6 | [Cp*Rh(MeCN)3](SbF6)2 | AcOH | DCE | 47 |
| 7 | — | AcOH | DCE | N.R. |
| 8 | [Cp*Rh(MeCN)3](SbF6)2 | PivOH | DCE | 48 |
| 9 | [Cp*Rh(MeCN) 3 ](SbF 6 )2 | 1-Ada-CO 2 H | DCE | 88 |
| 10 | [Cp*Rh(MeCN)3](SbF6)2 | NaOAc | DCE | 42 |
| 11 | [Cp*Rh(MeCN)3](SbF6)2 | LiOAc | DCE | 81 |
| 12 | [Cp*Rh(MeCN)3](SbF6)2 | — | DCE | 33 |
| 13 | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | Toluene | 51 |
| 14 | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | 1,4-Dioxane | Trace |
| 15 | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | TFE | 21 |
| 16c | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | DCE | 83 |
| 17d | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | DCE | 89 |
| 18e | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | DCE | 70 |
| 19f | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | DCE | 63 |
| 20g | [Cp*Rh(MeCN)3](SbF6)2 | 1-Ada-CO2H | DCE | 72 |
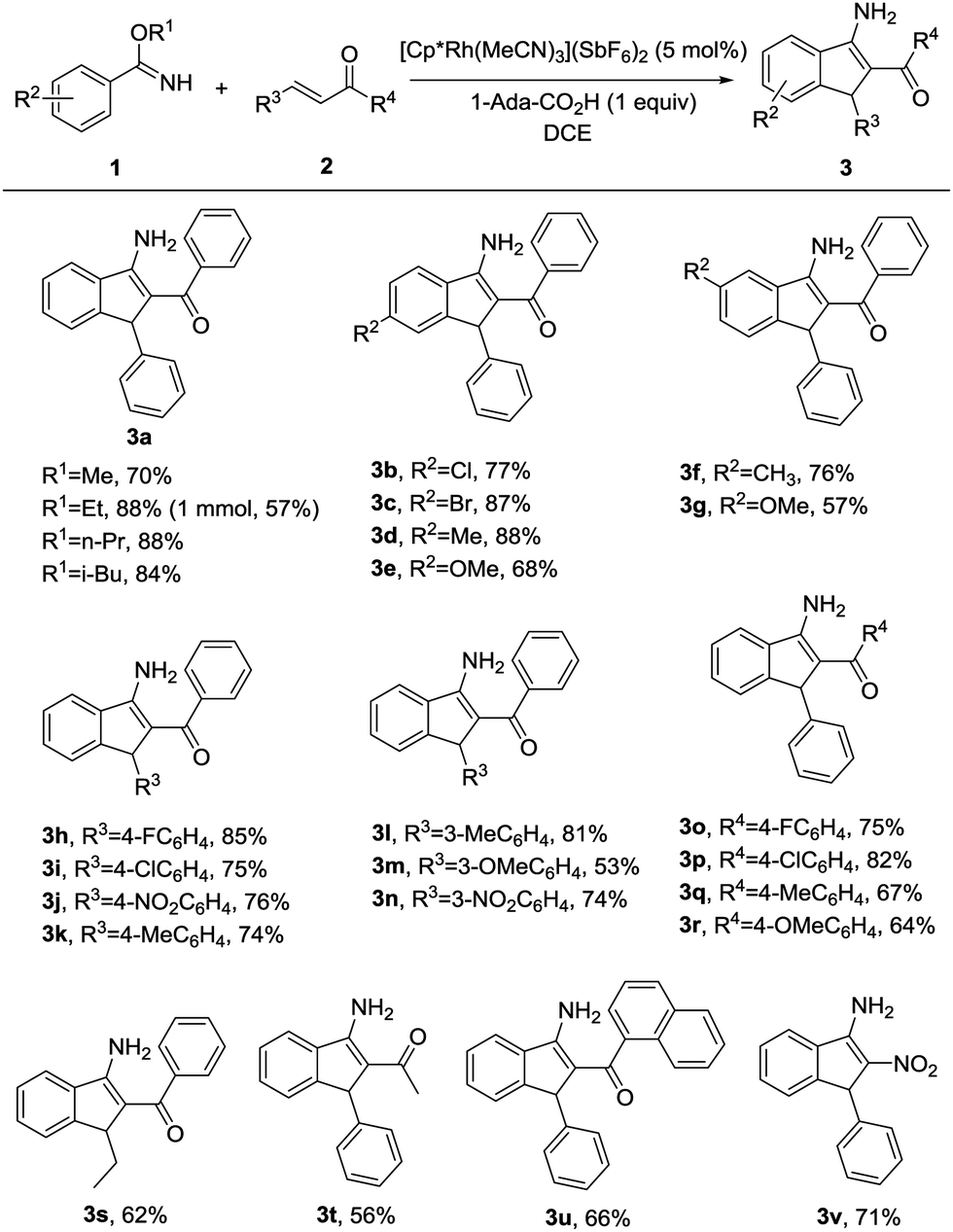
Under the optimal reaction conditions (entry 9, Table 1), we first studied the scope and generality with respect to aryl imidates as shown in Table 2. Different alkyl groups as R1 were used in the reaction and gave the intended product 3a in 70–88% yields. Ethyl and n-propyl groups gave superior results, whereas introducing a methyl group as R1 led to a relatively lower yield. Therefore, the ethyl group as R1 of aryl imidates was chosen for the next study. Significantly, the transformation could be carried out on a 1 mmol scale in a reasonable yield (57%). The molecular structure of 3a was confirmed by single-crystal X-ray diffraction analysis (Fig. 2 and ESI†). Aryl imidates bearing both electron-donating and electron-withdrawing groups at the para position of the phenyl ring all coupled smoothly with chalcone 2a to afford the annulated products (3b–3e) in 68–88% yields. In the case of meta-substituted arylimidates, the C–H activation occurred selectively and consistently at the less hindered site, furnishing the products in good yields (3f and 3g). Aryl imidates bearing a halogen atom (Cl and Br) were well tolerated under the optimized conditions to deliver the target products (3b and 3c).
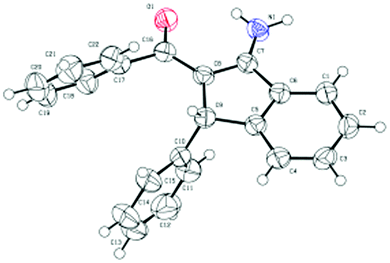 | ||
| Fig. 2 X-ray molecular structure of 3a. | ||
Subsequently, substituted α,β-unsaturated ketones were then tested using model benzyl imidate 1a under the optimized conditions (Table 2). When R3 and R4 were substituted by phenyl groups, a variety of electron-donating or electron-withdrawing groups in R3 or R4 were well-tolerated in this transformation, producing the corresponding 3-aminoindenes in moderate to excellent yields (3h–3r). These results revealed that the electron density on the moiety of R3 or R4 did not significantly influence the efficiency of the reaction. Moreover, the corresponding 3-aminoindene could be obtained in a good yield when R3 or R4 was an alkyl group (3s and 3t). The desired product was afforded in 66% yield when R4 was 1-naphthyl (3u). It is worth mentioning that nitroalkene could also be easily converted into the corresponding product in 71% yield (3v), whereas terminal olefins, such as 1-hexene or styrene and olefins with ester, amide, sulfone, or other electron-withdrawing groups do not work under the standard reaction conditions.
Furthermore, the resultant products obtained via our protocol are amenable to further chemical transformations. For example, compound 3a could be condensed with phenylhydrazine, leading to the corresponding pyrazole in excellent yield (Scheme 2).
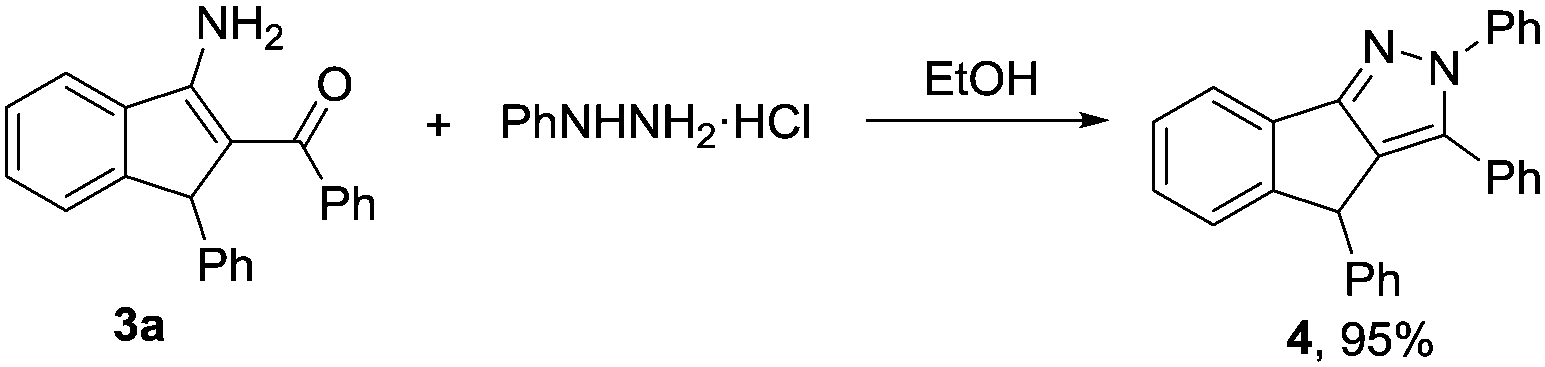 | ||
| Scheme 2 Conversion of 3a. | ||
To further investigate the reaction mechanism, we investigated the annulation of ethyl benzimidate (1ab) and 1,3-diphenyl-2-propen-1-one (2a) under the standard reaction conditions (Scheme 3). After 3 hours, intermediate E was generated and detected by HRMS. However, intermediate E could not be detected by HRMS after 36 hours. These results suggested that the final product 3a might be generated from intermediate E by the elimination of EtOH.
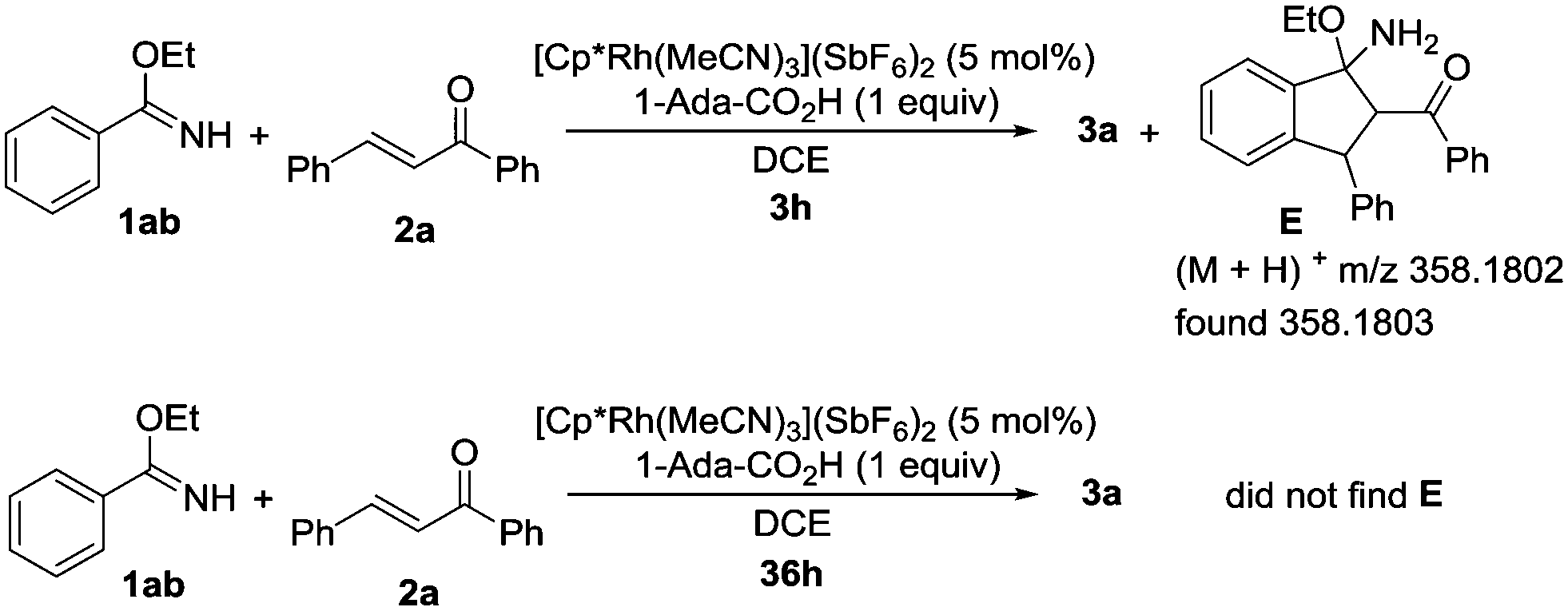 | ||
| Scheme 3 Control experiments. | ||
On the basis of the results obtained and previous reports,13 a plausible reaction pathway for this Rh(III)-catalysed cascade cyclization reaction is proposed and shown in Scheme 4. Firstly, coordination of the rhodium atom to the NH and the subsequent C–H activation result in the formation of the five-membered rhodacyclic intermediate A. Subsequently, the double bond of α,β-unsaturated ketone coordinates with the Rh atom in intermediate A to produce species B, which undergoes migratory insertion to generate seven-membered ring intermediate C. Then, an intramolecular nucleophilic addition leads to complex D. Protonation of intermediate D delivered the active Rh(III) species for the next catalytic cycle and intermediate E. Finally, elimination of EtOH as the only byproduct from E affords the final cyclization product 3a.
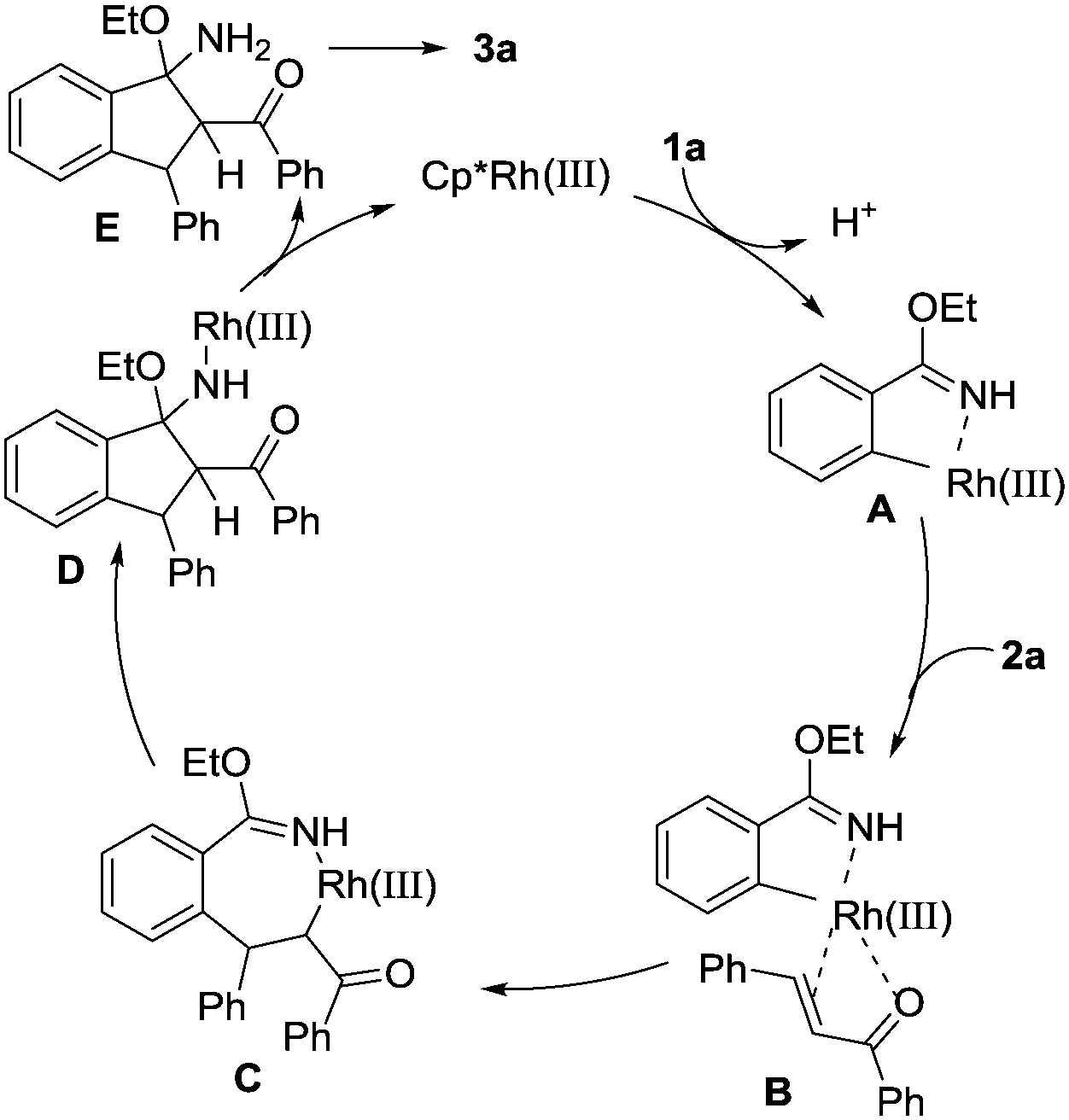 | ||
| Scheme 4 Proposed reaction mechanism. | ||
In summary, we have successfully developed a novel and efficient synthetic route for straightforward access to 1,2-disubstituted 3-aminoindenes in moderate to excellent yields via rhodium(III)-catalysed intermolecular cyclization of readily available benzimidates and α,β-unsaturated ketones or nitroalkenes. This operationally simple protocol features good functional-group tolerance and excellent regioselectivity. We believe that this methodology will be widely applied in the synthesis of complex molecules bearing an aminoindene motif. Furthermore, mechanistic studies are currently underway in our laboratory.
We gratefully acknowledge the NSF of China (21572072), the Xiamen Southern Oceanographic Center (15PYY052SF01), the Science and Technology Bureau of Xiamen City (3502Z20150054), the 111 project (BC2018061) and the Subsidized Project for Cultivating Postgraduates’ Innovative Ability in Scientific Research of Huaqiao University for the financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected examples, see: (a) T. George, R. Tahilramani, C. L. Kaul and R. S. Grewal, J. Pharm. Sci., 1969, 58, 47 CrossRef CAS PubMed; (b) D. T. Witiak, S. V. Kakodkar, G. E. Brunst, J. R. Baldwin and R. G. Rahwan, J. Med. Chem., 1978, 21, 1313 CrossRef CAS PubMed; (c) M. Pan, T. J. Mabry, J. M. Beale and B. M. Mamiya, Phytochemistry, 1997, 45, 517 CrossRef CAS PubMed; (d) M. Kameyama, F. A. Siqueira, M. Garcia-Mijares, L. F. Silva and M. T. A. Silva, Molecules, 2011, 16, 9421 CrossRef CAS PubMed; (e) O. Svendsen, J. Arnt, V. Boeck, K. P. Bogeso, A. V. Christensen, J. Hyttel and J.-J. Larsen, Drug Dev. Res., 1986, 7, 35 CrossRef CAS; (f) M. Suehiro, U. Scheffel, R. F. Dannals, A. A. Wilson, H. T. Ravert and H. N. Wagner, Nucl. Med. Biol., 1992, 19, 549 CAS; (g) J. Hyttel and J.-J. Larsen, J. Neurochem., 1985, 44, 1615 CrossRef CAS PubMed; (h) H. Minegishi, Y. Futamura, S. Fukashiro, M. Muroi, M. Kawatani, H. Osada and H. Nakamura, J. Med. Chem., 2015, 58, 4230 CrossRef CAS PubMed; (i) B. D. Dorsey, R. B. Levin, S. L. McDaniel, J. P. Vacca, J. P. Guare, P. L. Darke, J. A. Zugay, E. A. Emini, W. A. Schleif, J. C. Quintero, J. H. Lin, I.-W. Chen, M. K. Holloway, P. M. D. Fitzgerald, M. G. Axel, D. Ostovic, P. S. Anderson and J. R. Huff, J. Med. Chem., 1994, 37, 3443 CrossRef CAS PubMed; (j) D. G. Alonso, W. C. Koskinen, R. S. Oliveira, J. Constantin and S. Mislankar, J. Agric. Food Chem., 2011, 59, 13096 CrossRef CAS PubMed; (k) M. Lachia, H. C. Wolf, P. J. M. Jung, C. Screpanti and A. D. Mesmaeker, Bioorg. Med. Chem. Lett., 2015, 25, 2184 CrossRef CAS PubMed; (l) J. R. Courter, N. Madani, J. Sodroski, A. Schön, E. Freire, P. D. Kwong, W. A. Hendrickson, I. M. Chaiken, J. M. LaLonde and A. B. Smith III, Acc. Chem. Res., 2014, 47, 1228 CrossRef CAS PubMed; (m) R. Kunstmann, U. Lerch, H. Gerhards, M. Leven and U. Schacht, J. Med. Chem., 1984, 27, 432 CrossRef CAS PubMed.
- For selected examples, see: (a) K. P. Bogeso, A. V. Christensen, J. Hyttel and T. Liljefors, J. Med. Chem., 1985, 28, 1817 CrossRef CAS PubMed; (b) K. Andersen, T. Liljefors, K. Gundertofte, J. Perregaard and K. P. Bogeso, J. Med. Chem., 1994, 37, 950 CrossRef CAS PubMed; (c) J. G. A. Walton, D. C. Jones, P. Kiuru, A. J. Durie, N. J. Westwood and A. H. Fairlamb, ChemMedChem, 2011, 6, 321 CrossRef CAS PubMed; (d) E. S. Burstein, J. Ma, S. Wong, Y. Gao, E. Pham, A. E. Knapp, N. R. Nash, R. Olsson, R. E. Davis, U. Hacksell, D. M. Weiner and M. R. Brann, J. Pharmacol. Exp. Ther., 2005, 315, 1278 CrossRef CAS PubMed; (e) J. Parraga, S. A. Andújar, S. Rojas, L. J. Gutiérrez, N. E. Aouad, M. J. Sanz, R. D. Enriz, N. Cabedo and D. Cortes, Eur. J. Med. Chem., 2016, 122, 27 CrossRef CAS PubMed; (f) J. Augstein, A. L. Ham and P. R. Leeming, J. Med. Chem., 1972, 15, 466 CrossRef CAS PubMed; (g) K. P. Bogeso, J. Arnt, K. Frederiksen, H. O. Hansen, J. Hyttel and H. Pedersen, J. Med. Chem., 1995, 38, 4380 CrossRef CAS PubMed; (h) M. Froimowitz, K.-M. Wu, A. Moussa, R. M. Haidar, J. Jurayj, C. George and E. L. Gardner, J. Med. Chem., 2000, 43, 4981 CrossRef CAS PubMed; (i) J. Guillon, P. Dallemagne, J.-M. Léger, J. Sopkova, P. R. Bovy, C. Jarry and S. Rault, Bioorg. Med. Chem., 2002, 10, 1043 CrossRef CAS PubMed; (j) K. P. Bogeso, J. Med. Chem., 1983, 26, 935 CrossRef CAS PubMed.
- (a) C. G. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS PubMed; (b) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed; (c) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (d) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed; (e) J. Wencel-Delord, T. Drcge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (f) C. Sun, B. Li and Z. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (g) J. Mo, L. Wang, Y. Liu and X. Cui, Synthesis, 2015, 439 CAS; (h) M. Wei, L. Wang, Y. Li and X. Cui, Chin. Chem. Lett., 2015, 26, 1336 CrossRef CAS; (i) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481 CrossRef CAS PubMed; (j) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (k) L. Wang, D. Xiong, L. Jie, C. Yu and X. Cui, Chin. Chem. Lett., 2018, 29, 907 CrossRef CAS; (l) T. Yuan, C. Pi, C. You, X. Cui, S. Du, T. Wan and Y. Wu, Chem. Commun., 2019, 2, 163 RSC; (m) G. Mao, Q. Huang and C. Wang, Eur. J. Org. Chem., 2017, 3549 CrossRef CAS.
- (a) T. Miura, T. Harumashi and M. Murakami, Org. Lett., 2007, 9, 741 CrossRef CAS PubMed; (b) C.-C. Liu, R. P. Korivi and C.-H. Cheng, Chem. – Eur. J., 2008, 14, 9503 CrossRef CAS PubMed; (c) H. Tsukamoto, T. Ueno and Y. Kondo, Org. Lett., 2007, 9, 3033 CrossRef CAS PubMed; (d) U. Borthakur, M. Borah, M. J. Deka and A. K. Saikia, J. Org. Chem., 2016, 81, 8736 CrossRef CAS PubMed; (e) S. Janody, R. Jazzar, A. Comte, P. M. Holstein, J.-P. Vors, M. J. Ford and O. Baudoin, Chem. – Eur. J., 2014, 20, 11084 CrossRef CAS PubMed; (f) Z. Cao, H. Zhu, X. Meng, J. Guan, Q. Zhang, L. Tian, X. Sun, G. Chen and J. You, Chem. – Eur. J., 2016, 22, 16979 CrossRef CAS PubMed; (g) J. Paul, L. L. V. My, M. Behar-Pirès, C. Guillaume, E. Léonel, M. Presset and E. L. Gall, J. Org. Chem., 2018, 83, 4078 CrossRef CAS PubMed; (h) Y. Kuninobu, H. Ueda, A. Kawata and K. Takai, J. Org. Chem., 2007, 72, 6749 CrossRef CAS PubMed.
- Y. Kuninobu, A. Kawata and K. Takai, J. Am. Chem. Soc., 2005, 127, 13498 CrossRef CAS PubMed.
- Z.-M. Sun, S.-P. Chen and P. Zhao, Chem. – Eur. J., 2010, 16, 2619 CrossRef CAS PubMed.
- D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 11098 CrossRef CAS PubMed.
- (a) P. Zhao, F. Wang, K. Han and X. Li, Org. Lett., 2012, 14, 5506 CrossRef CAS PubMed; (b) Y. Chen, F. Wang, W. Zhen and X. Li, Adv. Synth. Catal., 2013, 355, 353 CAS.
- X. Jin, X. Yang, Y. Yang and C. Wang, Org. Chem. Front., 2016, 3, 268 RSC.
- W. Gong, Z. Zhou, J. Shi, B. Wu, B. Huang and W. Yi, Org. Lett., 2018, 20, 182 CrossRef CAS PubMed.
- (a) F. Wang, J. Shen, G. Cheng and X. Cui, RSC Adv., 2015, 5, 73180 RSC; (b) C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762 CrossRef CAS PubMed; (c) Y. Hu, S. Li, Z. Wang, Y. Yao, T. Li, C. Yu and C. Yao, J. Org. Chem., 2018, 83, 3361 CrossRef CAS PubMed; (d) H. Zhang, J. Shen, G. Cheng, B. Wu and X. Cui, Asian J. Org. Chem., 2018, 7, 1089 CrossRef CAS; (e) J.-T. Yu, R. Chen, H. Jia and C. Pan, J. Org. Chem., 2018, 83, 12086 CrossRef CAS PubMed.
- (a) A. G. M. Barrett and G. G. Graboski, Chem. Rev., 1986, 86, 751 CrossRef CAS; (b) T. J. Potter, D. N. Kamber, B. Q. Mercado and J. A. Ellman, ACS Catal., 2017, 7, 150 CrossRef CAS PubMed; (c) S. Tripathi, R. Kapoor and L. D. S. Yadav, Adv. Synth. Catal., 2018, 360, 1407 CrossRef CAS; (d) Y. Wang, Y. Du, X. Huang, X. Wu, Y. Zhang, S. Yang and Y. R. Chi, Org. Lett., 2017, 19, 632 CrossRef CAS PubMed; (e) S. Jana, A. Chakraborty, V. Z. Shirinian and A. Hajra, Adv. Synth. Catal., 2018, 360, 2402 CrossRef CAS; (f) X. Lei, L. Zheng, C. Zhang, X. Shi and Y. Chen, J. Org. Chem., 2018, 83, 1772 CrossRef CAS PubMed.
- (a) Y. Kuninobu, A. Kawata and K. Takai, J. Am. Chem. Soc., 2005, 127, 13498 CrossRef CAS PubMed; (b) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed; (c) S. Ye, K. Gao, H. Zhou, X. Yang and J. Wu, Chem. Commun., 2009, 5406 RSC; (d) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141 RSC; (e) Z.-M. Sun, S.-P. Chen and P. Zhao, Chem. – Eur. J., 2010, 16, 2619 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1889341. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc00567f |
| This journal is © The Royal Society of Chemistry 2019 |