
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-dispersed cellulose nanocrystals in hydrophobic polymers by in situ polymerization for synthesizing highly reinforced bio-nanocomposites†
Shiyu
Geng
 a,
Jiayuan
Wei
a,
Yvonne
Aitomäki
a,
Maxime
Noël
a and
Kristiina
Oksman
*ab
a,
Jiayuan
Wei
a,
Yvonne
Aitomäki
a,
Maxime
Noël
a and
Kristiina
Oksman
*ab
aDivision of Materials Science, Department of Engineering Sciences and Mathematics, Luleå University of Technology, SE-971 87, Luleå, Sweden. E-mail: kristiina.oksman@ltu.se
bFibre and Particle Engineering, University of Oulu, FI-900 14, Oulu, Finland
First published on 27th March 2018
Abstract
In nanocomposites, dispersing hydrophilic nanomaterials in a hydrophobic matrix using simple and environmentally friendly methods remains challenging. Herein, we report a method based on in situ polymerization to synthesize nanocomposites of well-dispersed cellulose nanocrystals (CNCs) and poly(vinyl acetate) (PVAc). We have also shown that by blending this PVAc/CNC nanocomposite with poly(lactic acid) (PLA), a good dispersion of the CNCs can be reached in PLA. The outstanding dispersion of CNCs in both PVAc and PLA/PVAc matrices was shown by different microscopy techniques and was further supported by the mechanical and rheological properties of the composites. The in situ PVAc/CNC nanocomposites exhibit enhanced mechanical properties compared to the materials produced by mechanical mixing, and a theoretical model based on the interphase effect and dispersion that reflects this behavior was developed. Comparison of the rheological and thermal behaviors of the mixed and in situ PVAc/CNC also confirmed the great improvement in the dispersion of nanocellulose in the latter. Furthermore, a synergistic effect was observed with only 0.1 wt% CNCs when the in situ PVAc/CNC was blended with PLA, as demonstrated by significant increases in elastic modulus, yield strength, elongation to break and glass transition temperature compared to the PLA/PVAc only material.
Introduction
Nanocomposites consisting of nano-size reinforcements and a polymer matrix have received tremendous interest during the past few decades. Various types of nanomaterials have been investigated as reinforcements, such as exfoliated clay,1–3 carbon nanotubes,4–6 graphene,7–9 and cellulose nanocrystals and nanofibers.10–14 Common to these widely different types of nanomaterials is the issue of the dispersion of the nanomaterials in the matrix. Poor dispersion – because of intermolecular interactions, like the van der Waals forces in the nanomaterials, and an incompatibility between the reinforcements and the matrix – limits the properties of nanocomposites.15,16 To address this, different methods have been studied to improve the dispersion, including silylation, esterification and grafting polymer chains to/from the nanomaterials to enhance the compatibility between the nanomaterials and hydrophobic polymers.17–21 The grafting methods can also provide a steric-stabilized effect that increases the stability of the nanomaterial suspensions.22 Some studies have also used the addition of small molecules together with nanomaterials as a plasticizer to avoid agglomeration.23,24 Improvements in the dispersion of nanomaterials arising from these methods have been found. However, different drawbacks accompany these methods, such as high energy consumption and relatively lengthy and toxic processing.25,26In addition to the modification methods mentioned above, a type of single-step method based on the in situ emulsion polymerization of producing nanocomposites reinforced by clay, carbon and metal oxide nanomaterials has been reported.27–32 It has been shown that laminate graphite oxide and clay can be penetrated by some monomers, such as vinyl acetate, methyl methacrylate and styrene, and be intercalated and expanded after the emulsion polymerization, resulting in exfoliated nanomaterial/polymer nanocomposites.27–30 Carbon nanotubes and metal oxide nanoparticles have also been investigated as reinforcements in nanocomposites produced via the in situ polymerization to achieve good dispersion of the nanomaterials.31,32 Cellulose nanocrystals (CNCs), a special nanomaterial with remarkable mechanical properties and biodegradability,33 are expected to be a promising reinforcement in environmentally friendly nanocomposites. However, the in situ emulsion polymerization has not been used to produce CNC reinforced nanocomposites until our previous study,34 although many earlier studies were conducted on preparing polymer/CNC composites by directly mixing the polymer latex with a CNC suspension followed by solvent casting or extrusion.35–39 Favier et al. firstly reported the method of mixing the CNC suspension and aqueous polymer latex followed by solvent casting to produce the CNC reinforced nanocomposites in the 1990s, and the thermal–mechanical properties of the nanocomposites were studied.35 De Rodriguez et al. prepared poly(vinyl acetate) (PVAc)/CNC composites using a similar method and investigated their water uptake and thermal behaviours.36 Gong et al. freeze-dried the mixed aqueous PVAc/CNC suspension as a master batch and then processed it in a twin-screw extruder to produce the composites, and the toughening effect of the CNCs on the PVAc was studied.37 Recently, Pracella et al. also prepared master batches by mixing the PVAc latex with the CNC suspension followed by drying, and then blended them with PLA in the Brabender Plasti-corder to produce the PLA/PVAc/CNC composites.38 They found that the dispersion of the CNCs in these composites was improved compared with that in the PLA/CNC composites, resulting in enhanced mechanical properties and thermal resistance.
Inspired by these previous studies, in our previous work, we developed a method based on in situ polymerization to prepare the CNC-reinforced PVAc nanocomposites in a simple and environmentally friendly manner, resulting in the improved dispersion of CNCs in the PVAc matrix compared with the direct mixing method.34 Basically, we in situ polymerized vinyl acetate in the presence of CNCs, and generated an in situ PVAc/CNC latex as the master batch to prepare nanocomposites by solvent casting. In the current study, this method is modified to generate in situ PVAc/CNC nanocomposites with a nanometer-scale homogeneous structure and a higher stability through the synthesis of a much smaller latex particle size of ca. 70 nm compared to the previous study where large latex particles were used (ca. 230 nm). Also, a larger solid content of the in situ latex (ca. 15 wt%) is generated for potential large-scale production compared to the previous study where the content was only ca. 3 wt%. As a comparison, nanocomposites of the same composition were prepared via direct mixing and solvent casting. The dispersion of the CNCs in both in situ and mixed nanocomposites is investigated using different microscopy techniques directly, and the results are further supported by the mechanical and rheological properties of the nanocomposites. We also demonstrate a modified Cox–Krenchel model with an additional term to include the effects of the interphase, which can be useful in theoretically estimating the mechanical properties of the nanocomposites with various reinforcement contents. The thermal behaviours of the materials that correspond with the mechanical properties are also presented.
Furthermore, polylactic acid (PLA) as one of the most promising polymer matrices in composites gained significant attention due to its high mechanical properties, high optical transparency and the ability of biodegradation.24,40 Producing PLA-based nanocomposites reinforced with well-dispersed cellulose nanomaterials is highly desired but still remains challenging.41 Therefore, blending the in situ PVAc/CNC with PLA is performed in this study to improve the dispersion of CNCs in the PLA matrix. Well-dispersed CNCs in PLA/PVAc/CNC nanocomposites are expected to influence the morphology and crystallinity of the PLA, and consequently improve the mechanical and thermal properties. In addition, the relationship between crack propagation and dispersion of the CNCs in the nanocomposites is discussed in this work.
Results and discussion
PVAc/CNC composite system
Fig. 1a illustrates the process of the in situ method used in this study, where the vinyl acetate monomer was emulsion polymerized in an aqueous CNC suspension consisting of nanocrystals with a diameter of 5.0 ± 1.5 nm and a length of 122.6 ± 53.3 nm (Fig. S1†), resulting in an in situ PVAc/CNC (weight ratio 80/20) latex with ca. 15 wt% of solid content. The viscosity of the obtained in situ latex (23.2 mPa s) is substantially higher than the native PVAc latex (1.4 mPa s) because of the presence of the CNCs (Fig. S2†). For reference, an equivalent suspension was prepared using a mechanical mixing method. This was done by directly stirring the CNC suspension and the PVAc latex together as shown in Fig. 1b.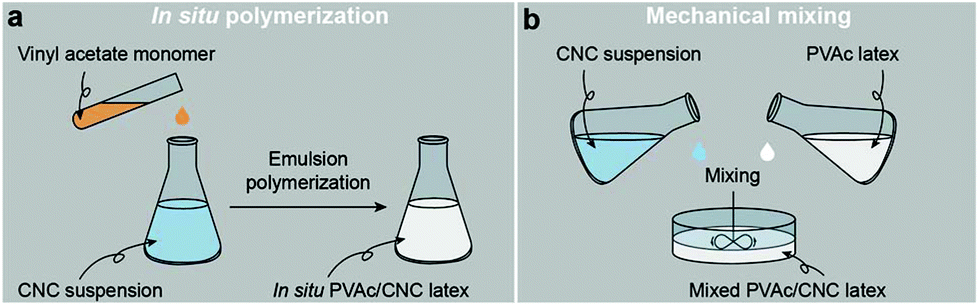 | ||
| Fig. 1 Illustrations of the process of (a) the in situ polymerization and (b) the mechanical mixing. | ||
In the liquid state, both the in situ and mixed PVAc/CNC latexes are electrostatically stable with zeta potentials significantly lower than −30 mV,42 as illustrated in Fig. 2-Ia and IIa, and the particle sizes of both are ca. 70 nm. However, substantial differences between the in situ and mixed PVAc/CNC samples are seen after drying. The unconsolidated in situ PVAc/CNC sample exhibits very good dispersion of both PVAc particles (∼70.5 nm, Fig. S3†) and CNCs (∼5 nm of diameter) after drying (Fig. 2-Ib and Ic), while substantially larger PVAc agglomerates (∼1 μm) and CNC aggregates (13.6 ± 6.2 nm of diameter and 324.8 ± 120.1 nm of length, Fig. S4 and S5†) are observed to have been generated in the mixed sample (Fig. 2-IIb and IIc). When the consolidated nanocomposite films were prepared using solvent casting from the latex, an investigation of the dispersion of the CNCs in the consolidated state was carried out using atomic force microscopy (AFM) phase images, and the results are shown in Fig. 2-Id and IId. Here, the phase angle shifts of the AFM probe oscillation are registered as brightness in the images and are correlated with the stiffness of the material regions.43 It can be seen that the stiffness of the cross-section of the in situ PVAc/CNC film is relatively homogeneous compared to that of the mixed film. It is thought that the localized areas of high stiffness in the mixed film (Fig. 2-IId) correspond to CNC aggregates. Overall, the microscopy results imply that the in situ method can increase the compatibility between the CNCs and PVAc, and consequently improve the dispersion of the CNCs in the PVAc matrix after the drying process, resulting in a more homogeneous material. The reason for the improved compatibility is thought to be that the CNCs in the in situ latex are coated with PVAc particles as confirmed in our previous study.34
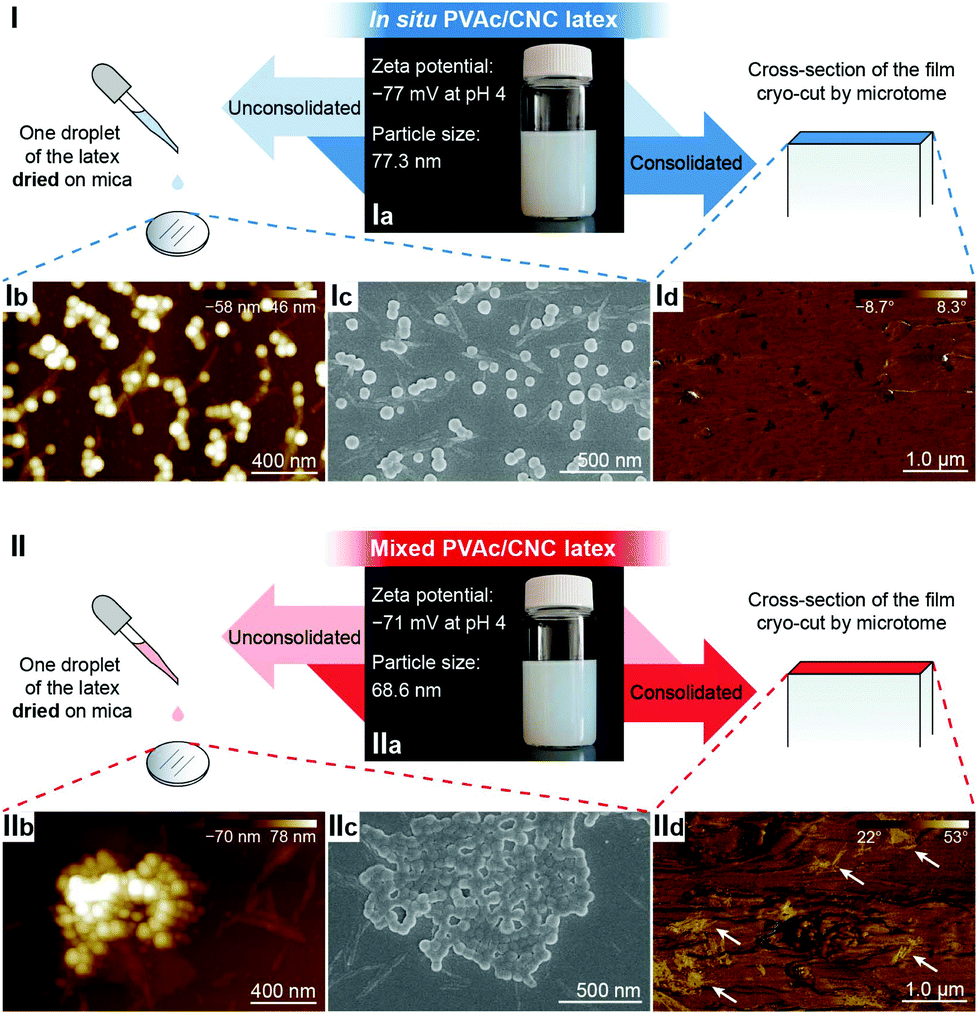 | ||
Fig. 2 Comparison of the dispersion of CNCs in the (I) in situ and (II) mixed PVAc/CNC samples in the latex, unconsolidated and consolidated states. (Ia, IIa) Illustrations of the in situ and mixed PVAc/CNC (weight ratio 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) latexes. (Ib, IIb) AFM height images and (Ic, IIc) SEM images of the unconsolidated in situ and mixed PVAc/CNC latexes (0.01 wt% of solid content) after drying. (Id, IId) AFM phase images of the cross-sections of the consolidated in situ and mixed PVAc/CNC films (weight ratio of PVAc to CNC is 80:20). Possible CNC aggregates are indicated by white arrows. :20) latexes. (Ib, IIb) AFM height images and (Ic, IIc) SEM images of the unconsolidated in situ and mixed PVAc/CNC latexes (0.01 wt% of solid content) after drying. (Id, IId) AFM phase images of the cross-sections of the consolidated in situ and mixed PVAc/CNC films (weight ratio of PVAc to CNC is 80:20). Possible CNC aggregates are indicated by white arrows. | ||
To further improve the interaction between PVAc and CNCs, the in situ latex was crosslinked by sodium tetraborate decahydrate (borax). The particle size of the in situ PVAc/CNC latex increases dramatically from 77.3 nm to 315 nm with the addition of borax (1.5 wt% of dry weight of the latex), but the zeta potential remains in the relatively stable region (Fig. S6†). The produced crosslinked latex is denoted as in situ XPVAc/CNC, in which “X” refers to crosslinking. To investigate the crosslinking effect of borax on both PVAc and CNCs, crosslinked PVAc (XPVAc), crosslinked CNCs (XCNC) and in situ XPVAc/CNC were characterized by Raman spectroscopy (Fig. 3), together with their references, PVAc, CNC and in situ PVAc/CNC, respectively. Fig. 3a shows that the intensities of Raman bands at 807 cm−1 and 932 cm−1 increased in XPVAc compared to that of native PVAc. The band at 932 cm−1 is assigned to the C–O stretching in polyvinyl alcohol, indicating that the degree of hydrolysis of PVAc was enhanced due to the crosslinking reaction. The increased intensity of the band at 807 cm−1 could be assigned to the newly formed borate ring including C–C bonds, as reported by Itou et al.,44 which implies that the crosslinks formed by tetra-coordinated borate ions were generated between the PVAc chains as shown in the chemical structure in Fig. 3a. Fig. 3b illustrates that the borate crosslinks can also be generated between CNCs, because the intensities of the Raman bands at 330 cm−1, 379 cm−1, 459 cm−1, and 519 cm−1, assigned to the heavy atom bending and stretching in cellulose,45 increased significantly in XCNC compared with native CNC. Furthermore, the bands at 664 cm−1, 768 cm−1 and 833 cm−1, correlated with different vibrational modes of the borate ring together with C–C bonds, also appeared in the spectrum of XCNC, which further supports the expectation. The Raman spectrum of the in situ XPVAc/CNC in Fig. 3c shows the evidence of crosslinking of both PVAc and CNC, including the appearance of the borate-ring band at 805 cm−1 and 833 cm−1, and the increased intensities of the bands at 330 cm−1 and 379 cm−1. Therefore, it is possible to form the borate crosslinks between the CNCs and PVAc chains as shown in the chemical structure in Fig. 3c, indicating that a stronger interaction between the CNCs and PVAc can be obtained in the crosslinked sample.
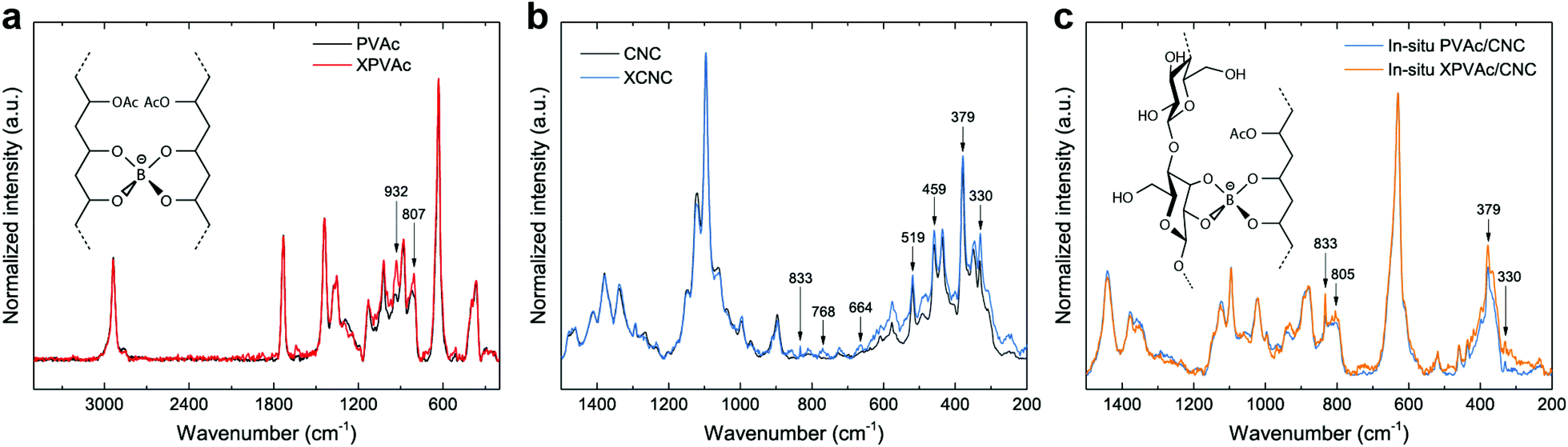 | ||
| Fig. 3 (a) Raman spectra of PVAc and XPVAc films in a spectral range of 200–3500 cm−1, and the chemical structure of borax-crosslinked PVAc chains. (b) Raman spectra of CNCs and XCNCs in a spectral range of 200–1500 cm−1. (c) Raman spectra of in situ PVAc/CNC and in situ XPVAc/CNC films (weight ratio of PVAc to CNCs is 80:20), and a possible chemical structure of borax-crosslinked PVAc-CNCs. | ||
Turning to the mechanical properties of the consolidated samples with various CNC amounts, the theoretical elastic modulus (Ec) of the in situ and mixed PVAc/CNC nanocomposites was obtained, based on a modified Cox–Krenchel model:46–48
| Ec = νfEfηoηl + (1 − νf)Em | (1) |
| Ec = νfEfηoηl + νiEi + (1 − νf − νi)Em | (2) |
000.52
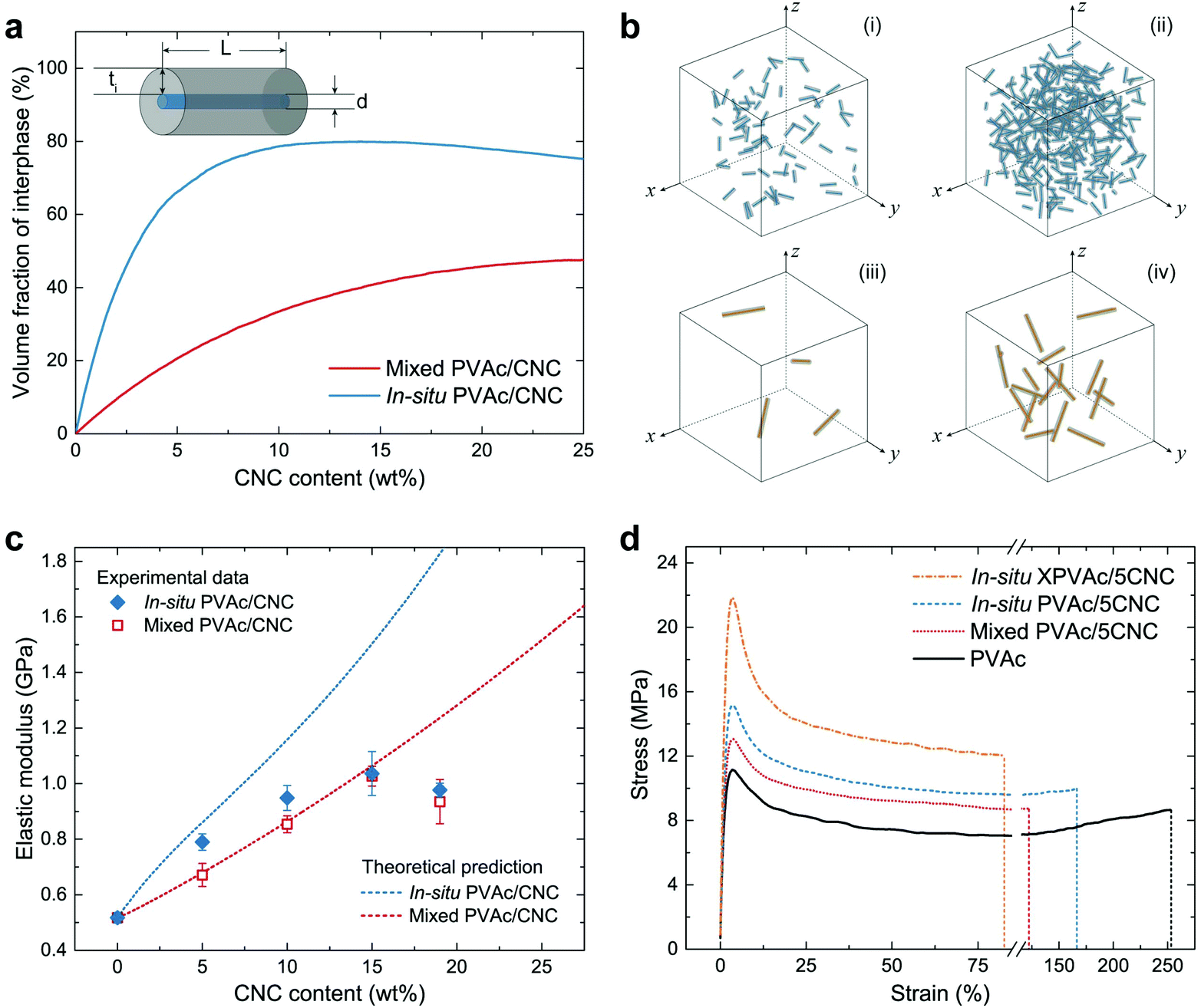 | ||
| Fig. 4 Volume fraction of the interphase and the mechanical properties of PVAc/CNC nanocomposites with various CNC contents. (a) Schematic of a CNC and its surrounding interphase, and the theoretical fraction of interphase volume (νi) in the in situ and mixed nanocomposites with a CNC content from 0 to 25 wt% simulated by MATLAB. (b) 3-Dimensional schematics of the in situ and mixed nanocomposites with the same CNC contents (i and iii: 0.02 wt%; ii and iv: 0.1 wt%). (c) Theoretical and experimental elastic moduli of the in situ and mixed nanocomposites. (d) Stress–strain curves of native PVAc and the nanocomposites with 5 wt% CNCs from tensile testing. | ||
The theoretical νi in the in situ and mixed nanocomposites were simulated by an in-house MATLAB script under the status of a uniformly random distribution of CNCs or CNC aggregates, and are shown in Fig. 4a as a function of the CNC content. The results indicate that the in situ nanocomposite possesses a much larger νi than the mixed sample does with the same CNC content, arising from the difference in the size of the CNCs and the CNC aggregates. The schematics of the in situ samples (Fig. 4b-i and ii) also clearly show more massive interphase area compared to those of the mixed samples (Fig. 4b-iii and iv) with the same CNC amounts. Fig. 4c demonstrates both theoretical and experimental Ec of the in situ and mixed nanocomposites. To obtain the experimental data from tensile testing, the in situ and mixed PVAc/CNC (weight ratio 80/20) latex as master batches were diluted by the native PVAc latex, and nanocomposite films with various CNC contents (from 5 wt% to 19 wt%) were generated after solvent casting and compression moulding. The compositions of all the samples are shown in Table S1.† As illustrated in Fig. 4c, the experimental Ec of the mixed samples fit the theoretical calculation very well, except the one with 19 wt% of CNCs, probably due to its higher degree of aggregation than that of the samples with lower CNC contents.53 The in situ PVAc/CNC nanocomposites with low CNC loadings (5 and 10 wt%) indicate improved Ec compared to the mixed samples, which is caused by a larger interphase volume because of the smaller nano-reinforcements, arising from a better dispersion. However, the improvements become negligible with increasing CNC loading, which indicates that the aggregation also occurred in the in situ samples. Furthermore, the experimental data of the in situ samples are lower than the theoretical prediction, implying that the CNCs are not individually dispersed in the in situ nanocomposites.
The experimental results from the tensile testing of all consolidated samples are presented in Table S2.† The in situ XPVAc/CNC nanocomposite films prepared using the same method show substantially higher Ec than the uncrosslinked samples do. This can be attributed to the crosslinks in the PVAc matrix that decrease the mobility of the PVAc chains,54 and the strengthened interaction between the CNCs and PVAc, as is shown in the Raman spectra (Fig. 3c). The stress–strain curves of the samples with 5 wt% CNCs (coded as PVAc/5CNC) illustrated in Fig. 4d also confirm the improved dispersion of the CNCs generated in the in situ PVAc/5CNC, as it has a strength and elongation to break higher than those of the mixed PVAc/5CNC. Moreover, the strength of the in situ XPVAc/5CNC increases even more while the elongation significantly reduces due to the crosslinking effect.
In addition to the mechanical properties, the rheological and thermal–mechanical behaviours of the nanocomposites produced from the different methods were investigated. The reference material, native PVAc melt, demonstrates a nonlinear viscoelastic behaviour in Fig. 5a, i.e., its shear viscosity is constant at low shear rates, and decreases at relatively high shear rates because of the breakdown of the entanglement of the PVAc chains.55,56 This behaviour is fully reversible, as shown by the ascending and descending flows. In contrast, the nanocomposite melts in Fig. 5b exhibit an additional thixotropic behaviour arising from the presence of the CNCs, resulting in significantly higher viscosities, and irreversible changes between ascending and descending flows caused by the rupture of the hydrogen bonds formed among the CNCs and PVAc chains.57 Moreover, during the ascending flow, the viscosity of the in situ PVAc/5CNC melt is significantly higher than that of the mixed PVAc/5CNC because of the better dispersion of CNCs,58 and that of the in situ XPVAc/5CNC melt increases further at high shear rates.
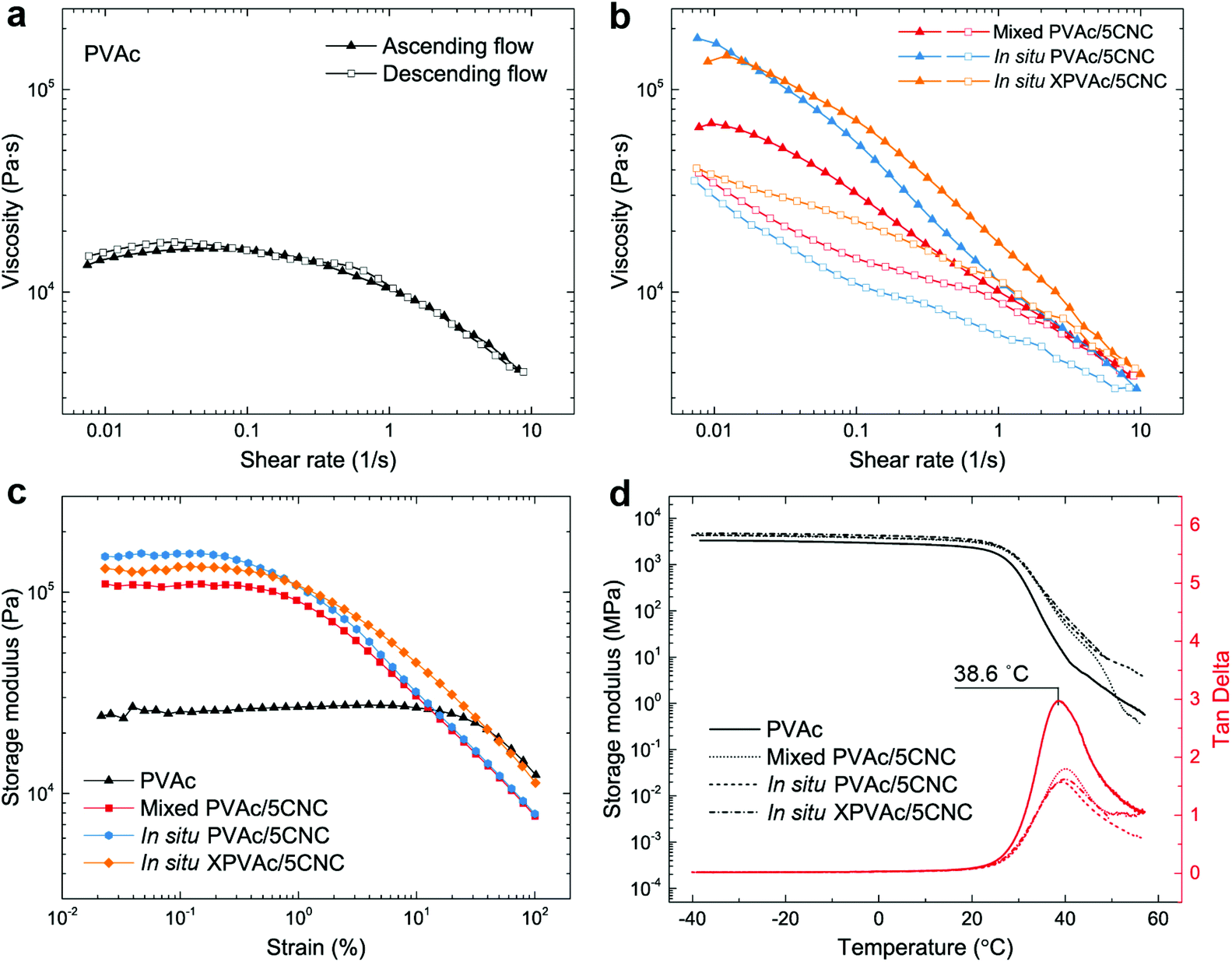 | ||
| Fig. 5 Rheological and thermal-mechanical behaviors of the native PVAc and the PVAc/CNC nanocomposites with 5 wt% of CNCs. (a, b) Steady shear viscosities of the native PVAc melt and the nanocomposite melts with an ascending flow (shear rate from 0.01 to 10 s−1) followed by a descending flow (shear rate from 10 to 0.01 s−1) at 120 °C. (c) Dynamic strain sweep tests of the melts at 120 °C. (d) Dynamic mechanical analyses of the nanocomposite films. | ||
Similarly, the plateau storage modulus (G′) of the nanocomposite melts is greater than that of the native PVAc melt in the dynamic strain sweeps (Fig. 5c), while the declines of G′ occur at lower strains because of the disruption of the hydrogen bonds. Notably, the in situ XPVAc/5CNC melt has a lower plateau G′ than the in situ PVAc/5CNC, which corresponds to the initial viscosities at low shear rates in Fig. 5b. The possible reasons are that the crosslinking performed in this study cannot generate a bulky 3D network,59 and a number of CNCs in the in situ XPVAc/5CNC are arrested by the crosslinking reaction, as indicated in the Raman spectra (Fig. 3c), resulting in their inability to form the hydrogen bonds.60 In the dynamic mechanical analysis (DMA) measurements (Fig. 5d), the storage moduli of the nanocomposite films are also higher than those of the native PVAc, however, the tan delta peak positions are nearly the same. This can be explained by the fact that the large free volume in PVAc caused by the large acetate side groups decreases the restrictive effects of CNCs on PVAc chains, especially at a relatively high temperature.61,62
PLA/PVAc/CNC composite system
To demonstrate the influence of the in situ method on the dispersion of the CNCs in PLA, a nanocomposite with 0.1 wt% of CNCs was prepared by blending the in situ PVAc/CNC with PLA (coded as in situ PLA/PVAc/0.1CNC, see the compositions in Table S1†). Compared to PLA/PVAc (Fig. 6a) and mixed PLA/PVAc/0.1CNC (Fig. 6b) films, the in situ sample demonstrates a significantly more homogeneous and ductile fracture surface after tensile testing (Fig. 6c). This unique morphology could be caused by the massive crazing effect, which indicates that the CNCs are homogeneously distributed and dispersed in the in situ sample and provide the bridging effect for the crazing.24,63 Network structures can be observed in both mixed and in situ samples, as shown in magnified SEM images (Fig. 6b and c), which are caused by the presence of CNCs. Small cavities on the fracture surface of the PLA/PVAc (magnified image in Fig. 6a) could be the result of a small amount of salt from the PVAc latex unexpectedly introduced into the sample during the sample preparation.64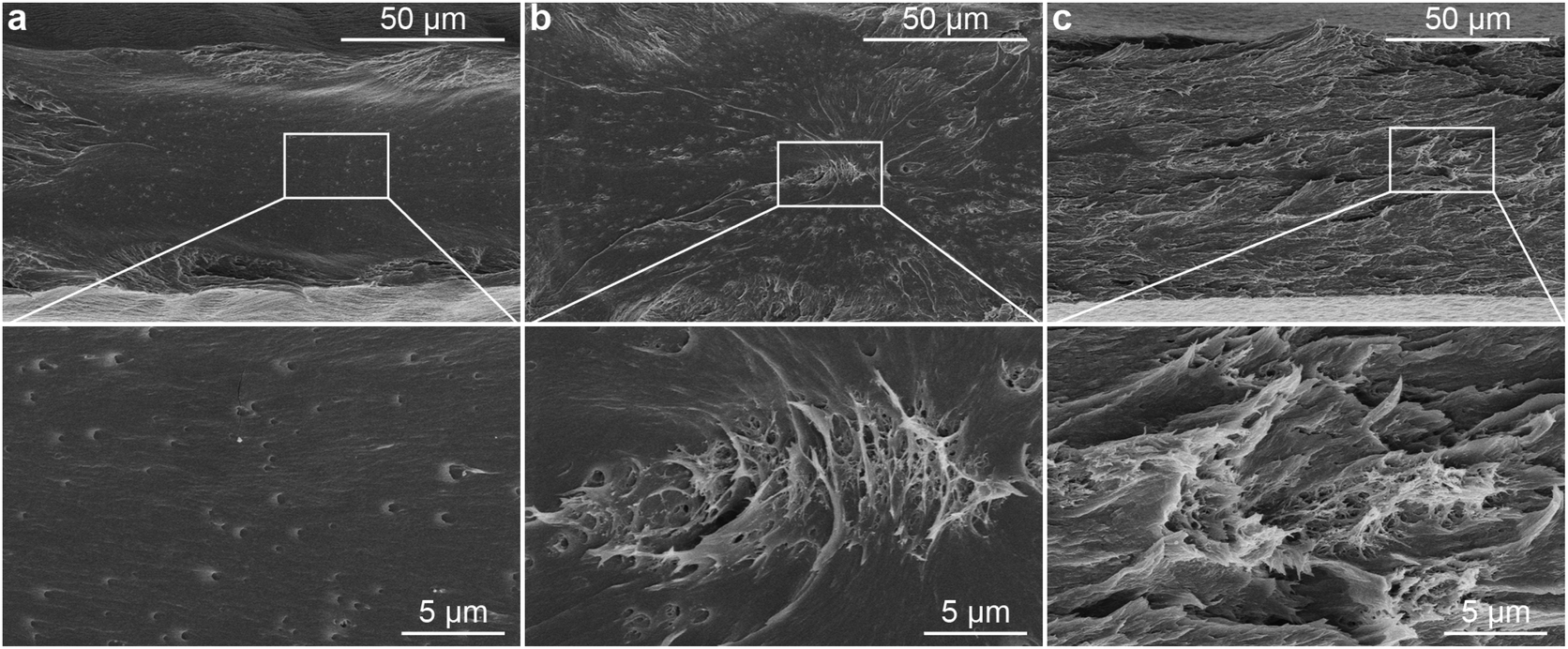 | ||
| Fig. 6 SEM images of the fracture surfaces of the (a) PLA/PVAc (weight ratio 90/10), (b) mixed PLA/PVAc/0.1CNC and (c) in situ PLA/PVAc/0.1CNC after tensile testing. | ||
The crystallization of PLA in the nanocomposites was investigated by polarized optical microscopy (POM, Fig. 7 and Fig. S7†), differential scanning calorimetry (DSC, Fig. 9a) and X-ray diffraction (XRD, Fig. S8–S10†). Fig. 7 illustrates that after 3 min isothermal annealing at 110 °C, a large number of fine PLA spherulites are present in the in situ PLA/PVAc/0.1CNC, while fewer of them appear in the mixed PLA/PVAc/0.1CNC and only few spherulites exist in the PLA/PVAc. This can be explained by the nucleation effect of the CNCs and shows furthermore that well-dispersed CNCs are more effective in causing nucleation.65 Similarly, after 10 min annealing, both the in situ and mixed samples are filled with spherulites; their size in the in situ sample is smaller than those in the mixed sample, and there are substantially fewer spherulites present in the PLA/PVAc. This is consistent with the relative crystallinity (Xc) of the samples calculated from the DSC and XRD results (Table S3†), in which the Xc of the in situ and mixed PLA/PVAc/0.1CNC according to DSC are 45.5% and 42.2% (43.3% and 35.8% according to XRD), respectively, and that of the PLA/PVAc is significantly lower (21.2% from DSC and 30.9% from XRD).
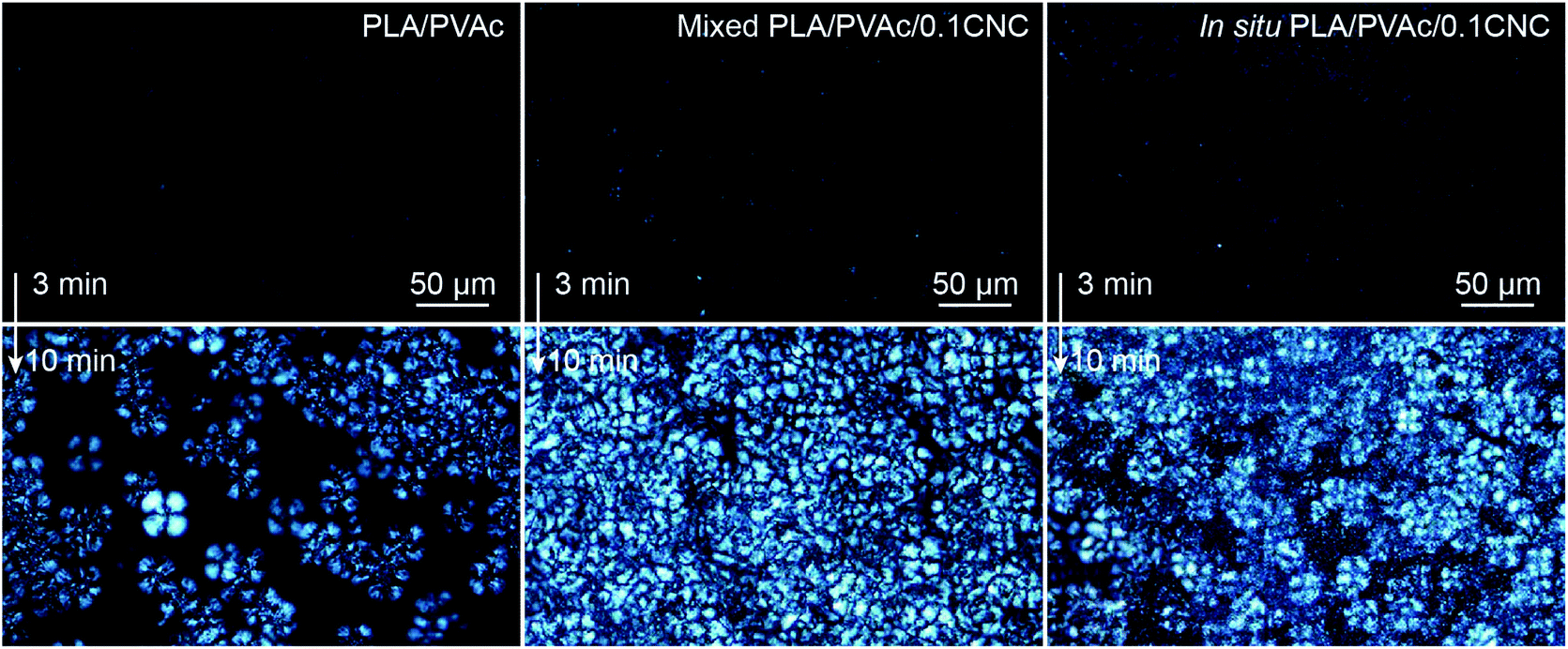 | ||
| Fig. 7 POM images of the PLA/PVAc, mixed PLA/PVAc/0.1CNC and in situ PLA/PVAc/0.1CNC after melting at 190 °C followed by isothermal annealing at 110 °C for 3 min (up) and 10 min (bottom). | ||
From the stress–strain curves obtained from tensile testing (Fig. 8a), appreciable increases in strength and elastic modulus are observed for both the in situ and mixed PLA/PVAc/0.1CNC relative to the PLA/PVAc, which is caused by a synergistic effect of the addition of 0.1 wt% of CNCs, i.e., the CNC reinforcing effect together with the increased crystallinity of the PLA caused by the CNC nucleation in the nanocomposites. The strength and elongation to break of the in situ sample exhibit prominent increases compared to those of the mixed sample, which is attributed to the enhanced dispersion of the CNCs and the massive crazing behaviour. The possible mechanism of the massive crazing is illustrated in the schematic in Fig. 8a. Instead of generating a macro-crack causing material failure as shown in the mixed sample, many micro-cracks were evolved in the in situ sample because of the bridging effect of the well-dispersed CNCs. This contributes to the extended elongation to break of the in situ sample, and is consistent with the microscopy result shown in Fig. 6c. Compared to other studies on nanocomposites with a low reinforcement content (≤1 wt%) as shown in Fig. 8b,15,24,65–75 our in situ PLA/PVAc/0.1CNC demonstrates remarkable improvements on both strength (23%) and toughness (153%) by adding only 0.1 wt% CNCs to the PLA/PVAc matrix (see Table S2†), indicating that the in situ method we developed is promising for producing low-cost and bio-based nanocomposites in the future.
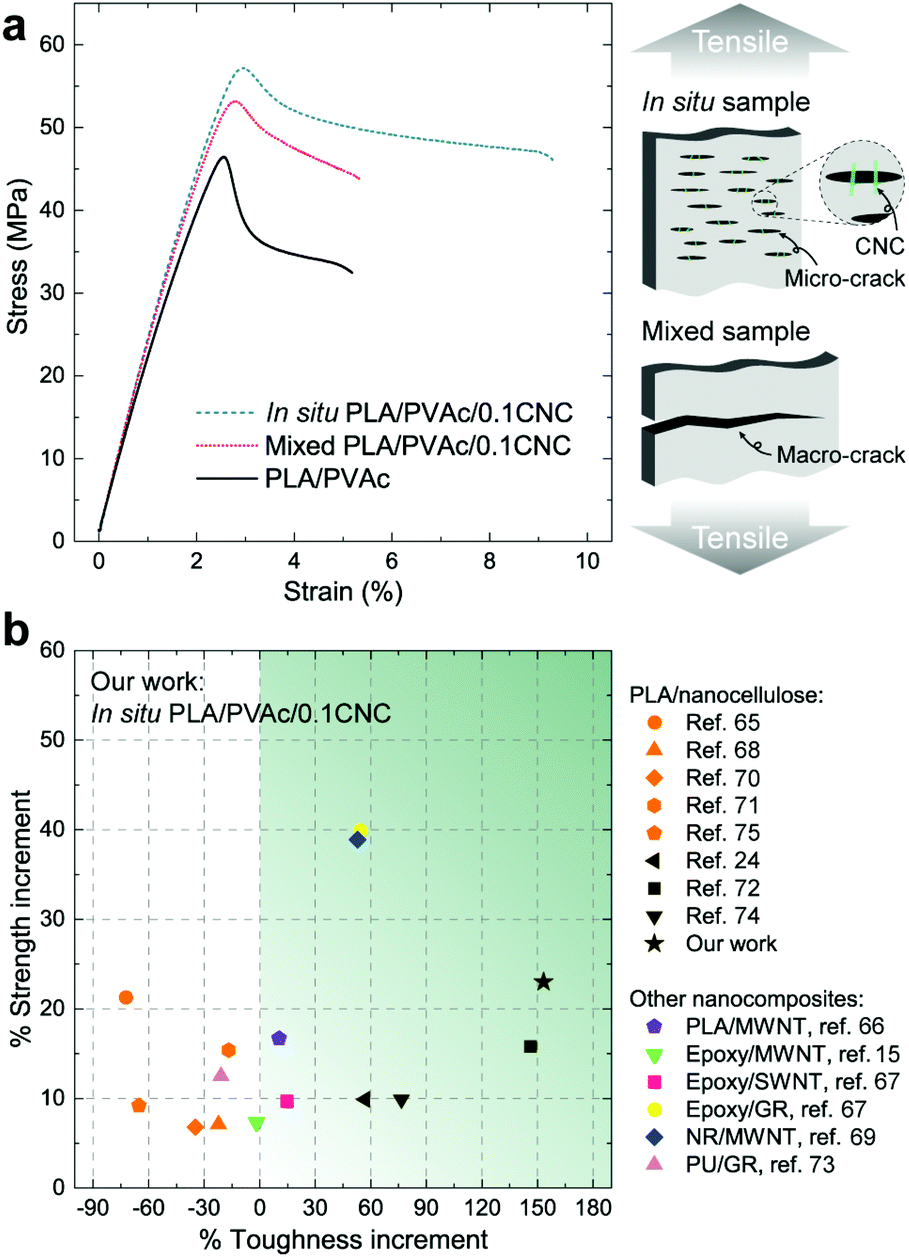 | ||
| Fig. 8 Mechanical properties of PLA/PVAc/CNC nanocomposites. (a) Stress–strain curves of the samples from tensile testing at 50% humidity, and a schematic indicating two types of cracks propagated in the in situ and mixed samples during the tensile testing. (b) Percent increments in the toughness and strength of the in situ PLA/PVAc/0.1CNC compared to other PLA/nanocellulose composites, and nanocomposites with multiwalled carbon nanotubes (MWNTs), single-walled carbon nanotubes (SWNTs) and graphene (GR) as reinforcements and PLA, epoxy, natural rubber (NR) and polyurethane (PU) as matrices. The reinforcement content in all the nanocomposites is ≤1 wt%. | ||
Good dispersion of CNCs also contributes to the improved thermal properties of the in situ PLA/PVAc/0.1CNC. The DMA results in Fig. 9a show that the tan delta peak, related to the glass transition temperature, of the in situ PLA/PVAc/0.1CNC shifts to a high temperature (57.2 °C) compared to those of the mixed sample (55.4 °C) and the PLA/PVAc (53.8 °C), which correspond to the glass transitions shown in the DSC thermograms in Fig. 9b. In addition, Fig. 9b indicates that a cold crystallization peak is observed in the PLA/PVAc rather than the in situ and mixed nanocomposites, and there are two melting peaks in all three samples at approximately 160 °C and 166 °C referring to the α′ and α crystalline phases of PLA, respectively.76
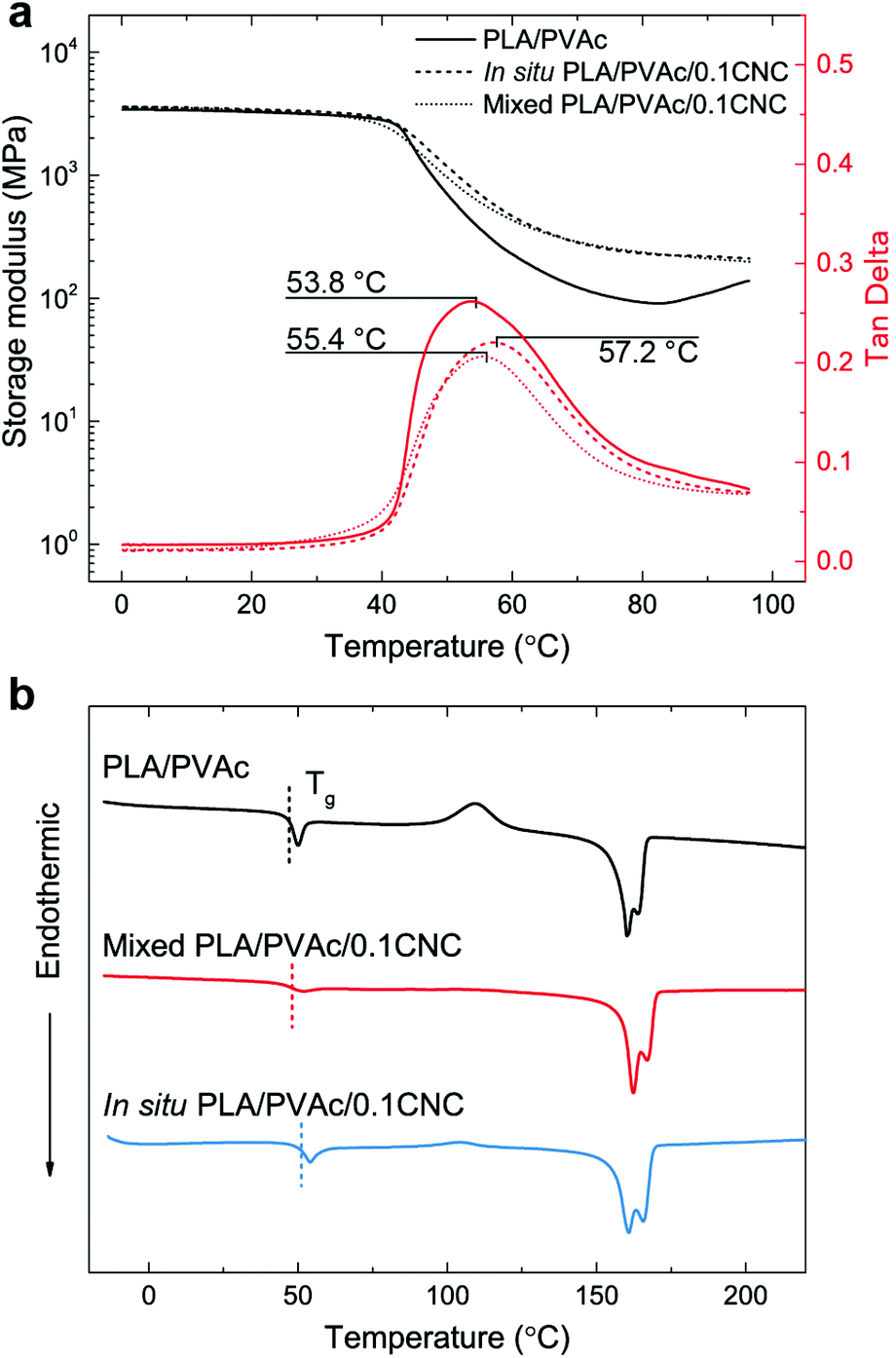 | ||
| Fig. 9 (a) Dynamic mechanical analyses and (b) DSC thermograms of the PLA/PVAc, mixed PLA/PVAc/0.1CNC and in situ PLA/PVAc/0.1CNC. | ||
Conclusions
In conclusion, we investigated the effects of the in situ polymerization on the dispersion of CNCs in hydrophobic PVAc and PLA/PVAc matrices and the properties of the prepared nanocomposites. The results for the in situ PVAc/CNC nanocomposites show that a superior dispersion of CNCs can be achieved using the in situ method compared to the traditional mechanical mixing method, therefore imparting increased strength, elastic modulus, and elongation to break at relatively low CNC loadings (5 and 10 wt%). Nevertheless, their elastic moduli are still lower than the theoretical prediction based on the interphase effect, revealing that the CNCs are not individually dispersed in the PVAc matrix. The crosslinking reaction improves the interaction between the CNCs and PVAc after the in situ polymerization, further enhancing the mechanical properties of the nanocomposites. Turning to the rheological and thermal properties, the in situ PVAc/CNC exhibits a viscosity and storage modulus that are higher than those of the mechanically mixed sample, which correlates with their mechanical properties. In the in situ PLA/PVAc/CNC nanocomposite, an excellent dispersion of CNCs in PLA can be achieved due to the dispersive action of the in situ polymerized PVAc. This leads to high crystallinity in PLA and increased mechanical and thermal properties compared to those of the mixed sample. We can conclude that the reported in situ method is a convenient, designable and generalizable process, which could be used for dispersing hydrophilic nanomaterials in different types of hydrophobic polymer matrices. This method has the potential to be widely used to produce biodegradable nanocomposites with remarkable dispersion and properties.Experimental
Materials
An aqueous cellulose nanocrystal (CNC) suspension (1.02 wt% of sulphur on dry CNC) was kindly supplied by the USDA Forest Service, Forest Products Laboratory, Madison, USA. Vinyl acetate monomer (99%, Alfa Aesar), docusate sodium salt (≥96%, Sigma-Aldrich), potassium peroxodisulfate (KPS) (VWR International), sodium bicarbonate (≥99.7%, Sigma-Aldrich), sodium tetraborate decahydrate (borax) (≥99.5%, Sigma-Aldrich), sodium hydroxide (pure pellets, Merck KGaA), glyceryl triacetate (GTA) (≥99%, Sigma-Aldrich) and N,N-dimethylformamide (DMF) (anhydrous, 99.8%, Sigma-Aldrich) were used without any further purification. Polylactic acid (Ingeo 4032D PLA) was purchased from NatureWorks, Nebraska, USA.Sample preparation
A poly(vinyl acetate) (PVAc) aqueous latex was synthesized by emulsion polymerization, and the method has been described in detail in our previous work.59 Briefly, 2.25 g of docusate sodium salt, 0.135 g of sodium bicarbonate and 255 g of distilled water were added to a three-necked flask with a water-cooled condenser, and the solution was stirred continuously and heated to 80 °C. After this, 0.225 g of KPS and 2.25 g of vinyl acetate monomer were added, and the pre-reaction lasted for 20 min. 42.75 g of vinyl acetate monomer was then fed dropwise in the flask over 3 h, followed by a 30 min post-reaction at 80 °C. Finally, a PVAc latex with approximately 15 wt% of solid content was obtained. To synthesize the in situ PVAc/CNC latex, 115.6 g of CNC suspension (10.3 wt% of solid content) was dispersed in 220.8 g of distilled water by stirring followed by 5 min sonication, and thereafter transferred to the flask for in situ emulsion polymerization. The polymerization procedure and the amount of the ingredients were the same as those of the PVAc latex mentioned above. In the final in situ PVAc/CNC latex, the solid content was approximately 15 wt%, and the ratio of PVAc to CNC was 80:20 in weight. The mixed PVAc/CNC reference latex was produced by the mechanical mixing of a CNC suspension and the PVAc latex for 2 h at room temperature, and the weight ratio of PVAc to CNC was also kept as 80:20.
To prepare consolidated PVAc/CNC composite films for this study, the in situ or mixed PVAc/CNC latex was diluted by the PVAc latex to reach the required CNC concentration for each sample. GTA (weight ratio of GTA to polymer is 5:95 in the dry state) was added in as a plasticizer, and the suspension was kept stirring for 1 h. After that, the suspension was poured into a Teflon Petri dish (12 cm in diameter) and dried in an oven with a fan at 40 °C for 30 h. The obtained cast film was then peeled off and compression moulded using a laboratory press (LabEcon 300, Fontijne Grotnes, Vlaardingen, The Netherlands) under a pressure of 1.1 MPa at 150 °C for 1 min with 2 min of preheating.
In situ XPVAc/CNC films were prepared using the same procedure mentioned above but with an additional crosslinking reaction. Briefly, a saturated borax solution (4.71 wt% at 20 °C) was added dropwise into the in situ PVAc/CNC latex and the pH of the suspension was tuned to 11 using a sodium hydroxide solution (0.1 M). Later, the suspension was heated to 80 °C and kept stirring for 1 h. The final in situ XPVAc/CNC films were obtained after following the steps including adding GTA, solvent casting and hot pressing. The concentration of borax in dried composites was kept as 1.5 wt%. The XCNC film was prepared via the same procedure but using a CNC suspension (5 wt%) instead of the in situ latex.
To prepare PLA/PVAc/CNC films, the in situ or mixed PVAc/CNC latex and PLA were dissolved in DMF at 80 °C, and then the solution was poured into a Teflon Petri dish and dried at 80 °C in an oven with a fan for 24 h. The obtained films were further compression moulded using a pressure of 2.2 MPa at 190 °C for 1 min with 2 min of pre-heating and then annealed at 110 °C for 10 min. The compositions of all the samples in this study are shown in Table S1.† The native PVAc, XPVAc and PLA/PVAc films without CNCs were prepared using the same procedures described above but using the PVAc latex instead of the in situ or mixed PVAc/CNC latex.
Characterization
Atomic force microscopy (AFM) was used to characterize the topography of the in situ and mixed PVAc/CNC latexes and the stiffness of the cross-sections of the in situ and mixed PVAc/19CNC films. The samples were scanned using the AFM tapping mode using a Veeco MultiMode scanning probe (Santa Barbara, USA) with Bruker TESPA tips (Camarillo, USA). To prepare the unconsolidated samples, the in situ or mixed PVAc/CNC latex was diluted to 0.01 wt% of solid content, and then one droplet of the suspension was deposited on a piece of freshly cleaved mica stuck on an atomic force microscope holder. Lastly, the sample was dried at room temperature overnight, and then the AFM height images were captured. To investigate the cross-sections of the consolidated films, the casted films were cryo-cut using a Leica EM UC7 microtome (Wetzlar, Germany) at −60 °C and then analysed by AFM phase images.Scanning electron microscopy (SEM) was used to investigate the morphology of the in situ and mixed PVAc/CNC latexes and the fracture surfaces of the PLA/PVAc and PLA/PVAc/CNC films after tensile testing. An FEI Magellan 400 XHR-SEM (Hillsboro, USA) was used and the secondary electron images were captured. The in situ and mixed PVAc/CNC latex samples for SEM were prepared in the same manner as for AFM and then coated with tungsten using a Bal-Tec MED 020 coating system. The fractured film generated from tensile testing was held vertically by two pieces of copper tape on a scanning electron microscope holder and coated with tungsten, and then the fracture surface was investigated.
The zeta potentials and particle sizes of the PVAc, in situ PVAc/CNC, mixed PVAc/CNC and in situ XPVAc/CNC latexes (the weight ratio of PVAc to CNC in each latex is 80:20 except the PVAc latex) were measured by using a Zeta sizer nano ZS (Malvern, UK). To measure the zeta potential of each sample, the latex was diluted to 0.2 wt% of solid content, and then the pH was tuned to 4 (the original pH of the PVAc latex after emulsion polymerization, for the PVAc, in situ and mixed PVAc/CNC latexes) and 11 (the pH during the crosslinking reaction, for the in situ XPVAc latex). The particle size measurements were performed after the zeta potential measurements. The viscosities of the PVAc and in situ PVAc/CNC latexes with 15 wt% of solid content were tested by using a sine-wave Vibro viscometer SV-10/SV-100 (A&D Company, Japan) at room temperature.
Raman spectroscopy measurements were executed on a Bruker Senterra Dispersive Raman microscope, Bruker Optik GmbH (Germany) equipped with an Olympus 10× objective. Spectra were obtained with a 785 nm laser at a power of 50 mW with a resolution of 3–5 cm−1. To assure the reproducibility of the measurements, and thus present typical spectra, several spots of each sample were probed.
The mechanical properties of the samples were measured by tensile testing using a universal testing machine (Shimadzu AG-X, Japan) with a SLBL-1kN load cell. At least 5 specimens of each sample were tested, and the average values of the elastic modulus, ultimate strength and elongation to break were calculated. For the PVAc and PVAc/CNC films, a 20 mm gauge length and a 5 mm min−1 crosshead speed were used, and the specimens were tested at 25% humidity and 20 °C, as controlled by using a thermostatic chamber (THC1-200SP, Shimadzu, Japan). For the PLA/PVAc and PLA/PVAc/CNC films, the specimens were kept in a chamber with 50 ± 5% humidity for 48 h and then tested with a 30 mm gauge length and a 5 mm min−1 crosshead speed.
Rheology measurements for the PVAc and PVAc/CNC melts were performed on a Discovery Hybrid rheometer (DHR-1, TA Instruments, USA) with a parallel plate configuration (plate diameter 25 mm). Before each measurement, the sample was heated to 120 °C in an environmental test chamber (ETC, TA Instruments, USA) and soaked for 360 s. The shear viscosity of the sample was determined by the steady state sweep at 120 °C with an ascending flow (shear rate from 0.01 to 10 s−1) and a descending flow (shear rate from 10 to 0.01 s−1). The dynamic shear properties (the storage modulus (G′) and the loss modulus (G′′)) were determined by the dynamic strain sweep at 120 °C with a strain range from 0.01% to 100%.
The thermo-mechanical properties of the samples were characterized by dynamic mechanical analysis (DMA) using a Q800 analyser (TA Instruments, New Jersey, USA) with a tension clamp configuration. For the PVAc and PVAc/CNC films, the temperature ramps were executed in a range from −40 to 60 °C with a 1 °C min−1 scanning rate and at a frequency of 1 Hz. For the PLA/PVAc and PLA/PVAc/CNC films, the temperature range was from 0 to 100 °C and the scanning rate was 3 °C min−1.
Differential scanning calorimetry (DSC) was performed on a Mettler Toledo DSC 821e with ME-27331 aluminium crucibles under a nitrogen atmosphere. A blank curve was firstly run to remove the background noise, and then the samples were measured from −20 to 220 °C with a scanning rate of 10 °C min−1. The crystallinity (Xc) of the samples was calculated from the DSC thermograms using the equation:
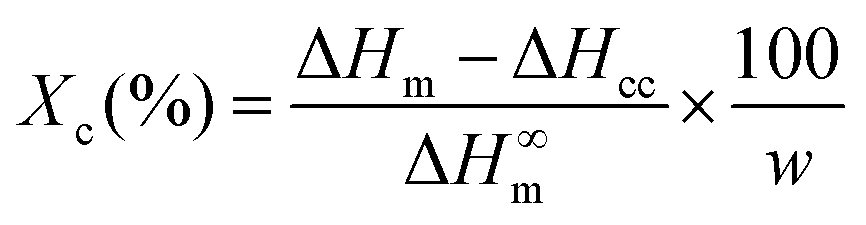 | (3) |
To investigate the crystallization morphology of PLA in the PLA/PVAc and PLA/PVAc/CNC samples, polarized optical microscopy (POM) was executed on a Nikon Eclipse LV100 POL polarizing optical microscope (Kanagawa, Japan) with a Linkam TH600 heating stage (Tadworth, UK). The sample was heated to 190 °C at first and then annealed at 110 °C for 10 min. The images were captured under crossed polarized light at 0, 3, 5, 7 and 10 min after the annealing started.
X-ray diffraction (XRD) measurements were performed on an Empyrean X-ray diffractometer (PANalytical, UK). The samples were scanned using CuKα radiation (λ = 1.5418 Å) at room temperature in a 2θ angular range from 5 to 40° with a scan speed of 0.01° s−1. The obtained XRD scattering curves were then analysed by using the software PANalytical X'Pert HighScore Plus. The Xc of the samples was calculated using the XRD scattering curves (see Fig. S8–S10†) from the ratio of the integrals of the crystalline peaks to the total integral under the scattering curve.78
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Md. Minhaz-Ul Haque and Prof. Aji P. Mathew for valuable discussions, Dr Peng Liu for technical support of zeta potential and particle size measurements, M.Sc. Hanzhu Zhang for technical support of XRD, and financial support from Knut och Alice Wallenberg Stiftelsen (WWSC), SNS Nordic Forest Research (WOOD-PRO), Bio4Energy and Kempe Foundation.Notes and references
- E. P. Giannelis, Adv. Mater., 1996, 8, 29–35 CrossRef CAS.
- S. S. Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef CAS.
- X. Su, S. Mahalingam, M. Edirisinghe and B. Chen, ACS Appl. Mater. Interfaces, 2017, 9(27), 22223–22234 CAS.
- M. S. Shaffer and A. H. Windle, Adv. Mater., 1999, 11, 937–941 CrossRef CAS.
- M. Moniruzzaman and K. I. Winey, Macromolecules, 2006, 39, 5194–5205 CrossRef CAS.
- J.-W. Kim, E. J. Siochi, J. Carpena-Núñez, K. E. Wise, J. W. Connell, Y. Lin and R. A. Wincheski, ACS Appl. Mater. Interfaces, 2013, 5, 8597–8606 CAS.
- T. Ramanathan, A. Abdala, S. Stankovich, D. Dikin, M. Herrera-Alonso, R. Piner, D. Adamson, H. Schniepp, X. Chen and R. Ruoff, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS PubMed.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- A. Asthana, T. Maitra, R. Büchel, M. K. Tiwari and D. Poulikakos, ACS Appl. Mater. Interfaces, 2014, 6, 8859–8867 CAS.
- G. Chauve, L. Heux, R. Arouini and K. Mazeau, Biomacromolecules, 2005, 6, 2025–2031 CrossRef CAS PubMed.
- K. Oksman, A. Mathew, D. Bondeson and I. Kvien, Compos. Sci. Technol., 2006, 66, 2776–2784 CrossRef CAS.
- K.-Y. Lee, Y. Aitomäki, L. A. Berglund, K. Oksman and A. Bismarck, Compos. Sci. Technol., 2014, 105, 15–27 CrossRef CAS.
- K. Yao, S. Huang, H. Tang, Y. Xu, G. Buntkowsky, L. A. Berglund and Q. Zhou, ACS Appl. Mater. Interfaces, 2017, 9(23), 20169–20178 CAS.
- Y. Xue, Z. Mou and H. Xiao, Nanoscale, 2017, 9, 14758–14781 RSC.
- Y. S. Song and J. R. Youn, Carbon, 2005, 43, 1378–1385 CrossRef CAS.
- P. M. Ajayan, L. S. Schadler and P. V. Braun, Nanocomposite science and technology, John Wiley & Sons, 2006 Search PubMed.
- A. Akelah and A. Moet, J. Mater. Sci., 1996, 31, 3589–3596 CrossRef CAS.
- C. Goussé, H. Chanzy, G. Excoffier, L. Soubeyrand and E. Fleury, Polymer, 2002, 43, 2645–2651 CrossRef.
- X. Fan, C. Xia and R. C. Advincula, Langmuir, 2003, 19, 4381–4389 CrossRef CAS.
- B. Braun and J. R. Dorgan, Biomacromolecules, 2008, 10, 334–341 CrossRef PubMed.
- F. Ansari, M. Salajková, Q. Zhou and L. A. Berglund, Biomacromolecules, 2015, 16, 3916–3924 CrossRef CAS PubMed.
- J. Araki, M. Wada and S. Kuga, Langmuir, 2001, 17, 21–27 CrossRef CAS.
- N. Ljungberg, C. Bonini, F. Bortolussi, C. Boisson, L. Heux and J.-Y. Cavaillé, Biomacromolecules, 2005, 6, 2732–2739 CrossRef CAS PubMed.
- N. Herrera, A. P. Mathew and K. Oksman, Compos. Sci. Technol., 2015, 106, 149–155 CrossRef CAS.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- S. M. Hahn, F. J. Sullivan, A. M. DeLuca, J. D. Bacher, J. Liebmann, M. C. Krishna, D. Coffin and J. B. Mitchell, Free Radical Biol. Med., 1999, 27, 529–535 CrossRef CAS PubMed.
- D. C. Lee and L. W. Jang, J. Appl. Polym. Sci., 1996, 61, 1117–1122 CrossRef CAS.
- D. C. Lee and L. W. Jang, J. Appl. Polym. Sci., 1998, 68, 1997–2005 CrossRef CAS.
- M. Okamoto, S. Morita, H. Taguchi, Y. H. Kim, T. Kotaka and H. Tateyama, Polymer, 2000, 41, 3887–3890 CrossRef CAS.
- P. Liu, K. Gong, P. Xiao and M. Xiao, J. Mater. Chem., 2000, 10, 933–935 RSC.
- H. Kong, C. Gao and D. Yan, J. Am. Chem. Soc., 2004, 126, 412–413 CrossRef CAS PubMed.
- M. M. Demir, M. Memesa, P. Castignolles and G. Wegner, Macromol. Rapid Commun., 2006, 27, 763–770 CrossRef CAS.
- S. Iwamoto, W. Kai, A. Isogai and T. Iwata, Biomacromolecules, 2009, 10, 2571–2576 CrossRef CAS PubMed.
- S. Geng, M. M.-U. Haque and K. Oksman, Compos. Sci. Technol., 2016, 126, 35–42 CrossRef CAS.
- V. Favier, H. Chanzy and J. Cavaille, Macromolecules, 1995, 28, 6365–6367 CrossRef CAS.
- N. L. G. De Rodriguez, W. Thielemans and A. Dufresne, Cellulose, 2006, 13, 261–270 CrossRef.
- G. Gong, A. P. Mathew and K. Oksman, Polym. Compos., 2011, 32, 1492–1498 CrossRef CAS.
- M. Pracella, M. M.-U. Haque and D. Puglia, Polymer, 2014, 55, 3720–3728 CrossRef CAS.
- M. M.-U. Haque, D. Puglia, E. Fortunati and M. Pracella, React. Funct. Polym., 2017, 110, 1–9 CrossRef CAS.
- K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef PubMed.
- J.-M. Raquez, Y. Habibi, M. Murariu and P. Dubois, Prog. Polym. Sci., 2013, 38, 1504–1542 CrossRef CAS.
- C. Freitas and R. H. Müller, Int. J. Pharm., 1998, 168, 221–229 CrossRef CAS.
- S. Magonov, V. Elings and M.-H. Whangbo, Surf. Sci., 1997, 375, L385–L391 CrossRef CAS.
- T. Itou, H. Kitai, A. Shimazu, T. Miyazaki and K. Tashiro, J. Phys. Chem. B, 2014, 118, 6032–6037 CrossRef CAS PubMed.
- J. H. Wiley and R. H. Atalla, Carbohydr. Res., 1987, 160, 113–129 CrossRef CAS.
- H. Cox, Br. J. Appl. Phys., 1952, 3, 72 CrossRef.
- H. Krenchel, Fibre reinforcement; theoretical and practical investigations of the elasticity and strength of fibre-reinforced materials, 1964 Search PubMed.
- J. Andersons, E. Spārniņš and R. Joffe, Polym. Compos., 2006, 27, 221–229 CrossRef CAS.
- A. Bansal, H. Yang, C. Li, K. Cho, B. C. Benicewicz, S. K. Kumar and L. S. Schadler, Nat. Mater., 2005, 4, 693–698 CrossRef CAS PubMed.
- H. Liu and L. C. Brinson, Compos. Sci. Technol., 2008, 68, 1502–1512 CrossRef CAS.
- T. Saito, R. Kuramae, J. Wohlert, L. A. Berglund and A. Isogai, Biomacromolecules, 2012, 14, 248–253 CrossRef PubMed.
- M. Matsumoto and Y. Ohyanagi, J. Polym. Sci., 1960, 46, 441–454 CrossRef CAS.
- G. Keledi, J. Hári and B. Pukánszky, Nanoscale, 2012, 4, 1919–1938 RSC.
- L. E. Nielsen, J. Macromol. Sci., Part C, 1969, 3(1), 69–103 CrossRef CAS.
- W. W. Graessley, in The Entanglement Concept in Polymer Rheology, Springer, 1974, pp. 1–179 Search PubMed.
- L. Utracki and M. Kanial, Polym. Eng. Sci., 1982, 22, 96–114 Search PubMed.
- J. Mewis and N. J. Wagner, Adv. Colloid Interface Sci., 2009, 147, 214–227 CrossRef PubMed.
- J. Vermant, S. Ceccia, M. Dolgovskij, P. Maffettone and C. Macosko, J. Rheol., 2007, 51, 429–450 CrossRef CAS.
- S. Geng, F. U. Shah, P. Liu, O. N. Antzutkin and K. Oksman, RSC Adv., 2017, 7, 7483–7491 RSC.
- A. Durmus, A. Kasgoz and C. W. Macosko, Polymer, 2007, 48, 4492–4502 CrossRef CAS.
- P. Meares, Trans. Faraday Soc., 1957, 53, 31–40 RSC.
- A. Kovacs, R. A. Stratton and J. D. Ferry, J. Phys. Chem., 1963, 67, 152–161 CrossRef CAS.
- L. Zhang, Y. Li, H. Wang, Y. Qiao, J. Chen and S. Cao, Chem. Eng. J., 2015, 264, 538–546 CrossRef CAS.
- G. W. Barnett and T. T. Chen, Small particle size latex based on vinyl acetate polymers, US Pat., 4812510, 1989 Search PubMed.
- A. Pei, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2010, 70, 815–821 CrossRef CAS.
- C.-S. Wu and H.-T. Liao, Polymer, 2007, 48, 4449–4458 CrossRef CAS.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef CAS PubMed.
- M. Jonoobi, J. Harun, A. P. Mathew and K. Oksman, Compos. Sci. Technol., 2010, 70, 1742–1747 CrossRef CAS.
- F. Deng, M. Ito, T. Noguchi, L. Wang, H. Ueki, K.-I. Niihara, Y. A. Kim, M. Endo and Q.-S. Zheng, ACS Nano, 2011, 5, 3858–3866 CrossRef CAS PubMed.
- N. Lin, J. Huang, P. R. Chang, J. Feng and J. Yu, Carbohydr. Polym., 2011, 83, 1834–1842 CrossRef CAS.
- L. C. Tomé, R. J. Pinto, E. Trovatti, C. S. Freire, A. J. Silvestre, C. P. Neto and A. Gandini, Green Chem., 2011, 13, 419–427 RSC.
- E. Fortunati, I. Armentano, Q. Zhou, D. Puglia, A. Terenzi, L. A. Berglund and J. Kenny, Polym. Degrad. Stab., 2012, 97, 2027–2036 CrossRef CAS.
- C. Xiang, P. J. Cox, A. Kukovecz, B. Genorio, D. P. Hashim, Z. Yan, Z. Peng, C.-C. Hwang, G. Ruan and E. L. Samuel, ACS Nano, 2013, 7, 10380–10386 CrossRef CAS PubMed.
- A. Abdulkhani, J. Hosseinzadeh, A. Ashori, S. Dadashi and Z. Takzare, Polym. Test., 2014, 35, 73–79 CrossRef CAS.
- N. Herrera, A. M. Salaberria, A. P. Mathew and K. Oksman, Composites, Part A, 2016, 83, 89–97 CrossRef CAS.
- T. Tabi, I. Sajó, F. Szabó, A. Luyt and J. Kovács, eXPRESS Polym. Lett., 2010, 4, 659–668 CrossRef CAS.
- E. Fischer, H. J. Sterzel and G. Wegner, Kolloid Z. Z. Polym., 1973, 251, 980–990 CAS.
- N. Murthy and H. Minor, Polymer, 1990, 31, 996–1002 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: AFM images and size distribution of the original CNC; Viscosity measurements of the PVAc and in situ PVAc/CNC latexes; an additional SEM image of the unconsolidated in situ PVAc/CNC; an additional AFM image of the unconsolidated mixed PVAc/CNC; size distribution of the CNC aggregates; zeta potentials of all latexes; calculation of the length efficiency used in the mechanical model; sample coding and compositions of all consolidated samples; mechanical properties of all samples; additional POM images of the crystallization of PLA in different nanocomposites; XRD scattering curves of PLA/PVAc/CNC nanocomposites. See DOI: 10.1039/c7nr09080c |
| This journal is © The Royal Society of Chemistry 2018 |