
MOF-derived porous N–Co3O4@N–C nanododecahedra wrapped with reduced graphene oxide as a high capacity cathode for lithium–sulfur batteries†
Jing
Xu
a,
Wenxue
Zhang
b,
Yi
Chen
a,
Hongbo
Fan
c,
Dawei
Su
 *a and
Guoxiu
Wang
*a
*a and
Guoxiu
Wang
*a
aCentre for Clean Energy Technology, School of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, NSW 2007, Australia. E-mail: Dawei.Su@uts.edu.au; Guoxiu.Wang@uts.edu.au
bSchool of Materials Science and Engineering, Chang'an University, Xi'an 710064, China
cSchool of Environment and Civil Engineering, Dongguan University of Technology, China
First published on 11th January 2018
Abstract
The lithium–sulfur (Li–S) battery has been regarded as a highly promising rechargeable energy-storage system due to its high energy density of 2567 W h kg−1. However, moderating the dissolution of lithium polysulfides (LiPSs) and enhancing the conductivity of the sulfur cathode are the main limitations for its successful application. Herein, we demonstrate an approach to simultaneously tackle these two barriers by designing a porous N–Co3O4@N–C nanododecahedral composite. This composite was derived from ZIF-67 via a facile pyrolysis method, which realizes the effective doping of nitrogen into both Co3O4 and the carbon framework, simultaneously achieving a well-defined porous structure. After wrapping with reduced graphene oxide (rGO), this porous N–Co3O4@N–C/rGO cathode supported a high sulfur loading (5.89 mg cm−2) and exhibited excellent stability (611 mA h g−1 at 2C after 1000 cycles). Furthermore, ex situ Raman spectroscopy, ex situ X-ray photoelectron spectroscopy, UV-vis absorption spectroscopy and first-principles calculations confirm that the N–Co3O4@N–C/rGO nanododecahedra effectively bind LiPSs in the electrode over multiple cycles. This proved that the cobalt oxides in the porous N–Co3O4@N–C nanododecahedra have strong affinity for binding LiPSs. The simultaneous doping of nitrogen both into the cobalt oxides and carbon framework not only strengthened the binding energy for LiPSs absorption, but also improved the overall conductivity of the nanododecahedra. Moreover, the interconnected porous structure contributes to the electron transfer and alleviates the volume changes of active materials during cycling.
Introduction
Booming developments in electric vehicles, grid energy storage systems and portable electronic devices have accelerated the demand for more powerful, durable batteries with high energy density and long cycle life.1–3 The development of lithium-ion batteries, which currently dominate the portable electronics markets, is hampered by their limited theoretical energy density. In this regard, the Li–S system is one of the most promising candidates due to the high theoretical capacity of the sulfur electrode (1672 mA h g−1) and energy density of the Li–S battery (2567 W h kg−1), which is at least 3 to 5 times that of conventional lithium-ion batteries.4,5 Additionally, sulfur is abundant, inexpensive and environmentally benign, making Li–S batteries even more commercially attractive compared with lithium-ion batteries. Despite their promising prospects, Li–S batteries face tough challenges including the insulating property of sulfur and lithium sulfides, volume changes in sulfur (∼80%) during cycling, dissolution of intermediate polysulfides (Li2Sx, 4 ≤ x ≤ 8) in organic electrolytes and the LiPSs shuttle effect.6–16 Among them, restricting the shuttle effect of LiPSs and improving the electronic conductivity are the primary tasks for the advanced development of Li–S batteries.It is generally acknowledged that an ideal sulfur host is expected to endow a highly porous interconnected architecture to accommodate sulfur, strong capability for confining soluble LiPSs, sufficient electronic conductivity to ensure high sulfur utilization as well as robust framework to withstand the volume expansion of sulfur. From this perspective, infiltrating molten sulfur into well-defined porous conductive carbon materials such as carbon nanotubes, graphene oxide and their mixtures seems to be meaningful in the beginning of cycling.17–29 However, the rather weak intermolecular interactions between non-polar hydrophobic carbonaceous materials and polar hydrophobic LiPSs species are not sufficient to prevent LiPSs diffusion and the shuttle effect over long-term cycling, resulting in serious capacity degradation.30–36 Considering that LiPSs are intrinsically polar species with their terminal sulfur bearing most of the negative charge, polar host materials with well-defined porous structures have been recently developed. In this regard, metal compounds endowed with ample polar active sites have been proven to strongly bind with LiPSs via polar–polar interactions, Lewis acid–base interactions and sulfur-chain catenation, thus effectively entrapping them within or on the surface of the hosts.37 Good examples include metal–organic frameworks (MOFs), metal oxides,38 metal carbides (MXene) and metal sulfides which exhibit superior electrochemical performances as cathode materials for Li–S batteries due to their unique chemisorption nature.31,39–42
Recently, polar metal composites derived from MOFs have attracted wide attention in the application of Li–S batteries due to the unique advantages of these MOF-derived metal composites including their abundant metal atoms, which serve as polar active sites, show strong affinity to LiPSs anions. In addition, the heteroatom dopant sites (N, S and P) derived from MOFs can also provide additional affinity for LiPSs, which can form multiple strength chemical interactions with LiPSs, thus balancing the chemical interactions that occur with LiPSs and also benefit LiPSs mobility.40 On the other hand, it has been proven that nitrogen doped metal-based materials with unique vacancies and good conductivity have great potential for application in energy storage systems due to their high density active sites for the surface reaction.55,56 Meanwhile, nitrogen doped carbon materials are also reported to effectively improve the polar surface properties54 because nitrogen dopants can promote electrochemical reactions and electronic conductivity.
Inspired by these ideas, herein, we report an MOF-derived metal composite that realizes the effective doping of nitrogen both into the metal oxides and carbon framework. For the first time, a nitrogen doped Co3O4 inlaid nitrogen doped carbon framework coated with reduced graphene oxide (denoted as N–Co3O4@N–C/rGO) is prepared and used as a cathode host in the Li–S battery. The as-prepared N–Co3O4@N–C/rGO nanododecahedra are synthesized via a facile pyrolysis process that introduces nitrogen dopant both into Co3O4 and carbon nanododecahedra simultaneously, achieving a porous structure. It uses a metal organic framework (ZIF-67) as a precursor, which provides the advantages of abundant Co–N moieties and unique dodecahedral morphology. In the as-prepared N–Co3O4@N–C/rGO nanododecahedral composite, its well-defined porous structure coupled with rGO can effectively accommodate sulfur molecules and alleviate the volume expansion during sulfur lithiation. Moreover, both metal atoms serving as polar active sites and the nitrogen dopant sites in the N–Co3O4@N–C/rGO nanododecahedra show strong affinity to LiPSs anions, which can form multiple strength chemical interactions with sulfur and LiPSs. Ex situ Raman, ex situ X-ray photoelectron spectroscopy (XPS) and UV-vis absorption spectroscopy results and first-principle calculations further confirm that the N–Co3O4@N–C/rGO nanododecahedra can effectively bind LiPSs in the electrode over cycles and exhibit strong binding energy. Therefore, this N–Co3O4@N–C/rGO–S cathode generates a high reversible capacity (1223 mA h g−1 at 0.2C) and excellent stability (611 mA h g−1 at 2C after 1000 cycles). Remarkably, when the sulfur loading is 5.89 mg cm−2, it can still retain a capacity of 568 mA h g−1 after 500 cycles.
Experimental section
Material synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mS = 1:4. After magnetic stirring for 12 h in an ice water bath. The CS2 and ethanol were completely evaporated. Following filtration and drying for 20 h, the as-synthesized N–Co3O4@N–C/GO–S composite was sealed in a glass vessel under nitrogen protection and heated at 230 °C for 3 h in a quartz tube furnace at a heating rate of 3 °C min−1. In this heating treatment, the GO nanosheets transformed into reduced graphene oxide (rGO) and simultaneously the extra sulfur on the outside of N–Co3O4@N–C/rGO–S evaporated. Hence N–Co3O4@N–C/rGO–S was obtained. The N–Co3O4@N–C–S and rGO–S composites were prepared using the same method.
:mS = 1:4. After magnetic stirring for 12 h in an ice water bath. The CS2 and ethanol were completely evaporated. Following filtration and drying for 20 h, the as-synthesized N–Co3O4@N–C/GO–S composite was sealed in a glass vessel under nitrogen protection and heated at 230 °C for 3 h in a quartz tube furnace at a heating rate of 3 °C min−1. In this heating treatment, the GO nanosheets transformed into reduced graphene oxide (rGO) and simultaneously the extra sulfur on the outside of N–Co3O4@N–C/rGO–S evaporated. Hence N–Co3O4@N–C/rGO–S was obtained. The N–Co3O4@N–C–S and rGO–S composites were prepared using the same method.
Characterization of materials
The morphology of the materials was investigated via field emission scanning electron microscopy (FESEM, Zeiss Supra 55VP), and their detailed crystal structure was further evaluated via transmission electron microscopy (TEM, Model JEM-2011, JEOL). The crystal structures and phases of the as-prepared materials were collected via XRD (Siemens D5000) using Cu Kα radiation at a very slow scanning step of 0.01° s−1. Raman spectra were obtained on a Renishaw inVia Raman spectrometer system (Gloucestershire, UK). XPS analysis was carried on an ESCALAB MK II X-ray photoelectron spectrometer in a JEOL JSM-6700F electron microscope with an accelerating voltage of 10 kV. Nitrogen adsorption–desorption measurements were conducted on a 3 Flex Surface Characterization Analyzer to determine the Brunauer–Emmett–Teller (BET) specific surface areas, which were calculated using the isothermal points at a relative pressure of P/P0 = 0.05–0.25. Thermal gravimetric analysis (TGA) was conducted in nitrogen atmosphere at a heating rate of 5 °C min−1 to determine the sulfur content (wt%) in the composites.Electrochemical characterization
Electrodes were prepared from 80 wt% of active materials, 10 wt% of conductive agent (carbon black) and 10 wt% of binder (polyvinylidene difluoride). The mass loading of sulfur on the electrode was around 2.13 mg cm−2. CR2032 coin cells were assembled in an argon-filled glove box (Mbraun, Unilab, Germany) and then used for electrochemical evaluation. The electrolyte contained 1 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and 1 wt% lithium nitrate (LiNO3) in 1,3-dioxolane and 1,2-dimethoxy-ethane (volume ratio 1:1). Porous polypropylene (Celgard 2300) was used as the separator. About 30 μL electrolyte was added to each coin cell. Electrochemical measurements were conducted using a LAND-CT2001C battery test system. The cells were discharged and charged galvanostatically in a fixed voltage range of 1.7–2.7 V. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed on a CHI660C electrochemistry workstation. CV was performed in the voltage range of 1.5 to 3.0 V (vs. Li+/Li) at a scan rate of 0.5 mV s−1. For the EIS measurement, an AC potential of 5 mV amplitude and frequency of 10 kHz to 100 MHz were applied.
Computational methods
Simulations were based on first-principles density functional theory (DFT) provided by the CASTEP package. The generalized gradient approximation (GGA) and Perdew–Burke–Ernzerhof scheme (PBE) were applied for the exchange–correlation potential to optimize geometrical structures and calculate properties. Ultrasoft pseudopotentials and a plane-wave expansion of the wave functions were chosen for computations. The Brillouin zone was sampled by 2 × 1 × 1 k-points and an energy cutoff of 340 eV was chosen in the geometry optimization calculations. To evaluate the contributions of the van der Waals (vdW) interactions between different layers, the DFT-D (D stands for dispersion) approach within the Grimme scheme was adopted for the vdW correction.5 These setups were proven to be accurate enough to describe the results via careful test calculations. A three-layer Co3O4 (110) surface model was constructed, in which the bottom layer of atoms were fixed, while the other two layers of atoms were fully relaxed. The nearest distance between nanosheets in neighboring cells was greater than 18 Å to ensure that no interactions exit between different layers. For geometric optimization, the atomic positions of all structures were allowed to relax until the convergence tolerances of energy, maximum force, and displacement of 1 × 10−5 eV, 3 × 10−2 eV Å, and 1 × 10−3 Å were reached, respectively. Periodic boundary conditions were adopted for all the models utilized in this work. The adsorption energies (Eads) are defined as: Eads = Etotal − Especies − Esubstrate, where, Etotal is the total energy of the adsorbed system, Especies is the energy of the adsorbate in vacuum and Esubstrate is the energy of the Co3O4 (110) surface. According to this definition, a more negative value represents a more energetically favorable (exothermic) reaction between the adsorbate and Co3O4 (110) surface.Results and discussion
Scheme 1 shows the formation of the nitrogen doped Co3O4 inlaid nitrogen doped carbon framework wrapped with reduced graphene oxide (denoted as N–Co3O4@N–C/rGO). First, ZIF-67 was synthesized via a novel method at room temperature (Fig. S1, ESI†).44 Through a pyrolysis process, ZIF-67 was converted into porous N–Co3O4@N–C nanododecahedra. Then via electrostatic interactions, the negatively charged graphene oxide (GO) nanosheets were effectively wrapped on the surface of the positively charged N–Co3O4@N–C nanododecahedra. Fig. S2 (ESI†) shows the field-emission scanning electron microscopy (FESEM) images of the as-synthesized ZIF-67, which exhibits a regular dodecahedral morphology with a uniform particle size of approximately 2 μm. Each dodecahedral has two types of vertices which share three and four edges, respectively. After pyrolysis treatment, the N–Co3O4@N–C nanododecahedra preserve the polyhedral morphology of ZIF-67, as characterised by the FESEM image in Fig. 1a and S3 (ESI†). In the transmission electron microscopy (TEM) image in Fig. 1b, it can be seen that N–Co3O4@N–C nanododecahedra consist of numerous secondary nanoparticles and between them pores exist, which demonstrate the well-defined porous structure of N–Co3O4@N–C nanododecahedra. The HRTEM image in Fig. 1c further discloses that the N–Co3O4@N–C nanododecahedra have a 3D dimensional porous structure with interstices 10–20 nm in size. The Brunauer–Emmett–Teller (BET) surface area of the N–Co3O4@N–C nanododecahedra was measured to be 237.4 m2 g−1, the pore size distribution is around 10 to 25 nm, and the pore volume is 0.93 cm3 g−1 (Fig. 1d). To confirm the crystal structure of the as-prepared material, X-ray diffraction (XRD) was conducted, as shown in Fig. 1e. The XRD pattern can be indexed to the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry of Co3O4 (JCPDS card number 01-074-2120). The sharp peaks in the XRD patterns indicate the well-defined crystalline structure of the material. Moreover, the energy-dispersive X-ray spectroscopy (EDS) spectrum in Fig. 1f, further illustrates the presence of C, N, Co and O elements in the porous N–Co3O4@N–C composite with the corresponding element content of 9.2 wt%, 4.8 wt%, 32.9 wt% and 53.0 wt%, respectively. The EDS line scan results in Fig. S4 (ESI†) reveal the spatial distribution of the C, N, Co and O elements in the N–Co3O4@N–C nanododecahedra. In addition, the XPS spectrum reveals the successful doping of nitrogen both into the carbon framework and Co3O4. As shown in Fig. 1g, the N 1s spectrum of N–Co3O4@N–C exhibits four peaks at 402.1, 400.2, 399.2 and 398.2 eV, which correspond to graphitic N, pyrrolic N, Co–N45–47 and pyridinic N, respectively. The element content of nitrogen in the N–Co3O4@N–C composite was around 5.8 at% (4.3 wt%), which is consistent with the EDS result in Fig. 1f. The Co 2p spectrum of N–Co3O4@N–C shows two main broad and asymmetric peaks with a spin–orbit splitting of 14.1 eV, as shown in Fig. S5 (ESI†). However, this value is found to be lower than that of pure Co3O4 which should be around 15.1 eV.48 This observation suggests the formation of heteroatoms in the Co3O4 matrix, which results in additional charge transfer from cobalt to the heteroatoms.49 This is further verified by the peak at 781.6 eV in the Co 2p3/2 spectrum (Fig. 1h), which is assigned to the Co–N binding.50,51 In addition, some researchers proposed that Co not only propels the conversion of polysulfide to soluble long-chain polysulfides, but also catalyses long-chain polysulfides to Li2S2 and even to Li2S, thereby promoting the reaction kinetics and yielding a high specific capacity.57 The C 1s spectrum (Fig. 1i) shows a primary peak at around 284.8 eV, which corresponds to sp2 carbon with C
m symmetry of Co3O4 (JCPDS card number 01-074-2120). The sharp peaks in the XRD patterns indicate the well-defined crystalline structure of the material. Moreover, the energy-dispersive X-ray spectroscopy (EDS) spectrum in Fig. 1f, further illustrates the presence of C, N, Co and O elements in the porous N–Co3O4@N–C composite with the corresponding element content of 9.2 wt%, 4.8 wt%, 32.9 wt% and 53.0 wt%, respectively. The EDS line scan results in Fig. S4 (ESI†) reveal the spatial distribution of the C, N, Co and O elements in the N–Co3O4@N–C nanododecahedra. In addition, the XPS spectrum reveals the successful doping of nitrogen both into the carbon framework and Co3O4. As shown in Fig. 1g, the N 1s spectrum of N–Co3O4@N–C exhibits four peaks at 402.1, 400.2, 399.2 and 398.2 eV, which correspond to graphitic N, pyrrolic N, Co–N45–47 and pyridinic N, respectively. The element content of nitrogen in the N–Co3O4@N–C composite was around 5.8 at% (4.3 wt%), which is consistent with the EDS result in Fig. 1f. The Co 2p spectrum of N–Co3O4@N–C shows two main broad and asymmetric peaks with a spin–orbit splitting of 14.1 eV, as shown in Fig. S5 (ESI†). However, this value is found to be lower than that of pure Co3O4 which should be around 15.1 eV.48 This observation suggests the formation of heteroatoms in the Co3O4 matrix, which results in additional charge transfer from cobalt to the heteroatoms.49 This is further verified by the peak at 781.6 eV in the Co 2p3/2 spectrum (Fig. 1h), which is assigned to the Co–N binding.50,51 In addition, some researchers proposed that Co not only propels the conversion of polysulfide to soluble long-chain polysulfides, but also catalyses long-chain polysulfides to Li2S2 and even to Li2S, thereby promoting the reaction kinetics and yielding a high specific capacity.57 The C 1s spectrum (Fig. 1i) shows a primary peak at around 284.8 eV, which corresponds to sp2 carbon with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, and the peak at 285.7 eV is assigned to C–N bonds.43 The N-doped carbon surface is reported to act as a conductive Lewis base “catalyst” matrix to improve the chemisorption of Li2Sn (n = 4–8), which promotes the oxidization of Li2S6 → Li2S8 → S8, and thus improves the sulfur utilization and cycling stability.52 All these results confirm the formation of Co–N and C–N bonding configurations in the N–Co3O4@N–C composite. The synergistic behavior of Co–N and C–N has a significant effect in enhancing the electrochemical performance of Li–S batteries.53
C bonds, and the peak at 285.7 eV is assigned to C–N bonds.43 The N-doped carbon surface is reported to act as a conductive Lewis base “catalyst” matrix to improve the chemisorption of Li2Sn (n = 4–8), which promotes the oxidization of Li2S6 → Li2S8 → S8, and thus improves the sulfur utilization and cycling stability.52 All these results confirm the formation of Co–N and C–N bonding configurations in the N–Co3O4@N–C composite. The synergistic behavior of Co–N and C–N has a significant effect in enhancing the electrochemical performance of Li–S batteries.53
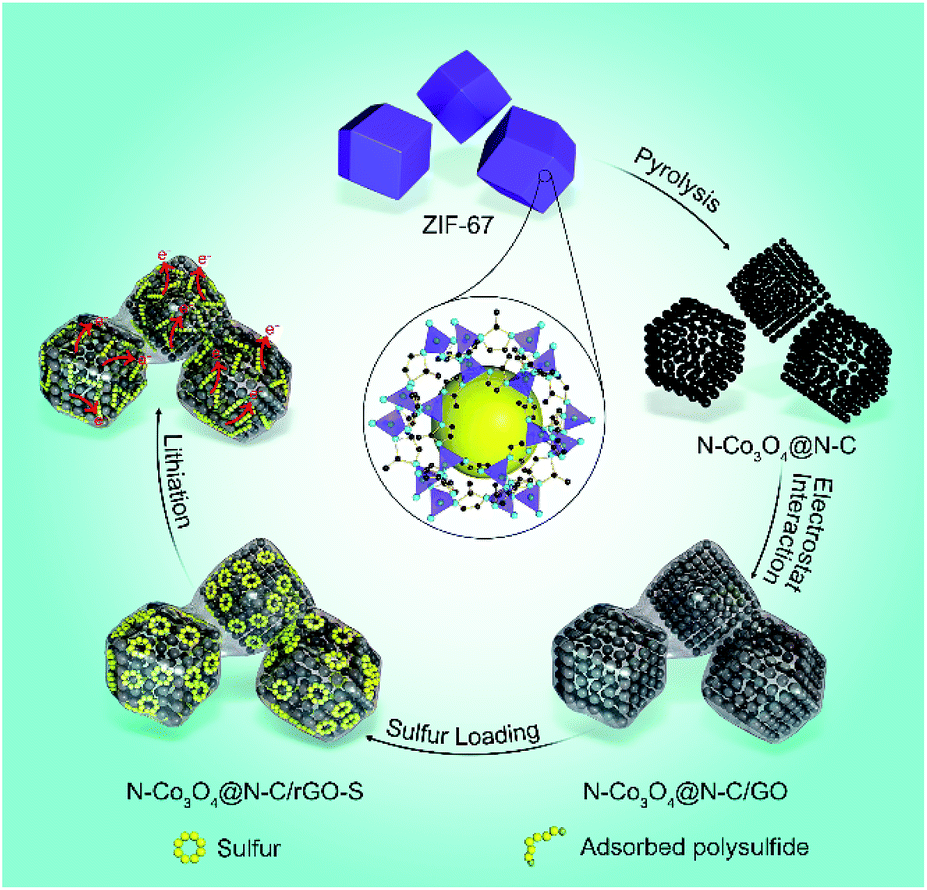 | ||
| Scheme 1 Schematic illustration of the formation of N–Co3O4@N–C/rGO–S. | ||
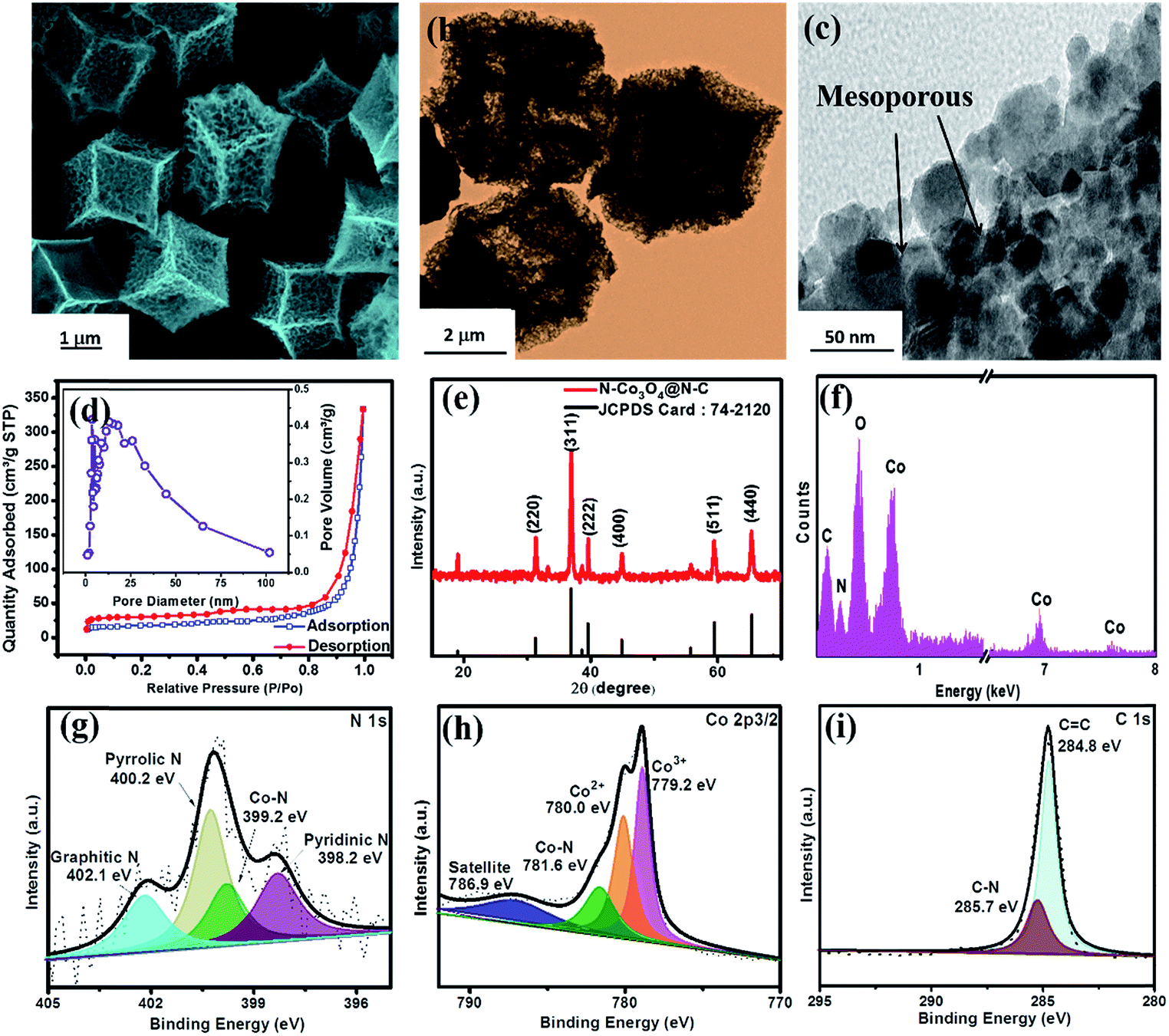 | ||
| Fig. 1 (a) FESEM, (b) TEM and (c) HRTEM images of N–Co3O4@N–C. (d) N2 adsorption–desorption isotherm curves of N–Co3O4@N–C. (e) XRD patterns of N–Co3O4@N–C. (f) EDS spectrum mapping of N–Co3O4@N–C. (g) N 1s, (h) Co 2p3/2 and (i) C 1s spectra of the N–Co3O4@N–C composite. | ||
Since the high surface area and abundant mesopores of the N–Co3O4@N–C framework are beneficial for a high amount of sulfur accommodation, here, we further used the rGO wrap to prevent the dissolution of the sulfur species in the electrolyte. Fig. 2a and S6b (ESI†) show the morphology of the N–Co3O4@N–C framework after sulfur impregnation and rGO wrapping (denoted as N–Co3O4@N–C/rGO–S), where we can see that no extra sulfur exists outside the cathode host. As evidenced by the TEM image in Fig. S7,† the inner spaces of N–Co3O4@N–C/rGO–S are much darker than that of Co3O4@N–C/GO, which confirms that sulfur was present inside the N–Co3O4@N–C/rGO host. Meanwhile, the HRTEM image (Fig. 2c) taken near the dodecahedra edges shows distinct lattice fringes with d spacings of 0.29 and 0.24 nm, which correspond with the (220) and (311) lattice planes of Co3O4, respectively. The effective encapsulation of sulfur particles is also confirmed, as circled by dotted lines. The EDS mappings in Fig. 2g and S8d (ESI†) illustrate the uniform distribution of sulfur element in the porous N–Co3O4@N–C/rGO–S composite. The XRD patterns of the porous N–Co3O4@N–C/rGO–S composite prove the presence of sulfur (Fig. S9, ESI†). All these results confirm that sulfur successfully permeated the N–Co3O4@N–C/rGO composite. It should also be noted that after sulfur impregnation, the textural properties reveal a tremendous change, as shown in Fig. S10 (ESI†). The specific surface area and pore volume decrease to 14.3 m2 g−1 and 0.09 cm3 g−1, respectively, which indicates that the majority of pores are occupied by sulfur. No obvious peak appears in the pore size distribution plot (inset of Fig. S10, ESI†), which verifies that sulfur occupied the mesopores of N–Co3O4@N–C/rGO. The thermal gravimetric analysis (TGA) demonstrates that the sulfur content in the N–Co3O4@N–C/rGO–S composite was around 75 wt% (Fig. 2j). The surface composition and the valence states of N–Co3O4@N–C/rGO–S were revealed by XPS (Fig. S11, ESI†). The S 2p spectrum for N–Co3O4@N–C/rGO–S has 2p3/2 and 2p1/2 spin–orbit levels at 163.8 and 165.0 eV (Fig. 2k), respectively. The slightly lower binding energy of S 2p3/2 (163.8 eV) compared with that of pure elemental sulfur suggests the possible chemical bonding of S with other species.30 This was confirmed by the peak located at 170.0 eV, which should be ascribed to the chemical bond formation of Co–S.58–60 Meanwhile, in the Co 2p spectrum of N–Co3O4@N–C/rGO–S, as shown in Fig. S12 (ESI†), the Co 2p3/2 and Co 2p1/2 peaks shift to lower binding energies of 779.5 eV and 794.7 eV, respectively, compared with that of N–Co3O4@N–C/rGO. This can be ascribed to the electron transfer from the sulfur molecules to the cobalt atoms,54–56 which indicates the strong chemical interaction between cobalt and sulfur.
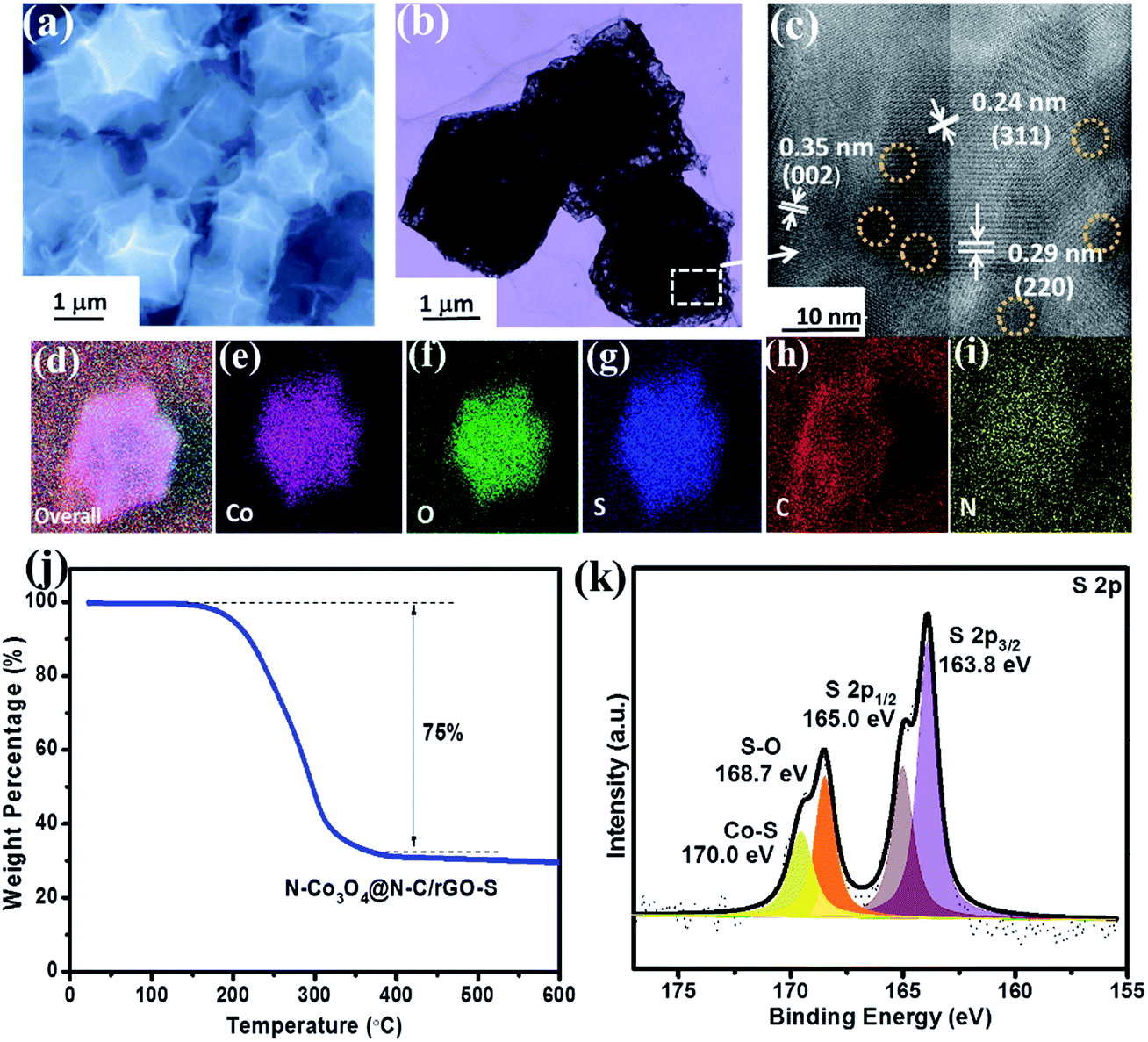 | ||
| Fig. 2 (a) FESEM, (b) TEM and (c) HRTEM image of N–Co3O4@N–C/rGO–S. EDX mapping results of (d) the overall image, (e) cobalt, (f) oxygen, (g) sulfur, (h) carbon and (i) nitrogen. (j) Thermogravimetric curves of N–Co3O4@N–C/rGO–S in an N2 atmosphere. (k) S 2p spectrum of the N–Co3O4@N–C/rGO–S composite. | ||
Cyclic voltammetry (CV) was performed in the voltage range of 1.5–3.0 V at a scan rate of 0.1 mV s−1 to illustrate the electrochemical processes during discharging and charging. Fig. 3a shows the typical CV curves of the N–Co3O4@N–C/rGO–S electrode. The two cathodic peaks at 2.35 V and 2.05 V are consistent with the voltage plateaus in Fig. 3c. The upper plateau at 2.35 V is related to the transformation of cyclo-S8 to long-chain soluble LiPSs (Li2Sn, n ≥ 4). The lower plateau at 2.05 V is associated with the decomposition of these LiPSs to insoluble lithium sulfides (Li2S2 and Li2S), which accounts for the major capacity of the electrode. It can be seen that the capacity of the N–Co3O4@N–C/rGO–S electrode did not significantly decrease during the first three cycles, which suggests good electrochemical stability. Fig. 3b presents the Nyquist plots of the fresh N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S electrodes before cycling. The equivalent circuit was established in Fig. S13 (ESI†). Table S1 (ESI†) shows the parameter data. It can be observed that the N–Co3O4@N–C/rGO–S electrode exhibits the lowest charge transfer resistance (Rct) compared with the N–Co3O4@N–C–S and rGO–S electrodes. Since all the test cathodes possessed nearly the same sulfur loading and electrolyte content, the different charge transfer resistances can be attributed to the interfacial kinetic property of the host materials. Moreover, the charge transfer resistance value for the N–Co3O4@N–C–S electrode is almost the same as that of the N–Co3O4@N–C/rGO–S electrode, but obviously smaller than that of rGO–S. This suggests that the framework of N–Co3O4@N–C can facilitate the Li+/e− transport, which is beneficial for the interfacial kinetics because the nitrogen doping significantly improves the electronic conductivity of the N–Co3O4@N–C composites. Previous studies have shown that nitrogen-doped carbon materials have metallic properties, which are characterized by the presence of a donor state close to the Fermi level, and thus increase the electronic conductivity of materials.57,58 Meanwhile, nitrogen doping into Co3O4 obviously narrows the bandgap, which is beneficial for the enhanced excitation of charge carriers to the conduction band, leading to an increase in electronic conductivity compared with pure Co3O4.49,59 Fig. S14 (ESI†) further confirms that nitrogen doping improved the electronic conductivity of N–Co3O4@N–C compared with that of Co3O4@C.
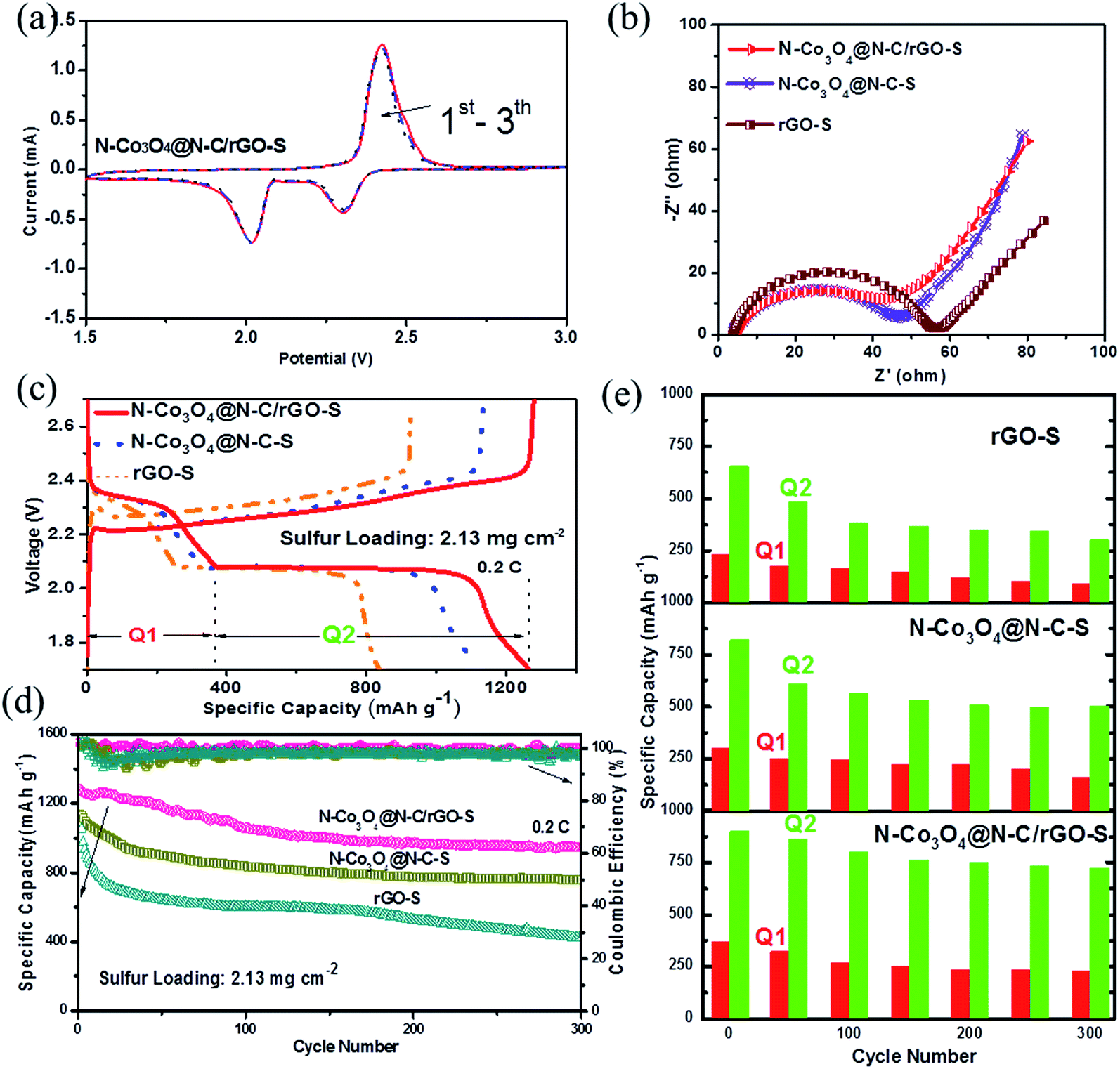 | ||
| Fig. 3 (a) Cyclic voltammetry (CV) tested between 1.5 and 3 V at a sweep rate of 0.1 mV s−1 for N–Co3O4@N–C/rGO–S. (b) Nyquist plots of the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S cathodes. (c) First-cycle galvanostatic charge/discharge voltage profiles of the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S cathodes at 0.2C. (d) Prolonged cycle life and coulombic efficiency of the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S electrodes at 0.2C. (e) Discharge capacities accumulated at the high and low voltage plateaus of N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S cathodes. | ||
The galvanostatic charge/discharge tests were performed at a current rate of 0.2C for the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S electrodes. The first cycling charge/discharge profiles are shown in Fig. 3c. Obviously, all three electrodes display two typical discharge voltage plateaus, which represent the multistep reduction of sulfur in the discharge process. However, both overpotentials (denoted as ΔE, voltage gap between the anodic and cathodic plateaus) the N–Co3O4@N–C/rGO–S and N–Co3O4@N–C–S electrodes are smaller than that of the rGO@S electrode, which is also consistent with the results in Fig. 3b. This confirms that the effective doping of nitrogen into N–Co3O4@N–C/rGO greatly contributes to the excellent interfacial kinetics of the N–Co3O4@N–C/rGO electrode. Fig. 3d shows the cycling performance of the three electrodes at a 0.2C current rate. The N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S electrodes delivered an initial capacity of 1223, 1129 and 1003 mA h g−1, respectively. After 300 cycles, the capacity of rGO–S dropped to only 358 mA h g−1. In contrast, due to their strong affinity for LiPSs, the N–Co3O4@N–C/rGO–S and N–Co3O4@N–C–S electrodes showed much better cycling stability with retained capacities of 945 and 743 mA h g−1 after 300 cycles, respectively. The corresponding charge/discharge profiles for the 100th and 300th cycles are provided in Fig. S15 (ESI†). The N–Co3O4@N–C/rGO–S electrode retained distinct voltage plateaus after 300 cycles and the overpotential change was smaller than that in the others. Fig. 3e compares the discharge capacities (Q1 and Q2) accumulated along the high and low discharge voltage plateaus within the 300 cycles for each electrode. The high plateau capacity Q1 corresponding to the formation of soluble LiPSs was well maintained for the N–Co3O4@N–C/rGO electrode in the first few cycles, which suggests that the enormous LiPSs were effectively entrapped by the ample polar sites in N–Co3O4@N–C/rGO.6 Meanwhile, it is obvious that Q2 experienced the least capacity degradation during cycling for the N–Co3O4@N–C/rGO–S electrode and low capacity degradation was observed for the N–Co3O4@N–C–S electrode. However, a dramatic capacity loss was observed in Q2 for the rGO–S electrode. These demonstrate that N–Co3O4@N–C/rGO–S and N–Co3O4@N–C–S with abundant polar sites could effectively suppress the shuttle effect of LiPSs during cycling,6 which greatly benefit the cycling stability of the electrodes. In addition, the rGO coating also positively contributes to prevent the dissolution of high-ordered LiPSs, which is observed from the comparison of the overall stage sulfur utilization between N–Co3O4@N–C/rGO–S and N–Co3O4@N–C–S electrodes, as shown in Fig. 3e.
Next, the rate capabilities and electrode kinetics of the cathode materials were evaluated at various current densities (Fig. 4a). We first tested the electrodes at increasing current rates from 0.1 to 0.5, 1, 2 and 3C (1C = 1672 mA g−1). The N–Co3O4@N–C/rGO–S electrode delivered high capacities of 1301, 1169, 927, 756 and 652 mA h g−1, respectively. When the current rate decreased back to 2, 1, 0.5 and 0.1C, the electrode recovered capacities of 679, 840, 989 and 1081 mA h g−1, respectively, showing excellent reversible stability. The corresponding charge/discharge profiles of N–Co3O4@N–C/rGO–S at various rates are provided in Fig. 4b. It is remarkable that even at a current rate of 3C, this electrode still preserved the typical discharge voltage plateau. In contrast, due to the insufficient LiPSs confinement of carbon based materials, the rGO–S electrode showed the worst rate capability (merely 130 mA h g−1) and sluggish voltage plateaus, especially, at the high current rate (3C), as shown in Fig. S16 (ESI†). On the other hand, the rate capability of N–Co3O4@N–C–S was inferior to that of the N–Co3O4@N–C/rGO–S electrode due to the lack of physical confinement induced by rGO. The calculated potential differences between the charge/discharge voltage plateaus of these three electrodes at current rates of 0.1C, 0.5C, 1C, 2C and 3C (1C = 1672 mA g−1) are also shown in Fig. 4c. These results further confirm that N–Co3O4@N–C/rGO–S possesses much less polarization and better reaction kinetics than the electrodes with single physical (rGO–S) or chemical confinement (N–Co3O4@N–C–S) at a wide range of current rates (0.1–3C).
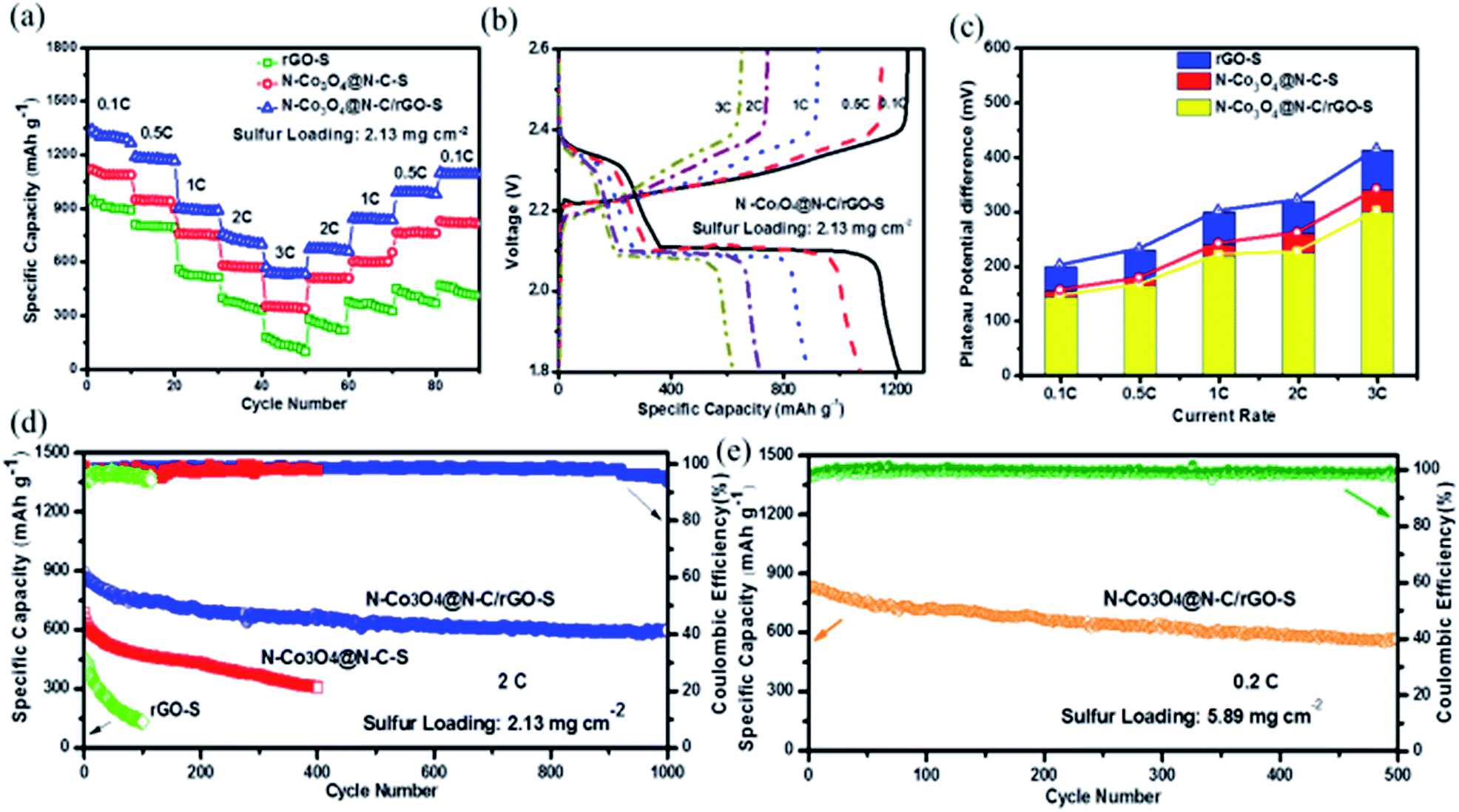 | ||
| Fig. 4 (a) Discharge capacity of the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S electrodes cycled at various rates spanning 0.1C, 0.5C, 1C, 2C and 3C. (b) Galvanostatic charge/discharge voltage profiles of the N–Co3O4@N–C/rGO–S cathode at step current densities from 0.1C to 3C. (c) Potential difference changes in the charge and discharge plateaus for the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C/rGO–S and rGO–S cathodes between the charge discharge plateaus at various current densities. (d) Cycling performance test of the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C/rGO–S and rGO–S electrodes at a 2C discharge rate and corresponding coulombic efficiency (sulfur loading: 2.13 mg cm−2). (e) Cycling performance test of the N–Co3O4@N–C/rGO–S electrode at a 0.2C discharge rate and corresponding Coulombic efficiency (sulfur loading: 5.89 mg cm−2). | ||
To demonstrate the potential of the sulfur cathode for practical Li–S batteries, electrochemical evaluation of the long-term cycling performance at a high areal sulfur loading and high rate is necessary. The cycling performance of the N–Co3O4@N–C/rGO–S, N–Co3O4@N–C–S and rGO–S electrodes at the current rate of 2C are shown in Fig. 4d. A reversible capacity of 611 mA h g−1 was successfully maintained for the N–Co3O4@N–C/rGO–S electrode after 1000 cycles, which maintained a stable cycling trend. In contrast, the rGO–S electrode could not deliver a capacity higher than 155 mA h g−1 after 100 cycles at 2C, which implies sufficient chemisorption of LiPSs is important for a high rate cycling performance. In addition, although the cycling performance of the N–Co3O4@N–C–S electrode was better than the rGO–S electrode, it still exhibited a distinct degradation trend and only maintained around 316 mA h g−1 after 400 cycles compared with the very stable cycling performance of the N–Co3O4@N–C/rGO–S electrode after 1000 cycles. This comparison further demonstrates that the synergistic effect between the chemisorption of LiPSs and physical confinement is of great significance for the high rate, long term, stable cycling performance. Moreover, we further tested the N–Co3O4@N–C/rGO–S electrode at a high sulfur loading of 5.89 mg cm−2 at 0.2C. This electrode maintained a stable capacity of 568 mA h g−1 after 500 cycles at 0.2C. Besides, the charge transfer resistance of this electrode did not remarkably increase with a 2.13 mg cm−2 sulfur loading, as shown in Fig. S17 (ESI†). It is generally acknowledged that the interfacial electrochemical kinetics are primarily dominated by the binding affinity and the efficient charge transfer between LiPSs and the whole electrode. The strong interaction with LiPSs induced by Co3O4 and the nitrogen dopant allow sufficient surface adsorption. Meanwhile, the improved electronic conductivity generated by the nitrogen doped into both Co3O4 and the carbon framework could facilitate the transport of electrons generated during the redox reactions of the LiPSs. Consequently, the synergism of these two factors result in the excellent cycling stability of the N–Co3O4@N–C/rGO–S electrode at a high current rate and high sulfur loading.
To verify the strong adsorption capability of the N–Co3O4@N–C/rGO composites towards LiPSs, we compared the LiPSs adsorption ability of the N–Co3O4@N–C/rGO–S and rGO–S electrodes, as shown in the inset of Fig. 5a. We dissembled the cells of the N–Co3O4@N–C/rGO–S and rGO–S electrodes under the same discharge state after 50 cycles in a glovebox, and the dissembled batteries are shown in Fig. S18 (ESI†). Then both cathode electrodes were immersed in a mixture of 1,3-dioxolane/1,2-dimethoxyethane (DOL/DME, 1:1 v/v) electrolyte for 6 hours. Obviously, the colour of the solution with the N–Co3O4@N–C/rGO–S electrode became slightly light yellow and it was difficult to observe as shown in Fig. 5a(1). This suggests that soluble LiPSs were effectively entrapped in the N–Co3O4@N–C/rGO–S electrode during cycling. In contrast, the solution with the rGO@S electrode exhibited a distinct colour change from colourless to bright yellow, as shown in Fig. 5a(2), which indicates that a large amount of soluble LiPSs detached from the rGO–S electrode during cycling. Meanwhile, UV-vis absorption measurements were also conducted to explore the concentration changes in the pure electrolyte after immersing the cycled rGO@S and N–Co3O4@N–C/rGO–S cathode electrodes, as shown in Fig. 5a. For the electrolyte with the cycled rGO–S electrode, the adsorption peak observed at around 280 nm is attributed to S8 and S62− species. The small adsorption peak at 310 nm should be assigned to S62− or S42− species, while the noticeable adsorption peak located at 420 nm is associated with S42− species,60–62 which demonstrate that serious detachment of the LiPSs from the rGO–S cathode occurred. However, only a negligible amount of LiPSs was observed in the electrolyte with the N–Co3O4@N–C/rGO–S electrode, which reveals that the N–Co3O4@N–C/rGO cathode electrode can effectively suppress the soluble LiPSs into the electrode instead of dissolving them into the electrolyte.
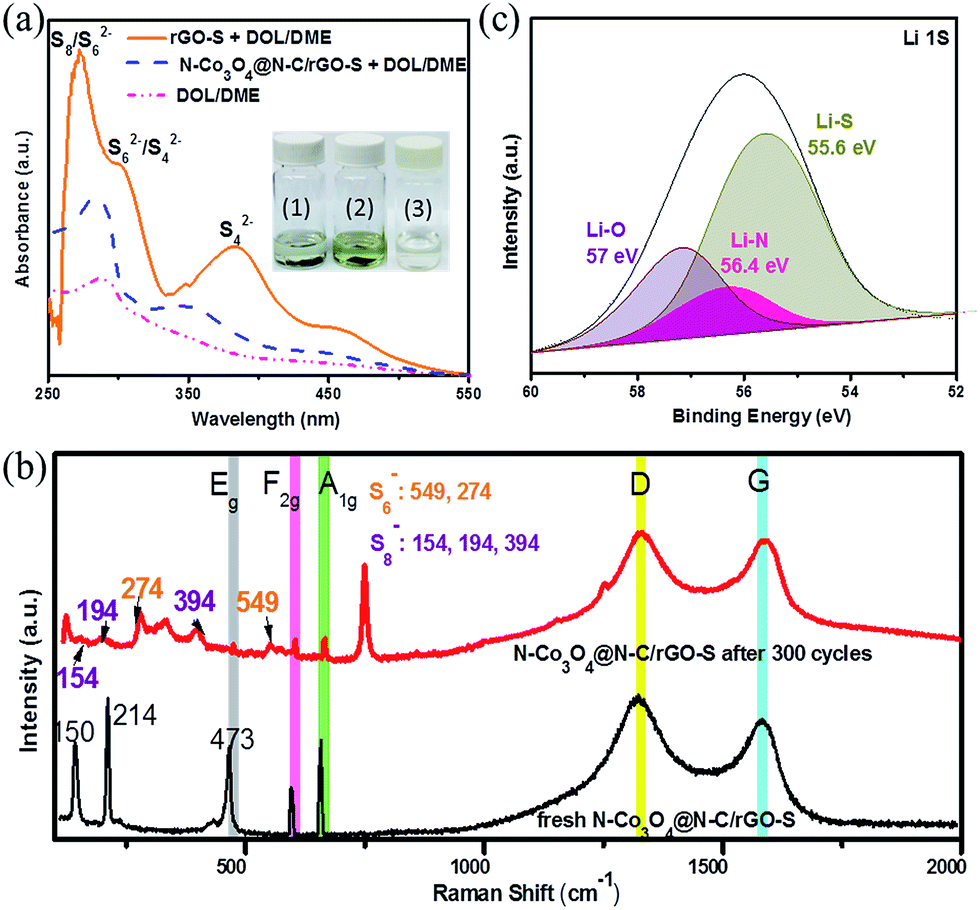 | ||
| Fig. 5 (a) UV-vis absorption spectra of the solution obtained by immersing the cycled rGO–S and N–Co3O4@N–C/rGO–S cathodes in a mixture of DOL/DME electrolyte. The inset images are visualized colour changes after the N–Co3O4@N–C/rGO–S and rGO–S cathode electrodes were immersed in (1) and (2) solutions for 6 hours, respectively, and (3) is DOL/DME solvent only as a reference. (b) Ex situ Raman spectra of bare N–Co3O4@N–C/rGO–S and the electrode after 300 cycles (discharged to 2.1 V). (c) XPS spectra of Li 1s for the N–Co3O4@N–C/rGO–S electrode after discharging to 2.1 V. | ||
Moreover, the ex situ Raman spectroscopy measurements of the N–Co3O4@N–C/rGO–S electrode before and after 300 cycles (discharged to 2.1 V, which is an appropriate voltage to generate LiPSs) also further evidence the strong LiPSs affinity of the N–Co3O4@N–C/rGO composites during cycling. As shown in Fig. 5b, the D and G bands observed in the Raman spectrum of the N–Co3O4@N–C/rGO electrode are attributed to rGO, while the Eg, F2g and A1g bands correspond to Co3O4. It can be seen that after 300 cycles, the peaks at 274 and 549 cm−1 can be assigned to S6−, while the peaks located at around 154, 194, and 394 cm−1 should be S8−. However, these peaks were not detected in the fresh electrode before cycling. In addition, the XPS spectrum of the N–Co3O4@N–C/rGO–S electrode after 300 cycles (discharged to 2.1 V) further confirms the strong polar interactions due to LiPSs chemisorption in the N–Co3O4@N–C/rGO–S electrode. The Li 1s spectrum in Fig. 5c exhibits a single asymmetric peak at around 55.9 eV, which can be fitted to three peaks at around 55.6, 57, 56.4 eV.
These three peaks correspond to the Li–S bond resulting from LiPSs, the Li–O bond from the surface group of Co–O and the Li–N bond assigned to Li in the LiPSs interacting with doped nitrogen (Li–N),63,64 respectively, which is consistent with previous reports.65 Therefore, these results sufficiently verify that the N–Co3O4@N–C/rGO electrode is endowed with multiple types of polar interactions, which have great capability to entrap LiPSs species into the electrode during cycling.
The enhancement of adsorption strength can be further understood by comparing the LiPSs adsorption energy of the Co3O4 nanocrystals and rGO nanosheets. First-principle calculations were performed to reveal the corresponding adsorption energies (Fig. 6). For simplicity, Li2S and Li2S4 were employed as the representative lithium polysulfide and sulfide, respectively. The lowest-energy adsorption configurations of Li2S and Li2S4 on Co3O4 are presented in Fig. 6a along with their Mulliken populations. Obviously, there is specific bonding between Li2S or Li2S4 with Co3O4, resulting in much higher adsorption energies of −3.551 and −1.398 eV, respectively. These values are obviously lower than the adsorption energy (−0.94/0.48 eV) between Li2S/Li2S4 and rGO.66 These interactions exist mainly in the form of Li–O and Co–S bonds. For Li2S and Li2S4 with the Co3O4 system, the closest distances between Li and O are approximately 1.967 Å and 1.916 Å, respectively, which imply strong ionic bonds between Li and O atoms. The strong Li–O bonding interaction is the main cause of the strong binding energy between lithium polysulfide/sulfide and the Co3O4 surface. The closest contact between cobalt and sulfur is approximately 2.13–2.3 nm, implying weak ionic bonds and van der Waals attractions, which can also partially enhance the Li2Sx attractions. Furthermore, as shown in Fig. 6b, there are 1.38 e and 1.48 e transferred from Li2S and Li2S4 to the Co3O4 surface, respectively. Whereas, only 0.227 and 0.019 electrons transfer from Li2S and Li2S4 to pristine rGO, which suggests that more charge migrates to the Co3O4 substrate. Therefore, stronger chemical binding between Li2Sx–Co3O4 is expected, which demonstrates that the presence of Co3O4 nanocrystals enhances the retention of LiPSs.
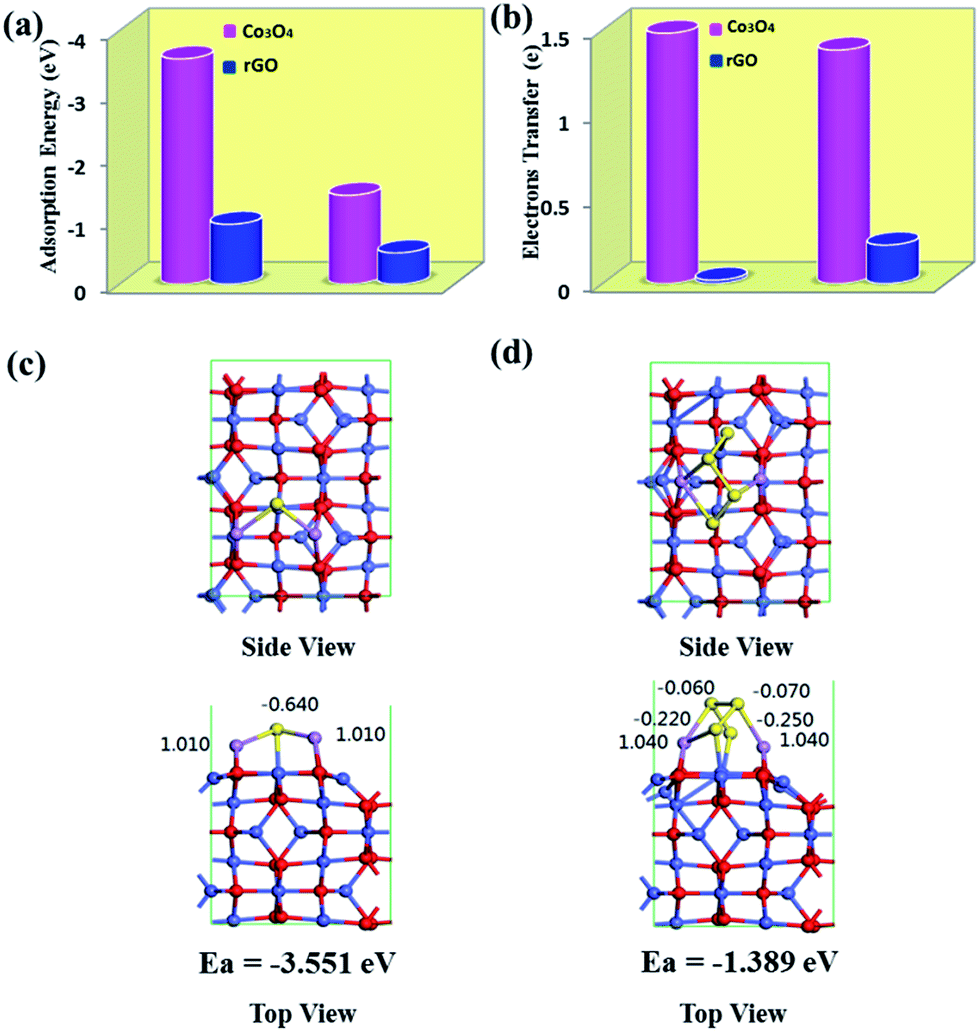 | ||
| Fig. 6 (a) Adsorption energies and (b) electrons transferred for Li2S and Li2S4 compounds on the rGO and Co3O4 surfaces. Schematic diagram showing the crystal structure of Co3O4 with top and side views and after adsorption of (c) Li2S and (d) Li2S4 with the corresponding atom Mulliken charge. Blue, red, yellow, and purple balls represent Co, O, S, and Li atoms, respectively. | ||
The excellent electrochemical performance of the MOF-derived N–Co3O4@N–C/rGO–S composites can be attributed to the following aspects: (I) the multiple interactions with LiPSs induced by Co3O4 and the nitrogen dopant allow sufficient surface adsorption of LiPSs. The high electronic conductivity generated by the nitrogen doped into both Co3O4 and the carbon framework facilitates the transport of electrons during the redox reactions of the LiPSs. The synergistic effect of these two factors result in excellent interfacial electrochemical kinetics. (II) The well-defined porous structure in the MOF-derived polar hosts is beneficial to accommodate sulfur and LiPSs, and rGO acts as a physical sulfur barrier, which reduces the LiPSs dissolution into the electrolyte and maximizes the utilization of sulfur. All these factors enable the N–Co3O4@N–C/rGO–S electrode to have better reaction kinetics than single physical or chemical confinement electrodes even at a high sulfur loading.
Conclusion
We synthesized N–Co3O4@N–C/rGO nanododecahedra via a facile pyrolysis method, which uses a metal organic framework (ZIF-67) as the precursor. Comprehensive physical characterisations confirmed that the N–Co3O4@N–C/rGO nanododecahedra had a well-defined porous structure, excellent conductivity and unique chemisorptive nature, which is an ideal cathode host for Li–S batteries. When applied in an Li–S battery, the composite exhibited excellent cycling stability at a high current rate (611 mA h g−1 at 2C after 1000 cycles) and retained a reversible capacity of 568 mA h g−1 after 500 cycles at a high sulfur loading of 5.89 mg cm−2. The excellent electrochemical performance could be attributed to the synergistic effect between the physical confinement and chemical binding in the N–Co3O4@N–C/rGO host. This cathode host can maximize the effectiveness of moderating LiPSs diffusion and simultaneously enhance the reaction kinetics of sulfur species. Ex situ Raman spectroscopy, ex situ X-ray photoelectron spectroscopy, UV-vis absorption spectroscopy and first-principle calculations further confirmed that the N–Co3O4@N–C/rGO nanoparticles could effectively bind LiPSs in the electrode over cycles, thus rendering good electronic contact for the sustainable utilization of sulfur and favorable cycle stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This original research was supported by the Australia Research Council, Commonwealth of Australia, the Australian Renewable Energy Agency (ARENA), University of Technology Sydney (UTS), through the Discovery Early Career Researcher Award (DECRA DE170101009), ARC Discovery Project (DP170100436), ARENA 2014/RND106, UTS Chancellor's Postdoctoral Research Fellowship project (PRO16-1893), and UTS Early Career Researcher Grants ECRGS PRO16-1304. The first author Jing Xu gratefully acknowledges support from the Chinese Scholarship Council (CSC, No. 201307090011) to undertake this research.References
- Z. Khan, S. Park, S. M. Hwang, J. Yang, Y. Lee, H.-K. Song, Y. Kim and H. Ko, NPG Asia Mater., 2016, 8, e294 CrossRef.
- Z. Khan, N. Parveen, S. A. Ansari, S. T. Senthilkumar, S. Park, Y. Kim, M. H. Cho and H. Ko, Electrochim. Acta, 2017, 257, 328–334 CrossRef CAS.
- Z. Khan, B. Senthilkumar, S. O. Park, S. Park, J. Yang, J. H. Lee, H.-K. Song, Y. Kim, S. K. Kwak and H. Ko, J. Mater. Chem. A, 2017, 5, 2037–2044 CAS.
- A. Manthiram, S. H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- J.-G. Wang, K. Xie and B. Wei, Nano Energy, 2015, 15, 413–444 CrossRef CAS.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- Q. Zhang, Y. Wang, Z. W. Seh, Z. Fu, R. Zhang and Y. Cui, Nano Lett., 2015, 15, 3780–3786 CrossRef CAS PubMed.
- Z. Yuan, H. J. Peng, T. Z. Hou, J. Q. Huang, C. M. Chen, D. W. Wang, X. B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed.
- J. Song, Z. Yu, M. L. Gordin and D. Wang, Nano Lett., 2016, 16, 864–870 CrossRef CAS PubMed.
- B. Cao, D. Li, B. Hou, Y. Mo, L. Yin and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 27795–27802 CAS.
- J. Cao, C. Chen, Q. Zhao, N. Zhang, Q. Lu, X. Wang, Z. Niu and J. Chen, Adv. Mater., 2016, 28, 9629–9636 CrossRef CAS PubMed.
- J. Sun, Y. Sun, M. Pasta, G. Zhou, Y. Li, W. Liu, F. Xiong and Y. Cui, Adv. Mater., 2016, 28, 9797–9803 CrossRef CAS PubMed.
- H. J. Peng, Z. W. Zhang, J. Q. Huang, G. Zhang, J. Xie, W. T. Xu, J. L. Shi, X. Chen, X. B. Cheng and Q. Zhang, Adv. Mater., 2016, 28, 9551–9558 CrossRef CAS PubMed.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
- J. Zhou, N. Lin, W. l. Cai, C. Guo, K. Zhang, J. Zhou, Y. Zhu and Y. Qian, Electrochim. Acta, 2016, 218, 243–251 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- J. Wang, Y. S. He and J. Yang, Adv. Mater., 2015, 27, 569–575 CrossRef CAS PubMed.
- M. Agostini, B. Scrosati and J. Hassoun, Adv. Energy Mater., 2015, 5, 1500481 CrossRef.
- J. Yan, X. Liu, M. Yao, X. Wang, T. K. Wafle and B. Li, Chem. Mater., 2015, 150701102311000, DOI:10.1021/acs.chemmater.5b01780.
- R. Cao, W. Xu, D. Lv, J. Xiao and J.-G. Zhang, Adv. Energy Mater., 2015, 5, 1402273 CrossRef.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- T. Yang, T. Qian, M. Wang, X. Shen, N. Xu, Z. Sun and C. Yan, Adv. Mater., 2016, 28, 539–545 CrossRef CAS PubMed.
- L. Qie, C. Zu and A. Manthiram, Adv. Energy Mater., 2016, 6, 1502459 CrossRef.
- J. Liang, L. C. Yin, X. Tang, H. Yang, W. Yan, L. Song, H. M. Cheng and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 25193–25201 CAS.
- J. He, Y. Chen, W. Lv, K. Wen, C. Xu, W. Zhang, W. Qin and W. He, ACS Energy Lett., 2016, 820–826, DOI:10.1021/acsenergylett.6b00272.
- R. Fang, S. Zhao, P. Hou, M. Cheng, S. Wang, H. M. Cheng, C. Liu and F. Li, Adv. Mater., 2016, 28, 3374–3382 CrossRef CAS PubMed.
- H.-J. Peng, T.-Z. Hou, Q. Zhang, J.-Q. Huang, X.-B. Cheng, M.-Q. Guo, Z. Yuan, L.-Y. He and F. Wei, Adv. Mater. Interfaces, 2014, 1, 1400227 CrossRef.
- X. Gu, C.-j. Tong, C. Lai, J. Qiu, X. Huang, W. Yang, B. Wen, L.-m. Liu, Y. Hou and S. Zhang, J. Mater. Chem. A, 2015, 3, 16670–16678 CAS.
- Z. Li, J. Zhang, B. Guan, D. Wang, L. M. Liu and X. W. Lou, Nat. Commun., 2016, 7, 13065 CrossRef CAS PubMed.
- M. Zhang, C. Yu, C. Zhao, X. Song, X. Han, S. Liu, C. Hao and J. Qiu, Energy Storage Materials, 2016, 5, 223–229 CrossRef.
- Q. Fan, W. Liu, Z. Weng, Y. Sun and H. Wang, J. Am. Chem. Soc., 2015, 137, 12946–12953 CrossRef CAS PubMed.
- Z. Li, C. Li, X. Ge, J. Ma, Z. Zhang, Q. Li, C. Wang and L. Yin, Nano Energy, 2016, 23, 15–26 CrossRef CAS.
- X.-q. Niu, X.-l. Wang, D.-h. Wang, Y. Li, Y.-j. Zhang, Y.-d. Zhang, T. Yang, T. Yu and J.-p. Tu, J. Mater. Chem. A, 2015, 3, 17106–17112 CAS.
- H. Wang, T. Zhou, D. Li, H. Gao, G. Gao, A. Du, H. Liu and Z. Guo, ACS Appl. Mater. Interfaces, 2016, 9, 4320–4325 Search PubMed.
- L. Jia, T. Wu, J. Lu, L. Ma, W. Zhu and X. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 30248–30255 CAS.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- L. J. Zhen, Zhang and X. W. (David) Lou, Angew. Chem., 2015, 54, 12886–12890 CrossRef PubMed.
- J. He, Y. Chen, W. Lv, K. Wen, C. Xu, W. Zhang, Y. Li, W. Qin and W. He, ACS Nano, 2016, 10, 10981–10987 CrossRef CAS PubMed.
- J. Zhang, H. Hu, Z. Li and X. W. Lou, Angew. Chem., 2016, 55, 3982–3986 CrossRef CAS PubMed.
- Z. Wang, B. Wang, Y. Yang, Y. Cui, Z. Wang, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2015, 7, 20999–21004 CAS.
- X. Li, Q. Sun, J. Liu, B. Xiao, R. Li and X. Sun, J. Power Sources, 2016, 302, 174–179 CrossRef CAS.
- S. Zheng, F. Yi, Z. Li, Y. Zhu, Y. Xu, C. Luo, J. Yang and C. Wang, Adv. Funct. Mater., 2014, 24, 4156–4163 CrossRef CAS.
- R. Wu, X. Qian, X. Rui, H. Liu, B. Yadian, K. Zhou, J. Wei, Q. Yan, X. Q. Feng, Y. Long, L. Wang and Y. Huang, Small, 2014, 10, 1932–1938 CrossRef CAS PubMed.
- F. Jaouen, J. Herranz, M. Lefevre, J. P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. Olson, S. Pylypenko, P. Atanassov and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CAS.
- K. Artyushkova, B. Kiefer, B. Halevi, A. Knop-Gericke, R. Schlogl and P. Atanassov, Chem. Commun., 2013, 49, 2539–2541 RSC.
- R. L. Arechederra, K. Artyushkova, P. Atanassov and S. D. Minteer, ACS Appl. Mater. Interfaces, 2010, 2, 3295–3302 CAS.
- R. Berenguer, T. Valdés-Solís, A. B. Fuertes, C. Quijada and E. Morallón, J. Electrochem. Soc., 2008, 155, K110 CrossRef CAS.
- M. Yu, Z. Wang, C. Hou, Z. Wang, C. Liang, C. Zhao, Y. Tong, X. Lu and S. Yang, Adv. Mater., 2017, 29, 1602868 CrossRef PubMed.
- A. Morozan, P. Jegou, B. Jousselme and S. Palacin, Phys. Chem. Chem. Phys., 2011, 13, 21600–21607 RSC.
- J. Li, Z. Zhou, K. Liu, F. Li, Z. Peng, Y. Tang and H. Wang, J. Power Sources, 2017, 343, 30–38 CrossRef CAS.
- J.-J. Chen, R.-M. Yuan, J.-M. Feng, Q. Zhang, J.-X. Huang, G. Fu, M.-S. Zheng, B. Ren and Q.-F. Dong, Chem. Mater., 2015, 27, 2048–2055 CrossRef CAS.
- Y.-J. Li, J.-M. Fan, M.-S. Zheng and Q.-F. Dong, Energy Environ. Sci., 2016, 9, 1998–2004 CAS.
- X. Wang, G. Li, J. Li, Y. Zhang, A. Wook, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 2533–2538 CAS.
- Y. Tao, Y. Wei, Y. Liu, J. Wang, W. Qiao, L. Ling and D. Long, Energy Environ. Sci., 2016, 9, 3230–3239 CAS.
- D. Su, M. Cortie, H. Fan and G. Wang, Adv. Mater., 2017, 1700587 CrossRef PubMed.
- R. Czerw, M. Terrones, J.-C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, D. Tekleab, P. M. Ajayan, W. Blau, M. Ruhle and D. L. Carroll, Nano Lett., 2001, 1, 457–460 CrossRef CAS.
- J. D. Wiggins-Camacho and K. J. Stevenson, J. Phys. Chem. C, 2009, 113, 19082–19090 CAS.
- L. Xu, Z. Wang, J. Wang, Z. Xiao, X. Huang, Z. Liu and S. Wang, Nanotechnology, 2017, 28, 165402 CrossRef PubMed.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891–2898 CrossRef CAS PubMed.
- C. Barchasz, F. Molton, C. Duboc, J. C. Lepretre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84, 3973–3980 CrossRef CAS PubMed.
- Y. Li, H. Zhan, S. Liu, K. Huang and Y. Zhou, J. Power Sources, 2010, 195, 2945–2949 CrossRef CAS.
- H. Chen, C. Wang, Y. Dai, S. Qiu, J. Yang, W. Lu and L. Chen, Nano Lett., 2015, 15, 5443–5448 CrossRef CAS PubMed.
- L. Ma, H. Zhuang, Y. Lu, S. S. Moganty, R. G. Hennig and L. A. Archer, Adv. Energy Mater., 2014, 4, 1400390 CrossRef.
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef CAS PubMed.
- J. Xu, D. Su, W. Zhang, W. Bao and G. Wang, J. Mater. Chem. A, 2016, 4, 17381–17393 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta10272k |
| This journal is © The Royal Society of Chemistry 2018 |