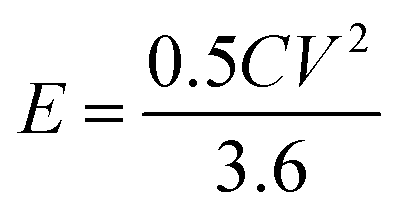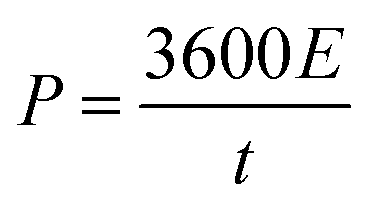DOI:
10.1039/C7NR07879J
(Paper)
Nanoscale, 2018,
10, 1779-1787
Mesoporous layered hexagonal platelets of Co3O4 nanoparticles with (111) facets for battery applications: high performance and ultra-high rate capability†
Received
23rd October 2017
, Accepted 7th December 2017
First published on 8th December 2017
Abstract
The thermally stable and crystalline 2D layered mesoporous hexagonal platelets of cobalt oxide (Co3O4) with (111) facets were prepared by using the template-free wet chemical synthesis approach. The high surface energy (111) facets known for a highly electroactive surface are expected to enhance the electrochemical properties, especially the rate capability. The highly crystalline Co3O4 with an average particle size of 25 nm formed a 2D mesoporous layered structure, with an average thickness of ∼40 nm, a pore size of 8–10 nm, and a specific surface area of 45.68 m2 g−1 promoting large surface confined electrochemical reaction. The 2D layered mesoporous Co3O4 exhibits a maximum specific capacity of 305 mA h g−1 at a scan rate of 5 mV s−1 and 137.6 mA h g−1 at a current density of 434.8 mA g−1. The maximum energy and power densities of 32.03 W h kg−1 and 9.36 kW kg−1, respectively, are achieved from the 2D hexagonal platelets of mesoporous Co3O4 nanoparticles with (111) facets. An excellent ultra-high rate capability of ∼62% capacity retention was observed after increasing the discharge current density from ∼434.8 mA g−1 to 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480 mA g−1. Furthermore, a cycling stability of 81.25% was achieved even after 2020 charge–discharge cycles at a current density of 12170 mA g−1. This high performance and ultra-high rate capability could be attributed to the (111) facets ‘crystal plane’ effect of Co3O4. Our results presented here confirm that the 2D mesoporous layered hexagonal platelets of Co3O4 exhibit “battery-mimic” behaviour in an aqueous electrolyte of KOH.
480 mA g−1. Furthermore, a cycling stability of 81.25% was achieved even after 2020 charge–discharge cycles at a current density of 12170 mA g−1. This high performance and ultra-high rate capability could be attributed to the (111) facets ‘crystal plane’ effect of Co3O4. Our results presented here confirm that the 2D mesoporous layered hexagonal platelets of Co3O4 exhibit “battery-mimic” behaviour in an aqueous electrolyte of KOH.
Introduction
One of the objectives of modern synthetic chemistry is to design materials possessing well-defined morphology with controlled shape and size, as these materials are considered to be promising candidates for a wide variety of technological applications. The electrical energy storage systems are incapable of satisfying the increasing demand for energy for large-scale applications, especially those related to high energy and power requirements. Hence, the research and development on high-performance electrochemical energy storage devices as an alternative energy storage system has become a commanding mission for researchers.1 Owing to many critical problems of the energy crisis and greenhouse effect derived from the use of fossil fuels, several alternative energy technologies have been developed to address the above problems. Among numerous energy storage devices, supercapacitors attracted considerable attention due to their high power density, fast charge–discharge capability, high rate capability, exceptionally long cycle life and safety.2 Several attempts have been made to investigate carbon-based materials and also transition metal oxides or hydroxides such as RuO2,3 MnO2,4 Co3O4,5 Ni(OH)2,6 Co(OH)2,7 NiCo2O4,8etc. along with their hybrid derivatives for improvement in both energy and power densities. However, in the presence of Li or Na ion electrolytes and aqueous electrolyte, transition metal oxides and hydroxides have behaved as battery-active materials.9,10
In batteries, the Li+/Na+ ion intercalation/deintercalation enables the redox reaction to the bulk of the electrode material in a sequential manner which is slow and diffusion controlled leading to high energy density. Supercapacitors have surface confined reaction and faster diffusion than batteries; that's why they possess high power density. The cubic spinel cobalt oxide (Co3O4) stands as a vital multifunctional material because of its vast applications in catalysis, pigments, sensors, electrochemistry, magnetism, and energy storage (e.g., intercalation compounds for battery materials).11 The spinel Co3O4 is a promising electrode material for electrochemical capacitors due to its environmentally friendly property, high theoretical specific capacitance of 3560 F g−1,12 good rate capability, high charge–discharge stability, low cost, and abundance in nature. However, it suffers from poor capacity retention and also limits the lifespan of many charge–discharge cycles by either agglomeration of Li/Li-alloying or the growth of passive layers, which in turn prevents the fully reversible insertion of Li ions to the electrode material. Consequently, several approaches have been adopted to improve the electrochemical performance of Co3O4, such as the use of nanoscale Co3O4,13 micron size Co3O4,14 mesoporous Co3O4,15 Co3O4/graphene nanocomposites and Co3O4 nanowire arrays.16 Multi-shelled Co-oxide hollow structures offering small diffusion lengths and sufficient void space could boost the cyclic stability and rate capacity. Moreover, an enhanced electrochemical performance was observed in porous Co3O4 nanotube structures that consist of nanoparticles of size ∼5–10 nm.17 However, theoretical studies on Co3O4 {111} facets have shown fairly greater adsorption energy, more adsorption sites, and lower transition state barrier than those of Co3O4 {001} facets, which are responsible for the facet-dependent electrochemical behaviour.18 Co based oxide/hydroxide shows a pair of redox peaks, which are separated by a gap of 0.066 V (= ΔEp), that point out a so-called “battery-mimic” mechanism.19 In a true sense, it can be a hybrid energy storage device combining both the supercapacitor and battery. Therefore, it has the capability to combine the high energy storage capability of conventional batteries with the high power delivery capability of the supercapacitor.20
In this study, we have endeavoured to obtain mesoporous hexagonal platelets consisting of Co3O4 nanoparticles using simple wet chemical bath synthesis. The (111) faceted 2D layered hexagonal structure of mesoporous Co3O4 composed of a number of nanoparticles with an average size of ∼25 nm exhibited a battery-type electrochemical performance in an aqueous (2 M KOH) electrolyte. Mesoporous Co3O4 delivered a specific capacity of 305 mA h g−1 at a scan rate of 5 mV s−1 and 137 mA h g−1 at a current density of 434.8 mA h g−1, an ultra-high rate capability of 62%, and 81.25% capacity retention after 2020 cycles at 12170 mA h g−1 current density. The 2D multi-layered hexagonal platelets of mesoporous Co3O4 nanoparticles played an important role in enhancing the electrochemical performance of pristine Co3O4 several orders of magnitude higher than the previously reported Co3O4 material.
Experimental
Synthesis of layered 2D hexagonal platelets of Co3O4 nanoparticles
We have already established a simple wet chemical synthesis approach for the preparation of well-controlled nanostructures21–24 for ZnO. The 2D mesoporous layered hexagonal platelets of Co3O4 with (111) facets were synthesized using the wet chemical synthetic approach from high purity cobalt nitrate hexahydrate (Co(NO3)2·6H2O) (0.050 M concentration). An aqueous solution of Co(NO3)2·6H2O was prepared in 100 mL deionized water (DI water) and stirred for 30 min. The reaction medium was made alkaline by adding ammonium hydroxide (NH4OH) solution to promote the growth of 2D mesoporous layered hexagonal platelets of Co3O4. The pH of the reaction was maintained at ∼12, and then the reaction mixture was heated at 80 °C for ∼3 hours, which resulted in a brownish-black precipitate. Later, the precipitate was filtered by using Whatman filter paper, and the product was washed several times with DI water and ethanol followed by air drying and then annealing at 450 °C for 4 hours in a muffle furnace to obtain mesoporous Co3O4. The growth mechanism of the hexagonal layered structure is represented in Scheme 1 in the ESI.†
Materials characterization
The phase purity and structural and morphological identification of spinel Co3O4 was carried out using X-ray diffraction (XRD; D2-phaser, Bruker, 30 kV/10 mA, Cu Kα irradiation (λ = 1.54 Å)), field emission scanning electron microscopy (FESEM, Karl Zeiss JEOL), and high resolution transmission electron microscopy (HRTEM, Tecnai G2 F20 S-Twin) techniques. The elemental composition was investigated by energy dispersive X-ray (EDX). Surface area analysis and pore size distribution analysis were carried out using the N2 adsorption–desorption (Brunauer–Emmett–Teller (BET)) and Barrett–Joyner–Halenda (BJH) methods, respectively, with an automated gas sorption analyser (Quantchrome Autosorb iQ2). Electrochemical measurements like cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), electrochemical impedance spectroscopy (EIS) and stability test were carried out using an electrochemical workstation (Autolab PGSTAT302N).
Electrode preparation
The working electrode was prepared as follows: 80% Co3O4 powder as an active material, 10% acetylene black as a conductive material and 10% polyvinylidene difluoride (PVDF) as a binder were homogeneously mixed in a few drops of ethanol and the mixture was ultrasonicated in a vessel until a homogeneous slurry was achieved. The slurry was drop casted on a Ni-foam with a geometric surface area of 1 cm2 (total mass loading ∼2.3 mg), followed by drying at 80 °C for 12 hours and pressing with a hydraulic press under 10 MPa pressure. The electrochemical performance of the device was measured by CV, GCD, and EIS in a 2 M KOH electrolyte solution. EIS measurement was performed in the frequency range from 1 MHz to 0.01 Hz by applying a sinusoidal waveform with an amplitude of 10 mV. The specific capacity was calculated from the CV and GCD curves using relations (1) and (2), respectively:8| | 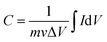 | (1) |
| | 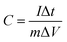 | (2) |
where C is the specific capacity (mA h g−1), 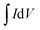 is the area under the CV curve, m is the mass of the active electrode material (g), v is the scan rate (mV s−1), ΔV is the potential window (V), I is the discharge current (mA g−1), and Δt is the discharge time (s).
is the area under the CV curve, m is the mass of the active electrode material (g), v is the scan rate (mV s−1), ΔV is the potential window (V), I is the discharge current (mA g−1), and Δt is the discharge time (s).
The energy density (E in W h kg−1) and power density (P in W kg−1) were calculated by using relations (3) and (4), respectively:8
| | 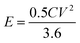 | (3) |
| | 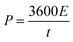 | (4) |
where
C is the specific capacity (mA h g
−1),
V is the potential range (V),
E (W h kg
−1) is the energy density and
t (s) is the elapsed time during the discharge period.
Results and discussion
The surface morphological features and structural analysis of the synthesized sample are shown in Fig. 1. The FESEM image in Fig. 1(a) shows a uniform distribution of hexagonal plates, composed of a number of nanoparticles. The periodic arrangement of nanoparticles leads to the 2D layered structure of hexagonal plates (Fig. 1(b)). The high magnification FESEM image in Fig. 1(b) depicts the highly dense 2D layered structure with hexagonal morphological features as schematically illustrated in the inset of Fig. 1(b). These hexagonal plates are ∼2 μm wide along their edges and ∼40–50 nm thick. However, the number of layers formed is not uniform in each of the hexagonal plates despite their layer-by-layer appearance. The hexagonal facets of the plates make an angle of 120° (Fig. 1(c)). TEM analysis (Fig. 1(d)) confirmed that the hexagonal plates are composed of uneven morphology nanoparticles with an average diameter of 25 nm (Fig. 1(d)). Each particle is distinct with clearly visible textural boundaries. The high-magnification TEM image (Fig. 1(e)) corroborates the inter-planer spacing of 0.288 nm between two successive fringes corresponding to the (220) crystallographic plane, which evidenced the formation of the cubic spinel phase of Co3O4. It was further confirmed by selected area electron diffraction (SAED) analysis. The presence of a number of nanoparticles in the hexagonal plate is reflected in the appearance of periodic hexagonal diffraction. The diffraction at the edge of the hexagonal plates showed a major exposed crystal plane of the spinel cubic crystal structure with a space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (227) along the [111] zone axis (Fig. 1(f)). The energy-dispersive X-ray spectra (EDS) in Fig. 1(g) show the presence of Co and O in the mesoporous layered structure, with an average atomic ratio of Co:O of ∼3:4. This confirms the hexagonal assembling of Co3O4.
m (227) along the [111] zone axis (Fig. 1(f)). The energy-dispersive X-ray spectra (EDS) in Fig. 1(g) show the presence of Co and O in the mesoporous layered structure, with an average atomic ratio of Co:O of ∼3:4. This confirms the hexagonal assembling of Co3O4.
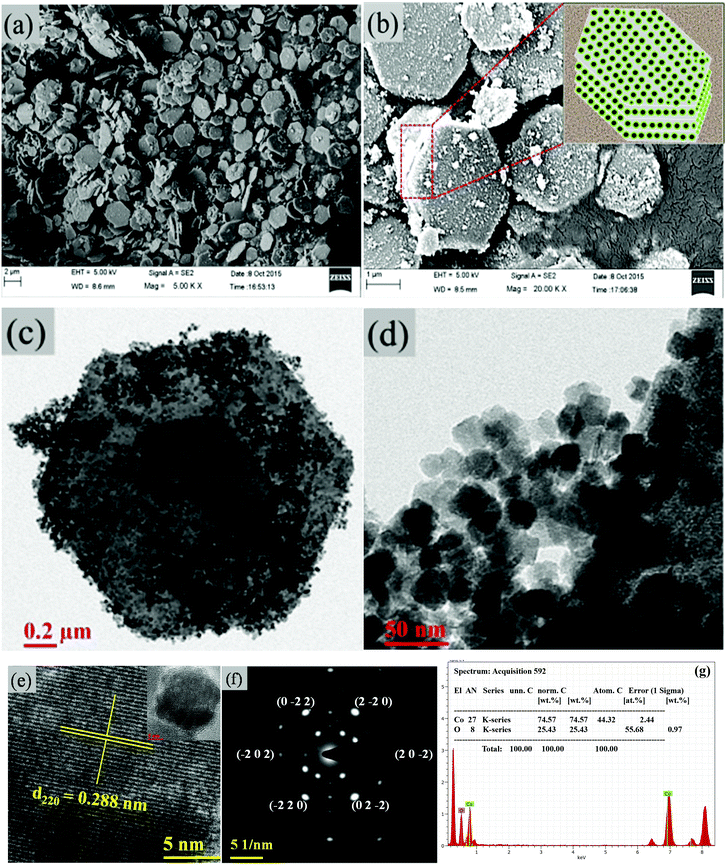 |
| | Fig. 1 (a, b) FESEM images of the hexagonal platelets of Co3O4. The inset in (b) shows the schematic of the layered hexagonal platelets. (c,d) TEM images. (e) Lattice resolved HRTEM image. (f) SAED pattern of Co3O4. (g) EDS of Co3O4. | |
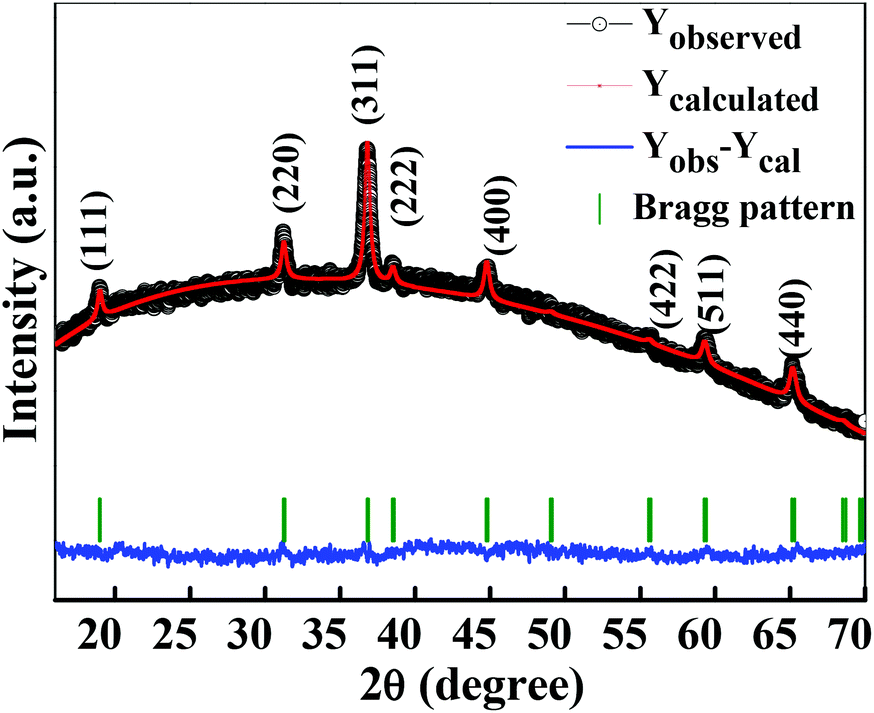
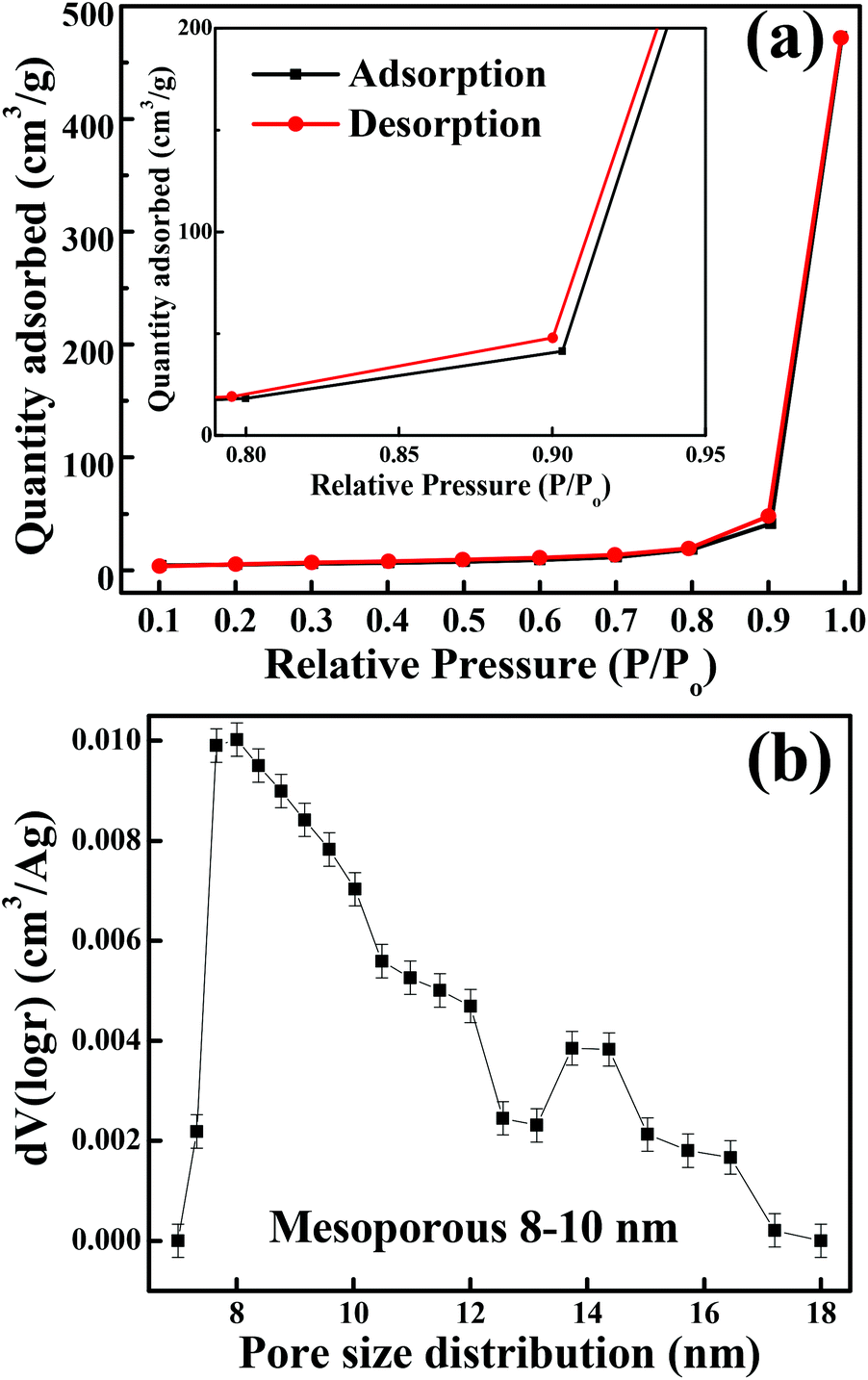
The crystalline structure and phase purity of as-prepared Co3O4 and annealed Co3O4 were investigated by powder XRD. Fig. S1 (ESI†) showing the XRD pattern of as-prepared Co3O4 confirms the formation of β-Co(OH)2. Fig. 2 shows the XRD pattern of Co3O4 annealed at 450 °C for 4 h. The XRD pattern of Co3O4 annealed at 450 °C (Fig. 2) shows the presence of diffraction peaks at 2θ of 18.96°, 31.20°, 38.46°, 44.79°, 59.34° and 64.14° assigned to (111), (220), (311), (222), (400), (511) and (440) diffraction peaks of the cubic spinel Co3O4 phase (JCPDS file 009-0418) in the space group Fdm (227). No other impurity phase was identified. The intensive diffraction peaks imply good crystallinity of Co3O4. The lattice parameters of a = b = c = 8.093 Å obtained from the Rietveld refinement are in a good agreement with that of JCPDS file 009-0418. In general, Co3O4 is known to be enclosed by low indexed facets such as (100) or (110),11 whereas mesoporous Co3O4 has (111) faceted crystal planes.25 The predominantly exposed (111) crystal plane is a highly active surface25 which leads to the facet-dependent properties of nanomaterials and helps the fast ion transfer between the surface and the interior of the structure.26 Highly dense metal oxide nanomaterials exposed with high-energy surfaces/facets favour fast ion transfer at both the surface and the interior.27 Typically, 111-faceted transition-metal oxides with high surface reactivity28 provide more reactive sites for redox reaction, which facilitate a fast surface reaction27 during the charge and discharge processes. Moreover, theoretical studies have confirmed that Co3O4 {111} facets exhibit a fairly greater adsorption energy, more adsorption sites, and a comparatively lower transition state barrier than that of Co3O4 {001} facets, responsible for the facet-dependent electrochemical behaviour.18 Overall, Co3O4 (111) faceted surfaces hold the potential for applications in highly efficient electrochemical activity,18 sensing,18 heterogeneous catalysis,29etc. Despite the intense interest in the associated structural details, porosity modifications have been almost exclusively described in terms of primitive bulk descriptors (surface area, pore volume, and average pore size), providing limited insight into the pore architecture. N2 adsorption–desorption isotherm measurements examined the porous nature of mesoporous Co3O4 at 77 K. Fig. 3(a) shows irreversible type IV isotherm, confirming the porous nature and a distinct hysteresis loop on the IUPAC classification. The specific surface area is found to be 45.68 m2 g−1. The mesoporous nature of the material was confirmed by pore size distribution analysis (Fig. 3(b)). Co3O4 with an average pore size of 8–10 nm is expected to be good for the diffusion of ions and electrons within the electrode. The dense packing of Co3O4 particles resulted in larger pores (mesopores) with a pore volume of 1.42 cm3 g−1. The surface area and mesoporosity of Co3O4 with layered hexagonal platelet nanostructures not only increase the electrode/electrolyte contact area but also provide abundant active sites for the accumulating ions. This leads to an efficient charge/ion transport, resulting in the enhancement of electrochemical properties. Very large surface area and mesoporous appearance of Co3O4 are expected to deliver an enhanced specific capacitance value.
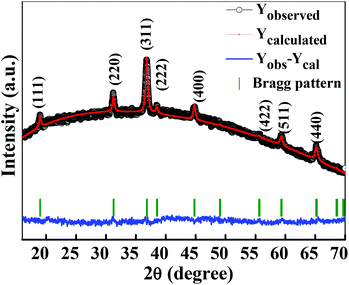 |
| | Fig. 2 Rietveld refined XRD pattern of Co3O4 (annealed at 450 °C for 4 h). | |
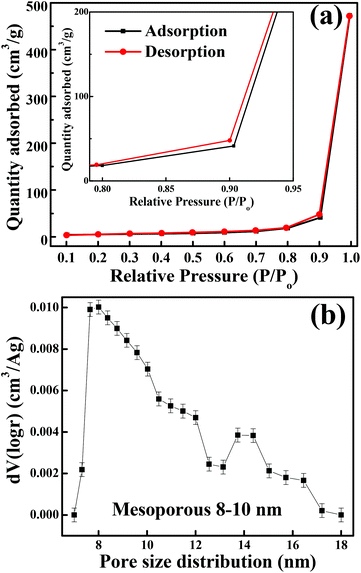 |
| | Fig. 3 (a) N2 adsorption–desorption isotherm (inset shows the magnified graph). (b) BJH pore size distribution. | |
The cyclic voltammogram (CV) measurements were carried out to investigate the redox behaviour and electrochemical nature of the electrode material. The CV measurements shown in Fig. 4(a) were carried out in a 2 M KOH aqueous electrolyte in the voltage range of −0.1 to 0.6 V at various sweep rates. The non-rectangular shapes of the CV curves reveal the battery nature originating from the reversible redox reactions. The anodic peak (at a positive potential) and cathodic peak (negative potential) in the CV curves represent the oxidation and reduction processes, respectively. At a scan rate of 5 mV s−1, the oxidation and reduction redox pair is observed at Epa = 0.33 V (anodic) and Epc = 0.15 V (cathodic), respectively. It indicates that layered hexagonal platelets composed of mesoporous Co3O4 nanoparticles undergo the electrochemical charge-transfer reactions Co(II) ↔ Co(III) ↔ Co(IV) in 2 M KOH.30 However, the literature reports three redox couples of Co3O4/Co(OH)2, Co(OH)2/CoOOH and CoOOH/CoO2. In the present case of Co3O4, in the first oxidation step, Co3O4 is reduced to the Co(OH)2 species during the CV scan. The peaks corresponding to each redox couple are not evidently distinguishable, and some are elusive, which might be due to the surface modification of the layered mesoporous Co3O4.31 The redox couple of P1/P2 is an indication of Co2+ → Co3+ redox reactions.32 As the scan rate increases, the anodic and cathodic peaks shift towards the positive and negative potential, respectively, on account of the redox process due to the polarization and the ohmic resistance during the faradaic process at the interface between the electrolyte and the electrode.8,33 It proposes a typical battery type behaviour, originating from the fast reversible redox reaction. The structural alterations at the atomic level are expected in the layered appearance of a hexagonal platelet of mesoporous Co3O4 during the electrochemical redox reaction.19 The variation in the valency of Co from +2 to +3 during charging (increases) and discharging (decreases) results in the transformation of Co(OH)2 → CoOOH19 as shown in eqn (5) and (6). The density functional theoretical analysis by Deng et al.19 further supported the vital role of lattice relaxation in the stabilization of the transition states and the removal of one H atom per formula unit from Co(OH)2 → CoOOH during this process, and therefore, the structural transformation towards CoOOH ↔ Co(OH)2. The deprotonation involves the rearrangements of the atoms of both O and OH. Co(OH)2 and CoOOH adjust themselves such that they transform mutually into each other (reaction (6) and (9)). These transformation reactions offer the advantage of very fast switching rates which contribute to high power density, ultra-high rate capability, and long cycling stability. The more prominent asymmetric nature at higher scan rates demonstrates a better high-rate response of the mesoporous layered hexagonal platelets of Co3O4.34 In the charge storage mechanism, the redox peaks P1 and P2 are related to reaction (5–7) and (8–10), respectively.35
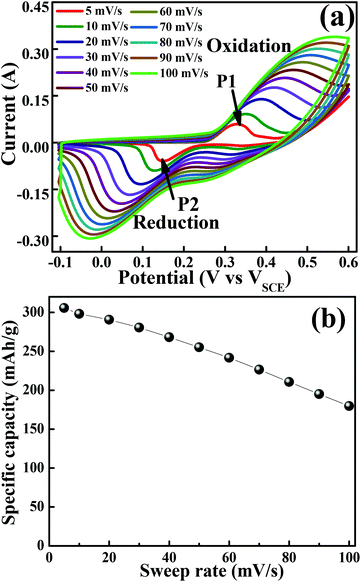 |
| | Fig. 4 Cyclic voltammogram of the layered mesoporous Co3O4 hexagonal platelet electrode. (a) CV curves obtained at different sweep rates. (b) Graph of specific capacity versus sweep rate. | |
Anodic scan (positive potential):
| | | Co3O4 + 4H2O + 2e− ↔ 3Co(OH)2 + 2OH− | (5) |
| | | 3Co(OH)2 + 2OH− ↔ 3CoOOH + 2H2O + e− | (6) |
| | | CoOOH + OH− ↔ CoO2 + H2O + e− | (7) |
Cathodic scan (negative potential):
| | | CoO2 + H2O + e− ↔ CoOOH + OH− | (8) |
| | | 3CoOOH + 2H2O + e− ↔ 3Co(OH)2 + 2OH− | (9) |
| | | 3Co(OH)2 + 2OH− ↔ Co3O4 + 4H2O + 2e− | (10) |
Fig. 4(b) illustrates specific capacity as a function of sweep rates. At a lower scan rate of 5 mV s−1, the electrolyte ions diffused to almost all the effective pores of the electrode material and resulted in the highest specific capacity of 305 mA h g−1. However, it is further reduced with an increase in the scan rate. At higher scan rates (typically 50–100 mV s−1), the effective interaction between the electrolyte ions and the electrode is significantly reduced due to inadequate time and leads to lower specific capacitance at higher scan rates.24 In general, the electrochemical properties are governed by the electronic structure, and a number of active sites (such as Co2+ and Co3+ in Co3O4) and many electrons/ions are exchanged during the electrochemical process.36 However, in the present study, the outperformed unique hexagonal layered structure of mesoporous Co3O4 exhibits an excellent battery-mimic performance over the other reported ones and not the supercapacitor.37 The origin of this unique electrochemical performance can be improved by the electrochemical surface area available for interfacial redox reactions38 due to optimized grain size, morphology, and porosity. The effective porosity induces accessible electrochemical sites for the intercalation of electrolyte ions, which is proportional to capacitance.39 Moreover, the observed mean pore size is higher than the size of solvated ions in typical aqueous electrolytes of KOH (size of Kþ ∼ 3.31 Å), reduces the kinetic charge transfer resistance at the electrode/electrolyte interface and increases the overall capacitance.40 The enhancement in the electrochemical performance can be attributed further to the combined effect of the 2D layered hexagonal platelet structure, (111) faceted surface and mesoporous morphology of Co3O4 nanoparticles. The layered materials with a fine crystallized structure store energy through ion intercalation–deintercalation between the layers of hexagonal platelets to exhibit high rate performance and good stability.41 Furthermore, the open space between/among layered hexagonal platelets also allows fast redox reactions, and thus improves the charge–discharge rate.42 Hence, the obtained morphology of Co3O4 has played an enormous role in enhancing the specific capacity.
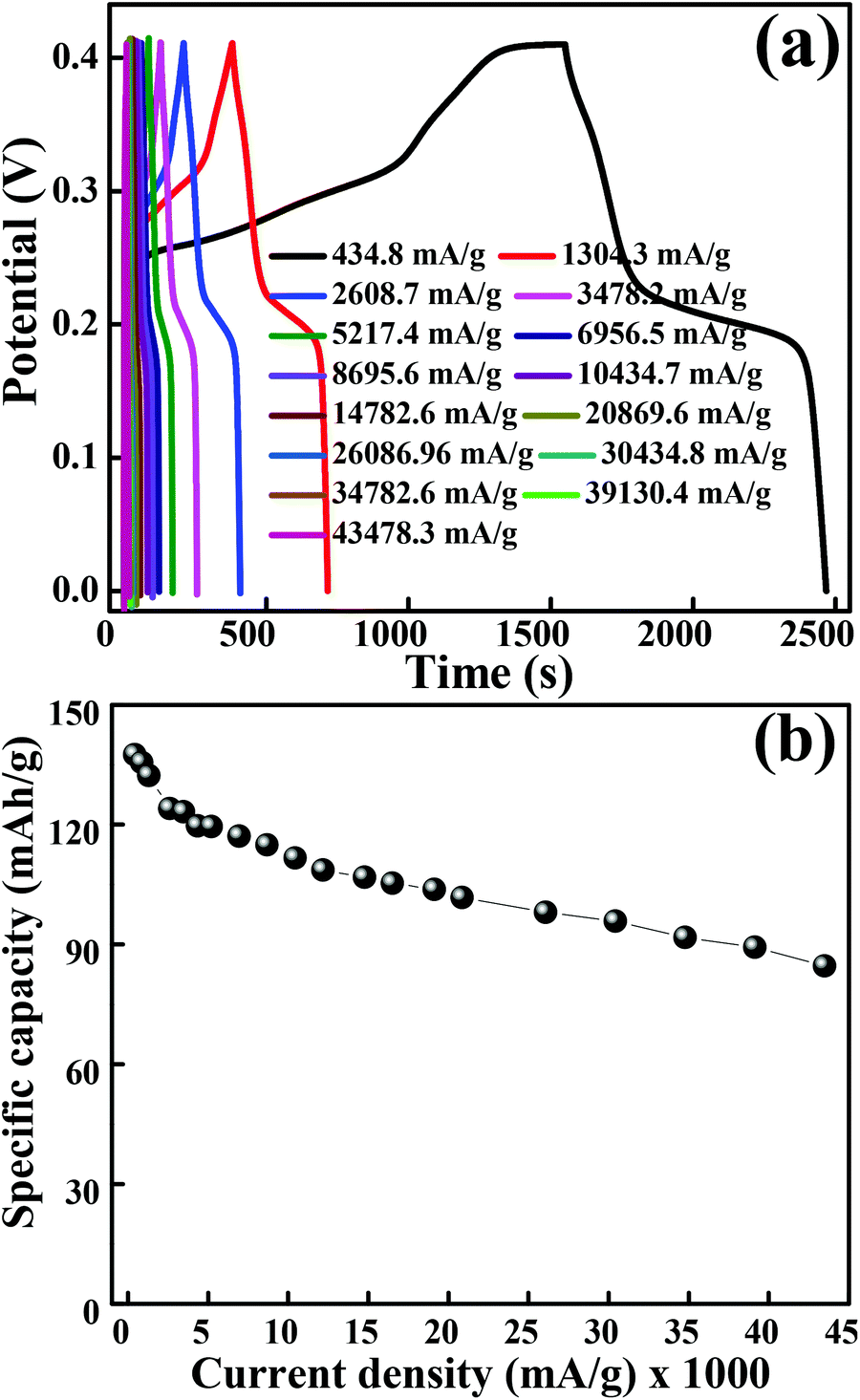
The galvanostatic charge–discharge measurements were performed at various current densities in the potential range of 0 to 0.41 V for layered hexagonal platelets of mesoporous Co3O4. The GCD curves are shown in Fig. 5(a). We have observed a deviation in the GCD curves from the ideal triangular behaviour reported in case of EDLCs. The GCD curves possess two distinct slopes/plateaus corresponding to the redox nature of the electrode material which is a battery-mimic behaviour. The charge–discharge curves at various current densities indicate the typical battery-type behaviour without IR drop. The values of specific capacity were derived from the discharge curves using eqn (2). Fig. 5(b) illustrates the specific capacity as a function of current density and Table S1† shows the specific capacity obtained at various current densities. The specific capacity of 137.6 mA h g−1 is observed at a current density of 434.8 mA g−1 which is further reduced to 84.6 mA h g−1 at a current density of 43480 mA g−1. At lower current densities, electrolytic ions ensure more time to penetrate into the innermost layers of the electrode material, hence accessing almost all available surface area of the electrode. However, at higher current densities, an efficient utilization of the electrode material is restricted only to the outer surface of the electrodes due to the lesser time taken by ion intercalation.43 The specific capacity values of mesoporous Co3O4 obtained from GCD are lower than those obtained from the CV measurement. This difference is due to insufficient faradaic redox reactions resulting from the reduced surface access by OH− ions under higher discharge current densities.30 Generally, the ions are stored on the surface of the material, lying within the pores, which causes an active diffusion within the bulk of the material. The surface area/pore size distribution results suggest the synthesized material to be mesoporous, so the ions are not only stored at the surface but also in the interiors of the pores. Therefore, there is an active diffusion from the pores; the diffusion is fast because the majority of the charges stored are surface confined.44 This indicates that, at higher current rates, the surface confined redox process as well as the in-pore diffusion of ions might take place, limiting the diffusion of ions depending on factors like pore size distribution, tortuosity of its pores, size of the electrolyte ions, electrolyte ion mobility, etc.44 which specifies the limitation arising from the charge transfer kinetics.45 This resulted in the reduction of effective specific capacity with an increase in current densities. Comprehensibly, the better utilization of numerous nanometric electroactive sites from the layered hexagonal appearance of mesoporous Co3O4 has considerably boosted the overall electrochemical performance. Even though Sun et al.46 reported capacities of 969 mA h g−1 for Co3O4 nanocubes, 1023 mA h g−1 for Co3O4 truncated octahedra and 1098 mA h g−1 for Co3O4 octahedra, one cannot neglect the utilization of a Li-based electrolyte to achieve these high values. Co3O4 with the {111} plane may be more beneficial to Li+ transport than the other low index planes like {001}.46 Therefore, it has shown the highest reversible capacity and excellent rate capability. Although various reports in the literature show a higher electrochemical performance after the Li+ ion intercalation/deintercalation, the performance in the present study is reported at a substantial current density (434.8 mA g−1) in the aqueous electrolyte of KOH and not in the Li ion electrolyte. The ultra-high rate capability was observed, and 38% reduction in specific capacity was observed after an increase in the initial current density of 434.8 mA g−1 to 43480 mA g−1. The crystal structure of the electrode material has a significant impact on the electrochemical performance; in particular, (111) faceted Co3O4 exhibits not only high charge–discharge capacity but also shows a high-rate capability compared to that of (001) or (110) facets.47 The ultra-high rate capability could be due to the (111) planes which possess the highest charge/discharge capacity not only at the low rate of 434.8 mA g−1 but also at the very high rate of 43480 mA g−1, which can be largely due to the “crystal plane” effect. It enhances the electroactive surface area due to the unique layered hexagonal structure of Co3O4 providing enormous active sites in its layered structure, and individual layers take part in the electrochemical activity. A more detailed comparison is provided in Table S2.†
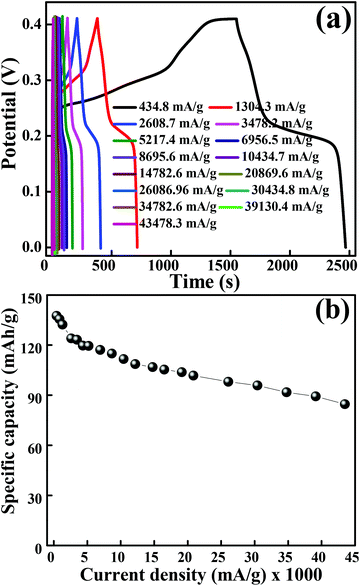 |
| | Fig. 5 (a) GCD curves obtained at different current densities. (b) Graph of specific capacity versus current densities. | |
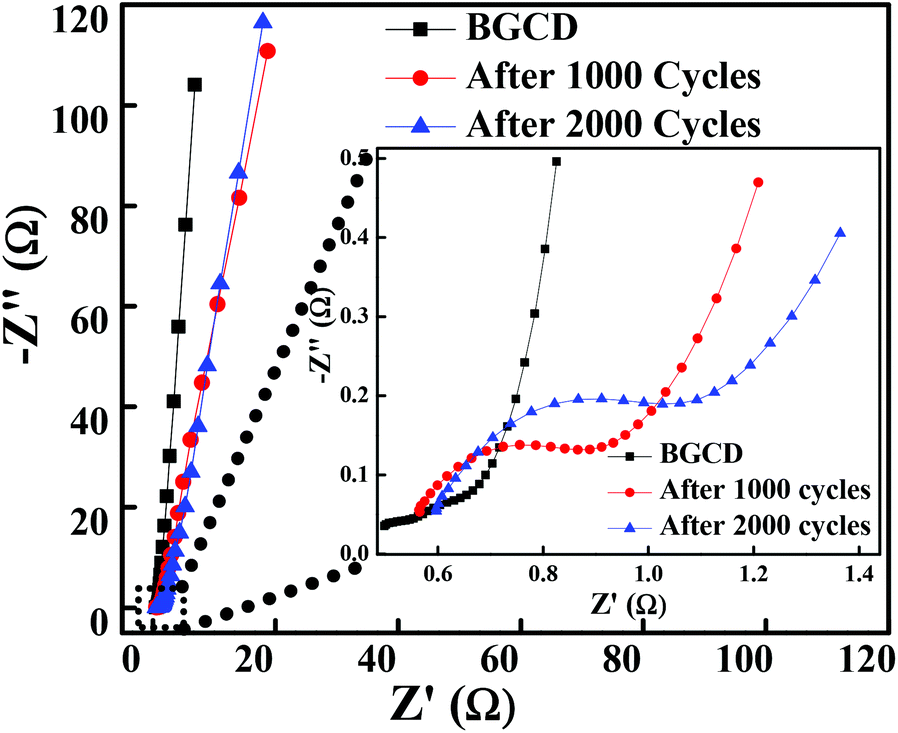
To further explore the electrical conductivity of the electrode, the EIS spectrum was recorded in the frequency range of 1 MHz to 0.01 Hz by applying a 10 mV sinusoidal perturbation. Fig. 6 illustrates the Nyquist plot for the Co3O4 electrode material, which consists of a semicircle accompanied by a straight line in the low-frequency region, which correspond to the electrochemical process and mass transfer process, respectively.48 The EIS spectra is composed of three distinct regions. The first region is the intercept on the real axis in a high-frequency region which provides the solution resistance (Rs); this is due to the ionic resistance of the electrolyte, intrinsic resistance of electrodes and contact resistance at the electrode/electrolyte interface. The second region is the charge transfer resistance (Rct) which can be calculated from the semicircle diameter in the high-frequency region; this is due to the diffusion of electrons. The third region is the Warburg resistance; the slope of the EIS curve in the low-frequency region describes the diffusion and fast kinetics of the redox species in the electrolyte.49 Here, the electrochemical process is diffusion controlled because of various parameters like electrolyte concentration, porous structure (pore size distribution), ion dynamics (ion movement in and out from the pores), etc., which take part in the electrochemical process.50 The vertical line in the low-frequency region parallel to the imaginary impedance axis indicates an ideal capacitor behaviour. The inset of Fig. 6 illustrates the EIS plot before and after 1000 and 2000 GCD cycles test and the inset shows the high-frequency region of the EIS spectra. It is observed that the magnitude of Rs and Rct is significantly low even after 1000 and 2000 cycles. Table 1 shows the Rs and Rct values, and it is evident from the results that there is a negligible change in the Rs and Rct values. The low ESR value indicates that the morphology obtained in the present study has a high impact on enhancing the electrochemical performance.
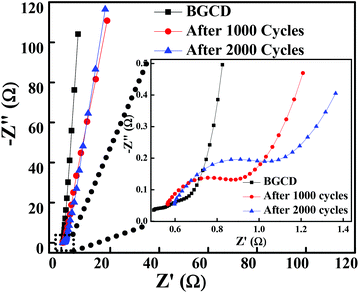 |
| | Fig. 6 Nyquist plot of the layered hexagonal mesoporous Co3O4 at different cycles before the GCD test and then after 1000 and 2000 GCD cycles (inset shows the expanded high-frequency region of the EIS spectra). | |
Table 1 Solution resistance (Rs) and charge transfer resistance (Rs) before and after stability tests (1000 and 2000 GCD cycles)
| |
R
s (Ω) |
R
ct (Ω) |
| Before test |
0.499 |
0.78 |
| After 1000 GCD cycles |
0.5627 |
1.5178 |
| After 2000 GCD cycles |
0.5971 |
1.735 |
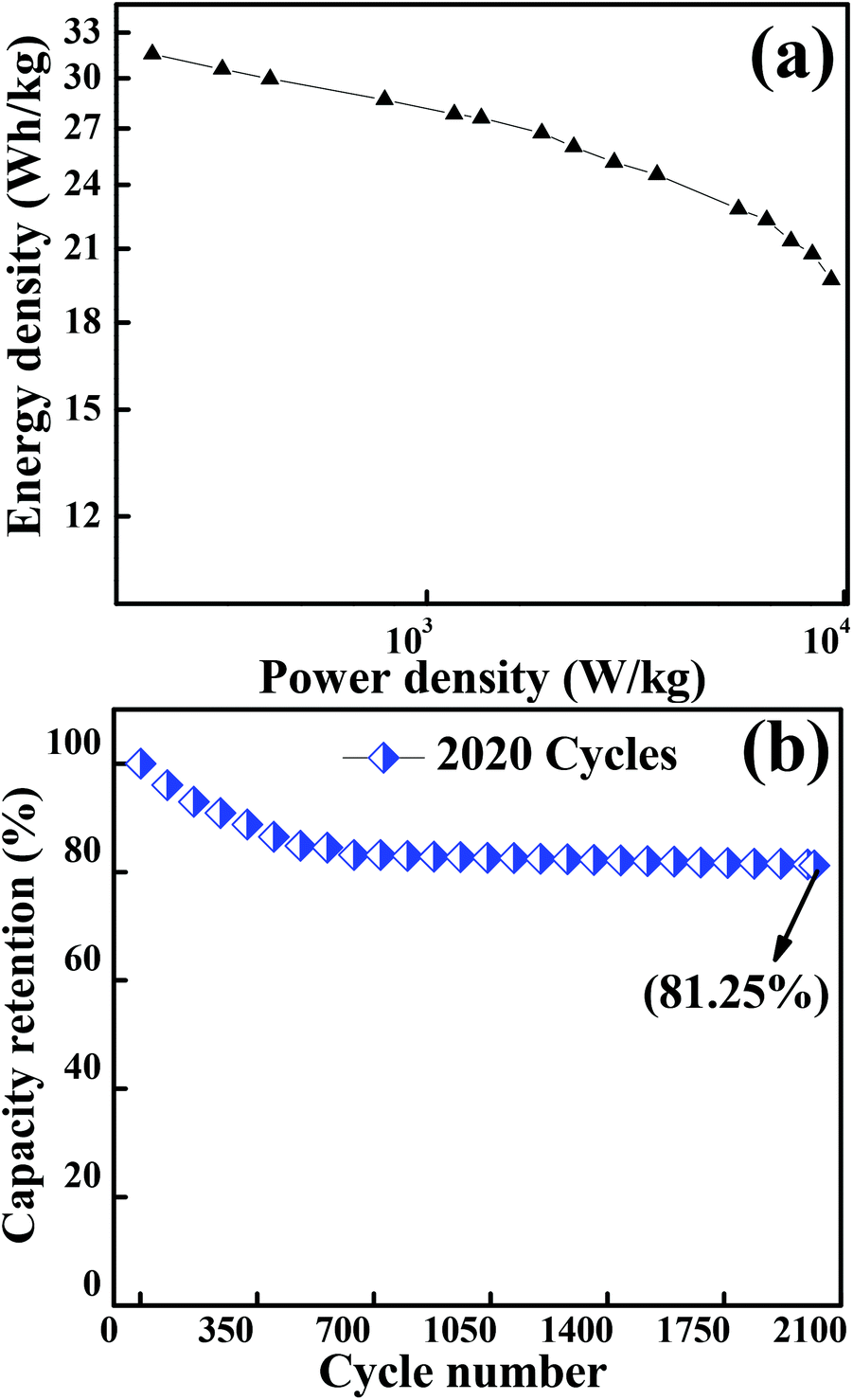
Fig. 7(a) depicts the Ragone plot (energy density as a function of power density at different current densities) of Co3O4. The energy and power densities of mesoporous Co3O4 were calculated from relations (3) and (4), respectively. With the increase in discharge current density from 434.8 mA g−1 to 43480 mA g−1, the specific energy density decreased from 32.03 W h kg−1 to 19.69 W h kg−1 while the specific power increased from 0.125 kW kg−1 to 9.326 kW kg−1, respectively. The present study demonstrated that the overall electrochemical performance of 2D layered hexagonal platelets of the Co3O4 mesoporous structure has a dramatic impact on the electrochemical performance and it has proved to be an excellent electrode material. A comparison of the energy density and power density with previous reports is presented in Table S3.†
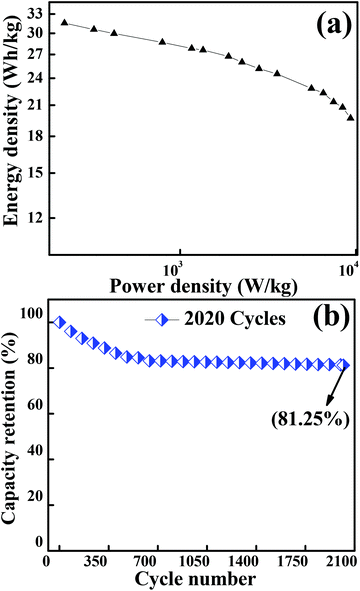 |
| | Fig. 7 (a) Graph showing the Ragone plot of the Co3O4 electrode material. (b) Plot for the capacity retention of 2020 cycles of Co3O4 operated at a current density of 12170 mA g−1. | |
A long-term charge/discharge cycling stability test was carried out to know the long-term capacity retention behaviour as is shown in Fig. 7(b). A capacity retention of 81.25% was observed after 2020 cycles at a very high current density of 12170 mA g−1. The diffusion might lead to the blockage of pores when accessed for a longer period of time, especially when the long-term stability is reduced to ∼81% (might be one of the reasons for capacitance fade). The overall enhancement in the electrochemical performance of the mesoporous layered hexagonal platelets of Co3O4 nanoparticles can be attributed to (1) highly dense mesoporous 2D layered hexagonal platelets for providing a short diffusion path, reducing internal resistance and faster kinetics, (2) the crystal plane effect and orientation of Co3O4 (111) facets for delivering enormous active sites, and (3) the layered stacking between the platelets for easier electrolyte penetration in the inner region of the electrode material.
Conclusions
In conclusion, the layered 2D hexagonal platelets were synthesized by using a simple wet chemical method. The synthesized electrode material showed a unique layered hexagonal platelet-like morphology (FESEM) with a large number of particles in the nanometre scale (∼25 nm). XRD and HRTEM confirm the pure cubic spinel phase of Co3O4 with no amorphous phases; the SAED pattern confirms the predominantly exposed (111) faceted crystal plane, which is considered to be a highly active surface and helpful for enhancing redox reaction and fast ion transport. The N2 adsorption–desorption isotherm confirms the irreversible type IV isotherm with a large surface area of 45.68 m2 g−1 and a pore size distribution between 8–10 nm. Co3O4 showed a battery-type behavior in the aqueous electrolyte of 2 M KOH. Co3O4 delivered a high specific capacity of 305 mA h g−1 at 5 mV s−1 and 137.6 mA h g−1 at ∼434.8 mA g−1 from CV and GCD, respectively. It is noteworthy that only 38% of the capacitance reduction was detected when the current density was increased by almost 100 times (i.e., from 434.8 mA g−1 to 43480 mA g−1). The capacity retention was found to be 81.25% after 2020 GCD cycles at a current density of 12170 mA g−1. The 2D layered hexagonal structure of mesoporous Co3O4 demonstrated a power density of 9.33 kW kg−1 and an energy density of 32.03 W h kg−1. The overall electrochemical performance of mesoporous Co3O4 has proved to be a potential candidate for electrochemical energy storage applications such as in batteries, with ultra-high rate capability, long cycling stability, and good power and energy density.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the SERB-DST (Department of Science and Technology, India) by awarding ‘Ramanujan Fellowship’ to PMS (SR/S2/RJN-121/2012). PMS also acknowledges CSIR research grant No. 03(1349)/16/EMR-II. We are thankful to Prof. Dr Pradeep Mathur, Director, IIT Indore, for encouraging the research and SIC-IIT Indore for providing the basic characterization facilities.
References
- J. Miller and P. Simon, Sci. Mag., 2008, 321, 651–652 CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- C. Hu, K. Chang, M. Lin and Y. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS PubMed.
- S. Shi, C. Xu, C. Yang, Y. Chen, J. Liu and F. Kang, Sci. Rep., 2013, 3, 2598–2604 CrossRef PubMed.
- S. Xiong, C. Yuan, X. Zhang and B. Xi, Chem. – Eur. J., 2009, 15, 5320–5326 CrossRef CAS PubMed.
- Z. Lu, Z. Chang, W. Zhu and X. Sun, Chem. Commun., 2011, 47, 9651–9653 RSC.
- L. Wang, Z. H. Dong, Z. G. Wang, F. X. Zhang and J. Jin, Adv. Funct. Mater., 2013, 23, 2758–2764 CrossRef CAS.
- P. Bhojane, S. Sen and P. M. Shirage, Appl. Surf. Sci., 2016, 377, 376–384 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- T. Brousse, D. Belanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- J. Feng and H. C. Zeng, Chem. Mater., 2003, 15, 2829–2835 CrossRef CAS.
- H. Cheng, Z. G. Lu, J. Q. Deng, C. Y. Chung, K. Zhang and Y. Y. Li, Nano Res., 2010, 3, 895–901 CrossRef CAS.
- C. C. Li, X. M. Yin, Q. H. Li, L. B. Chen and T. H. Wang, Chem. – Eur. J., 2011, 17, 1596–1604 CrossRef CAS PubMed.
- K. Deori, S. K. Ujjain, R. K. Sharma and S. Deka, ACS Appl. Mater. Interfaces, 2013, 5, 10665 CAS.
- L. Tian, H. Zou, J. Fu, X. Yang, Y. Wang, H. Guo, X. Fu, C. Liang, M. Wu, P. K. Shen and Q. Gao, Adv. Funct. Mater., 2010, 20, 617–623 CrossRef CAS.
- J. Liu, J. Jiang, C. Cheng, H. Li, J. Zhang, H. Gong and H. J. Fan, Adv. Mater., 2011, 23, 2076–2081 CrossRef CAS PubMed.
- N. Du, H. Zhang, B. Chen, J. Wu, X. Ma, Z. Liu, Y. Zhang, D. Yang, X. Huang and J. Tu, Adv. Mater., 2007, 19, 4505–4509 CrossRef CAS.
- X.-Y. Yu, Q.-Q. Meng, T. Luo, Y. Jia, B. Sun, Q.-X. Li, J.-H. Liu and X.-J. Huang, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- T. Deng, W. Zhang, O. Arcelus, J.-G. Kim, J. Carrasco, S. J. Yoo, W. Zheng, J. Wang, H. Tian, H. Zhang, X. Cui and T. Rojo, Nat. Commun., 2017, 8, 15194 CrossRef CAS PubMed.
- H. Shao, N. Padmanathan, D. McNulty, C. O'Dwyer and K. M. Razeeb, ACS Appl. Mater. Interfaces, 2016, 8, 28592–28598 CAS.
- R. Das, A. Kumar and Y. Kumar, RSC Adv., 2015, 5, 60365–60372 RSC.
- A. K. Rana, Y. Kumar, N. Saxena, R. Das, S. Sen and P. M. Shirage, AIP Adv., 2015, 5, 097118–097127 CrossRef.
- Y. Kumar, A. K. Rana, P. Bhojane, M. Pusty and V. Bagwe, Mater. Res. Express, 2015, 2, 105017–105026 CrossRef.
- P. Bhojane, A. Sharma, M. Pusty, Y. Kumar, S. Sen and P. Shirage, J. Nanosci. Nanotechnol., 2017, 17, 1387–1392 CrossRef CAS.
- Z.-Y. Zhou, N. Tian, J.-T. Li, I. Broadwell and S.-G. Sun, Chem. Soc. Rev., 2011, 40, 4167–4185 RSC.
- D. Zhang, M. Wen, P. Zhang, J. Zhu, G. Li and H. Li, Langmuir, 2012, 28, 4543–4547 CrossRef CAS PubMed.
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC.
- H. Xu, R. Pakpoom, S. Ouyang, H. Tong, N. Umezawa, T. Kako and J. Ye, Chem. Mater., 2013, 25, 405–411 CrossRef CAS.
- R. V. Jagadeesh, H. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776–10782 CrossRef CAS PubMed.
- S. K. Meher and G. R. Rao, J. Phys. Chem. C, 2011, 115, 15646–15654 CAS.
- E. B. Castro, C. A. Gervasi and J. R. Vilche, J. Appl. Electrochem., 1998, 28, 835–841 CrossRef CAS.
- R. Li, S. Wang, Z. Huang, F. Lu and T. He, J. Power Sources, 2016, 312, 156–165 CrossRef CAS.
- H. Wang, L. Zhang, X. Tan, C. M. B. Holt, B. Zahiri, B. C. Olsen and D. Mitlin, J. Phys. Chem. C, 2011, 115, 17599–17605 CAS.
- T.-Y. Wei, C. Chen, H. Chien, S. Lu and C.-C. Hu, Adv. Mater., 2010, 22, 347–351 CrossRef CAS PubMed.
- M. Pal, R. Rakshit, A. K. Singh and K. Mandal, Energy, 2016, 103, 481–486 CrossRef CAS.
- B. Cui, H. Lin, J. B. Li, X. Li, J. Yang and J. Tao, Adv. Funct. Mater., 2008, 18, 1441–1447 Search PubMed.
- G. R. R. Sumanta Kumar Meher, J. Phys. Chem. C, 2011, 115, 15646–15654 Search PubMed.
- Y.-K. Hsu, Y.-C. Chen, Y.-G. Lin, L.-C. Chen and K.-H. Chen, Chem. Commun., 2011, 47, 1252–1254 RSC.
- S. K. Meher, P. Justin and G. R. Rao, Nanoscale, 2011, 3, 683–692 RSC.
- I. I. Misnon, R. A. Aziz, N. K. M. Zain, B. Vidhyadharan, S. G. Krishnan and R. Jose, Mater. Res. Bull., 2014, 57, 221–230 CrossRef CAS.
- H. Zhou, D. Li, M. Hibino and I. Honma, Angew. Chem., Int. Ed., 2005, 44, 797–802 CrossRef CAS PubMed.
- Y. Li, B. Tan and Y. Wu, Nano Lett., 2008, 8, 265–270 CrossRef CAS PubMed.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, Nano Lett., 2012, 12, 2559–2567 CrossRef CAS PubMed.
- A. C. Forse, J. M. Griffin, C. Merlet, J. Carretero-Gonzalez, A.-R. O. Raji, N. M. Trease and C. P. Grey, Nat. Energy, 2017, 2, 16216 CrossRef.
- U. M. Patil, R. V. Ghorpade, M. S. Nam, A. C. Nalawade, S. Lee, H. Han and S. C. Jun, Sci. Rep., 2016, 6, 35490 CrossRef CAS PubMed.
- H. Sun, H. M. Ang, M. O. Tadé and S. Wang, J. Mater. Chem. A, 2013, 1, 14427–14442 CAS.
- X. Xiao, X. Liu, H. Zhao, D. Chen, F. Liu, J. Xiang, Z. Hu and Y. Li, Adv. Mater., 2012, 24, 5762–5766 CrossRef CAS PubMed.
- A. D. Jagadale, V. S. Kumbhar, D. S. Dhawale and C. D. Lokhande, Electrochim. Acta, 2013, 98, 32–38 CrossRef CAS.
- R. S. Devan, Y.-R. Ma, R. A. Patil and S.-M. Lukas, RSC Adv., 2016, 6, 62218–62225 RSC.
- M. Noked, E. Avraham, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2009, 113, 21319–21327 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr07879j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Lichchhavi
Sinha
a,
Rupesh S.
Devan
a,
Lichchhavi
Sinha
a,
Rupesh S.
Devan
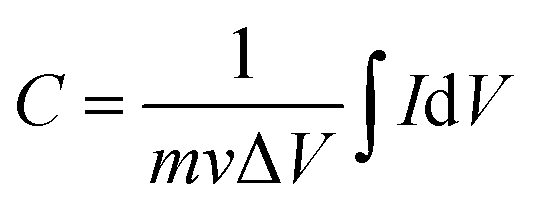
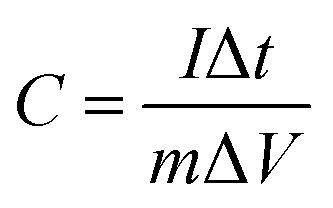
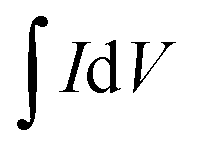 is the area under the CV curve, m is the mass of the active electrode material (g), v is the scan rate (mV s−1), ΔV is the potential window (V), I is the discharge current (mA g−1), and Δt is the discharge time (s).
is the area under the CV curve, m is the mass of the active electrode material (g), v is the scan rate (mV s−1), ΔV is the potential window (V), I is the discharge current (mA g−1), and Δt is the discharge time (s).