
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA long-lived cuprous bis-phenanthroline complex for the photodynamic therapy of cancer†
Cynthia
Al Hageh
,
Majd
Al Assaad
,
Zeinab
El Masri
,
Nawar
Samaan
,
Mirvat
El-Sibai
,
Christian
Khalil
 and
Rony S.
Khnayzer
*
and
Rony S.
Khnayzer
*
Department of Natural Sciences, Lebanese American University, Chouran, Beirut 1102-2801, Lebanon. E-mail: rony.khnayzer@lau.edu.lb
First published on 27th February 2018
Abstract
Copper is an earth-abundant and a biologically essential metal that offers a promising alternative to noble metals in photochemistry and photobiology. In this work, a series of sterically encumbered Cu(I) bis-phenanthroline complexes were investigated for their use in photochemotherapy (PCT). It was found that Cu(dsbtmp)2+ [dsbtmp = 2,9-disec-butyl-3,4,7,8-tetramethyl-1,10-phenanthroline] (compound 3), which possessed the longest excited state lifetime, exhibited significant in vitro photocytotoxicity on A375 (human malignant melanoma) and A549 (human lung carcinoma) cell lines. Fluorescence imaging demonstrated the significant uptake and localization of compound 3 in a perinuclear fashion. A comet assay indicated the induction of DNA damage in the dark. The DNA breaks were significantly amplified upon photoactivation. The light-induced enhancement of cytotoxicity was associated with the formation of reactive oxygen species (ROS), a known intermediate in photodynamic therapy (PDT). This successful demonstration of photocytotoxicity using long-lived cuprous phenanthroline paves the way to exploit this class of photosensitizers for PDT applications.
Introduction
Cancer is currently associated with large mortality rates due to its late prognosis.1 Though significant improvements have been achieved in the prevention, early detection and treatment of patients, cancer is still one of the leading causes of death worldwide.1 Cisplatin is a common chemotherapeutic drug with a platinum noble metal centre.2 Nevertheless, its inherent cytotoxicity and the lack of selectivity lead to side effects and the development of cellular resistivity.3 Since its discovery and effective use in clinical settings, research on cisplatin analogues and their mechanism of action is on the rise.4,5 The undesirable characteristics of cisplatin have triggered interest to investigate new generations of more specific anticancer agents. On the one hand, photodynamic therapy (PDT) entails the use of light energy to catalytically activate drugs with spatial and temporal control.6,7 On the other hand photoactivatable chemotherapy (PACT) relies on the photochemical transformation of a prodrug into a drug.8,9 In this perspective, targeting tumours with PDT or PACT minimizes damage to healthy tissues and organs. In PDT and PACT, a variety of transition metal complexes have been investigated especially ones based on noble metal centres such as Ru, Os and Pt.7–9 However, very few studies were aimed at using metal complexes based on the earth-abundant copper core for PDT applications. Copper is a mineral present in the body with important physiological functions.10 Cupric and cuprous complexes have shown anticancer activities, and were a topic of extensive review articles by Santini and coworkers.11,12 Despite the fact that Cu(I) is accepted to be the form internalized by cells via a copper transporter (CTR) protein,13 research on Cu(II) complexes had significantly outnumbered Cu(I) as anticancer agents.12 Cu(II) complexes bearing semicarbazones,14 pyrenyl-terpy,15 dipyrido[3,2-d:20,30-f]quinoxaline,16 dipyrido[3,2-a:20,30-c]phenazine,16 and ferrocenyl-terpy17 ligands were successfully exploited for PDT. The change of the redox properties of cupric compounds through the introduction of redox active ligands was proven to be advantageous for PDT application.18 The lack of detailed studies on Cu(I) could be due to the instability of the majority of four coordinate complexes in basic solvents such as water.12 The redox properties of a number of Cu(I) complexes were related to their ability to produce reactive oxygen species (ROS) in the dark.19,20 Notably, some Cu(I) complexes showed selectivity to specific cancer cells with no activity detected on healthy cells.21 Importantly, previously investigated cuprous phenanthroline complexes showed the capacity to interact with and sometimes cleave extracted DNA.22,23 Moreover, [Cu(phen)2]+ (phen = 1,10-phenanthroline) was found to act as an artificial nuclease,23–26 and [Cu(dmp)2]+ (dmp = 2,9-dimethyl-1,10-phenanthroline) demonstrated the ability to bind with DNA through surface association at the minor groove of DNA.22,27,28 Additionally, [Cu(bcp)2]+ (bcp = 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline) was believed to intercalate via the major groove.22,27,28 However, to the best of our knowledge, these Cu(I) bis-phenanthroline complexes were not tested on living cells, and their study was limited to interaction with extracted DNA.22,27–30 In this study we aim at studying in vitro cytotoxicity under dark and photoactivated conditions of these complexes among others on cancer cell lines.In general, Cu(I) bis-phenanthroline complexes absorb visible light that is attributed to metal-to-ligand charge transfer (MLCT) transition.22,31 Upon photoexcitation, an electron is promoted from a d10 HOMO ground state to a π* LUMO orbital residing on one of the ligands, formally resulting in a transient Cu(II) d9 electronic configuration. In the excited state, molecules can undergo a “flattening” distortion towards a D2 configuration. In this flattened geometry the transient Cu(II) centre is coordinatively unsaturated and prone to Lewis base (solvent) attack.32 This excited state pseudo Jahn–Teller distortion decreases the lifetime of Cu(I) complexes and limits their use for bimolecular photochemistry. One of the most efficient strategies used to prevent this Cu(I) geometrical distortion flattening of complexes in solutions is the introduction of steric hindrance in the coordination sphere using bulky ligands.29 Sterically congested Cu(I) complexes bearing diimine and/or phosphine based ligands possessed improved excited state lifetimes and photoluminescence quantum yields.33 The 2,9 substituents on the phenanthroline have been proven to extend the lifetime of the excited state of Cu(I) complexes with record values recently reported by Castellano and coworkers at the microsecond time scale.34 Substituents at the 2,9 positions were also found to increase the longevity of the complexes by protecting the Cu(I) core in a coordinating solvent environment.33 In this work, a series of Cu(I) bis-phenanthrolines was investigated and photocytotoxicity was assessed in vitro. The compounds used here possessed excited state lifetimes ranging from a nanosecond up to a microsecond, allowing a correlation between the photophysical properties and their application in PDT.
Experimental methods
Materials
DMEM culture media were used with 10% FBS (fetal bovine serum) and 1% antibiotics. The DCFDA (2′,7′-dichlorofluorescin diacetate) cellular ROS detection assay kit for a microplate assay was purchased from Abcam. The comet assay kit was purchased from Trevigen. 2,9-Diphenyl-1,10-phenanthroline was synthesized according to a published procedure.35 The remaining ligands, salts, and solvents (HPLC grade) were purchased from Aldrich and used without further purification.Synthesis and characterization
In this study four sterically encumbered cationic Cu(I) phenanthroline derivatives were synthesized and characterized: [Cu(dmphen)2]+ (1) (dmphen = 2,9-dimethyl-1,10-phenanthroline), [Cu(BC)2]+ (2) (BC = bathocuproine or 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline), [Cu(dsbtmp)2]+ (3) (dsbtmp = 2,9-disec-butyl-3,4,7,8-tetramethyl-1,10-phenanthroline), and [Cu(dpphen)2]+ (4) (dpphen = 2,9-diphenyl-1,10-phenanthroline), Scheme 1. The hexafluorophosphate salts of cuprous compounds 1, 2, and 4 were synthesised using a slightly modified literature method36 with yields ranging from 82 to 93%. Two equivalents of phenanthroline derivative ligands were dissolved in 10 mL of methanol. The resultant solution was degassed with argon for 1 h then added using a cannula while stirring to solid Cu(CH3CN)4PF6 under an argon atmosphere. An orange color was immediately observed. The solution was left stirring under argon for 1 h. Precipitation was done by adding the resultant solution to a vigorously stirring 50 mL of diethyl ether. The solid was filtered through a glass frit then washed with diethyl ether. The products were recrystallized from methanol by the slow addition of diethyl ether. The solids were stored in air until use. The hexafluorophosphate salts of compound 3 were synthesized according to a literature method.34 | ||
| Scheme 1 Chemical structures of compounds 1–4. | ||
Compound 1 , [Cu(dmphen) 2 ](PF 6 ), yield 93%. 1H NMR (CDCl3, 300 MHz), δ: 2.43 (s, 12H), 7.78 (d, 4H), 8.02 (s, 4H), 8.49 (d, 4H). Anal calcd for C28H24N4-CuPF6: C, 53.81; H, 3.87; N, 8.96. Found: C, 53.90; H, 3.86; N, 8.81.
Compound 2 , [Cu(BC) 2 ](PF 6 ), yield 82%. 1H NMR (CDCl3, 300 MHz), δ: 2.60 (s, 12H), 7.60 (m, 20H), 7.75 (d, 4H), 8.04 (d, 4H). Anal calcd for C52H40N4CuPF6: C, 67.20; H, 4.34; N, 6.03. Found: C, 67.46; H, 4.48; N, 5.97.
Compound 3 , [Cu(dsbtmp) 2 ](PF 6 ), yield 80%. 1H NMR (CDCl3, 300 MHz) 1H δ 8.20 (s, 2H), 3.68–3.10 (m, 2H), 2.78 (t, J = 6 Hz, 6H), 2.52 (t, J = 12 Hz, 6H), 1.76–1.22 (m, 4H), 1.12–0.76 (m, 6H), 0.21 to −0.18 (m, 6H).
Compound 4 , [Cu(dpphen) 2 ](PF 6 ), yield 86%. 1H NMR (CDCl3, 300 MHz), δ: 6.53 (t, 8H), 6.77 (t, 4H), 7.39 (d, 8H), 7.88 (d, 4H), 8.01 (s, 4H), 8.51 (d, 4H). Anal calcd for C48H32N4CuPF6: C, 66.02; H, 3.69; N, 6.42. Found: C, 65.99; H, 3.72; N, 6.31.
The water-compatible Cu(I) complex chloride salts of compounds 1, 2 and 4 were obtained by dissolving 20 mg of Cu(NN)2PF6 in 10 mL of water and a methanol mixture of a different ratio (between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2) for each reaction to obtain a saturated solution. 5 g of pre-washed Dowex-22 chloride form beads were added to the resultant solution. The mixture was left stirring for 2 h at room temperature. The solution was filtered on a glass frit and the filtrate was completely dried using a rotary evaporator. The resulting orange solid was re-dissolved in 10:1 water:methanol solution then filtered on a glass frit of medium porosity. The filtrate was then dried using a rotary evaporator followed by a vacuum oven. The orange solid was then dissolved in a minimum amount of methanol and purified using a Sephadex LH-20 column. The solvent of the collected orange band solution was then removed using the rotary evaporator and the solid was dried using a vacuum oven. The solid products were stored in air until further use. Attempts to convert compound 3 from the hexafluorophosphate to a chloride salt were not successful using the same strategy used for the remaining compounds. The water-compatible triflate salt of compound 3 was synthesized using a published procedure34 but using tetrakis(acetonitrile)copper(I) triflate instead of the hexafluorophosphate salt. The ESI-MS analysis of the water-compatible complexes matched the predicted M+ for each compound: compound 1 ESI-MS: m/z 479.13 (M+), compound 2 ESI-MS: m/z 783.44 (M+), compound 3 ESI-MS: m/z 759.56 (M)+ and compound 4 ESI-MS: m/z 727.33 (M)+.
:1 and 1:2) for each reaction to obtain a saturated solution. 5 g of pre-washed Dowex-22 chloride form beads were added to the resultant solution. The mixture was left stirring for 2 h at room temperature. The solution was filtered on a glass frit and the filtrate was completely dried using a rotary evaporator. The resulting orange solid was re-dissolved in 10:1 water:methanol solution then filtered on a glass frit of medium porosity. The filtrate was then dried using a rotary evaporator followed by a vacuum oven. The orange solid was then dissolved in a minimum amount of methanol and purified using a Sephadex LH-20 column. The solvent of the collected orange band solution was then removed using the rotary evaporator and the solid was dried using a vacuum oven. The solid products were stored in air until further use. Attempts to convert compound 3 from the hexafluorophosphate to a chloride salt were not successful using the same strategy used for the remaining compounds. The water-compatible triflate salt of compound 3 was synthesized using a published procedure34 but using tetrakis(acetonitrile)copper(I) triflate instead of the hexafluorophosphate salt. The ESI-MS analysis of the water-compatible complexes matched the predicted M+ for each compound: compound 1 ESI-MS: m/z 479.13 (M+), compound 2 ESI-MS: m/z 783.44 (M+), compound 3 ESI-MS: m/z 759.56 (M)+ and compound 4 ESI-MS: m/z 727.33 (M)+.
UV-vis titration
Compound 3 was dissolved in 50% DMSO in Tris-HCl buffer (5 mM, pH = 7.2). The absorbance of the compound in 50% DMSO was measured using UV-vis spectrophotometer (Cary 60) with an initial absorbance of 0.47 at λ = 450 nm.1.5 mg of activated calf thymus DNA type XV (Sigma-Aldrich) was dissolved to 1 mg/mL in Tris-HCl overnight at 4 °C to ensure complete dissolution. The DNA concentration was determined using UV-vis spectroscopy. The DNA was premixed with a solution of compound 3 to achieve a stock solution of 50% DMSO in Tris-HCl buffer (5 mM, pH = 7.2) containing a DNA concentration of 472 μg mL−1 and compound 3 with an absorbance of 0.47. The same concentration of compound 3 was used in the DNA stock and the initial solution used in the titration to avoid dilution effects on the UV-vis spectra. The DNA stock solution was added gradually onto the solution containing compound 3 and after each addition, the solution was homogenised and allowed to equilibrate for 2 min before the absorbance was measured.
Lipophilicity
LogP values, where P is the water–octanol partition coefficient, were measured using a slightly modified literature procedure37 while taking into consideration the EPA guidelines (EPA 712–C–96–038). 2 mg of the compounds were prepared in 2 mL Eppendorf tubes and dissolved in 1 mL of octanol saturated with water. The compounds were dissolved for 30 min in a sonicator bath set at room temperature and the solution was subjected to centrifugation for 1 min at 4300 rpm, followed by pipetting out the supernatant. Then, 1 mL of water saturated with octanol was added and the mixtures were shaken at 2200 rpm for 1 h at room temperature. After that, two phase solutions were formed, an upper phase with an octanol solvent and a lower phase with water solvent. The syringe was washed with octanol-saturated water prior to the aqueous phase aspiration, and water-saturated octanol prior to the octanol phase aspiration. The aqueous phase was aspirated using a glass syringe and air was expelled while the needle was passed through the octanol phase to prevent any traces of octanol inserting into the needle. Then, the octanol phase was aspirated. The UV-Vis spectra of the solution placed in a micro-cuvette was then obtained using a Cary 60 instrument. The octanol phases were appropriately diluted to keep the absorbance within the linear range. The logP values were calculated based on the following formula:where the ratio of concentrations was photometrically determined.

Alkaline comet assay
The comet assay was performed according to a previously published method38 using a Trevigen comet assay silver staining kit.Quantitative measurement of cellular reactive oxygen species (ROS) in cells
20 mM DCFDA (in DMSO), 10× buffer, 55 mM tert-butyl hydroperoxide (TBHP) were included in the kit. 1× supplemented buffer made up of 10% FBS in 1× buffer was considered as a negative control. 200 μM TBHP in the 1× supplemented buffer was considered as a positive control. 3 μM of compound 3 were prepared in 1× supplemented buffer. Two adherent cell lines, A375 and A549, were used. The cells were cultured in media without phenol red and were incubated overnight. Then the cells were harvested, seeded in two dark sided 96-well microplates with 100 μL containing ∼25000 cells per well and incubated overnight. The following day, the cells were washed with 1× buffer then incubated with 100 μL of 25 μM DCFDA solution for 45 min at 37 °C in the dark and then washed with 1× buffer. The cells were treated in triplicates with (a) 100 μL of 1× supplemented buffer, (b) 100 μL of 200 μM TBHP and (c) 100 μL of 3 μM compound 3. The data were measured on a fluorescence plate reader Varioskan Flash (Thermo Scientific) at Ex/Em of 485/535 nm immediately after treatment for baseline assessment. The light-exposed plate was subjected to blue LED light illumination for 30 min after 3 h of incubation. Both the light-exposed and dark plates were then analysed to furnish the final data. In all photobiological experiments, excitation was performed using a home-built blue LED setup as previously described.39,40
Cell survival assay
The cytotoxicity assay was performed using an MTS cell proliferation assay kit. Briefly, the assay measures mitochondrial dehydrogenase activity in cell systems. In the current investigations, human derived immortalized cell cultures (A549 and A375) were exposed to a serial dilution of the compounds tested under light and dark conditions. The compounds were dissolved in DMSO and diluted in media. The highest concentration of DMSO was 3% which did not lead to cell death under our experimental setting. The investigation in the dark was conducted in 96-well plates and post compound exposure cells were further incubated for 24 h prior to the addition of the MTS reagents. Photoexcitation was performed 6 h post incubation for 30 min using the blue-LED system and incubation was resumed for a total of 24 h. The resulting coloured solution was then collected and read at 492 nm for mitochondrial activity determination. The data were then used to generate dose response curves using Graphpad Prism software. The experiments were performed in triplicates in each run and runs were repeated three times for error determination.Fluorescence imaging and nuclear localization
These studies were performed as previously described.39 Briefly, the A549 and A375 cells were cultured in DMEM medium supplemented with 10% FBS and 100 U penicillin/streptomycin at 37 °C and 5% CO2. The cells were plated on cover slips 24 h prior to the experiment. The cells were then treated with compound 3 for 2 h and 6 h. After treatment, the cells were fixed with 4% paraformaldehyde for 10 min and blocked with 1% BSA in PBS for 1 h. Samples were then stained with DAPI for 10 min. Fluorescence images were taken using a 60× objective on an Axio observer Z1 fluorescence microscope (from Zeiss).Results and discussion
In this study four sterically encumbered cationic Cu(I) phenanthroline derivatives (compounds 1–4) were synthesized based on slightly modified literature methods, characterized and tested for potential photocytotoxicity. The hexafluorophosphate salts of Cu(I) bis-phenanthroline complexes were not water soluble and precipitated out of solution at a high percentage of water that is necessary for biological testing. Compounds 1, 2 and 4 were converted to chloride salts through a counter-ion exchange procedure40 (see the Experimental section for more details) whereas compound 3 was used as a triflate salt to afford water-compatible complexes. The UV-vis spectra (Fig. 1) of the hexafluorophosphate salts in acetonitrile and the chloride or triflate salts in DMSO were compared and matched to the reported data.33,34 The absorption spectra show a distinct metal-to-ligand charge transfer (MLCT) absorption band between 400 and 500 nm and an intra-ligand (π to π* transition) band between 250 and 325 nm. | ||
| Fig. 1 Normalized UV-vis spectra of compounds 1–4 of the hexafluorophosphate salts in acetonitrile (straight lines) and the water-compatible chloride/triflate salts in DMSO (dashed lines). The data were normalized to the intra-ligand band of each compound. | ||
The cytotoxicity of water-compatible compounds 1–4 was assessed by MTS cell proliferation assays. Stock solutions of Cu(I) compounds were prepared in DMSO and diluted in cell culture media to achieve the desired concentrations. At the highest concentration of the compound reported in this study, DMSO was not cytotoxic and its percentage did not exceed 3%. The IC50 values of all Cu(I) bis-phenanthroline complexes have been acquired in the dark after 24 h incubation on 2 different cell lines: A375 (human malignant melanoma) and A549 (human lung carcinoma). Control experiments with cisplatin showed marginal toxicity after 24 h incubation; hence this control was acquired after 48 h of incubation, Fig. S1.†
DMSO was not added to the cisplatin samples to avoid the deactivation of the drug. Each value reported in Table 1 is the average of three independent experiments along with the standard error on the mean. The cationic compounds 1–4 exhibited dark toxicity with IC50 value ranging from ∼2 to 26 μM (Fig. S2†, Table 1 and Fig. 2). Among the most lipophilic compounds, complexes 2 and 3 (logP = 2.21 and 2.64 respectively), with methyl or aryl groups at the 4,7 and/or 3,8 positions of the 2,9-disubstituted phenanthroline, exhibited lower cytotoxicity on both cell lines when compared to compounds 1 and 4. Compound 1, the least lipophilic (logP = 1.02) of the tested Cu(I) compounds, was the most potent (IC50 ∼ 1–2 μM). Compound 4, albeit displaying a high logP value of 2.18, has phenyl groups that are involved in intramolecular π-stacking interactions.41 The variation in the dark cytotoxicity of compounds 1–4 could not be solely correlated to lipophilicity since cellular localization and the mechanisms of action are expected to change with the substituents, as previously reported in Ru(II) complexes bearing the 4,7-diphenyl-1,10-phenanthroline ligand framework,39,42 and Ru(II) arene complexes.43
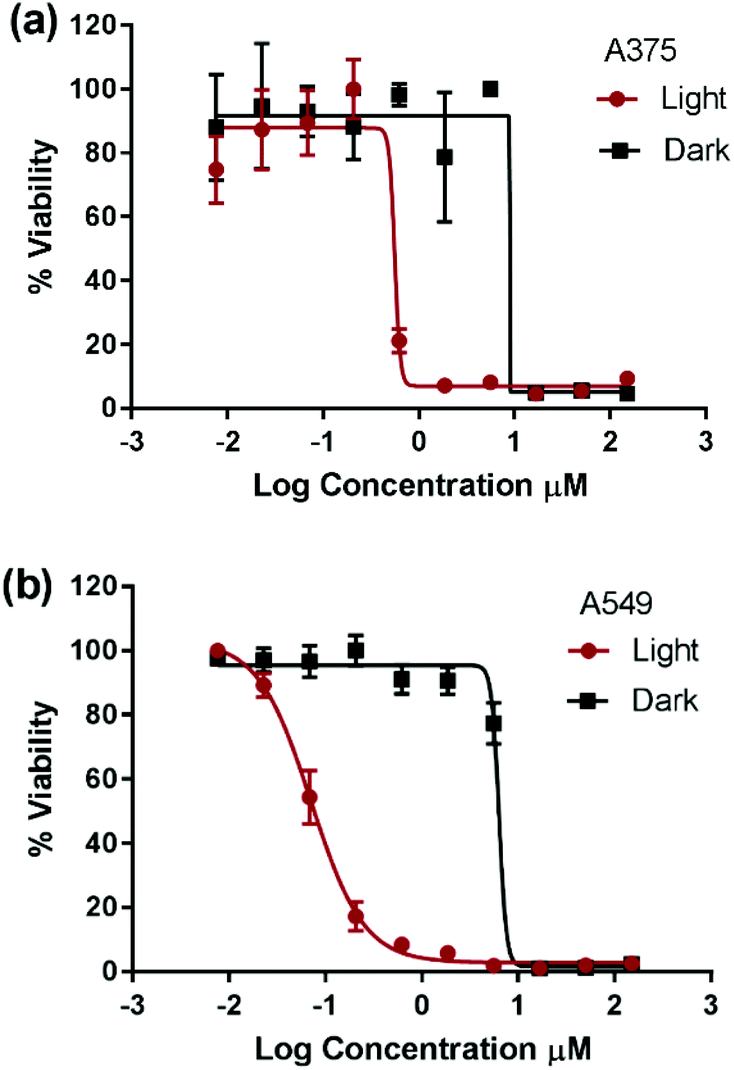 | ||
| Fig. 2 Representative cytotoxicity measurement of compound 3 on A375 (human malignant melanoma) (a) and A549 (human lung carcinoma) (b) cancer cell lines plotted as percent viability versus log of the compound concentration in μM in the dark (black squares) and upon light activation (red circles). The cells were incubated with the compound for 24 h prior to cytotoxicity assessment. Light activation occurred 6 h post incubation and lasted for 30 min using blue-LED. IC50 values were obtained using GraphPad Prism software by fitting the data into a sigmoidal dose response function. | ||
| Compounds in the dark | 1 | 2 | 3 | 4 | Cisplatin |
|---|---|---|---|---|---|
| a Value obtained from the literature.44 b Values for free ligands calculated from ChemDraw Professional (v15.0, CambridgeSoft). | |||||
| IC50 values on A375 (μM) | 1.8 ± 0.5 | 25.5 ± 0.2 | 7.0 ± 0.3 | 3.1 ± 0.1 | 7.7 ± 0.4 |
| IC50 values on A549 (μM) | 0.9 ± 0.1 | 21.8 ± 0.2 | 6.3 ± 0.1 | 1.8 ± 0.1 | 21.9 ± 0.4 |
| LogP of complexes |
1.02 ± 0.08 | 2.21 ± 0.19 | 2.64 ± 0.16 | 2.18 ± 0.15 | −2.21a |
| LogP of ligands |
3.61b | 6.96b | 8.16b | 6.40b | |
Compounds 1 and 4 have previously demonstrated different types of interactions with extracted DNA, and the former was found to associate at the minor groove of DNA and the latter to intercalate at the major groove.22,27,28 However, the detailed structure–activity relationship in the dark is beyond the scope of this work. The photocytotoxicity of compounds 1–4 was assessed on A375 and A549 cells. The cells were incubated with each compound for 6 h, photoactivated for 30 min under 460 nm blue LED illumination, and then incubated until cell viability was assessed. The total incubation time with each compound was 24 h allowing direct comparison between data. The IC50 value was measured and a phototoxicity index (PI = IC50 dark/IC50 light) was calculated for each complex, Fig. 2, Fig. S2† and Table 2.
| Compounds upon light activation | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
|
a Measured in CH3CN from ref. 29 and 33.
b In 1:1 CH3CN:H2O from ref. 34.
c In H2O for the sulfonated analogue from ref. 33.
|
||||
| IC50 values on A375 (μM) | 2.1 ± 1.0 | 38.9 ± 3.3 | 0.56 ± 0.01 | 2.63 ± 0.03 |
| PI values on A375 | 0.9 | 0.7 | 12.5 | 1.2 |
| IC50 values on A549 (μM) | 0.65 ± 0.01 | 21.3 ± 0.3 | 0.075 ± 0.003 | 1.33 ± 0.03 |
| PI on A549 | 1.3 | 1 | 82.9 | 1.3 |
| Excited state lifetime (ns) | <10a | <10a | 1200b | 70c |
Due to plate-to-plate variability, PI below 3 was assumed to indicate non-photoactivatable compounds. The cytotoxicity of complexes 1, 2, and 4 was not enhanced upon photoexcitation whereas complex 3 possessed significant phototoxicity indices (PI = IC50 dark/IC50 light) of 13 and 83 on A375 and A549 cells, respectively. To the best of our knowledge, complex 3 represents the first example of a cuprous phenanthroline complex that displays significant in vitro photocytotoxicity. The improved potency of complex 3 upon light activation was attributed to its extended excited state lifetime. Compounds 1–2 possessed an excited state lifetime of 100 ns in CH2Cl2 which was significantly reduced to few ns in weakly coordination solvents such as CH3CN.29,33 On the other hand, compound 4 displayed a lifetime of 310 ns in CH2Cl2 and its sulfonated analogue a lifetime of 70 ns in an aqueous medium at room temperature.29
Compound 3 exhibited a remarkable lifetime of 2.8 μs in CH2Cl2 and 1.2 μs in 1:1 CH3CN:H2O.34 The significant extension of lifetime in the weakly basic solvents of compound 3 relative to the others can be attributed to shielding the metal from the approach of coordinating solvents. This protection was enhanced moving from the methyl to phenyl to sec-butyl substituents at the 2,9-positions of the Cu(I) complex.33,45 Consequently, it can be inferred that efficient bimolecular energy/electron transfer,31,46,47 a prerequisite of PDT, was successfully achieved by extending the triplet excited state lifetime of Cu(I) bis-phenanthroline complexes. Importantly, upon light activation, compound 3 displayed an IC50 value that is 1–2 orders of magnitude smaller than that of the prototypical cisplatin. Since only compound 3 displayed high PI, further experiments were performed to elucidate its mechanism of action.
The level of DNA damage was assessed by an in vitro comet assay. In these gel electrophoresis experiments, DNA damage is evaluated at the cellular level. Upon the application of an electric field, the fragmented DNA migrates outside the nucleoid. However, the non-denatured DNA stays within the nucleoid and subsequently migrates slower on the gel. The DNA tail shape, migration pattern, and % tail DNA content correlate to DNA damages in the cells.
The alkaline comet assay is sensitive, and is used to detect small amounts of damage including single and double-stranded breaks. Negative controls contained cells without drug whereas, positive controls contained KMnO4 which is a known oxidant that induces DNA damage at the cellular level.48 The comet assay data of the A375 and A549 cancer cells incubated with compound 3 in the dark (Fig. 3) was suggestive that the latter operated, at least partly, through the induction of DNA damage. Importantly, the level of DNA single and/or double-stranded breaks increased in both the A375 and A549 cancer cells incubated with compound 3 upon light exposure, Fig. 3. The positive and negative controls showed identical results in the absence or presence of light irradiation confirming that blue light by itself did not change the level of DNA damage. These results are in line with the measured ability of cuprous phenanthroline complexes to interact with DNA.22,27,28 To corroborate the results from the comet assay on compound 3, a spectrophotometric titration experiment in 50% DMSO in Tris-HCl buffer at pH = 7.2 was done with calf-thymus DNA as a titrant. It was found that incrementing amounts of DNA induced a bathochromic shift in the absorbance of compound 3 which is typically observed upon binding to DNA base pairs,49,50Fig. 4. Using eqn (1),50 the intrinsic binding constant of compound 3 to DNA base pairs was found to be Kb = 3.3 ± 2.5 × 104 M−1 which is comparable to the values obtained for transition metal complexes known to bind to DNA.49,50
| [DNA]/(εA − εF) = [DNA]/(εB − εF) + 1/Kb(εB − εF) | (1) |
 | ||
| Fig. 3 Bar graph depicting comet assay results expressed in terms of tail DNA content (%) of A375 (red) and A549 (blue) cancer cells under the following conditions: negative control (cells alone), positive control (cells incubated with KMnO4), cells incubated with compound 3 in the dark and upon exposure to light. | ||
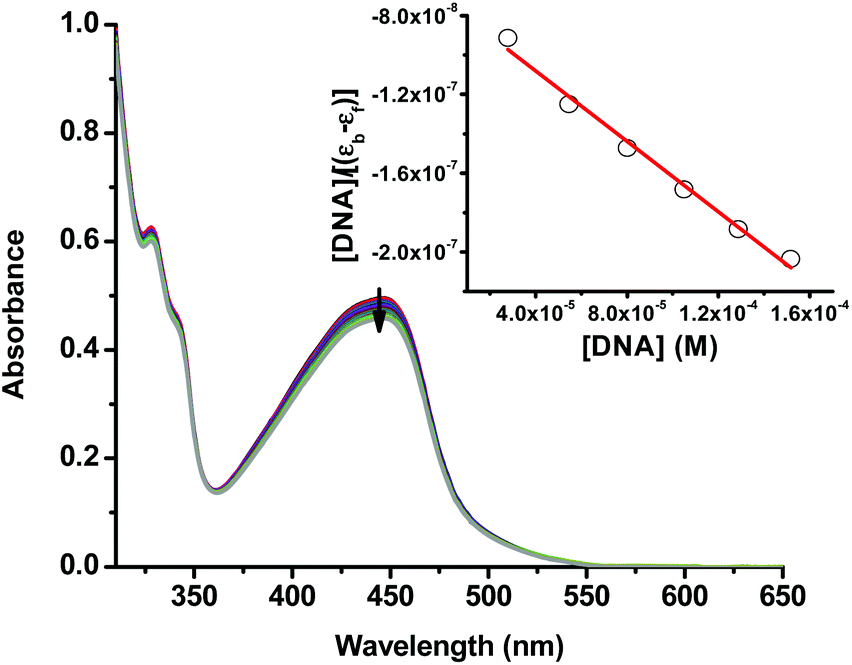 | ||
| Fig. 4 UV-vis spectra of compound 3 (50% DMSO in Tris-HCl buffer at pH = 7.2) with an incrementing concentration of calf thymus DNA base pairs. The inset shows a linear fit of the first 6 data points into eqn (1) to extract the intrinsic binding constant (Kb) of compound 3 to DNA base pairs. | ||
The qualitative uptake and localization of compound 3 was performed using fluorescence microscopy, Fig. 5.39,42 Since this complex is emissive at room temperature even in the presence of coordinative solvents,34 this experiment yielded important information on the uptake and cellular localization of compound 3. The compound (12 μM) was incubated with A375 and A549 cells for 2, 6 and 12 h and then visualized using Rhodamine filter on a fluorescence microscope. The cells were also stained with DAPI (4′,6-diamidine-2′-phenylindole dihydrochloride) which is a known fluorescence marker for DNA. After 2 h incubation (Fig. 5a and b on A375 and A549, respectively) compound 3 showed detectable uptake into cells and diffuse cytosolic localization. At that time the cells were still relatively healthy and the compound was not concentrated in the nucleus. At 6 h of incubation, compound 3 (Fig. 5c and d on A375 and A549, respectively) induced the loss of the membrane integrity and was partly found in the nucleus and predominantly concentrated in the cytosol in a perinuclear fashion. After 12 h, the cells completely detached from the microscope slides indicating complete cell death at the concentration used.
 | ||
| Fig. 5 Fluorescence imaging following the treatment of A375 and A549 cells for 2 h (a and b, respectively) and 6 h (c and d, respectively) with 12 μM of compound 3. The orange signal emanated from the intrinsic emission of compound 3 (Rhodamine filter/RH) and the blue emission is from DAPI which stains the nucleus. The cells were fixed with 4% paraformaldehyde for 10 min, and blocked with 1% BSA in PBS for 1 h. The samples were stained with DAPI for 10 min before taking fluorescence images using a 60× objective on an Axio observer Z1 fluorescence microscope (from Zeiss). | ||
Collectively the comet assay, spectrophotometric titration with DNA and fluorescence microscopy experiments indicate that the main target of compound 3 is the DNA of cells. This was not surprising given that metal complexes bearing phenanthroline based ligand(s) have demonstrated high affinity to DNA.39,50
Based on the cytotoxicity results in this work (Tables 1 and 2), the mechanism leading to the photoactivation of compound 3 was postulated to be through quenching of the long-lived triplet state to produce reactive oxygen species (ROS) which induce oxidative stress in cells and ultimately cell death, Fig. 6.51
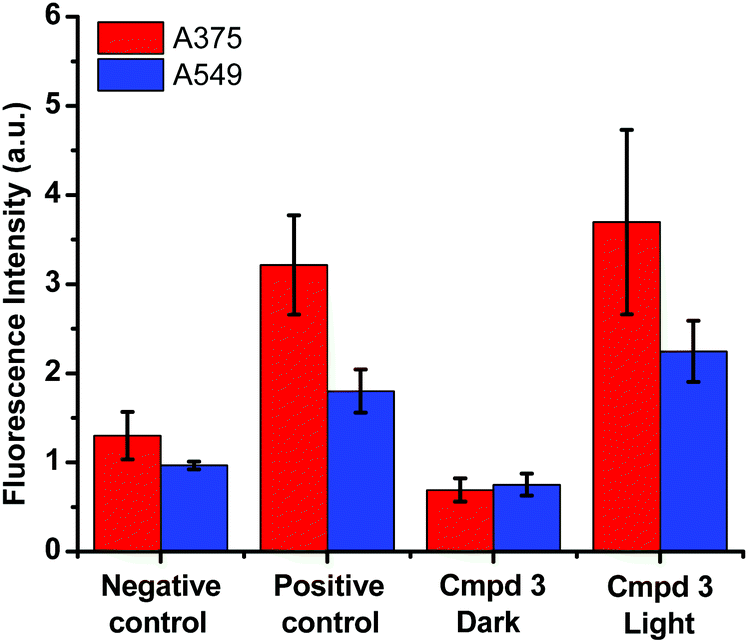 | ||
| Fig. 6 DCFDA (2′,7′-dichlorofluorescin diacetate) fluorescence assay for the detection of ROS in A375 (red) and A549 (blue) cancer cells performed on buffer (negative control), tert-butyl hydroperoxide (positive control) and compound 3 in the dark and upon photoactivation. | ||
The production of ROS was assessed using the 2′,7′-dichlorofluorescin diacetate (DCFDA) fluorescence assay which allows the measurement of hydroxyl, peroxyl and other ROS activities within the cell. Briefly, the cells were incubated with the non-fluorescent DCFDA. After diffusion into the cell, DCFDA is deacetylated by cellular esterases to a non-fluorescent compound. If ROS are formed, the latter gets oxidized into 2′,7′-dichlorofluorescein (DCF). DCF is a highly fluorogenic dye which can be detected by fluorescence spectroscopy with excitation and emission signals at 495 nm and 529 nm, respectively. Compound 3 was added at 3 μM concentration and tested in the dark and upon light activation following 4 h incubation. Control experiments were performed in the absence of drug (negative control) or upon incubation with TBHP (tert-butyl hydroperoxide), a known ROS forming agent.52 When compared to the controls for relative fluorescence intensity, a turn-on fluorescence was displayed in cells incubated with the photoactivated compound 3, Fig. 6. The blue light did not cause any increase of ROS level. Thus, it can be inferred that compound 3 induced the formation of ROS only upon light activation in both A375 and A549 cells. These results correlate well with the high phototoxicity indices of compound 3 on these cancer cell lines, Fig. 2 and Table 2. Importantly, the induction of ROS formation upon light activation in the targeted tumours offers an advantage over conventional chemotherapy.
Conclusions
The following sterically congested Cu(I) complexes were synthesized and characterized: [Cu(dmphen)2]+ (1), [Cu(BC)2]+ (2), [Cu(dsbtmp)2]+ (3), and [Cu(dpphen)2]+ (4). The cytotoxicity of these homoleptic Cu(I) bis-phenanthroline complexes was screened on A375 and A549 cancer cells. IC50 values were obtained in the dark and upon blue light MLCT excitation. Compounds 1–4 were potent in the dark with IC50 ranging from 2 to 25 μM. However, only compound 3, which exhibited the longest excited state lifetime, displayed enhanced potency upon photoactivation. In the latter case, high phototoxicity indices of ∼13 on A375 cells and ∼83 on A549 cancer cells were obtained which are comparable to ruthenium polypyridine congeners. This provides a proof-of-principle that cuprous phenanthroline photosensitizers can display in vitro photocytotoxicity and may provide a viable alternative to noble metal complexes in photobiology. The comet assay performed on compound 3 was strongly suggestive of DNA being a possible biological target. DNA damages were detected upon the incubation of the cells with compound 3 in the dark and were further elevated upon visible light excitation. Furthermore, the UV-vis titration of compound 3 with calf-thymus DNA revealed a hypochromic shift which is suggestive of DNA binding. An intrinsic binding constant Kb between complex 3 and DNA base pairs was found to be Kb = 3.3 ± 2.5 × 104 M−1 that is on the same order of magnitude to known DNA binding complexes.49,50 Owing to the photoluminescence properties of complex 3, fluorescence imaging experiments successfully indicated an intracellular uptake and perinuclear localization in A375 and A549 cells. A loss of membrane integrity was observed 6 h post-incubation and it proceeded to cell death. Finally, a DCFDA fluorescence assay revealed that the photocytoxicity of complex 3 was likely due to the formation of ROS following light activation. These results highlight the advantageous utilization of sterically encumbered Cu(I) bis-phenanthroline complexes for the PDT of cancer and their potential use for cancer imaging.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Felix N. Castellano and Dr Catherine E. McCusker for useful discussions and help with the initial synthesis of Cu(I) complexes. We would like to acknowledge the School Research and Development Council at the Lebanese American University for funding this work.Notes and references
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, CA-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- A. S. Abu-Surrah and M. Kettunen, Curr. Med. Chem., 2006, 13, 1337–1357 CrossRef CAS PubMed.
- M. A. Fuertes, C. Alonso and J. M. Perez, Chem. Rev., 2003, 103, 645–662 CrossRef CAS PubMed.
- V. Cepeda, M. A. Fuertes, J. Castilla, C. Alonso, C. Quevedo and J. M. Perez, Anti-Cancer Agents Med. Chem., 2007, 3–18 CrossRef CAS.
- T. N. Singh and C. Turro, Inorg. Chem., 2004, 43, 7260–7262 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- N. A. Smith and P. J. Sadler, Philos. Trans. R. Soc., A, 2013, 371, 20120519 CrossRef PubMed.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690–10701 RSC.
- J. K. White, R. H. Schmehl and C. Turro, Inorg. Chim. Acta, 2017, 454, 7–20 CrossRef CAS PubMed.
- M. C. Linder, Biochemistry of Copper, Springer US, Plenum Press, New York, 1991 Search PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- S. Puig, J. Lee, M. Lau and D. J. Thiele, J. Biol. Chem., 2002, 277, 26021–26030 CrossRef CAS PubMed.
- A. M. Thomas, A. D. Naik, M. Nethaji and A. R. Chakravarty, Inorg. Chim. Acta, 2004, 357, 2315–2323 CrossRef CAS.
- S. Roy, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Polyhedron, 2010, 29, 3251–3256 CrossRef CAS.
- G.-J. Chen, X. Qiao, P.-Q. Qiao, G.-J. Xu, J.-Y. Xu, J.-L. Tian, W. Gu, X. Liu and S.-P. Yan, J. Inorg. Biochem., 2011, 105, 119–126 CrossRef CAS PubMed.
- B. Maity, S. Gadadhar, T. K. Goswami, A. A. Karande and A. R. Chakravarty, Dalton Trans., 2011, 40, 11904–11913 RSC.
- D. Lahiri, R. Majumdar, D. Mallick, T. K. Goswami, R. R. Dighe and A. R. Chakravarty, J. Inorg. Biochem., 2011, 105, 1086–1094 CrossRef CAS PubMed.
- D. Galaris and A. Evangelou, Crit. Rev. Oncol. Hematol., 2002, 42, 93–103 CrossRef PubMed.
- M. Valko, H. Morris and M. T. D. Cronin, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS PubMed.
- V. Gandin, M. Pellei, F. Tisato, M. Porchia, C. Santini and C. Marzano, J. Cell. Mol. Med., 2012, 16, 142–151 CrossRef CAS PubMed.
- D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201–1220 CrossRef PubMed.
- D. S. Sigman, Acc. Chem. Res., 1986, 19, 180–186 CrossRef CAS.
- L. E. Pope and D. S. Sigman, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 3–7 CrossRef CAS.
- D. S. Sigman, A. Mazumder and D. M. Perrin, Chem. Rev., 1993, 93, 2295–2316 CrossRef CAS.
- T. B. Thederahn, M. D. Kuwabara, T. A. Larsen and D. S. Sigman, J. Am. Chem. Soc., 1989, 111, 4941–4946 CrossRef CAS.
- F. Liu, K. A. Meadows and D. R. McMillin, J. Am. Chem. Soc., 1993, 115, 6699–6704 CrossRef CAS.
- R. Tamilarasan, D. R. McMillin and F. Liu, in Metal-DNA Chemistry, American Chemical Society, 1989, vol. 402, ch. 3, pp. 48–58 Search PubMed.
- C. O. Dietrich-Buchecker, P. A. Marnot, J.-P. Sauvage, J. R. Kirchhoff and D. R. McMillin, J. Chem. Soc., Chem. Commun., 1983, 513–515 RSC.
- R. Tamilarasan and D. R. McMillin, Inorg. Chem., 1990, 29, 2798–2802 CrossRef CAS.
- M. S. Lazorski and F. N. Castellano, Polyhedron, 2014, 82, 57–70 CrossRef CAS.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 CrossRef CAS.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef CAS.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2013, 52, 8114–8120 CrossRef CAS PubMed.
- C. O. Dietrick-Buchecker, P. A. Marnot and J. P. Sauvage, Tetrahedron Lett., 1982, 23, 5291–5294 CrossRef.
- M. Ruthkosky, F. N. Castellano and G. J. Meyer, Inorg. Chem., 1996, 35, 6406–6412 CrossRef CAS PubMed.
- J.-A. Cuello-Garibo, M. S. Meijer and S. Bonnet, Chem. Commun., 2017, 53, 6768–6771 RSC.
- C. Khalil and W. Shebaby, Toxicol. Rep., 2017, 4, 441–449 CrossRef CAS PubMed.
- H. Audi, D. F. Azar, F. Mahjoub, S. Farhat, Z. El Masri, M. El-Sibai, R. J. Abi-Habib and R. S. Khnayzer, J. Photochem. Photobiol., A, 2018, 351, 59–68 CrossRef CAS.
- D. F. Azar, H. Audi, S. Farhat, M. El-Sibai, R. J. Abi-Habib and R. S. Khnayzer, Dalton Trans., 2017, 46, 11529–11532 RSC.
- M. T. Miller, P. K. Gantzel and T. B. Karpishin, Inorg. Chem., 1998, 37, 2285–2290 CrossRef CAS PubMed.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed.
- A. Pastuszko, K. Majchrzak, M. Czyz, B. Kupcewicz and E. Budzisz, J. Inorg. Biochem., 2016, 159, 133–141 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, J. Med. Chem., 2012, 55, 5326–5336 CrossRef CAS PubMed.
- M. K. Eggleston, D. R. McMillin, K. S. Koenig and A. J. Pallenberg, Inorg. Chem., 1997, 36, 172–176 CrossRef CAS.
- R. S. Khnayzer, C. E. McCusker, B. S. Olaiya and F. N. Castellano, J. Am. Chem. Soc., 2013, 135, 14068–14070 CrossRef CAS PubMed.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2015, 54, 6035–6042 CrossRef CAS PubMed.
- C. T. Bui, K. Rees and R. G. Cotton, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 1835–1855 CAS.
- C. N. Sudhamani, H. S. Bhojya Naik, K. R. S. Gowda, M. Giridhar, D. Girija and P. N. P. Kumar, Med. Chem. Res., 2017, 26, 1160–1169 CrossRef CAS.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS PubMed.
- O. Kučera, R. Endlicher, T. Roušar, H. Lotková, T. Garnol, Z. Drahota and Z. Červinková, Oxid. Med. Cell. Longevity, 2014, 2014, 752506 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt00140e |
| This journal is © The Royal Society of Chemistry 2018 |