
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Energetic insights into two electron transfer pathways in light-driven energy-converting enzymes†
Keisuke
Kawashima
a and
Hiroshi
Ishikita
 *ab
*ab
aDepartment of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8654, Japan. E-mail: hiro@appchem.t.u-tokyo.ac.jp
bResearch Center for Advanced Science and Technology, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8904, Japan. Fax: +81-3-5452-5083; Tel: +81-3-5452-5056
First published on 28th March 2018
Abstract
We report redox potentials (Em) for one-electron reduction for all chlorophylls in the two electron-transfer branches of water-oxidizing enzyme photosystem II (PSII), photosystem I (PSI), and purple bacterial photosynthetic reaction centers (PbRC). In PSI, Em values for the accessory chlorophylls were similar in both electron-transfer branches. In PbRC, the corresponding Em value was 170 mV less negative in the active L-branch (BL) than in the inactive M-branch (BM), favoring BL˙− formation. This contrasted with the corresponding chlorophylls, ChlD1 and ChlD2, in PSII, where Em(ChlD1) was 120 mV more negative than Em(ChlD2), implying that to rationalize electron transfer in the D1-branch, ChlD1 would need to serve as the primary electron donor. Residues that contributed to Em(ChlD1) < Em(ChlD2) simultaneously played a key role in (i) releasing protons from the substrate water molecules and (ii) contributing to the larger cationic population on the chlorophyll closest to the Mn4CaO5 cluster (PD1), favoring electron transfer from water molecules. These features seem to be the nature of PSII, which needs to possess the proton-exit pathway to use a protonated electron source—water molecules.
The crystal structures of photosystem II (PSII), photosystem I (PSI) and purple bacterial photosynthetic reaction centers from Rhodobacter sphaeroides (PbRC) show a pseudo-twofold axis of symmetry, forming the following heterodimeric protein subunit pairs: D1/D2 in PSII, PsaA/PsaB in PSI, and L/M in PbRC.1–6 In PbRC, electron-transfer branches (L- and M-branches) proceed from a pair of bacteriochlorophyll a (BChla) (PL and PM) via accessory BChla (BL and BM), bacteriopheophytin a (BPheoa) (HL and HM), and ubiquinone (QA and QB). In PSII, the corresponding cofactors are the pair of chlorophyll a (Chla) (PD1 and PD2), accessory Chla (ChlD1 and ChlD2), pheophytin a (Pheoa) (PheoD1 and PheoD2), and plastoquinone (QA and QB) of D1- and D2-branches, and in PSI, the pair of Chla and the 132 epimer7 (PA and PB), accessory Chla (A–1A and A–1B), acceptor Chla (A0A and A0B), and phylloquinone (A1A and A1B) of A- and B-branches (Fig. 1). In PSI (i.e., a type-I reaction center), electron transfer occurs in both A- and B-branches,8 whereas in PbRC and PSII (i.e., type-II reaction centers) electron transfer predominantly occurs along L- and D1-branches, respectively. In PbRC and PSII, excitation of BChla and Chla leads to charge separation on the L- and D1-branches and formation of the cationic [PL/PM]˙+ and [PD1/PD2]˙+ states, respectively (e.g.,9). Regardless of the structural similarities between the two reaction centers,1 many features are different.9 The [PL/PM]˙+ state has a redox potential (Em) of 500 mV for one-electron oxidation10 and accepts an electron from an outer protein subunit, cytochrome c2 (or tetraheme cytochrome in PbRC from Blastochloris viridis). The [PD1/PD2]˙+ state has a high Em (>1100 mV)11–13 and ultimately abstracts electrons from the substrate water molecules at the catalytic Mn4CaO5 moiety in D1 via redox-active D1-Tyr161 (TyrZ). Redox-active D2-Tyr160 (TyrD) exists at the symmetrical position in D2. Basic D2-Arg180 and D2-His61 near TyrD on the D2 side contribute to the larger PD1˙+ population than PD2˙+ in [PD1/PD2]˙+,13i.e., electrostatically pushing the cation onto PD1,14 thereby favoring electron transfer from the substrate water molecules in D1.15 Unlike PbRC, which only requires an electron transfer pathway, PSII also requires a proton transfer pathway from the substrate water molecules because the water molecules are protonated electron sources. In PSII, the release of protons (H+) has been observed in response to changes in the oxidation state (Sn) of the oxygen-evolving complex, and it occurs with a typical stoichiometry of 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:1:2 for the S0 → S1 → S2 → S3 → S0 transitions, respectively.16 Proton transfer may proceed via different pathways depending on the S-state transitions.17–19 The nature of the proton-conducting O4-water chain,20 which is composed exclusively of water molecules, is consistent with and may explain the pH-independence of proton transfer in the S0 to S1 transition.16 On the other hand, the pH-dependent rate constant for the S2-to-S3 transition16 indicates the involvement of ionizable groups in the proton transfer pathway (e.g., the pathway via D1-Asp6121,22). Acquirement of the Mn4CaO5 cluster seems to induce a polar protein environment near the electron acceptor [PD1/PD2]˙+ and may alter the energetics of electron transfer along the D1-branch with respect to that along the L-branch in PbRC.
:0:1:2 for the S0 → S1 → S2 → S3 → S0 transitions, respectively.16 Proton transfer may proceed via different pathways depending on the S-state transitions.17–19 The nature of the proton-conducting O4-water chain,20 which is composed exclusively of water molecules, is consistent with and may explain the pH-independence of proton transfer in the S0 to S1 transition.16 On the other hand, the pH-dependent rate constant for the S2-to-S3 transition16 indicates the involvement of ionizable groups in the proton transfer pathway (e.g., the pathway via D1-Asp6121,22). Acquirement of the Mn4CaO5 cluster seems to induce a polar protein environment near the electron acceptor [PD1/PD2]˙+ and may alter the energetics of electron transfer along the D1-branch with respect to that along the L-branch in PbRC.
The free energy difference between cofactors (e.g., electronically excited [PL/PM]* and [PL/PM]˙+HL˙−) was discussed experimentally (e.g.,23) and theoretically (e.g.,24). However, detailed Em values of the cofactors for one-electron reduction in active L- and D1-branches as well as inactive M- and D2-branches have not yet been experimentally determined and are a matter of debate (e.g.,25). Although we reported calculated Em values for one-electron oxidation in PSI, PbRC, and PSII,13,26 these Em values are more associated with distributions of the cationic states over the (B)Chla pairs (e.g., [PA/PB]˙+ (ref. 27) and [PD1/PD2]˙+ (ref. 13)). Due to a lack of Em values of the cofactors for one-electron reduction in PbRC, PSI, and PSII, it remains still unclear why electron transfer occurs in both A- and B-branches in PSI, whereas in PbRC and PSII electron transfer predominantly occurs along L- and D1-branches.
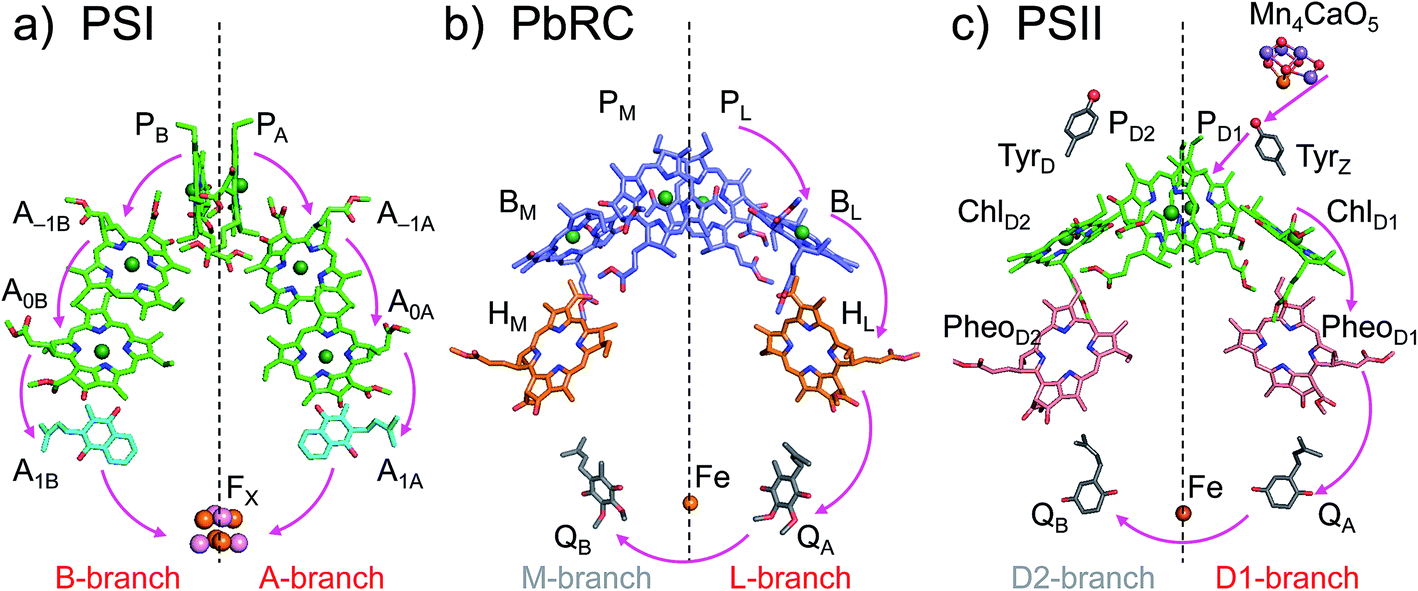 | ||
| Fig. 1 Electron transfer chains in photosynthetic reaction centers of (a) PSI (PDB code 1JB0), (b) PbRC (PDB code 3I4D), and (c) PSII (PDB code 3ARC). Pink arrows indicate electron transfer. Dotted lines indicate pseudo-C2 axes. Electron-transfer active branches are red labeled, whereas inactive branches are gray labeled. | ||
Here, we present Em values of Chla, Pheoa, BChla, and BPheoa for one-electron reduction in both electron-transfer branches of PbRC, PSI, and PSII; the Em values were calculated using the crystal structures, solving the linear Poisson–Boltzmann equation, and considering the protonation states of all titratable sites in the entire proteins.
Results
E m for accessory chlorophylls
In PSI, Em(A–1A) and Em(A–1B) as well as Em(A0A) and Em(A0B) are at essentially the same level in the cyanobacterial2 (Fig. 2a) and plant6 (Fig. S1†) PSI crystal structures. Em(A0A) = −1042 mV and Em(A0B) = −1023 mV (Fig. 2a), obtained using the cyanobacterial2 PSI crystal structure, are consistent with the experimentally estimated values, e.g., Em(A0) = −1050 mV28 and −1040 mV.29
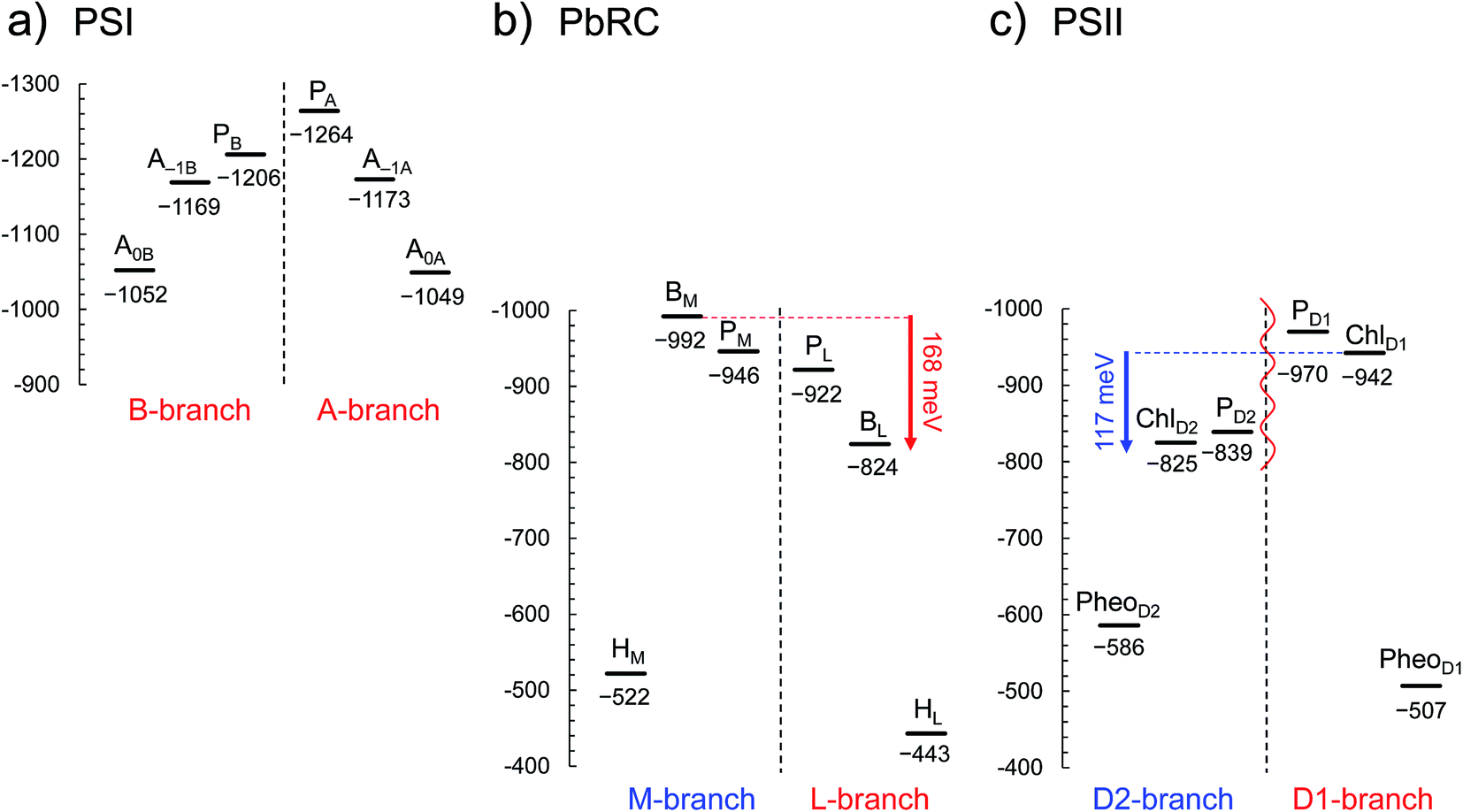 | ||
| Fig. 2 E m for one-electron reduction in the electron transfer chains in photosynthetic reaction centers of (a) PSI (PDB code 1JB0), (b) PbRC (PDB code 3I4D), and (c) PSII (PDB code 3ARC) in mV. Red and blue arrows indicate the Em difference between accessory (B)Chla cofactors in PbRC and PSII, respectively. The red wavy line indicates the weak electronic coupling (i.e., uncoupling) between PD1 and PD2.49,50 Dotted lines indicate pseudo-C2 axes. Electron-transfer active branches are red labeled, whereas inactive branches are blue labeled. See ref. 13 and 26 for calculated Em values for one-electron oxidation in PSI, PbRC, and PSII. | ||
In PbRC, Em(BL) is ∼170 mV less negative than Em(BM) based on the crystal structure analyzed at 2.01 Å resolution (Protein Data Bank (PDB) code 3I4D) (Fig. 2b). Em(BL) and Em(BM) were also calculated based on other PbRC crystal structures (e.g., PDB codes, 1M3X3 and 1EYS;30 Fig. S2†) and show the same tendency.
In sharp contrast to PbRC, Em(ChlD1) is 120 mV more negative than Em(ChlD2) in the 1.9 Å PSII crystal structure5 (Fig. 2c). At the ChlD1 and ChlD2 binding sites, “the PSII protein dielectric volume” (i.e., “uncharged protein volume”, which is ultimately comprised of van der Waals radii of all protein atoms) decreases the solvation of the Chla cofactors, destabilizes Chla˙−, and thus lowers the Em(ChlD1) and Em(ChlD2) values. On the other hand, “the atomic charges of proteins” (i.e., “protein charges”) also affect Em(Chla); e.g., negatively charged groups destabilize Chla˙− and lower the Em(ChlD1) and Em(ChlD2) values. To identify the factors that differentiate between Em(ChlD1) and Em(ChlD2) in PSII, we analyzed contributions of “protein atomic charges” and “loss of solvation” to Em(ChlD1) and Em(ChlD2). Contributions of the protein atomic charges are predominantly responsible for the difference in the Em values for accessory chlorophylls between PbRC and PSII, whereas contributions to Em from the protein volume, which prevents the solvation of reduced accessory chlorophylls and thus lowers Em, are much smaller (Table 1).
E
m(PheoD1) is −507 mV (Fig. 2c), which is consistent with the value of −499 mV31 obtained using the 3.0 Å PSII crystal structure (PDB code 2AXT)32 and the spectroelectrochemically determined value of −505 mV.33
| Protein | PSI | PbRC | PSII | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Accessory chlorophyll | A–1B | A–1A | PM | PL | ChlD2 | ChlD1 | |||
| a Calculated in the absence of all atomic partial charges of the proteins. | |||||||||
| E m | −1169 | −1173 | (−4) | −992 | −824 | (168) | −825 | −942 | (−117) |
| In uncharged proteina | −1085 | −1071 | (14) | −851 | −813 | (38) | −1047 | −1058 | (−11) |
| In water | −798 | −798 | (0) | −641 | −641 | (0) | −798 | −798 | (0) |
| E m shift (water to protein) | −371 | −375 | (−4) | −351 | −183 | (168) | −27 | −144 | (−117) |
| Due to protein charge | −84 | −102 | (−18) | −141 | −11 | (130) | 222 | 116 | (−106) |
| Due to loss of solvation | −287 | −273 | (14) | −210 | −172 | (38) | −249 | −260 | (−11) |
BL˙− stabilization in PbRC
In PbRC, electronic excitation of BChla leads to the formation of the (PL/PM)˙+BL˙− state.34Fig. 2 shows that Em(BL) is 170 mV less negative than Em(BM), facilitating electron transfer along the L-branch. The asymmetry of the electron-transfer energetics is caused by the different contributions of charges on the residues and cofactors, not the different shapes of the proteins (e.g., solvent accessibility near each BChla) (Table 1). In particular, loop a–b and helix cd in the periplasm region and helix d in the transmembrane region, which are structurally conserved in PSII (Fig. 3), helped to stabilize BL˙− with respect to BM˙− (Table 2). Among the L/M-residue pairs, Phe-L181/Tyr-M210 in helix d (Em(BL) − Em(BM) = 26 mV), Tyr-L67/Glu-M95 in loop a–b (20 mV), and Asp-L155/Asp-M184 in helix cd (22 mV) provide the greatest contribution to Em(BL) > Em(BM) (Table 3). | ||
| Fig. 3 Structural components of type-II reaction centers (e.g., PSII). | ||
| Region | Component | E m(BL) − Em(BM) in PbRC | E m(ChlD1) − Em(ChlD2) in PSII | Difference |
|---|---|---|---|---|
| a Including ligand groups. | ||||
| Periplasm/lumen | Mn4CaO5a | — | 56 | — |
| 2Cl− | — | −66 | — | |
| Loop a–b | 47 | −86 | −133 | |
| Helix cd | 40 | −91 | −131 | |
| Others | −7 | 55 | 48 | |
| Transmembrane | Helix a | −7 | 7 | 14 |
| Helix b | −3 | 15 | 18 | |
| Helix c | 15 | −50 | −65 | |
| Helix d | 50 | 17 | −33 | |
| Helix e | −5 | 26 | 31 | |
| Cofactors | 17 | −6 | −23 | |
| Cytoplasm/stroma | Subunit H | −2 | — | — |
| Others | 9 | 9 | 0 | |
| E m(BL) | E m(BM) | E m(BL) | E m(BM) | E m(BL) − Em(BM) | ||
|---|---|---|---|---|---|---|
| Phe-L181 | 0 | 22 | Tyr-M210 | 44 | −4 | 26 |
| Val-L157 | 19 | 0 | Thr-M186 | −1 | −4 | 22 |
| Tyr-L67 | 0 | 0 | Glu-M95 | −2 | −22 | 20 |
| Ser-L178 | −1 | −21 | Ala-M207 | −7 | −2 | 16 |
| Asp-L155 | −21 | −5 | Asp-M184 | −6 | −37 | 15 |
(i) Asp-M184 and Glu-M95 at the binding interface of cytochrome c2, decreasing Em(BM).Table 3 shows that Glu-M95 and Asp-M184 contribute to decreasing Em(BM) (22 mV and 37 mV, respectively) and thus stabilizing BL˙− with respect to BM˙−. From the observation of the PbRC–cytochrome c2 co-crystal structure, Axelrod et al. concluded that Glu-M95 and Asp-M184 provide the largest electrostatic interaction with cytochrome c2 (Fig. S3†).35 Notably, among the 17 PbRC mutants, mutation of Asp-M184 to Lys exhibits the largest change, with a decrease in the binding constant with cytochrome c2 by a factor of 800.36 Thus, negatively charged Asp-M184 likely contributes not only to binding of the one-electron donor of PbRC (i.e., cytochrome c2) but also to electron transfer along the L-branch.
(ii) How Tyr-M210 facilitates L-branch electron transfer. Among all L/M-residue pairs in PbRC, the difference in the Phe-L181/Tyr-M210 pair in helix d contributes to the Em difference the most, i.e., increasing Em(BL) with respect to Em(BM), as suggested in theoretical analysis by Parson et al.37 (Table 3). Indeed, mutations of Tyr-M210 to phenylalanine decreased the initial electron transfer with a time constant from 3.5 ps to 16 ps.38 The PbRC crystal structure analyzed at 2.01 Å (PDB code 3I4D) shows that the polar –OH group of Tyr-M210 is oriented toward BL, thus stabilizing BL˙− and increasing Em(BL). The –OH group cannot be oriented toward the methyl-keto (acetyl) group of PM because the methyl site, rather than the keto site, is near the –OH group of Tyr-M210 (Fig. 4a).
 | ||
| Fig. 4 (a) Orientations of the methyl-keto group in PM (yellow ball for methyl C and red ball for keto O) and the hydroxyl group in Tyr-M210 (red ball for hydroxyl O) in the 2.01 Å-PbRC structure (PDB code 3I4D). (b) The methyl-keto groups in BChla and BPheoa (yellow balls for methyl C and red balls for keto O) in the 1.87 Å-PbRC structure (PDB code 2J8C),4 whose assignments of the keto O atom and the methyl C atom are opposite to those in the 2.01 Å-PbRC structure (PDB code 3I4D). (c) The original assignment of the methyl-keto group of BL in the 1.87 Å-PbRC structure. The density is too low when the keto O atom is assumed (red mesh), whereas too much when the methyl C atom is assumed (green mesh). (d) The swapped assignment of the methyl-keto group of BL in the 1.87 Å-PbRC structure. | ||
In contrast to the 2.01 Å structure, the assignment of the methyl C and keto O atoms of PM is opposite in the PbRC crystal structure analyzed at 1.87 Å (PDB code 2J8C);4 the methyl-keto orientation allows the –OH group of Tyr-M210 to form an H-bond with the keto O atom of PM (OPM–OTyrM210 = 3.4 Å; Fig. 4b). Thus, the methyl-keto orientation of PM in the 1.87 Å structure cannot stabilize BL˙− (Fig. S4†). However, the electron density map of all BChla and Pheoa in the 1.87 Å structure,4 except for PL, indicates that the density is too low for the keto O atoms (red mesh in Fig. 4c), but too high for the methyl C atoms (green mesh in Fig. 4c) in the original assignment. Remarkably, the swapped assignment of the methyl-keto O and C atoms in PM, BL, BM, HL, and HM in the 1.87 Å structure (refined 1.87 Å structure), which is consistent with the original assignment in the 2.01 Å structure, is in better agreement with the density with a decrease in an R-factor by 0.01% (Fig. 4d). Just by rotating the methyl-keto groups (forming the refined 1.87 Å structure), the Em difference, i.e., Em(PL) − Em(BL), can be altered from −8 mV to −72 mV (Fig. S4†).
Hence, the methyl-keto orientations assigned in the 2.01 Å structure (PDB code 3I4D) appear to be relevant to the PbRC conformation; the –OH group of Tyr-M210 is predominantly oriented toward BL, stabilizing BL˙− and increasing Em(BL).
(iii) Low dielectric volume near BMprovided by spheroidene and the QBside chain. Around the Glu-M95/Asp-M184 moiety, approximately 30 hydrophobic residues from subunit M are in van der Waals contact with the carotenoid spheroidene (Fig. S5†).39 The electrostatic influence of the negative charges at the Glu-M95/Asp-M184 moiety is likely to be less screened at BM with respect to BL, thus destabilizing BM˙−. For the same reason, the cluster of hydrophobic residues seems also to enhance the polar –OH group of Tyr-M210 to stabilize BL˙−. Hence, spheroidene, the cluster of hydrophobic residues, and the QB isoprene side chain (see below) may be the origin of the low effective dielectric constant reported near BM with respect to BL in the Stark effect spectrum40 or the significantly small electric field along the M-branch suggested in electrostatic calculations.24 It should be noted that there are no water channels identified near ChlD1 and ChlD2 in the PSII crystal structures.41,42
(iv) The QBisoprene side chain, decreasing specifically Em(BM). The isoprene side chain of QB is oriented toward BM and is partly in van der Waals contact with spheroidene, whereas that of QA is oriented away from BL (Fig. S5†).
The isoprene side chain of QB in the PbRC crystal structure analyzed at 2.01 Å (PDB code 3I4D) is comprised of 56 C atoms. When the side chain of QB is shortened to 16 C atoms, as identified in the 1.87 Å PbRC crystal structure (PDB code 2J8C),4 and the corresponding inner space is filled by water (represented implicitly with the dielectric constant εw = 80), changes in Em are predominantly observed at Em(BM) with an increase of 57 mV (Fig. S6b†); this suggests that the isoprene chain of QB also contributes to the hydrophobic protein environment specifically for BM, enhancing electrostatic interactions and destabilizing BM˙−.
Factors that are responsible for Em(ChlD1) < Em(ChlD2) in PSII
Table 2 shows that loop a–b (86 mV) and helix cd (91 mV) in the lumen region (Fig. 3) are responsible for Em(ChlD1) < Em(ChlD2) in PSII. Below we describe the key components that contribute to Em(ChlD1) < Em(ChlD2).(i) D1-Asp61/D2-His61 pair in loop a–b. In the stromal/lumen region, loop a–b (that connects helices a and b) and helix cd (Fig. 3) seem most likely to characterize PSII with respect to PbRC (Table 2). Loop a–b is comprised of 55 residues in D1 (D1: 55–109) and 54 residues in D2 (D2: 55–108), which are almost twice as long as that in PbRC (26 residues in subunit L (L: 57–82) and 34 residues in subunit M (M: 79–112)). The region D2-Val55–Ser66 in PSII is structurally absent in PbRC (Fig. S7†). The insertion in PSII involves key residues for water oxidation, e.g., D1-Ile60 (O2-exiting pathway43), D1-Asp61 (proton transfer pathway22,44), D1-Glu65 (proton transfer pathway22,45 and water channel42), and D2-His61 (proton transfer pathway for TyrD44,46,47). In particular, the D1-Asp61/D2-His61 pair decreases Em(ChlD1) by 98 mV (Table 4). The corresponding residues and proton transfer pathways are absent in PbRC.
| E m(ChlD1) | E m(ChlD2) | E m(ChlD1) | E m(ChlD2) | E m(ChlD1) − Em(ChlD2) | ||
|---|---|---|---|---|---|---|
| a Cl-1 and D1-Asn181 interact directly (Cl−⋯ND1-Asn181 = 3.31 Å5). b D1-Tyr161 and D1-His190 form an H-bond, sharing a proton. D2-Tyr160 and D2-His189 form an H-bond, sharing a proton. c D1-Glu65 and D2-Glu312 form an H-bond, sharing a proton. | ||||||
| D1-Asp61 | −72 | −26 | D2-His61 | 23 | 75 | −98 |
| D1-Asn181a | 5 | −1 | D2-Arg180 | 44 | 123 | −73 |
| Cl-1a | −91 | −38 | — | — | — | −53 |
| D1-Asp170 | −75 | −30 | D2-Phe169 | −2 | −6 | −42 |
| D1-Tyr161 | −8 | 12 | D2-Tyr160 | 5 | 24 | −39 |
| +D1-His190b | +D2-His189b | |||||
| D1-Glu65 | −11 | −2 | D2-Ser65 | −36 | −15 | −30 |
| +D1-Asn315c | +D2-Glu312c | |||||
| D1-Glu189 | −47 | −26 | D2-Phe188 | 2 | 5 | −24 |
| D1-Ser305 | 1 | 3 | D2-Glu302 | −43 | −22 | −23 |
| D1-Asp59 | −32 | −11 | D2-Tyr59 | 0 | 2 | −23 |
| D1-Asn301 | 2 | 0 | D2-Asp297 | −50 | −29 | −20 |
(ii) D2-Arg180 in helix cd, specifically increasing Em(ChlD2). In PSII, lumenal helix cd (D1: 176–190/D2: 176–188 for PSII, Fig. 3) decreases Em(ChlD1) with respect to Em(ChlD2) by 131 mV, whereas in PbRC, lumenal helix cd (and L: 152–162/M: 179–192 for PbRC) increases Em(BL) with respect to Em(BM) by 40 mV (Table 2).
In particular, the D1-Asn181/D2-Arg180 pair in helix cd decreases Em(ChlD1) by 73 mV with respect to Em(ChlD2) (Table 4). D1-Asn181 also serves as the Cl-1 binding site5 in the proton-conducting E65/E312 water channel.42 D2-Arg180 is located at the entrance of the proton transfer pathway for TyrD44,46 and provides the driving force.47 Furthermore, the D1-Asn181/A2-Arg180 pair is responsible for a larger PD1˙+ population than PD2˙+ (ref. 13), which facilitates electron transfer from substrate water molecules at the Mn4CaO5 moiety in D1.
(iii) Influence of Mn4CaO5. Em values calculated using the Mn-depleted PSII crystal structure48 are similar to those obtained using the 1.9 Å PSII crystal structure (Fig. S8†). Calculated protonation states in the Mn-depleted PSII crystal structure show that the ligand residues (D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, the carboxy-terminal D1-Ala344, and CP43-Glu354) and the H-bond partner (D1-His337) are fully protonated, which could compensate for loss of the cationic Mn4CaO5 cluster (Table S1†). Hence, the inorganic Mn4CaO5 component itself is not a main factor that determines Em and the energetics of electron transfer.13,26
Discussion
Different mechanism of single-branch electron transfer between PbRC and PSII
E m(BL) is 170 mV less negative than Em(BM) in PbRC. In contrast, the corresponding Em(ChlD1) value is 120 mV more negative than Em(ChlD2) in PSII. These controversial Em profiles imply that the mechanisms of single-branch electron transfer are different between PbRC and PSII even though both are type-II reaction centers. The initial electron transfer from the PL/PM pair to BL is 100 meV downhill in the L-branch and 50 meV uphill in the M-branch (Fig. 2). This energy difference should facilitate L-branch electron transfer. If PD1 and PD2 could form the strongly coupled PD1/PD2 special pair and function as an initial electron donor, electronic excitation of the PD1/PD2 pair might possibly have led to electron transfer in the D2-branch, since Em(PheoD2) is sufficiently higher than Em(ChlD2) (Fig. 2). However, the electronic coupling between PD1 and PD2 (85 to 150 cm−1 (ref. 49 and 50)) in PSII is much weaker than that between PL and PM (500 to 1000 cm−1 (ref. 51)) in PbRC. In addition, the longest wavelength pigment is thought to be ChlD1 in PSII.50,52 Given that ChlD1 is the primary electron donor (i.e., Chla, where excitation occurs due to the lowest site-energy) in PSII (e.g.,53), the calculated Em values indicate that electron transfer can occur in the D1-branch because of the sufficiently high Em(PheoD1) value (−500 mV31,33 and Fig. 2).
Hence, it seems likely that PSII activates electron transfer in the D1-branch (i) by uncoupling the PD1/PD2 pair (i.e., making both electron-transfer branches electronically completely isolated) and (ii) by employing ChlD1 as the primary electron donor; in contrast, PbRC activates electron transfer in the L-branch by increasing Em(BL) with respect to Em(BM) in the presence of the strongly coupled PL/PM pair.
Influence of the periplasm/lumen region on Em for accessory (B)Chla
In PbRC, among the total Em difference of 168 mV between BL and BM (where Em(BL) > Em(BM)), 80 mV originates from the periplasm region, namely loop a–b (47 mV) and helix cd (40 mV); in PSII, among the total Em difference of −117 mV between ChlD1 and ChlD2 (where Em(ChlD1) < Em(ChlD2)), −132 mV originates from the corresponding lumen region, namely loop a–b (−86 mV) and helix cd (−91 mV) (Table 2). Thus, loop a–b and helix cd in the periplasm/lumen region primarily contribute to the different Em profiles (Fig. 2) for PbRC and PSII.In PbRC, acidic residues Asp-M184 in helix cd and Glu-M95 in loop a–b, which contribute to Em(BL) > Em(BM) (Table 3), serve as an H-bond network for the binding of cytochrome c2, the source of electrons for [PL/PM]˙+. In PSII, basic residues D2-Arg180 in helix cd and D2-His61 in loop a–b, which contribute to Em(ChlD1) < Em(ChlD2) (Table 4), serve as a proton-conducting H-bond network proceeding from TyrD44,46,47 and also increase the PD1˙+ population with respect to PD2˙+ in [PD1/PD2]˙+.13
Intriguingly, Asp-M184 in helix cd and Glu-M95 in loop a–b in PbRC correspond to D2-Arg180 in helix cd and D2-His61 in loop a–b in PSII, respectively (Fig. 5 and S8†). Furthermore, even water molecules in the proton transfer pathway from TyrD in PSII seem to be structurally conserved on the binding surface near Asp-M184 and Glu-M95 in PbRC (Fig. 5b). These structural features imply that the cytochrome c2 binding network in PbRC and the proton transfer pathway from TyrD in PSII have a common origin, which differentiate the mechanism of single-branch electron transfer between PbRC and PSII.
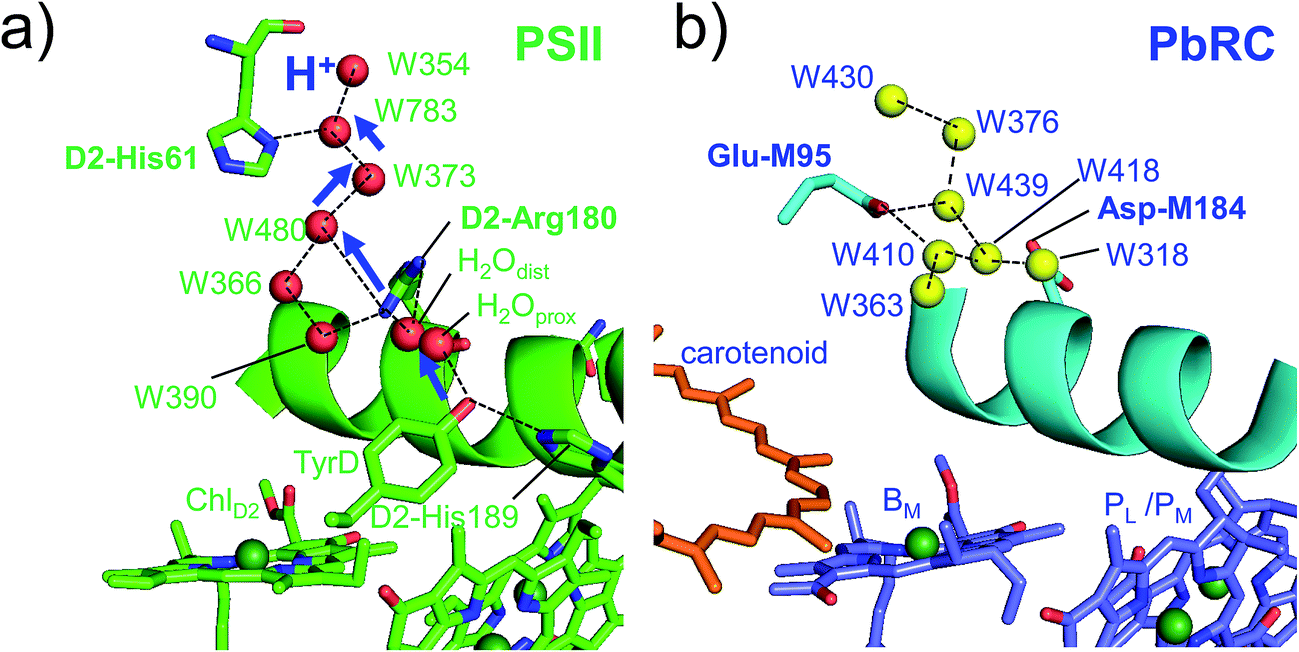 | ||
| Fig. 5 (a) H-bond network of water molecules (red balls) near ChlD2 in PSII (green), serving as a proton transfer pathway from TyrD (blue arrows)44,47 and (b) the corresponding H-bond network of water molecules (yellow balls) near BM in PbRC (cyan). The carotenoid molecule (spheroidene) exists only in PbRC. | ||
From the involvement of Asp-M184 in the binding interface with cytochrome c2 and correspondence of Asp-M184 to D2-Arg180 (Fig. S9†), the electrostatic differences in the periplasm/lumen regions are likely associated with the difference in sources of electrons–cytochrome c2/H2O.
Type-I reaction centers with respect to type-II reaction centers
In PSII, residues that increase the Em difference between ChlD1 and ChlD2 (Table 2) are mostly identical to those that have been identified to increase the Em difference between PD1 and PD2 significantly13 (e.g., D1-Asp61/D2-His61, D1-Asn181/D2-Arg180, D1-Asp170/D2-Phe169, and D1-Glu189/D2-Phe188). These results suggest that the same PSII protein electrostatic environment (discussed above) is responsible for asymmetry in energetics of the electron transfer branches (Fig. 2) as well as the cationic state distribution over the [PD1/PD2]˙+.13In PSI, the protein electrostatic environments of PsaA and PsaB are quite similar and no residues have been identified to induce the Em difference between PA and PB significantly.27 Indeed, Em(A–1A) and Em(A–1B) are also similar (Fig. 2a and S1†) and there are no residues that induce the Em difference between A–1A and A–1B. It seems likely that the similar protein electrostatic environment of PsaA and PsaB is a main factor that plays a role in keeping both branches open for electron transfer in PSI.
Concluding remarks
In PSII, substrate water molecules need to release protons when acting as an electron donor; thus, both electron and proton transfer pathways are expected to proceed from the substrate water molecules. The proton transfer pathway from O4 and the electron transfer pathway toward PD1˙+ go along the same axis in the opposite directions (Fig. 6), which allows PSII to use the common protein electrostatic environment for both transfer of electrons (e−) and protons (H+) without competing. It seems plausible that Em(ChlD1) < Em(ChlD2) in PSII, which is obviously inconsistent with Em(BL) > Em(BM) in PbRC, is due to a compromise between release of protons and release of electrons from the substrate water molecules using the common protein electrostatic environment and could have been overcome (i) by uncoupling the PD1/PD2 pair and (ii) by employing ChlD1 as the primary electron donor.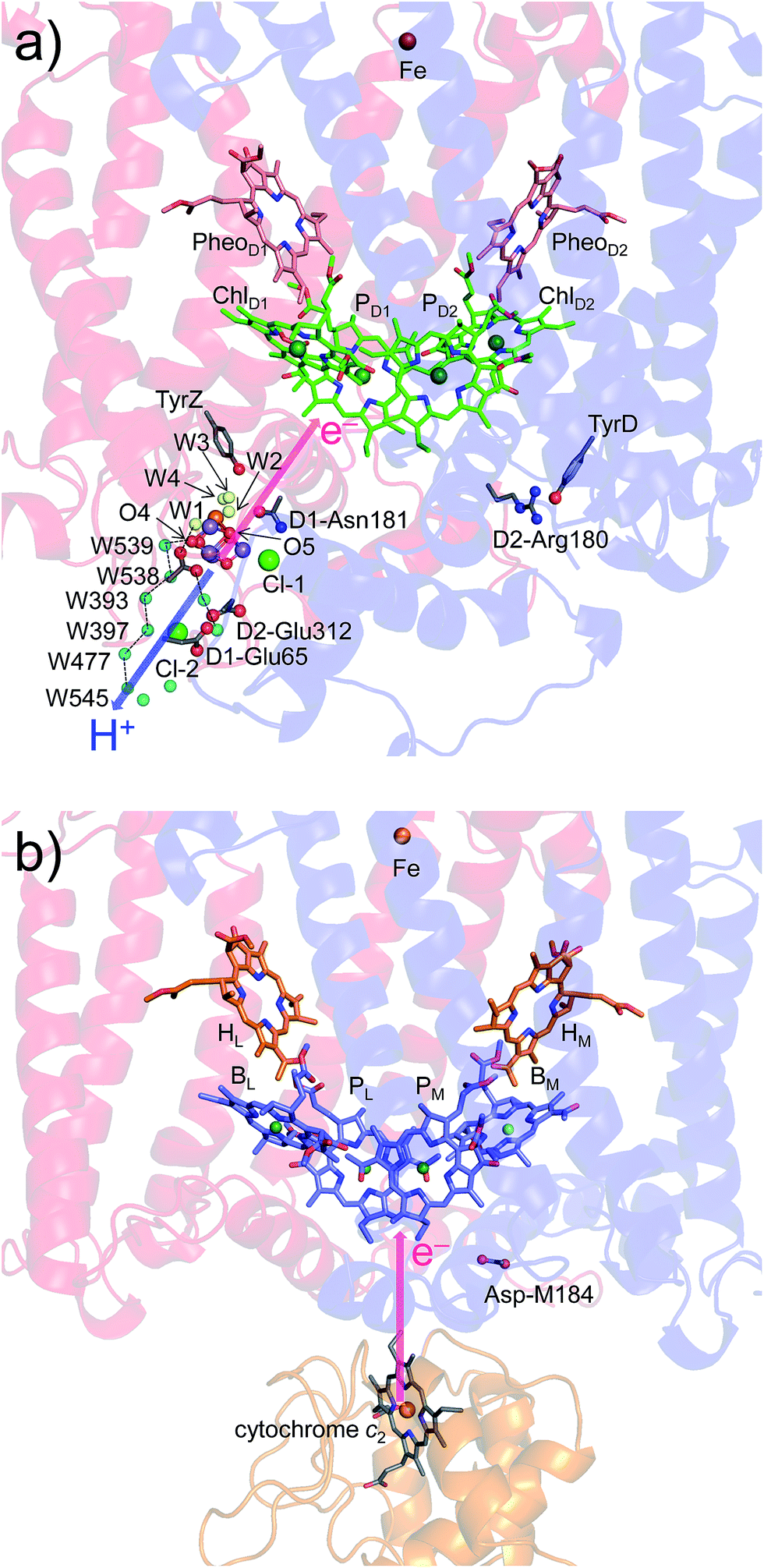 | ||
| Fig. 6 Locations of the electron transfer (pink arrows) and proton transfer (blue arrow) pathways in (a) PSII and (b) PbRC. Ligand water molecules are indicated by yellow balls and other water molecules by cyan balls. PbRCs from Rhodobacter sphaeroides and Blastochloris viridis have cytochrome c2 and a bound tetraheme cytochrome as the source of electrons, respectively. In both PbRCs, the sources of electrons are at an equidistance of 20–21 Å from PL and PM. In PSII, W539 (21.4 Å), O4 (19.7 Å), and W1 (18.5 Å) are at similar distances (∼20 Å) from the electron acceptor (monomeric PD1˙+). | ||
Hence, it is likely not a coincidence that the D1/D2 residue pairs, which are responsible for Em(ChlD1) < Em(ChlD2) (e.g., D1-Asn181/D2-Arg180 and D1-Asp61/D2-His61), can also serve as (i) electrostatically pushing the cation onto PD1 [basic residues in D2],14 providing a larger PD1˙+ population than PD2˙+ (ref. 13) and thereby facilitating electron transfer from substrate water molecules in D1.15 The presence of low pKa groups (i.e., acidic residues) near the proton releasing Mn4CaO5 site [acidic residues in D1] also (ii) facilitates release of protons from the substrate water molecules. These features seem to be the nature of PSII, which uses a protonated electron source—a pair of water molecules.
Methods
Coordinates and atomic partial charges
The atomic coordinates were taken from the X-ray structures; cyanobacterial PSI from Thermosynechococcus elongatus at 2.5 Å resolution (PDB code, 1JB0);2 plant PSI from Pisum sativum at 2.8 Å resolution (PDB code, 4XK8); PbRC from Rhodobacter sphaeroides at 2.01 Å resolution (PDB code, 3I4D), 1.87 Å resolution (PDB code, 2J8C),4 and 2.55 Å resolution (PDB code, 1M3X);3 PbRC from Thermochromatium tepidum at 2.2 Å resolution (PDB code, 1EYS);30 the PSII monomer unit (designated monomer A) of the PSII complexes from Thermosynechococcus vulcanus at 1.9 Å resolution (PDB code, 3ARC).5 Hydrogen atoms were generated and energetically optimized with CHARMM.54 Atomic partial charges of the amino acids were adopted from the all-atom CHARMM22) parameter set.55 For PSI, the atomic charges of cofactors were taken from previous studies (Chla, phylloquinone, β-carotene,56 and the Fe4S4 cluster57). The atomic charges of the other cofactors ((B)Chla, including (B)Chla˙+ and (B)Chla˙−, (B)Pheoa, ubiquinone, plastoquinone, spheroidene, sulfoquinovosyl diacylglycerol, heptyl 1-thiohexopyranoside, and the Fe complex) were determined by fitting the electrostatic potential in the neighborhood of these molecules using the RESP procedure58 (Tables S2–S11†). To obtain the atomic charges of the Mn4CaO5 cluster or the Fe complex, backbone atoms are not included in the RESP procedure (except for D1-Ala344) (Table S11†). The electronic wave functions were calculated after geometry optimization by the DFT method with the B3LYP functional and 6-31G** basis sets, using the JAGUAR program.59 For the atomic charges of the non-polar CHn groups in cofactors (e.g., the phytol chains of (B)Chla and (B)Pheoa and the isoprene side-chains of quinones), the value of +0.09 was assigned for non-polar H atoms. We considered the Mn4CaO5 cluster to be fully deprotonated in S1.The protein inner spaces were represented implicitly with the dielectric constant εw = 80, whereas the following water molecules were represented explicitly; (i) for PSII, ligand water molecules of the Mn4CaO5 cluster (W1 to W4), a diamond-shaped cluster of water molecules near TyrZ (W5 to W7)60, the water molecule distal to TyrD44, ligand water molecules of ChlD1 (A1003 and D424), ChlD2 (A1009 and A359), and other Chla (B1001, B1007, B1027, C816, and C1004); (ii) for PSI, clusters of water molecules near A1A (A5007, A5015, A5022, A5043, and A5049) and A1B (B5018, B5019, B5030, B5055, B5056, and B5058), ligand water molecules of A–1A (B5005), A–1B (A5005), and other Chla (A5004, A5010, A5012, A5024, A5032, A5051, B5006, B5010, B5022, B5036, B5053, B5054, J127, L4023, and M155).
E m calculation: solving the linear Poisson–Boltzmann equation
To obtain the Em values in the proteins, we calculated the electrostatic energy difference between the two redox states in a reference model system by solving the linear Poisson–Boltzmann equation with the MEAD program61 and using Em(BChla) = −641 mV, Em(BPheoa) = −384 mV (based on Em(BChla) = −830 mV and Em(BPheoa) = −600 mV for one-electron reduction measured in tetrahydrofuran,62 considering the solvation energy difference), Em(Chla) = −798 mV, and Em(Pheoa) = −641 mV (based on Em(Chla) = −910 mV and Em(Pheoa) = −700 mV for one-electron reduction measured in butyronitrile63). The difference in the Em value of the protein relative to the reference system was added to the known Em value. The ensemble of the protonation patterns was sampled by the Monte Carlo method with Karlsberg.64 The linear Poisson–Boltzmann equation was solved using a three-step grid-focusing procedure at resolutions of 2.5 Å, 1.0 Å, and 0.3 Å. Monte Carlo sampling yielded the probabilities [Aox] and [Ared] of the two redox states of molecule A. Em was evaluated using the Nernst equation. A bias potential was applied to obtain an equal amount of both redox states ([Aox] = [Ared]), thereby yielding the redox midpoint potential as the resulting bias potential. To facilitate direct comparisons with previous computational results (e.g.,13,26), identical computational conditions and parameters were used; all computations were performed at 300 K, pH 7.0, and an ionic strength of 100 mM; the dielectric constants were set to εp = 4 inside the protein and εw = 80 for water.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JST CREST (JPMJCR1656), JSPS KAKENHI (JP16H06560 and JP26105012), Japan Agency for Medical Research and Development (AMED), Materials Integration for engineering polymers of Cross-ministerial Strategic Innovation Promotion Program (SIP), and the Interdisciplinary Computational Science Program in CCS, University of Tsukuba.References
- H. Michel and J. Deisenhofer, Biochemistry, 1988, 27, 1–7 CrossRef CAS.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauss, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- A. Camara-Artigas, D. Brune and J. P. Allen, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11055–11060 CrossRef CAS PubMed.
- J. Koepke, E. M. Krammer, A. R. Klingen, P. Sebban, G. M. Ullmann and G. Fritzsch, J. Mol. Biol., 2007, 371, 396–409 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- X. Qin, M. Suga, T. Kuang and J. R. Shen, Science, 2015, 348, 989–995 CrossRef CAS PubMed.
- T. Watanabe, M. Kobayashi, A. Hongu, M. Nakazato, T. Hiyama and N. Murata, FEBS Lett., 1985, 191, 252–256 CrossRef CAS.
- P. Joliot and A. Joliot, Biochemistry, 1999, 38, 11130–11136 CrossRef CAS PubMed.
- T. Cardona, A. Sedoud, N. Cox and A. W. Rutherford, Biochim. Biophys. Acta, 2012, 1817, 26–43 CrossRef CAS PubMed.
- J. C. Williams, R. G. Alden, H. A. Murchison, J. M. Peloquin, N. W. Woodbury and J. P. Allen, Biochemistry, 1992, 31, 11029–11037 CrossRef CAS PubMed.
- V. V. Klimov, S. I. Allakhverdiev, S. Demeter and A. A. Krasnovskii, Dokl. Akad. Nauk SSSR, 1979, 249, 227–230 CAS.
- A. W. Rutherford, J. E. Mullet and A. R. Crofts, FEBS Lett., 1981, 123, 235–237 CrossRef CAS.
- K. Saito, T. Ishida, M. Sugiura, K. Kawakami, Y. Umena, N. Kamiya, J.-R. Shen and H. Ishikita, J. Am. Chem. Soc., 2011, 133, 14379–14388 CrossRef CAS PubMed.
- A. W. Rutherford, A. Boussac and P. Faller, Biochim. Biophys. Acta, 2004, 1655, 222–230 CrossRef CAS PubMed.
- P. Faller, R. J. Debus, K. Brettel, M. Sugiura, A. W. Rutherford and A. Boussac, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14368–14373 CrossRef CAS.
- H. Dau and M. Haumann, Coord. Chem. Rev., 2008, 252, 273–295 CrossRef CAS.
- M. Haumann and W. Junge, Biochim. Biophys. Acta, 1999, 1411, 86–91 CrossRef CAS.
- J. S. Vrettos, J. Limburg and G. W. Brudvig, Biochim. Biophys. Acta, 2001, 1503, 229–245 CrossRef CAS.
- G. Renger, Biochim. Biophys. Acta, 2001, 1503, 210–228 CrossRef CAS.
- K. Saito, A. W. Rutherford and H. Ishikita, Nat. Commun., 2015, 6, 8488 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- H. Ishikita, W. Saenger, B. Loll, J. Biesiadka and E.-W. Knapp, Biochemistry, 2006, 45, 2063–2071 CrossRef CAS PubMed.
- N. W. Woodbury and W. W. Parson, Biochim. Biophys. Acta, 1984, 767, 345–361 CrossRef CAS.
- M. R. Gunner, A. Nicholls and B. Honig, J. Phys. Chem., 1996, 100, 4277–4291 CrossRef CAS.
- A. W. Rutherford, A. Osyczka and F. Rappaport, FEBS Lett., 2012, 586, 603–616 CrossRef CAS PubMed.
- H. Ishikita, W. Saenger, J. Biesiadka, B. Loll and E.-W. Knapp, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9855–9860 CrossRef CAS PubMed.
- K. Saito and H. Ishikita, Biophys. J., 2011, 101, 2018–2025 CrossRef CAS PubMed.
- F. A. M. Kleinherenbrink, G. Hastings, B. P. Wittmershaus and R. E. Blankenship, Biochemistry, 1994, 33, 3096–3105 CrossRef CAS PubMed.
- S. Kumazaki, M. Iwaki, I. Ikegami, H. Kandori, K. Yoshihara and S. Itoh, J. Phys. Chem., 1994, 98, 11220–11225 CrossRef CAS.
- T. Nogi, I. Fathir, M. Kobayashi, T. Nozawa and K. Miki, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13561–13566 CrossRef CAS PubMed.
- H. Ishikita, J. Biesiadka, B. Loll, W. Saenger and E.-W. Knapp, Angew. Chem., Int. Ed., 2006, 45, 1964–1965 CrossRef CAS PubMed.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed.
- Y. Kato, M. Sugiura, A. Oda and T. Watanabe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17365–17370 CrossRef CAS PubMed.
- W. Holzapfel, U. Finkele, W. Kaiser, D. Oesterhelt, H. Scheer, H. U. Stilz and W. Zinth, Chem. Phys. Lett., 1989, 160, 1–7 CrossRef CAS.
- H. L. Axelrod, E. C. Abresch, M. Y. Okamura, A. P. Yeh, D. C. Rees and G. Feher, J. Mol. Biol., 2002, 319, 501–515 CrossRef CAS PubMed.
- M. Tetreault, S. H. Rongey, G. Feher and M. Y. Okamura, Biochemistry, 2001, 40, 8452–8462 CrossRef CAS PubMed.
- W. W. Parson, Z.-T. Chu and A. Warshel, Biochim. Biophys. Acta, 1990, 1017, 251–272 CrossRef CAS.
- U. Finkele, C. Lauterwasser, W. Zinth, K. A. Gray and D. Oesterhelt, Biochemistry, 1990, 29, 8517–8521 CrossRef CAS PubMed.
- A. W. Roszak, K. McKendrick, A. T. Gardiner, I. A. Mitchell, N. W. Isaacs, R. J. Cogdell, H. Hashimoto and H. A. Frank, Structure, 2004, 12, 765–773 CrossRef CAS PubMed.
- M. A. Steffen, K. Lao and S. G. Boxer, Science, 1994, 264, 810–816 CAS.
- N. Sakashita, H. C. Watanabe, T. Ikeda and H. Ishikita, Photosynth. Res., 2017, 133, 75–85 CrossRef CAS PubMed.
- N. Sakashita, H. C. Watanabe, T. Ikeda, K. Saito and H. Ishikita, Biochemistry, 2017, 56, 3049–3057 CrossRef CAS PubMed.
- A. G. Gabdulkhakov, V. G. Kljashtorny and M. V. Dontsova, Crystallogr. Rep., 2015, 60, 884–888 CrossRef CAS.
- K. Saito, A. W. Rutherford and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7690–7695 CrossRef CAS PubMed.
- R. J. Service, W. Hillier and R. J. Debus, Biochemistry, 2010, 49, 6655–6669 CrossRef CAS PubMed.
- S. Nakamura and T. Noguchi, Biochemistry, 2015, 54, 5045–5053 CrossRef CAS PubMed.
- K. Saito, N. Sakashita and H. Ishikita, Aust. J. Chem., 2016, 69, 991–998 CrossRef CAS.
- M. Zhang, M. Bommer, R. Chatterjee, R. Hussein, J. Yano, H. Dau, J. Kern, H. Dobbek and A. Zouni, eLife, 2017, 6, e26933 Search PubMed.
- J. R. Durrant, D. R. Klug, S. L. Kwa, R. van Grondelle, G. Porter and J. P. Dekker, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4798–4802 CrossRef CAS.
- G. Raszewski, B. A. Diner, E. Schlodder and T. Renger, Biophys. J., 2008, 95, 105–119 CrossRef CAS PubMed.
- N. W. Woodbury and J. P. Allen, in Anoxygenic photosynthetic bacteria: advances in photosynthesis, ed. R. E. Blankenship, M. T. Madigan and C. E. Bauer, Kluwer Academic Publishers, Dordrecht, 1995, pp. 527–557 Search PubMed.
- N. Cox, J. L. Hughes, R. Steffen, P. J. Smith, A. W. Rutherford, R. J. Pace and E. Krausz, J. Phys. Chem. B, 2009, 113, 12364–12374 CrossRef CAS PubMed.
- B. A. Diner and F. Rappaport, Annu. Rev. Plant Biol., 2002, 53, 551–580 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- A. D. MacKerell Jr, D. Bashford, R. L. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- K. Kawashima and H. Ishikita, Biochemistry, 2017, 56, 3019–3028 CrossRef CAS PubMed.
- J. Mouesca, J. L. Chen, L. Noodleeman, D. Bashford and D. A. Case, J. Am. Chem. Soc., 1994, 116, 11898–11914 CrossRef CAS.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- Jaguar, version 7.9, Schrödinger, LLC, New York, NY, 2011 Search PubMed.
- K. Saito, J.-R. Shen, T. Ishida and H. Ishikita, Biochemistry, 2011, 50, 9836–9844 CrossRef CAS PubMed.
- D. Bashford and M. Karplus, Biochemistry, 1990, 29, 10219–10225 CrossRef CAS PubMed.
- T. M. Cotton and R. P. van Duyne, J. Am. Chem. Soc., 1979, 101, 7605–7612 CrossRef CAS.
- T. Watanabe and M. Kobayashi, in Chlorophylls, ed. H. Scheer, CRC Press, Boca Raton, FL, 1991, pp. 287–303 Search PubMed.
- B. Rabenstein and E. W. Knapp, Biophys. J., 2001, 80, 1141–1150 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00424b |
| This journal is © The Royal Society of Chemistry 2018 |