
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequencing of human genomes extracted from single cancer cells isolated in a valveless microfluidic device†
Rodolphe
Marie
 *a,
Marie
Pødenphant‡
a,
Kamila
Koprowska‡
b,
Loic
Bærlocher‡
c,
Roland C. M.
Vulders‡
d,
Jennifer
Wilding
b,
Neil
Ashley
b,
Simon J.
McGowan
b,
Dianne
van Strijp
d,
Freek
van Hemert
d,
Tom
Olesen
e,
Niels
Agersnap
e,
Brian
Bilenberg
f,
Celine
Sabatel
g,
Julien
Schira
c,
Anders
Kristensen
a,
Walter
Bodmer
b,
Pieter J.
van der Zaag
d and
Kalim U.
Mir
h
*a,
Marie
Pødenphant‡
a,
Kamila
Koprowska‡
b,
Loic
Bærlocher‡
c,
Roland C. M.
Vulders‡
d,
Jennifer
Wilding
b,
Neil
Ashley
b,
Simon J.
McGowan
b,
Dianne
van Strijp
d,
Freek
van Hemert
d,
Tom
Olesen
e,
Niels
Agersnap
e,
Brian
Bilenberg
f,
Celine
Sabatel
g,
Julien
Schira
c,
Anders
Kristensen
a,
Walter
Bodmer
b,
Pieter J.
van der Zaag
d and
Kalim U.
Mir
h
aDepartment for Micro and Nanotechnology, Technical University of Denmark, Ørsteds Plads Building 345C, 2800 Kgs. Lyngby, Denmark. E-mail: rodolphe.marie@nanotech.dtu.dk; Fax: +45 45 88 77 62; Tel: +45 45 25 57 00
bWeatherall Institute of Molecular Medicine, Department of Oncology, John Radcliffe Hospital, Headington, Oxford OX3 9DS, UK
cFasteris SA, Chemin du Pont-du-Centenaire 109, CH-1228 Plan-les-Ouates, Switzerland
dPhilips Research Laboratories, High Tech Campus, 11 5656 AE Eindhoven, The Netherlands
ePhilips Biocell, Gydevang 42, 3450 Lillerød, Denmark
fNIL Technology ApS, Diplomvej 381, 2800 Kgs. Lyngby, Denmark
gDiagenode SA, Liege Science Park, Rue Bois Saint-Jean, 3, 4102 Seraing, Belgium
hXGenomes, Pagliuca Harvard Life Lab, 127 Western Ave, Boston, MA 02134, USA
First published on 6th June 2018
Abstract
Sequencing the genomes of individual cells enables the direct determination of genetic heterogeneity amongst cells within a population. We have developed an injection-moulded valveless microfluidic device in which single cells from colorectal cancer derived cell lines (LS174T, LS180 and RKO) and fresh colorectal tumors have been individually trapped, their genomes extracted and prepared for sequencing using multiple displacement amplification (MDA). Ninety nine percent of the DNA sequences obtained mapped to a reference human genome, indicating that there was effectively no contamination of these samples from non-human sources. In addition, most of the reads are correctly paired, with a low percentage of singletons (0.17 ± 0.06%) and we obtain genome coverages approaching 90%. To achieve this high quality, our device design and process shows that amplification can be conducted in microliter volumes as long as the lysis is in sub-nanoliter volumes. Our data thus demonstrates that high quality whole genome sequencing of single cells can be achieved using a relatively simple, inexpensive and scalable device. Detection of genetic heterogeneity at the single cell level, as we have demonstrated for freshly obtained single cancer cells, could soon become available as a clinical tool to precisely match treatment with the properties of a patient's own tumor.
1 Introduction
Standard molecular methods that analyse DNA sequences in populations of cells need sufficiently deep sequencing to detect heterogeneity at any given location of the genome and do not clearly define the co-occurrence of mutations in a given cell, and so do not define precisely the genetic heterogeneity in a tissue, especially cancers. Cancers arise from a somatic evolutionary process in which mutations, or relatively stable epigenetic changes, are successively selected for and therefore occur with relatively high frequency in the population of cancer cells. It is these genetic and epigenetic changes that determine the properties of a cancer and so are the major determinants of prognosis and of the responses to different treatments. With the extraordinary development of DNA sequencing technology, there is now extensive data on the types and frequencies of the major, so called ‘driver’, mutations found in a wide variety of cancers, and in some cases very clear evidence of the relationship between the mutational content of a cancer, and its response to therapy. As a result, there is now great interest in DNA sequencing of single cancer cells to determine the nature and extent of clonal genetic heterogeneity in a given cancer.Treatment can then be directed at the different clones that co-exist in the cancer and thus single cell DNA sequencing1 becomes an extremely important tool for matching the treatment of a cancer to its genetic make up. This is the essence of precision medicine as applied to cancer treatment. There is also an interest in single cell mRNA analysis,2 which can help to identify gene expression differences, due mainly to DNA methylation as well as interest in examining methylation directly.3,4 There has therefore been increasing focus on the development of methods that obtain molecular information from single cells by isolating and sequencing their DNA and mRNA content.5–7 Similarly, in metagenomics, where bacteria, fungi and other microbes may not be culturable, a robust single cell analysis is important to evaluate the genomes of the distinct microbes present in a sample.8 In addition to untangling heterogeneity, the single cell methods are relevant to cases when only a small number of cells is available, for example in the analysis of circulating tumour cells9,10 and circulating fetal cells in maternal blood.11 A single diploid human cell contains around 7 picograms of genomic DNA and some form of amplification is therefore needed to obtain the amounts of material necessary for current sequencing methods. Amplification by multiple displacement amplification (MDA)12 and PCR based methods such as DOP-PCR,13 Picoplex14 and, multiple annealing and looping-based amplification cycles (MALBAC)15 have been used relatively effectively to do single cell whole genome DNA sequencing. Genome coverage of >90% has been claimed to be routinely obtained from single cells using MDA and MALBAC, with a MDA kit adapted for single cells reportedly giving superior all round performance.13 However, issues around uniformity of coverage, allelic dropout, false positives (amplification and/or sequencing errors) and unmappable reads (e.g. from primer-dimers), remain.16
The MDA process results in uneven coverage across the genome. Some of the amplification biases are presumed to be a result of stochastic effects due to the sampling of a small number of molecules. In MALBAC amplification bias can be corrected by normalizing the GC content.17 Another approach to dealing with this problem is the use of barcoding or identification tags.18–20
Contamination, if not carefully controlled can lead to difficulty in interpreting results and limits the sequencing capacity that is available for a single cell of interest. To combat this, single cell genomics is preferably conducted in a clean room,16 in microwells,21 or in a microfluidic device.8,12,17 Contamination can also be assessed by the use of appropriate known genetic markers. Using a microfluidic device provides the containment of cells and their immediate lysis products, and the controlled amplification within the device limits the loss of material when handling small volumes.
Existing microfluidic devices8,17 requiring multiple-PDMS layers,22 one layer for the passage of fluids and another for valves to control the fluids through the device, are difficult and expensive to manufacture because PDMS casting is not a scalable industrial process. We introduce a novel valve-less microfluidic device for single cell genomics that is manufactured in a thermoplastic material by injection moulding, a process that is scalable at low cost.23 Our chip design is based on a hydrodynamic cell trap24 derived from a previously described device for cell culture.25 Our device design enables the process to be carried out on an optical microscope or in “Cell-O-Matic” a specially built single cell processing instrument (Philips BioCell), in either case allowing us to monitor both cell trapping and genome extraction.
Our single cell sequencing data obtained using the Cell-O-Matic instrument show that we can achieve reasonable levels of whole genome coverage in a significant proportion of cells. Our results compare well with other reported whole genome sequencing from single cells using instrumentation in which the amplification is performed in nanoliter-reaction chambers. In our device only the DNA extraction occurs in a sub-nanoliter volume of solution, while the amplification is performed by adding microliter volumes of reagents in the device outlet. We conclude that the critical step in single cell whole genome amplification with regard to sequence allelic dropout, contamination and genome coverage is to extract DNA in sub-nanoliter volumes in the confinement of the microfluidic device, while performing the amplification in such small volumes may only be required for reduction of reagent consumption, but at the cost of higher device complexity and cost.
2 Material and methods
2.1 Device fabrication
The device is fabricated by injection moulding of TOPAS 5013, a cyclic olefin copolymer with a glass temperature of 130 °C, as described elsewhere26 but with some modifications. First, the microchannel network is defined to have a depth of 30 μm in a silicon substrate by UV lithography and reactive ion etching. All dimensions of the microchannel network can be found in Fig. S1a–f.† Next, a Ni/V seed layer is deposited on the silicon master and nickel is electroplated at a final thickness of 300 μm. Silicon is removed by KOH etching. The nickel shim is cut to fit in the mould of the injection moulder (Engel, Germany). The mould creates a 2 mm-thick, 50 mm diameter disc replica of the shim and 12 holes through the disc used to connect the microfluidics. It also creates for each hole, a female LUER connector on the side opposite the microfluidics side. The temperature of the mould is regulated such that injection moulding is performed using a variotherm process27 allowing the polymer part to be moulded and removed from the mould at different temperatures. The shim is replicated using a mould temperature of 115 °C, a shim temperature of 155 °C, a holding pressure of 1150 bar and a cooling time of 14 seconds so the demoulding temperature is below 126 °C. The microfluidics chips are sealed with a 150 μm-thick TOPAS 5015 foil using UV assisted thermal bonding. The initial roughness of the foil is reduced by a hot embossing process on a hydraulic press (P/O/Weber, Germany) at 140 °C and 5.1 MPa using two smooth nickel discs electroplated from silicon wafers. The glass transition temperature (Tg) of TOPAS 5013 is 130 °C. The devices and lids are exposed to UV-light from a mercury arc lamp for 30 s and then placed in an aluminium holder that fits to the LUER fittings. A smooth nickel disc and a thin PDMS plate are placed on top of the lid to ensure a uniform pressure across the device during bonding at 125 °C and 1.5 MPa for 3 minutes.2.2 Instrument
The microfluidic device can either be mounted on a conventional epi-fluorescence microscope or on a custom designed instrument. The instrument is a modified Philips BioCell Fluidscope7 (Fig. S1g and S2†). In brief, the single use microfluidic chip is mounted on a stage equipped with translation (y-axis) as well as temperature control via three Peltier elements. The temperature inside the wells of the chip is calibrated and regulated by a proportional–integral–derivative (PID) controller using the temperature of the stage as input. The lid closing the microfluidic chip wells is connected to a multi-channel pressure controller (MFCS-EZ, 300 mbar range, Fluigent). The chip is imaged using an objective (10×, NA 0.45 Wild Heerbrugg) mounted on a translation stage (x-axis) and focusing by z-translation. Epi-fluorescence imaging is performed with an excitation at 470 nm (LED, Thorlabs model M470L3) equipped with collimator lenses. A dual band filter cube (Semrock, excitation filter model 733-495/605-Di01-25 × 36, dichroic mirror model 733-474/23-25, emission filter model 733-527/645-25) allows imaging of green fluorescence and bright field. The bright-field illuminator delivers light from the top side of the device through a window in the lid using a LED with center wavelength of 505 nm and a 20 nm bandwidth. A CMOS imaging sensor (Fairchild, CIS1910) with 1920 × 1080 pixels, and pixel size 6.5 μm, is used with a home-built image board. Finally, all the elements are controlled through software which guides the user through a pre-established workflow for priming, cell capture, lysis and amplification.2.3 Device operation (see Protocol S3† for the detailed operation procedure)
Solutions are pipetted into the LUER connectors used as reservoirs containing up to 50 μL of solution. Air pressure supplied by the pressure controller attached to the wells drives the flow inside the device. We apply pressure to the cell inlet as well as the buffer inlets B1 and B2 (Fig. 1) while the waste and the trap outlets are always left at atmospheric pressure. For cell capture, pressures in the range of 5 to 10 mbar were applied, corresponding to sample flow rates of 2.9–5.7 μL h−1. Details of the pressure settings used are given in the Protocol S3.†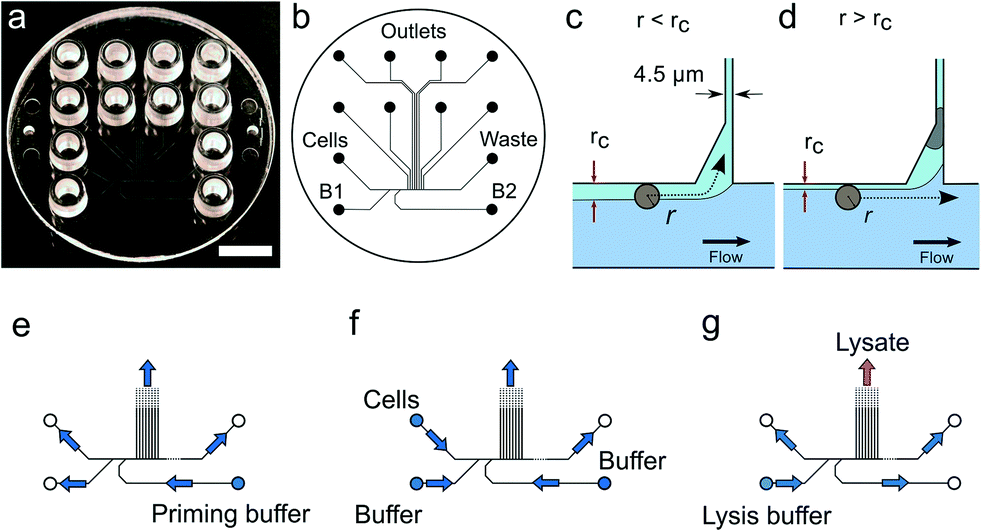 | ||
| Fig. 1 Device design and operation. (a) Image of the single use polymer device. Scale bar is 1 cm. (b) Microfluidics layout. (c) Conditions for cell trapping an unoccupied trap r < rc. (d) The trapped cell reduces the flow through the trap such that for the next incoming cell, r > rc. (e–g) Flow directions in the device under priming, cell capture and lysis. | ||
2.4 Wet lab
The colorectal cell lines used in this study were provided by the Department of Oncology located at the Weatherall Institute of Molecular Medicine (Oxford). Experiments were conducted across two sites, Oxford and Eindhoven where the LS174T, LS180 and RKO cells were maintained in the same fashion. Briefly, the LS174T and LS180 cells were cultured in DMEM (Gibco, Life Technologies). RKO was cultured in RPMI (Gibco, Life Technologies) and all media were supplemented with 10% fetal bovine serum (FBS), (Gibco, Life Technologies), 2 mM Glutamax (Gibco Life Technologies), 1% penicillin/streptomycin (Gibco, Life Technologies). Cells were grown at 37 °C in a humidified incubator containing 10% CO2 in air. When cells reached sub-confluency they were routinely passaged 2–3 times per week via dissociation with 0.25% trypsin containing 0.04% EDTA (Gibco, Life Technologies) upon two rinse steps with phosphate buffered saline (PBS) (Gibco, Life Technologies). In addition the cells were periodically tested for Mycoplasma spp. infection (Agilent 302107).Cells from colorectal cancer cell lines LS174T, LS180 or RKO (concentration: 6 × 105 cells per mL) were stained with 1 mM calcein AM and suspended in BD FACSFlow buffer (Becton Dickinson). After cell capture, the trap occupancy was checked by bright field and fluorescence imaging of the calcein signal. After trapping cells, the B1 and B2 inlets were emptied leaving negligible volumes in the outlets.
2.5 Library preparation and sequencing
Samples for sequencing were quantified using a Qubit fluorometer instrument before starting the library preparation procedure. Depending on the initial sample concentration, the library preparation was done using the Illumina Nextera or the Illumina Nextera XT kits. Sequencing reactions were performed for all samples using Illumina HiSeq technology as 2 × 125 base pairs high-output runs.2.6 Bioinformatics
All libraries sequenced were then mapped against the human genome GRCh37. We report on the percentage of reads mapped to the human genome (Fig. 2a) and the total number of reads (Fig. 2b). We also use the cumulative fraction of bases covered to generate Lorenz plots (Fig. 3) and the coverage plots (Fig. 4). For all samples, allelic dropout estimates (Fig. 5 and 6) were based on a set of validated heterozygous variants, selected for each cell line: 12 single nucleotide polymorphisms (SNPs) were used for the LS174T and LS180 cells, 13 for the RKO cells (see Table S2†). The known variants were searched for in the raw variant results, namely without applying any filter on the variant quality. All selected variants were correctly detected in the respective bulk samples as heterozygous variants. For single cells, a drop of a heterozygous variant to a homozygous call would reflect dropout of a single allele, while absence of any call would represent a dropout of both alleles (resulting in no coverage of the position). The procedure used for generating Fig. 5 and 6 is described in detail in the Results section. From the Lorenz and the coverage plot we calculate the Gini coefficient G and the evenness score E respectively. For the latter, the normalized coverage curve is used and scaled by the maximum fraction of genome covered in a bulk sequencing run of 10/9.136. This is done to account for the fact that in the bulk sequencing of LS174T, 8.36% of the bases remain uncovered.7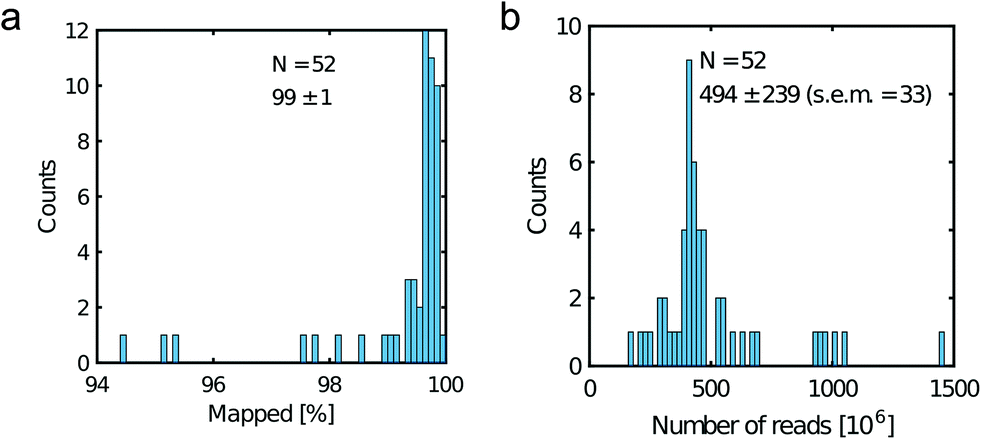 | ||
| Fig. 2 Read metrics. a) Percentage of mapped reads and b) total number of reads. Legend displays the mean, the standard deviation and the standard error of the mean (s.e.m). | ||
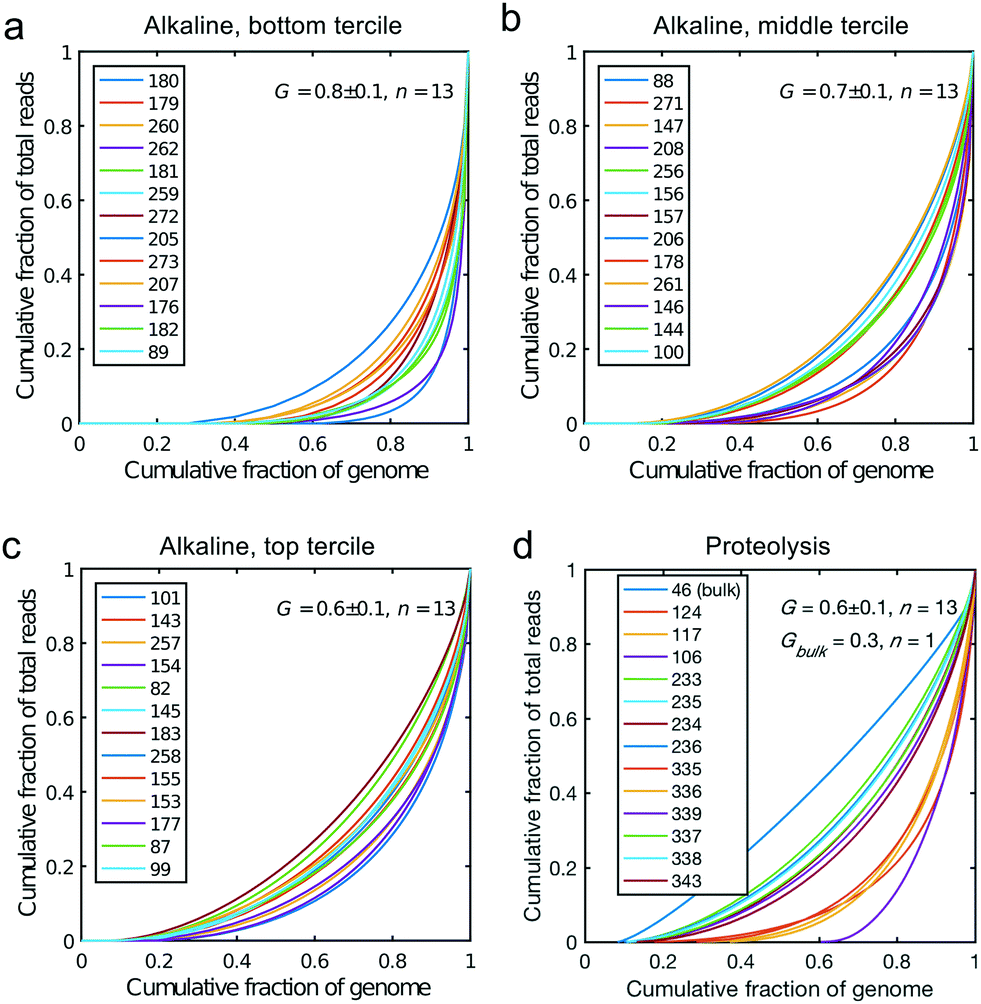 | ||
| Fig. 3 Lorenz plots for the cells processed by alkaline lysis in Oxford, in three groups of n = 13 cells: (a) bottom, (b) middle and (c) top tercile according to the percentage of non-covered bases in the genome. (d) Lorenz plot for the single cells processed by proteolysis in Eindhoven, n = 13 cells. Cells 124 to 236 are LS174T cells. Cells 335 to 343 are RKO cells. The bulk of LS174T is also shown (sample ID 46). We display the Gini coefficient G mean value and the standard deviation for each group. | ||
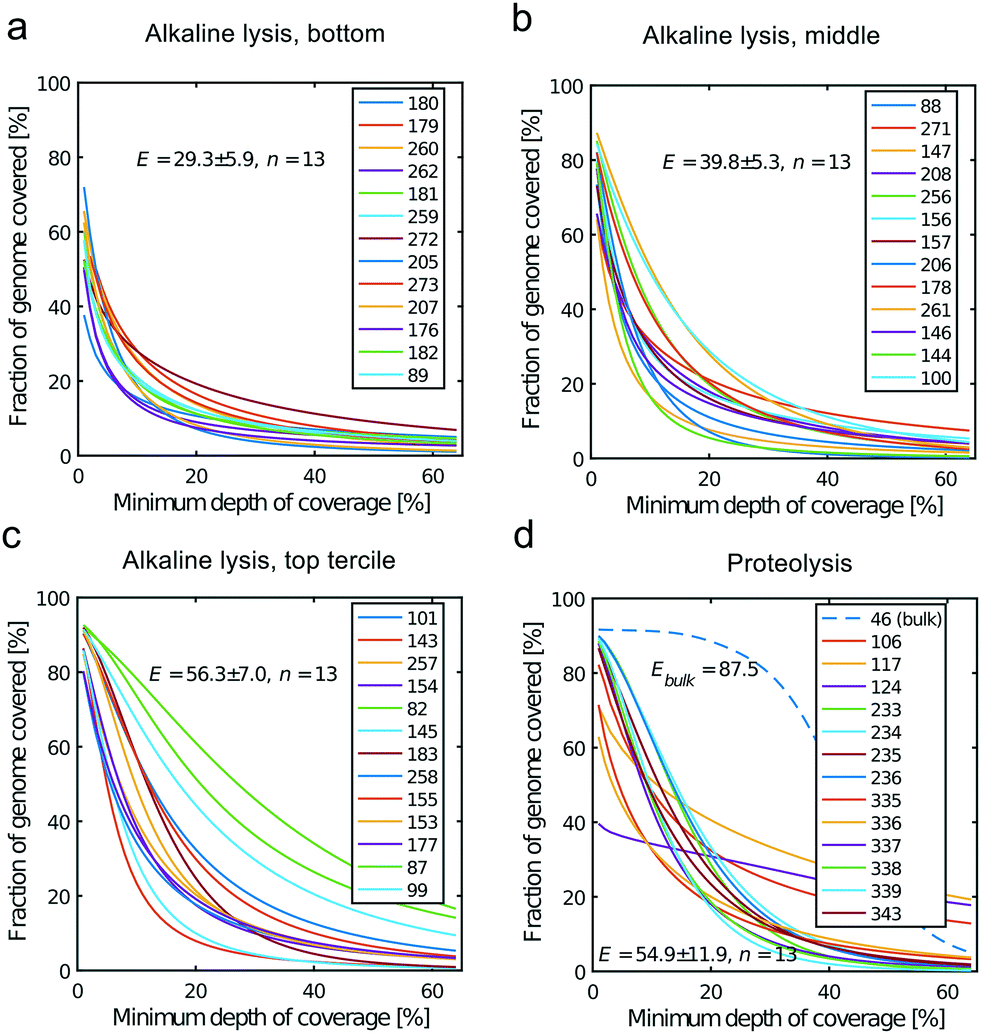 | ||
| Fig. 4 Coverage plots corresponding to the (a) bottom, (b) middle and (c) top tercile and (d) the single cells processed by proteolysis in Eindhoven. Cells 124 to 236 are LS174T cells. Cells 335 to 343 are RKO cells. The bulk of LS174T is also shown (sample ID 46). We display the E-score as mean value and the standard deviation for each group. The E-score is calculated from a normalized coverage curve as described in ref. 36. | ||
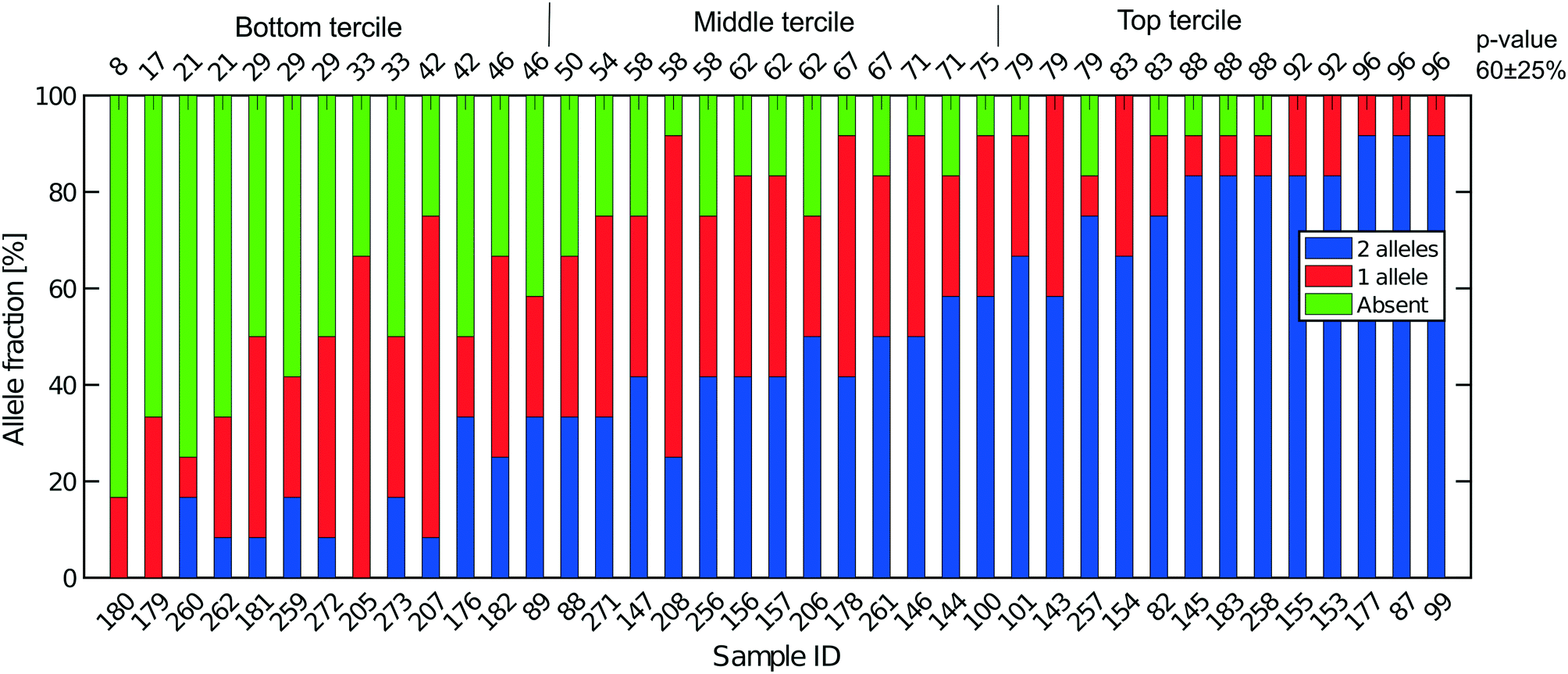 | ||
| Fig. 5 Extent of allelic dropout for 12 heterozygous SNPs selected for LS174T (36 cells) and LS180 cells (3 cells). On the x-axis, the sample ID for the cells analyzed. Cells are ordered by increasing p-value (in percent). The mean (SD) for all 39 cells processed in Oxford is 60 (25). | ||
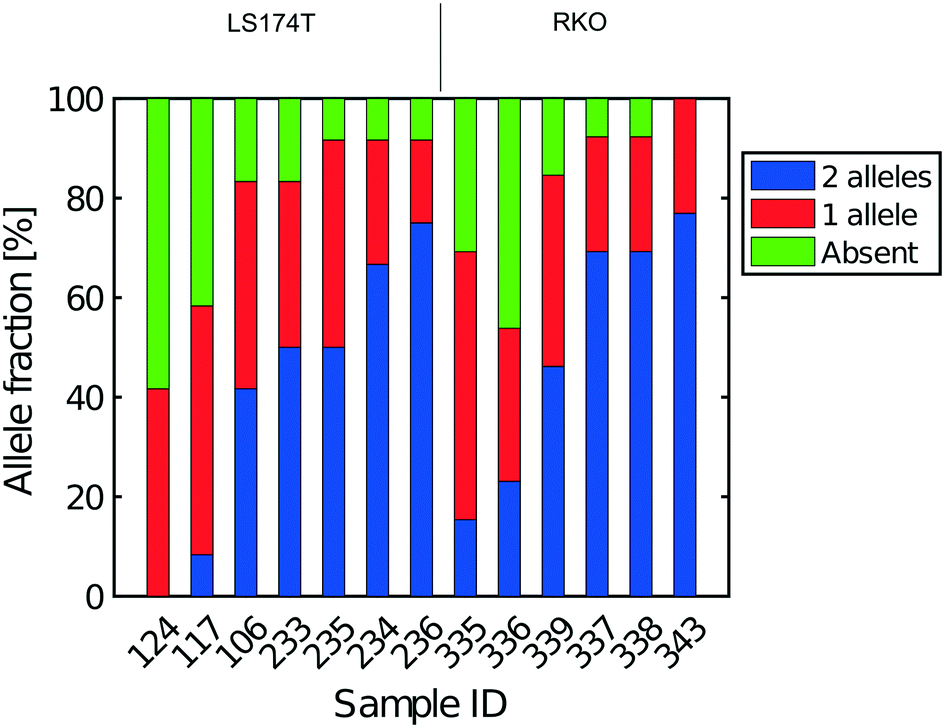 | ||
| Fig. 6 Extent of allelic dropout for heterozygous SNPs for cells processed in Eindhoven. Cells 124 to 236 are LS174T cells (12 heterozygous SNPs) and cells 335 to 343 are RKO cells (13 heterozygous SNPs). | ||
3 Results
We mainly used the LS174T cell line derived from a colorectal cancer (CRC) as a model system for single cell analysis. This is a very well characterized cell line (see e.g.ref. 28) that has been widely used for CRC cell characterization and drug response studies (see e.g.ref. 29 and 30). In some experiments, we also processed cells from the RKO and LS180 cell lines (the latter was alternatively derived from the same CRC as LS174T), and from cells obtained directly from fresh CRCs.For each experiment a single use microfluidic device (Fig. 1a and S1†) is placed in the instrument allowing bright field and fluorescence imaging, the control of the device temperature and connection of the device inlets (cell, B1 and B2 inlet, Fig. 1b) to a multi-channel air pressure controller7 (Fig. S1g†). Fluorescence imaging and the use of YOYO-1 intercalating DNA dye enabled monitoring of cell lysis. However, the dye may be omitted to avoid interference with the subsequent quality of the preparations with respect to their use for DNA, or RNA sequencing.7
3.1 Device design and fabrication
The microfluidic device is a single use passive device fabricated by injection moulding using a mould that creates 12 connectors placed at distances corresponding to a 96-well plate standard and can contain up to 50 μL of a solution26 (Fig. 1b). The microfluidic device has a single depth microchannel network with dimensions such that LS174T and similar colorectal epithelial cells are successfully transported and trapped (Fig. S4†). LS174T cells have a median diameter of 14 μm, which is in the characteristic size range for colorectal cancer cells, and so we designed the microfluidic network to be 30 μm-deep and at least 30 μm-wide except for the flow constrictions that are used as cell traps.Our design is the result of iterative optimization where we identified and improved three critical aspects of the device design and fabrication: i) the flow through the trap, which depends on its cross section and the flow resistance of the outlet channels, ii) the shape of the cell pocket and iii) the moulding quality of the cell inlet.
The trap cross section has to be smaller than the LS174T cells size to retain the cells, but also sufficiently large so that it collects a significant fraction of the main flow in the feeding channel for cells to be directed through the trap. As a boundary condition, our choice of a single depth design means that the trap depth remains the same throughout the chip, namely 30 μm. As a result, the trap has a high aspect ratio, within the limit achievable during the fabrication of the master in silicon by micromachining. Finally, the fabrication by polymer replication results in the channels and in particular the trap having tilted sidewalls (up to 3 degrees) to allow the separation of the polymer part from the mould during injection moulding. As a result, the cell traps have a cross section 30 μm-deep, 4.5 μm wide at the bottom and 7.5 μm wide at the top. The pocket receiving the cell has an asymmetric design (Fig. 1c and d). This is in contrast to previously reported devices based on hydrodynamic trapping where flow focusing is used to direct the cell to a microfluidic constriction that is a bypass in an otherwise symmetric flow profile. In our device, the flow focusing is asymmetric since cells are aligned against the wall of the feeding channel. A symmetric pocket creates a dead volume after the constriction (Fig. S4†) that is a spot where a cell decelerates and can settle just outside the cell trap. By making the pocket asymmetrical, we improve the flow profile such that cell trapping is more efficient. The optimized design gave the best results in terms of numbers of traps per chip having single cells.
Finally, the connection of the feeding channel and the well receiving the cells is a critical aspect of the design. The surface roughness at the inlet is of paramount importance since a sharp edge tends to stop cells entering the channel. The injection moulding parameters are therefore adjusted to produce a round edge. In addition, we ensured that the shim is mounted into the injection moulder only once. This greatly improved the quality of the final device since successive mounting of the shim increases the roughness at the connections with the inlets due to the alignment tolerance of the shim in the mould. On the optimized device (Fig. 1), single cells were trapped routinely, on average, in 3 to 6 out of 8 possible traps. On rare occasions, cell doublets are trapped and this may be because cell doublets enter the device in the first place. For this reason, cell traps are imaged in bright field and fluorescence after trapping to confirm the presence of, and then exclude, such cell doublets from further analysis.
3.2 Cell capture and lysis
For each experiment, solutions are loaded in the device wells and a single pressure is applied to the inlets either to wet the device, trap cells or lyse the trapped cells (Fig. 1e–g and Protocol S3†). After priming of the microfluidics, cells loaded in a cell inlet are pushed into the feeding channel where they become aligned against a sidewall of the channel by the incoming flow from the buffer inlets (B1 and B2 in Fig. 1b), similarly to a recently described cell-trapping device.31 Once aligned, a cell of radius r follows a streamline at a distance r from the sidewall. The cell traps are constrictions that connect the feeding channel with separate outlets. A trap collects a fraction of the flow from the feeding channel such that we call rc the position of the last streamline entering the trap. A cell of radius r enters the trap downstream if r < rc (Fig. 1c). Since a trap is only 4.5 μm-wide, a cell entering it is captured in the constriction. This cell then occupies a pocket recessed from the main flow through the feeding channel. A cell cannot block the flow through the trap completely since the channel depth (30 μm) is much larger than the cell. However, the flow resistance is sufficiently increased for the next incoming cells to pass by and be directed to the following free trap (Fig. 1d). Cells that are not trapped are collected in the waste outlet.Cellular DNA is eluted from the cell trap by introducing a lysis solution from the inlet B1 (Fig. 1b). In our study, we compared two lysis solutions. For one, a solution for proteolysis including proteinase K and Triton-X100 was used for the 13 cells (LS174T and RKO) whose results were obtained in Eindhoven. This lysis solution enables collecting the RNA prior to collecting the DNA of the trapped cell.7 Alternatively, an alkaline lysis buffer (D2, pH above 12) provided with the Repli-g UltraFast kit was used in Oxford for the analysis of the 39 single cells from the LS174T and LS180 cell lines, and from two fresh tumour samples (Fig. S4†). The alkaline lysis is the one adopted in commercially available kits for eluting DNA for sequencing. Both solutions successfully lyse the cells trapped and elute the DNA from the trap as observed in experiments where the DNA is labelled with an intercalating dye so it can be visualized by fluorescence microscopy. From the results of the single cell sequencing using the two different approaches, as discussed below, we conclude that both approaches to lysis were appropriate for MDA. This is, perhaps, surprising in the case of DNA extraction by proteolysis since proteinase K might be expected to digest the polymerase. However, there are six orders of magnitude difference between the volumes of lysate (pL) and the volumes of the reagents added to the well (μL), which thus makes the protease content in the MDA mix insignificant.7 The success of the amplification and sequencing is the best indication that the lysis is successful.
DNA samples that successfully amplified were passed through a quality control. For the samples processed in Oxford, we PCR amplified five genes from five different chromosomes to give five different sized fragments, and visualized them on an agarose gel (see Protocol S3† for details). Only samples which successfully displayed at least 4 of the 5 PCR products were used for library preparation and sequencing. Essentially all of the single cell lysates were successfully amplified for DNA and more than 90% of the Oxford samples passed the subsequent quality filter (i.e. quantification by a pico-green assay (Qubit)) before being passed on for DNA sequencing. For samples processed in Eindhoven, a quality check comprising PCR of RNase P was performed on some samples. Next, some of the samples were then checked by 1) quantification by Qubit and 2) a test run of sequencing performed at a low number of reads in order to assess the quality of the library before the actual sequencing presented in this paper. Sequencing libraries were successfully prepared from 97% of the samples that passed the initial quality control.
3.3 Non-human contamination
Fig. 2a shows the percentage of reads that mapped to the reference genome for 52 cells that were whole genome sequenced. Apart from 7 clear outlier cells (<99% mapped reads), 99% of the DNA sequences obtained mapped to a reference human genome, indicating that there was effectively no contamination of these samples from non-human sources. This is a significant achievement as there are several published reports of reagent-induced contamination.32–34 In addition, most of the reads are correctly paired, with a low percentage of singletons (0.17 ± 0.06%). Libraries prepared from single cell DNA show very different levels of representation of the human genome. In the following we summarise the metrics of our sequencing using Lorenz plots (Fig. 3), coverage plots (Fig. 4) and allelic dropout analysis (Fig. 5 and 6). For readability we display the alkaline lysis data set in three terciles where cells are grouped according to their allelic dropout p-value (Fig. 5). Bad representation of the human genome may be caused by the loss of DNA in the device as supported by the fact that increasing the depth of sequencing for a representative subset of samples did not result in significantly improved coverage. The distribution of the number of reads per cell is given in Fig. 2b.3.4 Coverage
We generated Lorenz plots displaying the fraction of the genome covered versus the fraction of the reads for 39 cells processed in Oxford (Fig. 3a–c) and 13 of the single cell sequencing sets obtained from the Eindhoven laboratory (Fig. 3d). There is good agreement between these plots and the coverage data shown in Fig. 2a. Thus, those plots farthest from the diagonal are for the cells with the poorest whole genome coverage. We follow Szulwach et al.35 and calculate G, the Gini coefficient for the Lorenz plots, to quantify the uniformity of the genome coverage. The Gini coefficient G is calculated as:| G = 1 − 2A. | (1) |
In which A is the area under the Lorenz curve. For an ideally uniform coverage of the genome, the Lorenz plot displays a diagonal and the area under the curve is 0.5. G = 0 indicates an ideally uniform coverage of the genome. In our study, G = 0.3 for the sequencing of the bulk of LS174T and many cells have a G = 0.5 (see Fig. 3d and S5†). In the top tercile of the cells processed in Oxford, corresponding to the highest coverage, G = 0.6 ± 0.1 (n = 13 cells). For comparison, using commercial instrumentation, Szulwach et al. report G = 0.36 ± 0.04 (n = 5) for GM12752 cells where the bulk sequencing gives a G just below 0.2, but also G = 0.6 for another cell type.35 Thus far most of the single cell sequencing studies only report the coverage results using the Lorenz plot.5,6,15,35 Although the Lorenz graph is effective in reporting which fraction on the genome is not covered, for reporting the distribution of the coverage the so-called coverage graph is more suited and used in (bulk) sequencing experiments. Previously we have reported a coverage graph of single cell sequencing experiments.7 Here, we report a more complete overview of the coverage of our results in Fig. 4. The evenness score E:
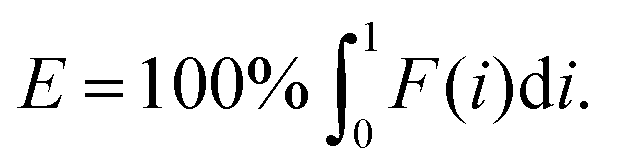 | (2) |
3.5 Allelic dropout
Next, SNP data were used to obtain estimates of the total recovery of genomic DNA taking into account the near diploid karyotype of LS174T, given that it is mismatch repair defective (Fig. 5). From bulk DNA sequencing of both LS174T and LS180 we can easily identify SNPs that are unequivocally heterozygous in both cell lines and which must represent germ line heterozygosity in the patient from whose cancer the two cell lines were derived. Full coverage of the near diploid genome present in LS174T in a single cell sequence would mean, that for such SNPs, both alleles must always be present and observed. If, in the presence of incomplete coverage, we assume that p is the probability that one allele of the SNP pair is observed, and that the probability of observing either allele is the same, then p2 is the probability of finding both SNP alleles, 2p(1 − p) is the probability of finding only one of the alleles, and (1 − p)2 is the probability that neither allele is found, namely a drop out from both genomes. This is the same binomial result as represented by the frequencies of homozygotes and heterozygotes in a random mating population according to the Hardy–Weinberg law. If a is the number of times both alleles are found, b the number of times one allele is found and c the number of times neither allele is found then the maximum likelihood estimate of p is:| p = 2a + b/2n. | (3) |
Fig. 5 shows the results of such an analysis for DNA prepared from the single cells in Oxford using a panel of 12 SNPs known to be heterozygous in the LS174T and LS180 cell lines. The different colours of the vertical bars for each single cell show the proportions of times 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or no alleles are found, and the cells are ordered from highest to lowest estimate of p. The corresponding Lorenz plots for these DNA sequences (Fig. 3a–c) show that there is a reasonable relationship between the coverage estimates from the Lorenz plots and the p value estimates. About 44% (17/39) of these single cell DNA sequences give p value estimates of around 0.7 or more, indicating total genomic coverage per cell of around 50%, while about 25% give total coverage of greater than 70%. The overall average p-value using the data on all 39 single cells is 0.60 ± 0.25 corresponding to complete coverage of just under 40% (the average p value for the Eindhoven data in Fig. 6 is 0.63 ± 0.22). Out of more than 10000 reads covering the 13 pairs of alleles for the SNPs, only 63 were ‘incorrect’ in the sense that they were not expected for either allele pair of a given SNP. This indicates a sequencing error rate of less than 1% and also the absence of any contaminating human DNA from external sources, namely other than the cells being analysed.
:1 or no alleles are found, and the cells are ordered from highest to lowest estimate of p. The corresponding Lorenz plots for these DNA sequences (Fig. 3a–c) show that there is a reasonable relationship between the coverage estimates from the Lorenz plots and the p value estimates. About 44% (17/39) of these single cell DNA sequences give p value estimates of around 0.7 or more, indicating total genomic coverage per cell of around 50%, while about 25% give total coverage of greater than 70%. The overall average p-value using the data on all 39 single cells is 0.60 ± 0.25 corresponding to complete coverage of just under 40% (the average p value for the Eindhoven data in Fig. 6 is 0.63 ± 0.22). Out of more than 10000 reads covering the 13 pairs of alleles for the SNPs, only 63 were ‘incorrect’ in the sense that they were not expected for either allele pair of a given SNP. This indicates a sequencing error rate of less than 1% and also the absence of any contaminating human DNA from external sources, namely other than the cells being analysed.
Additional evidence for the absence of contamination with exogenous DNA was obtained from the density of reads that mapped to male-specific genes on the Y-chromosome, see Table S3.† Since both the LS174T and the RKO cell lines are derived from female patients and the operators in the Eindhoven laboratory were male, lack of Y chromosome reads provides evidence that there was at least no contamination of Y chromosome reads from them. The male-specific genes used in this analysis are those for which there are no homologies on the X-chromosome as taken from the work of Page and co-workers.38 For almost all male-specific genes we found zero reads mapping to them whereas the mean number of reads per gene on the X-chromosome (taken over all genes listed in the Ensemble human genome annotation GTF file) is over 900 reads per gene on average for these all samples (Table S3†). This value is to be compared to average number of reads found for the male-specific genes which is 0.4 read per gene (Table S3†). This effectively rules out exogenous DNA contamination from the male operators in the Eindhoven laboratory to occur and suggests that these reads mapping to male specific genes found corresponds to amplification, sequencing and mapping errors. Since the error rate for sequencing on an Illumina HiSeq system is in the order of 1%. One usually refers to a minimum number of Q30 base (number of base where the error rate is below 1/1000). For 2 × 125 bp reads of HiSeq, we should have error rate below 1.5% (estimation using an indexed PhiX). The exact value depends on each run. Our data suggest a mapping error of 0.4/900 = 0.04%.
The heterogeneity of the frequencies of reads (data not shown) between SNPs within single cells suggests dropping out, namely absence of DNA in the initial single cell preparation, as the main reason for lack of complete coverage. Similarly, Fig. 6 gives an estimate of the allelic drop-out for the single cells processed in Eindhoven for which the Lorenz graph is shown in Fig. 3d. Note that for the RKO cells, 13 heterozygous SNPs across the genome where used. Again, the results are concordant with the measures of coverage and the Lorenz curves. The poorest cells, with the highest allelic drop-out are right in the far lower corner of the Lorenz plot in Fig. 3d, while the best cells, with 60–70% sharing, correspond to the curves nearest to the diagonal. In addition to the analysis of single cells from the colorectal cancer-derived cell lines, some single cell whole genome sequences were obtained directly from two fresh colorectal cancers. These were analysed following the same procedure described above using different appropriately chosen sets of SNP markers for each cancer. The results shown in Fig. S8† for a further total of 15 single cells demonstrate that at least comparable quality single cell whole genome DNA sequences can be obtained from fresh tumours as were obtained from the cell line cultures. Our overall results indicate that the independent analyses of singe cell DNA sequences using two different protocols in different laboratories, but using the same device and instrument, gave comparable results, with perhaps somewhat better coverage using the protocol with alkaline lysis compared to the protocol using proteolytic lysis. Moreover, the results obtained using our valve free devices which are simpler in design and manufacture are comparable with the best published results. For details of the experimental protocols and the use of the instrument see the methods section and the Protocol S3.†
4 Discussion
Experimental omics approaches39,40 to separately process multiple single cells are increasingly important. Four types of approaches are available for partitioning the molecular contents of one cell from another: 1) dilution and separately processing; 2) statistical dilution, tagging and pooling,41 2) droplet,42,43 3) micro/nanowell,21,44 4) microfluidic trapping.7,8,45–48We found that sequencing genomic DNA extracted from single cells inside our low-cost microfluidic device, gave single cell DNA sequencing results of comparable quality to those reported using more complex and expensive instruments. Our device is valve free and can thus be fabricated by injection moulding a polymer. It is also straightforward and can be operated on a commercial optical microscope or using a custom-built instrument, Cell-O-Matic.
Moreover, in this study the cell lysis is performed in the sub-nanoliter cell trap of the device while the amplification step is performed in the outlet wells in μL volumes. The representation of the genome in the sequencing data is similar to single cell sequencing obtained in devices where both DNA extraction and amplification take place in nL volumes.35 In addition, we also show that the genome representation of single cells processed in the microfluidic device is on average better than when both cell lysis and amplification are performed in μL volumes. Here we compare to sequencing of single cells sorted by FACS in individual PCR tubes and amplified using the unmodified MDA protocol, i.e. with an alkaline lysis (see Fig. S9†). In this case the Gini coefficient is higher (G = 0.9 ± 0.1, n = 7) and the evenness (E = 24.1 ± 16.9, n = 7) lower than for the lower tercile of the alkaline lysis data set (Fig. 3c and 4c). This shows that only the DNA extraction may be crucial to a good single cell sequencing. The main reason for a poor representation of the genome may be the loss of DNA and/or equally the loss of enzymatic activity. When amplification takes place in confinement of a nano-liter volume, enzyme may be lost on surfaces due to the high area-to-volume ratio and this may outbalance the benefit of maintaining a high template concentration.
Our device design focuses only on DNA extraction thus its design is not specific to any amplification protocol and the end-user may be free to implement any other amplification protocol. Moreover, the device design includes inlet wells that are placed in a grid matching that of a 96-well plate thus that standard lab robotics could be used to perform the amplification step.
Previously described microfluidic devices isolate single cells using either a physical valve8 or an oil phase21 at the time of the lysis and subsequently for the amplification. The use of valves to trap the cells necessitates two-layer devices which are complex and hard to manufacture. By contrast, our devices have no valves and are thus easier to design, manufacture and use. We are able to operate without valves because the liquid flow from different inlets is strictly controlled by air pressure with high accuracy. This, in particular, allows us to exchange reagents in the feeding channel while maintaining the cells trapped until they need to be lysed. The flow rate is minimal as too high flows would dislodge the cells from the traps. The laminar flow conditions in the feeding channel and through the traps ensure that the lysate is pushed through the trap. At a later stage, during the amplification, the solution is confined to the outlet since the loss of material from the outlet well through diffusion into the microfluidic channel is negligible (see ESI† and Fig. S10). The design of the traps, the mode of lysis and collection of the resulting DNA makes it unlikely that there is significant contamination between the cells trapped on the same chip. Preliminary data obtained by analysis of the LS174T cell line, which is a known mixture of two cell populations (unpublished observations), suggests that there is no major contamination between neighbouring traps on the same chip.
A further proof of the absence of contamination between neighbouring traps can be derived from a subset of data where mRNA was extracted from the captured cells.7 There, PCR of the AXIN2 and beta-actin genes was used to assess the presence of mRNA in the outlet wells. In those experiments, no mRNA was detected from empty traps adjacent to those where cells were successfully captured and lysed.
Finally, we also consider contamination by exogenous human DNA. The allele analysis of heterozygous SNPs from throughout the entire genome shown in Fig. 5 and 6. In this analysis, we detect only the alleles that we expect for the LS174T cell line which gives us a good indication that there is no contamination from extraneous human DNA. In addition, we also look at the presence of reads mapped to Y-chromosome genes knowing that the cell lines used in this study are female cell lines. Here, we find generally no reads mapping to those genes (see Table S2†). Some reads do map to a few Y-chromosome genes such as PCDH11Y for all samples but we show that this is due to homologies with genes on the X-chromosome. The density of reads that map to chromosome Y is below 3% and typically 0.05% of the input of a single cell, so should be attributed to amplification and sequencing errors. The absence of exogenous human contamination may be surprising at first since the devices are fabricated in a standard laboratory environment (i.e. not a clean room environment). The microfluidic chip is injection molded on an industrial equipment and assembled to a polymer foil. However, the assembly is realised by UV-assisted thermal bonding. The strong UV illumination during the bonding of the lid would destroy any foreign DNA before the microfluidic channels are sealed. Immediately after bonding the lid, the device connectors are covered by PCR tape.
When comparing our data to previously published DNA sequencing from single cells we see that the Gini coefficients (see Fig. 3) are similar to results obtained on a commercial system.35 Previously7 and here we have shown coverage graphs of our single-cell sequencing data. To our knowledge, this has not been done before for single cell sequencing data and we suggest that this should be incorporated in future single sequencing experiments as this gives a better insight in the read distribution in these experiments and to what extent reliable SNP calling can be performed. Finally, we have presented our results in terms of maximum likelihood estimate of allelic dropout p and find this value to be 0.60 ± 0.25.
The first commercial microfluidic device method for processing single cells for sequencing18 was known to suffer significantly from the capture of doublets rather than single cells. Our approach has the advantage that we can take an image of trapped cells to confirm the single cell occupancy of each trap before proceeding to sequencing.
The more recently emerging droplet-based single cell fluidics and dilution tagging and pooling approaches offer the highest throughput (up to 10000 s of cells) compared to 10–100 s of the microfluidic trapping approaches. However, an advantage of our approach, is that it can be used to extract and process RNA from the same cell as the DNA;7 such multi-omic characterization will be important for making the connection between genotype and molecular phenotype to gain a better understanding of cellular mechanisms and to better select the mutations that may be driving a cancer phenotype and which might be candidates for targeted therapy.
For such integrative omics applications it is important to know that the comparative performance metrics of our single cell processing devices are equivalent to other types of devices and approaches. We can conclude that our DNA sequencing results show that the output of our device is at least comparable to, if not better than the valve-based commercial devices and offers advantages over non-microfluidic approaches such as a very low contamination level.
5 Conclusions
We have developed a passive microfluidic device for individually isolating cancer cells and extracting their single cell whole genomes in sub-nanoliter volumes. The device is fabricated by injection moulding, mounted in a prototype instrument and used to prepare single cell DNA for pair-end Illumina sequencing. Using the sequencing metrics of more than 50 single cells, we compare our data to previous studies where both extraction and amplification steps are performed in nanoliter volumes inside microfluidic devices. From the high coverage, homogeneity and virtual elimination of contamination we obtain with our device, we conclude that only the extraction step needs to be done in sub-nanoliter volumes, while amplification can be done in larger volumes with a conventional MDA bench protocol. Since our device focuses on extracting DNA from isolated cells only, it provides the flexibility of using any DNA amplification protocol outside the device and reduces the complexity of the device and instrumentation.Author contribution
Conceptualization: KM, PZ, JS, RM, WB; data curation: RM, LB, JW, FH; formal analysis: RM, LB, JW, SM, FH, WB, PZ; funding acquisition: RM, AK, KM; investigation: RM, MP, KK, RV, NAs, DS, CS; methodology: LB, JS, WB, PZ, KM; project administration: JS, TO, BB, CS, AK, WB, PZ, KM; software: TO, NAg; supervision: RM, WB, AK, KM; visualization: RM, LB, JS, PZ; writing original draft: RM, WB, PZ, KM; writing review and editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the European Commission under the Seventh Framework Programme (FP7/2007-2013) under grant agreements number 278204 (Cell-O-Matic) and the Danish Council for Strategic Research Grant 10-092322 (PolyNano). The authors thank Charles Cantor and Wilhelm Ansorge for advice throughout the Cell-O-Matic project.References
- N. Navin, J. Kendall, J. Troge, P. Andrews, L. Rodgers, J. McIndoo, K. Cook, A. Stepansky, D. Levy, D. Esposito, L. Muthuswamy, A. Krasnitz, W. R. McCombie, J. Hicks and M. Wigler, Nature, 2011, 472, 90–94 CrossRef PubMed.
- F. Tang, C. Barbacioru, Y. Wang, E. Nordman, C. Lee, N. Xu, X. Wang, J. Bodeau, B. B. Tuch, A. Siddiqui, K. Lao and M. A. Surani, Nat. Methods, 2009, 6, 377–382 CrossRef PubMed.
- L. F. Cheow, E. T. Courtois, Y. Tan, R. Viswanathan, Q. Xing, R. Z. Tan, D. S. W. Tan, P. Robson, Y.-H. Loh, S. R. Quake and W. F. Burkholder, Nat. Methods, 2016, 13, 833–836 CrossRef PubMed.
- M. Farlik, N. C. Sheffield, A. Nuzzo, P. Datlinger, A. Schoenegger, J. Klughammer and C. Bock, Cell Rep., 2015, 10, 1386–1397 CrossRef PubMed.
- S. S. Dey, L. Kester, B. Spanjaard, M. Bienko and A. van Oudenaarden, Nat. Biotechnol., 2015, 33, 285–289 CrossRef PubMed.
- I. C. Macaulay, W. Haerty, P. Kumar, Y. I. Li, T. X. Hu, M. J. Teng, M. Goolam, N. Saurat, P. Coupland, L. M. Shirley, M. Smith, N. Van der Aa, R. Banerjee, P. D. Ellis, M. A. Quail, H. P. Swerdlow, M. Zernicka-Goetz, F. J. Livesey, C. P. Ponting and T. Voet, Nat. Methods, 2015, 12, 519–522 CrossRef PubMed.
- D. van Strijp, R. C. M. Vulders, N. Larsen, J. Schira, L. Baerlocher, M. A. van Driel, M. P. Jensen, T. S. Hansen, A. Kristensen, K. U. Mir, T. Olesen, W. F. J. Verhaegh, R. Marie and P. J. van der Zaag, Sci. Rep., 2017, 7, 11030 CrossRef PubMed.
- Y. Marcy, C. Ouverney, E. M. Bik, T. Loesekann, N. Ivanova, H. G. Martin, E. Szeto, D. Platt, P. Hugenholtz, D. A. Relman and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11889–11894 CrossRef PubMed.
- R. J. Hartmaier, L. A. Albacker, J. Chmielecki, M. Bailey, J. He, M. E. Goldberg, S. Ramkissoon, J. Suh, J. A. Elvin, S. Chiacchia, G. M. Frampton, J. S. Ross, V. Miller, P. J. Stephens and D. Lipson, Cancer Res., 2017, 77, 2464–2475 CrossRef PubMed.
- J. C. M. Wan, C. Massie, J. Garcia-Corbacho, F. Mouliere, J. D. Brenton, C. Caldas, S. Pacey, R. Baird and N. Rosenfeld, Nat. Rev. Cancer, 2017, 17, 223–238 CrossRef PubMed.
- A. M. Breman, J. C. Chow, L. U'Ren, E. A. Normand, S. Qdaisat, L. Zhao, D. M. Henke, R. Chen, C. A. Shaw, L. Jackson, Y. Yang, L. Vossaert, R. H. V. Needham, E. J. Chang, D. Campton, J. L. Werbin, R. C. Seubert, I. B. Van den Veyver, J. L. Stilwell, E. P. Kaldjian and A. L. Beaudet, Prenatal Diagn., 2016, 36, 1009–1019 CrossRef PubMed.
- R. S. Lasken, Biochem. Soc. Trans., 2009, 37, 450–453 CrossRef PubMed.
- Y. Hou, K. Wu, X. Shi, F. Li, L. Song, H. Wu, M. Dean, G. Li, S. Tsang, R. Jiang, X. Zhang, B. Li, G. Liu, N. Bedekar, N. Lu, G. Xie, H. Liang, L. Chang, T. Wang, J. Chen, Y. Li, X. Zhang, H. Yang, X. Xu, L. Wang and J. Wang, GigaScience, 2015, 4, 37 CrossRef PubMed.
- N. E. Navin, Genome Res., 2015, 25, 1499–1507 CrossRef PubMed.
- C. Zong, S. Lu, A. R. Chapman and X. S. Xie, Science, 2012, 338, 1622–1626 CrossRef PubMed.
- L. Huang, F. Ma, A. Chapman, S. Lu and X. S. Xie, Annu. Rev. Genomics Hum. Genet., 2015, 16, 79–102 CrossRef PubMed.
- Z. Yu, S. Lu and Y. Huang, Anal. Chem., 2014, 86, 9386–9390 CrossRef PubMed.
- B. A. Peters, B. G. Kermani, A. B. Sparks, O. Alferov, P. Hong, A. Alexeev, Y. Jiang, F. Dahl, Y. T. Tang, J. Haas, K. Robasky, A. W. Zaranek, J.-H. Lee, M. P. Ball, J. E. Peterson, H. Perazich, G. Yeung, J. Liu, L. Chen, M. I. Kennemer, K. Pothuraju, K. Konvicka, M. Tsoupko-Sitnikov, K. P. Pant, J. C. Ebert, G. B. Nilsen, J. Baccash, A. L. Halpern, G. M. Church and R. Drmanac, Nature, 2012, 487, 190–195 CrossRef PubMed.
- T. Kivioja, A. Vaharautio, K. Karlsson, M. Bonke, M. Enge, S. Linnarsson and J. Taipale, Nat. Methods, 2012, 9, 72–U183 CrossRef PubMed.
- E. Borgstroem, D. Redin, S. Lundin, E. Berglund, A. F. Andersson and A. Ahmadian, Nat. Commun., 2015, 6, 7173 CrossRef PubMed.
- J. Gole, A. Gore, A. Richards, Y.-J. Chiu, H.-L. Fung, D. Bushman, H.-I. Chiang, J. Chun, Y.-H. Lo and K. Zhang, Nat. Biotechnol., 2013, 31, 1126–1132 CrossRef PubMed.
- M. Unger, H. Chou, T. Thorsen, A. Scherer and S. Quake, Science, 2000, 288, 113–116 CrossRef PubMed.
- P. F. Østergaard, J. Lopacinska-Jørgensen, J. N. Pedersen, N. Tommerup, A. Kristensen, H. Flyvbjerg, A. Silahtaroglu, R. Marie and R. J. Taboryski, J. Micromech. Microeng., 2015, 25, 105002 CrossRef.
- W.-H. Tan and S. Takeuchi, Lab Chip, 2008, 8, 259–266 RSC.
- Z. Zhu, O. Frey, D. S. Ottoz, F. Rudolf and A. Hierlemann, Lab Chip, 2012, 12, 906–915 RSC.
- P. Utko, K. F. Persson, A. Kristensen and N. B. Larsen, Lab Chip, 2011, 11, 303–308 RSC.
- Q. Su, N. Zhang and M. D. Gilchrist, J. Appl. Polym. Sci., 2016, 133, 42962 Search PubMed.
- D. Mouradov, C. Sloggett, R. N. Jorissen, C. G. Love, S. Li, A. W. Burgess, D. Arango, R. L. Strausberg, D. Buchanan, S. Wormald, L. O'Connor, J. L. Wilding, D. Bicknell, I. P. M. Tomlinson, W. F. Bodmer, J. M. Mariadason and O. M. Sieber, Cancer Res., 2014, 74, 3238–3247 CrossRef PubMed.
- T. M. Yeung, S. C. Gandhi and W. F. Bodmer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4382–4387 CrossRef PubMed.
- M. Bacac, T. Fauti, J. Sam, S. Colombetti, T. Weinzierl, D. Ouaret, W. Bodmer, S. Lehmann, T. Hofer, R. J. Hosse, E. Moessner, O. Ast, P. Bruenker, S. Grau-Richards, T. Schaller, A. Seidl, C. Gerdes, M. Perro, V. Nicolini, N. Steinhoff, S. Dudal, S. Neumann, T. von Hirschheydt, C. Jaeger, J. Saro, V. Karanikas, C. Klein and P. Umana, Clin. Cancer Res., 2016, 22, 3286–3297 CrossRef PubMed.
- T. Yeo, S. J. Tan, C. L. Lim, D. P. X. Lau, Y. W. Chua, S. S. Krisna, G. Iyer, G. S. Tan, T. K. H. Lim, D. S. W. Tan, W.-T. Lim and C. T. Lim, Sci. Rep., 2016, 6, 22076 CrossRef PubMed.
- T. Woyke, D. Tighe, K. Mavromatis, A. Clum, A. Copeland, W. Schackwitz, A. Lapidus, D. Wu, J. P. McCutcheon, B. R. McDonald, N. A. Moran, J. Bristow and J.-F. Cheng, PLoS One, 2010, 5, e10314 Search PubMed.
- P. C. Blainey and S. R. Quake, Nucleic Acids Res., 2011, 39, e19 CrossRef PubMed.
- S. T. Motley, J. M. Picuri, C. D. Crowder, J. J. Minich, S. A. Hofstadler and M. W. Eshoo, BMC Genomics, 2014, 15, 443 CrossRef PubMed.
- K. E. Szulwach, P. Chen, X. Wang, J. Wang, L. S. Weaver, M. L. Gonzales, G. Sun, M. A. Unger and R. Ramakrishnan, PLoS One, 2015, 10, e0135007 Search PubMed.
- M. Mokry, H. Feitsma, I. J. Nijman, E. de Bruijn, P. J. van der Zaag, V. Guryev and E. Cuppen, Nucleic Acids Res., 2010, 38, e116 CrossRef PubMed.
- F. Mertes, A. ElSharawy, S. Sauer, J. M. L. M. van Helvoort, P. J. van der Zaag, A. Franke, M. Nilsson, H. Lehrach and A. J. Brookes, Briefings Funct. Genomics, 2011, 10, 374–386 CrossRef PubMed.
- H. Skaletsky, T. Kuroda-Kawaguchi, P. Minx, H. Cordum, L. Hillier, L. Brown, S. Repping, T. Pyntikova, J. Ali, T. Bieri, A. Chinwalla, A. Delehaunty, K. Delehaunty, H. Du, G. Fewell, L. Fulton, R. Fulton, T. Graves, S. Hou, P. Latrielle, S. Leonard, E. Mardis, R. Maupin, J. McPherson, T. Miner, W. Nash, C. Nguyen, P. Ozersky, K. Pepin, S. Rock, T. Rohlfing, K. Scott, B. Schultz, C. Strong, A. Tin-Wollam, S. Yang, R. Waterston, R. Wilson, S. Rozen and D. Page, Nature, 2003, 423, 825–837 CrossRef PubMed.
- D. Wang and S. Bodovitz, Trends Biotechnol., 2010, 28, 281–290 CrossRef PubMed.
- S. M. Prakadan, A. K. Shalek and D. A. Weitz, Nat. Rev. Genet., 2017, 18, 345–361 CrossRef PubMed.
- H. C. Fan, G. K. Fu and S. P. A. Fodor, Science, 2015, 347, 1258367 CrossRef PubMed.
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef PubMed.
- G. X. Y. Zheng, B. T. Lau, M. Schnall-Levin, M. Jarosz, J. M. Bell, C. M. Hindson, S. Kyriazopoulou-Panagiotopoulou, D. A. Masquelier, L. Merrill, J. M. Terry, P. A. Mudivarti, P. W. Wyatt, R. Bharadwaj, A. J. Makarewicz, Y. Li, P. Belgrader, A. D. Price, A. J. Lowe, P. Marks, G. M. Vurens, P. Hardenbol, L. Montesclaros, M. Luo, L. Greenfield, A. Wong, D. E. Birch, S. W. Short, K. P. Bjornson, P. Patel, E. S. Hopmans, C. Wood, S. Kaur, G. K. Lockwood, D. Stafford, J. P. Delaney, I. Wu, H. S. Ordonez, S. M. Grimes, S. Greer, J. Y. Lee, K. Belhocine, K. M. Giorda, W. H. Heaton, G. P. McDermott, Z. W. Bent, F. Meschi, N. O. Kondov, R. Wilson, J. A. Bernate, S. Gauby, A. Kindwall, C. Bermejo, A. N. Fehr, A. Chan, S. Saxonov, K. D. Ness, B. J. Hindson and H. P. Ji, Nat. Biotechnol., 2016, 34, 303–311 CrossRef PubMed.
- W. K. Chu, P. Edge, H. S. Lee, V. Bansal, K. Zhang, V. Bafna and X. Huang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12512–12517 CrossRef PubMed.
- D. Di Carlo, L. Y. Wu and L. P. Lee, Lab Chip, 2006, 6, 1445–1449 RSC.
- J. S. Marcus, W. F. Anderson and S. R. Quake, Anal. Chem., 2006, 78, 3084–3089 CrossRef PubMed.
- D. Irimia, R. Tompkins and M. Toner, Anal. Chem., 2004, 76, 6137–6143 CrossRef PubMed.
- N. M. Toriello, E. S. Douglas, N. Thaitrong, S. C. Hsiao, M. B. Francis, C. R. Bertozzi and R. A. Mathies, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20173–20178 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S9 and Tables S1–S3. See DOI: 10.1039/c8lc00169c |
| ‡ These authors contributed equally to this work. |
| § See http://www.sigmaaldrich.com/technical-documents/articles/life-science-innovations/qualitative-multiplex.html. |
| ¶ These 5 positions were chosen based on https://www.sigmaaldrich.com/technical-documents/articles/biology/ffpe-wga-poster.html. |
| This journal is © The Royal Society of Chemistry 2018 |