
Polymer blend-filled nanoparticle films via monomer-driven infiltration of polymer and photopolymerization†
Yiwei
Qiang
a,
Neha
Manohar
b,
Kathleen J.
Stebe
 b and
Daeyeon
Lee
*b
b and
Daeyeon
Lee
*b
aDepartment of Materials Science and Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
bDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. E-mail: daeyeon@seas.upenn.edu
First published on 18th October 2017
Abstract
Incorporation of nanoparticles into polymer blend films can lead to a synergistic combination of properties and functionalities. Adding a large concentration of nanoparticles into a polymer blend matrix via conventional melting or solution blending techniques, however, is challenging due to the tendency of particles to aggregate. Herein, we report a straightforward approach to generate polymer blend/nanoparticle ternary composite films with extremely high loadings of nanoparticles based on monomer-driven infiltration of polymer and photopolymerization. The fabrication process consists of three steps: (1) preparing a bilayer with a nanoparticle (NP) layer atop a polymer layer, (2) annealing of the bilayer with a vapour mixture of a monomer and a photoinitiator, which undergoes capillary condensation and imparts mobility to the polymer layer and (3) exposing this film to UV light to induce photopolymerization of the monomer. The monomer used in this process is chemically different from the repeat unit of the polymer in the bilayer and is a good solvent for the polymer. The second step leads to the infiltration of the plasticized polymer, and the third step results in a blend of two polymers in the interstices of the nanoparticle layer. By varying the thickness ratio of the polymer and nanoparticle layers in the initial bilayers and changing the UV exposure duration, the volume fraction of the two polymers in the composite films can be adjusted. This versatile approach enables the design and engineering of a new class of nanocomposite films that contain a nanoscale-blend of two polymers in the interstices of a nanoparticle film, which could have combinations of unique mechanical and transport properties desirable for advanced applications such as membrane separations, conductive composite films and solar cells. Moreover, these polymer blend-filled nanoparticle films could serve as model systems to study the effect of confinement on the miscibility and morphology of polymer blends.
Design, System, ApplicationMultiphasic nanocomposite films consisting of polymers and nanoparticles combine the functionalities of individual components and exhibit emergent properties desirable for advanced applications. However, incorporating large concentrations of nanoparticles into polymer blend films without using complicated methods remains a challenge. In this work, we present a method to fabricate polymer blend-filled nanoparticle films by annealing a bilayer of polymer and nanoparticles with the vapour of a monomer and a photoinitiator. Capillary condensation of the monomer in the interstices of a nanoparticle film leads to the infiltration of the polymer into the nanoparticle packing and subsequent photopolymerization results in the generation of polymer blend-filled nanoparticle films. By identifying the key control parameters such as the ratio of the thicknesses of the polymer and nanoparticle layers in the initial bilayer and the UV exposure time, the composition of these ternary nanocomposite films can be varied. This facile and versatile approach enables the design and engineering of novel nanocomposite films with unique mechanical and transport properties, which can be attractive for a variety of applications including sensors, electronic devices, and drug delivery as well as energy conversion and storage systems. |
Introduction
Films made of blends of polymers combine the functionalities and properties of individual polymers to generate novel high-performance materials.1–3 Depending on the properties of the polymers, blend composition and blending techniques, a wide range of physical and mechanical properties can be engineered and tailored to meet the needs of various applications.4–8 Incorporating nanoparticles in polymer blend films offers a versatile path to adding useful functionalities and is desirable in many applications.9–16 For example, conductive polymer composites based on high-density polyethylene and polybutylene terephthalate filled with silver nanoparticles exhibit a sharp and strong resistivity increase desirable for current limiting devices and temperature sensors.9 The addition of nanoparticles in polymer blend-based solar cells improves their power conversion efficiency due to enhanced transport properties.11–13 Incorporation of drug-containing nanoparticles in mucoadhesive polymer blend films has been shown to be a promising approach for buccal delivery of active drugs.16 Moreover, the presence of nanoparticles in polymer blend films has also been shown to stabilize the morphology of polymer blends that are prone to undergo coarsening and macroscopic phase separation due to the inherent immiscibility of polymers.17–21Despite these potential benefits of nanoparticle incorporation in polymer blend films, the manufacture of such multiphasic composite films presents significant challenges. Surface properties of nanoparticles have to be carefully tailored to enhance their dispersion and mixing during processing.22–24 Conventional blending techniques such as melt or solution-phase blending23–26 allow only a small amount of nanoparticles to be stably and uniformly dispersed in polymer matrices because of the strong tendency of nanoparticles to aggregate.27–33 Moreover, these techniques tend to be energy-intensive as high temperature and/or vigorous mechanical mixing is required. The production of highly filled polymer blend films (i.e., with a volume fraction of nanoparticles >0.5) without relying on complicated and energy-intensive processing is especially attractive for applications that require high mechanical strength and a percolating network of nanoparticles in the films.9,34
Recently, a new class of nanocomposite films with extremely high filler fractions has been introduced.35–37 Rather than by directly dispersing nanoparticles into a polymer matrix, these highly filled nanocomposite films are formed by preparing a bilayer consisting of a nanoparticle layer atop a polymer film, and by inducing infiltration of polymer into the interstices between the nanoparticles. We have shown that such polymer-infiltrated nanoparticle films can be fabricated by heating the bilayer above the glass transition temperature of the polymer or by exposing the bilayer to solvent vapour. The latter method leads to the capillary condensation of the solvent in the interstices of the nanoparticle packing and subsequent solvation and infiltration of the polymer into these interstitial voids. This solvent-driven infiltration of polymer (SIP) method takes advantage of molecular-scale interactions between the polymer and the solvent, thus the extent of polymer infiltration can be tailored by varying the quality of the solvent to design highly filled nanocomposite films.37
In this work, we present a method to produce polymer blend-filled nanoparticle films utilizing this SIP technique. In place of a typical solvent, we use a monomer which can undergo capillary condensation and plasticize the underlying polymer layer. Upon the infiltration of the solvated polymer, the monomer is photopolymerized to form polymer blend-filled nanoparticle films. Using in situ ellipsometry, we find that independent control of the volume fraction of each polymer can be achieved by tuning the ratio of the initial thicknesses of the polymer and NP layers as well as the duration of UV exposure during photopolymerization. This method enables the design and engineering of polymer blend/nanoparticle ternary composite films with high loadings of nanoparticles and tuneable compositions, which can be widely applicable to various nanoparticles and polymers for multiple applications.
Experimental methods
Materials
A 50 wt% suspension of 23 nm SiO2 nanoparticles (LUDOX® TM-50) was purchased from Sigma Aldrich. Polystyrene (PS) (MW = 8000 g mol−1, PDI = 1.10) was purchased from Polymer Source, Inc. Methyl methacrylate (MMA) and the photoinitiator, 2-hydroxy-2-methylpropiophenone (HMP), were obtained from Sigma-Aldrich. The UV light source (OmniCure® S1500) was purchased from Excelitas Technologies, and the alpha-SE spectroscopic ellipsometer was obtained from J.A. Woollam Co.Preparation of bilayer films
4 inch silicon wafers were cut to approximately 1 cm × 1 cm squares before film deposition. The wafers were rinsed with 2-propanol and deionized water, and dried with nitrogen. The wafers were treated with oxygen plasma for about 4 min to remove residual organic contaminants. The PS solution was prepared by dissolving PS in toluene, and SiO2 NPs were dispersed in water. The solutions were filtered using 0.45 μm filters prior to use. To generate a NP/polymer bilayer, a PS layer was first deposited onto a silicon substrate using a WS-400BZ-6NPP/Lite spin-coater from Laurell Technologies Corporation, followed by plasma treatment for 10 s to render the film surface hydrophilic. A SiO2 NP layer was then spin-coated on top of the PS layer. Film thicknesses were varied by changing the concentrations of PS and SiO2 NP solutions and/or the rotation speed. For PS films with a thickness between 30 and 230 nm, 1–7 wt% PS solutions were prepared and spin-coated at a rotation speed of 2250 rpm for 1.5 min. To generate a ∼250 nm SiO2 NP layer, 15 wt% SiO2 NP solutions were prepared and spin-coated at 2500 rpm for 2 min.Characterization
In situ monitoring of monomer-driven infiltration of PS into the interstices of a SiO2 nanoparticle packing and photopolymerization of the monomer was performed using a spectroscopic ellipsometer. To fabricate polymer blend-filled nanoparticle films, a bilayer film was placed in a trapezoidal chamber, which has one glass window attached to each side (see Fig. 1). Two containers, one containing 0.4 mL HMP, and another with 0.2 mL MMA, were placed in the chamber. The volume of the chamber is ∼16 mL. The chamber was immediately closed with a top glass slide and sealed with vacuum grease so that vapour could saturate the chamber. The sample was left in the chamber until polymer infiltration was completed and the voids were fully occupied by the solvent and infiltrated polymer (which can be determined from in situ ellipsometry). Subsequently, the sample was exposed to UV light (wavelength: 320–500 nm, intensity = 4 W m−2) for the desired duration (10 min, 30 min or 50 min). Finally, the top lid and the monomer/HMP containers were removed, and the sample was exposed to air for 10 min so that the unreacted monomer and photoinitiator could evaporate. The sample was later placed in a vacuum oven overnight for complete drying. Spectroscopic ellipsometry data was collected from 380 nm to 900 nm at an incident angle of 70° and was fitted using the CompleteEASE software package provided by J.A. Woollam.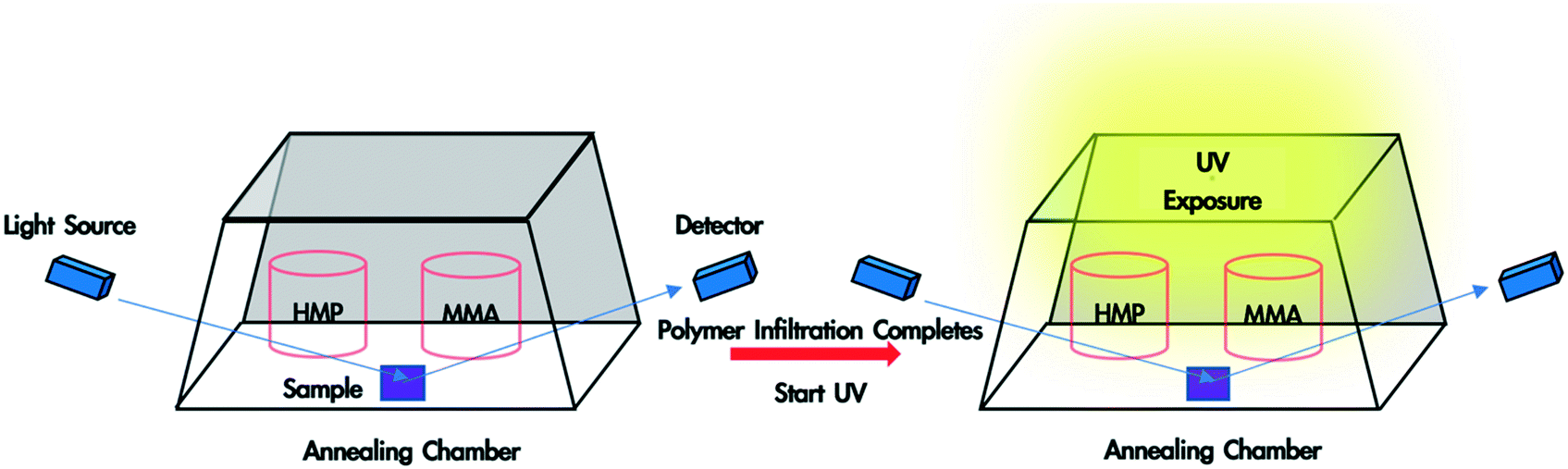 | ||
| Fig. 1 Schematic illustration of the in situ ellipsometry set up. The bilayer sample along with the solvent and photoinitiator is placed in the closed annealing chamber. When monomer-driven infiltration of polymer finishes (determined from dynamic data), the sample is exposed to UV light for a certain period. The upper glass slide and liquid containers are then removed so that unreacted liquids evaporate. | ||
Scanning electron microscopy (SEM) images were taken using a JEOL 7500F HRSEM to observe the morphology of the polymer blend-filled nanoparticle films and measure the film thickness. Before imaging, each sample was coated with a thin gold/palladium layer using a Cressington sputter coater 108 to prevent charging. The samples were imaged at an accelerating voltage of 5 kV, an emission current of 10 μA and a working distance of approximately 7 mm.
Results and discussion
A polymer blend-filled nanoparticle film is prepared by generating a bilayer consisting of a nanoparticle layer on top of a polymer layer, followed by annealing the bilayer in a chamber saturated with the vapour of the monomer (as the solvent) and photoinitiator (Fig. 1). The vapour containing the monomer and photoinitiator condenses within the nanoparticle layer, and in turn diffuses into the polymer layer and plasticizes it, subsequently leading to the infiltration of the polymer into the voids between nanoparticles. Once the monomer-driven infiltration of polymer has been achieved, the bilayer film is exposed to UV light, initiating the photopolymerization of the monomer. This process results in the formation of a blend of two polymers in the nanoparticle packing (Fig. 2).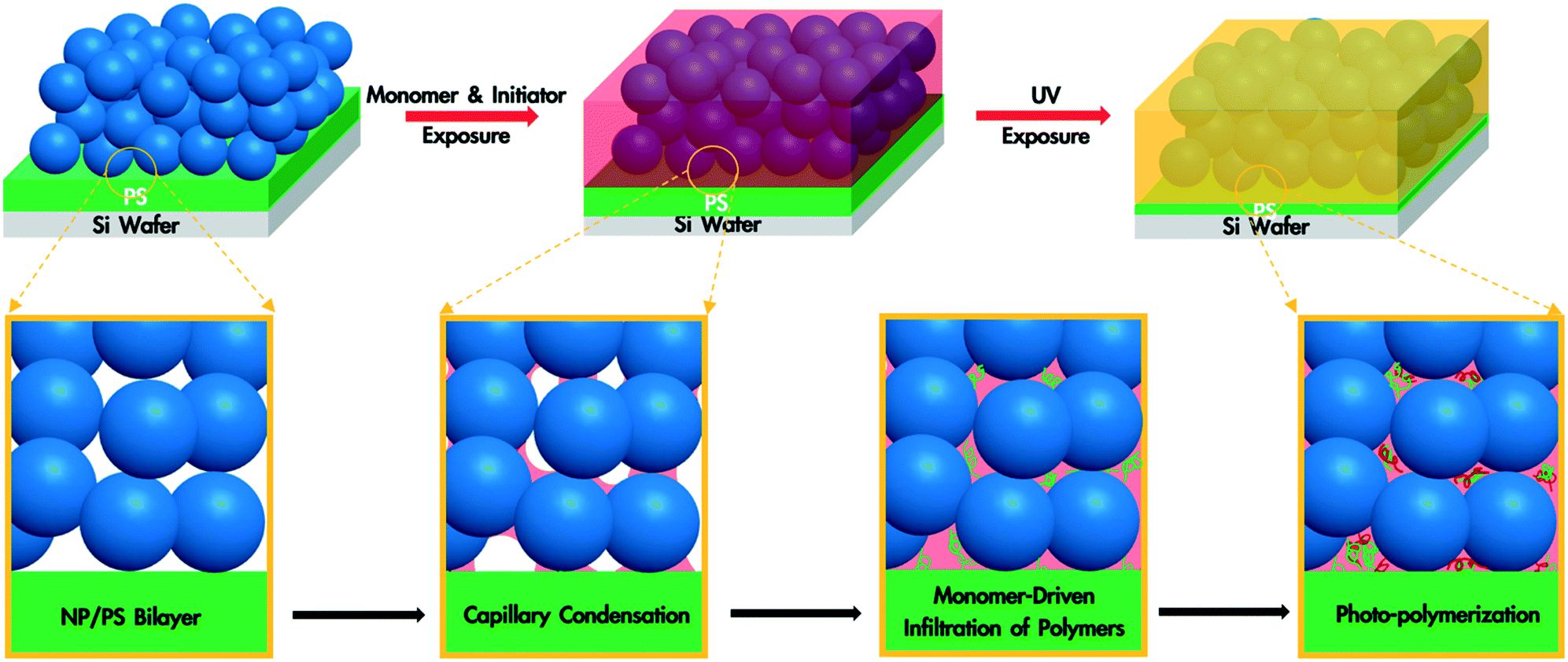 | ||
| Fig. 2 Schematic illustration of a NP/polymer bilayer being annealed with the monomer and photoinitiator vapour, leading to capillary condensation of the monomer and photoinitiator within the NP packing, followed by the infiltration of polymer. The subsequent UV exposure leads to the polymerization of the monomer and a blend of two polymers in the interstices of the packing. | ||
In this work, we use monodisperse polystyrene (PS, MW = 8000 g mol−1), 23 nm silicon dioxide (silica) nanoparticles (SiO2 NPs), methyl methacrylate (MMA) and 2-hydroxy-2-methylpropiophenone (HMP) as the polymer, nanoparticle, monomer and photoinitiator, respectively. MMA is chosen because it is a good solvent for PS (Flory–Huggins interaction parameter between MMA and PS, χMMA–PS < 0.5)38 and can be polymerized under UV exposure, forming a PS/PMMA blend in the nanoparticle packing. We will refer to the volume fraction of PS in the composite film by ∅PS = ΔhPS/hSiO2, where ΔhPS is the change in the thickness of the PS layer and hSiO2 is the thickness of the SiO2 NP layer before annealing. The average void fraction (∅void) of the densely-packed SiO2 NP film is roughly 0.34, as determined via ellipsometry (see the ESI†).
To understand the formation of polymer blend-filled nanoparticle films, we study the two key steps separately: monomer-driven infiltration of polymer and photopolymerization. To study the former process, we prepare bilayers with the thickness of the PS layer around 230 nm and the SiO2 NP layer around 250 nm via sequential spin coating. The volume of the PS layer is more than enough to completely fill the voids in the NP packing. The bilayer sample is placed in an ellipsometry cell that has small aliquots of MMA. After sealing the chamber, in situ spectroscopic ellipsometry is performed to monitor the condensation of the monomer vapour and PS infiltration into the SiO2 NP packing.35,36,39 We use a two-layer Cauchy model (see the ESI†) to show the changes in the refractive indices and thicknesses of the two layers. The bilayer is annealed in the annealing chamber for 120 min and then exposed to air for 10 min.
In the initial stage of annealing (the initial 10 min), we observe increases in the refractive index of the NP layer and in the thickness of the PS layer as shown in Fig. 3. These two phenomena, capillary condensation and swelling of the polymer, occur roughly simultaneously. Capillary condensation occurs when vapour condenses in a porous medium and forms a meniscus at the liquid–vapour interface, which allows equilibrium between the liquid and vapour phases below the saturated vapour pressure. Capillary condensation occurs to a greater extent as the size of the pore decreases.40–42 For 23 nm SiO2 NPs, the interstices of the NP packing are completely flooded with MMA (see the ESI†).
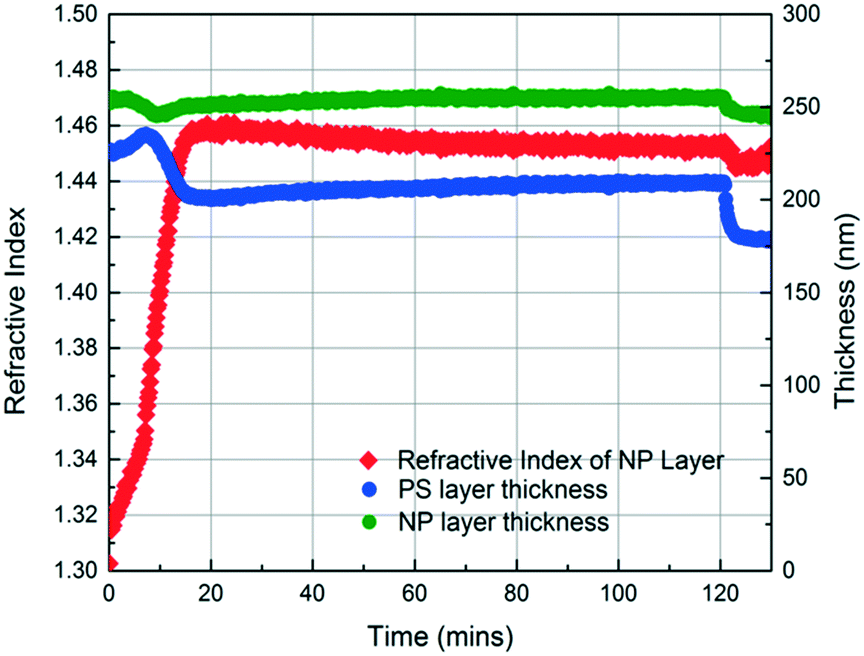 | ||
| Fig. 3 Thickness and refractive index profiles of the SiO2 NP layer and PS layer as a function of annealing time, obtained using in situ spectroscopic ellipsometry while annealing a 250 nm SiO2 NP/225 nm PS bilayer with MMA. The film is annealed with MMA for 120 min and then exposed to air for 10 min. | ||
The increase in the PS thickness indicates that a significant amount of swelling of the PS layer occurs while monomer condensation in the NP packing is taking place. The thickness of the PS layer decreases ∼8 min after exposure to the vapour, indicating the loss of polymer due to its infiltration into the interstices of the NP packing. Concomitantly, the increase in the refractive index of the nanoparticle layer shows a sudden jump, again consistent with the infiltration of the polymer into the interstices. As the polymer infiltrates into the nanoparticle packings, the refractive index of the nanoparticle layer increases because the refractive index of PS (1.58) is higher than that of MMA (1.41). Approximately 20 min after the exposure of the bilayer to the vapour, little change in the refractive index of the nanoparticle layer is observed, whereas the thickness of the PS layer increases gradually due to the diffusion of the condensed solvent from the nanoparticle packing into the polymer layer. The polymer layer continues to swell until the solvent is removed, and the film is exposed to air (∼120 min after the initial exposure). When the residual solvent in the film evaporates, the PS thickness decreases to ∼175 nm. When a trace amount of residual MMA is removed from the sample by drying under vacuum for 12 h, the PS layer has a thickness of 175 nm, indicating the loss of 51 nm from the initial state due to PS infiltration into the NP packing. We expose the bilayers to MMA vapour for 40, 120, and 150 min and find that the filling fractions of PS (∅PS) saturate around 0.21, which suggests that there is not much change after 40 min of exposure to MMA (see the ESI†). During the entire process, the thickness of the NP layer remains more or less constant, indicating that the nanoparticle layer does not undergo swelling and thus the changes in the interparticle spacing is negligible.
To investigate the photopolymerization of condensed MMA in the NP packings, we prepare a SiO2 NP film directly on a silicon wafer (i.e., without a PS sub-layer). To confirm that there is enough photoinitiator condensed in the NP film along with the monomer to initiate polymerization, we first expose the film to HMP without MMA in the annealing chamber. Fig. S3 in the ESI† shows that the refractive index of the NP layer gradually increases indicating capillary condensation of HMP despite its relatively low vapour pressure (0.01 kPa at 25°C). The volume fraction of HMP condensed in the NP layer (∅HMP) at 120 min is 0.12 based on the refractive index change.
Fig. 4a shows the refractive index profiles of a SiO2 NP film which is exposed to MMA and HMP for 120 min and subsequently exposed to UV light for 20 min. The increase in the refractive index of the film results from the condensation of MMA and HMP. The abrupt change in the curve at 120 min corresponds to the moment UV exposure starts, which we believe indicates the onset of MMA polymerization as the refractive index of PMMA (1.49) is higher than that of MMA (1.41). The refractive index continues to increase during the 20 min UV exposure. After drying the film under vacuum for 12 h, the volume fraction of PMMA in the NP layer (∅PMMA) is 0.11 based on the refractive index change. Fig. 4b shows that ∅PMMA can be varied by changing the UV exposure duration, and ∅PMMA can reach up to 0.16 when the film is exposed to UV for 110 min.
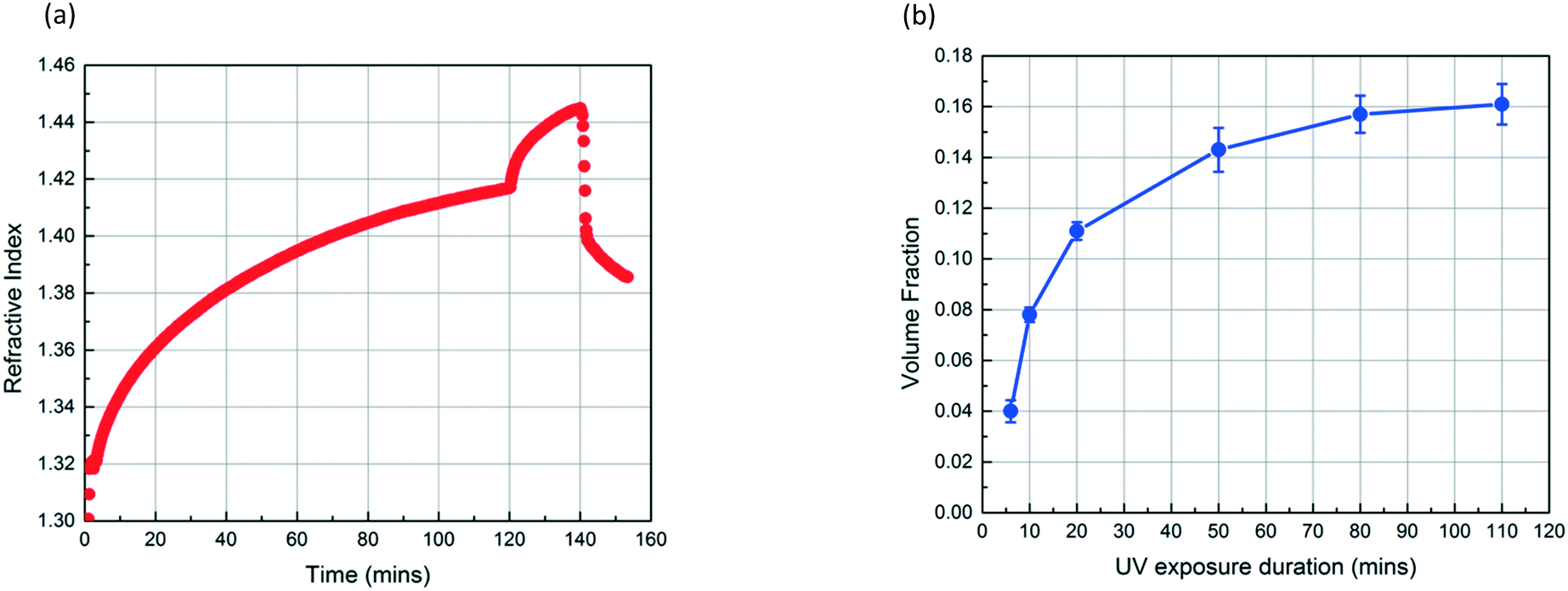 | ||
| Fig. 4 (a) Refractive index profile of a ∼300 nm SiO2 NP film without a PS layer as a function of time, obtained using in situ spectroscopic ellipsometry while exposing the NP film to MMA and HMP. UV exposure starts at 120 min and lasts for 20 min. Subsequently, the film is exposed to air for 10 min. (b) The volume fraction of PMMA in NP films which are exposed to UV for different periods. | ||
These results confirm that the PS infiltrates into the interstices of the NP packing upon annealing with MMA vapour and that, independently, MMA that is condensed within these interstices can be polymerized via exposure to UV in the presence of HMP as the photoinitiator. Based on these findings, we generate a PS/PMMA polymer blend in the interstices of a SiO2 nanoparticle packing. Inspired by the undersaturated capillary rise infiltration (UCaRI) method that demonstrated the fabrication of porous polymer-infiltrated NP films,36 we use thin PS films to control the amount of PS that infiltrates the NP layer and in turn the ratio of PS and PMMA in the final composite film. Fig. 5 shows the refractive index and PS thickness profiles of a 272 nm SiO2 NP/36 nm PS bilayer film exposed to MMA and HMP and subsequently to UV light. PS infiltration completes at around 20 min. During the first 20 min, the increase in the refractive index of the NP layer is due to the condensation of MMA and HMP as well as the infiltration of PS. After 20 min, the refractive index increases much slower and is solely due to vapour condensation. Similar to what is observed in Fig. 4a, when the UV exposure starts at 60 min, there is an abrupt increase in the refractive index which likely corresponds to the polymerization of MMA within the interstices. After exposing the film to air (at 90 min), there is a slight decrease in the refractive index of the NP layer due to the evaporation of residual MMA. In comparison, there is a sharp decrease in the refractive index of the NP layer if the bilayer film is not exposed to UV light as there is more unreacted monomer condensed in the films.
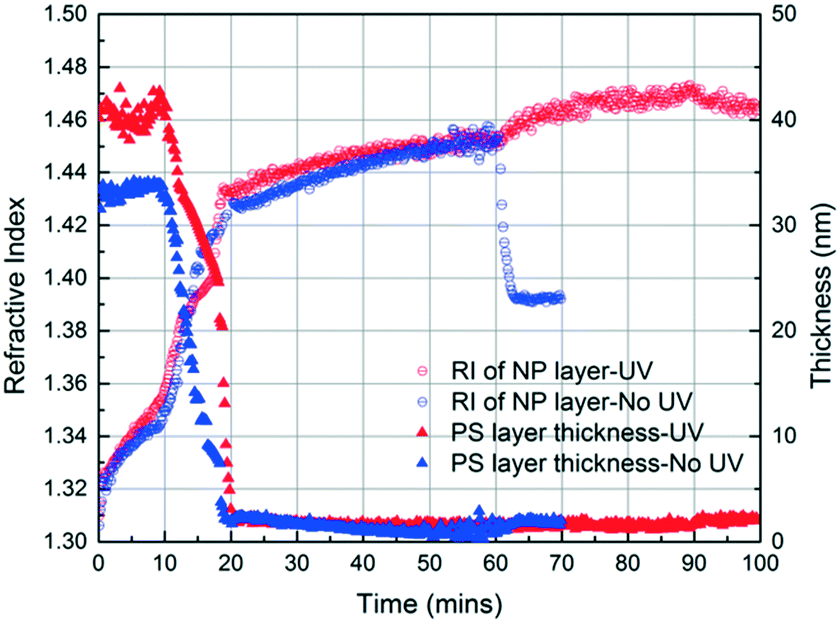 | ||
| Fig. 5 Refractive index and PS thickness of two SiO2 NP/PS bilayers as a function of time, obtained using in situ spectroscopic ellipsometry while exposing the films to MMA and HMP. One film is exposed to UV (UV exposure started at 60 min) for 30 min. Subsequently, this film is exposed to air for 10 min. The other is not exposed to UV and is exposed to air after 60 min of MMA and HMP exposure. | ||
The filling of the interstices by polymers can be further confirmed by SEM images. Compared to the as-prepared sample (Fig. 6a), which displays clear outlines of NPs, the polymers within the interstices of the NP layer in annealed samples (Fig. 6b and c) appear to cover the NPs, especially for the UV exposed sample. Furthermore, the decrease in the thickness of the PS layer (from ∼60 nm to ∼15 nm) in Fig. 6b and c indicates the infiltration of PS. The average distance between the NP centers (d ∼ 26 nm for the as-prepared samples) does not change significantly after PS infiltration (d ∼ 26 nm) and MMA polymerization (d ∼ 27 nm). Based on the ellipsometry results, the thickness of the SiO2 NP layer remains constant after PS infiltration and MMA polymerization. Thus, we believe that NPs do not undergo rearrangements and do not move into the polymer layer. Once fabricated, these polymer blend-infiltrated nanoparticle films are very stable under ambient conditions and do not show any significant changes in their morphology, likely due to the high glass transition temperature of the two polymers.
 | ||
| Fig. 6 Cross-sectional SEM images of 250 nm SiO2 NP/60 nm PS bilayers. (a) shows an as-prepared bilayer; (b) shows a bilayer after PS infiltration and (c) shows a bilayer after PS infiltration and UV exposure for 60 min. Inset: Top-down SEM images. All scale bars are 100 nm. | ||
Depending on the intended application of polymer blend-filled nanoparticle films, it is important to engineer the composition, namely the volume fraction of each polymer in the films. To vary the volume fraction of PS, bilayers of different ΔhPS/hSiO2 values are prepared and exposed to the vapour of MMA and HMP and subsequently irradiated with UV for 30 min. ∅PS is calculated from the thickness change of the PS layer, and ∅PMMA is calculated from the increase in the refractive index of the NP layer. ∅void is obtained by subtracting the volume fractions of PS and PMMA from the initial void fraction (see the ESI† for more details). As shown in Fig. 7a, when ΔhPS/hSiO2 is increased from 0.08 to 0.24, ∅PS increases from 0.065 to 0.205, and the ∅PMMA of these films ranges between 0.071 and 0.047. Correspondingly, the void fraction decreases from 0.213 to 0.075. These results clearly indicate that the amount of the infiltrated polymer can be changed by simply varying the ratio of the initial thicknesses of the PS layer and the NP layer, which also changes the ratio of PS to PMMA and the porosity of the final composite film.
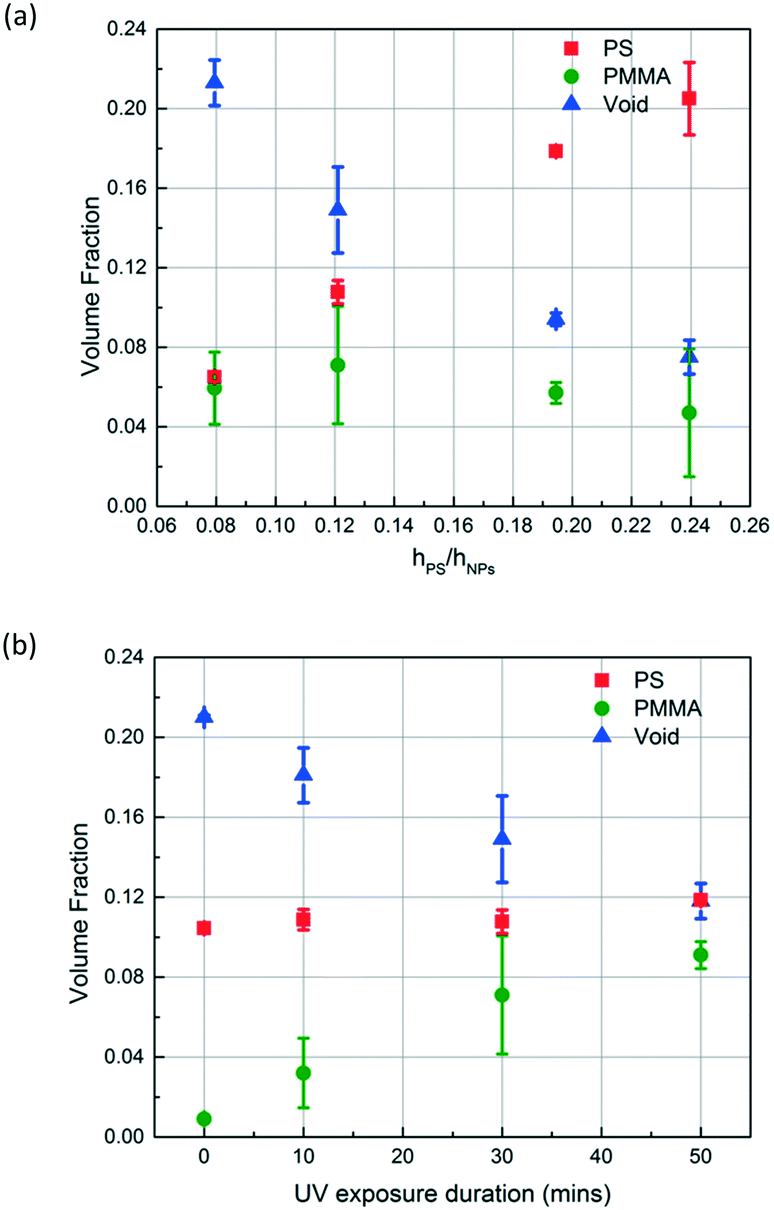 | ||
| Fig. 7 The volume fraction of each component (PS, PMMA and void) in polymer blend-filled nanoparticle films upon (a) varying the ratio of initial thicknesses of the PS layer and the NP layer in the initial bilayer (from ∼0.08 to ∼0.24). The bilayers are exposed to MMA and HMP and later exposed to UV for 30 min; (b) varying the UV exposure duration while keeping the ratio of the initial thicknesses of the PS layer and the NP layer in the prepared bilayer constant (∼0.12). | ||
To control the volume fraction of PMMA in the polymer blend composite (∅PMMA) while maintaining the concentration of PS in the composite (∅PS) constant, we prepare bilayers with similar ΔhPS/hSiO2 ratios (roughly 0.12) and expose them to MMA and HMP and subsequently to UV for different periods of time (10, 30, and 50 min) (Fig. 7b). As expected, ∅PMMA increases with the increase of UV exposure duration while ∅PS remains roughly constant at 0.11. Correspondingly, the void fraction decreases from 0.21 to 0.12.
Conclusions
We have successfully fabricated polymer blend-filled nanoparticle films with extremely high loadings of nanoparticles based on monomer-driven infiltration of polymer and photopolymerization. This facile and potentially scalable approach allows independent control over the volume fraction of each polymer by tuning the initial thickness ratio of the polymer layer to the NP layer and the duration of UV exposure. While this work focuses on the generation of PS/PMMA/SiO2 NP ternary composite systems, we believe this approach will be widely applicable to different sets of polymers and nanoparticles. Such composite films will have useful functionalities for various applications such as conductive polymer composites, drug delivery platforms, gas separation membranes and photovoltaics. Moreover, this unique approach presents opportunities to study how nano-confinement (i.e., in the interstitial voids in nanoparticle packings) affects the morphology and phase behaviour of macroscopically immiscible polymer blends. Our ongoing work also includes the investigation of the composition–structure–property relationship of composite films using different monomers, polymers and nanoparticles.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by NSF CBET-1449337 and CMMI-1662695. We also thank Jyo Lyn Hor (University of Pennsylvania) for her assistance with scanning electron microscopy.References
- P. Pötschke and D. R. Paul, J. Macromol. Sci., Polym. Rev., 2003, 43, 87–141 CrossRef.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Solid State Sci., 2013, 24, 36–43 CrossRef CAS.
- I. Prosycevas, S. Tamulevicius and A. Guobiene, Thin Solid Films, 2004, 453–454, 304–311 CrossRef CAS.
- D. Mori, H. Benten, I. Okada, H. Ohkita and S. Ito, Energy Environ. Sci., 2014, 7, 2939–2943 CAS.
- H. Li, Y. J. Hwang, T. Earmme, R. C. Huber, B. A. Courtright, C. O'Brien, S. H. Tolbert and S. A. Jenekhe, Macromolecules, 2015, 48, 1759–1766 CrossRef CAS.
- E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 12, 2799–2803 CrossRef PubMed.
- F. S. Li, Y. S. Wu, J. Chou, M. Winter and N. L. Wu, Adv. Mater., 2015, 27, 130–137 CrossRef CAS PubMed.
- H. Minehara, K. Dan, Y. Ito, H. Takabatake and M. Henmi, J. Membr. Sci., 2014, 466, 211–219 CrossRef CAS.
- A. Rybak, G. Boiteux, F. Melis and G. Seytre, Compos. Sci. Technol., 2010, 70, 410–416 CrossRef CAS.
- H. Dong, E. Frey, A. Gandelman and W. E. Jones, Chem. Mater., 2006, 18, 2008–2011 CrossRef CAS.
- S. Nam, J. Kim, H. Kim and Y. Kim, J. Nanosci. Nanotechnol., 2011, 11, 5733–5736 CrossRef CAS PubMed.
- J.-L. Wu, F.-C. Chen, Y.-S. Hsiao, F.-C. Chien, P. Chen, C.-H. Kuo, M. H. Huang and C.-S. Hsu, ACS Nano, 2011, 5, 959–967 CrossRef CAS PubMed.
- L. Lu, Z. Luo, T. Xu and L. Yu, Nano Lett., 2013, 13, 59–64 CrossRef CAS PubMed.
- H. Yan, L. Yang, Z. Yang, H. Yang, A. Li and R. Cheng, J. Hazard. Mater., 2012, 229–230, 371–380 CrossRef CAS PubMed.
- J. Lee, PhD thesis, The University of Akron, 2015 Search PubMed.
- P. Kraisit, S. Limmatvapirat, M. Luangtana-Anan and P. Sriamornsak, Asian J. Pharm. Sci. DOI:10.1016/j.ajps.2017.07.006 , in press.
- M. Salzano de Luna and G. Filippone, Eur. Polym. J., 2016, 79, 198–218 CrossRef CAS.
- K. C. Bryson, T. I. Löbling, A. H. E. Müller, T. P. Russell and R. C. Hayward, Macromolecules, 2015, 48, 4220–4227 CrossRef CAS.
- M. Kong, Y. Huang, Y. Lv, S. Wang, Q. Yang and G. Li, Polymer, 2014, 55, 4348–4357 CrossRef CAS.
- L. C. Costa, J. D. Ambrósio, M. A. Chinelatto and E. H. Junior, J. Appl. Polym. Sci., 2017, 134, 45030 CrossRef.
- V. Ojijo, S. S. Ray and R. Sadiku, ACS Appl. Mater. Interfaces, 2013, 5, 4266–4276 CAS.
- H. Wang, Z. Fu, X. Zhao, Y. Li and J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 14358–14370 CAS.
- S. Huang, L. Bai, M. Trifkovic, X. Cheng and C. W. Macosko, Macromolecules, 2016, 49, 3911–3918 CrossRef CAS.
- L. Elias, F. Fenouillot, J. C. Majeste and P. Cassagnau, Polymer, 2007, 48, 6029–6040 CrossRef CAS.
- S. P. Pawar, S. Stephen, S. Bose and V. Mittal, Phys. Chem. Chem. Phys., 2015, 17, 14922–14930 RSC.
- H.-J. Chung, J. Kim, K. Ohno and R. J. Composto, ACS Macro Lett., 2012, 1, 252–256 CrossRef CAS.
- J.-C. Huang, Adv. Polym. Technol., 2002, 21, 299–313 CrossRef CAS.
- E. Kontou, M. Niaounakis and P. Georgiopoulos, J. Appl. Polym. Sci., 2011, 122, 1519–1529 CrossRef CAS.
- N. Jouault, D. Lee, D. Zhao and S. K. Kumar, Adv. Mater., 2014, 26, 4031–4036 CrossRef CAS PubMed.
- N. Jouault, D. Zhao and S. K. Kumar, Macromolecules, 2014, 47, 5246–5255 CrossRef CAS.
- H. Chen and E. Ruckenstein, J. Chem. Phys., 2009, 131, 244904 CrossRef PubMed.
- H. Chen and E. Ruckenstein, Polymer, 2010, 51, 5869–5882 CrossRef CAS.
- H. Chen and E. Ruckenstein, J. Colloid Interface Sci., 2011, 363, 573–578 CrossRef CAS PubMed.
- S. Abdolmohammadi, S. Siyamak, N. A. Ibrahim, W. M. Yunus, M. Z. Rahman, S. Azizi and A. Fatehi, Int. J. Mol. Sci., 2012, 13, 4508–4522 CrossRef CAS PubMed.
- Y. R. Huang, Y. Jiang, J. L. Hor, R. Gupta, L. Zhang, K. J. Stebe, G. Feng, K. T. Turner and D. Lee, Nanoscale, 2015, 7, 798–805 RSC.
- J. L. Hor, Y. Jiang, D. J. Ring, R. A. Riggleman, K. T. Turner and D. Lee, ACS Nano, 2017, 11, 3229–3236 CrossRef CAS PubMed.
- N. Manohar, K. J. Stebe and D. Lee, ACS Macro Lett., 2017, 6, 1104–1108 CrossRef CAS.
- T. F. Tadros, in Emulsions: Formation, Stability, Industrial Applications, Walter de Gruyter GmbH & Co KG, Berlin, 2016, ch. 9, p. 119 Search PubMed.
- E. Langereis, S. B. S. Heil, H. C. M. Knoops, W. Keuning, M. C. M. van de Sanden and W. M. M. Kessels, J. Phys. D: Appl. Phys., 2009, 42, 073001 CrossRef.
- Z. Gemici, P. I. Schwachulla, E. H. Williamson, M. F. Rubner and R. E. Cohen, Nano Lett., 2009, 9, 1064–1070 CrossRef CAS PubMed.
- H. Yang, P. Jiang and B. Jiang, J. Colloid Interface Sci., 2012, 370, 11–18 CrossRef CAS PubMed.
- F. Li and A. Stein, J. Am. Chem. Soc., 2009, 131, 9920–9921 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic ellipsometry data fitting and modeling; calculating volume fraction of each component in composite films. See DOI: 10.1039/c7me00099e |
| This journal is © The Royal Society of Chemistry 2018 |