
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile molecularly imprinted polymer-based fluorometric assay for detection of histamine
Xiaotong Feng,
Jon Ashley,
Tongchang Zhou,
Arnab Halder and
Yi Sun *
*
Department of Micro- and Nanotechnology, Technical University of Denmark, Ørsteds Plads, DK-2800 Kgs, Lyngby, Denmark. E-mail: Sun.Yi@nanotech.dtu.dk; Tel: +45 4525 6319
First published on 9th January 2018
Abstract
Histamine is a biogenic amine naturally present in many body cells. It is also a contaminant that is mostly found in spoiled food. The consumption of foods containing high levels of histamine may lead to an allergy-like food poisoning. Analytical methods that can routinely screen histamine are thus urgently needed. In this paper, we developed a facile and cost-effective molecularly imprinted polymer (MIP)-based fluorometric assay to directly quantify histamine. Histamine-specific MIP nanoparticles (nanoMIPs) were synthesized using a modified solid-phase synthesis method. They were then immobilized in the wells of a microplate to bind the histamine in aqueous samples. After binding, o-phthaldialdehyde (OPA) was used to label the bound histamine, which converted the binding events into fluorescent signals. The obtained calibration curve of histamine showed a linear correlation ranging from 1.80 to 44.98 μM with the limit of detection of 1.80 μM. This method was successfully used to detect histamine in spiked diary milk with a recovery rate of more than 85%.
1. Introduction
Histamine is a biogenic amine that is involved in the immune response and regulation of physiological functions.1,2 High levels of histamine have been widely found in a number of food sources including fish, pork, spinach, cheese and nuts,3 which can trigger an intolerance within individuals, leading to symptoms such as itchy eyes, runny nose, congestion, asthma, eczema, hot flushes, migraines and in extreme cases death from anaphylactic shock.1,4 According to EU legislation, the maximum residual limit (MRL) of histamine is 2–20 mg L−1 in beverages and 50–100 mg kg−1 in fermented food.5,6 Therefore, monitoring the level of histamine in food is very important for food safety control.Currently, thin layer chromatography (TLC),7 gas chromatography (GC),8 high performance liquid chromatography (HPLC)9 and capillary zone electrophoresis (CZE)10 are among the most commonly used analytical methods to detect histamine. Although the sensitivity of these methods is high, they often require expensive, sophisticated instrumentation, as well as time-consuming sample preparation. Alternatively, colorimetric11 and enzyme-linked immunosorbent assay (ELISA)12 are the most commonly used methods for preliminary screening. These commercially available kits rely on either a dehydrogenase enzyme to catalyze oxidation of histamine or an antibody to selectively bind histamine and form tight complexes. Although the enzyme and antibody possess high specificity and affinity, these bioreceptors are expensive and inherently unstable due to the biological origins.
To overcome the limitations of natural receptors, great attention has been paid to develop biomimetic receptors using molecularly imprinted polymers (MIPs).13,14 The technique involves the formation of binding sites in a synthetic polymer matrix that are of a complementary functional and structural character to its “substrate” molecule. MIPs have been synthesized for diverse food relevant analytes ranging from small molecules to big proteins and pathogens. Attributed to their thermal stability, robustness and low cost, these so called “plastic antibodies” have shown great potential for use in bioassays.15,16 Molecular imprinting of histamine has been reported earlier.17–19 However, these histamine MIPs suffered from different drawbacks as they were synthesized mainly by bulk polymerization or precipitation polymerization. For bulk polymerization, the resultant MIP particles often display large size variations due to the formation of the monolith followed by grinding. Moreover, it is challenging to completely remove the residual templates in the MIPs.20 Precipitation polymerization is a simple one-step polymerization which results in nanoparticles with a narrower size distribution.21 However, the template, monomers and crosslinker need to be dissolved into the porogen solvent with high dilution factor, resulting in negative impact on the interaction between the monomers and the template. Additionally, both methods give high binding site heterogeneity (highly polyclonal), which can lead to high levels of non-specific binding.
To address the drawbacks of bulk polymerization and precipitation polymerization, Canfarotta et al. proposed a solid-phase approach to synthesize nano-sized MIPs (nanoMIPs) by attaching the analyte onto glass beads.22 These nanoparticles could be quickly formed and removed from the template without further purification, and nanoMIPs with uniform size and high binding site homogeneity were achieved. Solid-phase synthesis has been used to imprint molecules such as melamine, vancomycin and trypsin, and showed great promise for both small molecules and proteins.
Besides the synthesis of MIPs, another challenge associated with developing MIP-based assay is the signal transduction. Since MIPs usually contain no inherent signaling element, it is difficult to transform the binding events into measurable optical or electronic signals. So far, the MIPs have been used together with surface enhanced Raman spectroscopy,17 voltammetric sensor18 and UV-vis spectrometry19 for detection of histamine, but these methods are not suitable for routine high-throughput screening purpose. Recently, a so called “Pseudo-ELISA”23–26 has been developed, where the nanoMIPs were used as replace natural antibodies in ELISA. In the assay, the nanoMIPs were immobilized in a microplate, and the analyte was quantified through the competitive binding between the free analyte and horseradish peroxidase-conjugated analyte. Though the pseudo-ELISA was promising for high-throughput screening, it required pre-conjugation of analyte with the enzyme, as well as several rounds of blocking, incubation and washing steps, which made the assay costly and time-consuming.
In this work, we developed a new MIP-based fluorometric assay for the rapid detection of histamine in food samples. Histamine nanoMIPs were synthesized using the modified solid-phase synthesis (Fig. 1). The binding affinity of the nanoMIPs with histamine was determined to be 0.89 nM, similar to anti-histamine antibody. A simple and cost-effective bioassay using histamine nanoMIPs as artificial receptor was then developed. The concept is shown in Fig. 2. The nanoMIPs were immobilized in a 96-well microplate by physical absorption, and samples with different concentrations of histamine were added to the wells. After incubation for a short period, the wells were washed. Subsequently, o-phthaldialdehyde (OPA) solution which can react specifically with primary amines27 was added to the wells to label the bound histamine, and the fluorescent intensity at 440 nm was recorded and used for quantification. Unlike the pseudo-ELISA approach, there is no need to prepare pre-labeled analyte in this assay. Instead of the long incubation time and multiple steps needed to develop color with horseradish peroxidase, it only took 4 min for OPA to give fluorescence signal. The feasibility of using the newly developed assay to detect histamine in food matrices, such as dairy milk, was successfully demonstrated. A limit detection of 1.80 μM and a recovery rate of more than 85% were achieved. To our best knowledge, this is the first time that nanoMIP-based OPA fluorometric assay is reported.
 | ||
| Fig. 1 (a) Immobilization of histamine on the surface of glass beads. (b) Solid phase synthesis of histamine nanoMIPs. (c) Interaction between immobilized histamine and functional monomer MAA. | ||
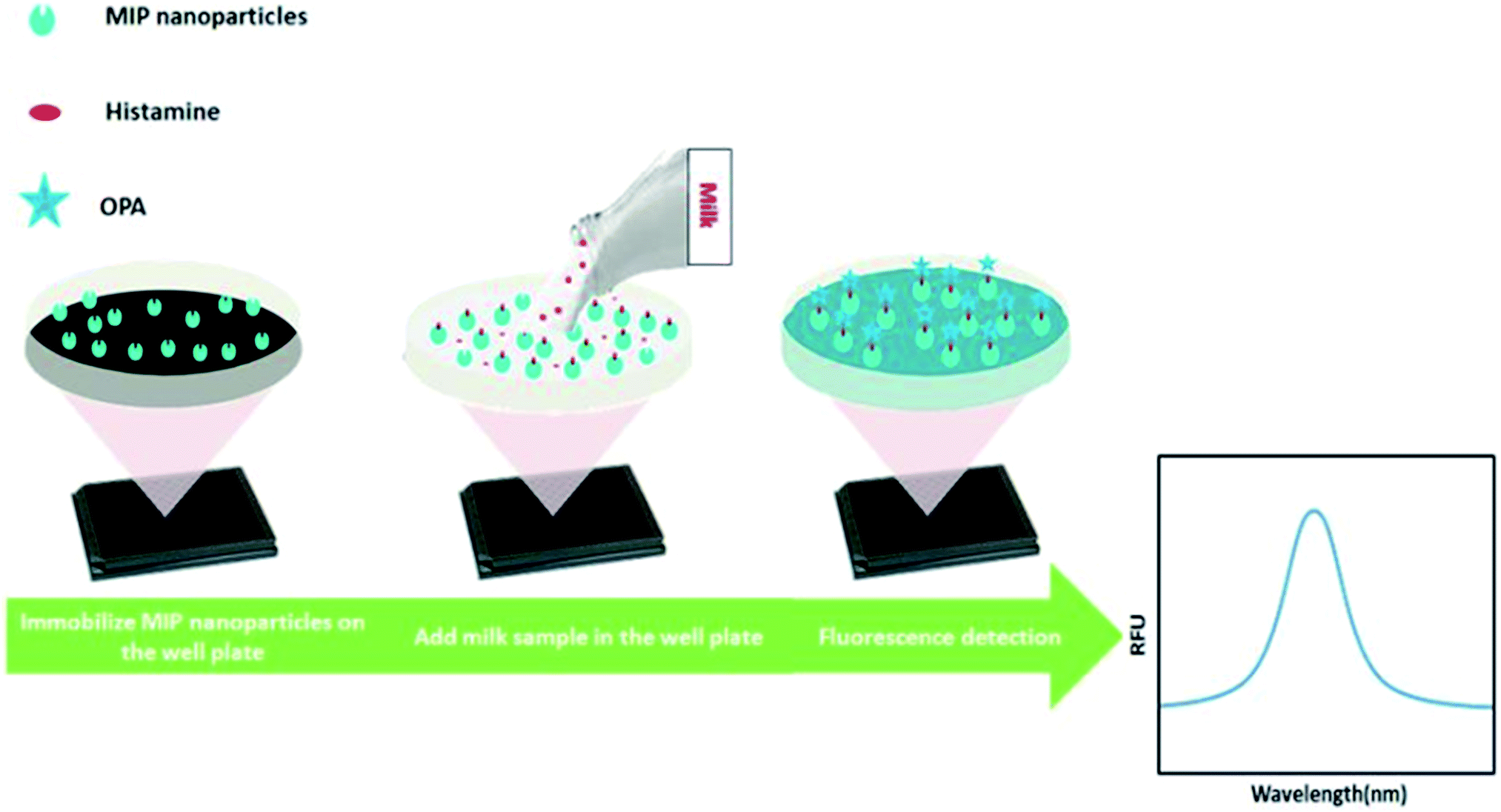 | ||
| Fig. 2 NanoMIP-based OPA fluorometric assay for detection of histamine. | ||
2. Experimental
2.1. Materials
Methacrylic acid (MAA), ethylene glycol dimethacrylate (EGDMA), trimethylolpropane trimethacrylate (TRIM), pentaerythritol-tetrakis-(3-mercaptopropionate) (PETMP), phosphate buffer solution (PBS) (pH 7.4), sodium hydroxide, glutaraldehyde (GA), histamine, histidine, melamine, (3-aminopropyl)triethoxysilane (APTES). Carbodiimide (EDC), N-hydroxysuccinimide (NHS) 98%, ethanolamine hydrochloride, hydrochloride acid (HCl), 11-mercaptoundecanoic acid (MUDA) 95%, hydrogen peroxide, acetonitrile (ACN), ethanol, acetone, phthaldialdehyde, 60 mL SPE tubes and 20 μm pore frits and glass beads (75 μm), were purchased from Sigma-Aldrich (DK). 2,4,6-Trinitrobenzene sulfonic acid (TNBSA) was purchased from Thermofisher Scientific (DK). N,N-Diethyldithiocarbamic acid benzyl ester (DABE) was obtained from TCI Europe (Belgium). Q-Max® Celluloseacetat (CA) syringe filters 0.45 μm were from FRISENETTE (DK). Gold chip was purchased from Bionavis, Sweden. Nunclon 96-well flat-bottom black microwell plates were purchased from Thermo scientific, DK. 0.1% milk (Arla) was bought from a local supermarket, Netto. Deionized water was obtained from a Millipore (MilliQ) purification system. All chemicals were analytical or HPLC grade and were used without further purification.2.2. Synthesis of histamine nanoMIPs
As shown in Fig. 1, the histamine nanoMIPs was synthesized by solid-phase approach. The protocol was adapted from the method developed by Canfarotta et al.222.3. Characterization of nanoMIPs
The yields of histamine nanoMIPs were measured by calculating the weight yield value from a 10 mL aliquot of nanoMIPs solution. In general, 10 mL of stock solution (in ACN) was evaporated under a stream of nitrogen in a pre-weighed empty vial. After evaporating, the glass vial was put into a vacuum oven at 80 °C for overnight.Nanoparticle size and quality were determined by dynamic light scattering (DLS) using a NanoBrook ZetaPALS Potential Analyzer (Brookhaven, USA). Ten milliliters of stock solution (in ACN) was evaporated under a stream of nitrogen. Two milliliters of MilliQ water was added and sonicated for 20 min. The dispersion was filtered through a 0.45 μm CA syringe filter to remove dust and possible aggregates. The filtered histamine nanoMIPs were analyzed by DLS at 25 °C in a 3 cm3 disposable polystyrene cuvette. The values were reported as an average of 5 measurements.
Scanning electron microscopy (SEM) analysis was carried out on a Quanta FEG 200 ESEM scanning electron microscopy (FEI). Five milliliters of stock solution (in ACN) was subjected to evaporation and replacement by 0.5 mL MilliQ water. The solution was sonicated for 20 min followed by filtering through a 0.45 μm CA syringe filter. A drop of histamine nanoMIPs was placed on a silica wafer and dried in a desiccator overnight. Prior to SEM, the sample was sputtered with a thin layer of gold using a Quorum Coater.
2.4. Surface plasma resonance (SPR)
Prior to SPR analysis, 25 mL of histamine nanoMIP stock solution was evaporated using a stream of nitrogen, 2 mL of PBS was added followed by 30 min of sonication. Aqueous stock suspensions of the histamine MIPs were filtered through CA syringe filters with 0.45 μm pore size and diluted in PBS for analysis followed by a series of 2× dilutions. Apparent molarity of nanoMIPs was calculated by approximating nanoMIPs as regular spheres, and the concentration of the nanoMIP was determined from the following equation.28
 | (1) |
SPR experiments were performed on a MP-SPR Navi™ 200 OTSO (Bionavis, Sweden). Au-coated chips were cleaned in Piranha solution (H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2, 3:1 v/v) for 5 min, and rinsed with MilliQ water. For immobilization of histamine, cleaned chips were dried under N2 and placed in a solution of 5 mM MUDA at 4 °C overnight to form a self-assembled layer (SAM). The resultant gold chip was promptly washed with water and ethanol, dried under nitrogen. The chip was docked in the MP-SPR Navi™ 200 OTSO and 10 mM PBS was pumped through the flow cell. The instrument was primed at a flow rate of 20 μL min−1. Upon obtaining a stable baseline, a mixture containing 100 μL 0.4 M EDC and 0.1 M NHS (100 μL) was injected followed by an injection of histamine (100 μL). Any remaining NHS ester groups were deactivated by injecting 1 M ethanolamine HCl (100 μL) in ethanol at a flow rate of 20 μL min−1. Hundred microliters of histamine nanoMIPs (0.05 nM to 5 nM) were injected sequentially over the immobilized histamine from the lowest to highest concentration, the responses were recorded and the signal from the reference channel was subtracted. The binding affinity dissociation constants (KD) were calculated from Advanced Analysis Software TraceDrawer™ by 1:1 binding model with drifting baseline fitting.
:H2O2, 3:1 v/v) for 5 min, and rinsed with MilliQ water. For immobilization of histamine, cleaned chips were dried under N2 and placed in a solution of 5 mM MUDA at 4 °C overnight to form a self-assembled layer (SAM). The resultant gold chip was promptly washed with water and ethanol, dried under nitrogen. The chip was docked in the MP-SPR Navi™ 200 OTSO and 10 mM PBS was pumped through the flow cell. The instrument was primed at a flow rate of 20 μL min−1. Upon obtaining a stable baseline, a mixture containing 100 μL 0.4 M EDC and 0.1 M NHS (100 μL) was injected followed by an injection of histamine (100 μL). Any remaining NHS ester groups were deactivated by injecting 1 M ethanolamine HCl (100 μL) in ethanol at a flow rate of 20 μL min−1. Hundred microliters of histamine nanoMIPs (0.05 nM to 5 nM) were injected sequentially over the immobilized histamine from the lowest to highest concentration, the responses were recorded and the signal from the reference channel was subtracted. The binding affinity dissociation constants (KD) were calculated from Advanced Analysis Software TraceDrawer™ by 1:1 binding model with drifting baseline fitting.
2.5. UV/vis binding assay
Binding properties of the synthesized nanoMIPs were tested in UV spectrophotometric analysis in PBS buffer. One milliliter of histamine nanoMIPs (5 mg mL−1) was mixed with 10 μL of various concentrations of histamine in glass vials followed by shaking for 20 h at room temperature. The used concentrations of histamine were 5 mg mL−1, 2.5 mg mL−1 and 1.25 mg mL−1, respectively. The same procedure was applied to histidine and melamine. After incubation, the solutions were centrifuged at 15000g for 30 min and the supernatant was collected (500 μL). The concentration of free analyte was measured by UV-2600 UV-vis spectrophotometer (Holm&Halby, Denmark). The absorbance maxima wavelength for UV/vis detection of histamine, histidine and melamine were 209 nm, 210 nm and 204 nm, respectively.
2.6. Development of OPA fluorometric assay for histamine detection
Fifty microliters of histamine nanoMIPs (2 μg μL−1) were dispensed into each well of Nunclon 96-well flat-bottom black microplate, and left to dry completely overnight at room temperature in the fume hood. To remove the unattached histamine nanoMIPs, 100 μL of 10 mM PBS was used to wash the well plate three times. Next, a series dilution of 100 μL histamine from 1.8 μM to 1799.4 μM was dispensed into the wells and incubated for 4 h. After incubation, histamine was removed and the plate was washed three times using 100 μL PBS. To each well, 90 μL of 0.1 M HCl was added followed by adding 18 μL of 1 M NaOH and 4.5 μL of OPA (in ethanol). Histamine was allowed to react with OPA in a dark room at room temperature for 4 min and the reaction was stopped by adding 9 μL of 3 M HCl. Finally, the fluorescence spectra were measured from 400 to 600 nm at an excitation wavelength of 360 nm, using a Spark® multimode microplate reader (Tecan, Sweden). The fluorescent intensity at 440 nm was used to build the calibration curve.2.7. Analysis of histamine in milk
0.1% milk (Arla) was diluted 10 folds by MilliQ water. The histamine with the concentrations of 0 μM, 4.5 μM, 13.5 μM, 27 μM and 40.48 μM was spiked into the milk. Histamine spiked milk was incubated into each nanoMIP-immobilized well for 4 h followed by washing using PBS for three times. OPA fluorometric assay was performed as described in Section 2.6. The concentration of histamine in the diluted milk was derived from the calibration curve.3. Results and discussion
3.1. Characterization of histamine nanoMIPs
With the solid-phase synthesis approach, nanoMIPs were produced within just a few minutes (depending on the power of UV sources). Furthermore, unlike in traditional methods such as suspension polymerization where 3 day-dialysis is required to remove the template, the nanoMIPs can be easily removed from the template since the binding sites are in the surface of nanoparticles.The nanoMIPs were synthesized via a UV-initiated living polymerization mechanism. Iniferter (initiator, transfer agent, terminator) was used instead of initiator because it allowed for a better control of polymer chain growing, thereby resulting in narrower molecular weight distribution of resultant polymers as compared to conventional free radical initiators.29 Since a custom-made light-tight UV photo reactor was used, the effect of UV exposure time on the properties of nanoMIPs was investigated. The photopolymerization was carried out at 10 s, 20 s, and 30 s, respectively. The size, yield and apparent dissociation constant KD of histamine nanoMIPs are summarized in Table 1. The results showed that there was a direct correlation between exposure time, yield and the size of the nanoMIPs produced. For instance, by increasing the exposure time from 10 s to 30 s, the yield of the nanoMIPs increased from 0.36 ± 0.05 mg g−1 glass beads to 0.56 ± 0.05 mg g−1 glass beads. At the same time, the hydrodynamic diameter of the nanoparticles increased from 119.47 ± 2.28 nm to 187.77 ± 3.35 nm. This may be because that the degree of polymerization was increased with longer exposure time, which in turn helped to produce larger nanoparticles, resulting in higher yields. SEM analysis of the synthesized histamine nanoMIPs was conducted. The image showed that the nanoparticles were quite homogenous in size (Fig. 3). Based on the SEM image scale, histamine nanoMIPs exhibited a dry size of 99–113 nm, which was smaller than that measured by DLS. This could be due to the fact that the nanoparticles shrank upon drying.
| Exposure time (s) | Yield (particle mass per g of glass beads, mg) | Size (nm) | KD (nM) |
|---|---|---|---|
| 10 | 0.36 ± 0.05 | 119.47 ± 2.28 | 11.30 |
| 20 | 0.45 ± 0.04 | 156.00 ± 2.69 | 8.01 |
| 30 | 0.56 ± 0.05 | 187.77 ± 3.35 | 0.89 |
 | ||
| Fig. 3 SEM image of the histamine nanoMIPs. | ||
The SPR sensorgram (Fig. 4) showed that the exposure time of 30 second gave the lowest value of apparent dissociation constant KD, which was 0.89 nM (Table 1). The reason could be that longer exposure time led to the formation of sufficiently rigid nanoparticles with well-formed recognition cavities, which allowed for stronger interactions between the immobilized template and the nanoMIPs.
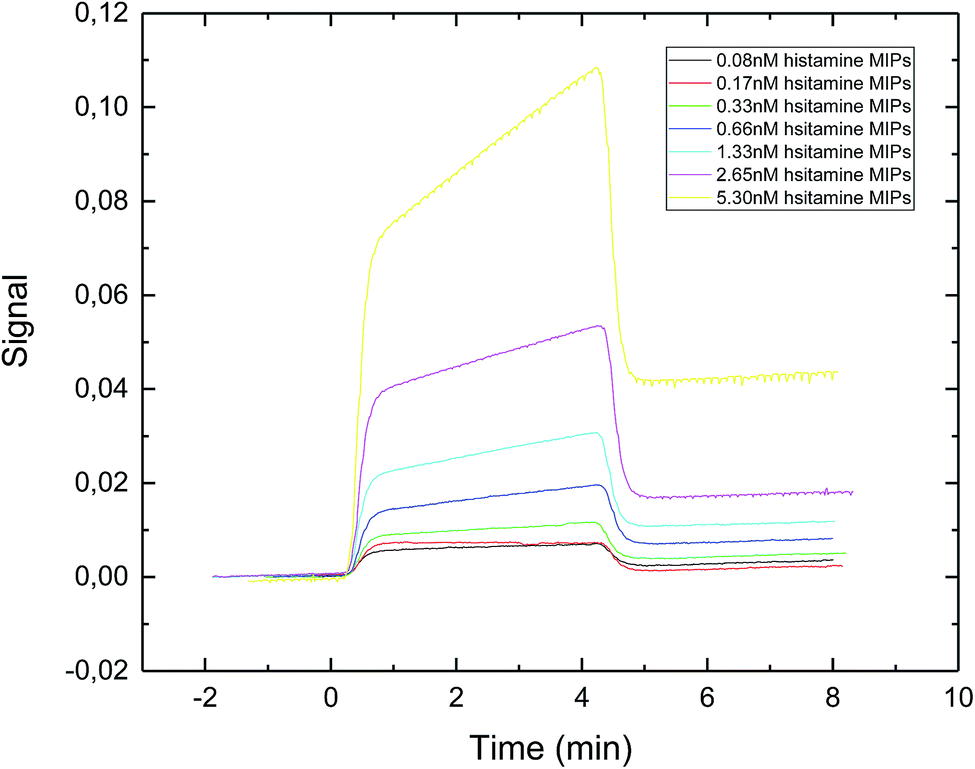 | ||
| Fig. 4 SPR sensorgram of nanoMIPs injected onto a specific histamine-coated sensor surface. The nanoMIPs were synthesized under UV exposure time of 30 s. | ||
Basozabal et al.30 utilized a similar approach to synthesize histamine nanoMIPs. The polymerization mixture was initiated using 2.5 min of UV exposure time. With this condition, the nanoparticles with the average hydrodynamic radius of 218.8 nm and the yield of 0.56 mg per gram of glass beads were obtained. It is worth to point out that due to the higher power output of our photoreactor, much shorter exposure time was adequate for the synthesis of the nanoMIPs. The apparent KD reported by Basozabol et al.30 was 0.099 nM while the apparent binding affinity in the current study was 0.890 nM. The difference in reported binding affinity were due to systematic errors in different SPR instrumentation, as well as the fact that we used an online amine coupling procedure whereas the chips prepared in the previous study were prepared offline using glutaraldehyde based conjugation. As such, in this study we were able to subtract the reference signal from the analyte signal.
Based on these results, histamine nanoMIPs with the highest binding affinity were synthesized using the exposure time of 30 second, and this exposure time was chosen for all further experiments. The main drawbacks of solid phase synthesis are low yield and typically there is only one binding site in each nanoparticle. This may result in low binding capacity, which may not suitable for sample preparation applications. However, the high-affinity (KD = 0.89 nM) of nanoMIPs and small size (∼200 nm) make them appropriate for use as receptors in biosensors. Moreover, the product yields can be increased by optimizing the morphology of the solid substrate, such as using high surface-to-volume supporting material as an alternative.
3.2. Selectivity of histamine nanoMIPs
Histamine nanoMIPs were synthesized using MAA as functional monomer. In organic solution, it can form strong hydrogen bond interactions with the nitrogen in the imidazole ring of histamine. When recognition occurred in aqueous solution, hydrogen bond interactions might still be involved to some extent, but it was found that electrostatic interactions served as the main channel to the recognition mechanism.19 In order to monitor the level of histamine in food samples, all selectivity experiments were conducted in aqueous PBS solution.To study the selectivity of histamine nanoMIPs, histidine and melamine were selected as an appropriate control. In solid-phase imprinting, non-imprinted polymers cannot be formed on the solid support, as they are immediately removed in the low temperature elution step. Furthermore, it is impractical to extract small fractions of non-imprinted polymer nanoparticles from high concentrations of residual monomers and oligomeric materials. Therefore, it is not possible to produce non-imprinted polymer as a control in this case.
Fig. 5a shows the structures of the three compounds. Histamine can be formed by decarboxylation of histidine. Fig. 5b showed the uptake concentrations of histamine, histidine and melamine by the nanoMIPs under the different concentrations. The results demonstrated that the nanoMIPs had higher (at least 2 fold) binding efficiency towards histamine than its structural analogs. Therefore, it can be concluded that the nanoMIP was selective for histamine.
 | ||
| Fig. 5 (a) Chemical structures of histamine, histidine and melamine. (b) Uptake of histamine, histidine and melamine at different concentrations by histamine nanoMIPs (5 mg) in 1 mL PBS buffer. | ||
3.3. OPA fluorometric assay for histamine
An OPA fluorometric assay was developed by using histamine nanoMIPs as artificial antibody. The histamine nanoMIPs were immobilized on the well plate by evaporation overnight. The physically adsorbed nanoMIPs were tightly bound to the surface of the plate and remained on the wells even after several washes with PBS buffer. As each well only required 100 μg nanoMIPs, histamine nanoMIPs obtained from one batch is sufficient for 50 reactions.OPA is a commonly used reagent which allows for the fluorescent detection and quantification of the primary amine in alkaline conditions, but this simple conjugation has not been used for MIP-based bioassays. In our method, histamine was firstly bound to the nanoMIPs, then OPA was added to form conjugation with the primary amine group of the histamine. The fluorescence signal clearly indicated that the histamine-nanoMIPs complex could be directly detected using the OPA conjugation. On the basis of this method, calibration curves were built as shown in Fig. 6. A good linear correlation between fluorescent intensity and histamine concentration was observed within the range of 1.80–44.98 μM.
 | ||
| Fig. 6 Saturation curve and calibration curve of histamine nanoMIP-based OPA fluorometric assay. Error bars were for experiments performed in duplicates. The R2 value of calibration curve was 0.992. | ||
Different incubation times were also tested (5 min, 10 min, 30 min, 1 h, 2 h, 4 h and overnight) in order to investigate the possibility of reducing the incubation time. The kinetic binding plot (Fig. 7) showed that the point of saturation was reached after 4 hours. Therefore, 4 hours of incubation time was chosen for all future experiments.
 | ||
| Fig. 7 Equilibrium binding of 2 μg mL−1 histamine to 100 μg histamine nanoMIPs. Error bars were for experiments performed in duplicates. | ||
Compared to the “pseudo-ELISA”, the process of OPA fluorometric assay was greatly simplified. “Pseudo-ELISA” needed preparation of horseradish peroxidase (HRP) conjugated target solution, as well as long development steps,25 whereas the OPA fluorometric assay required no pre-conjugation and only took 4 min to give fluorescent signals. Additionally, since there was no enzyme involved, the cost of analysis was significantly reduced.
3.4. Determination of histamine in milk
To test the feasibility of our method to detect histamine in food samples, histamine was spiked into milk with different concentrations (e.g. 0 μM, 4.5 μM, 13.5 μM, 27 μM and 40.48 μM). The concentrations of the spiked samples were then determined by interpolating the measured fluorescence intensity using the linear calibration plot. The results showed that the recovery rates of histamine in the spiked samples were in the range of 87.95–101.01% (Table 2). The RSD (n = 2) values were in the range of 3.13–7.70%. The limit of the detection was 1.8 μM (0.2 mg L−1), better than SERS- and UV-vis spectrometry-based methods, though not as low as voltammetry-based detection (Table 3). The sensitivity of our approach was much beyond the MRL of histamine in beverages (about 2–20 mg L−1).5 Furthermore, the recovery rate for non-spiked milk sample was nearly zero, suggesting that the matrix effects from the milk proteins and fats had no interferences on the analytical results. Therefore, this method could eliminate the need of sophisticated sample preparation.| Spiked (μM) | Found (μM) | Recovery (%) |
|---|---|---|
| 40.48 | 38.24 ± 3.12 | 94.45 ± 7.70 |
| 27.00 | 23.755 ± 2.23 | 87.95 ± 8.27 |
| 13.50 | 12.175 ± 0.54 | 90.15 ± 4.03 |
| 4.50 | 4.935 ± 0.42 | 101.01 ± 3.13 |
| 0 | −1.42 ± 1.48 | NA |
4. Conclusions
In conclusion, histamine nanoMIPs were successfully synthesized and characterized using solid-phase synthesis approach. The synthesized histamine nanoMIPs showed high binding affinity and high selectivity towards histamine. The nanoMIP-based fluorometric assay was developed in a microplate format. OPA was used to label the histamine, which enabled histamine to be quantified based on the fluorescent intensity. The assay has been employed to detect histamine in diary milk with detection limit of 1.8 μM and recovery efficiency of more than 85%. The simple, robust and cost-effective assay is very promising for routine high-throughput screening of histamine in complex food matrices.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Villum Fonden, Denmark, Project No. 13153.References
- I. San Mauro Martin, S. Brachero and E. Garicano Vilar, Allergol. Immunopathol., 2016, 44, 475–483 CrossRef CAS PubMed.
- R. Leurs, P. L. Chazot, F. C. Shenton, H. D. Lim and I. J. De Esch, Br. J. Pharmacol., 2009, 157, 14–23 CrossRef CAS PubMed.
- L. Maintz and N. Novak, Am. J. Clin. Nutr., 2014, 85, 832–833 Search PubMed.
- S. Wöhrl, W. Hemmer, M. Focke, K. Rappersberger and R. Jarisch, Allergy Asthma Proc., 2004, 25, 305–311 Search PubMed.
- T. Tang, K. Qian, T. Shi, F. Wang, J. Li, Y. Cao and Q. Hu, Food Control, 2011, 22, 1203–1208 CrossRef CAS.
- M. J. R. Nout, Food Res. Int., 1994, 27, 291–298 CrossRef CAS.
- C. A. L. De La Torre, Braz. J. Vet. Res. Anim. Sci., 2013, 50, 430 CrossRef.
- B. S. Hwang, J. T. Wang and Y. M. Choong, Food Chem., 2003, 82, 329–334 CrossRef CAS.
- T. Yoshitake, F. Ichinose, H. Yoshida, K. I. Todoroki, J. Kehr, O. Inoue, H. Nohta and M. Yamaguchi, Biomed. Chromatogr., 2003, 17, 509–516 CrossRef CAS PubMed.
- L. Y. Zhang and M. X. Sun, J. Chromatogr. A, 2004, 1040, 133–140 CrossRef CAS PubMed.
- P. Q. Leng, F. L. Zhao, B. C. Yin and B. C. Ye, Chem. Commun., 2015, 51, 8712–8714 RSC.
- A. Marcobal, M. C. Polo, P. J. Martín-Álvarez and M. V. Moreno-Arribas, Food Res. Int., 2005, 38, 387–394 CrossRef CAS.
- J. Wackerlig and R. Schirhagl, Anal. Chem., 2016, 88, 250–261 CrossRef CAS PubMed.
- L. Figueiredo, G. L. Erny, L. Santos and A. Alves, Talanta, 2016, 146, 754–765 CrossRef CAS PubMed.
- L. Uzun and A. P. F. Turner, Biosens. Bioelectron., 2016, 76, 131–144 CrossRef CAS PubMed.
- J. Ashley, M.-A. Shahbazi, K. Kant, V. A. Chidambara, A. Wolff, D. D. Bang and Y. Sun, Biosens. Bioelectron., 2017, 91, 606–615 CrossRef CAS PubMed.
- F. Gao, E. Grant and X. Lu, Anal. Chim. Acta, 2015, 901, 68–75 CrossRef CAS PubMed.
- M. Akhoundian, A. Rüter and S. Shinde, Sensors, 2017, 17, 645 CrossRef PubMed.
- F. A. Trikka, K. Yoshimatsu, L. Ye and D. A. Kyriakidis, Amino Acids, 2012, 43, 2113–2124 CrossRef CAS PubMed.
- N. Pérez-Moral and A. G. Mayes, Anal. Chim. Acta, 2004, 504, 15–21 CrossRef.
- C. He, Y. Long, J. Pan, K. Li and F. Liu, J. Biochem. Biophys. Methods, 2007, 70, 133–150 CrossRef CAS PubMed.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443 CrossRef CAS PubMed.
- J. Czulak, A. Guerreiro, K. Metran, F. Canfarotta, A. Goddard, R. H. Cowan, A. W. Trochimczuk and S. Piletsky, Nanoscale, 2016, 8, 11060–11066 RSC.
- Y. Garcia, K. S. Kempisty, E. Pereira, E. Piletska and S. Piletsky, Anal. Methods, 2017, 9, 4592–4598 RSC.
- S. Tang, F. Canfarotta, K. Smolinska-kempisty, E. Piletska, S. Piletsky and A. Guerreiroc, Anal. Methods, 2017, 9, 2853–2858 RSC.
- K. Smolinska-Kempisty, A. Guerreiro, F. Canfarotta, C. Cáceres, M. J. Whitcombe and S. Piletsky, Sci. Rep., 2016, 6, 37638 CrossRef CAS PubMed.
- C. C. Huang and W. L. Tseng, Analyst, 2009, 134, 1699–1705 RSC.
- Y. Hoshino, T. Kodama, Y. Okahata and K. J. Shea, J. Am. Chem. Soc., 2008, 130, 15242–15243 CrossRef CAS PubMed.
- E. V. Piletska, A. R. Guerreiro, M. J. Whitcombe and S. A. Piletsky, Macromolecules, 2009, 42, 4921–4928 CrossRef CAS.
- I. Basozabal, A. Guerreiro, A. Gomez-Caballero, M. Aranzazu Goicolea and R. J. Barrio, Biosens. Bioelectron., 2014, 58, 138–144 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |